Introduction
Tumor necrosis factor (TNF)-related
apoptosis-inducing ligand (TRAIL) is a member of the TNF
superfamily, which preferentially kills malignant cells over
nontransformed cells (1–4). TRAIL can induce extrinsic and
intrinsic death pathways by binding its specific receptors with
death domain TRAIL receptor (TRAIL-R)1/death receptor (DR)4 and
TRAIL-R2/DR5 (5,6). However, some cancer cell types are
inherently resistant to TRAIL, despite expressing death-inducing
receptors (7–11). Furthermore, some cell types acquire
considerable tolerance to TRAIL during prolonged treatment.
Accordingly, current clinical trials have been disappointing, and
the combined use of agents that overcome drug resistance is
necessary for efficient TRAIL therapy. Non-thermal (cold)
atmospheric plasma (CAP) has emerged as another promising means of
cancer treatment, since like TRAIL, it kills various cancer cells
while sparing nontransformed cells under optimal conditions
(12–15). Cold plasma-stimulated medium (PSM)
also exhibits vigorous and tumor-selective anticancer activities
(16–19) and has emerged as an alternative
method of direct CAP irradiation; PSM is better than direct CAP
irradiation for systematic or local administration to deep
tissues.
Cancer cells, including malignant melanoma (MM) and
osteosarcoma (OS) cells, are characterized by their intrinsic
resistance to apoptosis; in addition, they frequently become more
tolerant to numerous apoptosis-inducing antitumor drugs.
Nevertheless, the majority of conventional drugs primarily kill
cells by apoptosis. Accordingly, current chemotherapy toward these
cancers is severely compromised by intrinsic and acquired
resistance; therefore, induction of another mode of cell death may
be a useful approach for the treatment of apoptosis-resistant cells
(20,21). Autophagy is a primary catabolic
process that degrades cellular components and damaged organelles
via lysosomes; this process copes with cellular stressors, such as
starvation, and supplies energy and metabolic precursors. Autophagy
consists of numerous processes, including induction of cytoplasmic
double-layered membranes, which are known as phagophores,
phagophore elongation and autophagosome formation, a fusion of
autophagosomes with lysosomes, and degradation and recycling. All
processes, from the formation of autophagosomes to the degradation
of cellular components, are strictly regulated by autophagy-related
(Atg) proteins that are encoded by Atg genes (22). Autophagy is classified into three
different types: Macroautophagy (subsequently referred to as
autophagy), microautophagy and chaperone-mediated autophagy.
Autophagy is negatively regulated by mammalian target of rapamycin
complex I in response to insulin and amino acid signals, and is
driven transiently via removal of its suppression through the
depletion of these nutrients (23–25).
Therefore, autophagy is of particular importance for the survival
of constitutively proliferating cells, such as cancer cells, that
are regularly imposed to energy demands (20,26).
In addition, autophagy contributes to cancer cell survival by
removing damaged organelles, including mitochondria and endoplasmic
reticulum (ER) by microautophagy, which is also known as mitophagy
and ERphagy, respectively. These damaged organelles are degraded
via lysosomal enzymes following engulfment into autophagosomes;
such quality control is crucial for cell survival. Conversely,
autophagy is also characterized by a unique cell death pathway that
acts as a tumor suppressor when it leads to autophagic cell death
(ACD) (27–29).
Our previous study revealed that PSM prepared by CAP
irradiation of Dulbecco's modified Eagle's medium (DMEM) could kill
an array of MM, OS and lung cancer cells, while sparing
nontransformed melanocytes and fibroblasts (30). In addition, PSM led to increased
caspase-3/7 activity, and modest cleavage of caspase-9, caspase-3/7
and poly ADP-ribose polymerase; furthermore, caspase-3/7-specific
inhibitors failed to suppress cell death. Therefore, the present
study aimed to examine the possibility that PSM may induce another
cell death modality. The results demonstrated that PSM can trigger
ACD in MM and OS cells.
Materials and methods
Materials
Soluble recombinant human TRAIL was obtained from
Enzo Life Sciences, Inc. (Farmingdale, NY, USA). 3-Methyladenine
(3-MA), chloroquine (CQ) and bafilomycin A1 (Baf) were obtained
from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). The
pan-caspase-inhibitor z-VAD-fluorometheylketone (Z-VAD-FMK) was
purchased from Merck Ltd. (Tokyo, Japan). All insoluble reagents
were dissolved in dimethyl sulfoxide (DMSO) and diluted with high
glucose-containing DMEM supplemented with 10% fetal bovine serum
(FBS) (both from Sigma-Aldrich; Merck KGaA) or Hank's balanced salt
solution (HBSS; pH 7.4; Nissui Pharmaceutical Co., Ltd., Tokyo,
Japan) (final DMSO concentration, <0.1%) prior to use. The
manganese-porphyrin superoxide dismutase mimetic MnTBaP (Enzo Life
Sciences, Inc.) was dissolved in 1 mM NaOH (pH 8.0) and HBSS was
added to lower the pH to 7.4.
Cell culture
Human A375 MM cells [American Type Culture
Collection (ATCC)® cell number CRL-1619) were obtained
from ATCC (Manassas, VA, USA). A2058 MM cells (cell number IFO
50276) and human A549 lung adenocarcinoma cells (cell number
JCRB0076) were purchased from the JCRB Cell Bank of National
Institutes of Biomedical Innovation, Health, and Nutrition (Osaka,
Japan). Human HOS (TE85) OS cells (cell number RCB0992), SAOS-2 OS
cells (cell number RCB0428) and 143B cells (cell number RCB0701)
were obtained from the Riken BioResource Center (Tsukuba, Japan).
Human dermal fibroblast (HDF) cells from facial dermis were
obtained from Cell Applications (San Diego, CA, USA). All cells
were cultured in 10% FBS/DMEM supplemented with 100 U/ml penicillin
and 100 µg streptomycin (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) in a humidified atmosphere containing 95% air/5%
CO2 at 37°C. The cells were harvested by incubating with
0.25% trypsin-EDTA (Thermo Fisher Scientific, Inc.) for 5 min at
37°C. Throughout the study, various cell lines were used in each
experiment to determine whether the reactions observed were
specific for the cell line used or were general among the various
cell lines. A2058 and HOS (or SAOS-2) cells were used as the main
representatives of MM and OS cells, respectively. In some
experiments, autophagy was compared between normally grown A375
(confluence, 70–80%) cells and overgrown (confluent, partially
floating) cells.
PSM preparation
CAP was generated using an originally-developed
low-frequency plasma jet device equipped with an asymmetrical
dielectric barrier discharge, as previously described (30). The typical frequency was 20 kHz,
with a peak voltage of 8 kV, a current of 20 mA and a helium flow
rate of 300 ml/min. PSM was prepared by irradiating 1 ml FBS/DMEM
with CAP for 5 min once. The original PSM (100%) was diluted to a
final concentration of 25% with FBS/DMEM (for cell experiments) or
HBSS (for biochemical experiments), and indicated as PSM (25%).
Cell viability assay
Cell viability was measured according to the WST-8
assay using Cell Counting Reagent SF (Nacalai Tesque, Inc., Kyoto,
Japan) as previously described (31). This is a colorimetric assay that
detects viability based on the formation of a water-soluble
formazan product. Briefly, the cells were seeded at a density of
8×103 cells/well in 96-well plates (Corning
Incorporated, Corning, NY, USA) and cultured with PSM (12.5, 25,
50, and 100%), 100 ng/ml TRAIL, 3-MA (1.3–5 mM), CQ (100 and 300
µM), and Baf (100 and 300 nM) alone or in combination for 72
h at 37°C, prior to the addition of 10 µl cell counting
reagent SF for 1 h. For some experiments, the cells were cultured
with the aforementioned reagents in the presence of 10 µM
Z-VAD-FMK or 30 µM MnTBaP. Absorbance was measured at 450 nm
using an ARVO MX microplate reader (PerkinElmer Japan Co., Ltd.,
Yokohama, Japan).
Live-cell imaging
The mitochondrial network was analyzed as previously
described (32) with minor
modifications. Briefly, cells in FBS/DMEM (3×104/well)
adherent on an 8-well chambered coverslip with a glass bottom
(Imaging Chamber 8 CG; Zell-Kontakt GmbH, Nörten-Hardenberg,
Germany) were treated with PSM (25 and 100%), 100 ng/ml TRAIL, 0.5
or 1 µM rapamycin and 5 mM 3-MA alone or in combination for
24 h in a humidified atmosphere containing 95% air/5%
CO2 at 37°C. Following media removal by aspiration, the
cells were washed with fresh FBS/DMEM and stained with 20 nM
MitoTracker Red CMXRos for 1 h at 37°C in the dark. The nuclei were
counterstained with Hoechst 33342. The cells were then washed with
and immersed in FluoroBrite™ DMEM (Thermo Fisher Scientific, Inc.).
Images were obtained using a BZ X-700 Fluorescence Microscope
(Keyence Corporation, Osaka, Japan) equipped with a 100X, 1.40 n.a.
UPLSAPO super-apochromat, coverslip-corrected oil objective
(Olympus Corporation, Tokyo, Japan). Images were analyzed using
BZ-H3A application software (Keyence Corporation) and National
Institutes of Health (NIH) ImageJ software (bundled with 64-bit
Java 1.8.0_112; NIH, Bethesda, MD, USA). The formation of
autophagosomes was analyzed using the CYTO-ID® Autophagy
Detection kit (Enzo Life Sciences, Inc.) according to the
manufacturer's protocol. Briefly, cells were stained with CYTO-ID
for 1 h at 37°C in the dark and treated with the aforementioned
agents. For monitoring colocalization of mitochondria and
autophagosomes, cells were coincidently stained with MitoTracker
Red CMXRos and the CYTO-ID. Images were obtained using EVOS FL Cell
Imaging system (Thermo Fisher Scientific, Inc.) equipped with a
40X, 0.60 n.a. LUCPLFLN objective (Olympus Corporation) as
previously described (33).
Western blotting
Following treatment with PSM (25 and 100%) for 12,
24, 36, 48 and 72 h, cells were washed with Ca2+-,
Mg2+-free PBS, and were lysed during 30 min agitation
with CelLytic™ lysis buffer (Sigma-Aldrich; Merck KGaA) containing
a protease inhibitor cocktail and a phosphatase inhibitor cocktail
(both from Sigma-Aldrich; Merck KGaA). Cell debris was removed by
centrifugation at 20,000 × g for 15 min at 4°C. The supernatants
were collected and protein concentrations were analyzed using a
bicinchoninic acid protein assay (Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. Equal amounts of protein
(10 µg) were separated by 4–12% NuPage Bis-Tris acrylamide
gels (Invitrogen; Thermo Fisher Scientific, Inc.) and were
transferred to polyvinylidene difluoride membranes (Immobilon-P;
EMD Millipore, Bedford, MA, USA). Blots were blocked for 30 min in
Tris-buffered saline with 0.05% Tween-20 (TBST; Sigma-Aldrich;
Merck KGaA) containing 2% nonfat dry milk. The blots were then
incubated with primary antibodies against p62 (PM045, 1:1,000;
Medical & Biological Laboratories Co., Ltd., Nagoya, Japan),
microtubule-associated protein 1A/1B-light chain 3 (LC3; #12741,
1:1,000) and GAPDH (#5174, 1:1,000; Cell Signaling Technology,
Inc., Danvers, MA, USA) overnight at 4°C in TBST containing 2%
nonfat dry milk. After washing two times with TBST, membranes were
incubated with horseradish peroxidase-conjugated goat anti-rabbit
(#7074, 1:2,000; Cell Signaling Technology, Inc.) for 1 h at room
temperature. Subsequently, blots were washed three times with TBST
and immersed in enhanced chemiluminescence reagent (GE Healthcare,
Chicago, IL, USA) to enhance the signals. The signals were then
captured using an LAS-4000 Camera system (Fujifilm Corporation,
Tokyo, Japan).
Statistical analysis
Data are presented as the means ± standard
deviation. Data were analyzed by one-way analysis of variance
followed by Tukey's post hoc test using add-in software for Excel
2016 for Windows (Microsoft Corporation, Redmond, WA, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Human MM and OS cells exhibit substantial
basal autophagy
To gain insight into the possible role of autophagy
in the anti-tumor activity of PSM, the present study evaluated its
ability to modulate autophagic flux in MM and OS cells.
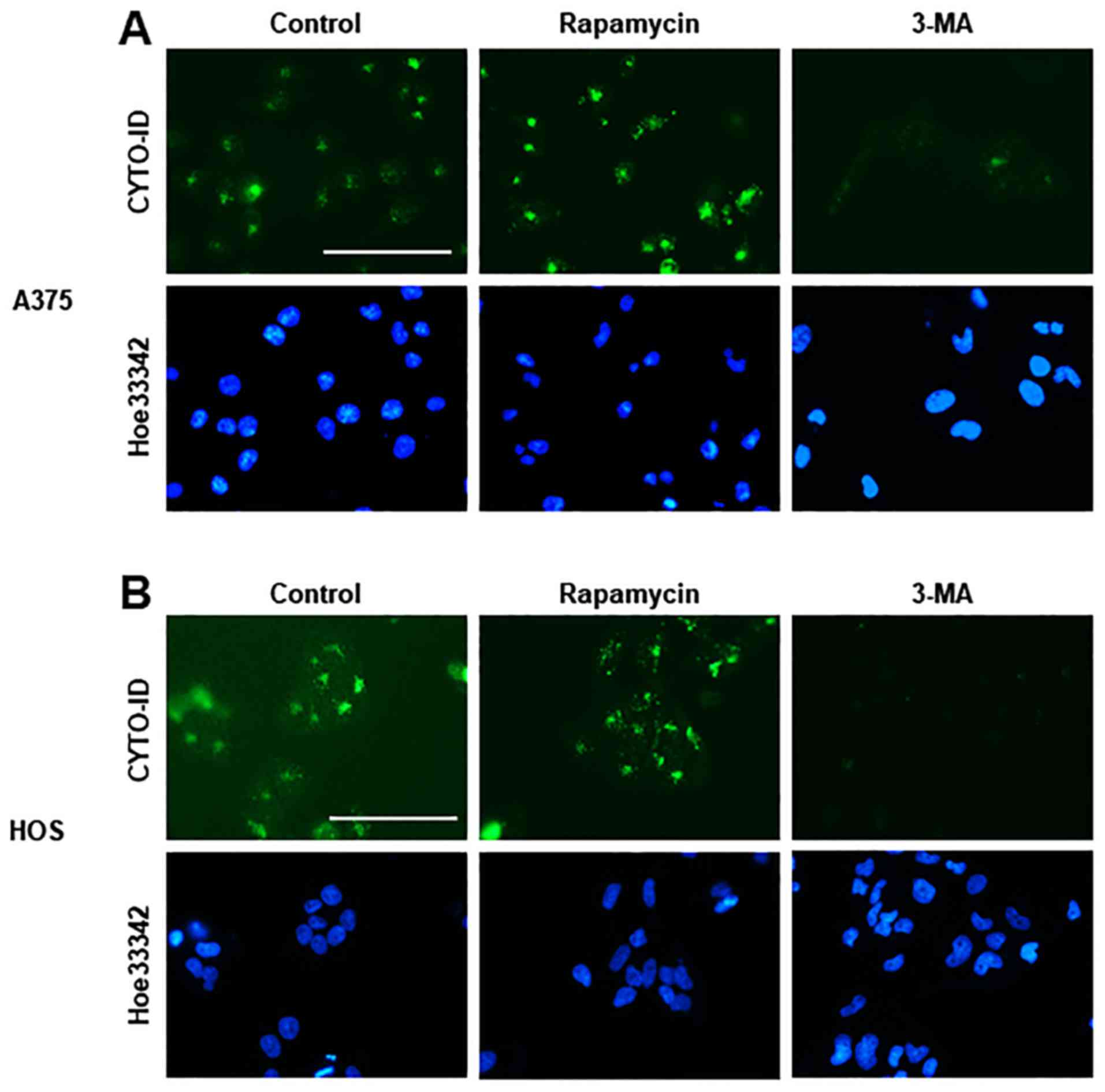
CYTO-ID® Green is a cationic amphiphilic dye that can
precisely monitor autophagic vacuoles without transfection.
Live-cell imaging revealed a considerable number of CYTO-ID puncta
in A375 and HOS cells in DMEM supplemented with 10% FBS. The
CYTO-ID puncta markedly increased in response to 1 µM
rapamycin, whereas these puncta were decreased almost entirely
following treatment with 5 mM 3-MA (Fig. 1), thus validating that staining
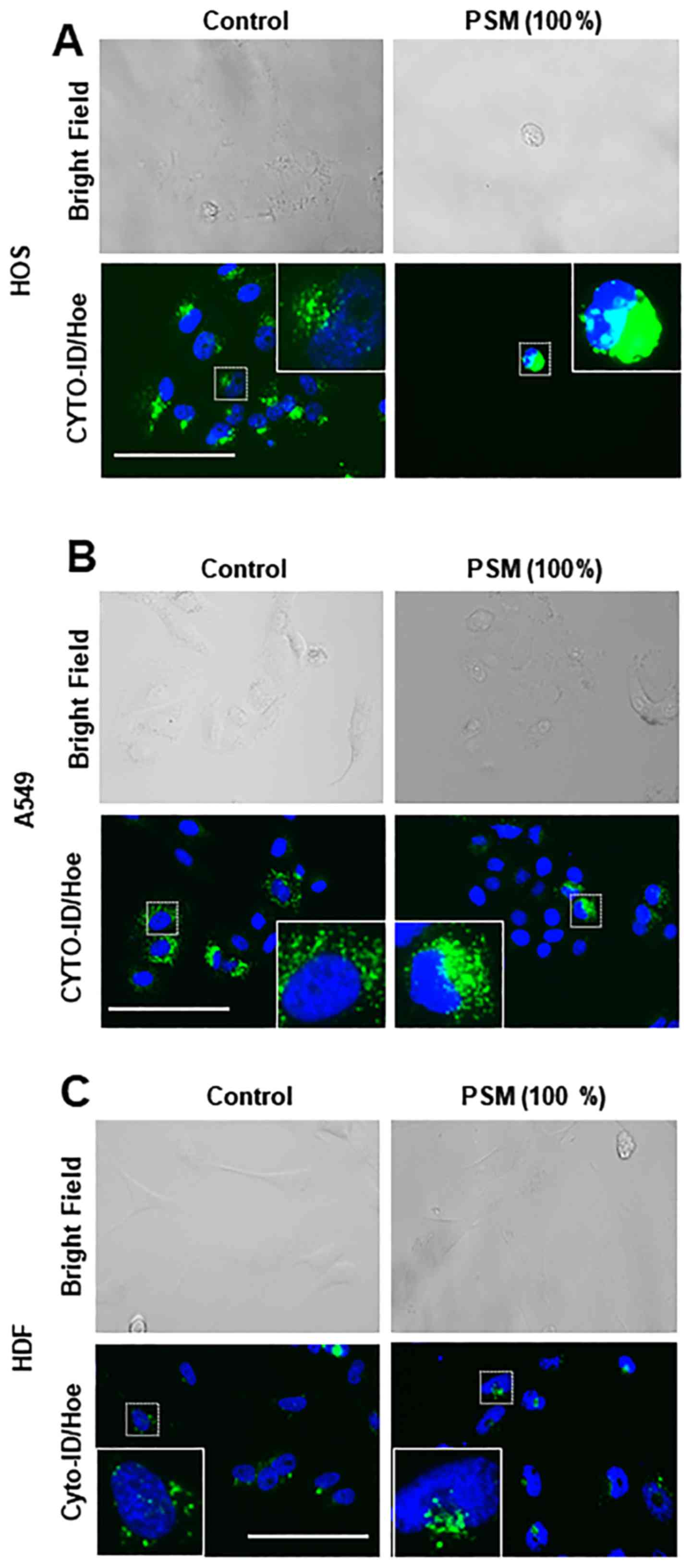
represents autophagosomes. Consistent with our previous study
(30), treatment with PSM for 24 h
resulted in robust cell damage. Accordingly, adherent spindle HOS
and A549 cells became round, and some cells lost their adherence
and integrity (Fig. 2A and B).
Concomitantly, CYTO-ID puncta became clustered in these cells.
Conversely, HDF cells possessed only modest basal CYTO-ID puncta
and minimal changes in CYTO-ID puncta were observed following PSM
treatment (Fig. 2C).
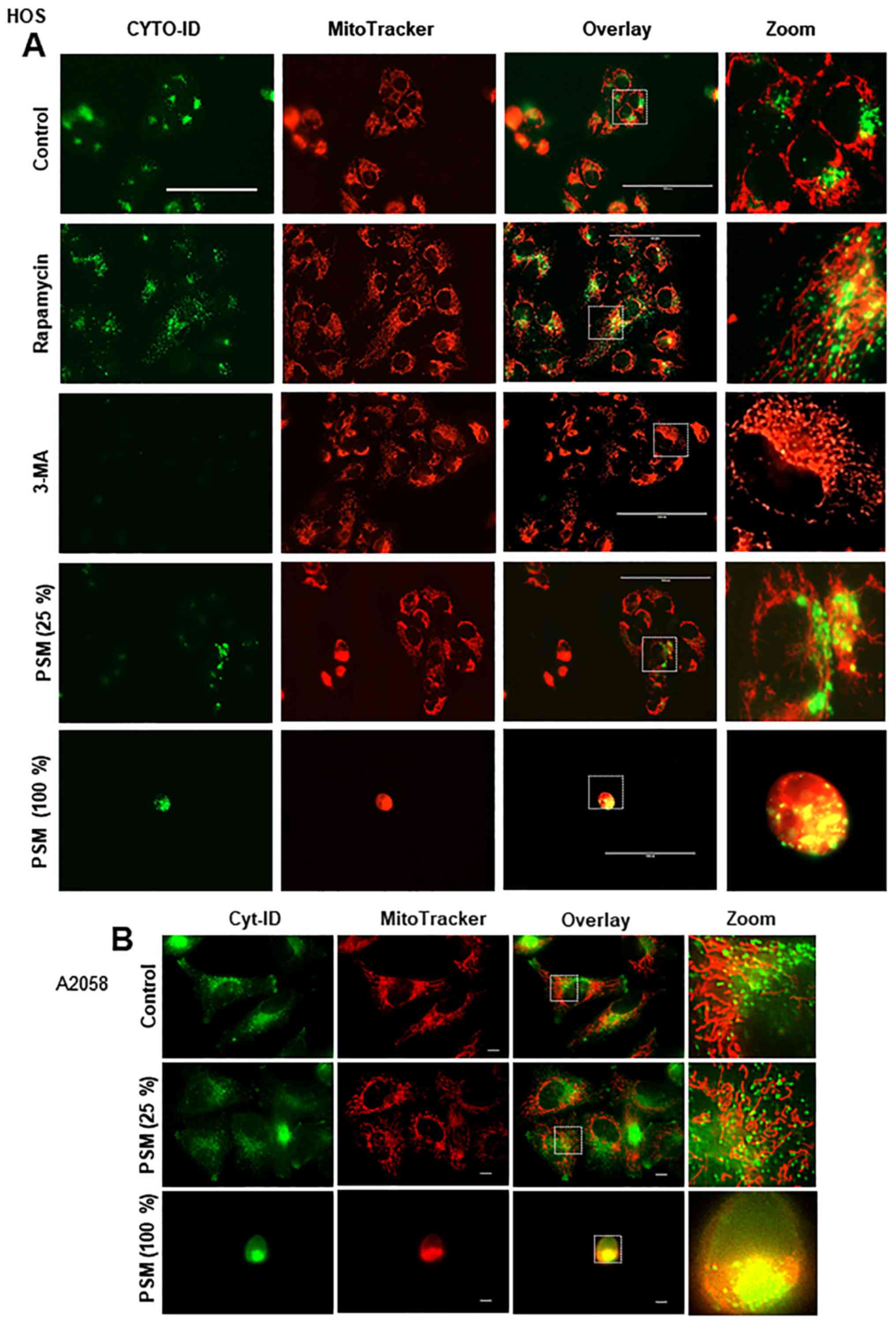
PSM induces colocalization of aggregated
mitochondria and autophagosomes
Damaged mitochondria are removed by autophagic
processes known as mitophagy. Accordingly, mitophagy controls the
quality of mitochondria, thereby facilitating cell survival,
whereas the excessive removal of mitochondria facilitates cell
death (34,35). Our previous study reported that PSM
could induce excessive mitochondrial fragmentation and aggregation,
alongside intensive mitochondrial damage (30). Therefore, the present study
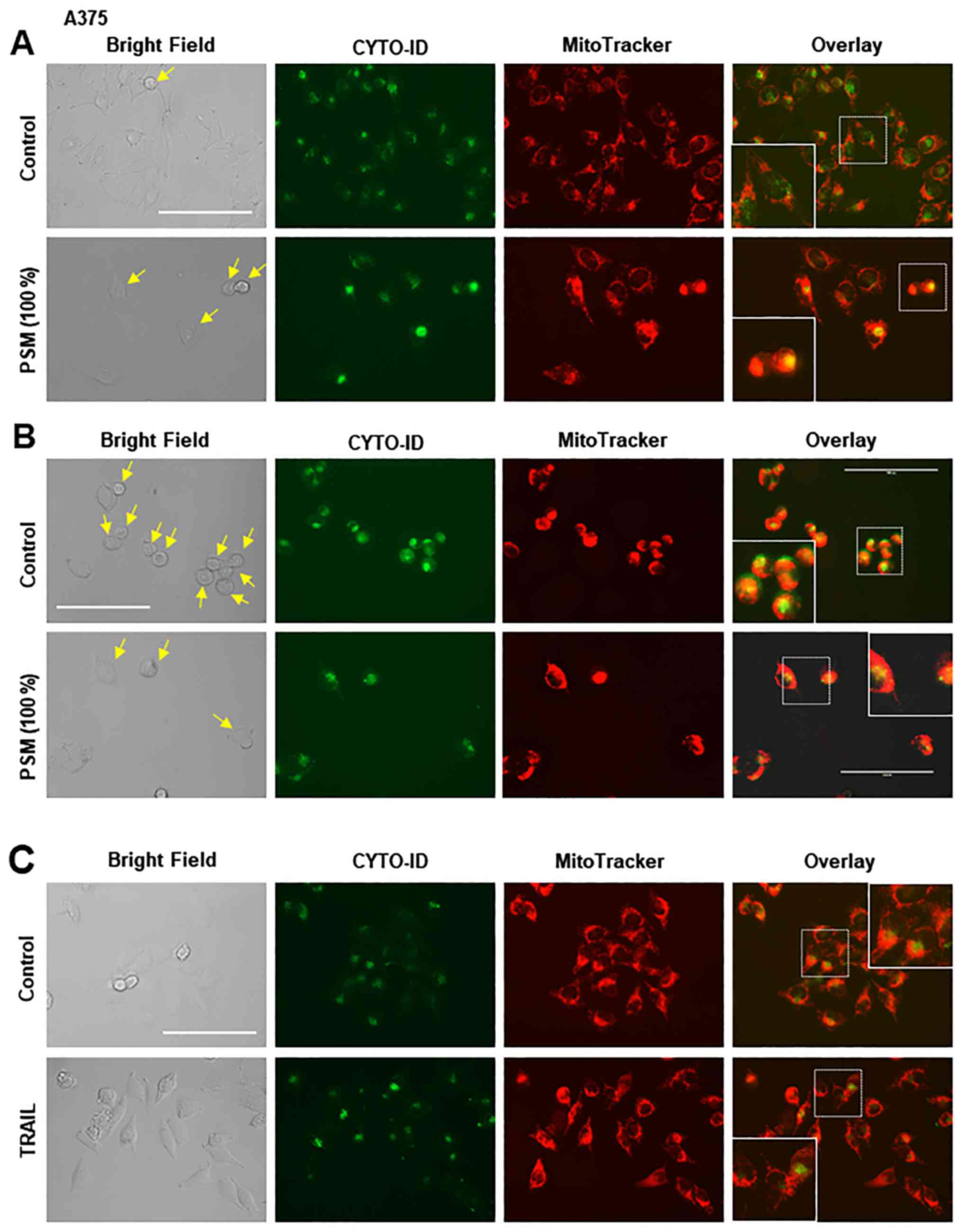
simultaneously monitored CYTO-ID puncta and mitochondrial
morphology in live cells. Unstimulated A375 cells possessed tubular
mitochondria and the CYTO-ID puncta that primarily diffused in the
cytoplasm. In this case, the puncta and the mitochondria were
separately located, as indicated by the minimal overlapping of
green and red signals (Fig. 3A,
upper panels). Following PSM treatment, the mitochondria became
fragmented, punctate and aggregated, as previously described
(30). Furthermore, some of the
damaged round cells possessed clustered CYTO-ID puncta, the
majority of which was colocalized with the aggregated mitochondria
(Fig. 3A, lower panels). Even in
the absence of insult, overgrown A375 cells became round and
damaged (Fig. 3B, upper panels).
Similarly, in PSM-treated cell images, damaged cells possessed
numerous clustered CYTO-ID puncta, which were colocalized with
aggregated mitochondria. To determine whether these effects were
specific for PSM, the effects of TRAIL were determined. The results
demonstrated that TRAIL increased CYTO-ID puncta in TRAIL-resistant
A375 cells; however, TRAIL led to modest mitochondrial
fragmentation, and caused minimal mitochondrial aggregation and
colocalization with autophagosomes (Fig. 3C, lower panels). These results
indicated that PSM may induce colocalization of aggregated
mitochondria and autophagosomes.
Toxic PSM specifically increases
autophagic flux
The present results indicated that PSM specifically
leads to colocalization of aggregated mitochondria and
autophagosomes. To determine whether this intrinsic event is
cytoprotective or cytotoxic, the study examined the association of
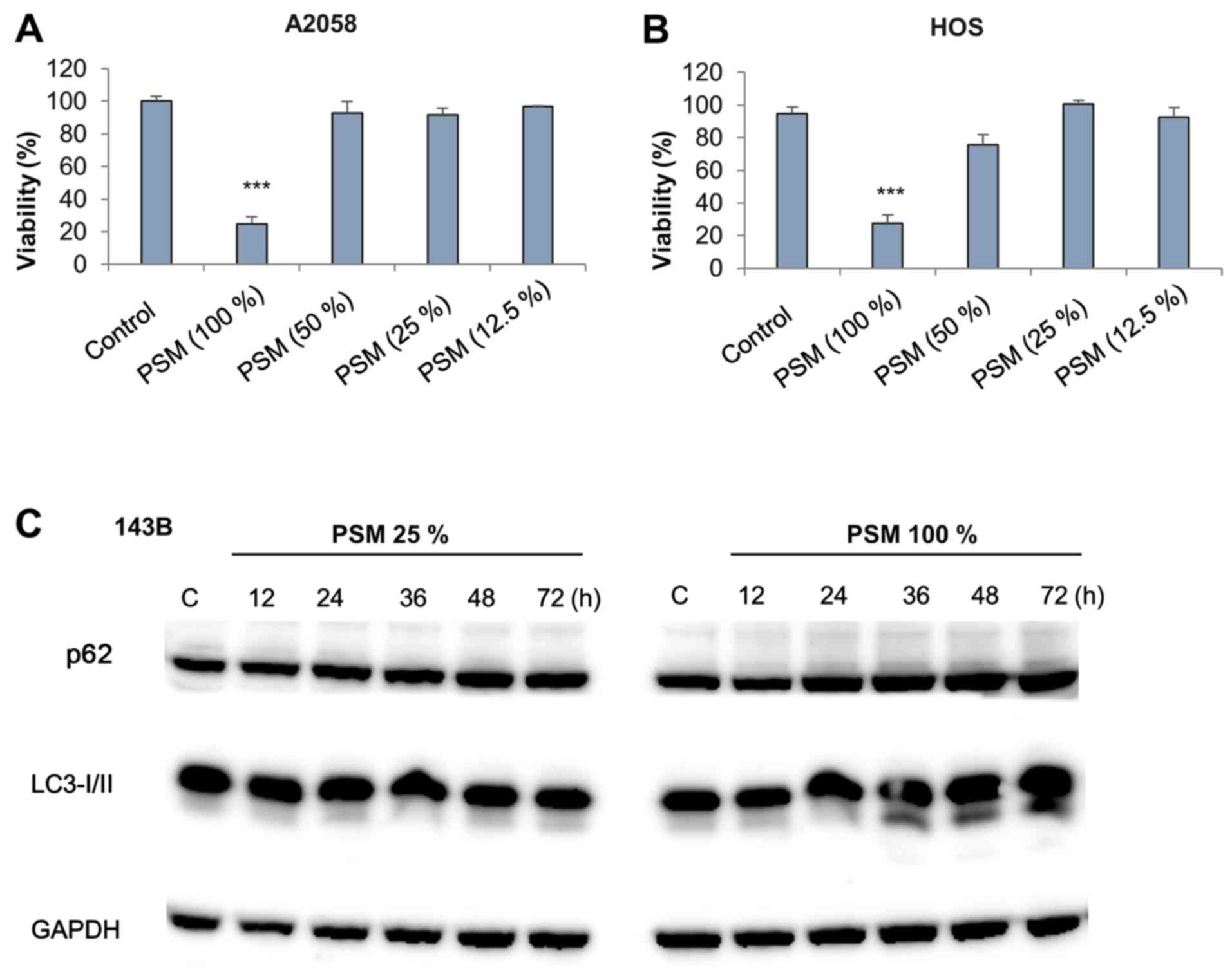
PSM with cell damage. PSM (100%) displayed significant cytotoxic
activity toward MM (A2058) and OS (HOS) cells, whereas it was
ineffective at lower concentrations (≤50%) (Fig. 4A and B). Subsequently, the present
study compared the effects of toxic and subtoxic PSM on autophagic
flux. The cytosolic protein LC3-I forms a complex with
phosphatidylethanolamine, thus resulting in the formation of
LC3-II, which directly associates with the autophagosome membrane.
LC3-II enables the binding of adaptor proteins, such as
p62/sequestome 1 (36). Therefore,
the present study analyzed the expression levels of p62 and LC3-II
following PSM treatment. Subtoxic PSM had minimal effects on the
expression levels of p62 and LC3-II up to 72 h post-stimulation
(Fig. 4C, left panels). However,
toxic PSM increased the expression levels of LC3-II, but not p62,
36–48 h post-stimulation (Fig. 4C,
right panels). These results suggested that toxic PSM may
specifically increase autophagic flux.
Colocalization of mitochondria and
autophagosomes is associated with autophagy induction and cell
death
To further explore the relationship between the
colocalization of mitochondria and autophagosomes and autophagy,
the present study examined the effects of autophagy modulators on
mitochondrial morphology and autophagosome location. Treatment with
the autophagy promoter rapamycin markedly increased CYTO-ID puncta
in HOS cells (Fig. 5A).
Concomitantly, some, but not all, of the puncta were colocalized
with the mitochondria, as shown by the appearance of yellow signals
in the overlay images. Conversely, 3-MA completely suppressed
CYTO-ID puncta and increased mitochondrial hyperfusion (Fig. 5A). In addition, subtoxic PSM did
not induce robust colocalization of the mitochondria and
autophagosomes (Fig. 5A). All of
these agents caused minimal mitochondrial fragmentation and
aggregation. Furthermore, toxic PSM strongly induced mitochondrial
fragmentation and aggregation, and colocalization of mitochondria
and autophagosomes (Fig. 5A).
Microscopic analysis with a higher resolution confirmed these
observations and indicated the colocalization of clustered CYTO-ID
puncta and aggregated mitochondria (Fig. 5B). These results suggested that the
colocalization of mitochondria and autophagosomes may be associated
with autophagy induction and cell damage.
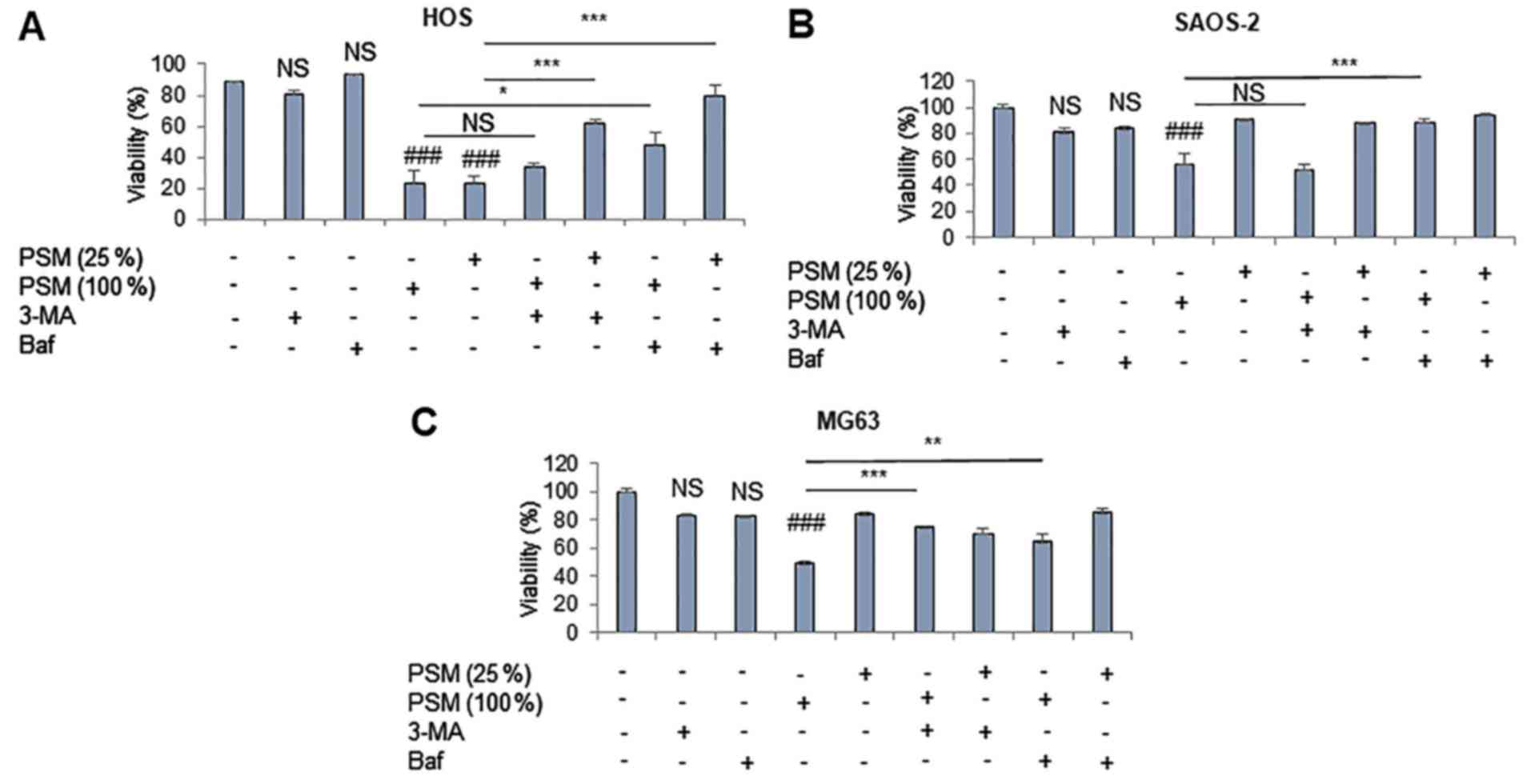
PSM induces ACD during the initial 24 h
post-stimulation
To further explore the role of autophagy in the
antitumor activity of PSM, the present study examined the effects
of numerous pharmacological autophagy inhibitors with various
mechanisms of action on PSM cytotoxicity. 3-MA inhibits
autophagosome formation by inhibiting the type III
phosphatidylinositol-3-kinase, whereas the antimalarial drug CQ
inhibits the fusion of autophagosomes and lysosomes. The antibiotic
Baf inhibits vacuolar-type H+-ATPases and increases
lysosomal pH, thus compromising the fusion of autophagosomes and
lysosomes. HOS cells were relatively susceptible to PSM
cytotoxicity. Accordingly, their viability often decreased
considerably (maximum reduction, 80%) at 24 h following treatment
with PSM (≥25%) (Fig. 6A). Baf
inhibited the cytotoxic effects of 25% PSM entirely, and those of
100% PSM partially. In addition, 3-MA significantly reduced the
effects of PSM (25%), but not those of PSM (100%). PSM (100%), but
not PSM (25%), also significantly decreased the viability of SAOS-2
and MG63 cells (Fig. 6B and C),
and 3-MA inhibited the cytotoxic effects of PSM in MG63 cells, but
not in SAOS-2 cells, whereas Baf blocked the effects in both cell
types. These results demonstrated that PSM may induce ACD during
the initial 24 h post-stimulation.
PSM induces another reactive oxygen
species (ROS)-dependent cell death modality upon prolonged
treatment
PSM exerted higher degrees of cytotoxicity toward MM
and OS cells following prolonged treatment (72 h). As a result, PSM
(≥25%) markedy decreased the viability of HOS, 143B and A2058 cells
(Fig. 7A–C). Notably, in all cell
lines tested, these effects were not blocked by any of the
autophagy inhibitors (3-MA, Baf and CQ) (Fig. 7A–C). In addition, 3-MA (5 mM) and
CQ (100 µM) alone significantly reduced the viability of
143B cells. Previously, it was demonstrated that ROS serve a vital
role in mediating PSM cytotoxicity (30,37).
As a result, the superoxide oxidase mimetic antioxidant MnTBaP has
been revealed to suppress the effects of PSM in HOS cells, whereas
the effects were more pronounced in cells treated with PSM (25%)
compared with in those treated with PSM (100%) (37). In agreement with our previous
observations (30), MnTBaP
significantly reduced the cytotoxicity of PSM in the present study,
with a higher potency toward the lower concentration of PSM
(Fig. 7D). Conversely, the
pan-caspase-inhibitor z-VAD-FMK exhibited minimal effects on PSM
cytotoxicity regardless of the PSM concentration and cell type
examined (data not shown). These results indicated that PSM may
also induce another ROS-dependent, non-apoptotic, non-autophagic
cell death upon prolonged treatment.
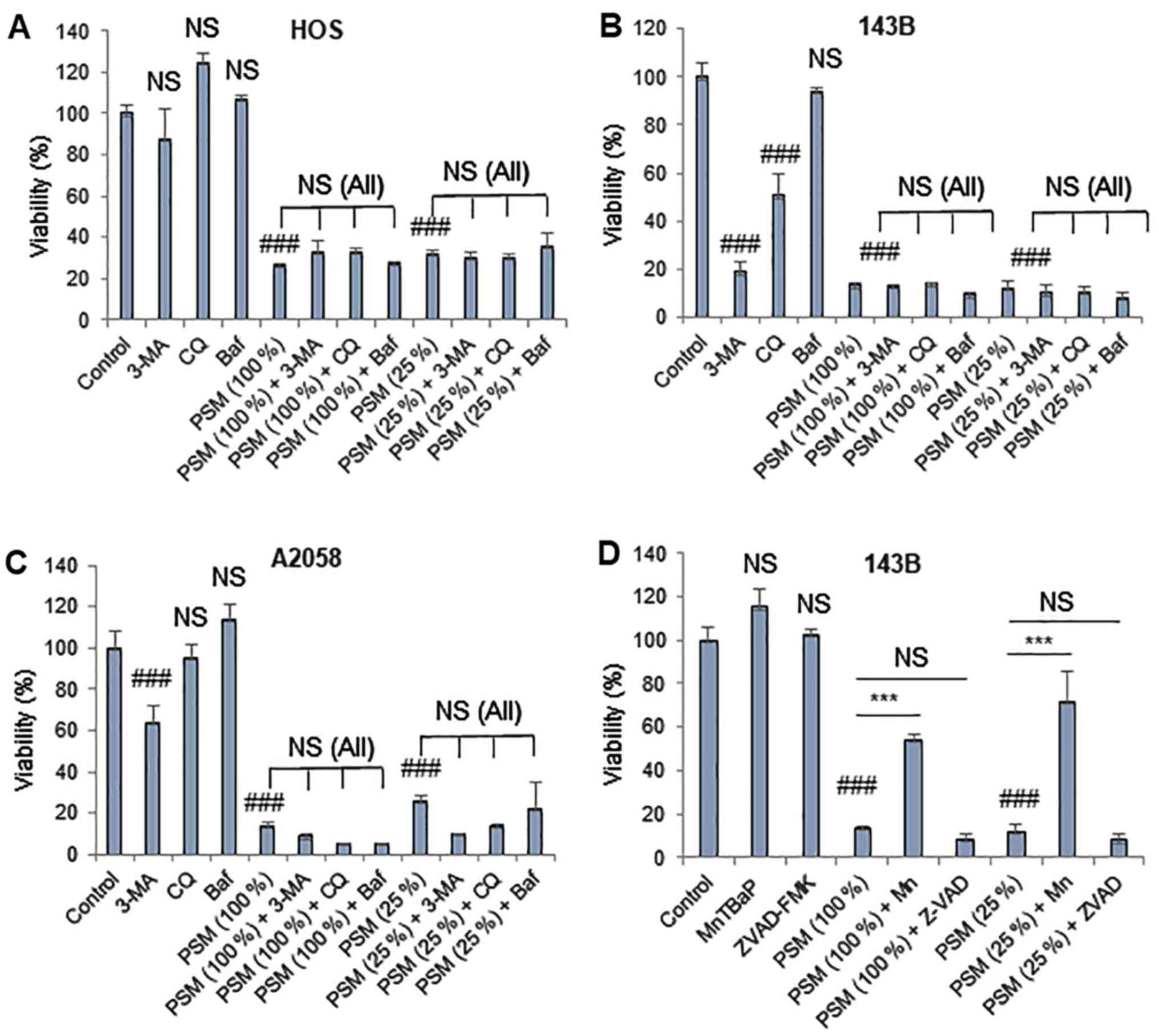 | Figure 7PSM induces another ROS-dependent
cell death modality upon prolonged treatment. (A) HOS, (B and D)
143B and (C) A2058 cells were treated with (A-C) PSM (25 and 100%)
and 5 mM 3-MA, 100 µM CQ and 100 nM Baf alone or in
combination for 72 h. (D) 143B cells were treated with PSM (25 and
100%) and 10 µM ZVAD and 30 µM Mn alone or in
combination for 72 h. Cell viability was analyzed using the WST-8
assay in triplicate. Data are presented as the means ± standard
deviation of a representative experiment (n=3). Data were analyzed
by analysis of variance followed by Tukey's post hoc test.
###P<0.001 vs. control; NS, not significant vs.
control; ***P<0.001. NS (All), no statistical
significance between PSM in combination with 3-MA, CQ and Baf
compared with PSM (25 and 100%) alone. 3-MA, 3-methyladenine; Baf,
bafilomycin A; CQ, chloroquine; Mn, MnTBaP; PSM, cold
plasma-stimulated medium; ZVAD, z-VAD-fluorometheylketone. |
TRAIL predominantly triggers
cytoprotective autophagy, which prevents apoptotic and
non-apoptotic cell death
Since TRAIL increased CYTO-ID puncta in cancer
cells, the present study explored the possible role of autophagic
flux in the antitumor activity of TRAIL. A 72-h-incubation protocol
was employed, because cells were highly TRAIL-resistant and TRAIL
treatment for 24 h had minimal cytotoxicity. The autophagy
inhibitors 3-MA (≥1.3 mM) and CQ (≥100 µM) alone
dose-dependently decreased the viability of A2058 and SAOS-2 cells.
Furthermore, the low concentration of 3-MA (1.3 mM) significantly
potentiated the cytotoxicity of TRAIL in the two cell lines
(Fig. 8A and B). Conversely, ≤300
nM Baf neither decreased cell viability nor enhanced the effect of
TRAIL, regardless of the cell type examined. The concentration of
3-MA suitable for TRAIL sensitization varied depending on the cell
lines tested (i.e., HOS and SAOS-2 cells), z-VAD-FMK entirely
suppressed the effect. Z-VAD-FMK significantly inhibited the
effects of 3-MA (2.5 mM) on HOS cells. In addition, z-VAD-FMK
completely blocked the effects of 3-MA (1.3 mM) and minimally
inhibited the effects of 3-MA (2.5 mM) on SAOS-2 cells (Fig. 8C and D). Furthermore, the present
study analyzed the effects of cotreatment with TRAIL and 3-MA on
autophagy and mitochondrial morphology. TRAIL alone increased
CYTO-ID puncta, whereas 3-MA alone abolished them in HOS cells
(Fig. 8E). Concomitantly,
mitochondria became hyperfused in response to TRAIL or 3-MA, as
shown by increased highly intra-connected mitochondria (Fig. 8E). The combined application of
TRAIL and 3-MA led to the formation of large clusters of CYTO-ID
puncta that colocalized with the mitochondria (Fig. 8E). Collectively, these results
indicated that TRAIL predominantly triggers cytoprotective
autophagy, the prevention of which may lead to colocalization of
the mitochondria and autophagosomes, apoptosis and another form of
non-apoptotic cell death.
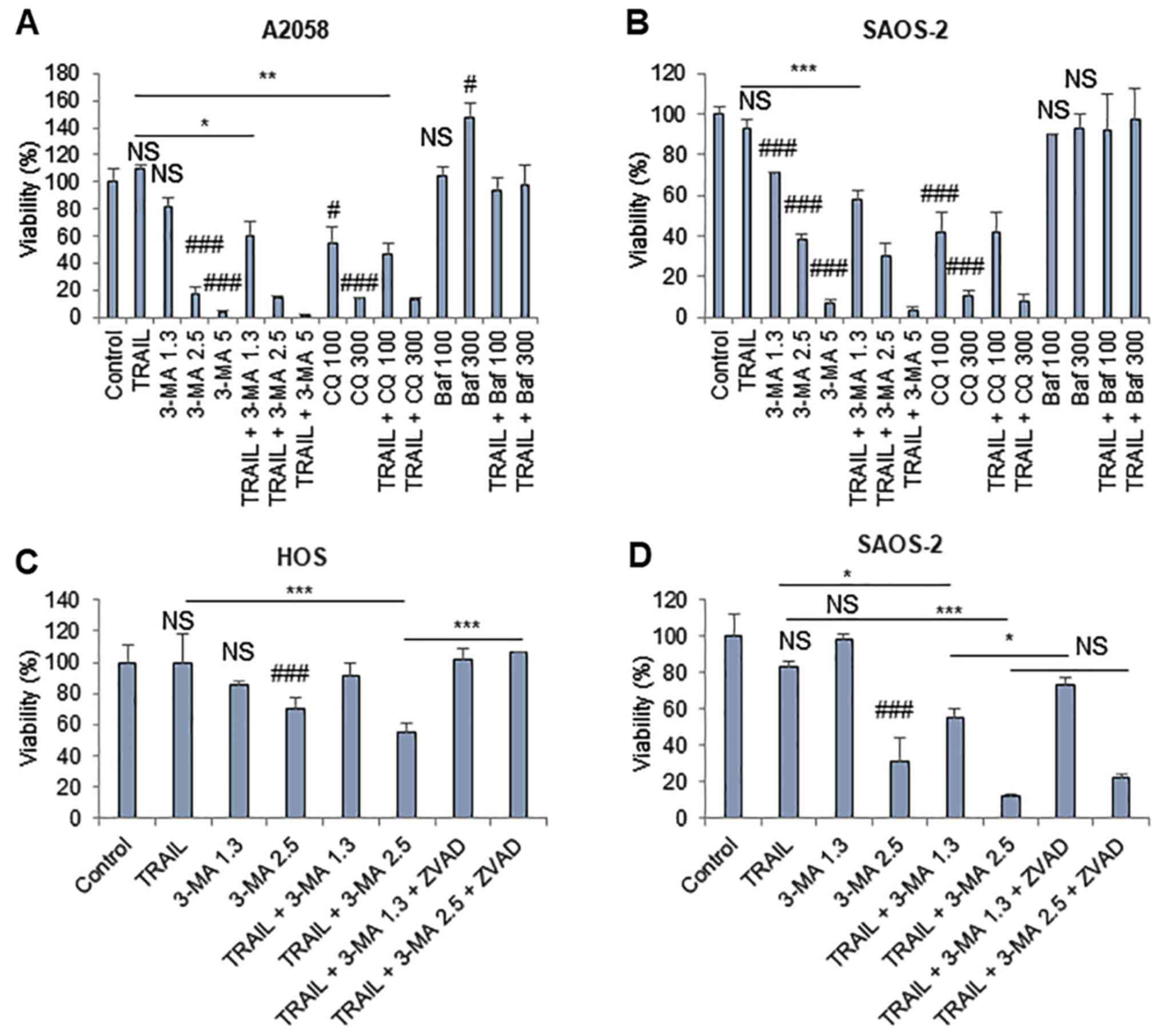 | Figure 8TRAIL predominantly triggers
cytoprotective autophagy. (A) A2058 and (B) SAOS-2 cells were
treated with 100 ng/ml TRAIL, 3-MA, CQ and Baf at the indicated
concentrations (3-MA, mM; CQ, µM; Baf, nM) alone or in
combination for 72 h. (C) HOS and (D) SAOS-2 cells were treated
with 100 ng/ml TRAIL and 3-MA at the indicated concentrations (mM)
for 72 h, in the absence or presence of 10 µM ZVAD. Cell
viability was analyzed using the WST-8 assay in triplicate. Data
are presented as the means ± standard deviation of a representative
experiment (n=3). Data were analyzed by analysis of variance
followed by Tukey's post hoc test. ###P<0.001;
#P<0.05 vs. control; NS, not significant.
***P<0.001; **P<0.01;
*P<0.05. (E) HOS cells were treated with 100 ng/ml
TRAIL and 5 mM 3-MA alone or in combination, and were stained with
CYTO-ID and MitoTracker Red CMXRos. Panels labeled 'Zoom' represent
the expansion of the small while boxes within 'Overlay' images.
Scale bar, 100 µm. 3-MA, 3-methyladenine; Baf, bafilomycin
A; CQ, chloroquine; Mn, MnTBaP; PSM, cold plasma-stimulated medium;
ZVAD, z-VAD-fluorometheylketone. |
Discussion
The present study aimed to determine whether PSM
modulated autophagy in human MM and OS cells. Western blotting
demonstrated that toxic, but not subtoxic, PSM increased LC3-II
expression, which is required for autophagosome formation.
Furthermore, the cytotoxic effects of PSM were strongly inhibited
following treatment with the pharmacological autophagy inhibitors
3-MA and Baf, thus indicating that autophagy is responsible for
cell death. These findings suggested that PSM may trigger ACD in
tumor cells. Notably, mitochondrial abnormalities accompanied the
induction of ACD. In agreement with our previous study (30), PSM induced mitochondrial
aberrations in a concentration-dependent manner. Toxic PSM led to
excessive mitochondrial fragmentation and aggregation, whereas
subtoxic PSM induced only modest mitochondrial fragmentation. In
addition, live-cell imaging of CYTO-ID puncta revealed marked
alterations in the status and location of autophagosomes in
response to PSM. When unstimulated, autophagosomes existed as small
diffuse particles, the majority of which were localized in the
cytoplasm separate from the mitochondria. However, following PSM
treatment, autophagosomes formed a cluster that colocalized with
the aggregated mitochondria. In mitophagy, damaged mitochondria are
engulfed by autophagosomes, so that mitochondria and autophagosomes
eventually colocalize. Therefore, the specific effects of toxic PSM
appear similar to mitophagy. Our previous study demonstrated that
mitochondria were heavily damaged, resulting in the loss of
mitochondrial membrane potential (ΔΨm) and integrity,
when they became punctate and aggregated (32,33).
Notably, dissipation of the ΔΨm is a primary trigger of
mitophagy (34). Diverse insults,
such as carbonyl cyanide m-chlorophenylhydrazone, carbonyl cyanide
p-triflouromethoxyphenylhydrazone and salinomycin, commonly induce
loss of the ΔΨm, thereby activating mitophagy (38,39).
Loss of the ΔΨm leads to the accumulation of phosphatase
and tensin homolog-induced putative kinase 1 (PINK1) on the outer
mitochondrial membrane. The localized PINK1 phosphorylates
E3-ubiquitin ligase Parkin and activates Parkin-mediated
ubiquitination, thus resulting in autophagic degradation of the
damaged organelles (34).
Therefore, it is possible that PSM injures the mitochondria,
thereby leading to mitophagy in the present cell system. p62 has
been reported to serve a vital role in mitophagy (40), whereas it is superfluous in some
circumstances (41). The present
study demonstrated that only marginal p62 accumulation occurred
following PSM treatment; therefore, the role of p62 in autophagic
flux appears to be minor in the present cell system. Several lines
of evidence indicated that the mitophagy-like event observed
promoted cell death and contributed to the antitumor activity of
PSM: i) The event spontaneously occurred in damaged or dying cancer
cells; ii) TRAIL, which had only a modest cytotoxic effect, did not
induce the event; iii) toxic, but not subtoxic PSM, caused the
event; (iv) TRAIL and 3-MA synergistically increased the event and
cell killing. It is widely accepted that controlled mitophagy is
cytoprotective, since it removes damaged mitochondria; however,
excessive mitophagy leads to cell damage by compromising the energy
supply, and Ca2+ and metabolic homeostasis. Therefore,
it is possible that PSM may induce excessive mitophagy, which is
responsible for the antitumor activity of PSM. However, further
studies are required to verify this hypothesis.
The present data indicated that a basal level of
autophagy operates in MM and OS cells; these cells possessed
substantial levels of autophagosomes even under nutritional and
stress-free conditions. Compared with the mitophagy-like event,
basal autophagy appeared to serve a cytoprotective role, since
autophagy inhibitors, including 3-MA and CQ alone exerted
significant cytotoxicity toward these cancer cells in the absence
of insult. Notably, the effects were more pronounced following 72 h
treatment compared with after 24 h. These observations indicated
that basal autophagy may be essential for cell survival,
particularly in response to hypo-nutritional conditions. The
present study suggested that in these cancer cells, TRAIL primarily
induced cytoprotective autophagy, since TRAIL increased
autophagosome formation, whereas its cytotoxicity was amplified,
rather than inhibited, following treatment with autophagy
inhibitors, including 3-MA. Notably, 3-MA/CQ and Baf had various
effects on cell viability and TRAIL cytotoxicity. At present, the
reason for the results are unclear, because they commonly serve as
autophagy inhibitors. However, it was previously demonstrated that
3-MA, but not Baf, interferes with mitochondrial Ca2+
loading, which is an essential process for energy production and
cell survival. These observations are intriguing, since our recent
study demonstrated that disturbance of mitochondrial
Ca2+ may lead to cancer cell death and sensitization to
TRAIL cytotoxicity (31).
Therefore, it is possible that the difference resides in
autophagy-independent effects. Further investigations are
required.
The results of the present study are consistent with
those previously reported in various cancer cell types, including
lung, bladder, prostate, colorectal cancer and hepatoma cells
(42–44). Notably, despite its ability to
trigger such cytoprotective autophagy, TRAIL minimally led to the
putative cytotoxic mitophagy-like event. Therefore, TRAIL may be
similar to various antitumor drugs, such as temozolomide,
epirubicin and sorafenib, which have been reported to engage
cytoprotective autophagy (45–48).
Furthermore, the blockade of basal autophagy by 3-MA enabled TRAIL
to increase the mitophagy-like event and kill TRAIL-resistant MM
and OS cells, whereas it promoted cell death, but not ACD. In
addition, the mitochondrial morphological alterations observed in
response to TRAIL + 3-MA appeared to be somewhat different from
those observed following toxic PSM treatment. Collectively, these
results suggested that suppression of basal cytoprotective
autophagy may promote the cytocidal mitophagy-like event, whereas
another fundamental cellular event is required for induction of the
full mitochondrial abnormalities leading to ACD induction. Further
studies are required to identify such events.
Recent evidence has indicated that autophagy and
apoptosis are closely associated, and that they may regulate one
another (49–51). This connection has a significant
impact on tumor cell sensitivity to antitumor chemotherapeutic
drugs. In particular, negative regulation of apoptosis by autophagy
is significant, since various antitumor drugs have been revealed to
induce cytoprotective autophagy, which allows cancer cells to cope
with apoptotic insults. Accordingly, the induction of autophagy in
cancer cells represents a double-edged sword, leading to cell
survival or ACD depending on the cell type and conditions.
Therefore, it is critical to understand the pathways controlling
context-dependent autophagy for the application of ACD in cancer
treatment.
In conclusion, the present study demonstrated that
PSM may trigger cytocidal autophagy, whereas TRAIL triggered
cytoprotective autophagy. To the best of our knowledge, the present
study is the first to demonstrate that PSM may induce ACD in human
cancer cells. These findings provide a rationale for the
development of PSM as a novel approach for the treatment of
apoptosis-resistant tumors, including MM and OS. In addition, PSM
may serve as a useful model for studying the mechanisms underlying
ACD induction in cancer cells.
Acknowledgments
The authors would like to thank the JCRB Cell Bank
of National Institutes of Biomedical Innovation, Health and
Nutrition (Osaka, Japan) and Riken BioResource Center (Tsukuba,
Japan) for providing cell lines.
Funding
This work was supported in part by JSPS KAKENHI
Grant Number 15K09750 to YSK and 15K09792 to TO.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request
Authors' contributions
TI performed experiments, analyzed data and wrote
the manuscript. TA performed experiments and wrote the manuscript.
MSK and TT performed experiments and analyzed data. YY, TO and YT
conceived and designed the study, performed the critical revision
of the manuscript, and provided analytical tools. YSK conceived and
designed the study, performed experiments and wrote the manuscript.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare that they have no competing
interests.
References
|
1
|
Almasan A and Ashkenazi A: Apo2L/TRAIL:
Apoptosis signaling, biology, and potential for cancer therapy.
Cytokine Growth Factor Rev. 14:337–348. 2003. View Article : Google Scholar
|
|
2
|
Johnstone RW, Frew AJ and Smyth MJ: The
TRAIL apoptotic pathway in cancer onset, progression and therapy.
Nat Rev Cancer. 8:782–798. 2008. View
Article : Google Scholar
|
|
3
|
Wang S: The promise of cancer therapeutics
targeting the TNF-related apoptosis-inducing ligand and TRAIL
receptor pathway. Oncogene. 27:6207–6215. 2008. View Article : Google Scholar
|
|
4
|
Gonzalvez F and Ashkenazi A: New insights
into apoptosis signaling by Apo2L/TRAIL. Oncogene. 29:4752–4765.
2010. View Article : Google Scholar
|
|
5
|
Kischkel FC, Lawrence DA, Chuntharapai A,
Schow P, Kim KJ and Ashkenazi A: Apo2L/TRAIL-dependent recruitment
of endogenous FADD and caspase-8 to death receptors 4 and 5.
Immunity. 12:611–620. 2000. View Article : Google Scholar
|
|
6
|
LeBlanc HN and Ashkenazi A: Apo2L/TRAIL
and its death and decoy receptors. Cell Death Differ. 10:66–75.
2003. View Article : Google Scholar
|
|
7
|
Ivanov VN, Bhoumik A and Ronai Z: Death
receptors and melanoma resistance to apoptosis. Oncogene.
22:3152–3161. 2003. View Article : Google Scholar
|
|
8
|
Dyer MJ, MacFarlane M and Cohen GM:
Barriers to effective TRAIL-targeted therapy of malignancy. J Clin
Oncol 25. 25:4505–4506. 2007. View Article : Google Scholar
|
|
9
|
Dimberg LY, Anderson CK, Camidge R,
Behbakht K, Thorburn A and Ford HL: On the TRAIL to successful
cancer therapy? Predicting and counteracting resistance against
TRAIL-based therapeutics. Oncogene. 32:1341–1350. 2013. View Article : Google Scholar
|
|
10
|
Guiho R, Biteau K, Heymann D and Redini F:
TRAIL-based therapy in pediatric bone tumors: How to overcome
resistance. Future Oncol. 11:535–542. 2015. View Article : Google Scholar
|
|
11
|
de Miguel D, Lemke J, Anel A, Walczak H
and Martinez-Lostao L: Onto better TRAILs for cancer treatment.
Cell Death Differ. 23:733–747. 2016. View Article : Google Scholar
|
|
12
|
Keidar M, Walk R, Shashurin A, Srinivasan
P, Sandler A, Dasgupta S, Ravi R, Guerrero-Preston R and Trink B:
Cold plasma selectivity and the possibility of a paradigm shift in
cancer therapy. Br J Cancer. 105:1295–1301. 2011. View Article : Google Scholar
|
|
13
|
Zucker SN, Zirnheld J, Bagati A, DiSanto
TM, Des Soye B, Wawrzyniak JA, Etemadi K, Nikiforov M and Berezney
R: Preferential induction of apoptotic cell death in melanoma cells
as compared with normal keratinocytes using a non-thermal plasma
torch. Cancer Biol Ther. 13:1299–1306. 2012. View Article : Google Scholar
|
|
14
|
Arndt S, Unger P, Wacker E, Shimizu T,
Heinlin J, Li YF, Thomas HM, Morfill GE, Zimmermann JL, Bosserhoff
AK, et al: Cold atmospheric plasma (CAP) changes gene expression of
key molecules of the wound healing machinery and improves wound
healing in vitro and in vivo. PLoS One. 8:e793252013. View Article : Google Scholar
|
|
15
|
Ishaq M, Evans MM and Ostrikov KK: Effect
of atmospheric gas plasmas on cancer cell signaling. Int J Cancer.
134:1517–1528. 2014. View Article : Google Scholar
|
|
16
|
Utsumi F, Kajiyama H, Nakamura K, Tanaka
H, Mizuno M, Ishikawa K, Kondo H, Kano H, Hori M and Kikkawa F:
Effect of indirect nonequilibrium atmospheric pressure plasma on
anti-proliferative activity against chronic chemo-resistant ovarian
cancer cells in vitro and in vivo. PLoS One. 8:e815762013.
View Article : Google Scholar
|
|
17
|
Torii K, Yamada S, Nakamura K, Tanaka H,
Kajiyama H, Tanahashi K, Iwata N, Kanda M, Kobayashi D, Tanaka C,
et al: Effectiveness of plasma treatment on gastric cancer cells.
Gastric Cancer. 18:635–643. 2015. View Article : Google Scholar
|
|
18
|
Hattori N, Yamada S, Torii K, Takeda S,
Nakamura K, Tanaka H, Kajiyama H, Kanda M, Fujii T, Nakayama G, et
al: Effectiveness of plasma treatment on pancreatic cancer cells.
Int J Oncol. 47:1655–1662. 2015. View Article : Google Scholar
|
|
19
|
Adachi T, Tanaka H, Nonomura S, Hara H,
Kondo S and Hori M: Plasma-activated medium induces A549 cell
injury via a spiral apoptotic cascade involving the
mitochondrial-nuclear network. Free Radic Biol Med. 79:28–44. 2015.
View Article : Google Scholar
|
|
20
|
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT,
Liu B and Bao JK: Programmed cell death pathways in cancer: A
review of apoptosis, autophagy and programmed necrosis. Cell
Prolif. 45:487–498. 2012. View Article : Google Scholar
|
|
21
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar
|
|
22
|
Mizushima N, Yoshimori T and Ohsumi Y: The
role of Atg proteins in autophagosome formation. Annu Rev Cell Dev
Biol. 27:107–132. 2011. View Article : Google Scholar
|
|
23
|
Codogno P and Meijer AJ: Autophagy and
signaling: Their role in cell survival and cell death. Cell Death
Differ. 12(Suppl 2): 1509–1518. 2005. View Article : Google Scholar
|
|
24
|
Díaz-Troya S, Pérez-Pérez ME, Florencio FJ
and Crespo JL: The role of TOR in autophagy regulation from yeast
to plants and mammals. Autophagy. 4:851–865. 2008. View Article : Google Scholar
|
|
25
|
Dennis MD, Baum JI, Kimball SR and
Jefferson LS: Mechanisms involved in the coordinate regulation of
mTORC1 by insulin and amino acids. J Biol Chem. 286:8287–8296.
2011. View Article : Google Scholar
|
|
26
|
Bhutia SK, Mukhopadhyay S, Sinha N, Das
DN, Panda PK, Patra SK, Maiti TK, Mandal M, Dent P, Wang XY, et al:
Autophagy: Cancer's friend or foe? Adv Cancer Res. 118:61–95. 2013.
View Article : Google Scholar
|
|
27
|
Gozuacik D and Kimchi A: Autophagy as a
cell death and tumor suppressor mechanism. Oncogene. 23:2891–2906.
2004. View Article : Google Scholar
|
|
28
|
Fulda S and Kögel D: Cell death by
autophagy: Emerging molecular mechanisms and implications for
cancer therapy. Oncogene. 34:5105–5113. 2015. View Article : Google Scholar
|
|
29
|
Fulda S: Autophagy in cancer therapy.
Front Oncol. 7:1282017. View Article : Google Scholar
|
|
30
|
Saito K, Asai T, Fujiwara K, Sahara J,
Koguchi H, Fukuda N, Suzuki-Karasaki M, Soma M and Suzuki-Karasaki
Y: Tumor-selective mitochondrial network collapse induced by
atmospheric gas plasma-activated medium. Oncotarget. 7:19910–19927.
2016. View Article : Google Scholar
|
|
31
|
Takata N, Ohshima Y, Suzuki-Karasaki M,
Yoshida Y, Tokuhashi Y and Suzuki-Karasaki Y: Mitochondrial
Ca2+ removal amplifies TRAIL cytotoxicity toward
apoptosis-resistant tumor cells via promotion of multiple cell
death modalities. Int J Oncol. 51:193–203. 2017. View Article : Google Scholar
|
|
32
|
Suzuki-Karasaki Y, Fujiwara K, Saito K,
Suzuki-Karasaki M, Ochiai T and Soma M: Distinct effects of TRAIL
on the mitochondrial network in human cancer cells and normal
cells: Role of plasma membrane depolarization. Oncotarget.
6:21572–21588. 2015. View Article : Google Scholar
|
|
33
|
Akita M, Suzuki-Karasaki M, Fujiwara K,
Nakagawa C, Soma M, Yoshida Y, Ochiai T, Tokuhashi Y and
Suzuki-Karasaki Y: Mitochondrial division inhibitor-1 induces
mitochondrial hyperfusion and sensitizes human cancer cells to
TRAIL-induced apoptosis. Int J Oncol. 45:1901–1912. 2014.
View Article : Google Scholar
|
|
34
|
Kulikov AV, Luchkina EA, Gogvadze V and
Zhivotovsky B: Mitophagy: Link to cancer development and therapy.
Biochem Biophys Res Commun. 482:432–439. 2017. View Article : Google Scholar
|
|
35
|
Bordi M, Nazio F and Campello S: The close
interconnection between mitochondrial dynamics and mitophagy in
cancer. Front Oncol. 7:812017. View Article : Google Scholar
|
|
36
|
Lystad AH, Ichimura Y, Takagi K, Yang Y,
Pankiv S, Kanegae Y, Kageyama S, Suzuki M, Saito I, Mizushima T, et
al: Structural determinants in GABARAP required for the selective
binding and recruitment of ALFY to LC3B-positive structures. EMBO
Rep. 15:557–565. 2014. View Article : Google Scholar
|
|
37
|
Tokunaga T, Ando T, Suzuki-Karasaki M, Ito
T, Onoe-Takahashi A, Ochiai T, Soma M and Suzuki-Karasaki Y:
Plasma-stimulated medium kills TRAIL-resistant human malignant
cells by promoting caspase-independent cell death via membrane
potential and calcium dynamics modulation. Int J Oncol. 52:697–708.
2018.
|
|
38
|
Narendra DP, Jin SM, Tanaka A, Suen DF,
Gautier CA, Shen J, Cookson MR and Youle RJ: PINK1 is selectively
stabilized on impaired mitochondria to activate Parkin. PLoS Biol.
8:e10002982010. View Article : Google Scholar
|
|
39
|
Jangamreddy JR, Ghavami S, Grabarek J,
Kratz G, Wiechec E, Fredriksson BA, Rao Pariti RK, Cieślar-Pobuda
A, Panigrahi S and Łos MJ: Salinomycin induces activation of
autophagy, mitophagy and affects mitochondrial polarity:
Differences between primary and cancer cells. Biochim Biophys Acta.
1833:2057–2069. 2013. View Article : Google Scholar
|
|
40
|
Geisler S, Holmström KM, Skujat D, Fiesel
FC, Rothfuss OC, Kahle PJ and Springer W: PINK1/Parkin-mediated
mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol.
12:119–131. 2010. View Article : Google Scholar
|
|
41
|
Narendra D, Kane LA, Hauser DN, Fearnley
IM and Youle RJ: p62/SQSTM1 is required for Parkin-induced
mitochondrial clustering but not mitophagy; VDAC1 is dispensable
for both. Autophagy. 6:1090–1106. 2010. View Article : Google Scholar
|
|
42
|
Knoll G, Bittner S, Kurz M, Jantsch J and
Ehrenschwender M: Hypoxia regulates TRAIL sensitivity of colorectal
cancer cells through mitochondrial autophagy. Oncotarget.
7:41488–41504. 2016. View Article : Google Scholar
|
|
43
|
He W, Wang Q, Xu J, Xu X, Padilla MT, Ren
G, Gou X and Lin Y: Attenuation of TNFSF10/TRAIL-induced apoptosis
by an autophagic survival pathway involving TRAF2- and
RIPK1/RIP1-mediated MAPK8/JNK activation. Autophagy. 8:1811–1821.
2012. View Article : Google Scholar
|
|
44
|
Lim SC, Jeon HJ, Kee KH, Lee MJ, Hong R
and Han SI: Involvement of DR4/JNK pathway-mediated autophagy in
acquired TRAIL resistance in HepG2 cells. Int J Oncol.
49:1983–1990. 2016. View Article : Google Scholar
|
|
45
|
Kanzawa T, Germano IM, Komata T, Ito H,
Kondo Y and Kondo S: Role of autophagy in temozolomide-induced
cytotoxicity for malignant glioma cells. Cell Death Differ.
11:448–457. 2004. View Article : Google Scholar
|
|
46
|
Knizhnik AV, Roos WP, Nikolova T, Quiros
S, Tomaszowski KH, Christmann M and Kaina B: Survival and death
strategies in glioma cells: Autophagy, senescence and apoptosis
triggered by a single type of temozolomide-induced DNA damage. PLoS
One. 8:e556652013. View Article : Google Scholar
|
|
47
|
Guo W, Wang Y, Wang Z, Wang YP and Zheng
H: Inhibiting autophagy increases epirubicin's cytotoxicity in
breast cancer cells. Cancer Sci. 107:1610–1621. 2016. View Article : Google Scholar
|
|
48
|
Prieto-Domínguez N, Ordóñez R, Fernández
A, García-Palomo A, Muntané J, González-Gallego J and Mauriz JL:
Modulation of autophagy by sorafenib: Effects on treatment
response. Front Pharmacol. 7:1512016. View Article : Google Scholar
|
|
49
|
Lockshin RA and Zakeri Z: Apoptosis,
autophagy, and more. Int J Biochem Cell Biol. 36:2405–2419. 2004.
View Article : Google Scholar
|
|
50
|
Eisenberg-Lerner A, Bialik S, Simon HU and
Kimchi A: Life and death partners: Apoptosis, autophagy and the
cross-talk between them. Cell Death Differ. 16:966–975. 2009.
View Article : Google Scholar
|
|
51
|
Mukhopadhyay S, Panda PK, Sinha N, Das DN
and Bhutia SK: Autophagy and apoptosis: Where do they meet.
Apoptosis. 19:555–566. 2014. View Article : Google Scholar
|