Introduction
Cervical cancer (CC) development is associated with
infection with high-risk human papillomavirus (HR-HPV) types,
predominantly HPV16 and HPV18 (1).
CC accounts for nearly 300,000 mortalities worldwide every year
(2), representing a medical
challenge in developing countries, and HPV16 is the most prevalent
viral subtype that is responsible for ~50% of CC cases (3). The HR-HPV oncoproteins E6 and E7 are
involved in the induction and maintenance of CC; however, other
co-factors are necessary for CC development. For example, in
transgenic mice, it was found that E7 cooperates with
17β-oestradiol (E2) to induce high-grade dysplasia,
in situ carcinoma and CC (4). Additionally, it has been shown that
E2 contributes not only to the onset, but also to the
persistence and malignant progression of CC in K14E7 transgenic
mice (5); notably, E2
fails to promote dysplasia or CC in K14E7 mice that lack oestrogen
receptors(6).
These studies suggest that E2 has a
prominent role together with the E7 protein in cell transformation
and tumour development during cervical carcinogenesis (7). However, little is known about the
causal and temporal associations between genotypic and phenotypic
alterations, particularly associated with E7 plus E2, in
the induction of cervical malignant lesions, as well as the
potential role of the cellular stress response in the earliest
stages of cervical carcinogenesis (8).
In the present study, global gene expression
profiling and RT-qPCR analysis were performed to determine the mRNA
expression landscape of cervical tissues obtained from 2-month-old
mice, in order to identify genes regulated by HPV16-E7 and
E2 that may serve a role in early cervical
carcinogenesis. Differentially expressed cancer-related genes were
identified that have a synergistic effect at the transcriptional
level on factors associated with cellular movement, cancer,
metabolism, apoptosis, cellular growth and proliferation, as well
as the regulation of cell morphology and inflammatory responses. Of
particular note was the downregulation of the Granzyme B (GrB)
pathway as a possible mechanism of immune evasion.
GrB/perforin-induced cell death is one of the main mechanisms used
by cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells to
destroy allogenic, virus-infected and tumour cells (9). However, it is known that GrB has
extracellular functions and is expressed in non-immune cells,
including smooth muscle cells, chondrocytes and keratinocytes
(10,11), as well as in the prostate cancer
PC-3 and DU145 cell lines (12),
and that the activation of GrB could be potentially harmful to
cells producing GrB. In this regard, it has been shown that the
activation of GrB by ultraviolet A (UVA) radiation in keratinocytes
is partially responsible for the reduction of viable cells
(13). Furthermore, in prostate
cancer cells, resveratrol has been shown to induce the expression
of GrB, which is correlated with enhanced apoptosis and increased
radiosensitivity of the PCA cells (12). As GrB pathway components are often
elevated in chronic inflammation-associated and/or degenerative
pathologies (14), and as GrB
produced in keratinocytes and other non-immune cells is active and
partially responsible for the induction of death in these cells
(12,13), the inhibition of GrB pathway
components may suppress the activation of the GrB pathway in CC
cells, thereby inducing neoplasia. In addition, the upregulation of
the natural inhibitor of GrB, the serine protease inhibitor
SERPINB9 [also known as protease inhibitor-9 (PI-9)], can also
mediate the inhibition of endogenous and exogenous GrB secretion by
immune cells (15,16). The present study opens the
possibility of assessing and validating novel early-risk biomarkers
and therapeutic targets for CC, enabling a better understanding of
the molecular mechanisms by which HPV16-E7 and E2 can
lead to CC development.
Materials and methods
Mouse models and hormone treatment
The K14E7 and FVB mouse models have been previously
described (4). Briefly, mice were
obtained from the mouse repository located at National Cancer
Institute at Frederick campus (Frederick, MD, USA). The K14E7
transgenic mice were generated and maintained in the FVB/N inbred
strain and used as transgenic heterozygotes harbouring overlapping
HPV16 E6 and E7 open reading frames (ORFs) spanning nucleotides
79-883 with a TTL in the E6 gene to preclude E6 expression.
Persistent oestrogen administration and E7 expression were
sufficient to produce high-grade cervical dysplasia and invasive
cervical malignancies (17). For
this study, four groups were created: FVB (not transgenic, not
treated, control group), FVB+E2 (not transgenic, treated
group), K14E7 (transgenic, untreated group) and K14E7+E2
(transgenic, treated group). The mice were housed and treated
according to guidelines from the Association for Assessment and
Accreditation of Laboratory Animal Care International and the
National Institutes of Health Guide for the Care and Use of
Laboratory Animals (publication no. 8023, revised 1978). The
euthanasia procedure via use of a CO2 chamber was
followed according to the American Veterinary Medical Association's
Guidelines for the Euthanasia of Animals (2013 edition).
All experiments and procedures were approved by the
Research Unit for Laboratory Animal Care Committee
(NOM-062-ZOO-1999; Unit for the Production and Experimentation of
Laboratory Animals, The Centre for Research and Advanced Studies of
the National Polytechnic Institute, México City, México). The
environmental conditions for the mice included free access to
food/water with a standard diet of LabDiet®, a 12:12
light:dark cycle, a room temperature of 25°C and 30% humidity, with
housing in Super Mouse 750™ Micro-Isolator® cages in
groups of 4–5 mice. Female, 1-month-old, virgin transgenic and
non-transgenic mice, weighing 12–13 g, were dorsally implanted with
continuous release pellets under the skin, which delivered 0.05 mg
E2 (Innovative Research of America, Sarasota, FL, USA)
over 60 days. After 1 month of treatment, the mice, weighing 13-15
g, were sacrificed at 2 months of age. Control and treatment groups
were formed using 20 to 24 mice in each.
Tissue procurement and
histopathology
Untreated or treated K14E7 hemizygous and
non-transgenic FVB control virgin female mice were sacrificed by
CO2 chamber. All specimens were fixed in 4%
paraformaldehyde at 4°C overnight and embedded in paraffin.
Sections were deparaffinized and rehydrated as previously described
(18). Serial sections were cut
(at 5-μm thickness) and stained with haematoxylin and eosin
(H&E) at room temperature (10 min haematoxylin and 2 min
eosin).
Tissue procurement and microarray sample
processing
Untreated or treated K14E7 hemizygous and
non-transgenic FVB control virgin female mice were sacrificed by
CO2 chamber. Cervical biopsy samples were immediately
stored in RNAlater® solution (Thermo Fisher Scientific,
Inc., Waltham, MA, USA) at 4°C overnight. Tissues were recovered
from RNAlater solution with sterile forceps, quickly blotted to
remove excess solution and immediately snap-frozen in liquid
nitrogen. Total RNA was extracted from snap-frozen tissues using
standard procedures with TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). In total, 2 pooled samples from 3 mice were used
to avoid variability and to identify biomarkers for the early
stages of carcinogenesis in a population of K14E7 mice treated with
E2, as recommended by previous studies (19–21).
Total RNA collected from 6 female mice from each group was pooled.
RNA quantity and quality were assessed on an Agilent 2100
Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA, USA). RNA
samples with an RNA integrity score of >8.0 were further
processed for microarray analysis. cDNA synthesis, amplification
and gene expression profiling were performed according to the
manufacturer's protocols (Affymetrix® WT Sense Target
Labelling assay; Thermo Fisher Scientific, Inc.). Affymetrix
microarray experiments were performed in triplicate for each group
in the GeneChip Mouse Gene 1.0 ST Array platform.
Analysis of array data
Signal intensities from each array were analysed
using Partek Genomic Suite version 6.4 (Partek, Inc., Chesterfield,
MO, USA). Raw intensity values for the probes were normalized using
robust multiarray analysis background correction. Two-way analysis
of variance (ANOVA) was performed to identify differentially
expressed genes. Genes with statistically significant differences
in expression levels (P<0.05) and a fold change of ≥1.5 and
≤−1.5 were included in the final set of differentially expressed
genes. To identify the biological processes altered by E7 and/or
E2, Ingenuity Pathway Analysis (IPA; https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/;
Ingenuity Systems, Redwood City, CA, USA), a bioinformatics tool
for visualizing expression data in the context of Kyoto
Encyclopaedia of Genes and Genomes-defined biological pathways
(http://www.genome.jp/kegg/) was
used.
Relative mRNA quantification by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
data analysis using the 2−∆∆Cq method
RNA isolated by TRIzol reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) from 6 mice from each group (K14E7, FVB,
K14E7+E2 and FVB+E2) was purified, and RNA
quality determined by electrophoresis on a 2% agarose gel, with
visualization of ribosomal RNA by ethidium bromide staining. RNA
was quantified by spectrophotometric analysis at 260 and 280 nm.
cDNA synthesis was performed using the Maxima First Strand cDNA
Synthesis kit according to the manufacturer's protocols (Thermo
Fisher Scientific, Inc.). RT-qPCR was performed using an Applied
Biosystems 7300 instrument and a DNA Master SYBR Green I kit (both
Thermo Fisher Scientific, Inc.). Templates were amplified using 45
cycles of a 3-step PCR protocol, which included 30 sec of
denaturation at 95°C, 30 sec of primer-dependent annealing at 60°C
and 30 sec of template-dependent elongation at 72°C. Each
gene-specific RNA was quantified in triplicate by qPCR, and mRNA
ratios relative to the house-keeping gene GAPDH were calculated for
standardization of gene expression levels across samples using the
2−ΔΔCq method (22).
All primer sequences and product sizes are shown in Table I.
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Gene name | Forward
primers | Reverse
primers |
|---|
| Gapdh
(mouse) | 5′-GTG GAG TCA TAC
TGG AAC ATG TAG-3′ | 5′-AAT GGT GAA GGT
CGG TGT G-3′ |
| Gzmb
(mouse) | 5′-CAT GTC CCC CGA
TGA TCT C-3′ | 5′-AAG AGA GCA AGG
ACA ACA CTC-3′ |
| Gzmc
(mouse) | 5′-CTC CTC CTT AGC
CTT GAT GTT G-3′ | 5′-CGA GAC AAA TTC
GTG CTA ACA G-3′ |
| Gzmd
(mouse) | 5′-GAA GCC TCC ACA
GTA TAT CCT G-3′ | 5′-CCT GAT TCT CCT
GAC CCT ACT-3′ |
| Gzme
(mouse) | 5′-CCT CCA CAG TAT
CTC CTA TTA CCT-3′ | 5′-ACC AGT CCT GAT
TCT CCT GA-3′ |
| Gzmf
(mouse) | 5′-CCA TTA TCT TTC
ACA AAC CTC ACA C-3′ | 5′-TGG AGC AGA GGA
GAT CAT CG-3′ |
| Gzmg
(mouse) | 5′-ACT CCA TAA GCT
AGG TTG TCA C-3′ | 5′-CTA TTC CAA GAC
CAC GCA GAT-3′ |
| Gdpd3
(mouse) | 5′-GTT CAT CCA TCC
ACA GCG AA-3′ | 5′-CCT TTT GTC TCC
ATC CCT GA-3′ |
| Lrat
(mouse) | 5′-AAG ACA GCC GAA
GCA AGA C-3′ | 5′-GCG AAC ACT TTG
TGA CTT ACT G-3′ |
| Nppc
(mouse) | 5′-CAT TGC GTT GGA
GGT GTT TC-3′ | 5′-GGT CTG GGA TGT
TAG TGC AG-3′ |
| Mgat4c
(mouse) | 5′-ACT GAT GAA AGT
CCA ATT GTG AG-3 | 5′-CAC CAA CTT AAT
TCT GAA CGC T3′ |
| Cyp2e1
(mouse) | 5′-GCC TCA TTA CCC
TGT TTC CC-3 | 5′-TTC CAG GAG TAC
AAG AAC AAG G-3′ |
| Il1r2
(mouse) | 5′-CCT TCC AGC CTC
AAT TCA GAT-3 | 5′-TGC TTT CAC CAC
TCC AAC AG-3′ |
| Stat5a
(mouse) | 5′-GCT CTC ATC CAG
GTC AAA CTC-3 | 5′-TGC CCT CAA CCT
CAC TAC A-3′ |
| Eomes
(mouse) | 5′-CCA GAA CCA CTT
CCA CGA AA-3 | 5′-CGG CAC CAA ACT
GAG ATG A-3′ |
| Gxylt2
(mouse) | 5′-TGG CCC CAG AGC
ATG AAA -3′ | 5′-CCG GGC AAA GCG
ACT GTA-3′ |
| Serpinb9
(mouse) | 5′-GTG CCA TTT CCT
TCA GAC AG-3 | 5′-GAA GTC CCT GCC
TTG TAC AG-3′ |
| SERPINB9B
(human) | 5′-AGT GAG AAG CGA
CTG GAA AG-3′ | 5′-CTG TTC TCC TGT
GAG CAT CTC-3′ |
| GZMB
(human) | 5′-CAG AGA CTT CTG
ATC CCA GAT-3 | 5′-TCC TGA GAA GAT
GCA ACC AAT -3′ |
| B2M
(human) | 5′-ACC TCC ATG ATG
CTG CTT AC-3 | 5′-GGA CTG GTC TTT
CTA TCT CTT GT-3′ |
Cell culture and transfection
Human foreskin primary keratinocytes (HFKs) were
obtained from the American Type Culture Collection (Manassas, VA,
USA) and cultured at 37°C in a humidified atmosphere with 5%
CO2 in Dulbecco's modified Eagle's medium:F12
supplemented with 0.18 mM adenine (Sigma-Aldrich®; Merck
KGaA, Darmstadt, Germany), 0.1 μg/ml hydrocortisone
(Sigma-Aldrich; Merck KGaA), 100X Human Keratinocyte Growth
Supplement (Thermo Fisher Scientific, Inc.), 2% foetal bovine serum
(Gibco; Thermo Fisher Scientific, Inc.) and an
antibiotic-antimycotic mixture (Invitrogen; Thermo Fisher
Scientific, Inc.). The cells were transfected with the HPV16-E7
oncogene using Attractene® reagent (Qiagen GmbH, Hilden,
Germany) according to the manufacturer's recommended protocols. The
cells were seeded in 6-well plates (3×105 cells/well),
and the next day, the cells were transfected with a mixture of 4.5
μl Attractene/well and 1.2 μg plasmid (PCDNA3
HPV-16E7) per well. To obtain transient and stable transfected cell
lines, the expression of β2-microglobulin was used as an internal
control of gene expression in the qPCR assay. Cells were selected
and maintained in growth media containing 200 mg/ml geneticin
(G418; Invitrogen; Thermo Fisher Scientific, Inc.).
Immunohistochemistry and
immunofluorescence procedures
Cervical tissue sections were fixed in 4%
paraformaldehyde overnight at 4°C for immunohistochemical staining.
Tissues were cut into 5-μm sections with a microtome and
placed on electro-charged slides (Van-Wessel, Querétaro, México)
and preserved using GVA mount solution (Zymed®; Thermo
Fisher Scientific, Inc.). Protein detection for
immunohistochemistry was conducted using the Mouse/Rabbit
PolyDetector HRP/DAB Detection system® (Bio SB, Santa
Barbara, CA, USA), according to the manufacturer's protocols, and
HRP-conjugated anti-rat antibodies. The samples were incubated at
4°C overnight with primary antibodies against Ki-67 (cat. no. BSB
5713; 1:100 dilution; Bio SB), p16INK4a (cat. no.
sc-468; 1:100 dilution; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA), Granzyme B (GZMB; cat. no. AB4059; 1:100 dilution; Abcam,
Cambridge, MA, USA) or PI-9 (cat. no. sc-57531; 1:100 dilution;
Santa Cruz Biotechnology, Inc.). Anti-rabbit HRP-conjugated
antibodies (1:200 dilution; cat. no. A0545; Sigma-Aldrich; Merck
KGaA) were used as secondary antibodies for the detection of
proteins. Following immunohistochemical procedures, the tissues
were counterstained with haematoxylin (10 min haematoxylin and 2
min eosin) at room temperature and mounted using GVA mount solution
(Zymed; Thermo Fisher Scientific, Inc.). For immunofluorescence,
cervical tissue sections were rinsed in 1X PBS and blocked for 2 h
at 4°C with 1X PBS supplemented with 0.3% Triton X-100 and 1%
bovine serum albumin (Sigma-Aldrich; Merck KGaA). Next, the
sections were washed three times with 1X PBS and incubated for 1 h
at 37°C with the aforementioned primary antibodies against GZMB or
PI-9. The sections were then incubated with a fluorescein
isothiocyanate (FITC)-labelled secondary antibody (rabbit; 1:50
dilution; cat. no. 65-6111; Zymed; Thermo Fisher Scientific, Inc.)
for 30 min at room temperature. The sections were then rinsed as
aforementioned, counterstained with propidium iodide (30 min at
room temperature) and mounted using Vectashield® (Vector
Laboratories, Inc., Burlingame, CA, USA). The stained sections were
examined by confocal microscopy using a Leica TCS SP2 (Leica
Microsystems, Inc., Buffalo Grove, IL, USA). Captured images were
imported into Adobe Photoshop CS6 (Adobe Systems, Inc., San Jose,
CA, USA) to generate maximum projections.
UV irradiation of HFKs
UVC irradiation was performed with a 1300 Series
Class II, Type A2 Biological Safety Cabinet (Thermo Fisher
Scientific, Inc.). HFKs were grown at 37°C in a humidified
atmosphere with 5% CO2 in DMEM:F12 media supplemented
with 0.18 mM adenine (Sigma-Aldrich; Merck KGaA), 0.1 μg/ml
hydrocortisone (Sigma-Aldrich; Merck KGaA), 4 μg/ml insulin
(Gibco; Thermo Fisher Scientific, Inc.), 20 ng/ml recombinant
epidermal growth factor (PeproTech, Inc., Rocky Hill, NJ, USA), 5%
foetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.) and an
antibiotic-antimycotic mixture (Invitrogen®). HFKs were
irradiated with UVC radiation for 30 sec in the absence of medium.
Following the addition of DMEM:F12-HFK medium, HFKs were cultured
for 24 h prior to harvesting.
Statistical analysis
For all microarray data comparisons, ANOVA with
Tukey's honest significant difference post hoc test was performed
using Partek Genomics Suite® 6.6 Software (Affymetrix;
Thermo Fisher Scientific, Inc). For all RT-qPCR data comparisons a
one-way ANOVA with Bonferroni correction was used, performed in
GraphPad Prism 5 (Graphpad Software, Inc., La Jolla, CA, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
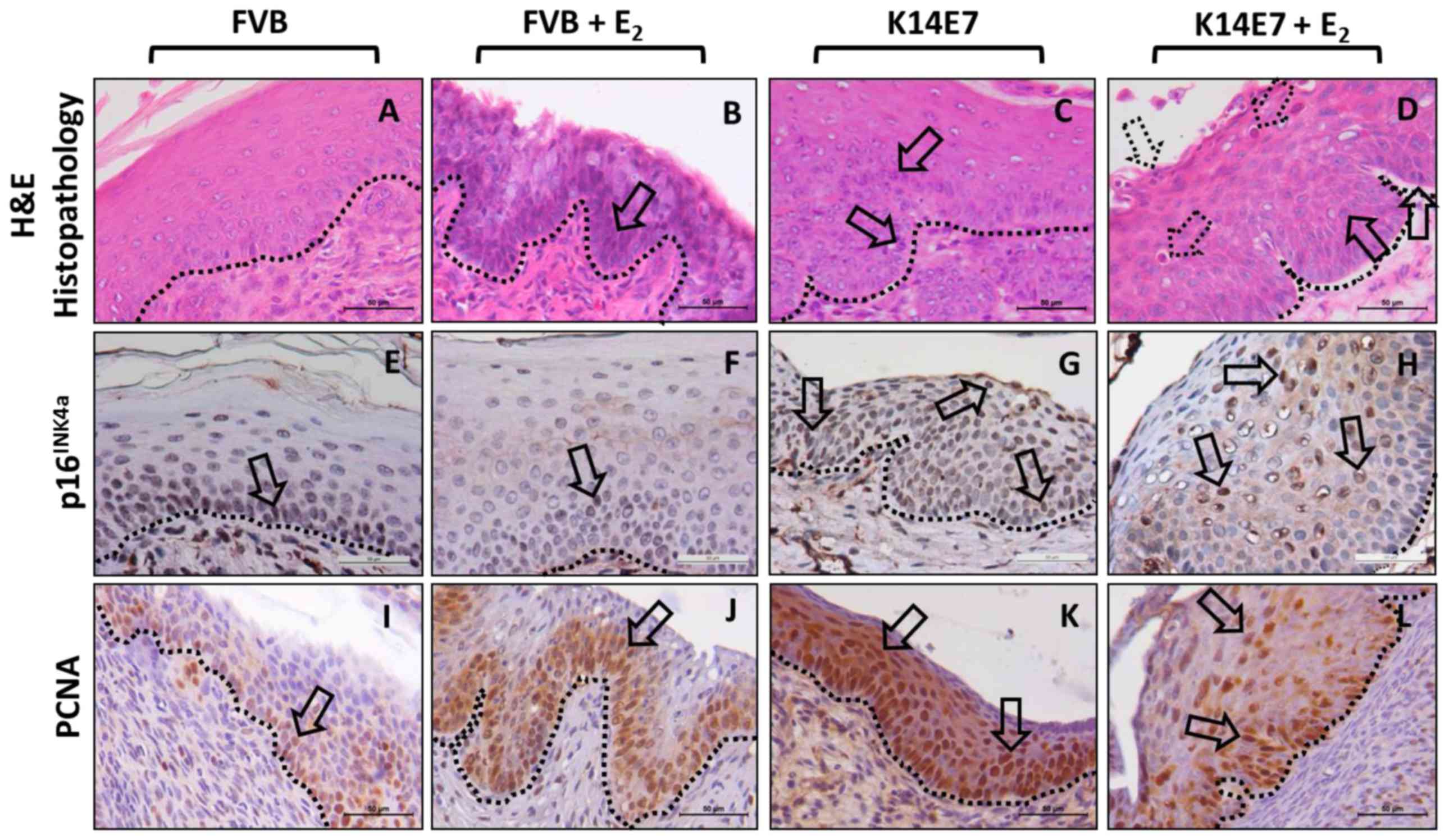
Histopathological characterization and
detection of p16INK4a and proliferating cell nuclear
antigen (PCNA) biomarker expression in low-grade cervical lesions
in K14E7 transgenic mice
H&E staining of cervical tissue sections from
2-month-old mice from the FVB, FVB+E2, K14E7 and
K14E7+E2 groups showed that only the K14E7+E2
group developed low-grade lesions corresponding to cervical
intraepithelial neoplasia grade I (CIN-I) (H&E; Fig. 1). Histopathology of the exocervical
tissues of FVB (non-transgenic) mice demonstrated normal stratified
squamous epithelium (H&E-FVB; Fig.
1), with sporadic expression of p16INK4a and basal
PCNA expression in the epithelial basal layer
(p16INK4a-FVB and PCNA-FVB; Fig. 1). Histopathology of the exocervical
tissues of the E2-treated FVB mice demonstrated
moderately hyperproliferative squamous epithelium (arrows,
H&E-FVB+E2; Fig.
1), with similar sporadic expression of p16INK4a as
the untreated FVB mice, but with a higher PCNA expression in the
epithelial basal layer, as expected with E2 treatment
(arrows, p16INK4a-FVB+E2 and
PCNA-FVB+E2; Fig. 1).
In K14E7 mice, wider stratified basal and middle epithelial layers
were observed (H&E-K14E7; Fig.
1). As expected with high-risk HPV-associated lesions (23–25),
p16INK4a expression was increased in these samples
compared with that in samples from FVB mice (arrows,
p16INK4a-K14E7; Fig.
1). Additionally, an even signal for PCNA expression along the
basal layer and a hyperproliferative stratified squamous epithelium
in the exocervical tissues was observed (arrows, PCNA-K14E7;
Fig. 1). Notably, in the
E2-treated K14E7 mice, the development of mild
dysplasia, indicated by the presence of epithelial cells with large
and extended nuclei in the basal and middle layers of the
exocervical tissues (arrows, Fig.
1), accompanied with infiltration of mixed inflammatory cells
(neutrophils and lymphocytes) was observed (dotted arrows,
H&E-K14E7+E2; Fig.
1). Similar to that in K14E7 mice, the expression of
p16INK4a in the E2-treated K14E7 mice was
increased relative to that in the FVB mice (arrows,
p16INK4a-K14E7+E2; Fig. 1). Finally, the expression of PCNA
in the E2-treated K14E7 mice was also detected in the
basal and suprabasal layers in the hyperproliferative stratified
squamous epithelium of the exocervix, consistent with the presence
of proliferating cells (arrows, PCNA-K14E7+E2; Fig. 1). All these traits are
characteristic of low-grade lesions corresponding to CIN-I.
Classification of cervical dysplastic lesions in 2-month-old
E2-treated K14E7 mice was determined according to the
'histopathological grading system for cervical squamous
carcinogenesis in transgenic mouse' (4).
Effect of the HPV16-E7 oncoprotein and
E2 on global gene expression in the initial stages of
carcinogenesis
To evaluate the global gene expression profiles in
the initial stages of cancer development, 2-month-old FVB and K14E7
mice treated for 1 month with E2 pellets were used and
compared against untreated FVB (control) or K14E7 mice. The 4
groups were examined with Whole Mice Genome Oligo Microarrays using
ANOVA, with a fold-change criterion of ≥1.5 and ≤-1.5 and a P-value
of <0.05. In summary, it was observed that the most marked
changes in gene expression occurred in the E2-treated
groups (FVB or K14E7). Compared with those in the FVB group, 685
differentially expressed genes (409 upregulated and 276
downregulated) were detected in the FVB+E2 group, 321
differentially expressed genes (181 upregulated and 140
downregulated) were detected in the K14E7 group and 933
differentially expressed genes (554 upregulated and 379
downregulated) were detected in the K14E7+E2 group
(Fig. 2A). A Venn diagram
(Fig. 2B) was constructed to
identify genes that were commonly and exclusively modulated by the
E7 oncoprotein and/or E2. The comparison between the
K14E7+E2 and FVB+E2 groups yielded 248
overlapping genes, and the comparison between the
K14E7+E2 and K14E7 groups yielded 50 overlapping genes.
In total, 108 genes were overlapping among all groups. Notably, 527
(K14E7+E2), 290 (FVB+E2) and 124 (K14E7)
genes were exclusively regulated in each group (data not shown;
dropbox data).
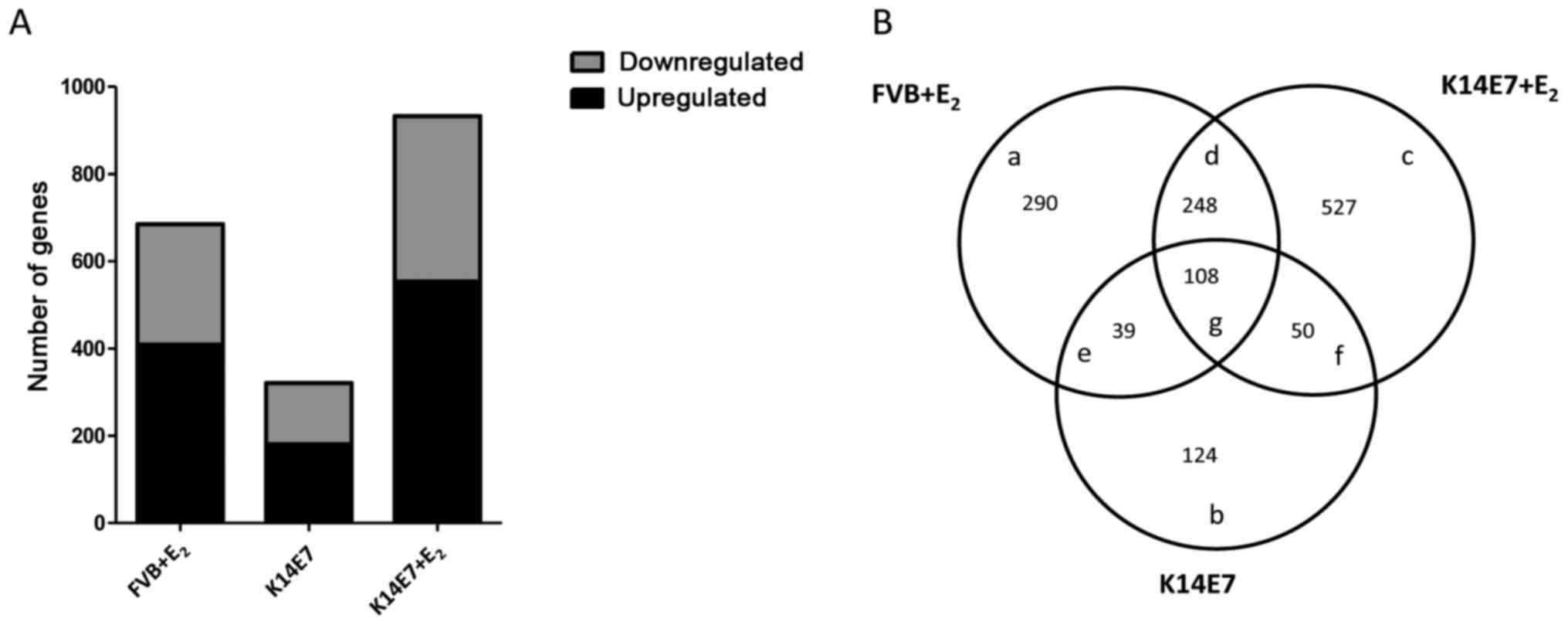 | Figure 2Global gene expression profile of the
control FVB group versus the FvB+E2, K14E7 and
K14E7+E2 groups. To determine the differentially
expressed genes, a fold change of ≥1.5 and ≤−1.5, and a P-value of
<0.001 were used. (A) In total, 409, 181 and 554 upregulated
genes and 276, 140 and 379 downregulated genes were detected in the
FVB+E2, K14E7 and K14E7+E2 groups,
respectively, relative to the FVB group. (B) Venn diagram showing
unique (a, b and c) and common (d, e, f and g) differentially
expressed genes among the FVB+E2, K14E7 and
K14E7+E2 groups. E2, 17β-oestradiol. |
We hypothesize that the 527 exclusively
downregulated or upregulated genes in the K14E7+E2 group
that lead to carcinogenesis are regulated by the cooperation
between E2 and E7 (Fig.
2B). To identify differentially expressed genes involved in
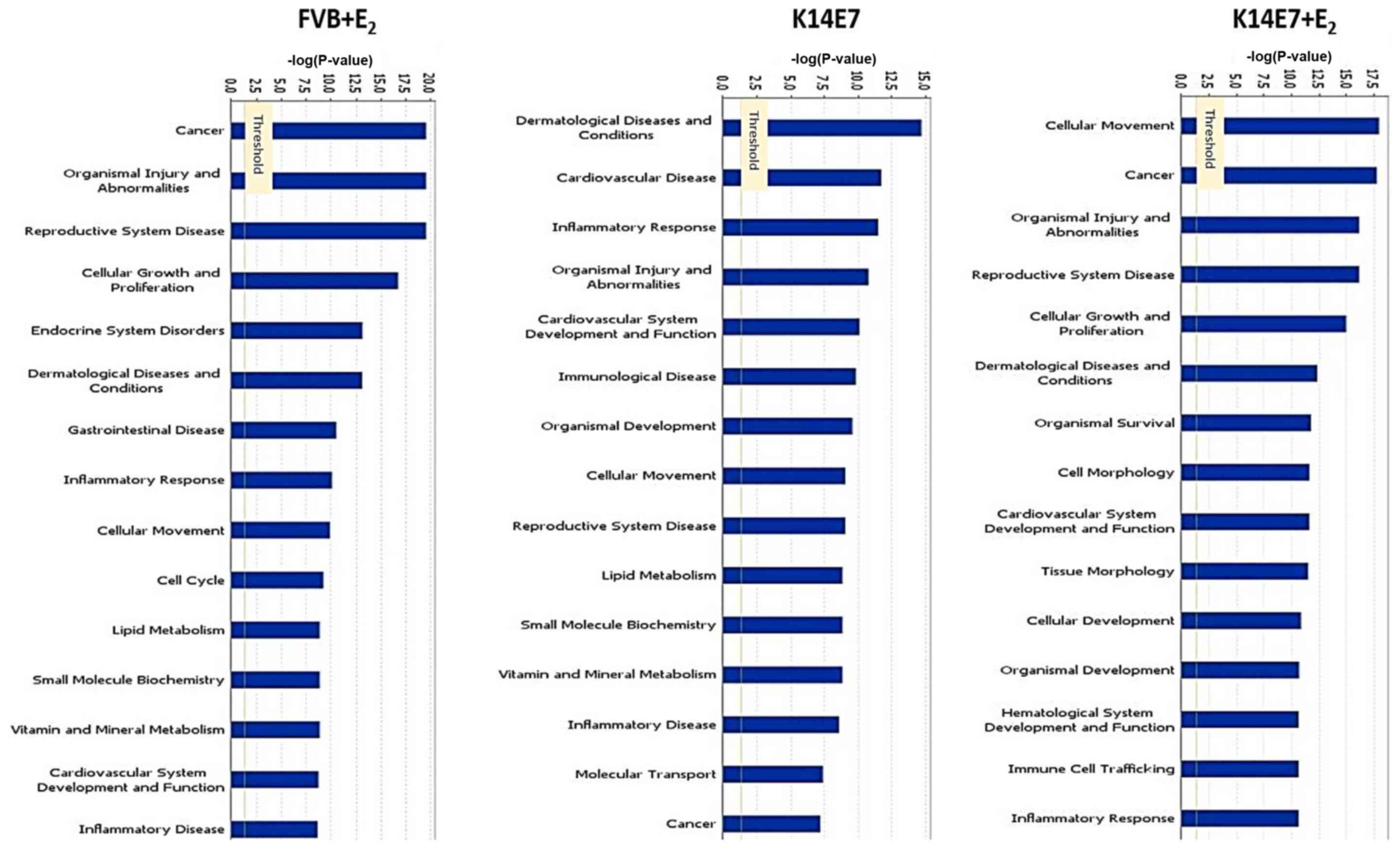
diverse cellular processes, a Gene Ontology analysis was performed
using the IPA software and it was observed that several processes
associated with cancer, including 'cellular movement', 'cancer' and
'cellular growth and proliferation', were mainly modified in the
K14E7+E2 group, and to a slightly lesser degree, in the
FVB+E2 group (Fig. 3),
whereas genes associated with 'inflammatory response', and
'dermatological diseases and conditions', were altered in the K14E7
group. Notably, even though the K14E7+E2 and
FVB+E2 groups shared differentially expressed genes
involved in several cellular processes, the gene profile for each
group was quite different (Fig.
3).
Validation of microarray data by
RT-qPCR
To validate the microarray data, genes involved in
different processes were selected (Fig. 3) and RT-qPCR analysis was
performed. A total of 15 specific genes that showed differential
expression in the microarray data obtained from the
K14E7+E2 group were validated. Among the upregulated
genes, cytochrome P450 family 2 subfamily e polypeptide 1 (Cyp2e1;
not synergistic), glycerophosphodiester phosphodiesterase domain
containing 3 (Gdpd3), interleukin 1 receptor type II
(Il1r2), lecithin-retinol acyltransferase
(phosphatidylcholine-retinol-O-acyltransferase) (Lrat),
MGAT4 family member C (Mgat4c), natriuretic peptide type C
(Nppc) and serine (or cysteine) peptidase inhibitor clade B
member 9 [Serpinb9b; (PI-9); synergistic] were validated,
and among the downregulated genes, eomesodermin (Eomes; not
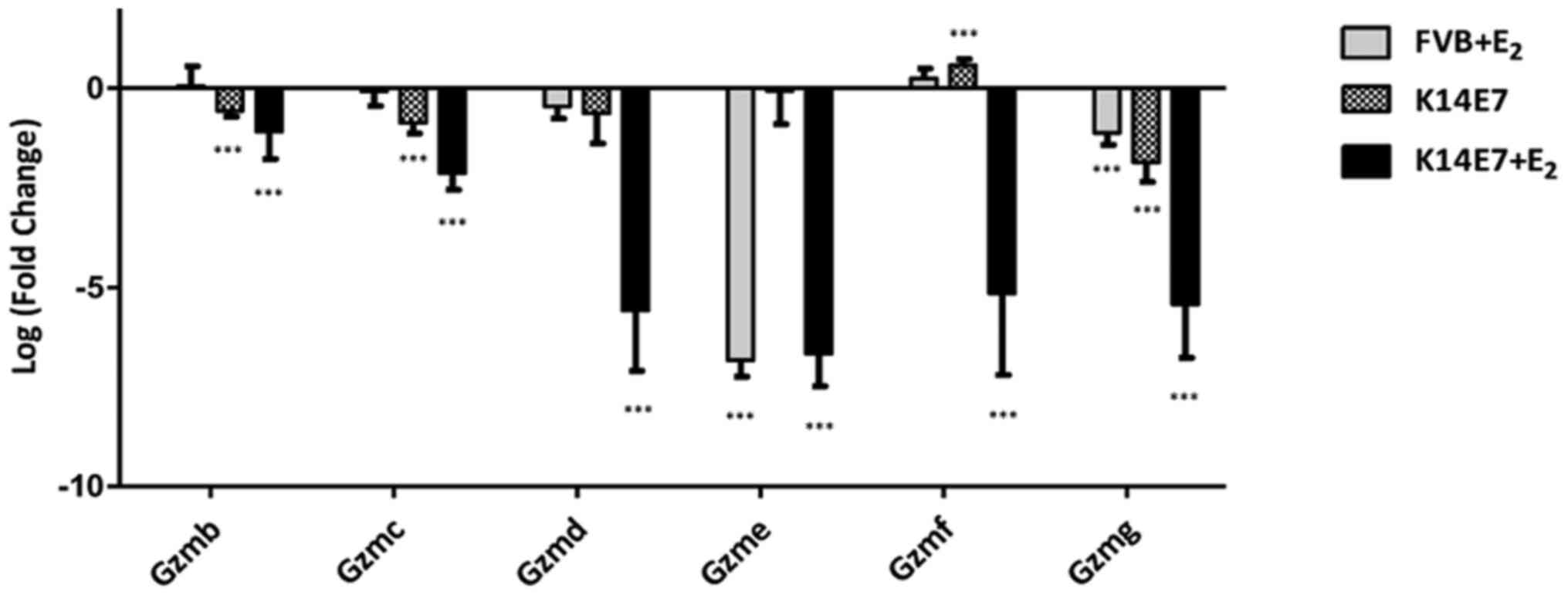
synergistic), Gzmb, Gzmc, Gzmd, Gzme, Gzmf, Gzmg and
glucoside xylosyltransferase 2 (synergistic) were validated. The
results for all the genes analysed by RT-qPCR were consistent with
the microarray data; the comparative results are shown in Table II. A synergistic effect at the
transcriptional level was found in 13 of the validated genes,
probably as a result of interactions between E2 and E7.
Another set of genes that showed differential expression in the
K14E7 group (Cyp2e1, Il1r2, Gzmd and Gzmg) and in the
FVB+E2 group (Gdpd3, Gzmd, Gzmg and Eomes)
was also validated. Validation of all groups was made using FVB
untreated mice as the control. The RT-qPCR results are summarized
in Figs. 4Figure 5–6.
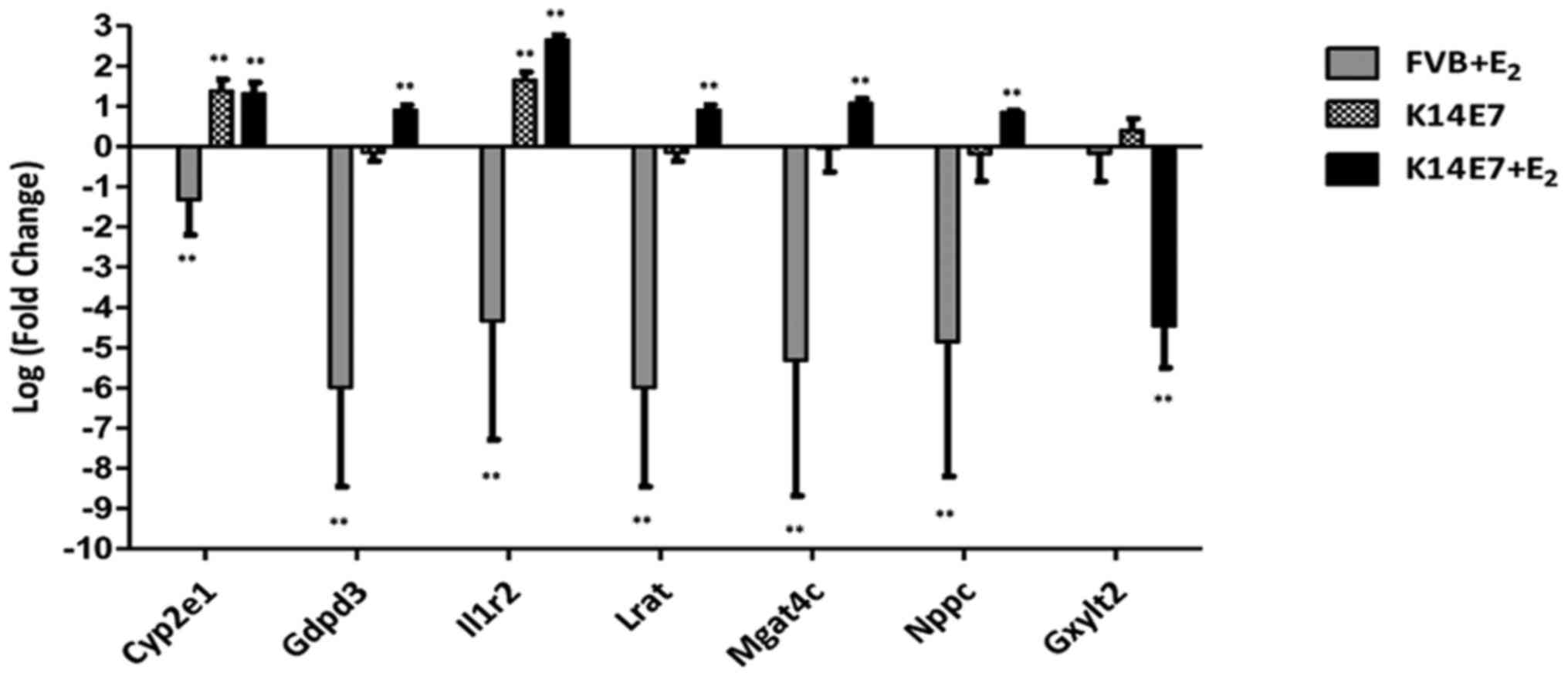 | Figure 4E2 and the E7 oncoprotein
synergistically regulate the expression of several cancer-related
genes. Reverse transcription-quantitative polymerase chain reaction
analysis results showed higher mRNA expression (or inhibition for
Gxylt2) in the K14E7+E2 group than in the
untreated K14E7 group or FVB+E2 when compared with the
control group FVB (base line). Bars (relative expression) represent
the mean ± standard deviation, normalized to Gapdh mRNA
levels, from six independent experiments (**P<0.01;
one-way analysis of variance with Bonferroni correction) compared
with the controls (untreated FVB mice). E2,
17β-oestradiol; Cyp2e1, cytochrome P450 family 2 subfamily e
polypeptide 1; Gdpd3, glycerophosphodiester
phosphodiesterase domain containing 3; Il1r2, interleukin 1
receptor type II; Lrat, lecithin-retinol acyltransferase
(phosphatidylcholine-retinol-O-acyltransferase); Mgat4c,
MGAT4 family member C; Nppc, natriuretic peptide type C;
Gxylt2, glucoside xylosyltransferase 2. |
| Table IIRT-qPCR gene validation for
microarray data of 2-month-old FVB and K14E7 mice, in
E2-treated or untreated mouse groups. |
Table II
RT-qPCR gene validation for
microarray data of 2-month-old FVB and K14E7 mice, in
E2-treated or untreated mouse groups.
| Symbol | FVB+E2
vs. FVB
| K14E7 vs. FVB
| K14E7+E2
vs. FVB
|
|---|
| Microarray
result | RT-qPCR result | Microarray
result | RT-qPCR result | Microarray
result | RT-qPCR result |
|---|
| Gxylt2 | | | | | −2.925 | −4.46 |
| Cyp2e1 | 2.47 | −1.32 | 6.278 | 1.38 | 5.179 | 1.31 |
| Gdpd3 | −23.674 | −5.99 | | −0.14 | | 0.90 |
| Il1r2 | 3.207 | −4.33 | 3.187 | 1.64 | 5.934 | 2.64 |
| Lrat | 3.567 | −5.99 | 2.191 | −0.14 | 6.533 | 0.9 |
| Mgat4c | 4.75 | −5.31 | | | 7.007 | 1.07 |
| Nppc | | −4.84 | | −0.17 | 9.527 | 0.84 |
| Gzmb | | 0.05 | | −0.56 | −2.15287 | −1.08 |
| Gzmc | | −0.07 | | −0.87 | −2.09209 | −2.12 |
| Gzmd | −2.4152 | −0.45 | −2.03554 | −0.62 | −9.45465 | −5.58 |
| Gzme | | −6.83 | | −0.06 | −2.59815 | −6.66 |
| Gzmf | −1.96428 | 0.24 | −2.0912 | 0.58 | −3.69359 | −5.14 |
| Gzmg | −1.84094 | −1.11 | −1.7933 | −1.86 | −3.48101 | −5.42 |
| Eomes | −1.509 | −3.62 | | −0.38 | −1.854 | −0.81 |
|
Serpinb9b | | −3.00 | | 2.15 | 2.085 | 4.24 |
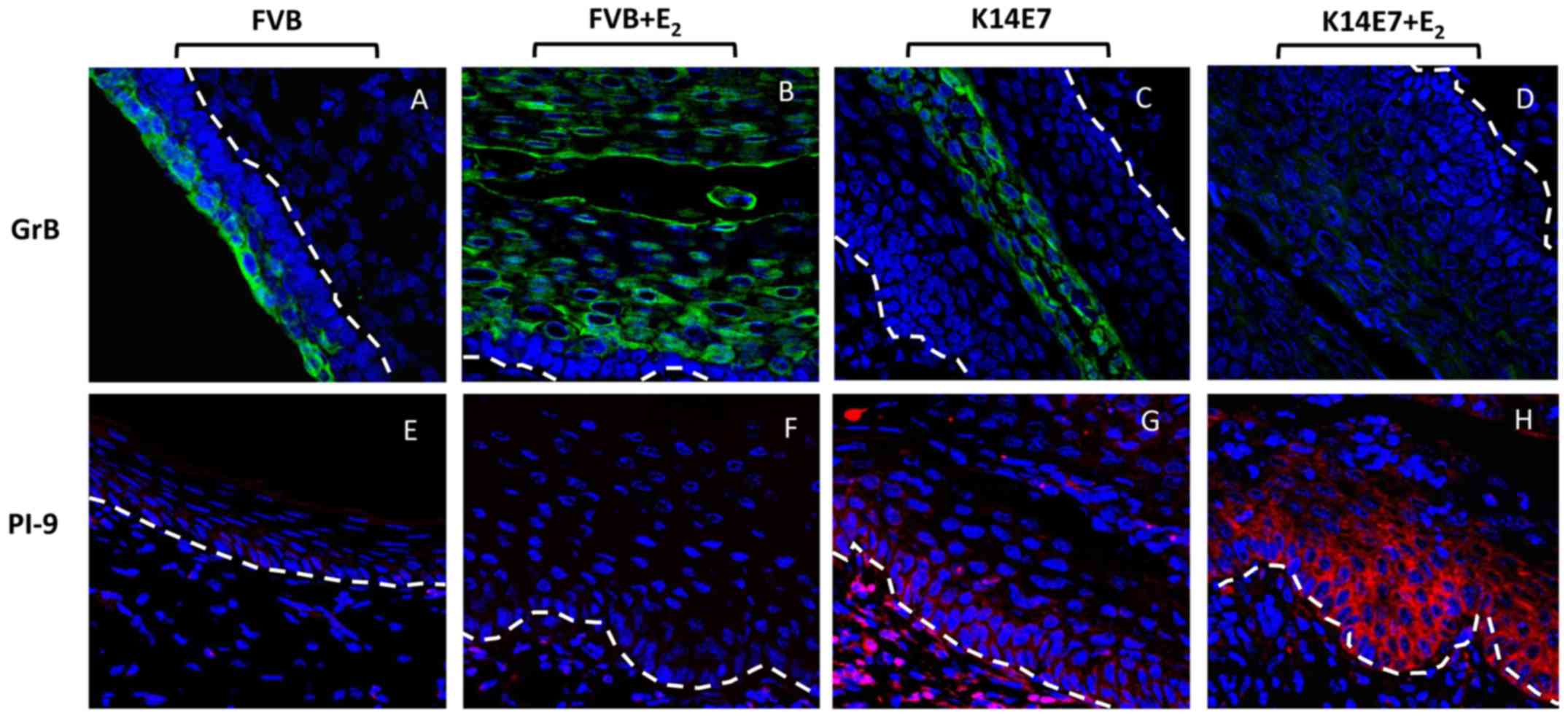
Protein expression of GrB and PI-9 in the
murine exocervix, as determined by immunofluorescence
To validate whether GrB and PI-9 protein expression
was associated with the mRNA expression, immunofluorescence assays
were performed for GrB and its inhibitor, PI-9, in tissue sections
of murine exocervix obtained from the FVB, FVB+E2, K14E7
and K14E7+E2 groups. Consistent with the mRNA expression
results, immunofluorescence assays demonstrated cytoplasmic GrB
protein expression in the suprabasal and granular layers of
exocervical tissues from the FVB control group (GrB-FVB; Fig. 7). For the FVB+E2 group,
high GrB expression in the hyperplasic epithelial cells of the
suprabasal and granular layers of the exocervix was also found
(GrB-FVB+E2; Fig. 7). A
low signal for GrB in the K14E7 group was found (GrB-K14E7;
Fig. 7), and as expected, a very
weak signal was observed in the K14E7+E2 mice,
indicating the downregulation of GrB at the protein level
(GrB-K14E7+E2; Fig. 7).
On the other hand, also in accordance with the microarray and
RT-qPCR results, the expression of PI-9 was very low in the FVB
group, and PI-9 expression was not observed in the
FVB+E2 group (PI-9-FVB and PI-9-FVB+E2;
Fig. 7). Moreover, a clear
cytoplasmic signal for PI-9 was observed in the basal and
suprabasal layers of the cervical epithelium and the stromal
tissues in the K14E7 group (PI-9-K14E7; Fig. 7), and a very high signal was
observed in the basal and suprabasal layers of the epithelial
tissues in the K14E7+E2 group (PI-9-K14E7+E2;
Fig. 7).
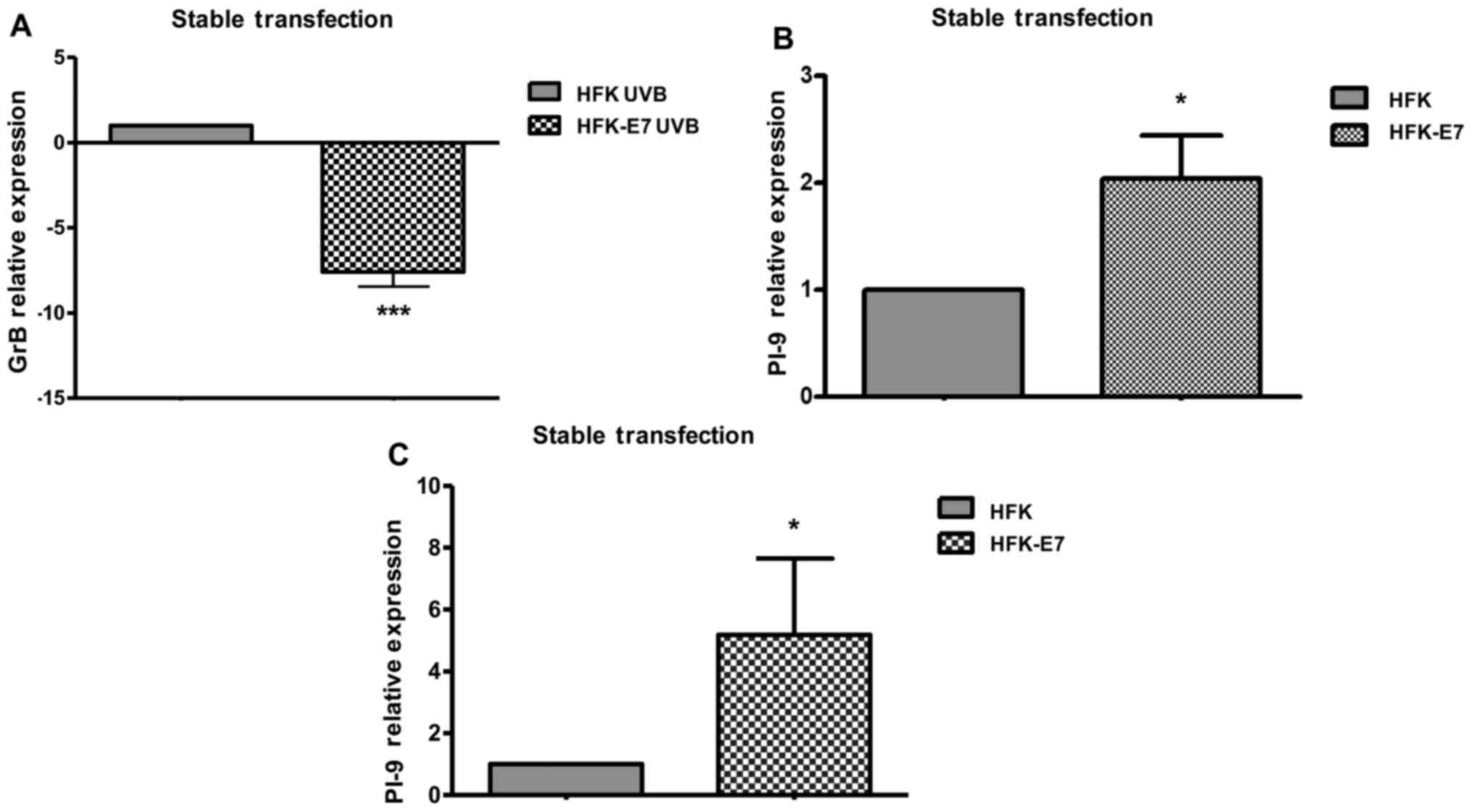
HPV16-E7 oncoprotein inhibits GrB
expression and enhances PI-9 expression in human keratinocytes
To verify the effect of the HPV16-E7 oncoprotein on
human GrB and PI-9 expression, HFK cell lines transiently or stably
transfected with plasmids carrying the ORF for the HPV16-E7
oncoprotein were generated. By RT-qPCR analysis, it was determined
that compared with that in the HFKs transfected with empty vector,
GrB expression in HFKs that were stably transfected with HPV16-E7
and exposed to UV was inhibited (Fig.
8A). PI-9 expression was upregulated by 2.0-fold when HFKs were
transiently transfected with the HPV16-E7 oncoprotein ORF (Fig. 8B) and by 5.2-fold when HFKs were
stably transfected (Fig. 8C).
These results suggest that the HPV16-E7 oncoprotein induces PI-9
expression in vivo and in vitro. The expression of E7
was determined by RT-qPCR in stably transfected HFKs (see figure S1
available in the following Dropbox location: https://www.dropbox.com/s/h8kbqty7umkbg60/Figure%20S1.pdf?dl=0).
The expression of E7 was determined by RT-qPCR in stably
transfected HFKs (data not shown; dropbox data).
Discussion
HR-HPV infection and E2 treatment
increase the risk of CC development through oestrogen receptor α
(6,26,27).
In addition, E2 cooperates with HPV oncogenes in mouse
models to induce premalignant lesions and CC, but little is known
about the molecular mechanisms by which E7 and E2
contribute to the early stages of cervical carcinogenesis.
Global gene expression studies are of great
importance in understanding various processes in human cancer
(28). In the present study,
global gene expression profiling was performed of cervical tissues
from young (2-month-old) K14E7 and FVB mice, which were either
treated with E2 or left untreated for 1 month to obtain
a better understanding of the transcriptional events occurring in
the early stages of cervical tumourigenesis. One important
observation was that the profiles of genes associated with cancer
were different between the FVB+E2 and
K14E7+E2 groups (Fig.
3). Gene Ontology analysis showed that the genes relevant to
'cancer' in the FVB+E2 group (Fig. 3) were mainly associated with cell
cycle regulation and cell morphology, whereas genes in 'cancer' in
the K14E7+E2 group (Fig.
3) were particularly associated with lipid metabolism and
endocrinological disorders (dropbox data). The overexpression of
the genes involved in lipid metabolism in the K14E7+E2
group is notable, as this group of mice eventually develop
high-grade lesions and CC. It is known that cancer cells must
reprogram their metabolic pathways to match their accelerated
proliferation and survival rates (29) by changing the expression and
activity of lipid metabolism-related enzymes that are directly
regulated by the activity of oncogenic signals (30). Altered lipid metabolism is
currently recognized as a hallmark of cancer, and the expression
and activity of numerous enzymes involved in fatty acid synthesis,
including ATP citrate lyase, acetyl-CoA carboxylase and fatty acid
synthase (FASN), are upregulated in a number of cancer types
(31). In the present study, the
microarray results from the K14E7+E2 group demonstrated
overexpression of acyl-CoA synthetase bubblegum family member 1,
acyl-CoA synthetase long-chain family member 1 and DDHD domain
containing 1 genes, which encode enzymes involved in the synthesis
of long-chain fatty acids in the FASN pathway. The upregulation of
the fatty acid biosynthetic pathway starts at a relatively early
stage in various types of tumours (32,33),
which is consistent with the present results. Notably, the
endocrine process was the second-most altered process, and it is
associated with genes encoding receptors of hormonal responses,
such as insulin (protein tyrosine phosphatase, non-receptor type
1), oestrogen (steroid 5 α-reductase 2, and gene regulated by
oestrogen in breast cancer protein) and progesterone (progesterone
receptor). This is not unexpected, as CC has been identified as a
hormone-related malignancy. In experimental animal models, the
incidence of neoplasia can be increased by excessive hormonal
stimulation of the target organ (34). In this type of hormone-related
neoplasia, the neoplasms produced are initially responsive to and
dependent on hormones, but eventually, these neoplasms become
autonomous (35).
In 1996, Arbeit et al (36) described a distinct synergism
between oestrogen and the HPV oncoprotein, which was independent of
the marked effect of E2 promotion of HPV transcription
of E6 and E7. A review also highlighted the presence of synergistic
effects in HPV mouse models that were treated with oestrogen to
induce cancer, although the full implications of this synergistic
collaboration remain unclear (26). The present study observed a
synergistic association between E7 and E2 at the level
transcriptional regulation, and these two factors enhanced or
repressed the expression of several genes and promoted the
expression of unique genes in each group of mice. Furthermore, this
synergistic interaction between E7 and E2 resulted in
distinct gene expression profiles even within the same ontological
processes compared with that in the groups that did not develop
cancer (FVB, FVB+E2 and K14E7). Although some of these
genes have already been implicated in cancer, a number are new in
the context of CC; for example, Nppc is expressed in prostate
cancer (37–39), GDPD3 is overexpressed in luminal B
type breast cancer (40), MGAT4C
is associated with prostate cancer risk (41), IL1R2 is observed in relation to
colon cancer and is associated with enhanced angiogenesis (42), and LRAT is involved in the
metabolism of retinoic acid, which when overexpressed, increases
the sensitivity of cells to carcinogens (43). Most notably, the synergistic effect
of the HPV16-E7 oncoprotein and E2 on the downregulation
of the Granzyme gene family was observed in the present study. The
expression of GrB by cytotoxic lymphocytes and natural killer cells
has been known for a considerable time, but it is now accepted that
GrB can also be expressed in other cell types of non-immune origin,
including smooth muscle cells, keratinocytes and chondrocytes
(14,44). For example, under certain
circumstances, such as exposure to UVB and UVA radiation,
keratinocytes can induce the expression and activation of GrB,
which exerts cytotoxic activities and the capacity to degrade
extracellular matrix (ECM) components, including collagen, elastin
and fibronectin (13,45). Thus, the present results in the
K14E7+E2 group suggest that the early inhibition of the
GrB pathway may prevent keratinocytes from acquiring several
characteristics that could be disadvantageous to CC development,
thereby promoting the survival of CC cells. It has been shown that
the increase in endogenous GrB levels in chondrocytes and
keratinocytes corresponds to the increase in the levels of
apoptosis (13,46). Thus, the downregulation of GrB
could prevent undesired activation of GrB, which may induce
self-regulated apoptosis in cancer cells. On the other hand, GrB
has non-apoptotic activities, including the degradation of ECM
components, which could assist in enhancing innate immune responses
(47). In this regard, it has been
shown that the GrB-mediated degradation of ECM components may
positively modulate signals for innate immune responses, and it has
also been shown that tumour-associated ECM hampers T-cell functions
(48,49). This suggests that collagen in the
tumour microenvironment can be a barrier against T-cell
penetration, and GrB collagen degradation may facilitate T-cell
migration into the tumour (50).
Finally, it has also been reported that collagen degradation can
produce a chemotactic signal that attracts immune cells (51,52).
Thus, GrB inhibition may aid in inhibiting or diminishing the host
immune responses. Although there is the possibility that GrB
downregulation can be induced by epigenetic mechanisms mediated by
E7 and E2 (53,54), there is also the possibility that
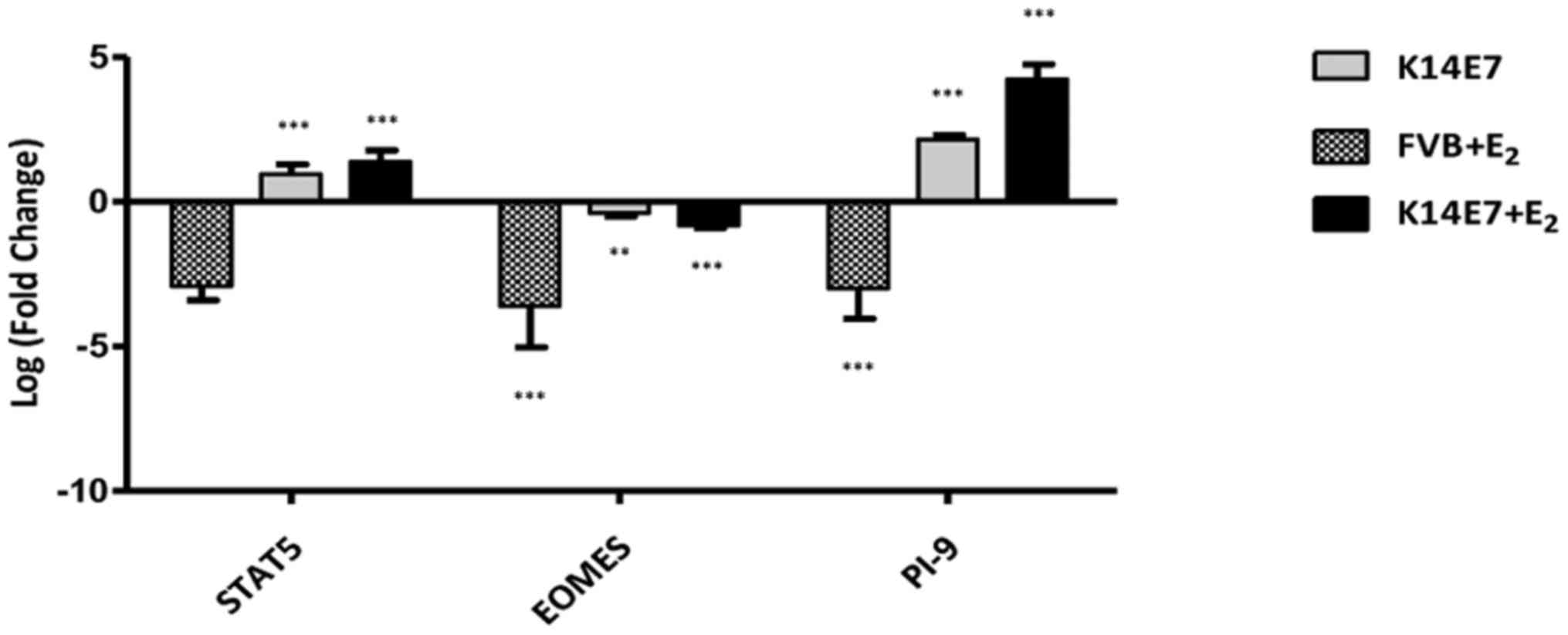
the transcriptional inhibition of GrB was likely due to a decrease
in the expression of specific transcription factors, including
T-cell-specific T-Box transcription factor and eomesodermin
(EOMES), which are regulated upstream by signal transducer
and activator of transcription 5A (STAT5). It has been shown that
these transcription factors cooperate to promote cytotoxic
lymphocyte activation by inducing the expression of perforin and
GrB (55). In this regard, it was
found that the expression of the transcription factor Eomes
was downregulated in the FVB+E2, K14E7 and
K14E7+E2 groups compared with that in the FVB control
group, regardless of the rise in the expression of Stat5 in the
K14E7 and K14E7+E2 groups compared with that in the FVB
control group, suggesting in this case that downregulation of Eomes
and GrB is independent of Stat5. Furthermore, it has previously
been shown that Eomes induces the expression of
interferon-γ, which is important for the initiation of antiviral
defence mechanisms in keratinocytes, suggesting that the inhibition
of Eomes could also be a mechanism of immune evasion
(56–58). However, there is little evidence to
support that the HPV-E7-mediated downregulation of GrB observed in
keratinocytes could also occur in immune cells, and if this process
does occur, it may be regulated by mechanisms distinct from those
regulating the downregulation of endogenous GrB in keratinocytes;
for example, these mechanisms may involve soluble components (e.g.,
cytokines) produced by infected cells. A previous study on lung
cancer cells demonstrated a reduction in the expression of GrB in
cluster of differentiation 8-positive T cells in response to
culture media from cancer cells, supporting the idea of an
inhibitory signal for GrB expression in immune cells (59). In addition, in the
K14E7+E2 group in the present study, an increase was
observed in the expression of PI-9, which is a potent inhibitor of
GrB (60) and a key regulator of
GrB activity. Endothelial cells, vascular smooth muscle cells and
hepatocytes have the ability to express PI-9 to protect themselves
from GrB-mediated cytotoxicity (61,62).
Notably, oestrogens have been shown to induce PI-9 expression in
hepatoblastoma cells to protect the cells from CTLs and NK
cell-mediated GrB-dependent apoptosis (63). Furthermore, the exposure of MCF7
cells to oestrogens (E2 and genistein) has been shown to
increase PI-9 protein level, and at the same time, to increase the
number and size of tumour spheres and to promote cell proliferation
(64). In addition, PI-9
transfection experiments in HeLa and CHO cells lines have also
shown that PI-9 expression aids in the evasion of cell death by
lymphocytes (65,66). In addition suppression of Granzyme
B initiated apoptosis in PI-9-expressing cells could contribute to
immune evasion (68). The present
study used in vitro assays to support the in vivo
observations, which confirmed that in HPV16-E7 transfected
keratinocytes, GrB expression was inhibited, while protease
inhibitor-9 (PI-9) expression was robustly enhanced. There are also
clinical studies in non-Hodgkin's and Hodgkin's lymphoma that
support these observations (67).
In conclusion, HPV16-E7 and oestrogens
(E2) negatively regulate GrB expression and activity,
and increase the expression of PI-9 in keratinocytes, indicating
the possibility that GrB downregulation prevents precancerous cells
from acquiring cytotoxic capacities, which may be detrimental to
cancer development, and indicating that the upregulation of PI-9
may contribute to immune evasion by cancer cells. These two factors
may thus be useful as predictive markers and as novel therapeutic
targets during the early stages of cancer (68). Considering the results of the
present study, in order to demonstrate in vivo that the
depletion of Granzyme B or the expression of PI-9 is essential for
the progression of low-grade lesions to carcinoma in situ, a
double transgenic mouse model that expresses E7 and GrB under K14
promoter control or a double transgenic system that expresses E7
and incorporates a PI-9-knockout, could be useful. Another possible
experiment would be to xenotransplant CC cells that express
constitutively GrB or PI-9 in a wild-type mouse model in order to
evaluate its ability to generate a tumour. Furthermore, an extended
histopathological analysis of GrB and PI-9 in clinical samples of
low-, medium- and high-grade lesion is necessary to establish a
positive correlation with CC progression.
Acknowledgments
The authors would like to thank to Mr. Raúl Mojica
(The National Institute of México City, México), Mr. Jaime
Escobar-Herrera from the Department of Cellular Biology (The Centre
for Research and Advanced Studies of the National Polytechnic
Institute, México City, México), Mr. Ricardo Águila Guadarrama (The
National Institute of Medical Sciences and Nutrition Salvador
Zubirán - Health Secretary, México City, México), Dr Ricardo
Gaxiola-Centeno, Dr Benjamín Chavez-Alvarez and Mr. Rafael Leyva
(Unit for the Production and Experimentation of Laboratory Animals,
The Centre for Research and Advanced Studies of the National
Polytechnic Institute) for their excellent technical support. The
authors would also like to thank the Cinvestav employees and Mr.
Lauro Macías González (The Centre for Research and Advanced Studies
of the National Polytechnic Institute) for providing technical
support and Miss Rosa Angélica Becerril Gutiérrez, Miss Altair
Munguia Torres and Mrs. Denisse Torres Hernandez (all affiliated to
The Centre for Research and Advanced Studies of the National
Polytechnic Institute) for their critical comments.
Funding
This study was supported by government grants from
the Institute of Science and technology of Mexico City (nos.
PICSA10-190 and 326/2011), from the National Council for Science
and Technology (no. 168896) and from the Sectorial Funding for
Research in Health and Social Security (no. 261875).
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request. The datasets generated during the current study
are also available in the following dropbox locations: https://www.dropbox.com/s/z55z32y738s54sq/TABLE%202%20venn%20diagram%20database%20.pdf?dl=0
and https://www.dropbox.com/s/h8kbqty7umkbg60/Figure%20S1.pdf?dl=0.
Authors' contributions
JAMM and JDC performed experiments, contributed to
the analysis and interpretation of the data, and were major
contributors in writing the manuscript. EGV, MEAS, ROD, JBD, AMF,
EAR and EMCM also performed experiments. DMV and AHM performed the
microarray experiments, analysis and interpretation of the data. AÜ
and HÇ achieved the E7 transfection in keratinocytes. PL developed
the K14E7 transgenic mice, and participated in the writing and
revision of the manuscript. PG contributed to the conception and
design of the study, the data analysis and interpretation, and the
writing and revision of the manuscript. All authors have read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
All experiments and procedures were approved by the
Research Unit for Laboratory Animal Care Committee
(NOM-062-ZOO-1999; Unit for the Production and Experimentation of
Laboratory Animals, The Centre for Research and Advanced Studies of
the National Polytechnic Institute, México City, México).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
zur Hausen H: Papillomaviruses in the
causation of human cancers - a brief historical account. Virology.
384:260–265. 2009. View Article : Google Scholar
|
|
2
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
3
|
Smith JS, Lindsay L, Hoots B, Keys J,
Franceschi S, Winer R and Clifford GM: Human papillomavirus type
distribution in invasive cervical cancer and high-grade cervical
lesions: A meta-analysis update. Int J Cancer. 121:621–632. 2007.
View Article : Google Scholar
|
|
4
|
Riley RR, Duensing S, Brake T, Münger K,
Lambert PF and Arbeit JM: Dissection of human papillomavirus E6 and
E7 function in transgenic mouse models of cervical carcinogenesis.
Cancer Res. 63:4862–4871. 2003.
|
|
5
|
Brake T and Lambert PF: Estrogen
contributes to the onset, persistence, and malignant progression of
cervical cancer in a human papillomavirus-transgenic mouse model.
Proc Natl Acad Sci USA. 102:2490–2495. 2005. View Article : Google Scholar
|
|
6
|
Chung SH, Wiedmeyer K, Shai A, Korach KS
and Lambert PF: Requirement for estrogen receptor alpha in a mouse
model for human papillomavirus-associated cervical cancer. Cancer
Res. 68:9928–9934. 2008. View Article : Google Scholar
|
|
7
|
Cortés-Malagón EM, Bonilla-Delgado J,
Díaz-Chávez J, Hidalgo-Miranda A, Romero-Cordoba S, Uren A, Celik
H, McCormick M, Munguía-Moreno JA, Ibarra-Sierra E, et al: Gene
expression profile regulated by the HPV16 E7 oncoprotein and
estradiol in cervical tissue. Virology. 447:155–165. 2013.
View Article : Google Scholar
|
|
8
|
Nickens KP, Han Y, Shandilya H, Larrimore
A, Gerard GF, Kaldjian E, Patierno SR and Ceryak S: Acquisition of
mitochondrial dysregulation and resistance to
mitochondrial-mediated apoptosis after genotoxic insult in normal
human fibroblasts: A possible model for early stage carcinogenesis.
Biochim Biophys Acta. 1823:264–272. 2012. View Article : Google Scholar
|
|
9
|
Rousalova I and Krepela E: Granzyme
B-induced apoptosis in cancer cells and its regulation (review).
Int J Oncol. 37:1361–1378. 2010.
|
|
10
|
Berthou C, Michel L, Soulié A, Jean-Louis
F, Flageul B, Dubertret L, Sigaux F, Zhang Y and Sasportes M:
Acquisition of granzyme B and Fas ligand proteins by human
keratinocytes contributes to epidermal cell defense. J Immunol.
159:5293–5300. 1997.
|
|
11
|
Hernandez-Pigeon H, Jean C, Charruyer A,
Haure MJ, Titeux M, Tonasso L, Quillet-Mary A, Baudouin C,
Charveron M and Laurent G: Human keratinocytes acquire cellular
cytotoxicity under UV-B irradiation. Implication of granzyme B and
perforin J Biol Chem. 281:13525–13532. 2006.
|
|
12
|
Fang Y, Herrick EJ and Nicholl MB: A
possible role for perforin and granzyme B in resveratrol-enhanced
radiosensitivity of prostate cancer. J Androl. 33:752–760. 2012.
View Article : Google Scholar
|
|
13
|
Hernandez-Pigeon H, Jean C, Charruyer A,
Haure MJ, Baudouin C, Charveron M, Quillet-Mary A and Laurent G:
UVA induces granzyme B in human keratinocytes through MIF:
Implication in extracellular matrix remodeling. J Biol Chem.
282:8157–8164. 2007. View Article : Google Scholar
|
|
14
|
Boivin WA, Cooper DM, Hiebert PR and
Granville DJ: Intracellular versus extracellular granzyme B in
immunity and disease: Challenging the dogma. Lab Invest.
89:1195–1220. 2009. View Article : Google Scholar
|
|
15
|
Khan MS, Singh P, Azhar A, Naseem A,
Rashid Q, Kabir MA and Jairajpuri MA: Serpin inhibition mechanism:
A delicate balance between native metastable state and
polymerization. J Amino Acids. 2011:6067972011. View Article : Google Scholar
|
|
16
|
Medema JP, de Jong J, Peltenburg LT,
Verdegaal EM, Gorter A, Bres SA, Franken KL, Hahne M, Albar JP,
Melief CJ, et al: Blockade of the granzyme B/perforin pathway
through overexpression of the serine protease inhibitor PI-9/SPI-6
constitutes a mechanism for immune escape by tumors. Proc Natl Acad
Sci USA. 98:11515–11520. 2001. View Article : Google Scholar
|
|
17
|
Herber R, Liem A, Pitot H and Lambert PF:
Squamous epithelial hyperplasia and carcinoma in mice transgenic
for the human papillomavirus type 16 E7 oncogene. J Virol.
70:1873–1881. 1996.
|
|
18
|
Ibarra Sierra E, Díaz Chávez J,
Cortés-Malagón EM, Uribe-Figueroa L, Hidalgo-Miranda A, Lambert PF
and Gariglio P: Differential gene expression between skin and
cervix induced by the E7 oncoprotein in a transgenic mouse model.
Virology. 433:337–345. 2012. View Article : Google Scholar
|
|
19
|
Kendziorski C, Irizarry RA, Chen KS, Haag
JD and Gould MN: On the utility of pooling biological samples in
microarray experiments. Proc Natl Acad Sci USA. 102:4252–4257.
2005. View Article : Google Scholar
|
|
20
|
Chabas D, Baranzini SE, Mitchell D,
Bernard CC, Rittling SR, Denhardt DT, Sobel RA, Lock C, Karpuj M,
Pedotti R, et al: The influence of the proinflammatory cytokine,
osteopontin, on autoimmune demyelinating disease. Science.
294:1731–1735. 2001. View Article : Google Scholar
|
|
21
|
Kainkaryam RM, Bruex A, Gilbert AC,
Schiefelbein J and Woolf PJ: poolMC: Smart pooling of mRNA samples
in microarray experiments. BMC Bioinformatics. 11:2992010.
View Article : Google Scholar
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
23
|
McLaughlin-Drubin ME, Crum CP and Münger
K: Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B
histone demethylase expression and causes epigenetic reprogramming.
Proc Natl Acad Sci USA. 108:2130–2135. 2011. View Article : Google Scholar
|
|
24
|
McLaughlin-Drubin ME, Park D and Munger K:
Tumor suppressor p16INK4A is necessary for survival of
cervical carcinoma cell lines. Proc Natl Acad Sci USA.
110:16175–16180. 2013. View Article : Google Scholar
|
|
25
|
Yim EK and Park JS: Biomarkers in cervical
cancer. Biomark Insights. 1:215–225. 2007.
|
|
26
|
Chung SH, Franceschi S and Lambert PF:
Estrogen and ERalpha: Culprits in cervical cancer. Trends
Endocrinol Metab. 21:504–511. 2010. View Article : Google Scholar
|
|
27
|
den Boon JA, Pyeon D, Wang SS, Horswill M,
Schiffman M, Sherman M, Zuna RE, Wang Z, Hewitt SM, Pearson R, et
al: Molecular transitions from papillomavirus infection to cervical
precancer and cancer: Role of stromal estrogen receptor signaling.
Proc Natl Acad Sci USA. 112:E3255–E3264. 2015. View Article : Google Scholar
|
|
28
|
Dozmorov MG, Giles CB and Wren JD:
Predicting gene ontology from a global meta-analysis of 1-color
microarray experiments. BMC Bioinformatics. 12(Suppl 10): S142011.
View Article : Google Scholar
|
|
29
|
Cairns RA, Harris IS and Mak TW:
Regulation of cancer cell metabolism. Nat Rev Cancer. 11:85–95.
2011. View Article : Google Scholar
|
|
30
|
Zhang F and Du G: Dysregulated lipid
metabolism in cancer. World J Biol Chem. 3:167–174. 2012.
View Article : Google Scholar
|
|
31
|
Menendez JA and Lupu R: Fatty acid
synthase and the lipogenic phenotype in cancer pathogenesis. Nat
Rev Cancer. 7:763–777. 2007. View Article : Google Scholar
|
|
32
|
Röhrig F and Schulze A: The multifaceted
roles of fatty acid synthesis in cancer. Nat Rev Cancer.
16:732–749. 2016. View Article : Google Scholar
|
|
33
|
Baenke F, Peck B, Miess H and Schulze A:
Hooked on fat: The role of lipid synthesis in cancer metabolism and
tumour development. Dis Model Mech. 6:1353–1363. 2013. View Article : Google Scholar
|
|
34
|
Henderson BE, Ross RK, Pike MC and
Casagrande JT: Endogenous hormones as a major factor in human
cancer. Cancer Res. 42:3232–3239. 1982.
|
|
35
|
Furth J: A meeting of ways in cancer
research: Thoughts on the evolution and nature of neoplasms. Cancer
Res. 19:241–258. 1959.
|
|
36
|
Arbeit JM, Howley PM and Hanahan D:
Chronic estrogen-induced cervical and vaginal squamous
carcinogenesis in human papillomavirus type 16 transgenic mice.
Proc Natl Acad Sci USA. 93:2930–2935. 1996. View Article : Google Scholar
|
|
37
|
Nielsen SJ, Gøtze JP, Jensen HL and
Rehfeld JF: ProCNP and CNP are expressed primarily in male genital
organs. Regul Pept. 146:204–212. 2008. View Article : Google Scholar
|
|
38
|
Nielsen SJ, Iversen P, Rehfeld JF, Jensen
HL and Goetze JP: C-type natriuretic peptide in prostate cancer.
APMIS. 117:60–67. 2009. View Article : Google Scholar
|
|
39
|
Scotland RS, Ahluwalia A and Hobbs AJ:
C-type natriuretic peptide in vascular physiology and disease.
Pharmacol Ther. 105:85–93. 2005. View Article : Google Scholar
|
|
40
|
Grinde MT, Skrbo N, Moestue SA, Rødland
EA, Borgan E, Kristian A, Sitter B, Bathen TF, Børresen-Dale AL,
Mælandsmo GM, et al: Interplay of choline metabolites and genes in
patient-derived breast cancer xenografts. Breast Cancer Res.
16:R52014. View Article : Google Scholar
|
|
41
|
Demichelis F, Setlur SR, Banerjee S,
Chakravarty D, Chen JY, Chen CX, Huang J, Beltran H, Oldridge DA,
Kitabayashi N, et al: Identification of functionally active, low
frequency copy number variants at 15q21.3 and 12q21.31 associated
with prostate cancer risk. Proc Natl Acad Sci USA. 109:6686–6691.
2012. View Article : Google Scholar
|
|
42
|
Mar AC, Chu CH, Lee HJ, Chien CW, Cheng
JJ, Yang SH, Jiang JK and Lee TC: Interleukin-1 receptor type 2
acts with c-Fos to enhance the expression of interleukin-6 and
vascular endothelial growth factor A in colon cancer cells and
induce angiogenesis. J Biol Chem. 290:22212–22224. 2015. View Article : Google Scholar
|
|
43
|
Amann PM, Czaja K, Bazhin AV, Rühl R,
Eichmüller SB, Merk HF and Baron JM: LRAT overexpression diminishes
intracellular levels of biologically active retinoids and reduces
retinoid antitumor efficacy in the murine melanoma B16F10 cell
line. Skin Pharmacol Physiol. 28:205–212. 2015. View Article : Google Scholar
|
|
44
|
Hiebert PR and Granville DJ: Granzyme B in
injury, inflammation, and repair. Trends Mol Med. 18:732–741. 2012.
View Article : Google Scholar
|
|
45
|
Provenzano PP, Inman DR, Eliceiri KW,
Knittel JG, Yan L, Rueden CT, White JG and Keely PJ: Collagen
density promotes mammary tumor initiation and progression. BMC Med.
6:112008. View Article : Google Scholar
|
|
46
|
Saito S, Murakoshi K, Kotake S, Kamatani N
and Tomatsu T: Granzyme B induces apoptosis of chondrocytes with
natural killer cell-like cytotoxicity in rheumatoid arthritis. J
Rheumatol. 35:1932–1943. 2008.
|
|
47
|
Parkinson LG, Toro A, Zhao H, Brown K,
Tebbutt SJ and Granville DJ: Granzyme B mediates both direct and
indirect cleavage of extracellular matrix in skin after chronic
low-dose ultraviolet light irradiation. Aging Cell. 14:67–77. 2015.
View Article : Google Scholar
|
|
48
|
Meyaard L: The inhibitory collagen
receptor LAIR-1 (CD305). J Leukoc Biol. 83:799–803. 2008.
View Article : Google Scholar
|
|
49
|
Vesely MD, Kershaw MH, Schreiber RD and
Smyth MJ: Natural innate and adaptive immunity to cancer. Annu Rev
Immunol. 29:235–271. 2011. View Article : Google Scholar
|
|
50
|
Salmon H, Franciszkiewicz K, Damotte D,
Dieu-Nosjean MC, Validire P, Trautmann A, Mami-Chouaib F and
Donnadieu E: Matrix architecture defines the preferential
localization and migration of T cells into the stroma of human lung
tumors. J Clin Invest. 122:899–910. 2012. View Article : Google Scholar
|
|
51
|
Sorokin L: The impact of the extracellular
matrix on inflammation. Nat Rev Immunol. 10:712–723. 2010.
View Article : Google Scholar
|
|
52
|
Weathington NM, van Houwelingen AH,
Noerager BD, Jackson PL, Kraneveld AD, Galin FS, Folkerts G,
Nijkamp FP and Blalock JE: A novel peptide CXCR ligand derived from
extracellular matrix degradation during airway inflammation. Nat
Med. 12:317–323. 2006. View
Article : Google Scholar
|
|
53
|
Jadhav RR, Ye Z, Huang RL, Liu J, Hsu PY,
Huang YW, Rangel LB, Lai HC, Roa JC, Kirma NB, et al: Genome-wide
DNA methylation analysis reveals estrogen-mediated epigenetic
repression of metallothionein-1 gene cluster in breast cancer. Clin
Epigenetics. 7:132015. View Article : Google Scholar
|
|
54
|
Laurson J, Khan S, Chung R, Cross K and
Raj K: Epigenetic repression of E-cadherin by human papillomavirus
16 E7 protein. Carcinogenesis. 31:918–926. 2010. View Article : Google Scholar
|
|
55
|
McLane LM, Banerjee PP, Cosma GL,
Makedonas G, Wherry EJ, Orange JS and Betts MR: Differential
localization of T-bet and Eomes in CD8 T cell memory populations. J
Immunol. 190:3207–3215. 2013. View Article : Google Scholar
|
|
56
|
Fukuoka N, Harada M, Nishida A, Ito Y,
Shiota H and Kataoka T: Eomesodermin promotes interferon-γ
expression and binds to multiple conserved noncoding sequences
across the Ifng locus in mouse thymoma cell lines. Genes Cells.
21:146–162. 2016. View Article : Google Scholar
|
|
57
|
Banno T, Adachi M, Mukkamala L and
Blumenberg M: Unique keratinocyte-specific effects of
interferon-gamma that protect skin from viruses, identified using
transcriptional profiling. Antivir Ther. 8:541–554. 2003.
|
|
58
|
Black AP, Ardern-Jones MR, Kasprowicz V,
Bowness P, Jones L, Bailey AS and Ogg GS: Human keratinocyte
induction of rapid effector function in antigen-specific memory
CD4+ and CD8+ T cells. Eur J Immunol.
37:1485–1493. 2007. View Article : Google Scholar
|
|
59
|
Soriano C, Mukaro V, Hodge G, Ahern J,
Holmes M, Jersmann H, Moffat D, Meredith D, Jurisevic C, Reynolds
PN, et al: Increased proteinase inhibitor-9 (PI-9) and reduced
granzyme B in lung cancer: Mechanism for immune evasion. Lung
Cancer. 77:38–45. 2012. View Article : Google Scholar
|
|
60
|
Bird CH, Sutton VR, Sun J, Hirst CE, Novak
A, Kumar S, Trapani JA, Bird PI, et al: Selective regulation of
apoptosis: The cytotoxic lymphocyte serpin proteinase inhibitor 9
protects against granzyme B-mediated apoptosis without perturbing
the Fas cell death pathway. Mol Cell Biol. 18:6387–6398. 1998.
View Article : Google Scholar
|
|
61
|
Buzza MS, Hirst CE, Bird CH, Hosking P,
McKendrick J and Bird PI: The granzyme B inhibitor, PI-9, is
present in endothelial and mesothelial cells, suggesting that it
protects bystander cells during immune responses. Cell Immunol.
210:21–29. 2001. View Article : Google Scholar
|
|
62
|
Barrie MB, Stout HW, Abougergi MS, Miller
BC and Thiele DL: Antiviral cytokines induce hepatic expression of
the granzyme B inhibitors, proteinase inhibitor 9 and serine
proteinase inhibitor 6. J Immunol. 172:6453–6459. 2004. View Article : Google Scholar
|
|
63
|
Jiang X, Orr BA, Kranz DM and Shapiro DJ:
Estrogen induction of the granzyme B inhibitor, proteinase
inhibitor 9, protects cells against apoptosis mediated by cytotoxic
T lymphocytes and natural killer cells. Endocrinology.
147:1419–1426. 2006. View Article : Google Scholar
|
|
64
|
Lauricella M, Carlisi D, Giuliano M,
Calvaruso G, Cernigliaro C, Vento R and D'Anneo A: The analysis of
estrogen receptor-α positive breast cancer stem-like cells unveils
a high expression of the serpin proteinase inhibitor PI-9: Possible
regulatory mechanisms. Int J Oncol. 49:352–360. 2016. View Article : Google Scholar
|
|
65
|
Cunningham TD, Jiang X and Shapiro DJ:
Expression of high levels of human proteinase inhibitor 9 blocks
both perforin/granzyme and Fas/Fas ligand-mediated cytotoxicity.
Cell Immunol. 245:32–41. 2007. View Article : Google Scholar
|
|
66
|
Deisting W, Raum T, Kufer P, Baeuerle PA
and Münz M: Impact of diverse immune evasion mechanisms of cancer
cells on T cells engaged by EpCAM/CD3-bispecific antibody construct
AMG 110. PLoS One. 10:e01416692015. View Article : Google Scholar
|
|
67
|
Bladergroen BA, Meijer CJ, ten Berge RL,
Hack CE, Muris JJ, Dukers DF, Chott A, Kazama Y, Oudejans JJ, van
Berkum O, et al: Expression of the granzyme B inhibitor, protease
inhibitor 9, by tumor cells in patients with non-Hodgkin and
Hodgkin lymphoma: A novel protective mechanism for tumor cells to
circumvent the immune system. Blood. 99:232–237. 2002. View Article : Google Scholar
|
|
68
|
Fritsch K, Finke J and Grüllich C:
Suppression of granzyme B activity and caspase-3 activation in
leukaemia cells constitutively expressing the protease inhibitor 9.
Ann Hematol. 92:1603–1609. 2013. View Article : Google Scholar
|