Introduction
Pancreatic cancer (PC) is a malignancy with one of
the poorest outcomes (1). It is
also the fourth most common cause of cancer mortality in the United
States (1). Although several risk
factors have been identified, diagnostic methods using specific
markers to track the occurrence and progression of PC are lacking
(2). Considering that the 5-year
overall survival of PC remains <30%, with a median survival of
18–24 months (3), it is urgent to
understand the pathogenesis of PC to aid the identification of
markers that are useful for developing innovative diagnostic and
therapeutic methods for treating this disease.
MicroRNAs (miRNAs/miRs) are small, noncoding RNAs
that are 18–25 nucleotides in length and are involved in the
regulation of cancer development and progression in various types
of cancer, acting as either oncogenes or tumor suppressor genes.
miRNAs regulate gene expression by binding to the 3′-untranslated
regions (3′-UTRs) of specific mRNAs, thus controlling mRNA
stability and the efficiency of translation (4,5).
Growing evidence suggests that miRNAs have an important role in
various biological processes, including cell proliferation,
apoptosis, and differentiation (6). A number of miRNAs, including miR-132,
miR-34, miR-506, and miR-21, have been reported to be associated
with PC via microarrays (7).
miR-125a is a novel miRNA that is located at chromosome 19q13
(8). miR-125a is frequently
downregulated in several types of human cancer, including breast
cancer (9), ovarian cancer
(10), lung cancer (11) and medulloblastoma (12). Low expression of miR-125a is
associated with potential malignant indicators of enhanced gastric
cancer, including tumor size and tumor invasion (13). Thus, it is important to establish
whether miR-125a is also involved in PC inhibition and, if so, what
molecules link miR-125a with cancer mortality.
Mitochondria are central to several cellular
physiological processes that range from regulation of bioenergetics
to maintenance of the cellular oxidation-reduction (redox) status
to the execution of apoptosis (14,15).
The mitochondrial network exists along a spectrum of morphologies,
from a highly interconnected, elongated network to a highly
fragmented, punctate morphology, which is considered to be the
mitochondrial dynamics (mitochondrial fission and fusion) (16,17).
Notably, the critical regulator of mitochondrial dynamics is
mitofusin 2 (Mfn2). Lower Mfn2 is associated with excessive
mitochondrial fission, an early event that occurs during cancer
cell proliferation, apoptosis, metabolism, cell motility and
migration via activation of the intrinsic (mitochondrial) apoptotic
pathway (18–20). However, a higher Mfn2 concentration
has been implicated in cellular survival, chemoresistance and
radiotherapy resistance via inhibition of mitochondrial fission
(21,22). However, whether miR-125a is able to
regulate PC cell death by modifying Mfn2-inhibited mitochondrial
fission remains unknown.
In the current study, it was demonstrated that
miR-125a is decreased in PANC-1 cells, accompanied by an increase
in the contents of Mfn2. In addition, reintroduction of miR-125a
triggered mitochondrial fission in PANC-1 cells via downregulation
of Mfn2 transcription and expression. Excessive mitochondrial
fission contributes to activation of mitochondria-dependent
apoptosis. Furthermore, extensive mitochondrial fission also
impairs cellular migration via induction of F-actin degradation.
These findings illustrate that miR-125a has a role in mitochondrial
fission and is a potential target to slow the development of PC
because it modulates cellular apoptosis, energy metabolism and
cellular migration.
Materials and methods
Cell culture
PANC-1 cell line was purchased from National
Infrastructure of Cell Line Resource (Beijing, China). PANC-1 cells
were cultured in RPMI-1640 medium (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS;
HyClone; GE Healthcare Life Sciences, Logan, UT, USA), 1%
L-glutamine and 0.5% gentamycin (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) at 37°C in an atmosphere of 5% CO2.
Hypoxic conditions were induced in hypoxia chamber in a humidified
atmosphere with 94% N2, 5% CO2 and 1%
O2 for 24 h (15). To
inhibit the mitochondrial fission, mitochondrial division inhibitor
1 (Mdivi1; 10 mM; Sigma-Aldrich; Merck KGaA) was used for 12 h at
37°C.
Transfection
The miR-125a mimic (agmir-125a), miR-control
(scramble miRNA), miR-125a inhibitor (antagomir), small interfering
RNA (siRNA) targeting hypoxia-inducible factor 1 (HIF1) and
negative control siRNA (siCtrl) were purchased from GenePharma Co.,
Ltd. (Shanghai, China). The oligonucleotides used in these studies
are as follows: Agmir-125a, 5′-CCACAUGAACGCCCAGAGAUU-3′; scramble
miRNA, 5′-GAACGGGAGUACAGAGAGAUU-3′; antagomir,
5′-UAACAAGACCAGAGAGCUGUU-3′; siHIF1, 5′-GAGGAAAAGGGAAAAUCUAUU-3′;
siCtrl, 5′-AAUUCUUAAAUUGGGCUGGUU-3′. siRNA (1 µg/ml) and
miRNA (2 µg/ml) were added to the media (per
3.5×104 cells/well) supplemented with
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.)
Media containing siRNAs were replaced with Dulbecco's modified
Eagle's medium (DMEM) 12 h after transfection. Transfection was
performed for 72 h and the knockdown of gene expression was
assessed by western blot analysis.
Western blot analysis
To determine the protein levels, 1×106
cells were lysed with radioimmunoprecipitation assay (RIPA) buffer
(Thermo Fisher Scientific, Inc.) supplemented with
phenylmethylsulfonyl fluoride. The protein concentration was
analyzed using the bicinchoninic acid protein assay. Protein (50
µg) was separated by 10% SDS-PAGE and then transferred to
polyvinylidene difluoride membranes. The membranes were blocked
with 5% nonfat milk for 1 h at room temperature and then incubated
with primary antibodies: Caspase-3 (cat. no. 9662; 1:2,000),
caspase-9 (cat. no. 9508; 1:2,000), Bcl-2 (cat. no. 3498; 1:2,000),
X-linked inhibitor of apoptosis (x-IAP; cat. no. 2042; 1:1,000) and
Mfn2 (cat. no. 11925; 1:1,000) from Cell Signaling Technology, Inc.
(Danvers, MA, USA); Bcl-2-associated agonist of cell death (Bad;
cat. no. ab32445; 1:1,000), Bcl-2 associated X, apoptosis regulator
(Bax; cat. no. ab32503; 1:2,000), HIF1 (cat. no. ab16066; 1:1,000),
poly(ADP-ribose) polymerase (cat. no. 32064; 1:2,000), complex II
(cat. no. ab110410; 1:1,000), complex IV subunit II (cat. no.
ab110268; 1:1,000), complex I subunit NDUFB8 (cat. no. ab110242;
1:1,000), density-regulated protein 1 (Drp1; cat. no. ab56788;
1:1,000), mitochondrial fission 1 protein (Fis1; cat. no. ab71498;
1:1,000), G-actin (cat. no. ab123034; 1:1,000) and F-actin (cat.
no. ab205; 1:1,000) from Abcam (Cambridge, MA, USA); and complex
III subunit core (1:1,000; cat. no. 459220) from Invitrogen (Thermo
Fisher Scientific, Inc.) overnight at 4°C. The membranes were
washed in TBS Tween-20 for 15 min and then incubated with a
horseradish peroxidase-conjugated secondary antibody (cat. nos.
sc-2004 and sc-2005; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA) for 1 h at room temperature. Blots were detected via an
enhanced chemiluminescence substrate kit (Thermo Fisher Scientific,
Inc.), and were analyzed using Quantity One 4.6 software (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) (23).
Construction of adenovirus for Mfn2
overexpression
To over-express Mfn2, the pDC316-mCMV-Mfn2 plasmid
(pDC316-mCMV-Mfn2; NheI-forward,
5′-TATCTCATCAGATTGAGCTCGTCCA-3′ and HindIII-reverse,
5′-CGCCTTAGATCCACTCACTGTAGATTCGA-3′) was purchased from Vigene
Biosciences, Inc. (Rockville, MD, USA). This pDC316-mCMV-Mfn2
plasmid (2.5 µg, per 3.5×104 cells/well) was
transfected into 293T cells (National Infrastructure of Cell Line
Resource) in RPMI-1640 medium supplemented with 10% FBS using
Lipofectamine® 2000 according to the manufacturer's
protocol. The viral supernatant was collected 48 h after
transfection. Supernatant was acquired again and filtered through a
0.45-µm filter to obtain the adenovirus-Mfn2 (Ad-Mfn2). A
total of 1×105 cells/well were infected with 100
multiplicity of infection adenovirus in serum-free DMEM for 6 h at
37°C, following which the media was replaced with DMEM supplemented
with 10% FBS.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted with TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and reverse
transcribed with a One-step RT-PCR kit (TransGen Biotech Co., Ltd.,
Beijing, China) at 42°C for 3 min according to the manufacturer's
instructions. The mRNA levels were determined by RT-qPCR in
triplicate for each of the independently prepared RNAs and were
normalized to the levels of GAPDH expression (24). Gene expression was determined 90
using cBNA SYBR-Green real-time PCR Master Mix (Takara Bio, Inc.,
Otsu, Japan) and qPCR. Primers for qPCR used were as follows:
miR-125a reverse transcription primer,
5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACTCACAGG-3′; miR-125a
PCR primers, sense, 5′-CTGGAGUCCCUGAGACCCUUUA-3′ and antisense,
5′-ACGCTTCACGAATTTGCGTGTC-3′; GAPDH, sense,
5′-TGAGTGCTGTCTCCATGTTTGA-3′ and antisense,
5′-TCTGCTCCCCACCTCTAAGTTG-3′. qPCR was performed at 94°C for 3 min
and 94°C for 30 sec for 38 cycles, and finally 51°C for 30 sec. The
mRNA ratio of the target genes to β-actin was calculated using the
2−ΔΔCq formula (25).
Terminal deoxynucleotidyl transferase
dUTP nick end label- ling (TUNEL) staining
A TUNEL assay was performed using a one-step TUNEL
kit (Beyotime Institute of Biotechnology, Haimen, China) according
to the manufacturer's instructions. TUNEL staining was performed
with fluorescein-dUTP (Invitrogen; Thermo Fisher Scientific, Inc.)
to stain apoptotic cell nuclei, and DAPI (5 mg/ml) was used to
stain all cell nuclei at room temperature for 3 min. Cells in which
the nucleus was stained with fluorescein-dUTP were defined as TUNEL
positive. The slides were then imaged under a confocal microscope
(26).
Hypoxia treatment
Hypoxic conditions were induced using fresh PBS
solution with 94% N2, 1% O2 and 5%
CO2. The pH was adjusted to pH 6.8 with lactate
(Sigma-Aldrich; Merck KGaA) to mimic ischemic conditions. The
dishes were placed into a hypoxia incubator that was equilibrated
with 94% N2, 1% O2 and 5% CO2.
Deferoxamine (DFX; 10 mg/ml) was used to treat cells for ~4 h to
induce the hypoxia condition at 37°C.
Cell viability assay
Cell proliferation was measured with a Cell Counting
Kit-8 (CCK-8) assay (Beyotime Institute of Biotechnology) (27). Briefly, 200 µl of the cell
suspension was seeded in 96-well cell culture plates at a density
of 1,000 cells/well and incubated at 37°C for 1–4 days as
previously described (28).
Cell migration assay
Following treatments, PANC-1 cells were seeded at a
density of 0.5×106 cells/well in 6-well plates and then
cultured overnight (90% confluence). A wound track was scored in
each dish with a pipette head. Debris was removed by washing with
PBS. After 0, 24 and 48 h of culturing, the migration distances
were visualized and imaged (Olympus IX71; Olympus Corporation,
Tokyo, Japan). Cell migration was also analyzed using a Transwell
chamber assay (24 wells, 8-µm pore size with a polycarbonate
membrane) as previously described (29).
Determination of caspase-3/9 activity,
glucose uptake and lactate production
A caspase-3 activity kit and a caspase-9 activity
kit (Beyotime Institute of Biotechnology) were used to detect the
activity of caspase-3 and caspase-9, respectively, according to the
manufacturer's protocol (30).
Following the appropriate treatments, cultured cells were lysed
with RIPA lysis buffer (Beyotime Institute of Biotechnology) for 30
min and centrifuged at 14,000 × g for 30 min at 4°C. The absorbance
was measured at a wavelength of 405 nm using a microplate reader
(BioTek Instruments, Inc., Winooski, VT, USA). The data are
expressed as the ratio of the optical density (OD) value of the
treated group to the OD of the control group. The extracellular
lactate was measured using the cell culture medium with lactate
assay kit (cat. no. K607-100; BioVision, Inc., Milpitas, CA, USA).
Intracellular glucose was measured using cell lysates with glucose
assay kit (cat. no. K606-100; BioVision, Inc.). The uptake of
glucose, the production of lactate and the levels of ATP were all
measured according to the manufacturer's instruction. The assay was
repeated three times.
Microchondrial DNA (mtDNA) strand breaks,
copy numbers and transcription level detection
mtDNA strand breaks were detected based on methods
described previously (11).
Briefly, a 200 µl cell (1×106) suspension was
centrifuged at 15,000 × g at 4°C for 20 min. The supernatant was
discarded and 400 μl solution (0.25 mmol/l inositol, 10 mmol/l
Na3PO4 and 1 mmol/l MgCl2, pH 7.2)
was added at 4°C for 30 min (31).
The relative amounts of mtDNA and nuclear DNA content were used to
assess the mtDNA copy numbers via qPCR, which was performed as
described above. The mtDNA and nuclear ampli-cons were generated
from a complex IV sequence and GAPDH segment, respectively. The
mtDNA primers were 5′-CTATGTCGTGTCCAGAG-3′ and
5′-CATGTTGTCCCGTGTCATG-3′. The GAPDH primers, chosen as the
internal standards, were 5′-CTCAGTCGTATTCGAGTGGTCCT-3′ and
5′-CCTGTGGAAGTCCACAACATGTC-3′. The transcript level of mtDNA was
reflected by two different components: NADH dehydroge-nase subunit
1 (ND1) and cytochrome c oxidase subunit I. The primers for
cytochrome c oxidase subunit I were
5′-ATCGTTCGGTGAGGTCGTG-3′ and 5′-CGCCGGTGTCATTATCGTATA-3′. The
primers for ND1 were 5′-TTGCCGTATATTCAGTATC-3′ and
5′-ATCCTGTTGCCCAGTCCAGT-3′. GAPDH was selected as the internal
standard (32).
ATP production, JC-1 staining,
mitochondrial permeability transition pore (mPTP) opening and
mitochondrial respiratory function
The cellular ATP levels were measured using a
firefly luciferase-based ATP assay kit (Beyotime Institute of
Biotechnology). The opening of mPTP was visualized as a rapid
dissipation of tetramethylrhodamine ethyl ester fluorescence as
described in a previous study (33). Mitochondrial respiration was
initiated by adding glutamate/malate to final concentrations of 5
and 2.5 mmol/l for 5 min, respectively. State 3 respiration was
initiated by adding ADP (150 nmol/l) for 5 min; state 4 was
measured as the rate of oxygen consumption after ADP
phosphorylation. The respiratory control ratio (state 3/state 4)
and ADP/O ratio (number of nmol ADP phosphorylated to atoms of
oxygen consumed) were calculated as previously described (34). Mitochondrial depolarization was
evaluated using MitoProbe™ JC-1 assay kit (Thermo Fisher Scientific
Inc.), according to the manufacturer's protocol.
Immunofluorescence staining
To determine cytochrome c (cyt-c)
localization and mitochondrial division, immunofluorescence
staining was used. Cells were fixed in 3.7% paraformaldehyde for 10
min at room temperature and permeabilized in 100% pre-chilled
acetone (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China).
Following blocking with 5% bovine serum albumin (Sigma-Aldrich;
Merck KGaA) in PBS for 1 h at room temperature, the cells were
incubated with primary antibodies for 4 h at room temperature.
Subsequently, the cells were incubated with Alexa-Fluor 116 488
donkey anti-rabbit secondary antibody (1:1,000; cat. no. A-21206;
Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C for 1 h in the
dark. Images were captured using a laser confocal microscope (TcS
SP5; Leica Microsystems, Inc., Buffalo Grove, IL, USA). The primary
antibodies used for cell immunofluorescence were cyt-c (1:500; cat.
no. ab90529), translocase of outer mitochondrial membrane 20
(1:500; cat. no. ab56783) and F-actin (1:500; cat. no. ab205) from
Abcam. DAPI (5 mg/ml; Sigma-Aldrich; Merck KGaA) was used to stain
the nucleus at room temperature for 3 min (35).
Transmission electron microscopy
Following treatment, cells were collected and fixed
with 3% glutaraldehyde in 100 mM cacodylate buffer at 4°C
overnight, post-fixed in 1% cacodylate-buffer osmium tetroxide for
2 h at room temperature, and dehydrated in a graded series of
ethanol (50, 70, 90 and 100% for 20 min each). Then, cells were
embedded in EponAradite. Ultrathin sections (60 nm) were cut with a
diamond knife on a Leica EM UC6rt (Leica Microsystems GmbH,
Wetzlar, Germany) and double-stained with uranyl acetate and lead
citrate. The ultrastructure of cells was observed with a Hitachi
H7650 transmission electron microscope (TEM; Hitachi, Ltd., Tokyo,
Japan) at 80 kV. Three slides were used in each experiments and the
TEM assay was repeated three times (36).
Luciferase activity assay
Wild-type Mfn2 3′-UTR (WT) and mutant Mfn2 3′-UTR
(MUT) containing the putative binding site of miR-125a were
chemically synthesized and cloned downstream of the firefly
luciferase gene in a pGL3-promoter vector (Promega Corporation,
Madison, WI, USA). PANC-1 cells were placed on a 48-well plate and
cultures until 80% confluence (37). Cells were then co-transfected with
luciferase plasmids (2.5 µg per 3.5×104
cells/well) and miR-125a or control miRNA in DMEM medium
supplemented with 10% FBS using Lipofectamine® 2000
according to the manufacturer's protocol. The pRL Renilla
control reporter vectors (Promega Corporation) was used as an
internal control to normalize the values of the experimental
reporter gene. At 48 h after transfection, the intensities were
measured with a Luciferase Reporter Assay System (Promega
Corporation).
Mitochondrial reactive oxygen species
(Mito-ROS) detection via flow cytometry and electron transport
chain complexes (ETCx) activity detection
Mito-ROS levels were measured using flow cytometry
with a MitoSOX red mitochondrial superoxide indicator (Molecular
Probes; Thermo Fisher Scientific, Inc.). Cells (3.5×106
cells/well) were plated in 6-well culture plates. Subsequently,
cells were incubated with MitoSOX (25 µM) in PBS at 37°C for
30 min, washed twice with PBS, and detached by treatment with
trypsin-EDTA. The detached cells were collected and resuspended in
PBS, and the fluorescence intensity of cells (3.5×106
cells/well) was measured using flow cytometry (BD FAcSVerse) and
analyzed via BD Paint-A-Gate™ Pro software (version 4.2) (both from
BD Biosciences) (38). ETCx
activities were analyzed via ELISA according to the manufacturer's
protocol. The ELISA assay kits for ETCx I, II, and V were purchased
from Beyotime Institute of Biotechnology (cat. nos. S0052, S0101
and S0052) (39).
Statistical analysis
All analyses were performed with SPSS 20.0 software
(IBM Corporation, Armonk, NY, USA). All experiments were repeated
three times. The data are presented as the mean ± standard
deviation and statistical significance for each variable was
estimated by a one-way analysis of variance followed by Tukey's
test for the post hoc analysis. P<0.05 was considered to
indicate a statistically significant difference.
Results
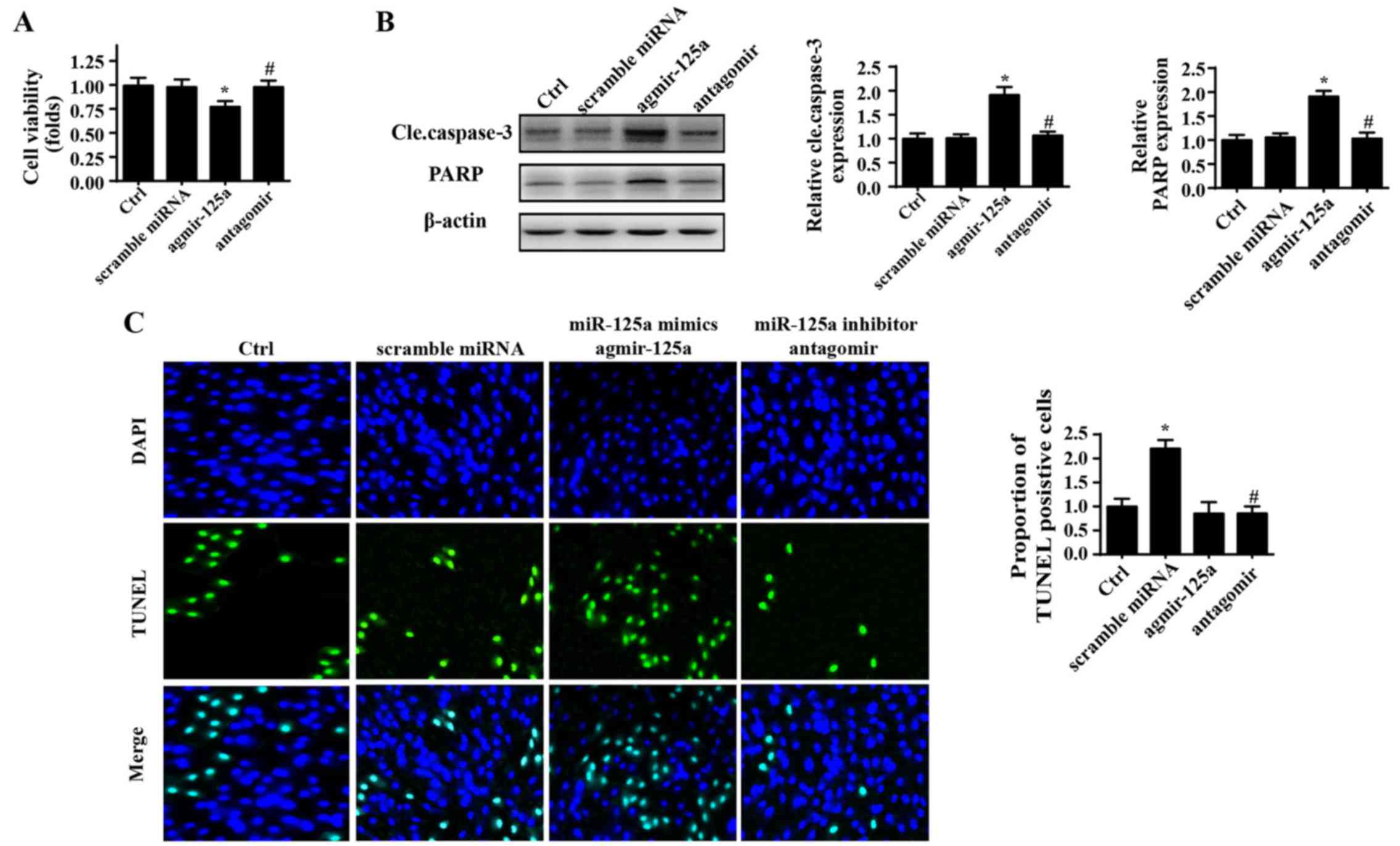
miR-125a enhances PANC-1 cell death
Initially, to verify the role of miR-125a in
regulation of the physiological processes of PANC-1 cells, a mimic
(agmir-125a) and inhibitor (antagomir) of miR-125a were used. A
CCK-8 assay was used to evaluate the growth capacity of PANC-1
cells. As shown in Fig. 1A,
agmir-125a reduced the viability of PANC-1 cells, whereas
application of antagomir marginally promoted cellular growth, as
evidenced by higher OD values in the CCK-8 assay. Furthermore,
expression of cleaved caspase-3 and its substrate, indicators of
cellular apoptosis, were also significantly elevated in response to
agmir-125a treatment in PANC-1 cells (Fig. 1B). To further understand whether
miR-125a modifies cellular survival, TUNEL staining was used
(Fig. 1C). Similarly, introduction
of miR-125a via agmir-125a augmented the ratio of TUNEL positive
cells. By contrast, inhibition of miR-125a reduced the percentage
of TUNEL positive cells. These data indicated that miR-125a is a
pro-apoptotic factor in PANC-1 cells.
Overexpression of miR-125a promotes cell
mitochondrial death by inducing mitochondrial fission
To explore the mechanism by which miR-125a regulates
PANC-1 cell death, mitochondrial damage was investigated. Recent
studies have suggested that mitochondrial fission is an early event
that triggers mitochondria-related apoptosis pathways (40,41).
Therefore, the change of mitochondrial morphology was evaluated. As
shown in Fig. 2A, compared to
spindle mitochondria in the control group, agmir-125a markedly
increased the amount of fragmented mitochondria, as evidenced by
more round and mitochondrial debris. However, application of the
mitochondrial fission inhibitor Mdivi1 blocked the effects of
agmir-125a on mitochondria fragmentation. By contrast, antagomir
treatment led to more mitochondria with a longer length compared
with the control group. These data indicated that miR-125a
activates mitochondrial fission. Additionally, to investigate
whether mitochondrial fission was associated with apoptosis,
proteins associated with mitochondrial damage were evaluated.
Introduction of agmir-125a increased Bax, Bad and caspase-9
expression, and reduced Bcl-2 and x-IAP expression, suggesting
activation of mitochondria-associated apoptosis pathways (Fig. 2B–H). However, the mitochondrial
fission inhibitor blocked the pro-apoptotic effects of agmir-125a.
These data indicated that the excessive fission activated by
miR-125a contributes to the initiation of mitochondria-associated
apoptosis pathways. Furthermore, mitochondrial apoptosis is induced
due to mitochondrial membrane potential dissipation, mPTP opening
and subsequent cyt-c leakage into the cytoplasm, which activates
caspase-9 and caspase-3. Therefore, we observed upstream changes in
the context of miR-125a-mediated fission. As shown in Fig. 2I, agmir-125a treatment destroyed
the membrane potential with evidence of decreased red fluorescence,
but increased green fluorescence. However, the mitochondrial
fission inhibitor reversed these changes. Furthermore, the mPTP
opening rate was increased upon application of agmir-125a, but
decreased when treated with fission inhibitors (Fig. 2J). The agmir-125a also caused more
cyt-c leakage from mitochondria into the cytoplasm, and some cyt-c
even migrated into the nucleus (Fig.
2K). However, these changes were blocked by the mitochondrial
fission inhibitor. Together, these data indicate that miR-125a
activates mitochondrial fission, which induces mitochondrial
potential collapse, mPTP opening and cyt-c leakage into the
cytoplasm and nucleus, leading to activation of caspase-9-dependent
mitochondria apoptosis pathways.
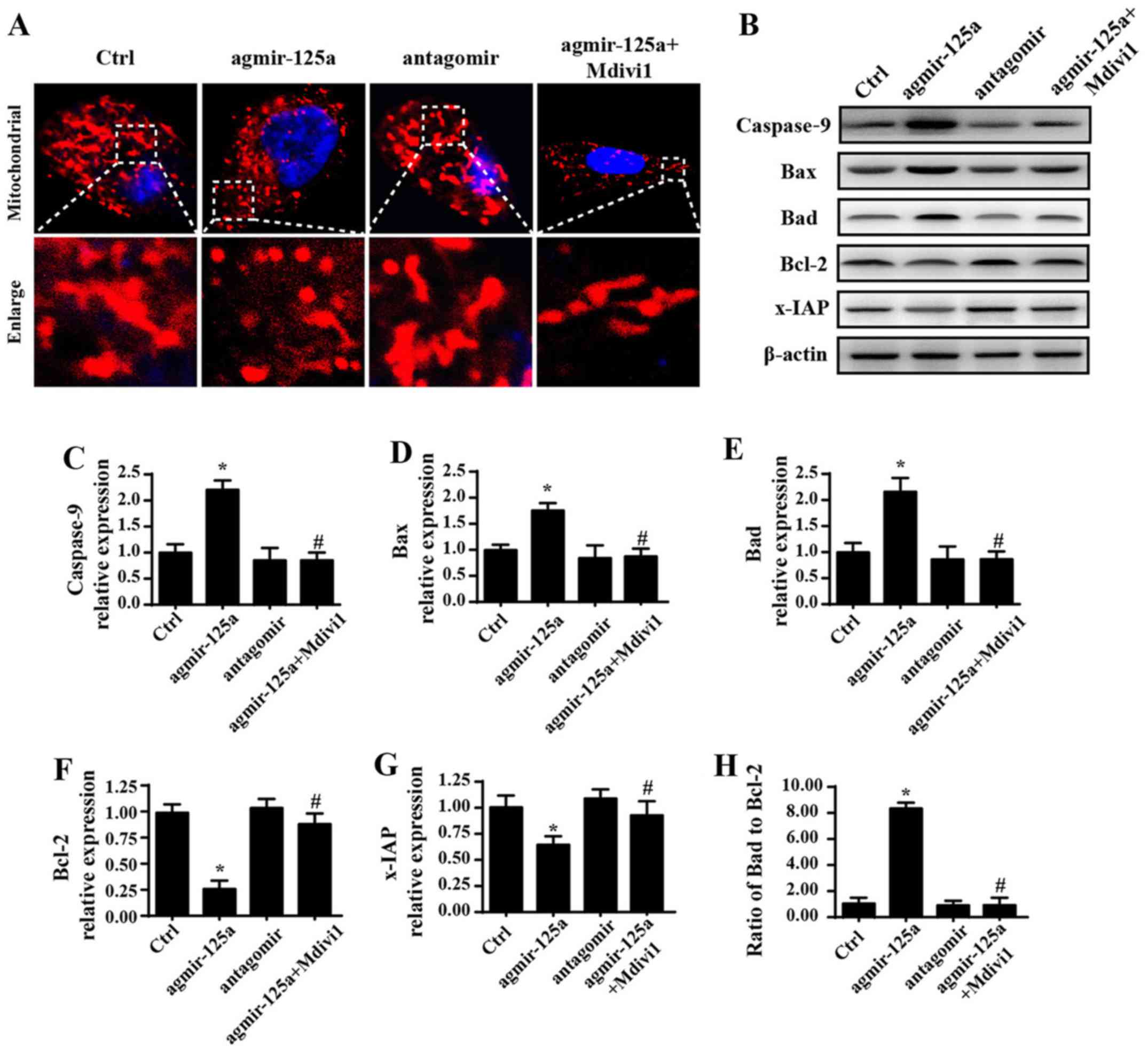 | Figure 2Overexpression of miR-125a promotes
cell death by inducing mitochondrial fission-related mitochondrial
apoptosis pathways. (A) Change of the mitochondrial morphology via
Tomm20 staining. (B) Western blot analysis was performed and
densitometry performed for expression of (C) caspase-9, (D) Bax,
(E) Bad, (F) Bcl-2 and (G) x-IAP, and (H) Bax/Bcl-2 ratio was
calculated. (I) Mitochondrial membrane potential was measured by
JC-1. (J) mPTP opening rate increased upon application of
agmir-125a, but decreased when treated with fission inhibitors. (K)
cyt-c immunofluorescence showed cyt-c leakage from mitochondria
into the cytoplasm induced by agmir-125a, and some cyt-c even
migrated into the nucleus. *P<0.05 vs. Ctrl group;
#P<0.05 vs. agmir-125a group. Tomm20, translocase of
outer mitochondrial membrane 20; Ctrl, control; miR, microRNA;
agmir-125a, miR-125a mimic; antagomir, miR-125a inhibitor; Mdivi-1,
mitochondrial division inhibitor 1; Bax, Bcl-2 associated X,
apoptosis regulator; Bad, Bcl-2-associated agonist of cell death;
x-IAP, X-linked inhibitor of apoptosis; mPTP, mitochondrial
permeability transition pore; cyt-c, cytochrome c. |
Mitochondrial fission causes
mitochondrial energy disorder
In addition to activating apoptosis, the central
role of mitochondria is to generate energy and regulate metabolism,
which are fundamental for tumor development and progression
(42,43). Therefore, we observed energy or
metabolism alterations upon miR-125a stimulation. As shown in
Fig. 3A, agmir-125a reduced the
contents of double-stranded mtDNA (Fig. 3A), mtDNA copy number (Fig. 3B), mtDNA transcripts (Fig. 3C) and ATP generation (Fig. 3D) in PANC-1 cells, which were
blocked by the fission inhibitor. Electron transport chain
complexes (ETCx) are mainly encoded by mtDNA and primarily
responsible for ATP generation via using hydrion, electron and
oxygen. ETCx expression and activity is vital to delivery hydrion
and electron to oxygen, favoring to the mitochondrial oxidative
phosphorylation. Accordingly, the downregulation and inactivation
of ETCx would predispose the energy undersupply. Based on this, the
influence of miR-125a on ETCx expression and activity was
evaluated. The results shown in Fig.
3E–J demonstrate that agmir-125a treatment significantly
reduced the ETCx concentration and activity, which are coupled to
the decrease of the state 3 respiratory rate. Furthermore,
agmir-125a-treated PANC-1 cells took up less glucose and therefore
produced less lactate. However, the fission inhibitor abolished the
inhibitory effects of agmir-125a on PANC-1 cells. Additionally, the
inhibitor of miR-125a slightly elevated ETCx activity (P>0.05)
and the subsequent mitochondrial respiratory function, which were
also associated with increased glucose consumption and lactate
production. Similar results were observed with mitochondrial
reactive oxygen species (Fig. 3K).
These data suggest that mitochondrial fission activated by miR-125a
impairs PANC-1 cell energy metabolism.
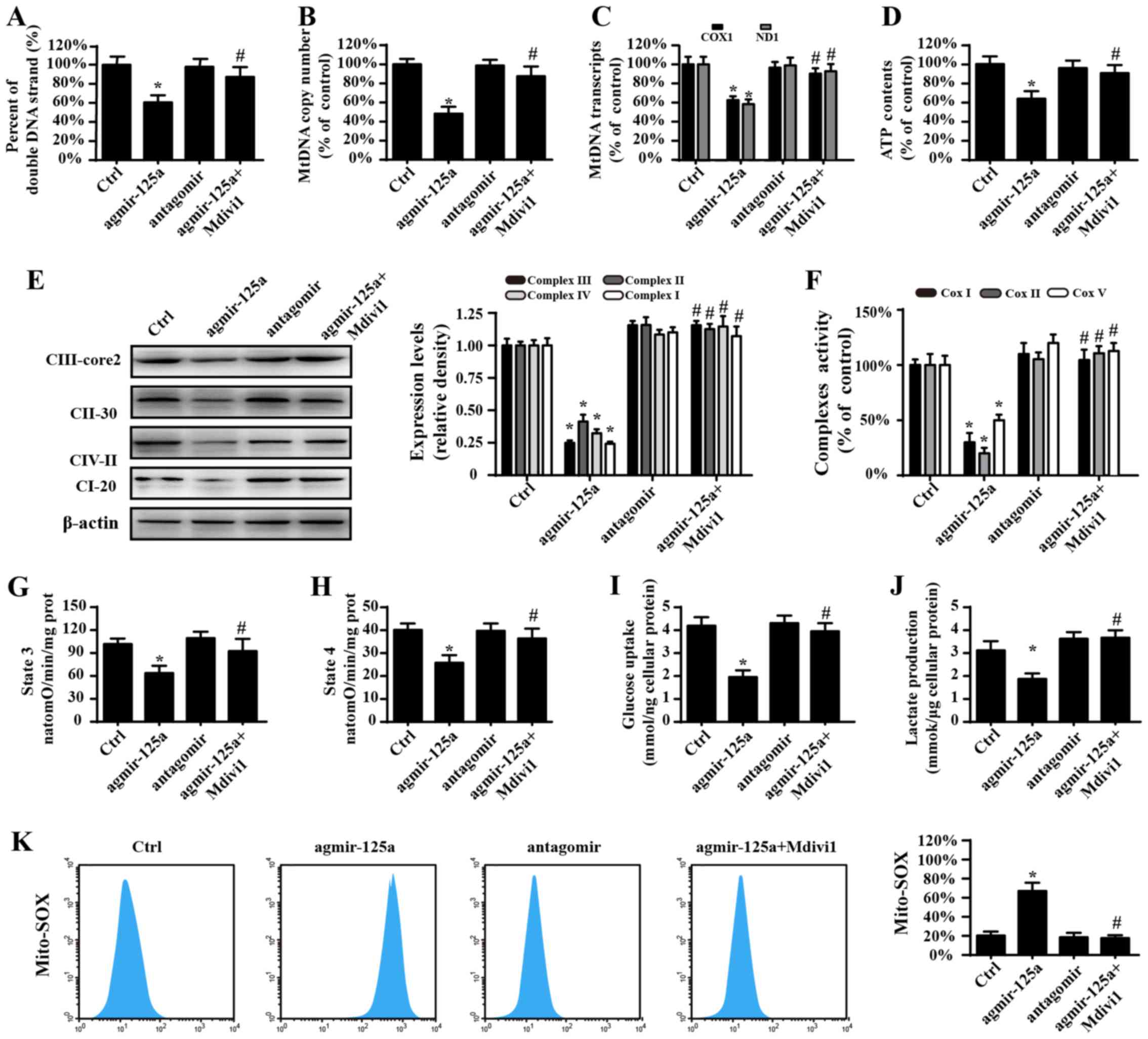 | Figure 3Mitochondrial fission causes
mitochondrial energy disorder. (A) The percentage of
double-stranded mtDNA indicates mtDNA strand breaks. (B) The mtDNA
copy number was assessed by complex IV segments. (C) The transcript
levels of mtDNA are reflected by two different components: ND1,
encoded by the light chain of mtDNA, and COX I, encoded by the
heavy chain of mtDNA. (D) Change in ATP contents. (E) Expression of
mitochondrial ETCxs. (F) Changes in the ETCx I, II and V activities
as measured via ELISA. Effects of agmir-125a on (G) state 3
respiration, (H) state 4 respiration, respiratory control ratio
(RCR [state 3/4]). (I) Glucose uptake. (J) Lactate production. (K)
Mito-ROS contents. The curve chart indicates the quantitative flow
cytometry results. *P<0.05 vs. Ctrl group;
#P<0.05 vs. agmir-125a group. Ctrl, control; miR,
microRNA; agmir-125a, miR-125a mimic; antagomir, miR-125a
inhibitor; Mdivi-1, mitochondrial division inhibitor 1; COX 1,
cytochrome c oxidase subunit I; ND1, NADH dehydrogenase
subunit 1; ETCxs, electron transport chain complexes; Mito-ROS,
mitochondrial reactive oxygen species. |
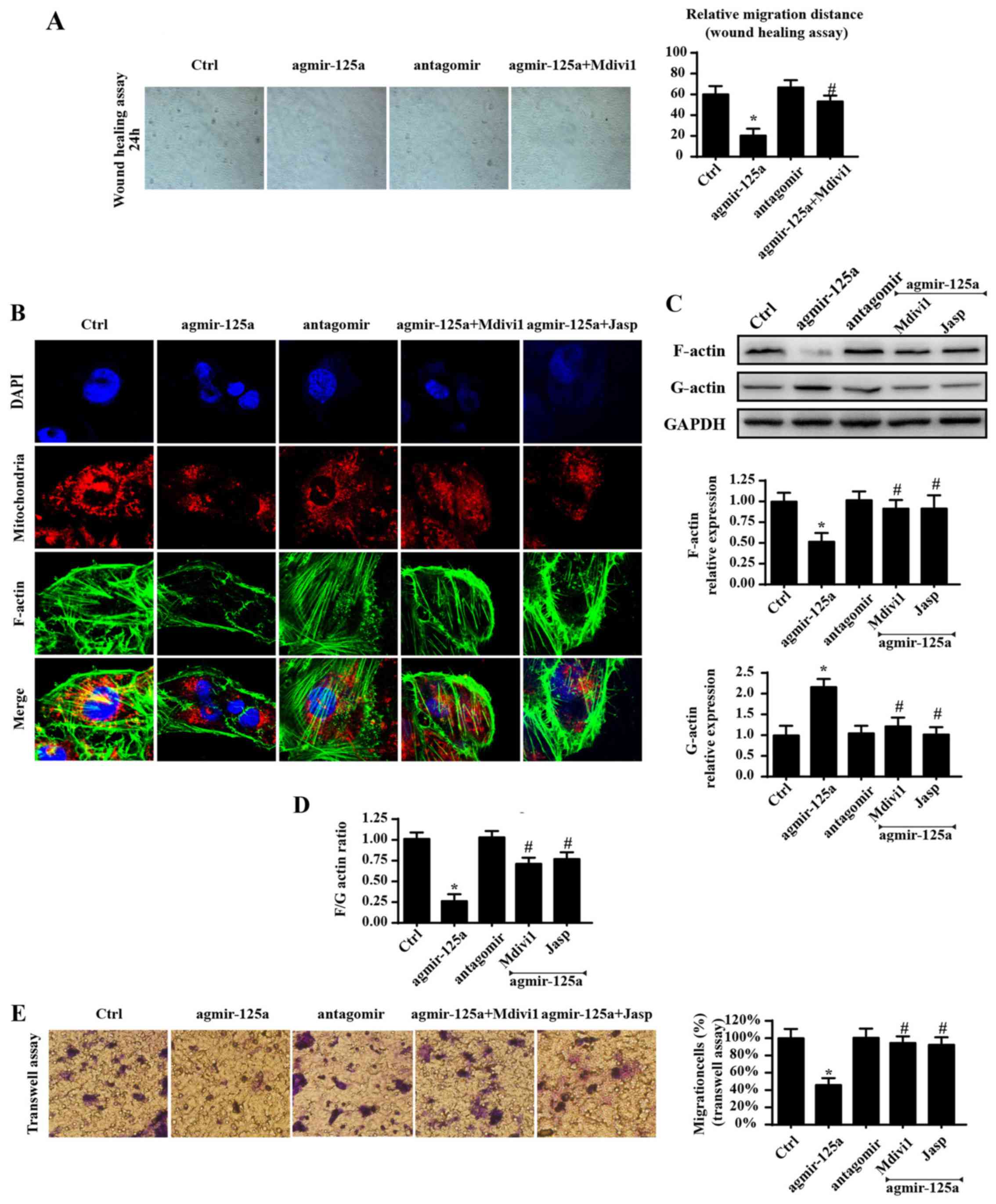
Excessive fission impaired the cellular
migration capacity by inducing F-actin depolymerization into
G-actin
Apart from cellular survival and energy metabolism,
cellular migration is another factor that contributes to
tumorigenesis (44). Therefore,
whether mitochondrial fission has a role in cellular migration was
investigated. Initially, a wound-healing assay was used to evaluate
the influence of miR-125a on the mobilization of PANC-1 cells. As
shown Fig. 4A, exogenously
administered agmir-125a reduced the migration of PANC-1 cells,
while the mitochondrial fission inhibitor blocked such changes.
These data indicated that miR-125a has a role in cellular migration
via fission. As F-actin is the key stress fiber that directly
regulates cellular mobilization, it was hypothesized that the
blunted migration was derived from mitochondrial fission-involved
F-actin dyshomeostasis. Fluorescence was also used to obverse
F-actin changes. As shown in Fig.
4B, agmir-125a significantly reduced the fluorescence intensity
of F-actin, but increased the amount of mitochondrial debris, in
PANC-1 cells, which were reversed by the mitochondrial fission
inhibitor and F-actin depolymerization inhibitors. As F-actin is
composed of G-actin, it was hypothesized that there was an
imbalance between F-actin and G-actin. As expected, agmir-125a
treatment accelerated F-actin division into G-actin, as evidenced
by increased G-actin and decreased F-actin protein expression in
the agmir-125a group (Fig. 4C and
D). Mdivi1 blocked these conformation alterations, suggesting
that mitochondrial fission is responsible for F-actin
depolymerization to G-actin. Furthermore, inhibition of F-actin
degradation via Jasplakinolide reversed migration under agmir-125a
treatment, which is similar to the results in the Mdivi1 group
(Fig. 4A–D). Similar results were
observed in Transwell assays (Fig.
4E). Together, these data indicated that miR-125a impairs
cellular migration by inducing F-actin depolymerization into
G-actin by activating mitochondrial fission.
miR-125a activates mitochondrial fission
via negative regulation of Mfn2
To determine the mechanism by which miR-125
regulates mitochondrial fission, the change of Mfn2, which prevents
excessive mitochondrial fission was investigation. Treatment with
agmir-125a reduced the contents of Mfn2 in PANC-1 cells. However,
introduction of miR-125a antagomir reversed expression of Mfn2 in
PANC-1 cells (Fig. 5A). The
similar results were observed for other mitochondrial fission
makers, including Fis1 and Drp1. These data indicated that miR-125a
negatively regulated the expression of Mfn2. Furthermore, through
analysis of the gene sequences, miR-125a was matched to Mfn2, which
is highly conserved among rat, human and mouse (Fig. 5B). To test whether miR-125a
directly targeted the 3′-UTR of Mfn2, luciferase assays were
performed using 3′-UTR sequence fragments containing the predicted
target of Mfn2 and its mutated version inserted downstream of a
luciferase reporter (Fig. 5C). The
results demonstrated that the luciferase activity was downregulated
in cells co-transfected with miR-125a mimics and the wild-type Mfn2
3′-UTR compared with the mutated type, suggesting that Mfn2 is a
target gene of miR-125a. These data indicate that miR-125a directly
regulates transcription and expression of Mfn2.
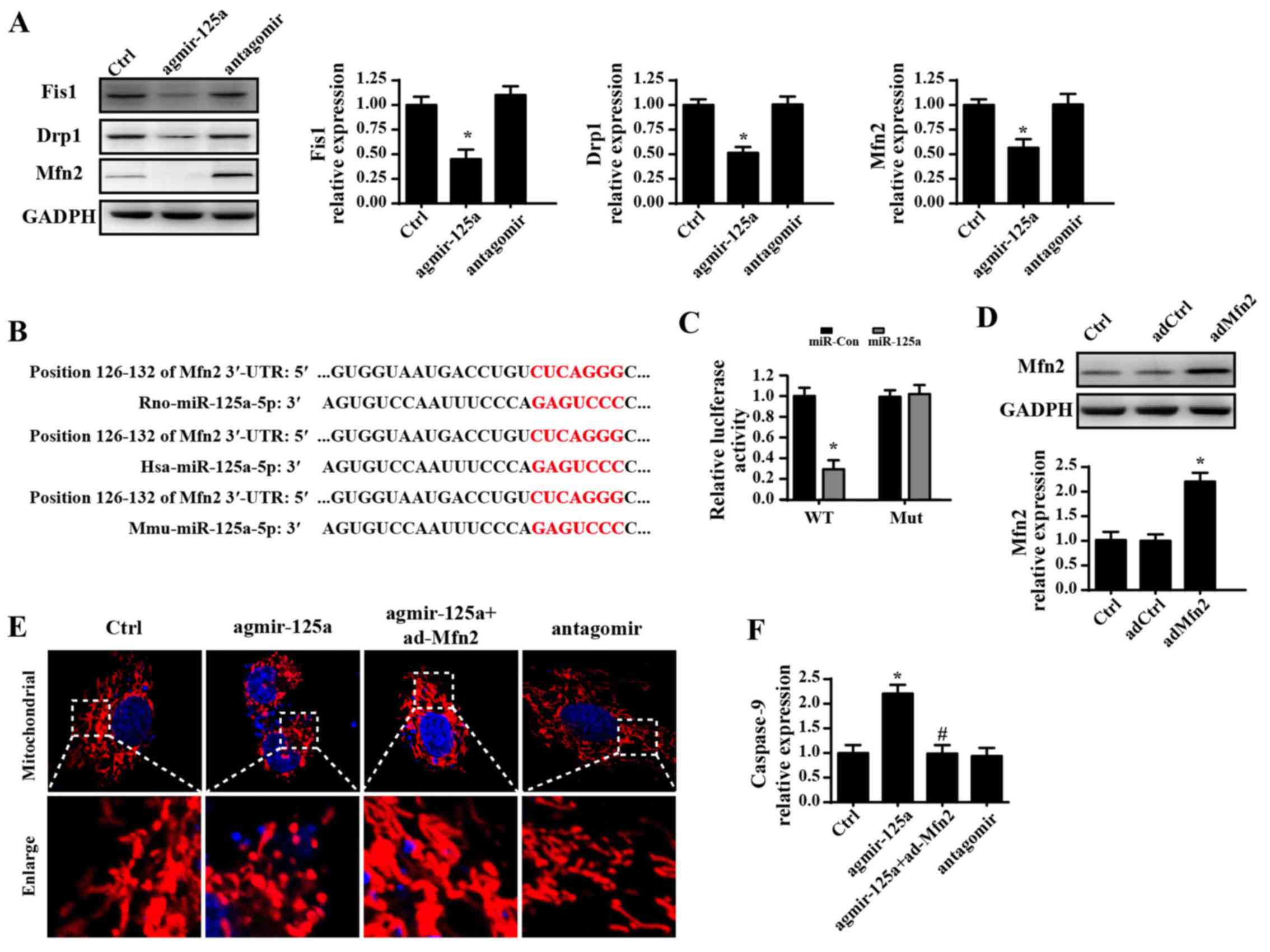 | Figure 5miR-125a activates mitochondrial
fission by negatively regulating Mfn2. (A) miR-125a antagomir
reversed expression of Fis1, Drp1 and Mfn2 in PANC-1 cells. (B)
Computational analysis was performed for miRNA seed sequences
complementary to the 3′-UTR of Mfn2 mRNA, which were conserved at
the putative binding sites in rat, human, and mouse. (C) Luciferase
assay for post-transcriptional repression of Mfn2. (D) The
transfection efficiency of Mfn2 evaluated by western blot analysis.
(E) The change of mitochondrial fragmentation or debris measured
via Tomm20 immunofluorescence after overexpression of Mfn2 via
ad-Mfn2. (F) The change of caspase-9 activity following
overexpression of Mfn2 via ad-Mfn2. *P<0.05 vs. Ctrl
group; #P<0.05 vs. agmir-125a group. Tomm20,
translocase of outer mitochondrial membrane 20; Ctrl, control; miR,
microRNA; agmir-125a, miR-125a mimic; antagomir, miR-125a
inhibitor; Fis1, mitochondrial fission 1 protein; Drp1,
density-regulated protein; Mfn2, mitofusin 2; ad-, adenovirus;
3′-UTR, 3′-untranslated region. |
To confirm whether Mfn2 was responsible for fission,
Mfn2 was overexpressed using an adenovirus (Ad-Mfn2) in PANC-1
cells. The transfection efficiency is shown in Fig. 5D. Mfn2 overexpression via Ad-Mfn2
blocked the promotive effects of agmir-125a on mitochondrial
fission, as demonstrated by less mitochondrial fragmentation or
debris in PANC-1 cells (Fig. 5E).
These data confirmed the hypothesis that miR-125a triggers
mitochondrial fission by inhibiting Mfn2 expression. Additionally,
transfection with Ad-Mfn2 also ablated caspase-9 activity, which
was upregulated in response to agmir-125a stimulation (Fig. 5F), suggesting that Mfn2 is also
associated with mitochondrial fission-mediated mitochondrial
apoptosis.
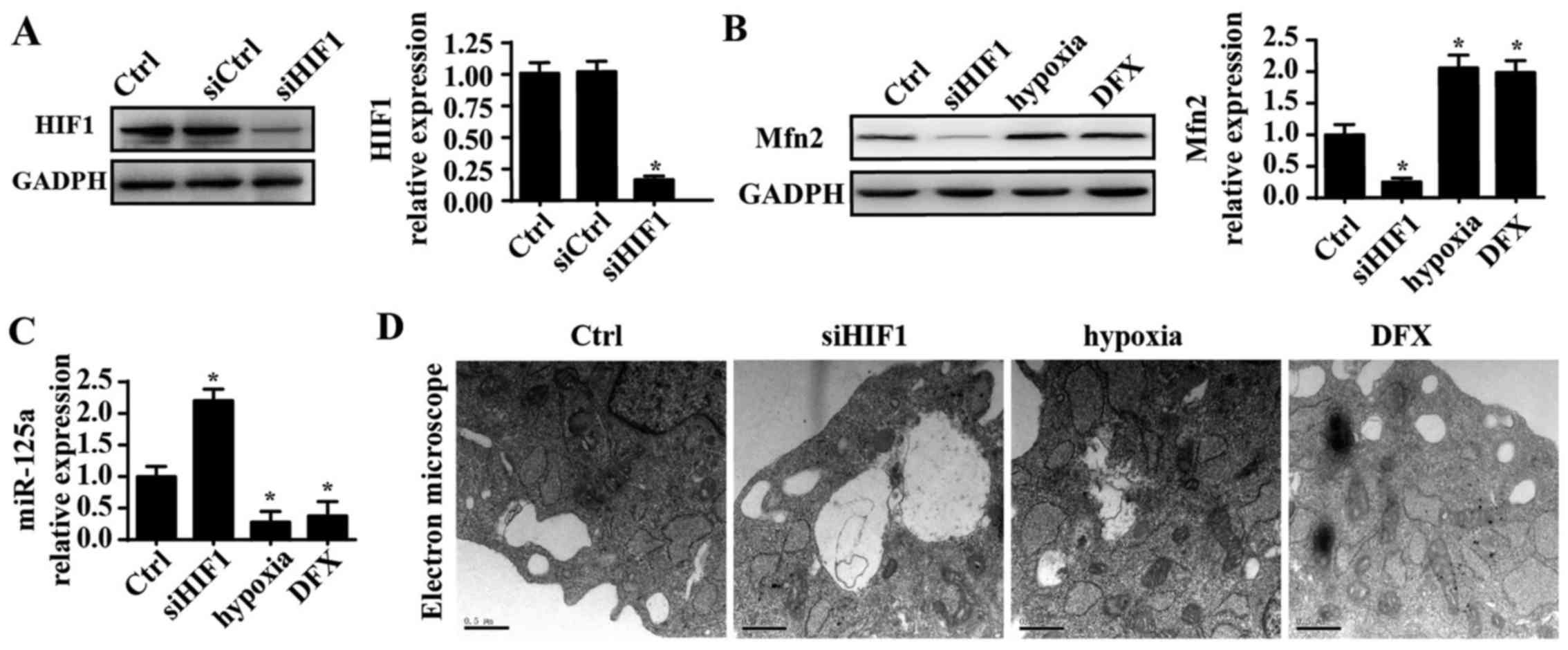
miR-125a is negatively regulated by
HIF1
Finally, to elucidate the mechanism by which
miR-125a is downregulated in PANC-1 cells, HIF1 was investigated.
Several studies have reported that miR-125a is negatively modified
by HIF1 and that hypoxic conditions further repress expression of
miR-125a (45,46). Thus, it was hypothesized that HIF1
is the upstream regulator of miR-125a. siRNA was used to knockdown
expression of HIF1 in PANC-1 cells, and the silencing efficiency of
the siRNA is shown in Fig. 6A.
Following HIF1 knockdown, mRNA expression of miR-125a was
significantly increased. Additionally, hypoxic conditions were
induced using a hypoxia chamber with 1% oxygen to enhance the HIF1
signals. Under hypoxic conditions, miR-125a was further
downregulated. Furthermore, DFX was also used to mimic hypoxic
conditions, and the results were in agreement with the above
findings. These data indicated that miR-125a is negatively
regulated by HIF1. To provide further evidence regarding the role
of HIF1 in fission, the influence of HIF1 on Mfn2 expression was
evaluated. Knockdown of HIF1 also reduced expression of Mfn2, which
was increased under hypoxic conditions, suggesting a regulatory
role for HIF1 in Mfn2 (Fig. 6B and
C). Furthermore, the TEM results indicated that more fragmented
mitochondria appeared in response to HIF1 silencing. However, upon
exposure to hypoxic conditions, mitochondrial fission was
inhibited, as demonstrated by longer mitochondria (Fig. 6D). Collectively, these data confirm
that HIF1 inhibits mitochondrial fission via the miR125a/Mfn2
pathways.
Discussion
The results of the demonstrated that miR-125a
targets Mfn2 and inhibits transcription and expression of Mfn2.
Furthermore, reduced Mfn2 contributed to excessive mitochondrial
fission in PANC-1 cells, which resulted in activation of
mitochondria-associated apoptosis pathways. Excessive fission
causes cellular energy disorder and migration impairment. Finally,
the results of the current study demonstrated that HIF1 is an
upstream regulator of miR-125a/Mfn1, and subsequent mitochondrial
fission. To the best of our knowledge, this is the first study to
describe the tumor-suppressive actions of HIF1/Mfn2/miR-125a in
PANC-1 cells, including regulation of cellular apoptosis, energy
metabolism and migration by inhibition of mitochondrial
fission.
Previous studies have reported that miR-125a has a
tumor-suppressive role in various cancer types by negatively
regulating tumor oncogenes (47,48).
For example, miR-125a is an independent prognostic factor and
inhibits the proliferation of gastric cancer (49). Additionally, miR-125a functions as
a tumor suppressor by regulating abnormal activity of sirtuin 7 in
hepatocellular carcinoma tumorigenesis (50). In the current study, miR-125a
enhances the mitochondrial fission that is involved in PANC-1 cell
apoptosis, metabolism and migration. At the time of PC diagnosis,
the majority of patients are determined to have unresectable
disease. Considerable efforts have been made to develop novel
therapeutic treatments for this disease; however, progress on this
issue is limited. Identifying the appropriate miRNAs and tapping
into the excellent potential of miRNAs will have a beneficial
impact on the treatment of PC. The present study provides evidence
that the concentration of miR-125a is positively associated with
PANC-1 cell apoptosis, energy disorders and migration impairment,
and may be used as a possible target to regulate PANC-1 cell
biological processes. However, more clinical data are required.
Mitochondrial homeostasis is known to be associated
with the pathogenesis of neurodegenerative diseases, diabetes and
myopathies, among other human diseases. In the present study,
overexpression of miR-125a induced mitochondrial fission, as
demonstrated by increased mitochondrial fragmentation. Furthermore,
excessive mitochondrial fission causes mitochondrial
depolarization, followed by cyt-c leakage into the cytoplasm
(51). As a consequence of cyt-c
release, mitochondria-associated apoptosis pathways are activated,
as evidenced by increased pro-apoptosis protein expression and
decreased anti-apoptosis protein expression (52). Other studies have reported that
mitochondrial fission leads to voltage-dependent anion channel 1
oligomerization and the separation of hexokinase 2 (HK2) from the
outer mitochondrial membrane, resulting in the opening of the mPTP
and reduced mitochondrial membrane potential (53,54).
These findings were similar those of the present study, which
demonstrated that mitochondrial fission involved in cell apop-tosis
by damaging the mitochondrial structure and function. Additionally,
miR125a-regulated mitochondrial fission is reported to be involved
in cellular energy disorder. Excessive mitochondrial fission
impaired mitochondrial ATP production by reducing ETCx activity
(55), followed by decreased
glucose consumption and lactate production. Notably, other studies
have indicated that miR-125a has a negative role in the regulation
of HK2 (56), a glycolytic
rate-limiting enzyme, and therefore has a negative role in the
modified Warburg effect in hepatocellular carcinoma. These findings
were similar to the data of the present study that demonstrated
that miR-125a is the key regulator of cancer cell energy
metabolism. Furthermore, in the current study, it was also
demonstrated that miR125a-induced mitochondrial fission is involved
in PANC-1 cell migration. Increased fragmentation was associated
with impaired cellular mobilization (57). As F-actin is the key stress fiber
that directly monitors cellular mobilization (58), it was hypothesized that blunted
migration arose from mitochondrial fission-involved F-actin
dyshomeostasis. The results of the current study indicated that
mitochondrial fission leads to F-actin depolymerization to G-actin,
eventually causing a considerable obstacle in cellular migration.
Successive mitochondrial fission is dependent on binding between
Drp1, the Drp1 receptor and stress fibers (59). Previously, studies have indicated
that intracellular F-actin accumulation on the surface of
mitochondria for short periods of time is a prerequisite for
subsequent mitochondrial division (60,61).
Under physiological conditions, F-actin is regularly distributed in
certain parts of the cytoplasm that control cellular migration to
direct cells to move in a particular direction. When mitochondrial
fission is initiated, F-actin decomposes into G-actin, which is
then reassembled into F-actin at the outer mitochondrial membrane
to promote the formation of a contractile ring with the help of
Drp1 and its receptor. Considering the indispensable nature of
F-actin in fission, excessive fission may consume large amounts of
cytoplasmic F-actin, causing an uneven distribution of F-actin
(62), ultimately leading to
dysregulated F-actin homeostasis and impaired migration. More
evidence is required to support this hypothesis. Together, the data
provide useful information about the tumor-suppressive role of
mitochondrial fission in PANC-1 cells, including apoptosis,
migration and energy metabolism. These findings provide a clear
target to inhibit PC development and progression.
Dysfunction of mitochondria is largely dependent on
Mfn2 activity, which preserves the mitochondrial dynamic balance
(fission and fusion) (63). The
findings of the current study confirmed that miR-125a can directly
target Mfn2 and inhibit Mfn2 transcription and expression.
Furthermore, decreased Mfn2 contents were associated with excessive
mitochondrial fission. Several studies have illustrated the
regulatory role of Mfn2 in mitochondrial fission by counteracting
Drp1 (64,65). Furthermore, studies have also
reported that Mfn2 is able to indirectly activate Parkin1-dependent
mitophagy, which removes fragmented mitochondria and therefore
blocks mitochondrial fission (66). This information describes the
comprehensive role of Mfn2 in mitochondrial homeostasis. Several
studies have also defined other elements of the mitochondrial
fission machinery, including mitochondrial fission factor, Drp1 and
Opa1 mitochondrial dynamin like GTPase (14,16,67).
However, whether these factors have anti-tumor properties remains
unclear. This may be the mechanism by which Mfn2 regulates
mitochondrial fission. Furthermore, the results of the current
study demonstrated that miR-125a/Mfn2 is regulated by HIF1, a
regulatory protein that was significantly increased under hypoxic
conditions. In fact, the normal pancreas has an abundant blood
flow, in contrast to pancreatic cancer, which is a hypovascular
tumor. This is partly because of fibrotic changes around the tumor
due to pancreatitis associated with cancer invasion. In the current
study, knockdown of HIF1 enhanced miR-125a expression and promoted
mitochondrial fission, suggesting that miR-125a is negatively
regulated by HIF1. Additionally, these data also indicated that
HIF1 has a tumor-promotive role in PC. In fact, hypoxia and HIF1
are essential components of the neoplastic microenvironment, often
allowing a selective advantage for tumor cells over otherwise
non-invasive cells that are more sensitive to a low oxygen state.
The evidence for hypoxia and HIF expression in pancreatic cancer is
in their characteristic avascular appearance on computed tomography
and from intra-tumoral oxygen tension measurements. Hypoxic
conditions and HIF1 expression in solid malignancies may confer
resistance to conventional radiation and chemotherapy. Therefore,
there is a focus on identifying the mechanism by which hypoxia/HIF1
contributes to PC development. The findings of the present study
suggest that HIF1 negatively affects miR-125a expression and
therefore augments Mfn2 expression, contributing to PANC-1 survival
and migration by inhibiting mitochondrial fission.
However, the present study aimed to explore the role
of mitochondrial fission in PANC-1 cells rather than in normal
pancreatic ductal epithelial cell lines. Thus, additional
experimental evidence is required.
In conclusion, the current study described the
critical roles of HIF1/miR-125a/Mfn2 in PANC-1 cell apoptosis,
migration and energy metabolism via mitochondrial fission.
Regulation of mitochondrial fission by HIF1/miR-125a/Mfn1 during PC
carcinogenesis may be a potential target to modify tumor growth and
promotion, which opens a novel avenue for PC treatment.
Acknowledgments
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LCP, LZ and WJY were involved in conception and
design, performance of experiments, data analysis and
interpretation, and manuscript writing. LCP, JB, and RL were
involved in data analysis and interpretation.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Xu Z, Pothula SP, Wilson JS and Apte MV:
Pancreatic cancer and its stroma: A conspiracy theory. World J
Gastroenterol. 20:11216–11229. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pinho AV, Chantrill L and Rooman I:
Chronic pancreatitis: A path to pancreatic cancer. Cancer Lett.
345:203–209. 2014. View Article : Google Scholar
|
|
3
|
Lin QJ, Yang F, Jin C and Fu DL: Current
status and progress of pancreatic cancer in China. World J
Gastroenterol. 21:7988–8003. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
He B, Zhao Y, Xu L, Gao L, Su Y, Lin N and
Pu J: The nuclear melatonin receptor RORα is a novel endogenous
defender against myocardial ischemia/reperfusion injury. J Pineal
Res. 60:313–326. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kleszczyński K, Zillikens D and Fischer
TW: Melatonin enhances mitochondrial ATP synthesis, reduces
reactive oxygen species formation, and mediates translocation of
the nuclear erythroid 2-related factor 2 resulting in activation of
phase-2 antioxidant enzymes (γ-GCS, HO-1, NQO1) in ultraviolet
radiation- treated normal human epidermal keratinocytes (NHEK). J
Pineal Res. 61:187–197. 2016. View Article : Google Scholar
|
|
6
|
Zhang R and Sun Y, Liu Z, Jin W and Sun Y:
Effects of melatonin on seedling growth, mineral nutrition, and
nitrogen metabolism in cucumber under nitrate stress. J Pineal Res.
62:e124032017. View Article : Google Scholar
|
|
7
|
Sandesc M, Dinu A, Rogobete AF, Bedreag
OH, Sandesc D, Papurica M, Bratu LM, Negoita S, Vernic C, Popovici
SE, et al: Circulating microRNAs expressions as genetic biomarkers
in pancreatic cancer patients continuous non-invasive monitoring.
Clin Lab. 63:1561–1566. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liang L, Wei DM, Li JJ, Luo DZ, Chen G,
Dang YW and Cai XY: Prognostic microRNAs and their potential
molecular mechanism in pancreatic cancer: A study based on The
Cancer Genome Atlas and bioinformatics investigation. Mol Med Rep.
17:939–951. 2018.
|
|
9
|
Shi C, Cai Y, Li Y, Li Y, Hu N, Ma S, Hu
S, Zhu P, Wang W and Zhou H: Yap promotes hepatocellular carcinoma
metastasis and mobilization via governing
cofilin/F-actin/lamellipodium axis by regulation of
JNK/Bnip3/SERCA/CaMKII pathways. Redox Biol. 14:59–71. 2018.
View Article : Google Scholar
|
|
10
|
Jiang L, Huang Q, Zhang S, Zhang Q, Chang
J, Qiu X and Wang E: Hsa-miR-125a-3p and hsa-miR-125a-5p are
downregulated in non-small cell lung cancer and have inverse
effects on invasion and migration of lung cancer cells. BMC Cancer.
10:3182010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nam EJ, Yoon H, Kim SW, Kim H, Kim YT, Kim
JH, Kim JW and Kim S: MicroRNA expression profiles in serous
ovarian carcinoma. Clin Cancer Res. 14:2690–2695. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ferretti E, De Smaele E, Po A, Di
Marcotullio L, Tosi E, Espinola MS, Di Rocco C, Riccardi R,
Giangaspero F, Farcomeni A, et al: MicroRNA profiling in human
medulloblastoma. Int J Cancer. 124:568–577. 2009. View Article : Google Scholar
|
|
13
|
Hashiguchi Y, Nishida N, Mimori K, Sudo T,
Tanaka F, Shibata K, Ishii H, Mochizuki H, Hase K, Doki Y, et al:
Downregulation of miR-125a-3p in human gastric cancer and its
clinicopathological significance. Int J Oncol. 40:1477–1482.
2012.PubMed/NCBI
|
|
14
|
Zhou H, Ma Q, Zhu P, Ren J, Reiter RJ and
Chen Y: Protective role of melatonin in cardiac
ischemia-reperfusion injury: From pathogenesis to targeted therapy.
J Pineal Res. 64:e124712018. View Article : Google Scholar
|
|
15
|
Das N, Mandala A, Naaz S, Giri S, Jain M,
Bandyopadhyay D, Reiter RJ and Roy SS: Melatonin protects against
lipid-induced mitochondrial dysfunction in hepatocytes and inhibits
stellate cell activation during hepatic fibrosis in mice. J Pineal
Res. 62:e124042017. View Article : Google Scholar
|
|
16
|
Jin Q, Li R, Hu N, Xin T, Zhu P, Hu S, Ma
S, Zhu H, Ren J and Zhou H: DUSP1 alleviates cardiac
ischemia/reperfusion injury by suppressing the Mff-required
mitochondrial fission and Bnip3-related mitophagy via the JNK
pathways. Redox Biol. 14:576–587. 2018. View Article : Google Scholar
|
|
17
|
Zhou H, Du W, Li Y, Shi C, Hu N, Ma S,
Wang W and Ren J: Effects of melatonin on fatty liver disease: The
role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission, and
mitophagy. J Pineal Res. 64:e124502018. View Article : Google Scholar
|
|
18
|
Zhu H, Jin Q, Li Y, Ma Q, Wang J, Li D,
Zhou H and Chen Y: Melatonin protected cardiac microvascular
endothelial cells against oxidative stress injury via suppression
of IP3R-[Ca2+] c/VDAC-[Ca2+]m axis by
activation of MAPK/ERK signaling pathway. Cell Stress Chaperones.
23:101–113. 2018. View Article : Google Scholar
|
|
19
|
Hu SY, Zhang Y, Zhu PJ, Zhou H and Chen
YD: Liraglutide directly protects cardiomyocytes against
reperfusion injury possibly via modulation of intracellular calcium
homeostasis. J Geriatr Cardiol. 14:57–66. 2017.PubMed/NCBI
|
|
20
|
Zhou H, Zhu P, Guo J, Hu N, Wang S, Li D,
Hu S, Ren J, Cao F and Chen Y: Ripk3 induces mitochondrial
apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury.
Redox Biol. 13:498–507. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou H, Li D, Zhu P, Hu S, Hu N, Ma S,
Zhang Y, Han T, Ren J, Cao F, et al: Melatonin suppresses platelet
activation and function against cardiac ischemia/reperfusion injury
via PPARgamma/FUNDC1/mitophagy pathways. J Pineal Res.
63:e124382017. View Article : Google Scholar
|
|
22
|
Reiter RJ, Mayo JC, Tan DX, Sainz RM,
Alatorre-Jimenez M and Qin L: Melatonin as an antioxidant: Under
promises but over delivers. J Pineal Res. 61:253–278. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fuhrmann DC and Brüne B: Mitochondrial
composition and function under the control of hypoxia. Redox Biol.
12:208–215. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dong X, Fu J, Yin X, Qu C, Yang C, He H
and Ni J: Induction of apoptosis in HepaRG cell line by aloe-emodin
through generation of reactive oxygen species and the mitochondrial
pathway. Cell Physiol Biochem. 42:685–696. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Brasacchio D, Alsop AE, Noori T, Lufti M,
Iyer S, Simpson KJ, Bird PI, Kluck RM, Johnstone RW and Trapani JA:
Epigenetic control of mitochondrial cell death through
PACS1-mediated regulation of BAX/BAK oligomerization. Cell Death
Differ. 24:961–970. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gao Y, Xiao X, Zhang C, Yu W, Guo W, Zhang
Z, Li Z, Feng X, Hao J, Zhang K, et al: Melatonin synergizes the
chemotherapeutic effect of 5-fluorouracil in colon cancer by
suppressing PI3K/AKT and NF-kappaB/iNOS signaling pathways. J
Pineal Res. 62:e123802018. View Article : Google Scholar
|
|
27
|
Dufour F, Rattier T, Shirley S, Picarda G,
Constantinescu AA, Morlé A, Zakaria AB, Marcion G, Causse S,
Szegezdi E, et al: N-glycosylation of mouse TRAIL-R and human
TRAIL-R1 enhances TRAIL-induced death. Cell Death Differ.
24:500–510. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Griffiths HR, Gao D and Pararasa C: Redox
regulation in metabolic programming and inflammation. Redox Biol.
12:50–57. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Iggena D, Winter Y and Steiner B:
Melatonin restores hippocampal neural precursor cell proliferation
and prevents cognitive deficits induced by jet lag simulation in
adult mice. J Pineal Res. 62:e123972017. View Article : Google Scholar
|
|
30
|
Hambright WS, Fonseca RS, Chen L, Na R and
Ran Q: Ablation of ferroptosis regulator glutathione peroxidase 4
in forebrain neurons promotes cognitive impairment and
neurodegeneration. Redox Biol. 12:8–17. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Murphy PS, Wang J, Bhagwat SP, Munger JC,
Janssen WJ, Wright TW and Elliott MR: CD73 regulates
anti-inflammatory signaling between apoptotic cells and
endotoxin-conditioned tissue macrophages. Cell Death Differ.
24:559–570. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhou H, Wang J, Zhu P, Hu S and Ren J:
Ripk3 regulates cardiac microvascular reperfusion injury: The role
of IP3R-dependent calcium overload, XO-mediated oxidative stress
and F-action/filopodia-based cellular migration. Cell Signal.
45:12–22. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li J, Chen L, Xiong Y, Zheng X, Xie Q,
Zhou Q, Shi L, Wu C, Jiang J and Wang H: Knockdown of PD-L1 in
human gastric cancer cells inhibits tumor progression and improves
the cytotoxic sensitivity to CIK therapy. Cell Physiol Biochem.
41:907–920. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lee K and Back K: Overexpression of rice
serotonin N-acetyltransferase 1 in transgenic rice plants confers
resistance to cadmium and senescence and increases grain yield. J
Pineal Res. 62:e123922017. View Article : Google Scholar
|
|
35
|
Jokinen R, Pirnes-Karhu S, Pietiläinen KH
and Pirinen E: Adipose tissue NAD+-homeostasis, sirtuins
and poly(ADP-ribose) polymerases-important players in mitochondrial
metabolism and metabolic health. Redox Biol. 12:246–263. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang HH, Chen Y, Gao CY, Cui ZT and Yao
JM: Protective Effects of microRNA-126 on human cardiac
microvascular endothelial cells against
hypoxia/reoxygenation-induced injury and inflammatory response by
activating PI3K/Akt/eNOS signaling pathway. Cell Physiol Biochem.
42:506–518. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu Z, Gan L, Xu Y, Luo D, Ren Q, Wu S and
Sun C: Melatonin alleviates inflammasome-induced pyroptosis through
inhibiting NF-kappaB/GSDMD signal in mice adipose tissue. J Pineal
Res. 63:e124142017. View Article : Google Scholar
|
|
38
|
Lee HJ, Jung YH, Choi GE, Ko SH, Lee SJ,
Lee SH and Han HJ: BNIP3 induction by hypoxia stimulates
FASN-dependent free fatty acid production enhancing therapeutic
potential of umbilical cord blood-derived human mesenchymal stem
cells. Redox Biol. 13:426–443. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yu S, Wang X, Geng P, Tang X, Xiang L, Lu
X, Li J, Ruan Z, Chen J, Xie G, et al: Melatonin regulates PARP1 to
control the senescence- associated secretory phenotype (SASP) in
human fetal lung fibroblast cells. J Pineal Res. 63:e124052017.
View Article : Google Scholar
|
|
40
|
Zhang Y, Zhou H, Wu W, Shi C, Hu S, Yin T,
Ma Q, Han T, Zhang Y, Tian F, et al: Liraglutide protects cardiac
microvascular endothelial cells against hypoxia/reoxygenation
injury through the suppression of the SR-Ca(2+)-XO-ROS axis via
activation of the GLP-1R/PI3K/Akt/survivin pathways. Free Radic
Biol Med. 95:278–292. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou H, Yang J, Xin T, Li D, Guo J, Hu S,
Zhou S, Zhang T, Zhang Y, Han T, et al: Exendin-4 protects
adipose-derived mesenchymal stem cells from apoptosis induced by
hydrogen peroxide through the PI3K/Akt-Sfrp2 pathways. Free Radic
Biol Med. 77:363–375. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhou H, Hu S, Jin Q, Shi C, Zhang Y, Zhu
P, Ma Q, Tian F and Chen Y: Mff-dependent mitochondrial fission
contributes to the pathogenesis of cardiac microvasculature
ischemia/reperfusion injury via induction of mROS-mediated
cardiolipin oxidation and HK2/VDAC1 disassociation-involved mPTP
opening. J Am Heart Assoc. 6:e0053282017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhou H, Yang J, Xin T, Zhang T, Hu S, Zhou
S, Chen G and Chen Y: Exendin-4 enhances the migration of
adipose-derived stem cells to neonatal rat ventricular
cardiomyocyte-derived conditioned medium via the phosphoinositide
3-kinase/Akt-stromal cell-derived factor-1α/CXC chemokine receptor
4 pathway. Mol Med Rep. 11:4063–4072. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Richard V, Kindt N and Saussez S:
Macrophage migration inhibitory factor involvement in breast cancer
(Review). Int J Oncol. 47:1627–1633. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ma C, Zhang C, Ma M, Zhang L, Zhang L,
Zhang F, Chen Y, Cao F, Li M, Wang G, et al: MiR-125a regulates
mitochondrial homeostasis through targeting mitofusin 1 to control
hypoxic pulmonary vascular remodeling. J Mol Med (Berl).
95:977–993. 2017. View Article : Google Scholar
|
|
46
|
Van Nostrand JL, Bowen ME, Vogel H, Barna
M and Attardi LD: The p53 family members have distinct roles during
mammalian embryonic development. Cell Death Differ. 24:575–579.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhao JL, Huang F, He F, Gao CC, Liang SQ,
Ma PF, Dong GY, Han H and Qin HY: Forced activation of notch in
macrophages represses tumor growth by upregulating miR-125a and
disabling tumor-associated macrophages. Cancer Res. 76:1403–1415.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fang R, Xiao T, Fang Z, Sun Y, Li F, Gao
Y, Feng Y, Li L, Wang Y, Liu X, et al: MicroRNA-143 (miR-143)
regulates cancer glycolysis via targeting hexokinase 2 gene. J Biol
Chem. 287:23227–23235. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Nishida N, Mimori K, Fabbri M, Yokobori T,
Sudo T, Tanaka F, Shibata K, Ishii H, Doki Y and Mori M:
MicroRNA-125a-5p is an independent prognostic factor in gastric
cancer and inhibits the proliferation of human gastric cancer cells
in combination with trastuzumab. Clin Cancer Res. 17:2725–2733.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kim JK, Noh JH, Jung KH, Eun JW, Bae HJ,
Kim MG, Chang YG, Shen Q, Park WS, Lee JY, et al: Sirtuin7
oncogenic potential in human hepatocellular carcinoma and its
regulation by the tumor suppressors miR-125a-5p and miR-125b.
Hepatology. 57:1055–1067. 2013. View Article : Google Scholar
|
|
51
|
Du K, Ramachandran A and Jaeschke H:
Oxidative stress during acetaminophen hepatotoxicity: Sources,
pathophysiological role and therapeutic potential. Redox Biol.
10:148–156. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kakimoto PA and Kowaltowski AJ: Effects of
high fat diets on rodent liver bioenergetics and oxidative
imbalance. Redox Biol. 8:216–225. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Xu S, Pi H, Zhang L, Zhang N, Li Y, Zhang
H, Tang J, Li H, Feng M, Deng P, et al: Melatonin prevents abnormal
mitochondrial dynamics resulting from the neurotoxicity of cadmium
by blocking calcium-dependent translocation of Drp1 to the
mitochondria. J Pineal Res. 60:291–302. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhou H, Zhang Y, Hu S, Shi C, Zhu P, Ma Q,
Jin Q, Cao F, Tian F and Chen Y: Melatonin protects cardiac
microvasculature against ischemia/reperfusion injury via
suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis.
J Pineal Res. 63:e124132017. View Article : Google Scholar
|
|
55
|
Perdiz D, Lorin S, Leroy-Gori I and Poüs
C: Stress-induced hyperacetylation of microtubule enhances
mitochondrial fission and modulates the phosphorylation of Drp1
at616Ser. Cell Signal. 39:32–43. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Jin F, Wang Y, Zhu Y, Li S, Liu Y, Chen C,
Wang X, Zen K and Li L: The miR-125a/HK2 axis regulates cancer cell
energy metabolism reprogramming in hepatocellular carcinoma. Sci
Rep. 7:30892017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Tallman KA, Kim HH, Korade Z,
Genaro-Mattos TC, Wages PA, Liu W and Porter NA: Probes for protein
adduction in cholesterol biosynthesis disorders: Alkynyl lanosterol
as a viable sterol precursor. Redox Biol. 12:182–190. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhou H, Wang S, Zhu P, Hu S, Chen Y and
Ren J: Empagliflozin rescues diabetic myocardial microvascular
injury via AMPK- mediated inhibition of mitochondrial fission.
Redox Biol. 15:335–346. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Oanh NT, Park YY and Cho H: Mitochondria
elongation is mediated through SIRT1-mediated MFN1 stabilization.
Cell Signal. 38:67–75. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Torres-Quesada O, Mayrhofer JE and Stefan
E: The many faces of compartmentalized PKA signalosomes. Cell
Signal. 37:1–11. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Schock SN, Chandra NV, Sun Y, Irie T,
Kitagawa Y, Gotoh B, Coscoy L and Winoto A: Induction of
necroptotic cell death by viral activation of the RIG-I or STING
pathway. Cell Death Differ. 24:615–625. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Salminen A, Kaarniranta K and Kauppinen A:
Integrated stress response stimulates FGF21 expression: Systemic
enhancer of longevity. Cell Signal. 40:10–21. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Park J, Tran Q, Mun K, Masuda K, Kwon SH,
Kim SH, Kim DH, Thomas G and Park J: Involvement of S6K1 in
mitochondria function and structure in HeLa cells. Cell Signal.
28:1904–1915. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Karbowski M, Lee YJ, Gaume B, Jeong SY,
Frank S, Nechushtan A, Santel A, Fuller M, Smith CL and Youle RJ:
Spatial and temporal association of Bax with mitochondrial fission
sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 159:931–938.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Song M, Franco A, Fleischer JA, Zhang L
and Dorn GW II: Abrogating mitochondrial dynamics in mouse hearts
accelerates mitochondrial senescence. Cell Metab. 26:872–883.e5.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Chen Y and Dorn GW II:
PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling
damaged mitochondria. Science. 340:471–475. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Banerjee K, Keasey MP, Razskazovskiy V,
Visavadiya NP, Jia C and Hagg T: Reduced FAK-STAT3 signaling
contributes to ER stress-induced mitochondrial dysfunction and
death in endothelial cells. Cell Signal. 36:154–162. 2017.
View Article : Google Scholar : PubMed/NCBI
|