Introduction
Colorectal cancer (CRC) is the third most prevalent
type of carcinoma and the second leading cause of cancer-associated
mortality in western countries (1). At present, >1 million people are
newly diagnosed with CRC every year and ~50% of patients with CRC
develop metastatic diseases. Although research has been conducted
on the pathogenesis of CRC, the molecular mechanisms underlying CRC
remain largely unknown (2).
Combinations of anticancer agents form the basis for treatment of
CRC and have improved the overall survival outcome; however,
despite the availability of novel classes of drugs, advanced
inoperable CRC remains incurable. Therefore, further research is
required to identify novel therapeutic agents and develop more
effective combination strategies for CRC treatment.
Histone deacetylase (HDAC)6 is a unique class IIb
HDAC with two deacetylase domains and a C-terminal zinc-finger
domain (3,4). HDAC6 is mainly localized in the
cytosol, where it performs diverse functions through deacetylation
of numerous targets, including α-tubulin, heat shock protein 90 and
cortactin (5). HDAC6 acts as a
master regulator of cellular protective responses, including
misfolded protein-induced aggresomes, autophagy and stress granules
(6–8). Selective inhibition of HDAC6 exerts
antitumor effects by inducing differentiation, cell cycle arrest,
apoptosis, responsiveness to chemotherapy, and inhibition of
migration and angiogenesis (5).
Recently, research focusing on the development of a HDAC6-selective
inhibitor has increased, since among the HDACs only HDAC6
inhibition does not cause major side effects (9). Therefore, HDAC6 is considered a
promising target in drug development for cancer therapy.
Numerous HDAC6-selective inhibitors have been
reported and tested as anticancer drugs (10–14).
Tubacin and tubastatin A have been reported to be the most
effective HDAC6-selective inhibitors (15,16);
however, both have non-druggable qualities, including non-drug-like
structure, high lipophilicity and complex synthesis, and are
therefore used primarily as research tools (17). ACY-1215, also known as
ricolinostat, is the only first-in-class clinically relevant HDAC6
inhibitor (13). Ricolinostat is
currently being studied in clinical trials (18), both as a monotherapy, and in
combination with dexamethasone and proteasome inhibitors
(bortezomib) (19), or
immunomodulatory drugs [lenalidomide (20) and pomalidomide] in multiple myeloma
(MM). Ricolinostat has also been evaluated in a clinical trial as a
monotherapy in relapsed/refractory (R/R) lymphoid malignancies
(NCT02091063) (21).
Despite these advances, the antitumor effects of
ACY-1215 on solid tumors remain to be evaluated, and its clinical
efficacy in solid tumors remains elusive. Therefore, the present
study aimed to determine whether similar interactions occurred with
the HDAC6-selective inhibitor ACY-1215 in solid tumors as in
hematological cancers. The present study investigated the
preclinical anticancer activity of ACY-1215 in CRC, either alone or
in combination with chemotherapeutic agents, including
5-fluorouracil (5-FU), oxaliplatin and irinotecan. The results
indicated that ACY-1215 may interact synergistically with
oxaliplatin in CRC, resulting in the induction of apoptosis, and
inhibition of numerous signaling pathways and antitumor immune
responses. In addition, ACY-1215 exhibited slight synergism with
irinotecan. Collectively, these findings suggested that targeting
HDAC6 activity using ACY-1215 may present a promising therapeutic
opportunity that improves the sensitivity of solid tumors to
conventional therapy.
Materials and methods
Cell culture and drug treatment
The human HCT116 and HT29 CRC cell lines were
purchased from American Type Culture Collection (Manassas, VA, USA)
and were cultured in Dulbecco's modified Eagle's medium (HCT116)
and RPMI-1640 medium (HT29) (HyClone; GE Healthcare, Logan, UT,
USA) containing 10% fetal bovine serum (HyClone; GE Healthcare),
100 U/ml penicillin and 100 µg/ml streptomycin (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) in a humidified
atmosphere containing 5% CO2 and 95% air at 37°C. Each
cell line was treated with dimethyl sulfoxide (DMSO),
suberoylanilide hydroxamic acid (SAHA; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) (5 µM), ACY-1215 (ricolinostat) (0.01,
0.05, 0.1, 0.2, 0.5, 1, 2, 5, 10 and 20 µM), 5-FU (1, 5 and
10 µM), oxaliplatin (2, 10 and 20 µM) or irinotecan
(1, 2, 5 and 10 µM) (all from Selleck Chemicals Houston, TX,
USA).
Cell growth and viability assay
Cell growth and viability were assessed by measuring
the dye absorbance of the water-soluble tetrazolium salt, WST-8
[Cell Counting kit (CCK)-8 kit, Dojindo Molecular Technologies,
Inc., Kumamoto, Japan], according to the manufacturer's protocol.
Cells were seeded in triplicate at a density of
0.5–1×104 cells in 200 µl medium in 96-well
plates. The drugs were added to the cells at the indicated
concentrations 24 h after seeding at 37°C for 72 h. Cells in each
well were pulsed with 20 µl WST-8 for the final 3 h of a
72-h incubation and absorbance was then measured at 450 nm using a
multimode microplate reader (Tecan Group, Ltd., Mannedorf,
Switzerland). For cell growth analysis, cell counts were indirectly
estimated from absorbance measurements, relative to a standard
curve generated using solutions of known cell counts
(0.5×104, 1×104, 2×104,
3×104, 4×104, 5×104,
6×104, 7×104 and 8×104
cells/well). Absorbance was normalized to that of the negative
control (no DMSO vehicle) at each time interval. To analyze cell
viability, the absorbance percentage was calculated relative to
negative control cultures. Results are presented from three
independent experiments performed in triplicate.
Inhibitory assays
The present study determined the drug concentrations
that inhibited 50% of cell growth (GI50) and 50% of cell
viability (IC50) using a CCK-8 assay. All cell lines
were treated for 72 h on day 2, unless otherwise stated.
GI50 and IC50 were determined using Prism
version 6.0 software (GraphPad Software, Inc., La Jolla, CA,
USA).
Acid extraction of histones
Acid extraction of histones was prepared from HCT116
(2×106) and HT29 (2×106) cells, as described
previously (8). Cells were washed
with PBS and suspended in 10 ml PBS, followed by centrifugation at
200 × g for 10 min at 4°C. Cells were then resuspended in 1 ml
hypotonic lysis buffer [10 mM Tris-Cl (pH 8.0), 1.5 mM
MgCl2, 1 mM KCl, 1 mM DTT and 1 mM phenylmethylsulfonyl
fluoride] and 0.4 N H2SO4 at a final
concentration of 0.2 M, and were lysed on ice for 30 min. After
centrifugation at 16,000 × g for 10 min at 4°C, the supernatant
fraction that contained the acid-soluble proteins was retained.
Trichloroacetic acid (132 µl) was added to the supernatant
and the samples were incubated on ice overnight. The proteins were
then pelleted by centrifugation at 16,000 × g for 10 min at 4°C,
and were washed four times with ice-cold acetone under
centrifugation at 16,000 × g for 5 min at 4°C. Finally, the histone
pellets were air-dried for 20 min at room temperature and were
dissolved in an appropriate volume of ddH2O. The
subsequent histone solution underwent western blotting.
Apoptosis assay
An apoptosis assay was performed using Annexin
V/propidium iodide (PI) double staining, according to the
manufacturer's protocol (BD Biosciences, Franklin Lakes, NJ, USA).
After treatment, the cells were trypsinized and stained with 0.5
mg/ml Annexin V in binding buffer (10 mM HEPES free acid, 0.14 M
NaCl and 2.5 mM CaCl2) for 30 min at room temperature.
Subsequently, PI (5 mg/ml final concentration) was added, and the
cells were incubated for a further 15 min at room temperature.
Cells were then analyzed using a flow cytometer and BD FACSDiva
software version 7 (both BD Biosciences).
Western blot analysis
Treated cells were collected and lysed with NP-40
lysis buffer [0.5% NP-40, 50 mM Tris-HCl (pH 7.4), 120 mM NaCl, 25
mM NaF, 25 mM glycerol phosphate, 1 mM EDTA, 5 mM EGTA, 1 mM PMSF
and 1 mM bezamidine]. Protein concentration was measured using a
bicinchoninic acid kit (Pierce; Thermo Fisher Scientific, Inc.).
Cell lysates containing 50–80 µg total protein were
subjected to 8–15% SDS-PAGE. Western blotting was performed as
described previously (8). The
membranes were blocked for 1 h in PBS containing 0.1% Tween-20 and
10% (v/v) skim milk (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
and incubated overnight at 4°C with primary antibodies
(1:500–1:2,000). The following primary antibodies were used.
Acetylated-α-tubulin (clone number, DM1A; T6793-2ML; 1:2,000) was
purchased from Sigma-Aldrich (Merck KGaA). Protein kinase B (AKT;
H-136, sc-8312; 1:1,000), phosphorylated (p)-AKT (Ser473, sc-7985;
1:500), B-cell lymphoma (Bcl)-2 homologous antagonist/killer (Bak;
G-23, sc-832; 1:1,000), extracellular signal-regulated kinase (ERK;
K-23, sc-94; 1:1,000), HDAC6 (H-300, sc-11420; 1:1,000) and
α-tubulin (sc-32293; 1:2,000) were obtained from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). Anti-poly (ADP-ribose)
polymerase (PARP; 551024; 1:1,000) was purchased from BD
Biosciences. Bcl-extra large protein (Bcl-xL; 2762; 1:500),
caspase-3 (9662S; 1:1,000), p-ERK (Thr202/Tyr204; 4376; 1:1,000),
signal transducer and activator of transcription 3 (STAT3; 12640;
1:1,000), and p-STAT3 (Y705, 9138; 1:500) were obtained from Cell
Signaling Technology, Inc. (Danvers, MA, USA). Acetylated-histone
H3 (06-599, 1:1,000) and histone H3 (06-755; 1:500) antibodies were
obtained from EMD Millipore (Billerica, MA, USA). Anti-programmed
death-ligand 1 (PD-L1) antibody (PA5-28115; 1:1,000) was purchased
from Invitrogen; Thermo Fisher Scientific, Inc. The membranes were
then washed with 0.1% Tween-20/PBS and incubated at room
temperature for 1 h with horseradish peroxidase-conjugated
anti-rabbit (111-035-003; 1:5,000) and anti-mouse (115-035-003;
1:10,000) secondary antibodies (Jackson ImmunoResearch
Laboratories, Inc., West Grove, PA, USA). The bound antibodies were
detected using an enhanced chemiluminescence western blot analysis
system (NCI4080KR, Thermo Fisher Scientific, Inc.) and the blots
were semi-quantified using FusionCapt software version 16.08a
(Viber Lourmat Sté, Collégien, France).
Drug combination analysis
For combined drug analysis, a constant ratio of
ACY-1215 and anticancer agents was evaluated. Drug dilutions and
combinations were prepared in media immediately prior to use. Cells
(3×103/well) in 96-well plates were incubated with the
drugs for 72 h at 37°C. A CCK-8 assay was performed to determine
cell viability. Drug interactions were determined according to the
combination index (CI) method described by Chou (22); CI>1 implies antagonism, CI=1 is
additive and CI<1 implies synergism. CIs for the combination
treatment groups were generated using CalcuSyn software version
2.11 (Biosoft, Cambridge, UK). The fraction affected (FA) was
calculated from the percent viability, as follows: FA = (100 −
percentage viability) / 100.
Statistical analysis
Statistical analyses were conducted using GraphPad
Prism software version 5.01 (GraphPad Software, Inc.). All data are
presented as the means ± standard deviation of more than three
independent experiments. Statistical differences were determined by
Student's t-test or one-way analysis of variance with post hoc
analysis using Tukey's multiple comparison test. P<0.05 was
considered to indicate a statistically significant difference.
Results
ACY-1215 selectively inhibits HDAC6 in
CRC cells
To the best of our knowledge, ACY-1215 is the first
oral selective HDAC6 inhibitor to be studied clinically for
hematological cancers (13). To
confirm the specific inhibitory effects of ACY-1215 on HDAC6
activity in solid tumors, the present study evaluated its effects
on the acetylation of α-tubulin (a substrate of HDAC6) and histone
H3 (a substrate of class I HDACs). Human CRC HCT116 and HT29 cells
were cultured with an increasing dose of ACY-1215 for 24 h. In
HCT116 cells, a dose-dependent increase in acetylated-α-tubulin was
observed following treatment with low doses (0.01 µM) of
ACY-1215, without affecting acetylation of histone (<1
µM), confirming a greater selective inhibitory effect on
HDAC6 activity (Fig. 1A). Higher
acetylation selectivity for α-tubulin was observed in HCT116 cells
(0.01 µM) compared with HT29 cells (0.2 µM) (Fig. 1B), indicating that ACY-1215 may
exert differential HDAC6-inhibitory effects in different CRC cells.
To determine the selectivity of ACY-1215 for HDAC6 over class I
HDACs, we compared the concentration required to increase
acetylated levels of α-tubulin compared with that required to
increase acetylated levels of histone H3. Levels of
acetylated-α-tubulin and acetylated-H3 were semi-quantified
relative to α-tubulin and histone H3, respectively, and a
>2-fold increase in acetylated levels was considered to indicate
a significant inhibitory effect. Based on western blotting,
ACY-1215 was ~5-fold (HDAC6 inhibitory concentration, 0.2
µM; vs. class I HDAC inhibitory concentration, 1 µM)
and 100-fold (0.01 vs. 1 µM) less active against class I
HDACs in HT29 and HCT116 cells, respectively (Fig. 1A and B). The pan-HDAC inhibitor,
SAHA, was used as a positive control for HDAC inhibition (23). These results indicated that
ACY-1215 may be a selective HDAC6 inhibitor with minimal class I
HDAC activity in CRC cells.
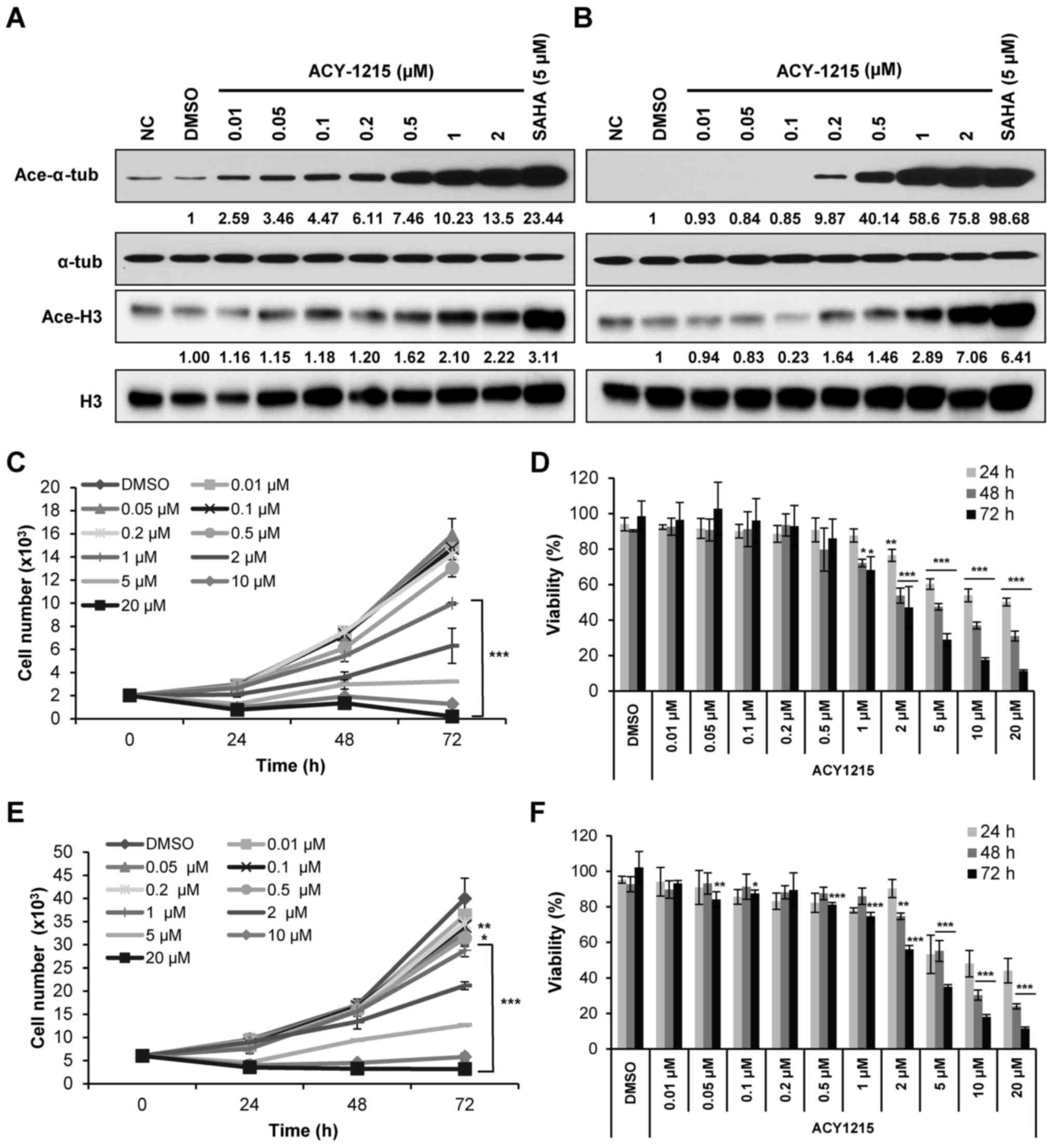 | Figure 1ACY-1215, an HDAC6 inhibitor with
minimal effects on class I HDACs, suppresses the growth and
viability of CRC cells. (A) HCT116 and (B) HT29 cells were treated
with ACY-1215 at the indicated concentrations for 24 h, and western
blot analysis was performed using antibodies against Ace-α-tub,
Ace-H3, α-tub and total H3. Levels of Ace-α-tub and Ace-H3 were
semi-quantified relative to α-tub and H3, respectively, and the
levels in the 0.1% DMSO-treated groups were set at 1. α-tub and
histone H3 were used as equal loading controls. The pan-HDAC
inhibitor, SAHA, was used as a positive control for HDAC
inhibition. Cell growth and viability of (C and E) HCT116 and (D
and F) HT29 cells cultured with 0.1% DMSO (control) or ACY-1215 at
the indicated concentrations (0.01, 0.05, 0.1, 0.2, 0.5, 1, 2, 5,
10 and 20 µM) for 72 h. Viable cell numbers and viability
were measured using CCK-8 assays. Cell counts were estimated
indirectly from a standard curve generated using solutions of known
cell counts. Absorbance was normalized to that of the negative
control at each time interval. Data are expressed as the means ±
standard deviation from three independent experiments.
*P<0.05, **P<0.01 and
***P<0.001 vs. the DMSO control or as indicated;
analysis of variance test. Ace-, acetylated; α-tub, α-tubulin;
CCK-8, Cell Counting kit-8; CRC, colorectal cancer; DMSO, dimethyl
sulfoxide; H3, histone H3; NC, negative control; SAHA,
suberoylanilide hydroxamic acid. |
ACY-1215 induces time- and dose-dependent
cytotoxicity in CRC cells
The present study examined the effects of ACY-1215
on cell growth and viability in HCT116 and HT29 CRC cells. Cells
were cultured with ACY-1215 for up to 72 h, and cell growth and
viability were assessed using CCK-8 assays. Compared with the
control cells, ACY-1215 treatment resulted in a time- and
dose-dependent decrease in the growth and viability of CRC cells,
with GI50 values between 1.37 and 5.67 µM, and
IC50 values between 1.16 and 5.05 µM (Fig. 1C–F and Table I). Taken together, these findings
suggested that selective inhibition of HDAC6 by ACY-1215 may result
in inhibition of the growth and viability of CRC cells.
 | Table IInhibitory effects of ACY-1215 on the
growth and viability of CRC cell lines. |
Table I
Inhibitory effects of ACY-1215 on the
growth and viability of CRC cell lines.
| Cell line | Time (h) | ACY-1215
(µM)
|
|---|
|
GI50 |
IC50 |
|---|
| HCT116 | 48 | 1.386 | 1.441 |
| 72 | 1.368 | 1.164 |
| HT29 | 48 | 5.668 | 5.051 |
| 72 | 3.804 | 3.683 |
Combined treatment of ACY-1215 with an
anticancer agent efficiently induces apoptosis of CRC cells
Based on prior evidence confirming the efficacy of
ACY-1215 and proteasome inhibitors or immunomodulatory drugs for
the treatment of hematopoietic malignancies (13,24–27),
the present study aimed to identify effective therapeutic
combinations of ACY-1215 with conventional therapeutic drugs in CRC
cells. CRC cells were treated with either ACY-1215 alone or in
combination with conventional anticancer drugs, including the
topoisomerase I inhibitor irinotecan, the DNA synthesis inhibitor
5-FU, and the DNA-damaging agent oxaliplatin. Cells then were
analyzed by western blotting to detect apoptosis. A previous study
demonstrated that in HCT116 and HT29 cells cultured with 2
µM ACY-1215, the expression levels of PARP and caspase-3,
and the Bak to Bcl-xL ratio, remained unchanged (27). In the present study, cells cultured
with a combination of ACY-1215 and oxaliplatin or irinotecan
exhibited a synergistic increase in cleaved PARP and active
caspase-3 expression compared with in the single agent-treated
groups (Fig. 2A and B). The
expression levels of the proapoptotic molecule Bak were additively
increased, whereas the expression levels of the anti-apoptotic
molecule Bcl-xL were additively decreased in both cell lines when
ACY-1215 was combined with either 5-FU, oxaliplatin or irinotecan
compared with the anticancer compound alone (Fig. 2A and B). Furthermore, Annexin V/PI
staining revealed that cell apoptosis was significantly increased
following combination treatment of cells with ACY-1215 and
anticancer drugs (Fig. 2C and D).
Overall, these results suggested that ACY-1215 alone may not
efficiently induce apoptosis in solid tumors; however, combination
treatment of ACY-1215 with anticancer drugs may trigger apoptosis
by activating caspases and downregulating the expression of
anti-apoptotic factors.
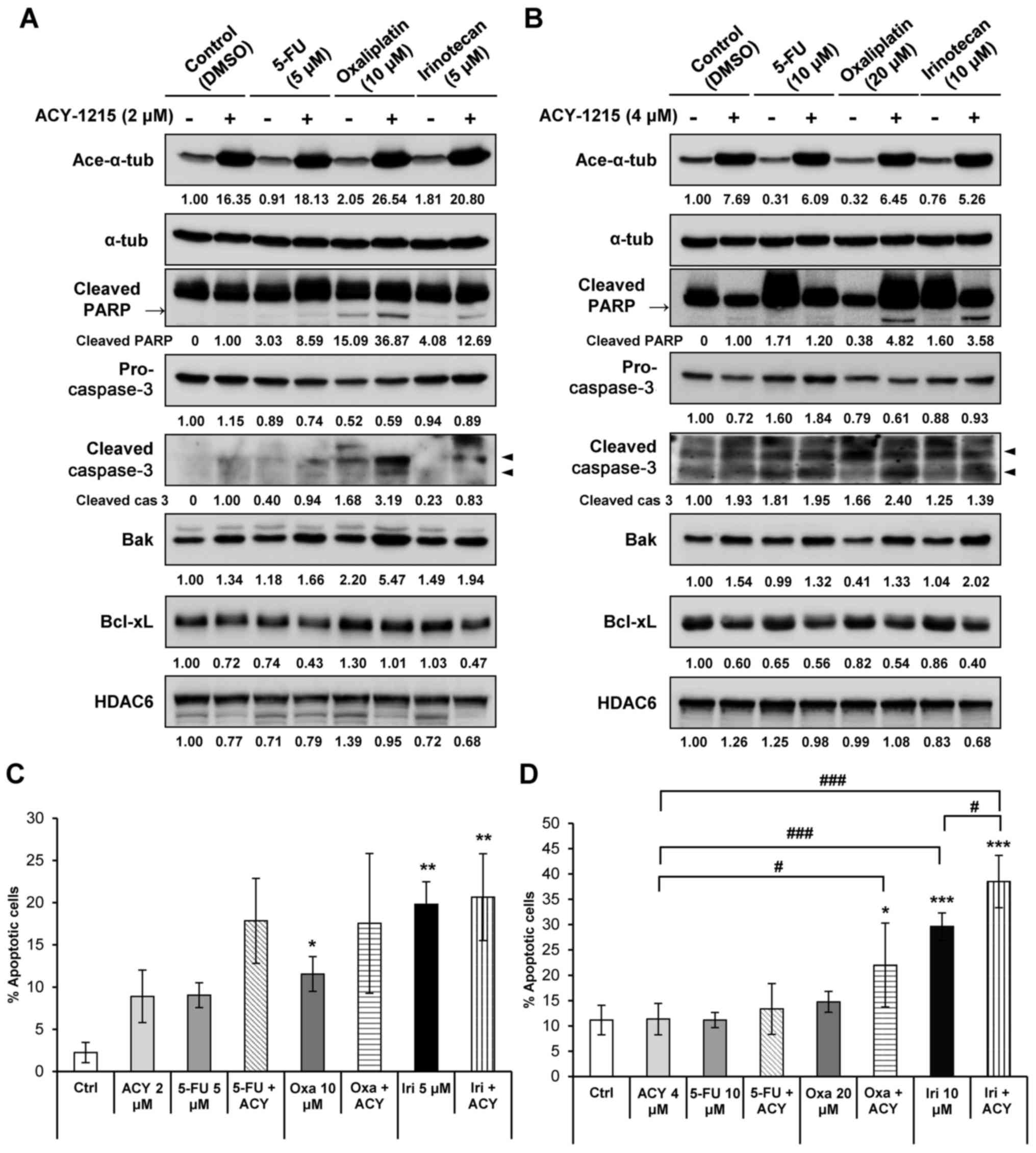 | Figure 2ACY-1215 synergistically increases
apoptosis induced by anticancer agents. (A) HCT116 and (B) HT29
cells were treated with 0.1% DMSO (control), ACY-1215, 5-FU,
oxaliplatin, irinotecan or a combination of these compounds for 24
h. Western blot analysis was performed using the indicated
antibodies. The protein expression levels were semi-quantified
relative to α-tub; the levels in the 0.1% DMSO group or
ACY-1215-treated group (cleaved PARP and cleaved caspase-3) were
set at 1. α-tub was used as a loading control. (C) HCT116 and (D)
HT29 cells were treated with the indicated compounds for 48 h and
stained with Annexin V and propidium iodide for 15 min. Apoptosis
induced by these compounds was assessed by flow cytometry (n=3).
Data are presented as the means ± standard deviation from three
independent experiments. *P<0.05,
**P<0.01 and ***P<0.001 vs. the DMSO
control; #P<0.05, ##P<0.01 and
###P<0.001 vs. single agent-treated group, analysis
of variance test. 5-FU, 5-fluorouracil; α-tub, α-tubulin; Ace-,
acetylated; ACY, ACY-1215; Bak, Bcl-2 homologous antagonist/killer;
Bcl, B-cell lymphoma; Bcl-xL, Bcl-extra large protein; Ctrl,
control; DMSO, dimethyl sulfoxide; HDAC6, histone deacetylase 6;
Iri, irinotecan; Oxa, oxaliplatin; PARP, poly (ADP-ribose)
polymerase. |
ACY-1215 in combination with anticancer
agents exhibits synergistic cytotoxicity
The present study also detected the combined effects
of ACY-1215 and conventional therapeutic drugs on CRC cytotoxicity.
Cells were treated with either ACY-1215 alone or in combination
with conventional anticancer drugs, and CCK-8 assays were performed
to measure cell growth and viability (Fig. 3A–D). Combined treatment resulted in
synergistic growth inhibition in both cell lines (Fig. 3A and C). The combination of
ACY-1215 with 5-FU, oxaliplatin or irinotecan increased cell death
compared with treatment with the anticancer agent alone in HCT116
and HT29 cells (Fig. 3B and D). To
further confirm the synergistic interaction between ACY-1215 and
anticancer drugs, synergism was evaluated according to the Chou and
Talalay method (22). The
combination of ACY-1215 and 5-FU, oxaliplatin or irinotecan
exhibited moderately synergistic anticancer activities with
CI<1.0 in HT29 cells. However, ACY-1215 had no detectable
synergistic effect on cell viability when combined with all three
anticancer agents in HCT116 cells (Fig. 3E and F; Table II). These data confirmed the
significant potentiation of anti-proliferative effects when two
agents were combined in HT29 cells, but not in HCT116 cells.
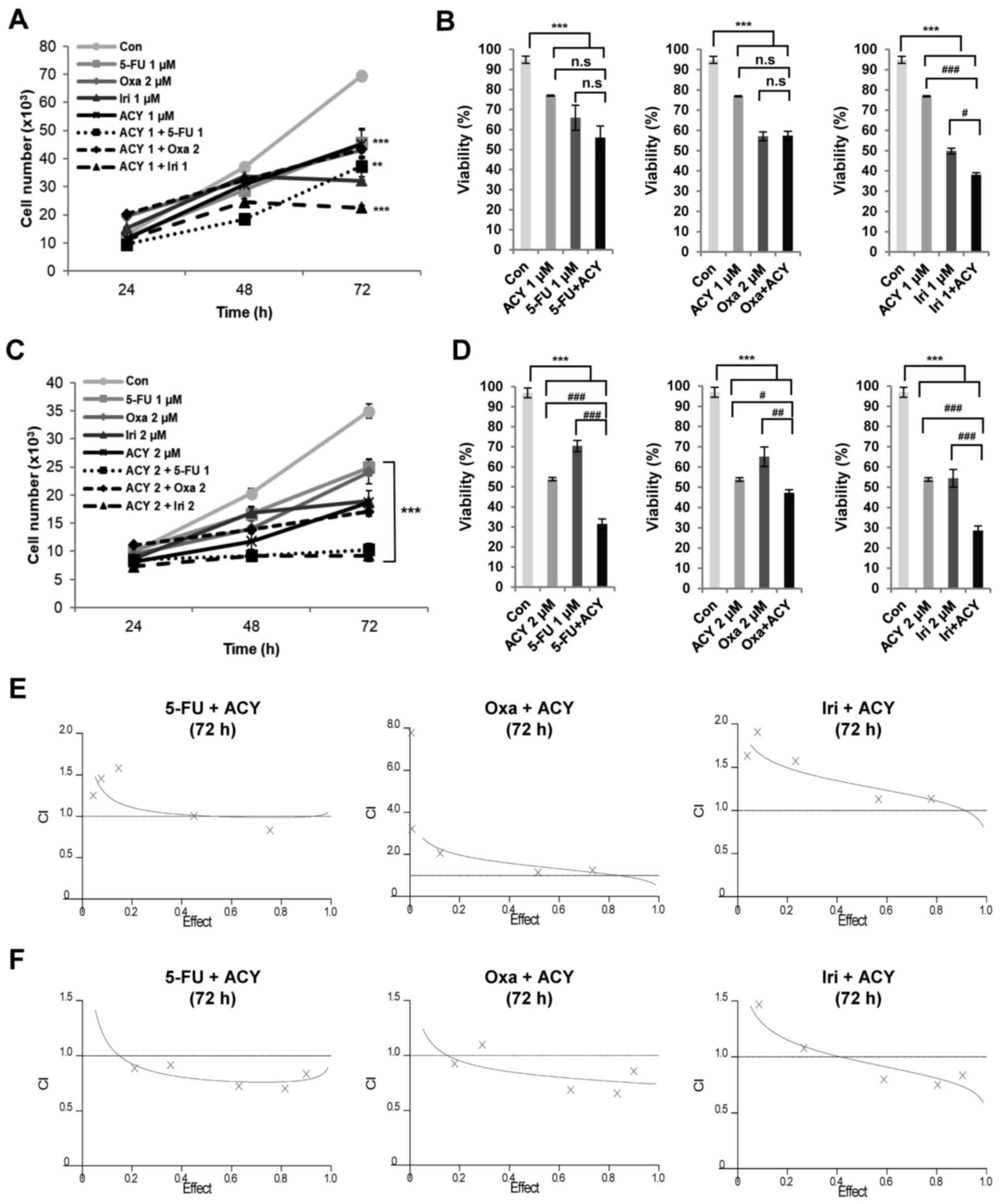 | Figure 3Combined treatment of ACY-1215 and
anticancer agents triggers synergistic cytotoxicity. (A and B)
HCT116 and (C and D) HT29 cells were treated with 0.1% DMSO
(control) or the indicated compounds alone or in combination for 72
h. Cell growth (24–72 h) and viability (72 h) were measured by
CCK-8 assays. Cell counts were indirectly estimated from a standard
curve generated using solutions of known cell counts. The CI and
FA of ACY-1215 and anticancer agents in (E) HCT116 and
(F) HT29 cells. Cells maintaining a constant ratio between the dose
of ACY-1215 and 5-FU, oxaliplatin or irinotecan, and cell viability
were assessed at 72 h using the CCK-8 assay. The CI value and the
relative FA were determined at each dose combination
(actual), and simulations were run to estimate the CI value and
confidence interval across the entire FA range
(simulation). CI<1, CI=1 and CI>1 indicate synergistic,
additive and antagonistic effects, respectively. Data are presented
as the means ± standard deviation from three independent
experiments. *P<0.05, **P<0.01 and
***P<0.001 vs. the DMSO control;
#P<0.05, ##P<0.01 and
###P<0.001 vs. the single agent-treated groups;
analysis of variance test. 5-FU, 5-fluorouracil; ACY, ACY-1215; CI,
combination index; Con, control; FA, fraction affected;
Iri, irinotecan; Oxa, oxaliplatin; n.s, not significant. |
| Table IIMean CI for cells exposed to various
drug combinations. |
Table II
Mean CI for cells exposed to various
drug combinations.
| Cell line | Drug | Mean CI value | CI range | Synergy |
|---|
| HCT116 | ACY + 5-FU | 1.2246 | 0.832 – 1.58 | No synergism |
| ACY + Oxa | 1.4758 | 1.133 – 1.908 | No synergism |
| ACY + Iri | 3.0902 | 1.139 – 7.789 | No synergism |
| HT29 | ACY + 5-FU | 0.812 | 0.701 – 0.915 | Moderate
synergism |
| ACY + Oxa | 0.8454 | 0.654 – 1.099 | Moderate
synergism |
| ACY + Iri | 0.9862 | 0.751 – 1.469 | Moderate
synergism |
Inhibition of AKT and ERK contributes to
ACY-1215-induced apoptosis
ACY-1215 treatment has previously been reported to
slightly decrease phosphorylation of AKT and ERK1/2, without
altering total AKT and ERK1/2 expression, in CRC cells (27). To assess synergism at the molecular
level, the present study evaluated the expression levels of
mitogen-activated protein kinases and AKT signaling pathways in CRC
cells (Fig. 4). Consistent with
increased apoptosis (Fig. 2),
combination treatment with ACY-1215 and 5-FU, oxaliplatin or
irinotecan synergistically decreased phosphorylation of ERK1/2 in
HCT116 and HT29 cells compared with treatment with the single
agents (Fig. 4A, B and D).
Combined treatment with ACY-1215 and oxaliplatin synergistically
decreased phosphorylation of AKT without changes in total AKT in
HT29 cells but not HCT116 cells (Fig.
4A–C). Together, these findings indicated that coadministration
of ACY-1215 and anticancer agents may suppress survival signaling
pathways in CRC cells.
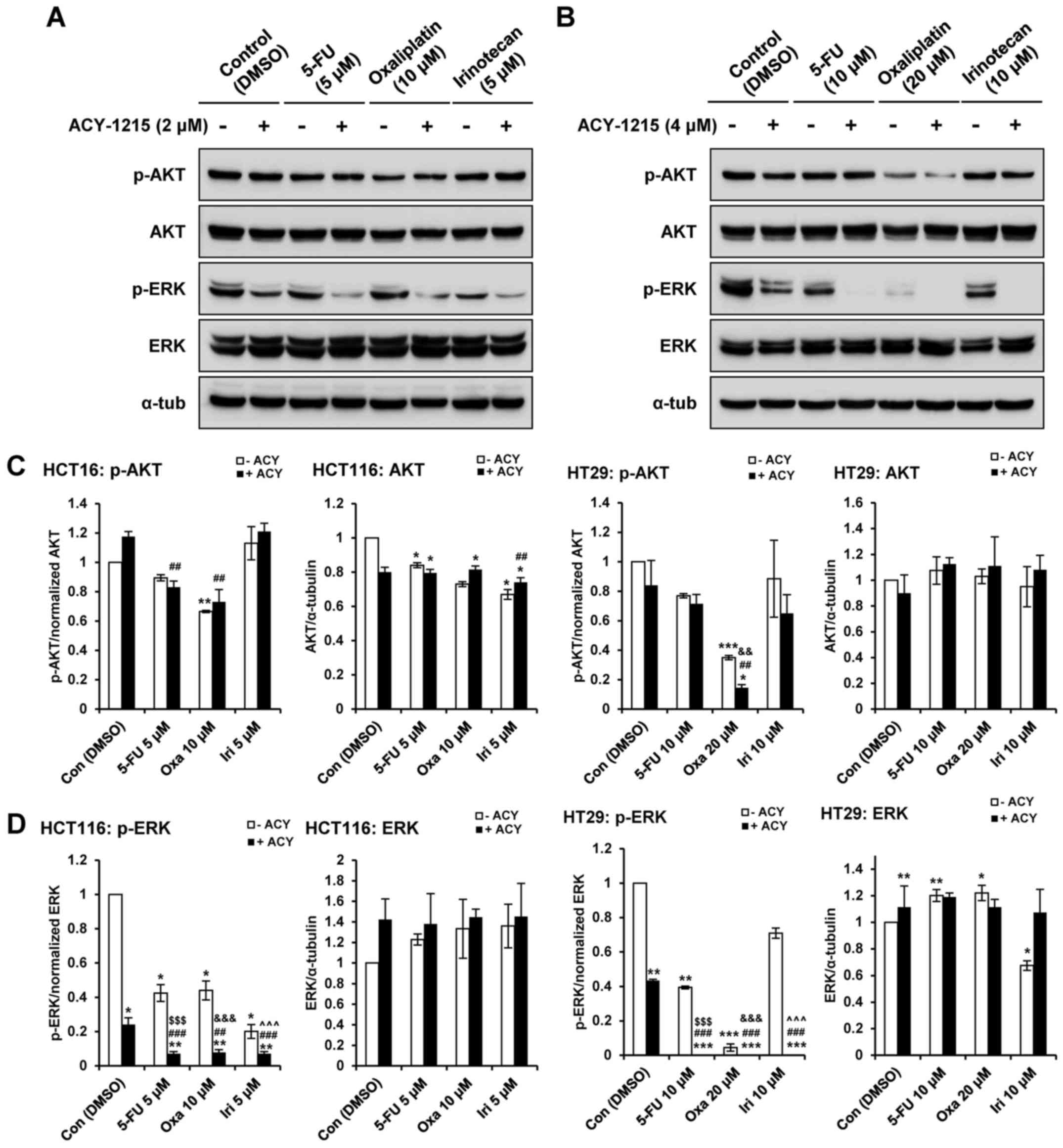 | Figure 4ACY-1215 regulates the AKT and
mitogen-activated protein kinases pathways. (A) HCT116 and (B) HT29
cells were treated with 0.1% DMSO (control), or ACY-1215, 5-FU,
oxaliplatin and irinotecan alone or in combination for 24 h.
Whole-cell lysates were subjected to western blotting with the
indicated antibodies (upper panel). Relative protein expression
levels were semi-quantified by densitometric analysis of the blots.
Expression levels of (C) p-AKT and (D) p-ERK were semi-quantified
relative to AKT and ERK. The abundance of the indicated proteins
was semi-quantified relative to α-tub; the levels in the 0.1% DMSO
group were set at 1. α-tub was used as a loading control.
*P<0.05, **P<0.01 and
***P<0.001 vs. the DMSO control;
##P<0.01 and ###P<0.001 vs. the
ACY-1215-treated group; $$$P<0.001 vs. the
5-FU-treated group; &&P<0.01 and
&&&P<0.001 vs. the oxaliplatin-treated
group; ^^^P<0.001 vs. the irinotecan-treated group;
analysis of variance test. 5-FU, 5-fluorouracil; α-tub, α-tubulin;
ACY, ACY-1215; AKT, protein kinase B; Con, control; DMSO, dimethyl
sulfoxide; ERK, extracellular signal-regulated kinase; Iri,
irinotecan; Oxa, oxaliplatin; p-, phosphorylated. |
ACY-1215 in combination with anticancer
agents upregulates PD-L1 in CRC cells
Our recent report demonstrated that single-agent
treatment with ACY-1215 resulted in increased levels of PD-L1, and
greater phosphorylation of STAT3 in CRC cells (27). Therefore, the present study aimed
to determine whether combined treatment with ACY-1215 and
anticancer agents could modulate the expression of PD-L1 and the
phosphorylation of STAT3 in CRC cells. Combination treatment with
ACY-1215 and 5-FU, oxaliplatin or irinotecan decreased the
phosphorylation of STAT3 in HT29, but not HCT116 cells, compared
with in the single agent-treated groups (Fig.. 5A–C). Combination treatment with
ACY-1215 and oxaliplatin or irinotecan enhanced the expression
levels of PD-L1 in HCT116 compared with in the single agent-treated
groups in a STAT3-independent manner in HCT116 cells (Fig. 5A–D). Treatment with the four single
agents caused an increase in PD-L1 levels irrespective of p-STAT3;
however, combination treatment did not induce synergistic effects
in HT29 cells. These results indicated that HDAC6 may regulate the
expression levels of PD-L1 and serve an important role in
immune-related pathways in CRC cells.
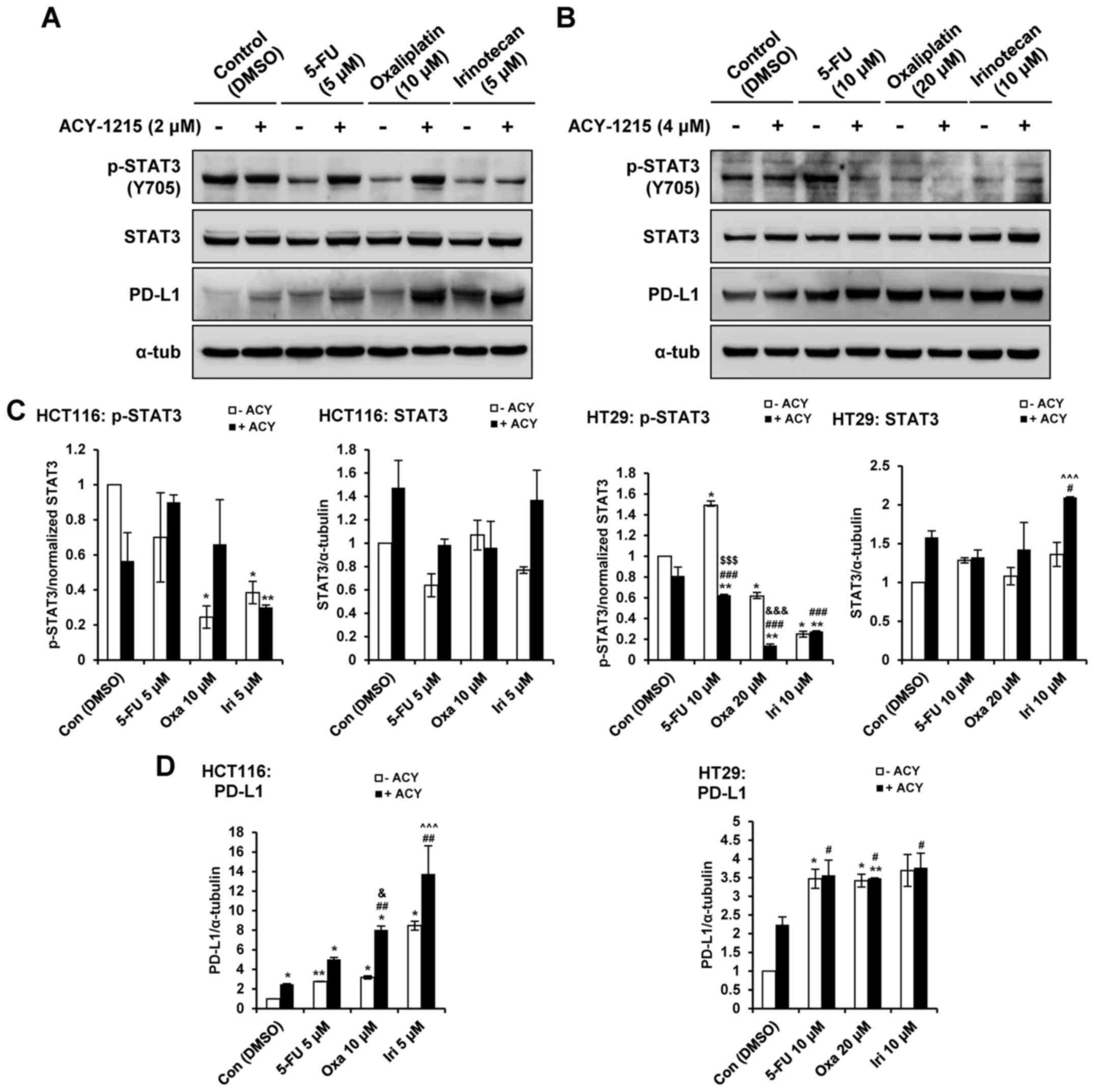 | Figure 5ACY-1215 regulates the STAT and PD-L1
pathway. (A) HCT116 and (B) HT29 cells were treated with 0.1% DMSO
(control), or ACY-1215, 5-FU, oxaliplatin and irinotecan alone or
in combination for 24 h. Whole-cell lysates were subjected to
western blotting with the indicated antibodies. Relative protein
expression levels were semi-quantified by densitometric analysis of
the blots. Expression levels of (C) p-STAT3 and (D) PD-L1 were
semi-quantified relative to STAT3 and α-tub, respectively; the
levels in the 0.1% DMSO group were set at 1. α-tub was used as a
loading control. *P<0.05 and **P<0.01
vs. the DMSO control; #P<0.05, ##P<0.01
and ###P<0.001 vs. the ACY-1215-treated group;
$$$P<0.001 vs. the 5-FU-treated group;
&P<0.05 and &&&P<0.001
vs. the oxaliplatin-treated group; ^^^P<0.001 vs. the
irinotecan-treated group; analysis of variance test. 5-FU,
5-fluorouracil; α-tub, α-tubulin; ACY, ACY-1215; Con, control;
DMSO, dimethyl sulfoxide; Iri, irinotecan; Oxa, oxaliplatin; p-,
phosphorylated; PD-L1, programmed death-ligand 1; STAT3, signal
transducer and activator of transcription 3. |
Discussion
HDACs are key epigenetic regulators that are
considered promising therapeutic targets for cancer treatment. HDAC
inhibitors (HDACis) kill tumor cells via numerous mechanisms,
including DNA repair inhibition, cell-cycle checkpoint disruption
and the induction of apoptosis (28,29).
HDACis represent a chemically diverse group of drugs that affect
cancer, which are currently at various stages of development. At
present, two HDACis, vorinostat and romidepsin, have been approved
by the United States Food and Drug Administration for the treatment
of cutaneous T cell lymphoma. In addition, belinostat has recently
been approved for the treatment of peripheral T cell lymphoma, and
panobinostat has been approved for the treatment of MM. In
addition, >100 clinical trials have been initiated with HDACis
used as a monotherapy or in combination therapy (30). However, pan-HDACis elicit profound
side effects, including fatigue, nausea, vomiting, diarrhea,
thrombocytopenia and neutropenia, because they target several HDAC
isoforms. Therefore, more tolerable HDACis are required, with
HDAC6-selective inhibitors considered promising agents that avoid
the toxicity that is associated with class I HDACis (9,31).
ACY-1215 (ricolinostat) is a leading HDAC6
inhibitor, which is currently being tested in advanced clinical
trials for hematological cancer (myeloma and lymphoid malignancies)
(13,18–21).
Ricolinostat is well tolerated and is the only clinically tested
HDAC6-selective inhibitor. In addition, ricolinostat has been
analyzed in a phase I/II clinical trial as a monotherapy, and in
combination with bortezomib and dexamethasone in MM (NCT01323751).
At the recommended phase II dose of 160 mg/day ricolinostat, the
combination with bortezomib and dexamethasone is safe,
well-tolerated and active in MM (19). Ricolinostat has also been evaluated
in a phase I/II clinical trial in combination with lenalidomide and
dexamethasone, in order to determine the maximum tolerated dose
when used in combination therapy (NCT01583283). The phase Ib study
provides preliminary evidence that ricolinostat in combination with
lenalidomide and dexamethasone is safe and well-tolerated in R/R MM
(20). An additional phase Ib/II
clinical trial aims to determine the optimal dosing of ricolinostat
when combined with pomalidomide and dexamethasone. The phase II
study among patients with R/R MM aims to determine the overall
response rate of this combination (NCT01997840). Finally, a phase
Ib study in patients with R/R MM aims to determine the maximum
tolerated dose and safety of a liquid formulation of ricolinostat
used in combination with pomalidomide and low-dose dexamethasone
(NCT02189343).
Unlike hematological malignancies, ricolinostat is a
single agent that exhibits limited anticancer activity in solid
tumors, as confirmed in our previous study, which indicated that
ricolinostat at low doses is unable to decrease viability and to
induce apoptosis (27). Although
HDACis alone may be clinically useful, they too are most often used
in combination with other anticancer agents for cancer treatment.
Synergistic interactions between oxaliplatin and the HDACis, MS275,
suberoyl bishydroxamic acid and SAHA, have been reported in CRC
cells (32,33). Oxaliplatin is a chemotherapeutic
agent approved for use in the treatment of numerous types of solid
tumor; however, incomplete clinical responses are commonly
observed. Therefore, there is a need to enhance therapeutic
activity in the form of novel drugs that are well tolerated in
combination with oxaliplatin. The present study demonstrated that
synergistic CRC cell cytotoxicity was triggered by ricolinostat
when used in combination with oxaliplatin; ricolinostat enhanced
the oxaliplatin-induced inhibition of cell viability. Furthermore,
the increased caspase and PARP cleavage observed following combined
drug treatment confirmed that the apoptotic response was enhanced
following combination therapy. To further confirm these in
vitro results, a future in vivo study is required.
Consistent with these results, Wang et al reported that
ricolinostat inhibits the proliferation of glioblastoma, induces
apoptosis at high doses and sensitizes glioblastoma cells to
temozolomide (34). ACY-241 is a
second-generation orally available selective HDAC6 inhibitor that
is structurally related to ACY-1215. Recently, ACY-241 treatment
with paclitaxel was revealed to suppress cancer cell proliferation
and increase cell death relative to either single agent (35). Overall, these findings provide a
direct rationale for the use of ricolinostat as a therapeutic agent
for the treatment of patients with solid tumors, in addition to
blood cancers.
In conclusion, the present study identified ACY-1215
as an HDAC6 inhibitor that potentiated the anticancer drug efficacy
of certain chemotherapeutic drugs; however, the use of ACY-1215 as
a single agent in CRC was not potent or efficient. A predominant
downregulation of p-ERK and p-AKT was detected in CRC cells
following combined treatment with ACY-1215 and anticancer drugs.
Furthermore, combined treatment effectively induced CRC cell
apoptosis via activation of caspase-3 and elevation of the Bak to
Bcl-xL ratio. Taken together, these data indicated that an
HDAC6-selective inhibitor may be able to significantly augment
chemotherapeutic drug-mediated anticancer effects. ACY-1215
increased the expression levels of PD-L1 independent of the STAT3
pathway. Although little is currently known regarding the molecular
mechanism by which HDAC6 regulates the expression and stability of
PD-L1, combination treatment with ACY-1215 and other anticancer
agents markedly upregulated PD-L1 expression in CRC cells. These
results strongly suggested that a combination of ACY-1215 and other
chemotherapeutic agents could be beneficial for treating CRC by
modulating PD-L1. Therefore, we aim to conduct further studies
regarding the underlying molecular mechanisms. The present results
suggested that ACY-1215 in combination with oxaliplatin may be
considered a valuable novel treatment for patients with CRC.
ACY-1215 is currently undergoing clinical trials, and data from
phase I studies exist with regards to maximum tolerated doses,
toxicity, and pharmacokinetic and pharmacodynamic information.
Collectively, the present findings may inform clinical trials of
the efficiency of combina tion chemotherapy containing a
HDAC6-selective inhibitor.
Abbreviations:
|
Bcl-xL
|
B-cell lymphoma-extra large
protein
|
|
CI
|
combination index
|
|
CRC
|
colorectal cancer
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
FA
|
fraction affected
|
|
5-FU
|
5-fluorouracil
|
|
HDAC
|
histone deacetylase
|
|
HDACi
|
histone deacetylase inhibitor
|
|
PARP
|
poly (ADP ribose) polymerase
|
|
PD-L1
|
programmed death-ligand 1
|
|
SAHA
|
suberoylanilide hydroxamic acid
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
Acknowledgments
Not applicable.
Funding
The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
funded by the Ministry of Education, Science and Technology (grant
no. 2016R1D1A1A02937071).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DHL designed and performed the experiments, and
analyzed the data. HRW and HWR performed the experiments. JMH
analyzed some data and helped to edit the manuscript. SHK conceived
the general design of the study, wrote the initial draft of the
manuscript, extensively edited the manuscript and supervised the
work.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interest.
References
|
1
|
Jemal A, Center MM, DeSantis C and Ward
EM: Global patterns of cancer incidence and mortality rates and
trends. Cancer Epidemiol Biomarkers Prev. 19:1893–1907. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hubbert C, Guardiola A, Shao R, Kawaguchi
Y, Ito A, Nixon A, Yoshida M, Wang XF and Yao TP: HDAC6 is a
micro-tubule-associated deacetylase. Nature. 417:455–458. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang Y, Gilquin B, Khochbin S and
Matthias P: Two catalytic domains are required for protein
deacetylation. J Biol Chem. 281:2401–2404. 2006. View Article : Google Scholar
|
|
5
|
Li Y, Shin D and Kwon SH: Histone
deacetylase 6 plays a role as a distinct regulator of diverse
cellular processes. FEBS J. 280:775–793. 2013.
|
|
6
|
Boyault C, Zhang Y, Fritah S, Caron C,
Gilquin B, Kwon SH, Garrido C, Yao TP, Vourc'h C, Matthias P, et
al: HDAC6 controls major cell response pathways to cytotoxic
accumulation of protein aggregates. Genes Dev. 21:2172–2181. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kawaguchi Y, Kovacs JJ, McLaurin A, Vance
JM, Ito A and Yao TP: The deacetylase HDAC6 regulates aggresome
formation and cell viability in response to misfolded protein
stress. Cell. 115:727–738. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kwon S, Zhang Y and Matthias P: The
deacetylase HDAC6 is a novel critical component of stress granules
involved in the stress response. Genes Dev. 21:3381–3394. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang Y, Kwon S, Yamaguchi T, Cubizolles
F, Rousseaux S, Kneissel M, Cao C, Li N, Cheng HL, Chua K, et al:
Mice lacking histone deacetylase 6 have hyperacetylated tubulin but
are viable and develop normally. Mol Cell Biol. 28:1688–1701. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bergman JA, Woan K, Perez-Villarroel P,
Villagra A, Sotomayor EM and Kozikowski AP: Selective histone
deacetylase 6 inhibitors bearing substituted urea linkers inhibit
melanoma cell growth. J Med Chem. 55:9891–9899. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Butler LM, Agus DB, Scher HI, Higgins B,
Rose A, Cordon-Cardo C, Thaler HT, Rifkind RA, Marks PA and Richon
VM: Suberoylanilide hydroxamic acid, an inhibitor of histone
deacetylase, suppresses the growth of prostate cancer cells in
vitro and in vivo. Cancer Res. 60:5165–5170. 2000.PubMed/NCBI
|
|
12
|
Inks ES, Josey BJ, Jesinkey SR and Chou
CJ: A novel class of small molecule inhibitors of HDAC6. ACS Chem
Biol. 7:331–339. 2012. View Article : Google Scholar :
|
|
13
|
Santo L, Hideshima T, Kung AL, Tseng JC,
Tamang D, Yang M, Jarpe M, van Duzer JH, Mazitschek R, Ogier WC, et
al: Preclinical activity, pharmacodynamic, and pharmacokinetic
properties of a selective HDAC6 inhibitor, ACY-1215, in combination
with bortezomib in multiple myeloma. Blood. 119:2579–2589. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Smil DV, Manku S, Chantigny YA, Leit S,
Wahhab A, Yan TP, Fournel M, Maroun C, Li Z, Lemieux AM, et al:
Novel HDAC6 isoform selective chiral small molecule histone
deacetylase inhibitors. Bioorg Med Chem Lett. 19:688–692. 2009.
View Article : Google Scholar
|
|
15
|
Estiu G, Greenberg E, Harrison CB,
Kwiatkowski NP, Mazitschek R, Bradner JE and Wiest O: Structural
origin of selectivity in class II-selective histone deacetylase
inhibitors. J Med Chem. 51:2898–2906. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Haggarty SJ, Koeller KM, Wong JC,
Grozinger CM and Schreiber SL: Domain-selective small-molecule
inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin
deacetylation. Proc Natl Acad Sci USA. 100:4389–4394. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dallavalle S, Pisano C and Zunino F:
Development and therapeutic impact of HDAC6-selective inhibitors.
Biochem Pharmacol. 84:756–765. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ashjian E and Redic K: Multiple myeloma:
Updates for pharmacists in the treatment of relapsed and refractory
disease. J Oncol Pharm Pract. 22:289–302. 2016. View Article : Google Scholar
|
|
19
|
Vogl DT, Raje N, Jagannath S, Richardson
P, Hari P, Orlowski R, Supko JG, Tamang D, Yang M, Jones SS, et al:
Ricolinostat, the first selective histone deacetylase 6 inhibitor,
in combination with bortezomib and dexamethasone for relapsed or
refractory multiple myeloma. Clin Cancer Res. 23:3307–3315. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yee AJ, Bensinger WI, Supko JG, Voorhees
PM, Berdeja JG, Richardson PG, Libby EN, Wallace EE, Birrer NE,
Burke JN, et al: Ricolinostat plus lenalidomide, and dexamethasone
in relapsed or refractory multiple myeloma: A multicentre phase 1b
trial. Lancet Oncol. 17:1569–1578. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dasmahapatra G, Patel H, Friedberg J,
Quayle SN, Jones SS and Grant S: In vitro and in vivo interactions
between the HDAC6 inhibitor ricolinostat (ACY1215) and the
irreversible proteasome inhibitor carfilzomib in non-Hodgkin
lymphoma cells. Mol Cancer Ther. 13:2886–2897. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Witter DJ, Harrington P, Wilson KJ,
Chenard M, Fleming JC, Haines B, Kral AM, Secrist JP and Miller TA:
Optimization of biaryl Selective HDAC1&2 Inhibitors (SHI-1:2).
Bioorg Med Chem Lett. 18:726–731. 2008. View Article : Google Scholar
|
|
24
|
Hideshima T, Cottini F, Ohguchi H,
Jakubikova J, Gorgun G, Mimura N, Tai YT, Munshi NC, Richardson PG
and Anderson KC: Rational combination treatment with histone
deacetylase inhibitors and immunomodulatory drugs in multiple
myeloma. Blood Cancer J. 5:e3122015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mishima Y, Santo L, Eda H, Cirstea D,
Nemani N, Yee AJ, O'Donnell E, Selig MK, Quayle SN, Arastu-Kapur S,
et al: Ricolinostat (ACY-1215) induced inhibition of aggresome
formation accelerates carfilzomib-induced multiple myeloma cell
death. Br J Haematol. 169:423–434. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Quayle SN and Jones SS: ACY-1215, a
first-in-class selective inhibitor of HDAC6, demonstrates
significant synergy with immunomodulatory drugs (IMiDs) in
preclinical models of multiple myeloma (MM). Blood.
122:19522013.
|
|
27
|
Ryu HW, Shin DH, Lee DH, Won HR and Kwon
SH: A potent hydroxamic acid-based, small-molecule inhibitor A452
preferentially inhibits HDAC6 activity and induces cytotoxicity
toward cancer cells irrespective of p53 status. Carcinogenesis.
39:72–83. 2018. View Article : Google Scholar
|
|
28
|
Falkenberg KJ and Johnstone RW: Histone
deacetylases and their inhibitors in cancer, neurological diseases
and immune disorders. Nat Rev Drug Discov. 13:673–691. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Marks PA and Xu WS: Histone deacetylase
inhibitors: Potential in cancer therapy. J Cell Biochem.
107:600–608. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bruserud O, Stapnes C, Ersvaer E, Gjertsen
BT and Ryningen A: Histone deacetylase inhibitors in cancer
treatment: A review of the clinical toxicity and the modulation of
gene expression in cancer cell. Curr Pharm Biotechnol. 8:388–400.
2007. View Article : Google Scholar
|
|
31
|
Kaliszczak M, Trousil S, Åberg O, Perumal
M, Nguyen QD and Aboagye EO: A novel small molecule hydroxamate
preferentially inhibits HDAC6 activity and tumour growth. Br J
Cancer. 108:342–350. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Alzoubi S, Brody L, Rahman S,
Mahul-Mellier AL, Mercado N, Ito K, El-Bahrawy M, Silver A, Boobis
A, Bell JD, et al: Synergy between histone deacetylase inhibitors
and DNA-damaging agents is mediated by histone deacetylase 2 in
colorectal cancer. Oncotarget. 7:44505–44521. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Flis S, Gnyszka A and Spławiński J: HDAC
inhibitors, MS275 and SBHA, enhances cytotoxicity induced by
oxaliplatin in the colorectal cancer cell lines. Biochem Biophys
Res Commun. 387:336–341. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang Z, Hu P, Tang F, Lian H, Chen X,
Zhang Y, He X, Liu W and Xie C: HDAC6 promotes cell proliferation
and confers resistance to temozolomide in glioblastoma. Cancer
Lett. 379:134–142. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Huang P, Almeciga-Pinto I, Jarpe M, van
Duzer JH, Mazitschek R, Yang M, Jones SS and Quayle SN: Selective
HDAC inhibition by ACY-241 enhances the activity of paclitaxel in
solid tumor models. Oncotarget. 8:2694–2707. 2017.
|














