Introduction
Lung cancer is one of the most severe forms of
cancer, with a 5-year survival rate of only 15% among patients with
all stages of the disease (1).
Certain subtypes of lung adenocarcinomas are treatable with
effective molecular targeted drugs. For example, tyrosine kinase
inhibitors against epidermal growth factor receptor (EGFR) and
anaplastic lymphoma kinase (ALK) are highly effective against
EGFR-mutant and ALK fusion-positive lung
adenocarcinomas, respectively (2).
Effective therapies for rare lung adenocarcinomas have also been
developed. For example, crizotinib for ROS proto-oncogene 1,
receptor tyrosine kinase (ROS-1) fusion-positive tumors, and
vandetanib for RET fusion-positive tumors (3,4).
However, there are still no effective treatment strategies for
KRAS-mutant lung adenocarcinomas, which account for ~8.5%
and 20–25% of lung adenocarcinoma cases in the Japanese and
Caucasian (European and North American) populations, respectively
(5–7). As a result, systemic chemotherapy is
still preferentially used to treat the majority of patients with
KRAS-mutant lung adenocarcinomas. Constitutively activated
mutant KRAS protein has been proven to be notoriously difficult to
target with small molecule inhibitors, and this has led to the use
of trametinib and selumetinib, two MEK (MAPK/ERK kinase)
inhibitors, as promising candidates in the treatment of
KRAS-mutant cancer (5,6).
Clinical trials have been conducted for KRAS-mutant lung
cancer using second line treatment with trametinib alone, or
combination therapy with selumetinib and docetaxel; however, these
treatments have failed to significantly improve progression-free
survival (8,9). Other effective therapeutic options
are currently available, including treatment with anti-vascular
endothelial growth factor (VEGF) antibody (bevacizumab), anti-VEGFR
antibody (ramucirumab) and anti-PD1 antibodies (nivolumab and
pembrolizumab) (10–13); however, each of these drugs has
shown only limited efficacy against KRAS-mutant lung
adenocarcinomas.
Survivin, which is encoded by the baculoviral IAP
repeat containing 5 (BIRC5) gene, is not expressed in normal
adult tissues, apart from the thymus, placenta, CD34-positive
hematopoietic stem cells and the colorectal mucosa. However, the
protein is clearly expressed in the majority of malignant tumors,
and is involved in carcinogenesis and therapeutic resistance
(14,15). Survivin is a member of the
inhibitor of apoptosis family of proteins, and it functions to
suppress the activity of caspase 3/7 (14). Moreover, it plays an important role
in chromosomal segregation and cytokinesis, and its expression
level increases mostly at the G2/M phase of the cell cycle
(15). The overexpression of
survivin has been reported to be associated with a poor outcome in
brain tumors, colorectal cancers, breast cancers and non-small-cell
lung cancers (16–19). Furthermore, its expression levels
are particularly high in tumors in which carcinoma cells are known
to evade apoptosis, despite treatment with radiation or cytotoxic
agents, including taxane and platinum (20–22).
Indeed, the inhibition of survivin by treating cells with
small-interfering RNAs (siRNAs) or ribozymes has been shown to
enhance the sensitivity of cancer cells to these treatments
(23). Thus, it has been proposed
that inhibiting the expression or function of survivin may be a
promising approach for overcoming resistance to therapy. However,
to date, to the best of our knowledge, studies on the role(s) of
survivin in KRAS-mutant lung adenocarcinomas are limited
(24). In this study, through
clinicopathological and molecular pathological analyses, we
demonstrate that survivin may be a potential therapeutic target in
KRAS-mutant lung adenocarcinomas.
The majority of invasive mucinous adenocarcinomas of
the lung bear KRAS mutations and are negative for thyroid
transcription factor 1 (TTF-1), an essential transcription factor
for lung development encoded by the NK2 homeobox 1 (NKX2-1)
gene. TTF-1 is expressed and is oncogenic in virtually all
EGFR-mutant lung adenocarcinomas (25); however, the expression rate and
role(s) of TTF-1 in KRAS-mutant lung adenocarcinomas remain
to be elucidated. In the present study, we aimed to determine the
role of survivin in KRAS-mutant lung adenocarcinomas, as
well as the role of TTF-1. Our data indicate that the majority of
KRAS-mutant lung adenocarcinomas analyzed were positive for
TTF-1; thus, TTF-1 may play a role in the proliferation of
KRAS-mutant cancer cells.
Materials and methods
Primary KRAS-mutant lung adenocarcinoma
tissues and immunohistochemistry
All the experimental procedures involving human
samples were approved by the Institutional Review Board at Sapporo
Medical University. The samples used in this study were obtained
from patients (n=208) who were operated on for lung adenocarcinoma
at the Sapporo Medical University Hospital, Sapporo, Japan between
January, 2005 and December, 2009. From these patients, we selected
28 patients (13%) whose tumors had a KRAS mutation (at codon
12 or 13) detected by the loop-hybrid mobility shift assay
(26). These patients included 20
males and 8 females, with a median age of 63 years (range, 31–85
years; Table I). We did not obtain
written informed consent from the patients for conducting this
retrospective study. Instead, the patients were informed of the
outline of this study through the website of Sapporo Medical
University so that they could 'opt out' from the study if they
wished. All pathological slides were reviewed and evaluated by two
of the authors (T.S. and Y.S.). Hematoxylin and eosin (H&E)
staining was performed using Tissue-Tek Hematoxylyn 3G (8657;
Sakura Finetek Japan, Tokyo, Japan) and Tissue-Tek Eosin (8660;
Sakuma FineTek Japan) according to the manufacturer's instructions.
Immunohistochemical analysis for the expression of survivin, TTF-1
and E-cadherin was also carried out on formalin-fixed,
paraffin-embedded tissue sections of the cancer specimens.
Whole-tissue sections were retrieved using Novocastra Epitope
Retrieval Solution 1 (pH 6.0) for E-cadherin expression or Solution
2 (pH 9.0) (both from Leica Biosystems, Nussloch, Germany) for the
other antigens at 100°C for 20 min. The primary antibodies used
were anti-survivin (sc-17779; D-8; 1:200; Santa Cruz Biotechnology,
Dallas, TX, USA), anti-TTF-1 (N1635; 8G7G3/1; pre-diluted; Dako
Japan, Tokyo, Japan) and anti-E-cadherin (#14472; 4A2; 1:100; Cell
Signaling Technology Japan, Tokyo, Japan). Immunohistochemical
staining was conducted using the Leica BOND-MAX (Leica Biosystems).
In this study, the cancer tissues were judged as positive for
survivin and TTF-1 expression when ≥10% and ≥50% of the cancer
cells exhibited positive nuclear staining, respectively. As for
E-cadherin, the tissues were judged as positive when membranous
staining was observed in ≥80% of the cancer cells. In addition, all
the cancer tissues were classified as terminal respiratory unit
(TRU) type or non-TRU type based on the definition in the
literature (27).
 | Table IClinicopathological findings of the
28 patients with KRAS-mutant lung adenocarcinoma. |
Table I
Clinicopathological findings of the
28 patients with KRAS-mutant lung adenocarcinoma.
| Pt | Age/sex | Smoking history
(pack year) | Dominant
history | TRU or non-TRU
type | Staging | Survivin | TTF-1 | E-cadherin | OS (months) | Deceased or
alive | DFS (months) | Recurrence |
|---|
| 1 | 74/M | 37.5 | Pap | TRU | 1B | + | + | + | 117 | Alive | 117 | − |
| 2 | 68/M | 34.5 | MIA | TRU | 1A | + | + | + | 55 | Alive | 55 | − |
| 3 | 62/M | 82 | MIA | TRU | 1A | − | + | + | 127 | Alive | 127 | − |
| 4 | 81/F | 50 | Pap | TRU | 2A | + | + | + | 25 | Deceased | 25 | − |
| 5 | 60/M | 80 | Pap | TRU | 2A | + | + | + | 120 | Alive | 120 | − |
| 6 | 58/M | 40 | Pap | TRU | 1B | + | − | + | 45 | Deceased | 28 | + |
| 7 | 64/M | 0 | MIA | TRU | 1A | − | + | + | 115 | Alive | 115 | − |
| 8 | 60/M | 69 | AIS | TRU | 1A | − | + | + | 51 | Alive | 51 | − |
| 9 | 49/F | 14 | Pap | Non−TRU | 2A | − | − | + | 97 | Alive | 97 | − |
| 10 | 65/M | 44 | AIS | TRU | 1A | − | + | + | 106 | Alive | 106 | − |
| 11 | 73/F | 50 | Pap | Non−TRU | 2A | + | − | + | 76 | Alive | 76 | − |
| 12 | 31/F | 0 | IMA | Non−TRU | 4 | + | − | + | 7 | Deceased | 1 | + |
| 13 | 73/M | 53 | Pap | Non−TRU | 3A | + | − | + | 11 | Deceased | 11 | + |
| 14 | 33/F | 0 | IMA | Non−TRU | 2B | − | − | + | 71 | Alive | 71 | − |
| 15 | 68/M | 48 | Pap | Non−TRU | 1B | + | − | + | 86 | Alive | 86 | − |
| 16 | 67/M | 0 | Acinar | TRU | 2A | + | + | + | 10 | Deceased | 9 | + |
| 17 | 59/M | 39 | Pap | TRU | 1A | + | + | + | 104 | Alive | 104 | − |
| 18 | 50/M | 40 | IMA | Non−TRU | 2B | + | + | + | 25 | Alive | 25 | − |
| 19 | 61/F | 0 | IMA | Non−TRU | 1A | + | − | + | 82 | Alive | 82 | − |
| 20 | 40/M | 13.5 | AIS | TRU | 1A | − | + | + | 99 | Alive | 99 | − |
| 21 | 62/M | 30 | Pap | TRU | 1A | + | + | + | 96 | Alive | 96 | − |
| 22 | 79/M | 0 | Pap | TRU | 1A | + | + | + | 53 | Deceased | 9 | + |
| 23 | 55/M | 37 | Pap | TRU | 1A | − | + | + | 91 | Alive | 91 | − |
| 24 | 71/F | 0 | Pap | TRU | 1A | − | + | + | 120 | Alive | 120 | − |
| 25 | 59/M | 38 | Solid | Non−TRU | 1A | + | + | + | 42 | Deceased | 43 | − |
| 26 | 85/F | 0 | IMA | Non−TRU | 2B | + | − | + | 26 | Deceased | 14 | + |
| 27 | 73/M | 40 | MIA | TRU | 1A | + | + | + | 57 | Deceased | 57 | − |
| 28 | 78/M | 0 | Pap | TRU | 3A | − | + | + | 64 | Deceased | 64 | − |
Kaplan-Meier Plotter database
We used the Kaplan-Meier Plotter online service
(http://kmplot.com/analysis/index.php?p=background) to
clarify whether a high BIRC5 mRNA expression was associated
with an unfavorable outcome in patients with lung adenocarcinomas
(28).
Cell culture and drugs used
Two KRAS-mutant lung adenocarcinoma cell
lines, NCI-H358 (G12C) and NCI-H441 (G12V), were obtained from the
American Type Culture Collection (ATCC, Manassas, VA, USA) and
maintained at 37°C in a humidified incubator with 5%
CO2. The cells were cultured in RPMI-1640 (Nacalai
Tesque, Kyoto, Japan) with 10% fetal bovine serum and antibiotics.
The MEK inhibitor, trametinib (AdooQ BioScience, Irvine, CA, USA)
and the Bcl-2 inhibitor (also known as a BH3 mimetic drug), ABT-263
(AdooQ BioScience), were also used in this study.
Immunofluorescence staining of the
cells
Immunofluorescence staining was conducted as
previously described for the expression of survivin (sc-17779; D-8;
1:200; Santa Cruz Biotechnology) and cytokeratin 7 (#4465; D1E4;
1:400; Cell Signaling Technology) in the H441 cells (29,30).
Briefly, cells grown on 35-mm glass bottom, collagen-coated dishes
(D11134H; Matsunami Glass, Osaka, Japan) were fixed with 4%
paraformaldehyde at room temperature for 15 min followed by
permeabilization with 0.5% Triton X-100 for 2 min. After the cells
were rinsed with tris-buffered saline (TBS), they were blocked
using 3% BSA/TBS for 60 min at room temperature. The samples were
then incubated overnight at 4°C with the two primary antibodies
mentioned above. The samples were subsequently incubated with two
secondary antibodies, Alexia Fluor 488-conjugated donkey anti-mouse
IgG (A-21202; 1:400; Thermo Fisher Scientific Japan, Yokohama,
Japan) and Alexa Fluor 594-conjugated goat anti-rabbit IgG
(A-11012; 1:400; Thermo Fisher Scientific). The cells were finally
observed under an inverted microscope (IX-71; Olympus, Tokyo,
Japan) and photographed (DP80; Olympus).
RNA interference assay
The cells (3×106) were plated in 94-mm
culture dishes and transfected with negative control (NC) siRNA
duplexes (1027281; Qiagen, Valencia, CA, USA) or siRNA duplexes
targeting BIRC5 and NKX2-1 using Lipofectamine
RNAiMAX reagent and OPTI-MEM I (Thermo Fisher Scientific), as
previously described (28-30). Two types of siRNA duplexes were
used for transient survivin (encoded by the BIRC5 gene) or
TTF-1 (encoded by the NKX2-1 gene) knockdown: Silencer
Select Validated siRNA (Ambion #s1457 and #s1458, termed
BIRC5 siRNA #1 and #2, respectively, in this study; Thermo
Fisher Scientific) and Silencer Select Pre-designed siRNA (Ambion
#s14152 and #s14153, termed NKX2-1 siRNA #1 and #2, respectively;
Thermo Fisher Scientific). The final concentration of the siRNA
used in each in vitro experiment was 10 nM. The
downregulation of the expression of the targeted genes was verified
by western blot analysis.
Assessment of cell viability and
apoptosis
The number of viable cells was estimated using a
CellTiter Glo 3D Cell Viability assay (Promega, Madison, WI, USA)
according to the manufacturer's instructions. Apoptosis was
assessed by western blot analysis of cleaved poly(ADP-ribose)
polymerase 1 (PARP-1) or Caspase-Glo 3/7 assay (Promega), as
previously described (30,31). The luminescence of viable or
apoptotic cells was measured with the Infinite 200 microplate
reader (Tecan Japan, Kawasaki, Japan). All results are presented as
the means ± SD.
Cell cycle analysis by flow
cytometry
The cells were reverse transfected with NC siRNA or
BIRC5 siRNA, and then grown for 48 h. The cells were then
harvested and fixed in 75% methanol at 4°C overnight. The cells
were washed twice with cold phosphate-buffered saline (PBS) and
stained with propidium iodide (PI) (0.5 mg/ml RNase, and 0.1 mg/ml
PI in PBS) for 30 min at room temperature. The stained cells were
characterized using a flow cytometer (Beckman Coulter Corp., Tokyo,
Japan), and the DNA content was analyzed using FlowJo software.
Senescence associated β-galactosidase
staining and the counting of multinuclear cells
The cells (5×105 cells) were plated in
60-mm culture dishes before staining. Cellular senescence was
detected using the Cellular Senescence kit (OZ Biosciences, San
Diego, CA, USA), according to the manufacturer's instructions.
Briefly, the cells were washed twice with PBS and then fixed in the
fixation buffer provided in the kit at room temperature for 15 min.
The cells were then washed twice with PBS, and stained using the
β-galactosidase staining solution provided with the kit for 18 h at
37°C in a humidified incubator. The cells were visualized under a
light microscope (IX-71; Olympus) and photographed (DP80; Olympus).
Granules stained blue within the cytoplasm were considered positive
for β-galactosidase staining. We also assessed the proportion of
multinuclear cells, which are cells with two or more nuclei in the
cytoplasm, by counting the number of multinuclear cells in at least
3 high-power fields under the same microscope mentioned above.
Western blot analysis
Western blot analysis for the expression of
E-cadherin, vimentin, TTF-1, PARP-1, γH2AX, Bim, total AKT,
phospho-AKT, total ERK1/2, phospho-ERK1/2 and β-actin was performed
as previously described (29–31).
Additional primary antibodies used in the present study were
anti-survivin (#2808; 71G4B7; 1:1,000 dilution; Cell Signaling
Technology) and anti-p21 (#2947; 12D1; 1:1,000 dilution; Cell
Signaling Technology). The cells were lysed in NuPAGE LDS Sample
Buffer (Thermo Fisher Scientific). The cell lysates (15 μg
of total protein in each well) were separated by SDS-PAGE (SuperSep
Ace, 5–20%, 13 wells; Wako Pure Chemicals, Osaka, Japan) and
transferred onto polyvinylidene difluoride (PVDF) membranes. The
membranes were blocked by incubation with 3% non-fat dry milk in
TBS for 1 h at room temperature and incubated overnight at 4°C with
the primary antibodies mentioned above. The membranes were then
washed 3 times and then incubated for 1 h at room temperature with
species-specific horseradish peroxidase-conjugated secondary
antibodies (NA931 or NA934; GE Healthcare, Buckinghamshire, UK).
Blots were visualized using Supersignal West Pico Chemiluminescent
Substrate (Thermo Fisher Scientific). Band intensity levels on
X-ray films were normalized to β-actin using ImageJ (National
Institutes of Health, Bethesda, MD, USA).
Crystal violet staining
The NC siRNA- or BIRC5 siRNA-transfected H358
cells and H441 cells were cultured for 48 h. The cells were then
plated at a density of 5×105 cells into the wells of a
6-well plate and treated with ABT-263 (1 μM) alone,
trametinib (25 nM) alone, both, or neither for 72 h. The plate was
placed on ice, and the cells were washed twice with cold PBS, and
then were fixed in ice-cold 100% methanol for 10 min. Following
fixation, the cells were moved to room temperature and stained with
0.5% crystal violet solution (Tokyo Chemical Industry, Tokyo,
Japan) diluted in 25% methanol for 10 min. Plates containing
stained cells were then photographed using a digital camera (D610;
Nikon, Tokyo, Japan).
Three-dimensional (3D) 'on-top'
culture
The cells were cultured above a thin layer of 100%
Matrigel (Corning, Corning, NY, USA) in RPMI-1640 medium, termed
'3D on-top culture', as previously described, with minor
modifications (32,33). Briefly, the Corning 96-well Flat
Clear White Polystyrene TC-Treated Microplates were coated with 30
μl/well of Matrigel and incubated at 37°C for 30 min to
allow the Matrigel to solidify. Subsequently, each microspheroid
[consisting of 50 cells in suspension for 24 h using PrimeSurface
96U plate (Sumitomo Bakelite, Tokyo, Japan)] was placed on the top
of the Matrigel in each well, and grown for 72 h in 100 μl
medium.
Mouse tumor xenograft model
All animal experimentation was conducted in
accordance with the protocol approved by the Animal Committee at
Sapporo Medical University. The experiments were performed using
6-8-week-old female mice (n=15) (KSN/Slc nude mice; Hokudo,
Sapporo, Japan). This study used a minimum of 5 mice per group. The
mice were kept throughout the experiment in pleasant conditions
(temperature, 20-26 °C; humidity, 40–60%) and were able to freely
access food and water. The weights of the mice upon purchase and
upon sacrifice were 23 g and 20 g on average, respectively. At 6 h
following NC siRNA or BIRC5 siRNA #1 transfection,
5×106 H358 cells were collected in 50 μl of
RPMI-1640 and then mixed with 50 μl of Matrigel. The
cell/Matrigel mixture was subcutaneously injected into the right
flanks of mice anesthetized by inhalation of isoflurane (3–5%;
Pfizer, New York, NY, USA). Each mouse received a single injection
of H358 cells, and thus developed a single tumor nodule later. At
10 days after the injection, we injected AteloGene (Koken, Tokyo,
Japan) containing NC siRNA (n=5) or BIRC5 siRNA (n=10) near
the tumor nodules to administer the siRNA continuously. Mice that
were injected with BIRC5 siRNA-transfected cells (n=10) were
administered the vehicle (n=5) or a combination of ATB-263 (50
mg/kg) and trametinib (0.6 mg/kg) (n=5) orally once daily for 22
days (days 10–32 post-injection of the cells) (34–36).
ABT-263 was dissolved in 10% ethanol, 30% polyethylene glycol 400,
and 60% Phosal 50 PG; trametinib was dissolved in 10% Cremophor EL,
10% PEG400, and 80% dH2O (Nacalai Tesque). The mice were
monitored daily for body weight and general condition. Following
any sign of deterioration in any mouse during the ABT-263 and
trametinib therapy, the administration to the mouse was terminated
immediately. Tumors were measured twice weekly using a caliper, and
the volume was calculated using the following formula: 0.5 ×
(width)2 × length. After the observation, mice were
anesthetized by inhalation of isoflurane and then sacrificed by
cervical dislocation. The tumor nodules were subsequently removed
for weight and histology.
Statistical analysis
The Kaplan-Meier method was used to estimate overall
survival and disease-free survival of the 28 patients with
KRAS-mutant lung adenocarcinoma. Differences in the survival curves
between patients with survivin-positive and -negative tumors were
compared using the log-rank test. Differences in caspase activity
or cell viability between the untreated and treated cells were
evaluated by Dunnett's test, a multiple comparison test as were
differences in the number of multinuclear cells or
senescence-associated β-galactosidase-positive cells between
untreated and treated cells. One-way analysis of variance (ANOVA)
followed by the Tukey-Kramer multiple comparison test was performed
to evaluate the effects of survivin knockdown, and survivin
depletion plus combined therapy with ATB-263 and trametinib, on the
cells or tumor xenografts. A P-value <0.05 were considered
significant. All statistical calculations were performed using JMP
software (JMP for Windows version 7, SAS Institute Japan, Tokyo,
Japan).
Results
Patients with survivin-positive tumors
have poorer outcomes
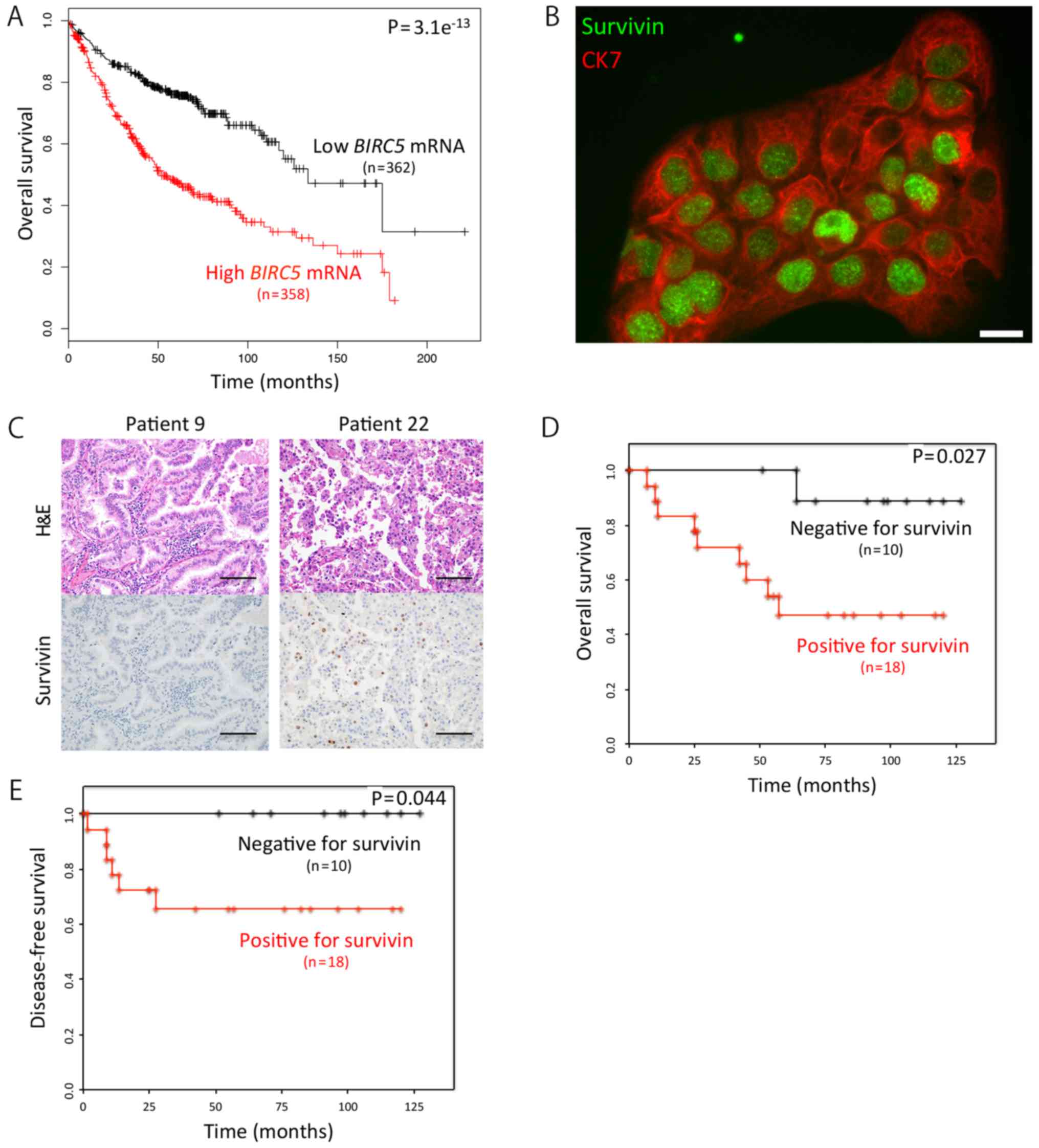
First, we searched the Kaplan-Meier Plotter for an
association between the mRNA expression levels of the BIRC5
and the outcome of patients with lung adenocarcinoma. We found that
the overall survival (OS) of the patients with a high BIRC5
expression (n=358) was markedly shorter than that of patients with
a low BIRC5 expression (n=362) (hazard ratio, 2.41;
P=3.1e−13) (Fig. 1A) by
utilizing the Kaplan-Meier Plotter (28). We then focused on survivin (BIRC5)
protein, not its mRNA, using immunocytochemistry, and found that it
was mainly localized to the nuclei of the cells (Fig. 1B). Subsequently, we examined 28
KRAS-mutant lung adenocarcinoma samples for survivin
expression using immunohistochemistry (Fig. 1C), and found that the OS rates of
the patients with survivin-positive adenocarcinoma (n=18) was
significantly shorter than that of patients with survivin-negative
tumors (n=10) (P=0.027) (Fig. 1D).
As expected, we obtained a similar result in the disease-free
survival data analyzed (Fig.
1E).
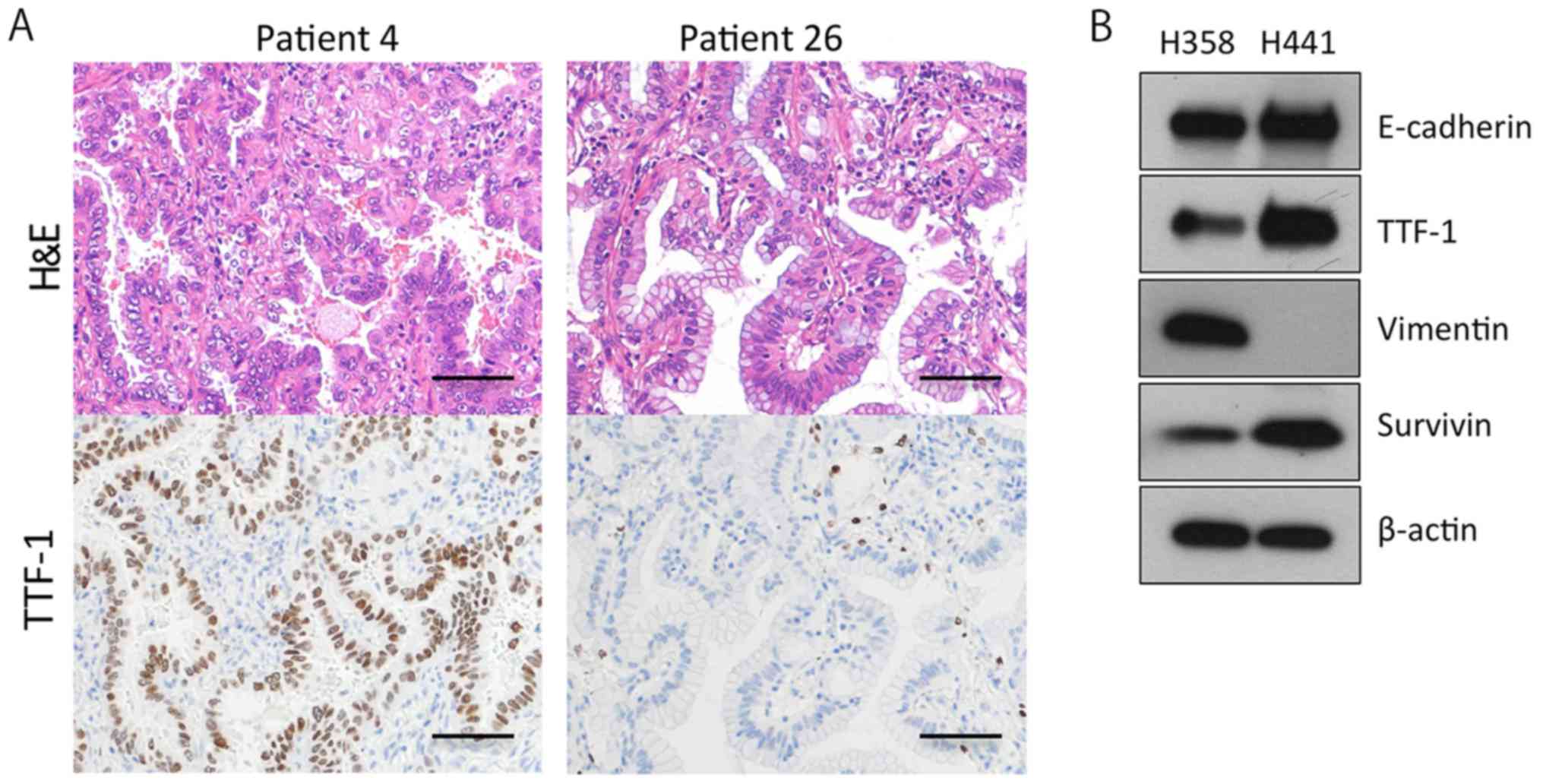
KRAS-mutant lung adenocarcinomas are
differentiated and positive for TTF-1 and E-cadherin
The majority of invasive mucinous adenocarcinomas of
the lung bear the KRAS mutation, and do not express TTF-1
(25). Furthermore, more than half
of KRAS-mutant lung adenocarcinoma cell lines (41/75, 55%)
are negative for E-cadherin and positive for vimentin, indicating
that the cell lines are more mesenchymal (36). In this study, however, the
KRAS-mutant lung adenocarcinoma tissues were mostly positive
for TTF-1 (19/28, 68%) and E-cadherin (28/28, 100%) (Table I). In addition, more than half of
the tumors (18/28, 64%) were classified as TRU-type, and 7 out of
the 28 (25%) tumors were diagnosed as adenocarcinoma in situ
(AIS) or minimally invasive adenocarcinoma (MIA) (Table I; Fig.
2A). These findings suggested that the majority of the operable
KRAS-mutant lung adenocarcinomas in Japan are of a
relatively well-differentiated type. We further confirmed that 4
out of the 5 invasive mucinous adenocarcinomas stained negative for
TTF-1 (Table I; Fig. 2A). Based on these findings, we then
sought to analyze two KRAS-mutant lung adenocarcinoma cell
lines, H358 cells and H441 cells, expressing TTF-1, E-cadherin and
survivin (Fig. 2B).
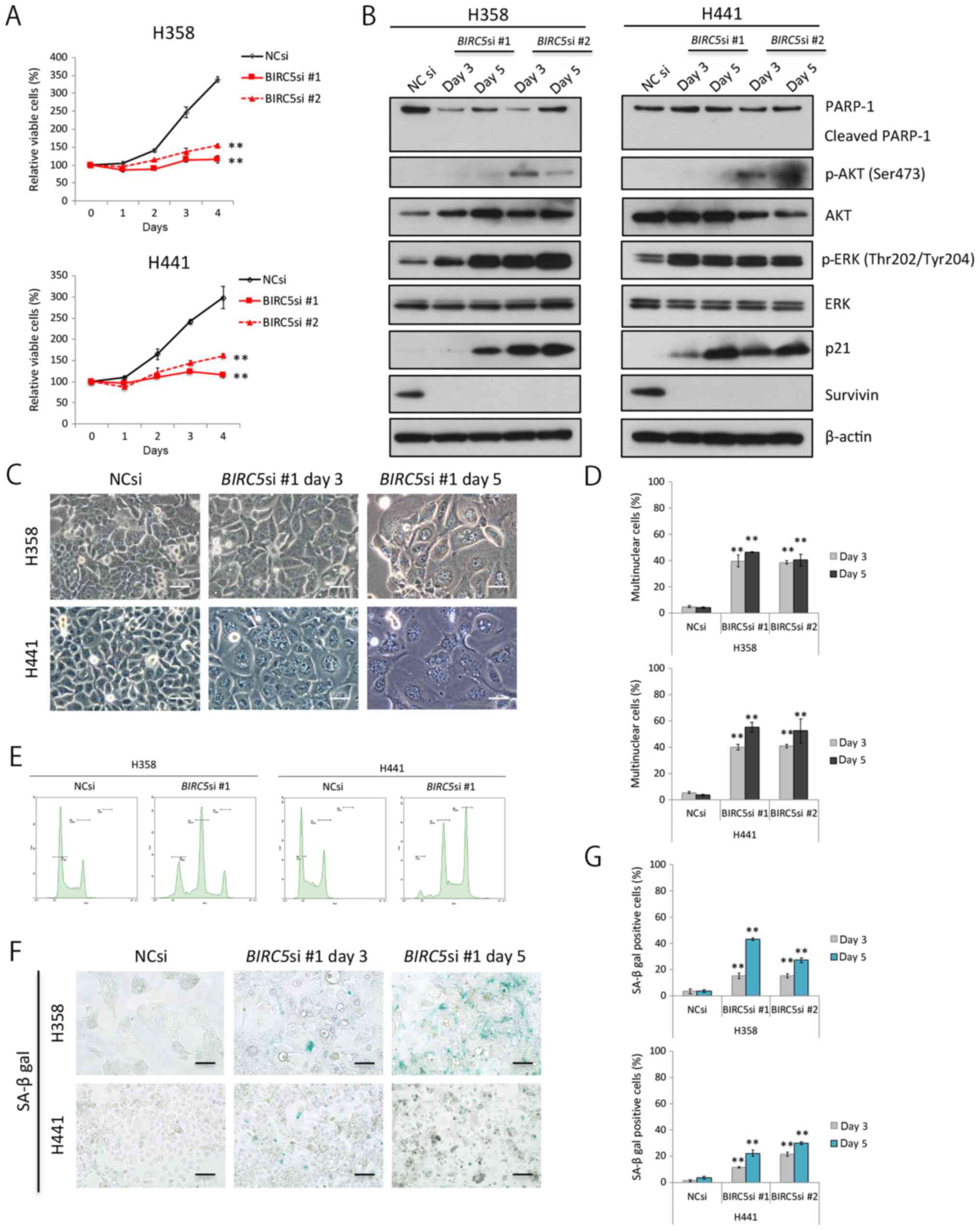
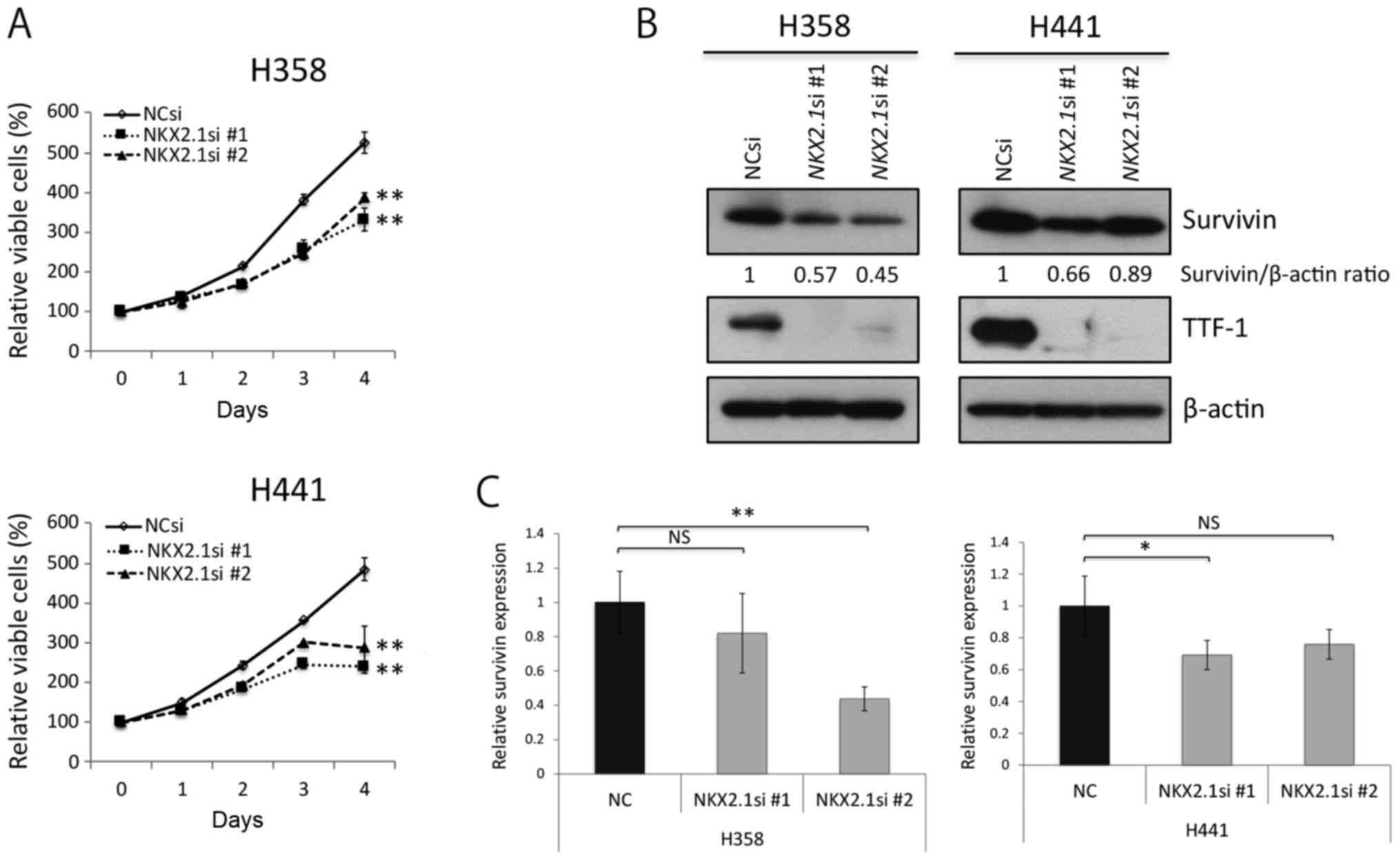
Survivin knockdown impairs cell division,
and induces senescence in KRAS-mutant lung adenocarcinoma
cells
We knocked down survivin expression through RNA
interference in the H358 cells and H441 cells in order to elucidate
whether and to what degree survivin affects the survival and/or
proliferation of these cells. Survivin knockdown clearly decreased
the proliferation of the two cell lines examined (Fig. 3A); however, it is unlikely that
this was a result of apoptosis in terms of PARP-1 cleavage analyses
(Fig. 3B). In addition, survivin
knockdown considerably upregulated the phosphorylation of ERK1/2
(the main proliferative signal downstream of KRAS); however, the
knockdown of survivin had a lesser effect on the PI3K-AKT pathway
(Fig. 3B).
Survivin knockdown significantly increased the ratio
of swelled to flattened cells, some of which had several nuclei
(Fig. 3C and D). Cell cycle
analyses using flow cytometry revealed an increase in the peak of
tetraploid cells, reflecting cell cycle arrest and an increase in
the number of binuclear cells. Furthermore, we observed a peak in
the number of octoploid cells, suggesting that the binuclear cells
were unable to divide under these conditions (Fig. 3E). The expression of p21, a
senescence-associated protein, increased gradually with time in the
cells in which survivin was knocked down (Fig. 3B), and senescence-associated
β-galactosidase activity was also elevated (Fig. 3F and G). These findings
collectively suggest that survivin knockdown inhibits cell
division, and induces senescence in KRAS-mutant lung
adenocarcinoma cells.
Combination therapy with survivin
knockdown, ABT-263 and trametinib, induces massive apoptosis of
KRAS-mutant lung adenocarcinoma cells
Given that survivin knockdown induced senescence,
not apoptosis of KRAS-mutant lung adenocarcinoma cells, we
then wished to determine whether ABT-263, a Bcl-2 inhibitor, and
trametinib, a MEK inhibitor, induced apoptosis of
KRAS-mutant lung cancer cells, as others have demonstrated
that ABT-263 preferentially induces apoptosis of senescent cells
(37), and MEK inhibitors are
particularly effective at inducing apoptosis of KRAS-mutant
cells when combined with ABT-263 (38). Thus, in this study, we treated the
H358 and H441 cells in which survivin was knocked down with ABT-263
alone, trametinib alone, or a combination of ABT-263 and trametinib
(hereafter referred to as AT therapy). As expected, AT therapy
clearly decreased cell viability (Fig.
4A and B), markedly elevated caspase 3/7 activation (Fig. 4C), and considerably increased the
expression levels of γH2AX and cleaved PARP-1 (Fig. 4D). Of note, selumetinib, another
MEK inhibitor, replaced trametinib and had similar results (data
not shown). Taken together, these findings suggest that the
combination treatment of survivin knockdown, ABT-263 and
trametinib, is an efficacious treatment for KRAS-mutant lung
adenocarcinomas.
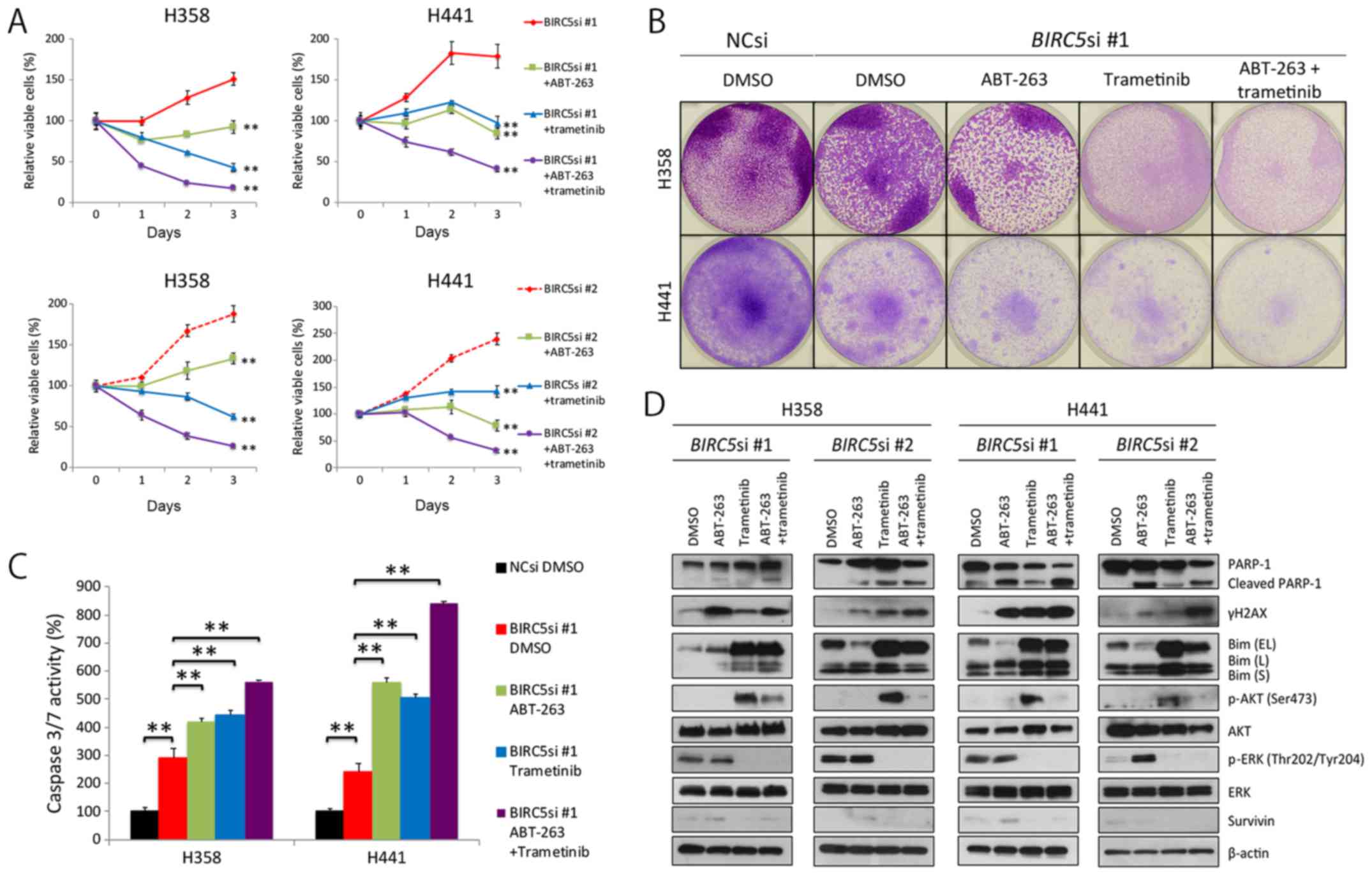 | Figure 4Combination of survivin knockdown
with ABT-263 and trametinib effectively induces apoptosis of
KRAS-mutant lung adenocarcinoma cells. (A) Effects of triple
combination therapy of survivin knockdown, ABT-263 and trametinib
on the viability of H358 and H441 cells. Cells were transfected
with BIRC5 siRNA #1 or #2 (10 nM each) and cultured for 48
h. Cells were then treated with the ABT-263 (1 μM) alone,
trametinib (25 nM) alone, both, or neither for a further 72 h.
Results are shown as the means ± SD; **P<0.01. (B)
Crystal violet staining of viable cells. NC siRNA or BIRC5
siRNA #1 (10 nM each)-transfected cells were grown for 48 h. A
total of 5×105 cells were then seeded in 6-well plates
and treated with ABT-263 (1 μM) alone, trametinib (25 nM)
alone, both, or neither for a further 72 h. Cells were then fixed
and stained with crystal violet. (C) The effects of triple
combination therapy of survivin knockdown, ABT-263 and trametinib
on caspase 3/7 activity in H358 and H441 cells. The NC siRNA- or
BIRC5 siRNA #1 (10 nM each)-transfected cells were cultured
for 48 h, and the cells were then treated with ABT-263 (1
μM) alone, trametinib (25 nM) alone, both, or neither for a
further 24 h before evaluating caspase activity. Caspase 3/7
activity was normalized to 100 for the mean of three control (NCsi)
dishes. Columns, mean (n=3); bars, SD; **P<0.01. (D)
Western blot analysis of the effects of the triple combination
therapy on H358 and H441 cells. Cells were treated in the same
manner as described in (A) or (B). Of note, Bim (EL) was
dephosphorylated by trametinib treatment, and then accumulated in
the cell. EL, extra-long; L, long; S, short. |
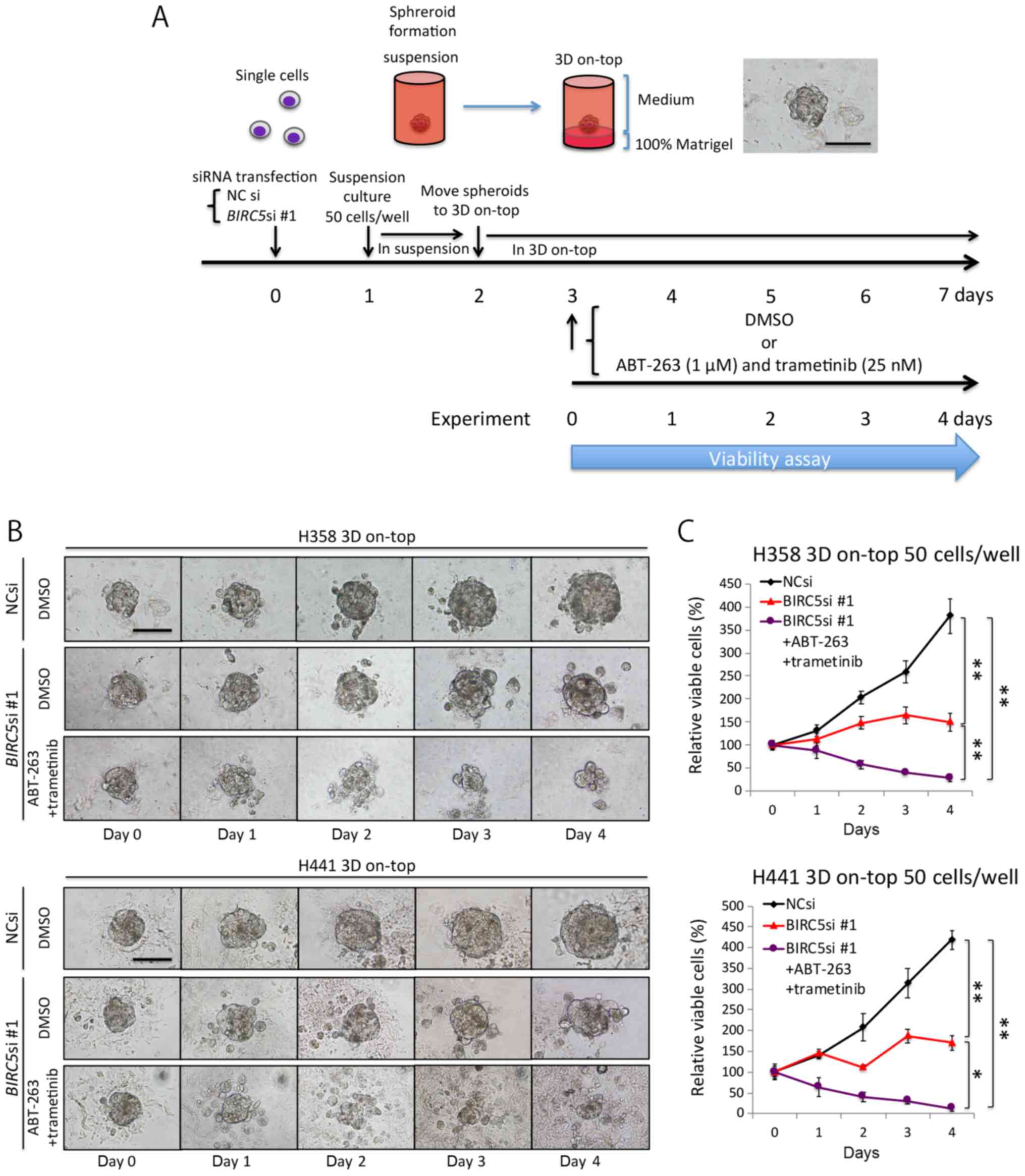
Triple combination therapy is also
effective against 3D-cultured KRAS-mutant lung adenocarcinomas
cells
Up to this point, our experiments were carried out
using conventional, monolayer culture conditions, which do not
truly reflect the in vivo environment (32,33).
We thus employed a 3D on-top culture condition to recapitulate
aspects of the in vivo environment (Fig. 5A), and then analyzed microspheroids
comprising ~50 cells (<100 μm in diameter), which we
believe mimic the micrometastases of lung adenocarcinoma. The NC
siRNA-transfected microspheroids steadily increased in size over
time, whereas the BIRC5 siRNA-transfected microspheroids
grew at a much slower rate (Fig.
5B). In addition, the microspheroids treated with the triple
combination of survivin knockdown and AT therapy were markedly
decreased in size (Fig. 5B), and
this was confirmed by a viability assay (Fig. 5C). These findings suggest that the
triple combination therapy could potentially eradicate the
micrometastases of KRAS-mutant lung adenocarcinoma cells
in vivo.
Triple combination therapy is also
effective for subcutaneous KRAS-mutant lung adenocarcinomas in
mice
To confirm the in vivo potency of the triple
combination therapy, we subcutaneously implanted NC siRNA- or
BIRC5 siRNA-transfected H358 cells into mice. Since the
efficacy of the pre-treated siRNAs was transient, we used the
AteloGene to administer siRNAs to subcutaneous tumors. The NC
siRNA-transfected cells grew steadily, whereas the BIRC5
siRNA-transfected cells exhibited minimal proliferation. Moreover,
AT therapy further reduced the size and weight of the tumor nodules
derived from BIRC5 siRNA-transfected cells, although the
effects of the therapy did not reach statistical significance (when
comparing the BIRC5 siRNA group to the triple combination
treatment group) (Fig. 6A–C).
Pathological analyses demonstrated that not only the NC
siRNA-transfected tumors, but also the BIRC5
siRNA-transfected nodules thrived in the absence of AT therapy,
with a positive survivin expression noted in the cancer cell nuclei
of both tumor types (Fig. 6D). We
surmised that the BIRC5 siRNA in the AteloGene had already
been consumed by day 11, as Fig.
6A indicates that the tumor nodules derived from the
BIRC5 siRNA-transfected cells slightly, but steadily
increased in size after day 11. However, Fig. 6D demonstrates that the triple
combination therapy did suppress the expression of survivin, and
induced apoptosis to a certain extent in the cells, indicating the
in vivo efficacy of the triple combination therapy.
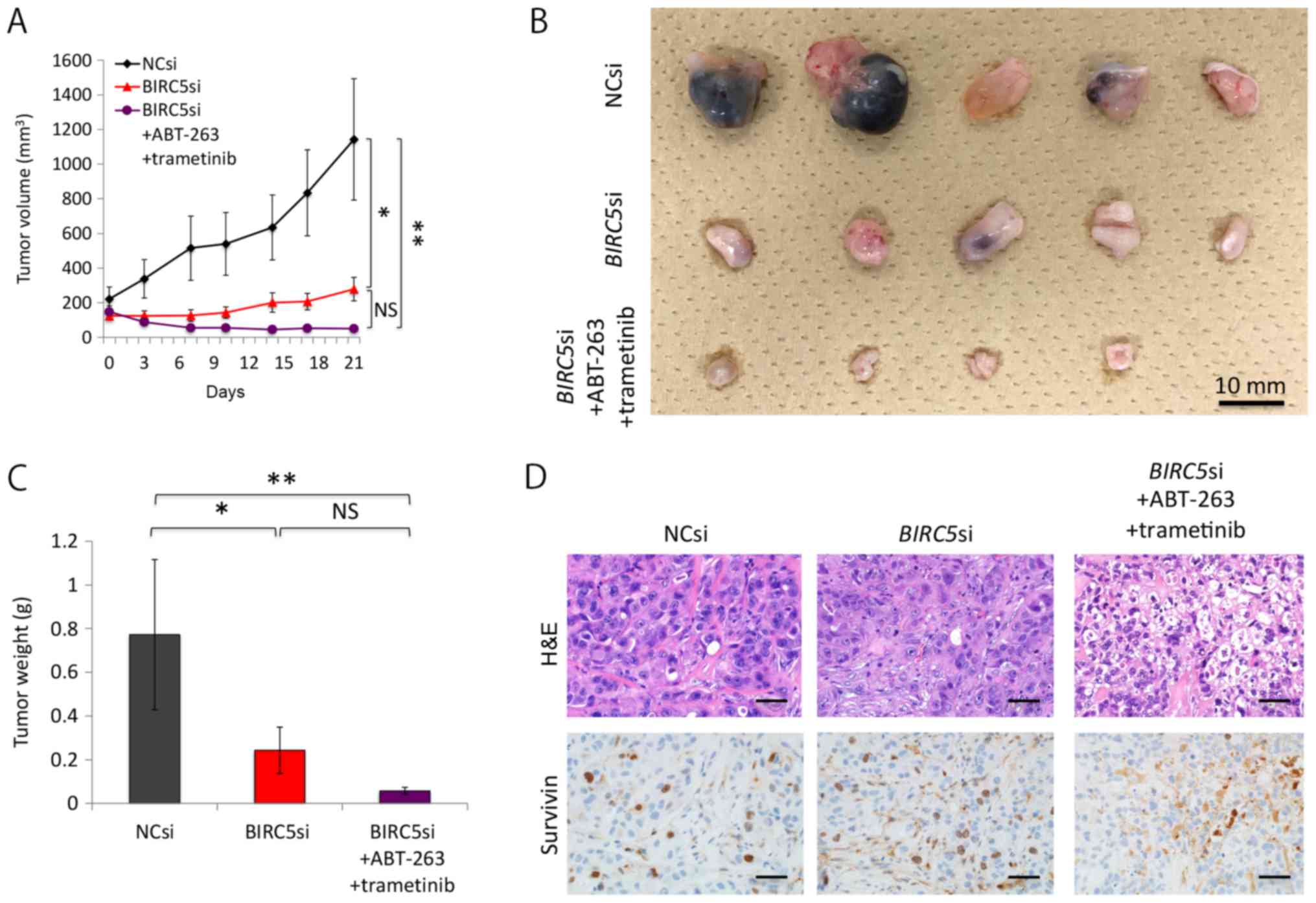 | Figure 6Triple combination of survivin
knockdown, ABT-263, and trametinib decreases the size of xenografts
of H358 cells. (A) Effects of the triple combination therapy on
xenografts of H358 cells. Mice with tumor xenografts derived from
BIRC5 siRNA-transfected cells were untreated (n=5) or
treated (n=5) with ABT-263 (50 mg/kg) and trametinib (0.6 mg/kg)
for 22 days. Mice with NC siRNA-transfected xenografts (n=5) were
treated with vehicle. Drugs were administered once daily by oral
gavage. Tumor volumes (means ± SD) were measured after the
initiation of the AT treatment (day 1). 'Day 1' in this figure
corresponds to the day 10 after implantation of the cells, when the
AteloGene was also administered. Of note, one of the five mice with
BIRC5 siRNA-transfected xenografts receiving AT treatment
died during the experiment. The death at day 13 was probably due to
the side-effects of trametinib used, as only this mouse developed a
severe skin rash from day 10, when the administration to the mouse
was terminated. As previously shown in clinical practice, the use
of trametinib can lead to the development of skin rash (55). *P<0.05,
**P<0.01. (B) Effects of the triple combination
therapy on xenografts derived from H358 cells. Macroscopic images
of the tumors resected from mice are represented. Fourteen tumor
nodules presented herein were derived from the 14 mice that
survived to the end of the therapy. (C) Effect of the triple
combination therapy on xenografts of H358 cells. The weight of each
tumor, shown in (B), was measured, and the results are presented as
the means ± SD. *P<0.05, **P<0.01. (D)
Pathological examination of xenografts. Hematoxylin and eosin
(H&E) staining and immunohistochemistry for survivin of
xenografts in each treatment condition. Of note, tumors derived
from both the NC si-transfected and BIRC5 si-transfected
cells without AT therapy expressed survivin in the nuclei, whereas
the staining observed in the tumors derived from BIRC5
si-transfected cells with AT therapy were likely to reflect
immunoglobulins derived from the mouse; i.e., non-specific
staining. |
TTF-1 knockdown suppresses the
proliferation of KRAS-mutant lung adenocarcinoma cells and induces
partial survivin knockdown
TTF-1 knockdown using NKX2-1 siRNA slightly,
but significantly suppressed the proliferation of the H358 cells
and H441 cells (Fig. 7A). Of note,
the expression levels of survivin appeared to decrease slightly
when TTF-1 was knocked down (Fig. 7B
and C). These observations suggest that TTF-1 probably enhances
the growth of TTF-1-positive, differentiated KRAS-mutant
lung adenocarcinoma cells, and that the expression levels of
survivin are possibly controlled, at least in part, by TTF-1.
Discussion
In this study, we demonstrated that in lung
adenocarcinoma patients, the OS of patients with tumors expressing
high mRNA levels of BIRC5 (survivin) is markedly shorter
than that of patients with low expression levels. Furthermore, the
OS of patients with KRAS-mutant lung adenocarcinomas
expressing survivin protein is also significantly shorter than that
of patients with survivin-negative tumors. Since the majority of
the KRAS-mutant lung adenocarcinoma tissues we analyzed were
well-differentiated and positive for TTF-1 and E-cadherin, we
selected and analyzed H358 and H441 cells, two TTF-1-positive,
differentiated KRAS-mutant lung adenocarcinoma cell lines,
to explore a treatment strategy that would be able to induce
extensive apoptosis of these cancer cells. We found that the
combination of survivin knockdown along with ABT-263 and trametinib
treatment induced massive apoptosis of these cancer cells.
Previous studies have indicated that
survivin-positive, small-sized lung adenocarcinomas and non-small
cell lung carcinomas are significantly associated with an
unfavorable outcome (39–42); however, little information is
available as to the roles of survivin in KRAS-mutant lung
adenocarcinoma. In this study, we demonstrated that
survivin-positive, KRAS-mutant lung adenocarcinomas are more
malignant than survivin-negative tumors in terms of survival data.
Although the knockdown of survivin in cells reportedly inhibits
cell division and causes multinucleation (43,44),
we confirmed that survivin knockdown renders the H358 and H441
cells multinucleated owing to a cytokinesis block. Furthermore, we
observed a gradual increase in p21 expression and an increase in
senescence-associated β-galactosidase activity in the
survivin-depleted cells. In other words, survivin knockdown alone
was not able to trigger apoptosis of the H358 and H441 cells, but
induced cellular senescence. However, the KRAS-mutant cells
in which survivin was knocked down in monolayer underwent massive
apoptosis following AT therapy, probably as trametinib
dephosphorylates Bim, a pro-apoptotic Bcl-2 family member, and
dephosphorylated Bim can avoid undergoing degradation through the
ubiquitin-proteasome system (37,38).
It has also been indicated that KRAS-mutant carcinomas,
particularly ones exhibiting an epithelial phenotype, usually
depend on Bcl-xL, an anti-apoptotic Bcl-2 family of proteins, for
survival (38). Although we
inhibited the anti-apoptotic function of Bcl-xL using ABT-263 in
this study, a recent study demonstrated that not only Bcl-xL, but
also Mcl-1, another anti-apoptotic Bcl-2 family member, plays a key
role in the survival of KRAS-mutated lung cancers (45). More importantly, the triple
combination of survivin knockdown, ABT-263 and trametinib,
substantially decreased the size of the microspheroids of
KRAS-mutant cancer cells in 3D on-top culture. Although the
3D 'on-top' culture model exploited in this study cannot fully
recapitulate all aspects of the in vivo microenvironment, it
does offer more benefits than a monolayer culture (32,33).
Distant metastases following surgery in clinical practice are
likely to result from the marked growth of pre-existing
micrometastatic foci, which cannot be detected using current
imaging diagnostics (46). Our
triple combination therapy clearly decreased the viability of the
cells in the 3D-cultured microspheroids, each of which consisted of
only 50 KRAS-mutant cancer cells as a model of
micrometastasis. This suggests that the triple combination could
eradicate micrometastatic foci, or at least prevent them from
growing. We further confirmed the effectiveness of the triple
combination therapy for H358 cells in vivo, and also found
that the treatment induced extensive apoptosis of the
EGFR-mutant H1975 cells in monolayer (data not shown),
suggesting that the triple combination could be applicable to lung
adenocarcinoma in general.
Almost all EGFR-mutant lung adenocarcinomas
express TTF-1, whereas invasive mucinous adenocarcinomas of the
lung, most of which harbor the KRAS mutation, do not express
TTF-1 (25,27,47).
Of note, the loss of TTF-1 expression in pulmonary
KRAS-mutated mucinous tumors is probably due to an
inactivating mutation and/or hypermethylation of the NKX2-1
gene (48,49). TTF-1 serves as a lineage-specific
oncogene in EGFR-mutant lung adenocarcinomas through the
induction of ROR1, a tyrosine kinase (50). By contrast, the TTF-1-negative,
KRAS-mutant lung adenocarcinoma cell line, A549 cells, are
sensitized to cisplatin by forced expression of the NKX2-1
gene, suggesting that KRAS mutation and TTF-1 expression
have negative effect(s) on each other (51). However, the majority of the primary
KRAS-mutant lung adenocarcinoma tissues analyzed in the
present study were relatively differentiated, TTF-1-positive
tumors. In addition, the H358 cells and H441 cells, exhibiting a
similar phenotype to that of the cancer tissues examined, are
partly dependent on TTF-1 for proliferation. This effect of TTF-1
on KRAS-mutant cancer cells may be regulated by, at least in
part, survivin. Collectively, these findings raise the possibility
that TTF-1 serves as an oncogene in KRAS-mutant,
well-differentiated lung adenocarcinomas through survivin induction
and other unknown mechanism(s).
More than half of the KRAS-mutated lung
adenocarcinomas analyzed in the present study were of the TRU-type,
and one fourth of them were classified as AIS or MIA. Although
these pathological findings may seem unexpected in that quite a few
KRAS-mutant lung adenocarcinomas are less aggressive than
might be expected, we would like to stress that the findings
mentioned above are consistent with those of previous reports
(27,52). This suggests that
KRAS-mutant lung adenocarcinomas, at least operable ones,
appear more differentiated and less aggressive than might be
thought.
This study has several limitations. First, although
the immunohistochemical expression of survivin in
KRAS-mutant lung adenocarcinomas was linked to an
unfavorable outcome, we analyzed the tissue samples from a limited
number of patients. We thus need to confirm our findings with a
larger selection of KRAS-mutant lung cancer tissue samples.
Second, although trametinib is available in clinical practice,
ABT-263 is currently undergoing clinical trials (https://clinicaltrials.gov/ct2/show/NCT02079740), and
has been reported to cause thrombocytopenia (53,54).
Finally, we limited our analysis of KRAS-mutant lung
adenocarcinoma cell lines to two types, and therefore we cannot
conclude that the majority of KRAS-mutant lung cancers will
be sensitive to the triple combination therapy.
In conclusion, KRAS-mutant lung
adenocarcinoma tissues examined in this study mostly express TTF-1,
and the cancer cells depend, at least in part, on TTF-1 for growth.
In addition, survivin-positive KRAS-mutant lung
adenocarcinomas are more malignant than survivin-negative
adenocarcinomas, and survivin knockdown induces senescence in
cancer cells. The triple combination therapy of survivin-depletion,
ABT-263, and trametinib can induce substantial apoptosis in
KRAS-mutant cancer cells both in vitro and in
vivo. It can thus be concluded that survivin is a promising
target for the treatment of KRAS-mutant lung
adenocarcinomas, with a triple combination therapy potentially a
valuable approach for treating such cancers.
Acknowledgments
The authors would like to thank Dr Rebecca Jackson,
from the Edanz Group (www.edanzediting.com/ac) for editing a draft of this
manuscript.
Funding
This study was supported in part by Grants-in-Aid
for Young Scientists from JSPS, Grant Number 16K19459 (to TS), and
funded by support from the Ono Cancer Research Fund (to YS).
Availability of data and materials
The analyzed datasets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
TS conducted most of the experiments in this study
with some assistance from YT and MT. SH, YM, and YS examined the
lung cancer tissues for KRAS mutation. MY carried out cell
cycle analyses. GY, TH, AW, and HT provided the tumor tissues and
analyzed the clinical data. TN, AW and HT provided helpful
discussions and critically reviewed the manuscript. YS designed and
conceived the study. TS and YS analyzed all the data, and wrote the
manuscript. All authors commented on and approved the
manuscript.
Ethics approval and consent to
participate
All the experimental procedures involving human
samples were approved by the Institutional Review Board at Sapporo
Medical University. Written informed consent was not obtained from
the patients for conducting this retrospective study. Instead, the
patients were informed of the outline of this study through the
website of Sapporo Medical University so that they could 'opt out'
from the study if they wished. All animal experimentation was
conducted in accordance with the protocol approved by the Animal
Committee at Sapporo Medical University.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chalela R, Curull V, Enríquez C, Pijuan L,
Bellosillo B and Gea J: Lung adenocarcinoma: From molecular basis
to genome-guided therapy and immunotherapy. J Thorac Dis.
9:2142–2158. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shaw AT, Ou SH, Bang YJ, Camidge DR,
Solomon BJ, Salgia R, Riely GJ, Varella-Garcia M, Shapiro GI, Costa
DB, et al: Crizotinib in ROS1-rearranged non-small-cell lung
cancer. N Engl J Med. 371:1963–1971. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yoh K, Seto T, Satouchi M, Nishio M,
Yamamoto N, Murakami H, Nogami N, Matsumoto S, Kohno T, Tsuta K, et
al: Vandetanib in patients with previously treated RET-rearranged
advanced non-small-cell lung cancer (LURET): An open-label,
multicentre phase 2 trial. Lancet Respir Med. 5:42–50. 2017.
View Article : Google Scholar
|
|
5
|
Serizawa M, Koh Y, Kenmotsu H, Isaka M,
Murakami H, Akamatsu H, Mori K, Abe M, Hayashi I, Taira T, et al:
Assessment of mutational profile of Japanese lung adenocarcinoma
patients by multitarget assays: A prospective, single-institute
study. Cancer. 120:1471–1481. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pylayeva-Gupta Y, Grabocka E and Bar-Sagi
D: RAS oncogenes: Weaving a tumorigenic web. Nat Rev Cancer.
11:761–774. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stephen AG, Esposito D, Bagni RK and
McCormick F: Dragging ras back in the ring. Cancer Cell.
25:272–281. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jänne PA, van den Heuvel MM, Barlesi F,
Cobo M, Mazieres J, Crinò L, Orlov S, Blackhall F, Wolf J, Garrido
P, et al: Selumetinib plus docetaxel compared with docetaxel alone
and progression-free survival in patients with KRAS-mutant advanced
non-small cell lung cancer: The SELECT-1 Randomized Clinical Trial.
JAMA. 317:1844–1853. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Blumenschein GR Jr, Smit EF, Planchard D,
Kim DW, Cadranel J, De Pas T, Dunphy F, Udud K, Ahn MJ, Hanna NH,
et al: A randomized phase II study of the MEK1/MEK2 inhibitor
trametinib (GSK1120212) compared with docetaxel in KRAS- mutant
advanced non-small-cell lung cancer (NSCLC). Ann Oncol. 26:894–901.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sandler A, Gray R, Perry MC, Brahmer J,
Schiller JH, Dowlati A, Lilenbaum R and Johnson DH:
Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell
lung cancer. N Engl J Med. 355:2542–2550. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Garon EB, Ciuleanu TE, Arrieta O, Prabhash
K, Syrigos KN, Goksel T, Park K, Gorbunova V, Kowalyszyn RD, Pikiel
J, et al: Ramucirumab plus docetaxel versus placebo plus docetaxel
for second-line treatment of stage IV non-small-cell lung cancer
after disease progression on platinum-based therapy (REVEL): A
multicentre, double-blind, randomised phase 3 trial. Lancet.
384:665–673. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Borghaei H, Paz-Ares L, Horn L, Spigel DR,
Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, et al:
Nivolumab versus docetaxel in advanced nonsquamous non-small-cell
lung cancer. N Engl J Med. 373:1627–1639. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Reck M, Rodríguez-Abreu D, Robinson AG,
Hui R, Csőszi T, Fülöp A, Gottfried M, Peled N, Tafreshi A, Cuffe
S, et al: KEYNOTE-024 Investigators: Pembrolizumab versus
chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl
J Med. 375:1823–1833. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Altieri DC: Survivin, cancer networks and
pathway-directed drug discovery. Nat Rev Cancer. 8:61–70. 2008.
View Article : Google Scholar
|
|
15
|
Mita AC, Mita MM, Nawrocki ST and Giles
FJ: Survivin: Key regulator of mitosis and apoptosis and novel
target for cancer therapeutics. Clin Cancer Res. 14:5000–5005.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Adida C, Berrebi D, Peuchmaur M,
Reyes-Mugica M and Altieri DC: Anti-apoptosis gene, survivin, and
prognosis of neuroblastoma. Lancet. 351:882–883. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kawasaki H, Altieri DC, Lu CD, Toyoda M,
Tenjo T and Tanigawa N: Inhibition of apoptosis by survivin
predicts shorter survival rates in colorectal cancer. Cancer Res.
58:5071–5074. 1998.PubMed/NCBI
|
|
18
|
Tanaka K, Iwamoto S, Gon G, Nohara T,
Iwamoto M and Tanigawa N: Expression of survivin and its
relationship to loss of apoptosis in breast carcinomas. Clin Cancer
Res. 6:127–134. 2000.PubMed/NCBI
|
|
19
|
Monzó M, Rosell R, Felip E, Astudillo J,
Sánchez JJ, Maestre J, Martín C, Font A, Barnadas A and Abad A: A
novel anti-apoptosis gene: Re-expression of survivin messenger RNA
as a prognosis marker in non-small-cell lung cancers. J Clin Oncol.
17:2100–2104. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chaopotong P, Kajita S, Hashimura M and
Saegusa M: Nuclear survivin is associated with cell proliferative
advantage in uterine cervical carcinomas during radiation therapy.
J Clin Pathol. 65:424–430. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Als AB, Dyrskjøt L, von der Maase H, Koed
K, Mansilla F, Toldbod HE, Jensen JL, Ulhøi BP, Sengeløv L, Jensen
KM, et al: Emmprin and survivin predict response and survival
following cisplatin-containing chemotherapy in patients with
advanced bladder cancer. Clin Cancer Res. 13:4407–4414. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang W, Mao Y, Zhan Y, Huang J, Wang X,
Luo P, Li LI, Mo D, Liu Q, Xu H, et al: Prognostic implications of
survivin and lung resistance protein in advanced non-small cell
lung cancer treated with platinum-based chemotherapy. Oncol Lett.
11:723–730. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zaffaroni N and Daidone MG: Survivin
expression and resistance to anticancer treatments: Perspectives
for new therapeutic interventions. Drug Resist Updat. 5:65–72.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yilmaz A, Mohamed N, Patterson KA, Tang Y,
Shilo K, Villalona-Calero MA, Davis ME, Zhou X, Frankel W, Otterson
GA, et al: Increased NQO1 but not c-MET and survivin expression in
non-small cell lung carcinoma with KRAS mutations. Int J Environ
Res Public Health. 11:9491–9502. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yamaguchi T, Hosono Y, Yanagisawa K and
Takahashi T: NKX2-1/TTF-1: An enigmatic oncogene that functions as
a double-edged sword for cancer cell survival and progression.
Cancer Cell. 23:718–723. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Matsukuma S, Yoshihara M, Suda T, Shiozawa
M, Akaike M, Ishikawa T, Koizume S, Sakuma Y and Miyagi Y:
Differential detection of KRAS mutations in codons 12 and 13 with a
modified loop-hybrid (LH) mobility shift assay using an insert-
type LH-generator. Clin Chim Acta. 412:1874–1878. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yatabe Y, Kosaka T, Takahashi T and
Mitsudomi T: EGFR mutation is specific for terminal respiratory
unit type adenocarcinoma. Am J Surg Pathol. 29:633–639. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Győrffy B, Surowiak P, Budczies J and
Lánczky A: Online survival analysis software to assess the
prognostic value of biomarkers using transcriptomic data in
non-small-cell lung cancer. PLoS One. 8:e822412013. View Article : Google Scholar
|
|
29
|
Yamaguchi M, Hirai S, Tanaka Y, Sumi T,
Miyajima M, Mishina T, Yamada G, Otsuka M, Hasegawa T, Kojima T, et
al: Fibroblastic foci, covered with alveolar epithelia exhibiting
epithelial-mesenchymal transition, destroy alveolar septa by
disrupting blood flow in idiopathic pulmonary fibrosis. Lab Invest.
97:232–242. 2017. View Article : Google Scholar
|
|
30
|
Sakuma Y, Nishikiori H, Hirai S, Yamaguchi
M, Yamada G, Watanabe A, Hasegawa T, Kojima T, Niki T and Takahashi
H: Prolyl isomerase Pin1 promotes survival in EGFR-mutant lung
adenocarcinoma cells with an epithelial-mesenchymal transition
phenotype. Lab Invest. 96:391–398. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sakuma Y, Matsukuma S, Nakamura Y,
Yoshihara M, Koizume S, Sekiguchi H, Saito H, Nakayama H, Kameda Y,
Yokose T, et al: Enhanced autophagy is required for survival in
EGFR-independent EGFR-mutant lung adenocarcinoma cells. Lab Invest.
93:1137–1146. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee GY, Kenny PA, Lee EH and Bissell MJ:
Three-dimensional culture models of normal and malignant breast
epithelial cells. Nat Methods. 4:359–365. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shibue T and Weinberg RA: Integrin
beta1-focal adhesion kinase signaling directs the proliferation of
metastatic cancer cells disseminated in the lungs. Proc Natl Acad
Sci USA. 106:10290–10295. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tse C, Shoemaker AR, Adickes J, Anderson
MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, et
al: ABT-263: A potent and orally bioavailable Bcl-2 family
inhibitor. Cancer Res. 68:3421–3428. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yamaguchi T, Kakefuda R, Tajima N, Sowa Y
and Sakai T: Antitumor activities of JTP-74057 (GSK1120212), a
novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro
and in vivo. Int J Oncol. 39:23–31. 2011.PubMed/NCBI
|
|
36
|
Kitai H, Ebi H, Tomida S, Floros KV,
Kotani H, Adachi Y, Oizumi S, Nishimura M, Faber AC and Yano S:
Epithelial-to-mesenchymal transition defines feedback activation of
receptor tyrosine kinase signaling induced by MEK inhibition in
KRAS-mutant lung cancer. Cancer Discov. 6:754–769. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chang J, Wang Y, Shao L, Laberge RM,
Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W,
et al: Clearance of senescent cells by ABT263 rejuvenates aged
hematopoietic stem cells in mice. Nat Med. 22:78–83. 2016.
View Article : Google Scholar :
|
|
38
|
Corcoran RB, Cheng KA, Hata AN, Faber AC,
Ebi H, Coffee EM, Greninger P, Brown RD, Godfrey JT, Cohoon TJ, et
al: Synthetic lethal interaction of combined BCL-XL and MEK
inhibition promotes tumor regressions in KRAS mutant cancer models.
Cancer Cell. 23:121–128. 2013. View Article : Google Scholar :
|
|
39
|
Ikehara M, Oshita F, Kameda Y, Ito H,
Ohgane N, Suzuki R, Saito H, Yamada K, Noda K and Mitsuda A:
Expression of survivin correlated with vessel invasion is a marker
of poor prognosis in small adenocarcinoma of the lung. Oncol Rep.
9:835–838. 2002.PubMed/NCBI
|
|
40
|
Wang M, Liu BG, Yang ZY, Hong X and Chen
GY: Significance of survivin expression: Prognostic value and
survival in stage III non-small cell lung cancer. Exp Ther Med.
3:983–988. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Oshita F, Ito H, Ikehara M, Ohgane N,
Hamanaka N, Nakayama H, Saito H, Yamada K, Noda K, Mitsuda A, et
al: Prognostic impact of survivin, cyclin D1, integrin beta1, and
VEGF in patients with small adenocarcinoma of stage I lung cancer.
Am J Clin Oncol. 27:425–428. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sun PL, Jin Y, Kim H, Seo AN, Jheon S, Lee
CT and Chung JH: Survivin expression is an independent poor
prognostic marker in lung adenocarcinoma but not in squamous cell
carcinoma. Virchows Arch. 463:427–436. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yang D, Welm A and Bishop JM: Cell
division and cell survival in the absence of survivin. Proc Natl
Acad Sci USA. 101:15100–15105. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dai D, Liang Y, Xie Z, Fu J, Zhang Y and
Zhang Z: Survivin deficiency induces apoptosis and cell cycle
arrest in HepG2 hepatocellular carcinoma cells. Oncol Rep.
27:621–627. 2012.
|
|
45
|
Yan X, Li P, Zhan Y, Qi M, Liu J, An Z,
Yang W, Xiao H, Wu H, Qi Y, et al: Dihydroartemisinin suppresses
STAT3 signaling and Mcl-1 and Survivin expression to potentiate
ABT-263-induced apoptosis in Non-small Cell Lung Cancer cells
harboring EGFR or RAS mutation. Biochem Pharmacol. 150:72–85. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Coello MC, Luketich JD, Litle VR and
Godfrey TE: Prognostic significance of micrometastasis in
non-small-cell lung cancer. Clin Lung Cancer. 5:214–225. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sakuma Y: Epithelial-to-mesenchymal
transition and its role in EGFR-mutant lung adenocarcinoma and
idiopathic pulmonary fibrosis. Pathol Int. 67:379–388. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hwang DH, Sholl LM, Rojas-Rudilla V, Hall
DL, Shivdasani P, Garcia EP, MacConaill LE, Vivero M, Hornick JL,
Kuo FC, et al: KRAS and NKX2-1 mutations in invasive mucinous
adenocarcinoma of the lung. J Thorac Oncol. 11:496–503. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Matsubara D, Soda M, Yoshimoto T, Amano Y,
Sakuma Y, Yamato A, Ueno T, Kojima S, Shibano T, Hosono Y, et al:
Inactivating mutations and hypermethylation of the NKX2-1/TTF-1
gene in non-terminal respiratory unit-type lung adenocarcinomas.
Cancer Sci. 108:1888–1896. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yamaguchi T, Yanagisawa K, Sugiyama R,
Hosono Y, Shimada Y, Arima C, Kato S, Tomida S, Suzuki M, Osada H,
et al: NKX2-1/TITF1/TTF-1-induced ROR1 is required to sustain EGFR
survival signaling in lung adenocarcinoma. Cancer Cell. 21:348–361.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Maeda Y, Tsuchiya T, Hao H, Tompkins DH,
Xu Y, Mucenski ML, Du L, Keiser AR, Fukazawa T, Naomoto Y, et al:
Kras(G12D) and Nkx2-1 haploinsufficiency induce mucinous
adenocarcinoma of the lung. J Clin Invest. 122:4388–4400. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang T, Zhang T, Han X, Liu XI, Zhou N and
Liu Y: Impact of the International Association for the Study of
Lung Cancer/American Thoracic Society/European Respiratory Society
classification of stage IA adenocarcinoma of the lung: Correlation
between computed tomography images and EGFR and KRAS gene
mutations. Exp Ther Med. 9:2095–2103. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Rudin CM, Hann CL, Garon EB, Ribeiro de
Oliveira M, Bonomi PD, Camidge DR, Chu Q, Giaccone G, Khaira D,
Ramalingam SS, et al: Phase II study of single-agent navitoclax
(ABT-263) and biomarker correlates in patients with relapsed small
cell lung cancer. Clin Cancer Res. 18:3163–3169. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kipps TJ, Eradat H, Grosicki S, Catalano
J, Cosolo W, Dyagil IS, Yalamanchili S, Chai A, Sahasranaman S,
Punnoose E, et al: A phase 2 study of the BH3 mimetic BCL2
inhibitor navitoclax (ABT-263) with or without rituximab, in
previously untreated B-cell chronic lymphocytic leukemia. Leuk
Lymphoma. 56:2826–2833. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Infante JR, Fecher LA, Falchook GS,
Nallapareddy S, Gordon MS, Becerra C, DeMarini DJ, Cox DS, Xu Y,
Morris SR, et al: Safety, pharmacokinetic, pharmacodynamic, and
efficacy data for the oral MEK inhibitor trametinib: A phase 1
dose-escalation trial. Lancet Oncol. 13:773–781. 2012. View Article : Google Scholar : PubMed/NCBI
|