1. The principle of the CRISPR genome
engineering tool
Over the past decades, genome editing technologies
have been composed of zinc-finger nucleases (ZFNs) and
transcriptional activator-like effector nucleases (TALENs),
empowering scientific results at both the basic and clinical level
(1,2). Despite the advances that have been
reported in the field of genomic engineering, the use of ZNF or
TALEN nucleases is associated with several obstacles. For example,
the design for genomic engineering techniques remains complex, and
therefore, these techniques cannot modulate the expression of
multiple target genes. The principle in using ZNFs and TALENs is
protein-based and the associated toxicity is very high (3) (Table
I), thus prompting researchers to uncover a novel genome
engineering tool.
 | Table IComparison of genome engineering
tools properties. |
Table I
Comparison of genome engineering
tools properties.
| Properties | ZNF | TALEN | CRISPR |
|---|
| DNA-binding
moiety | Protein | Protein | RNA |
| Target recognition
size | 18–36
nucleotides | 30–40
nucleotides | 22 nucleotides |
| Nuclease | FokI | FokI | Cas |
| Toxicity | Variable to
high | Low | Low |
| Complexity of
design | Very complex | Complex | Simple |
| Ease of targeting
multiple targets | Low | Low | High |
| Off-target
effects | Moderate | Low | Variable |
A novel RNA-guided endonuclease-relied genome
editing technology that was termed the clustered regularly
interspaced short palindromic repeats (CRISPR)-associated protein 9
(Cas9) system, markedly altered the landscape of genomic
engineering (4,5). The story began with the study of the
immune system in bacteria and archaea in an attempt to elucidate
the mechanisms through which these organisms combat viral
infection. In native context, it was found that CRISPR in
combination with Cas protein provide bacteria with immunity against
infections. Specifically, it was shown that the role of repeats was
to recognize mobile genetic elements (MGEs), and thus it was
possible to cut them into small sequences and integrate them as
spacers into the genome of bacteria. That approach was based on the
microbial immune system that used RNA-guided nuclease to recognize
and cleave foreign genetic elements (6,7). In
2012, an adaptation of the prokaryotic immune system in mammalian
cells as a gene editing tool was simultaneously reported for the
first time by four different research groups [Mali et al
(8), Wright et al (9), Jinek et al (14), Swiech et al (30)], causing a certain debate regarding
the intellectual rights of this innovative technique. The newly
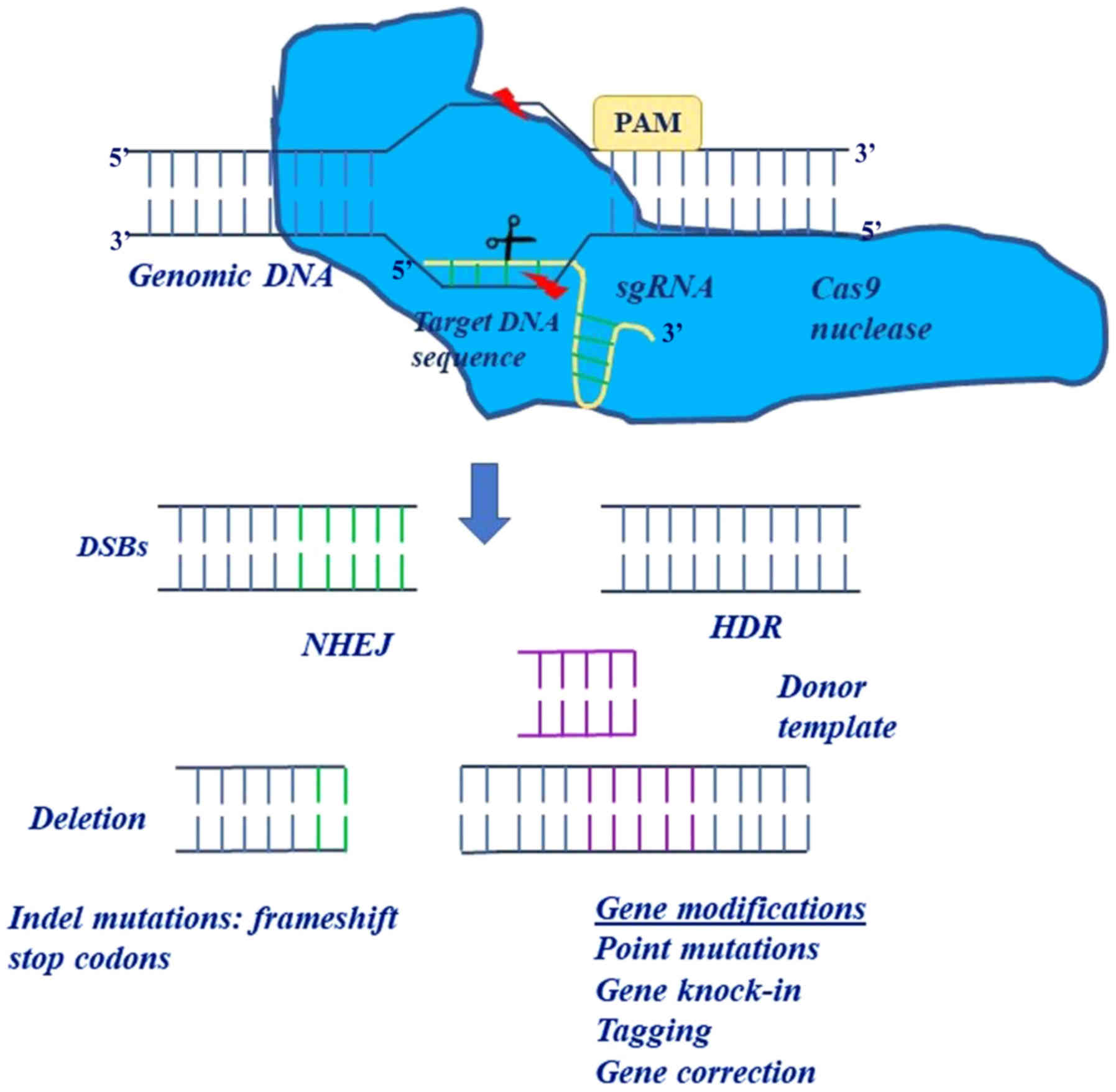
engineered CRISPR system consisted of two components: A chimeric
single-guide RNA (sgRNA) that provided target specificity and Cas9
that acted as a heli-case and a nuclease in order to unwind and cut
the target DNA (4,8). In this system, the only restriction
for the targeting of a specific locus was the protospacer adjacent
motif (PAM) sequence ('NGG' in the case of SpCas9) (6).
The CRISPR system was further simplified, based on
its ability to interfere with and participate in bacterial adaptive
immunity, comprising Cas nuclease and single-guide RNA (sgRNA). In
general, the CRISPR system main mechanism of action is mediated by
the Cas nuclease, which interacts with DNA and generates
double-strand breaks (DSBs) in the DNA sequence, and also matches
the broken genomic region with a sgRNA. The sgRNA is a chimeric
RNA, which consists of programmable CRISPR RNA (crRNA) and a
trans-activating RNA (tracrRNA) (9). Specifically, the CRISPR-Cas system
includes a cluster of proteins, categorized into Class 1 (Types I,
III and IV) and Class 2 (Types II, V, VI) (7), all of which constitute specific
RNA-guided DNA endonuclease proteins (Cas) (7,9–11).
Cas proteins are driven by RNA and not by other proteins, to
recognize the desired DNA sequence. The Class 2 subtype of the
CRISPR system, which generally exploits Cas9 nuclease, is usually
selected (9–11). The 100 bp sgRNA forms complementary
bonds with the target DNA sequence of 17–20 nucleotides, via
Watson-Crick base pairing, and the tracrRNA is the component which
Cas9 nuclease binds to. Specifically, the sgRNA recognizes the
target sequence, which is located upstream of the triplicate
sequence named PAM, given that the PAM motif recruits Cas9 nuclease
at site of DNA cleavage (12)
(Fig. 1). Of note, the PAM
sequence plays the determinant role in recognizing the correct DNA
sequence and in preventing the direction of RNA to self-targets and
non-specific sequences (13). This
is possible as repeats of the CRISPR system do not involve PAM and
the orientation of Cas9 depends on the PAM sequence (14). Overall, the genomic sequence of 14
nucleotides defines the target at which Cas9 nuclease exerts its
effects (15). More specifically,
this sequence is composed of 12 nucleotides of sgRNA in conjunction
with two nucleotides of protospacer adjacent motif. Notably, there
is a wide range of PAM sequences depending on their origin
(16). In the case of Cas9 derived
from Streptococcus pyogenes, the motif of the PAM sequence
may be composed of any base, followed by two additional guanine
bases (16).
The CRISPR system is sufficient on its own to
instigate double helical DNA breaks, which can be repaired by
non-homologous end joining (NHEJ) or homology directed repair
(HDR). However, the efficiency and specificity of the CRISPR system
are not based on DNA repair mechanisms (17). In the NHEJ repair mechanism, the
DNA ends are chemically ligated back together with a small
insertion or deletion at the site of the break. Thus, the NHEJ
mechanism is usually employed in cases of gene disruption (small
deletions or insertions), inversions, duplications or deletions,
whereas the HDR repair mechanism is used for large deletions, base
mutations, insertions and replacements (Fig. 2). In the HDR repair mechanism, a
donor DNA molecule matches with the genomic sequence flanking the
site of the DSB, thus introducing new genetic information into the
genome at the site of the break. The CRISPR technique can utilize
the HDR mechanism by using single-strand DNA oligonucleotides in
order to cause silent mutations, thus allowing us to monitor the
anticipated phenotype in a particular cell type (18,19).
Notably, the repair pathway is selected based on the phases of the
cell cycle; the NHEJ mechanism is employed in cells that are at the
G1, S and G2 phases, whereas the HDR mechanism is restricted to the
S and G2 phases (20).
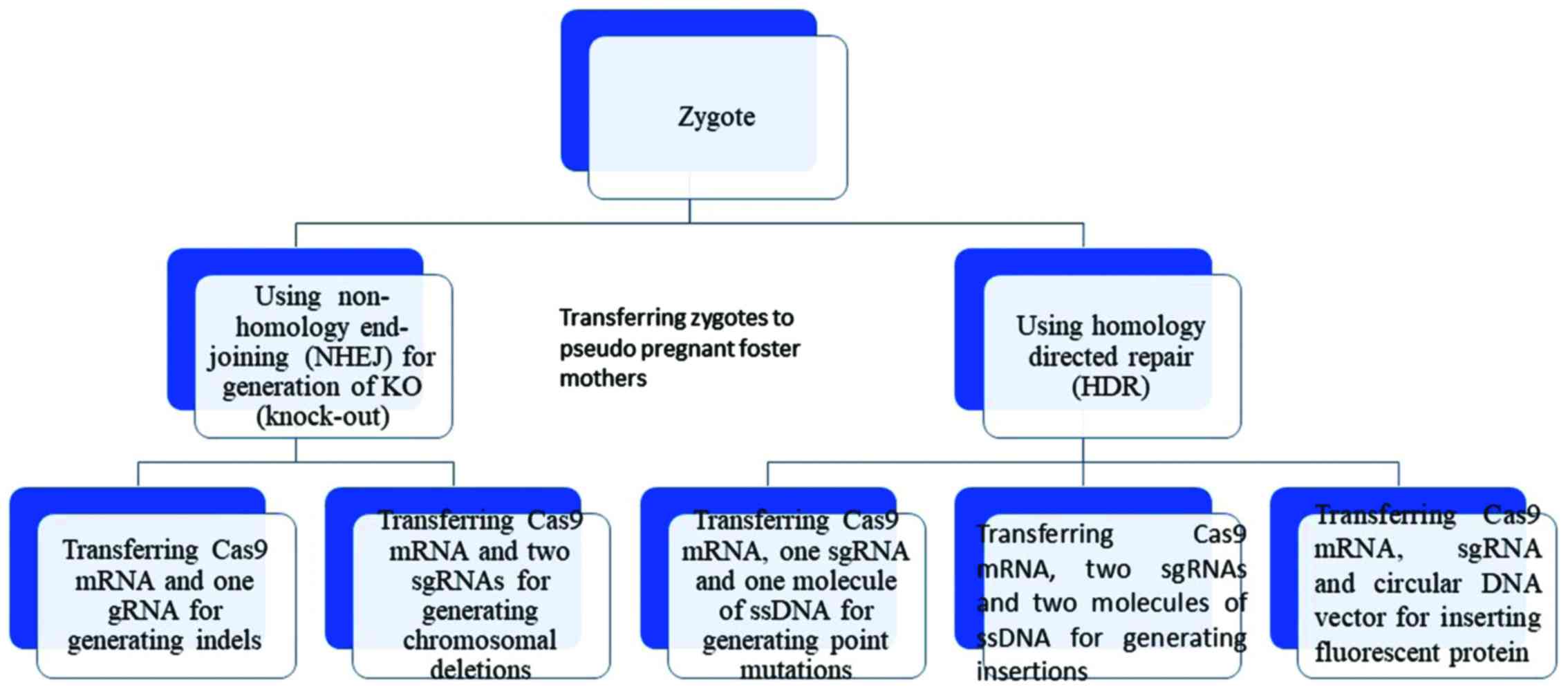 | Figure 2The use of two distinct repair
pathways in performing different modifications. In the NHEJ
mechanism, the ends of the DNA are chemically ligated back together
with a small insertion or deletion at the site of the break. The
NHEJ mechanism is usually employed in cases of gene disruption
(small deletions or insertions), inversions, duplications or
deletions whereas the HDR mechanism is used for deletions, base
mutations, insertions and replacements. In HDR, a donor DNA
molecule matches with the genomic sequence flanking the site of the
double-strand break and thus it can be integrated into the genome
at the site of the break, introducing new genetic information into
the genome. NHEJ, non-homologous end joining; HDR, homology
directed repair; sgRNA, single-chimeric guide RNA. |
Nonetheless, the major challenge when using both
DNA-repair mechanisms is the creation of DSBs, which can either
trigger signaling cascades mediated by DNA damage checkpoints or
cause the formation of gene translocations (21,22).
In the case of the NHEJ repair mechanism, most obstacles are
related to disrupting the open reading frames of genes, considering
that the ligation of two ends after DSBs is error-prone. The HDR
repair mechanism, on the other hand, is characterized by low
efficiency, particularly in non-dividing cells, despite its high
overall accuracy as a repair mechanism. Therefore, the CRISPR
method has been modified as an alternative to the above repair
mechanisms, using cytidine deaminases fused to Cas9 nickase, aiming
to circumvent the formation of DSBs and to implement the process in
non-diving cells. Specifically, it has been shown that the
association of Cas9-D10A nickases mutant with apolipoprotein B mRNA
editing enzyme catalytic polypeptide-like family protein 1
(APOBEC1) and uracil glycosylase inhibitor (UGI), leads to a 37%
increase in CRISPR efficiency (23).
The homology-independent targeted integration (HITI)
constitutes another advancement of the CRISPR system, as it
surpasses the limitations met in both repair mechanisms (NHEJ and
HDR). HITI takes advantage of the NHEJ mechanism and aims at
specific gene modifications (24).
Characteristically, it has been documented that the most effective
HITI rate is at 55.9% in neurons, as compared to HDR (1–3%
efficiency) (24). These examples
constitute irrefutable evidence of the advancements that have been
made in order to accommodate the 'in vivo' delivery of the
CRISPR system, including non-dividing cells.
The structural elements of the CRISPR system through
which Cas9 is assembled to RNA and DNA sequence include a T-shaped
configuration comprising the four stem cell loops, the linker
region and the repeat: anti-repeat binary complex (25). The formation of stem cell loops has
been reported to be crucial for the efficiency and stability of the
CRISPR-sgRNA complex (25).
In the field of functional studies, the CRISPR
system has rapidly revolutionized genetic engineering, allowing
researchers to easily alter a vast range of genomes. The mechanism
of the pioneer CRISPR approach is based on RNA-DNA interaction,
whereas previous genome editing tools (ZNFs and TALENs) were based
on protein-DNA associations (2,26)
(Table I). The properties of the
CRISPR system that render it amenable are as follows: its
simplicity in constructing the Cas9 nuclease and its capacity to
target many genomic loci simultaneously. Notably, the CRISPR system
has been distinguished over other approaches, as it enables the
simultaneous study of numerous genetic modifications in one step,
based on the method of multiplex target recognition, which uses
many sgRNAs at the cellular level (27). The multiplex capacity of the CRISPR
system is invaluable in studying the underlying molecular
mechanisms that are implicated in tumor progression, given that
cancer is a multistep procedure that involves the accumulation of
genetic changes, such as mutations, genome rearrangements and
epigenetic alterations (28,29).
Furthermore, the potential redundancy among several genes in a
functional output can be delineated using the CRISPR method. For
example, it has been shown that the Cas9-mediated elimination of
each DNA methyltransferase in mouse brains highlights the role of
any DNA methyltransferase in the memory compartment (30). The CRISPR system has proven to be
efficient in inducing a wide variety of genetic modifications,
ranging from the elimination and mutations of genes to genomic
insertions (4,8,31,32),
inversions (33,34) and translocations (21,32,35,36).
For example, the insertion of one specific DNA template can be
accomplished using HDR with duplex DNA templates (4,8,31,32)
or single-strand oligonucleotides (31,37–41)
or viral encoded templates (42,43).
In addition, the Cas9 nuclease appears to be superior to other
nucleases, as it has the ability not only to induce
gain-of-function and loss-of-function mutations, but also to cause
specific modifications.
To sum up, the CRISPR technology comes with a surge
of excitement, as it can be applied to a wide range of biological
models, including immortalized cancerous lines, primary cells
derived from mouse and human origins, xenografts, organoid
cultures, as well as the generation of genetically engineered
animal models. The CRISPR technology can be employed for the
comprehensive dissection of oncogenic signaling pathways via
sequential or multiplex gene editing. We envision a new era in
cancer biology during which CRISPR-based genome engineering will
serve as an important link between the bench and the bedside. The
successful implementation of sophisticated genetic technology aims
at the comprehensive characterization of tumors individually in
patients, thus paving the road for the development of tailored
cell-based or whole animal-based experimental systems.
2. The revolution in generating animal
models and cell lines
Cell and animal models play an essential role in
expanding our knowledge in the field of tumor biology. Undoubtedly,
the use of classical biological systems is crucial for evaluating
the efficacy of various potential therapeutic drugs. In this
review, we analyze the mechanisms of action of the CRISPR system,
compare it with other gene editing tools and discuss its
contribution to the generation of genetically engineered mouse
models (GEMMs), cell lines or organoids. The table below summarizes
the advances achieved to date with Cas9 nuclease in introducing
genetic changes that appear to have therapeutic potential in
several cancer subtypes (Table
II). The CRISPR system has been implemented not only in
classical biological models, but also in primary cells, such as
induced pluripotent stem cells (iPSCs), with the aim to identify
novel oncogenic pathways and consequently novel therapeutic options
against diverse cancer subtypes.
| Table IICancer therapeutics arising from the
CRISPR system. |
Table II
Cancer therapeutics arising from the
CRISPR system.
| Cancer type | Modification | Contribution to
therapy | Authors/(Refs.),
year | Journal |
|---|
| Breast cancer | Knock-out of
miR-644a | Inhibition of tumor
growth, metastasis, and drug resistance | Raza et al
(227), 2016 | Oncotarget |
| Breast cancer | Knock-out (KO)
BC200 lncRNA by CRISPR system | BC200 may serve as
a prognostic marker and possible target for attenuating deregulated
cell proliferation in estrogen-dependent breast cancer | Singh et al
(228), 2016 | Cell Death and
Disease |
| Endometrial
cancer | Knock-out of
MUC1 at cells by CRISPR system | Concomitant
decrease of MUC1 and EGFR can be prognostic markers in human
endometrial tumors | Engel et al
(229), 2016 | Oncotarget |
| Lung adenocarcinoma
and endometrial carcinoma | Deletion of
super-enhancers 3′ to MYC in cells by using CRISPR
system | Super-enhancers
stimulate cancer driver genes in diverse types of cancer | Zhang et al
(230), 2016 | Nature
Genetics |
| Endometrial
cancer | ERα-null
endometrial cancer cells | Inverse
relationship between the tumor suppressor PR and the oncogene Myc
in endometrial cancer | Kavlashvili et
al (231), 2016 | PLOS One |
| Prostate
cancer | NANOG and
NANOGP8 knockout DU145 prostate cancer cell lines | Attenuation of
malignant potential of prostate cancer | Kawamura et
al (232), 2015 | Oncotarget |
Genetically-engineered mouse models have been
extensively used in the study of tumorigenesis mechanisms and in
the design of drugs that confer tumor resistance (44,45).
Initially, embryonic stem cells were modified through Cre-LoxP
homologous recombination and injected in the pro-nucleus of
wild-type mouse blastocysts, thus rendering embryonic stem cells as
a necessary prerequisite for the generation of genetically
engineered mouse models. Consequently, sequential breedings were
required until the animals contained mutant alleles (46), supporting germ-line transmission.
It should be noted that the time for the generation of modified
mice was approximately 9–12 months, while the insertion of multiple
alterations was associated with a number of technical difficulties.
In other words, the entire process was time-consuming, costly and
in some cases, uncertain.
In contrast to classical methods, the CRISPR system
enables the elucidation of tumorigenesis networks and abolishes the
need for embryonic stem cells or time-consuming mouse breeding. The
CRISPR system emerges as a reliable and powerful tool for the
creation of mouse models that harbor multiple oncogenic alleles, at
a low cost. With this technological approach, various gene networks
can be targeted simultaneously, allowing researchers to study the
synergistic or antagonistic effects of genes in tumor initiation
and progression in an accurate and effective manner. The CRISPR
system appears to be a major contributor to the design of mouse
models (Table III), given that
CRISPR technology can simulate the genetic heterogeneity of the
cancer genome by creating indels, point mutations, large deletions,
large insertions and chromosomal rearrangements.
| Table IIIModeling of cancer mouse models
through the CRISPR system. |
Table III
Modeling of cancer mouse models
through the CRISPR system.
| Cancer type | Mouse models | Modifications | Authors/(Refs.),
year | Journal |
|---|
| Lung
adenocarcinoma | CD1 and C57BL/6J
(B6) | Eml4-Alk
translocation | Maddalo et
al (56), 2014 | Nature |
| Lung
adenocarcinoma |
p53+/− or
p53−/− | Eml4-Alk
translocation | Blasco et al
(55), 2014 | Cell Reports |
| Lung
adenocarcinoma |
KrasLSL-G12D−/+ | Nkx,
Pten, Apc | Sanchez-Rivera
et al (61), 2014 | Nature |
| Liver cancer | FVB/NJ mice | p53,
Pten, Ctnb1 | Xue et al
(47), 2014 | Nature |
| Pancreatic ductal
adenocarcinoma |
KrasLSL-G12D−/+;
R26LSL-Tom; H11LSL-Cas9−/+ | Lkb1 | Chiou et al
(62), 2015 | Genes and Dev. |
| Pancreatic ductal
adenocarcinoma |
Kras+/LSL-G12D;
Trp53loxP/loxP | p53,
Kras and p57 | Mazur et al
(233), 2015 | Nature
Medicine |
|
Medulloblastoma | C57BL/6N mice | Ptch1 | Zuckermann et
al (48), 2015 | Nature
Communications |
| Glioblastoma | Crl:CD1 (ICR)
mice | Trp53,
Pten, Nf1 | Zuckermann et
al (48), 2015 | Nature
Communications |
| Breast cancer | WapCre;
Cdh1F/F;
Col1a1invCAG-AktE17K-IRES-Luc/+ | Cdh1,
Akt-E17K or Pten | Annunziato et
al (63), 2016 |
Resource/Methodology |
| Breast cancer | Mammary stem cell
(MaSC) organoid-based approach | Inactivation of
Ptpn22 or Mll3 | Zhang et al
(234), 2016 | Cell Reports |
| Ovarian high-grade
serous carcinoma (HGSC) | Double
Trp53−/−; Brca2−/− mutant
mice | Deletion at
p53 and Brac2 genes | Walton et al
(235), 2016 | Cancer
Research |
| Invasive lobular
breast carcinoma (ILC) |
Cdh1F/F;
PtenF/F mice | Ablation of
Pten expression through CRISPR and lentivirus in mammary
glands of mice with loss of E-cadherin | Annunziato et
al (63), 2016 | Genes and
Development |
The CRISPR system is invaluable in mutating,
deleting, inserting or translocating genes. Several research groups
have produced extraordinary results in the field of mutagenesis via
the CRISPR approach. For example, in the modeling of hepatocellular
carcinoma, one group managed to create a frameshift truncation at
two genetic loci through the hydrodynamic injection of plasmids
encoding Cas9 nuclease and sgRNAs (47), while another research group managed
to generate oncogenic point mutations in the CTNNB1 gene
with the use of homology-directed repair at DSBs induced by Cas9
(39). Of note, the former
researchers demonstrated the desired modification of the
phosphatase and tensin homolog (Pten) or p53 gene
(tumor suppressor genes), alone or in combination, in 20 to 30% of
mouse hepatocytes. Following the inoculation of Cas9 and sgRNAs,
the authors demonstrated indel mutations in the Pten and
p53 genes at low efficiencies of 4 and 6.4%, respectively.
Thus, it was shown that it is possible to disrupt gene expression
in two major suppressor genes, causing hepatocellular carcinoma.
The study produced an accurate and reliable model of
hepatocarcinogenesis, equal to that provided by Cre-LoxP
recombination (47). Similarly,
gain of function mutations in oncogenes, such as the Catenin b1
gene (Ctnnb1) were conducted at 0.5% efficiency (47). Another example was illustrated
through the CRISPR system-mediated abnormal gene expression of
tumor suppressor genes (Ptch1, Trp53, Pten and
Nf1), ultimately causing medulloblastoma or glioblastoma
(48). Based on these results, it
appears that the CRISPR approach allows the genetic engineering of
oncogenes and tumor suppressor genes in specific somatic cells
simultaneously, despite the low efficiency.
In general, specific nucleotide modifications or
small deletions have been conducted with the use of the CRISPR
system (31,37), thus simulating the conditions that
characterize specific human diseases. The CRISPR system has been
confirmed to be invaluable in cancer research, due to its ability
to generate cancer mouse models harboring multiple mutations
simultaneously (49,50). For example, Findlay et al
exploited the properties of Cas9 nuclease to create mouse models
with distinct combinations of genetic alterations (51).
When it comes to introducing large deletions,
classical methods have, in most cases, proven to be insufficient
due to the many recombination events occurring in embryonic stem
cells (52). By contrast,
eliminating large chromosomal regions became very simple with the
use of the CRISPR system, as indicated by the results of two
research groups. Specifically, Yang et al introduced Cas9
mRNA and four sgRNAs into murine zygotes in one step, producing
mice with fluorescent tags into the following genes: Nanog,
Sox2, Oct4 (essential stem cell genes) and
Mecp2 (which causes Rett syndrome). When Cas9 nuclease mRNA
and two sgRNAs were specifically used against the Mecp2
gene, a 700 bp deletion was created (31). Additionally, the generation of
germline CRISPR mice was accelerated by injecting CRISPR components
in one-cell-stage embryos, as opposed to using embryonic stem
cells, as validated by Krishnaswamy et al (53). The CRISPR system also proved to be
very efficient inreplacing large ablated genomic region (exons
10–14) of dedicator of cytokinesis 8 (DOCK8) in Nlrp10
deficient mice (Nlrp10−/−), thus restoring the dynamics
of the immune cell cytoskeleton and the dendritic cell migration,
which in turn orchestrate the immune response (53). When it comes to introducing large
insertions at precise locations, insertion of large DNA sequences
has also been accomplished through homologous directed repair
mechanism in combination with the CRISPR system and fluorescent
tags (31).
Nonetheless, the CRISPR system does not only induce
targeted genetic alterations, but it can also be exploited for the
evaluation of nonsense mutations involved in tumorigenesis, as
indicated by Billon et al (54). Similarly, Billon et al
presented a further advancement of the CRISPR system, fusing Cas9
nickase to base editor and constructing specific sgSTOP to mediate
the transition of (CAA, CAG, CGA, TGG) codons located in the window
of PAM into stop codons. The whole process was evaluated by
restriction fragment length polymorphism (RFLP), through which the
disruption of restriction enzyme recognition sites was verified
(54). As a result, one can
monitor the presence of cancer-related nonsense mutations in a
considerable proportion (97–99%) of the eukaryotic genome in eight
species.
Furthermore, the CRISPR system has played a
fundamental role in the production of chromosomal rearrangements
that are implicated in cancer progression either as 'driver' or
'passenger' alterations. The tumorigenic process is influenced not
only by the presence of mutations in oncogenes or tumor suppressor
genes, but also indirectly by the presence of 'passenger changes'.
For example, Torres et al accomplished the introduction of
chromosomal translocation t(11;22)/ESWR1-FLI1 at
percentages of 1.76 and 0.15 in 293 cells and human primary
mesenchymal stem cells, respectively, using Cas9 nuclease and its
related sgRNAs (32). In addition,
the chromosomal translocation t(8;21)/RUNX1-ETO was
introduced into 293 cells and CD34+ human hematopoietic
progenitor cells with the use of the CRISPR system, successfully
recapitulating the phenotype of acute myeloid leukemia (AML). Three
other research teams used the CRISPR approach to introduce the lung
oncogenic gene rearrangement that results in the echinoderm
microtubule associated protein like 4-anaplastic lymphoma kinase
(Eml4-Alk fusion protein). Specifically, Blasco et al used
intratracheal or intrapulmonary lentiviral delivery of CRISPR
components to trigger the oncogenic rearrangement between the
Eml4 and Alk genes, located in chromosome 17
(55). Similarly, Maddalo et
al (56) and Nishio et
al (57) introduced the same
oncogenic rearrangement using adenoviral delivery. In all of these
studies, the experimental mice harboring the Eml4-Alk
inversion appeared to display all the symptoms of non-small-cell
lung cancer (NSCLC) (56) and
exhibited high sensitivity to ALK inhibitors, such as crizotinib
(57). In a similar setting, the
CRISPR system was used to introduce the KIF5B-RET or
EML4-ALK inversion (35), thus demonstrating that the
proximity of two loci, in which a chromosomal rearrangement takes
place, determines the capacity of the CRISPR system to reach its
maximum efficiency. Last but not least, Ghezraoui et al
successfully introduced the anaplastic large cell chromosomal
translocation t(2;5)/NPM-ALK using classical Cas9
nuclease or paired Cas9 nickases (21). Cas9 nickases are distinguished from
classical Cas9 nickases due to one of the endonuclease domains
being inactivated by a mutation, which in turn confers additional
efficiency.
Although the CRISPR system has been applied both to
'in vitro' and 'in vivo' biological systems, several
difficulties are encountered when the Cas9 nuclease is delivered to
the mitotic tissues of mice. The most common methods of
transferring Cas9 along with sgRNAs consist of viral vectors
(lentivirus, adenovirus and adeno-associated virus); even though
adenoviruses have high packaging capacity, thus being able to
deliver large genomic sequences (such as that of Cas9) (58), they may cause high immunogenic
reactions independently of cell type (59). This has led to a combination of the
CRISPR system and Cre-LoxP recombination technique in the study of
the networks implicated in tumorigenesis (42). Specifically, Cre-dependent Cas9
knock-in mice have been created via incorporation of a flanked Cas9
expression cassette (Cre-dependent CAG-LSL-Cas9) upon the exposure
of strong CAG promoter into the Rosa26 locus, without the
need to package Cas9 into viral particles and the accomplished
inducible expression of Cas9 mediated by Cre recombinase. The
generation of Cre dependent Cas9 knock-in mice was the result of
using the above-mentioned procedure in embryonic stem cells that
were transplanted into the blastocysts of C57BL6/J mice. In turn,
the Cas9 knock-in mice were transduced with a cassette containing
suitable sgRNAs, orientated towards specific yet multiple genetic
loci in conjunction with the sequence of Cre recombinase (42). This combination of Cre recombinase
and Cas9 nuclease was used to introduce loss of function mutations
in tumor suppressor genes p53 and Lkb1 and a gain of
function mutation (KRASG12D) in the Kras lung gene,
given that these genes are regarded 'the driver cancer genes' by
the CGA Network (60). At the same
time, it was shown that the introduction of loss of functional
mutations in NK2 homeobox 1 (Nkx2.1), Pten and
adenomatous polyposis coli (Apc) genes with the CRISPR
system allows for the creation of mouse models of lung
adenocarcinoma with deficient p53 expression or heterozygous
expression of Kras gene (KrasG12D−/+) through
Cre-LoxP recombination (61).
Thus, the CRISPR system was proven to be functionally significant
in elucidating the putative drivers of signal transduction pathways
in established mouse models of cancer. In addition, Chiou et
al constructed H11LSL-Cas9/+ mice, by
inserting the Cas9 cassette with a flanked stop region
(Loxp-STOP-Loxp) into the H11 locus of mice and they were
crossed with CMV-Cre deleter mice, demonstrating constitutive
expression of Cas9 due to the recombinase action of Cre. Following
this, H11LSL-Cas9/+ mice were crossed with
KrasLSL-G12D/+; R26LS (KT) mice,
generating KT; H11LSL-Cas9/+ mice. The latter
were infected with Lentivirus-sgLkb1/Cre with an ubiquitous
promoter, accomplishing the disruption of pancreatic Lkb1
expression in adult pancreatic cells 'in vivo', and thus
simulating stages of pancreatic cancer progression (62). Elsewhere, Annunziato et al
performed a Cdh1 gene deletion by encoding E-cadherin
through Cre-LoxP recombination in mammary epithelium, with the
concomitant disruption of Pten by the CRISPR system.
Unexpectedly, the authors observed an increased immune response
following the exposure to Cas9 (63).
3. Germ-line gene editing through the CRISPR
system
Apart from the wide spectrum of genetic alterations,
the CRISPR system holds considerable potential as a tool that can
be applied to either embryonic stem cells or other types of stem
cells, surpassing somatic mosaicism that is commonly found when
genetic modifications are performed in zygotes. Genetic mosaicism
has been attributed to the slow rate of Cas9 nuclease mutagenesis
and the discordance between transcription and translation.
Characteristically, it has been mentioned that the translation of
Cas9 mRNA occurs until the first cell division (64). Therefore, the CRISPR system has
been applied to embryonic stem cells for the generation of
conditional knock-out mice. For example, Flemr et al
transduced mouse embryonic stem cells with a vector expressing
bacterial BirA ligase in a constitutive manner, driven by the
promoter of the Rosa26 locus; at the same time, the cells
were enriched with Cas9, a single-strand oligonucleotide containing
the FLAG-AviTag sequence that could be biotinylated by BirA ligase
and a recombination reporter equipped with (pRR-Puro) selection
marker which was activated following homology recombination events
(65). Thus, they established a
straightforward and flexible method for accelerating the production
of conditional deficient mice inessential genes, tracing
endogenously the biallelic deficient cells without using selection
markers (65). More importantly,
they excluded the possibility that the phenotypes of deficient mice
could be the result of non-targeted system efficacy.
The targeted disruption of gene expression at both
alleles of genes Tet1, 2 and 3 in zygotes with
20% efficiency has been previously demonstrated (37). Following this, researchers
attempted to disrupt the expression of five genes (Tet1,
2, 3, Sry and Uty); however, the
elimination efficiency appeared to below, with the genetically
modified cells constituting only10% of the total population. On the
other hand, Wang et al (37) produced impressive results by
introducing Cas9 nuclease with the appropriate sgRNAs in the
germ-line of mice, thus managing to genetically manipulate
Tet genes without the need for embryonic stem cells.
Notably, the sgRNA can be delivered either as plasmid or
single-strand RNA (ssRNA), while Cas9 nuclease can be packaged as
plasmid, mRNA or protein. In the former approach, a concomitant
introduction of Cas9 and a single sgRNA for each Tet gene
produced 89% of mice harboring the anticipated genetic alterations
(37). In the second approach, the
targeting of Tet1-2 genes with the use of sgRNAs proved that
mutations in both genes occurred at a percentage of 70%. In the
third approach, a complex comprising Cas9 mRNA, sgRNA and
single-strand DNA harboring the desirable change was used,
resulting in 60% of the produced mice harboring one mutation and 7%
of the mice carrying a combination of two different genetic
alterations.
In the context of hematological malignancies, the
proposed methodology includes the editing of progenitor cells
'ex vivo' and their subsequent delivery into the syngeneic
recipient (66–68). Hematopoietic cells have the unique
capacity of expanding after being re-injected into the human body.
For example, Heckl et al generated mouse models of AML by
simultaneously altering a couple of genes in hematopoietic stem and
progenitor cells (67). In the
same context, another research team eliminated the tumor suppressor
gene, mixed lineage leukemia 3 (Mll3; also known as
Kmt2c), in primary mouse hematopoietic progenitor cells
(HSPCs) of the shNf1 genotype; Trp53−/−
cells that were transplanted in AML (68). Impressively, it was shown that
Mll3 haploinsufficiency acts as secondary determinant factor
in the progression of leukemogenesis (68). Mll3 mutant mice alone did
not exhibit any signs of leukemia, while mice harboring Mll3
mutations developed ureter epithelial tumors in
p53+/− genotype (68).
Other studies have also documented the therapeutic
efficiency of the CRISPR system 'ex vivo' in the setting of
Eμ-Myc lymphomas (66). Heckl
et al devised a series of sgRNAs against eight candidate
genes usually implicated in myeloid cancers, thereby recapitulating
the proposed genetic networks and the mutations responsible for
disease progression and outcome (67). Specifically, the primary HSPCs
harboring a knock-in Flt3 internal tandem duplication
(Flt3-ITD) were edited for five characteristic genes. These
genes were either epigenetic modifiers, transcription factors or
mediators of cytokine signaling and were as follows: Tet2,
Dnmt3a, Runx1, Nf1, Ezh2, Smc3,
p53 and Asxl1. The selected genes were modified to
simulate the genetic networks responsible for the phenotype of
myeloid malignancies (67).
Similarly, Zhong et al presented an
innovative method for genetically manipulating AG-haESCs harboring
a disruption at two distinct DNA methylated regions (H19 and
Gtl2), thus providing evidence for the generation of live
embryos following AG-haESC injection into mature oocytes. Of note,
the authors demonstrated that the CRISPR genome editing tool
successfully introduced genetic modifications in AG-haESCs, thus
allowing genetic screening in haploid cells in a simple and rapid
manner (69).
4. CRISPR system: A therapeutic tool in a
wide range of genetic diseases
With the prospect of personalized therapy, the gene
editing of iPSCs has drawn a surge of interest in a wide range of
diseases. The reasons behind this lie in the capacity of the cells
to divide unlimitedly, while maintaining their genome integrity and
their differentiation capacity into three different cell layers
(endoderm, ectoderm and mesoderm). Notably, the use of iPSCs for
the generation of modified cell lines appears to provide further
insight into the underlying molecular mechanisms of each disease.
Therefore, the autologous transplantation of iPSCs appears to be a
promising therapy in personalized medicine, and the use of iPSCs in
the CRISPR approach holds promise for the field of genetic diseases
(Table IV).
| Table IVTherapeutic approaches of the CRISPR
method in a wide range of genetic diseases. |
Table IV
Therapeutic approaches of the CRISPR
method in a wide range of genetic diseases.
| Disease gene | Target | Concept | Substrate | Authors/(Refs.),
year |
|---|
| Hemophilia A | hF8 | NHEJ-mediated
correction of inversion | Patient iPSCs | Park et al
(70), 2015 |
| β-thalassemia | HBB | Cleave the
endogenous β-globin gene (HBB) | Tripronuclear (3PN)
zygotes | Liang et al
(236), 2015 |
| β-thalassemia | HBB | HDR-mediated
correction | Patient iPSCs | Xie et al
(74), 2014 |
| Cysticfibrosis | CFTR | HDR-mediated
correction of CFTRdeltaF508 mutation | PatientiPSCs | Firth et al
(83), 2015 |
| Cysticfibrosis | CFTR | HDR-mediated cDNA
knock-in | Intestinal
organoid | Schwank et
al (82), 2013 |
| Cataract | Crygc | HDR-mediated
correction | Zygote, mouse
SSC | Wu et al
(84), 2013; Wu et al
(85), 2015 |
| Huntington
disease | HTT | NHEJ mediated
allele editing | | Monteys et
al (78), 2017 |
| Hereditary
tyrosinemia I | Fah | HDR mediated point
mutation of Fah gene | Adult tissue
cells | Yin et al
(39), 2014 |
|
Cardiovasculardisease | Pcsk9 | NHEJ-mediated
disruption of PCSK9 | Adult tissue
cells | Ding et al
(58), 2014 |
Initially, Park et al supported that the
CRISPR system can remodel the large inversions that are encountered
in introns of blood coagulation factor VIII in hemophilia. The
researchers corrected the mutations of the F8 gene in
patient endothelial cells which were propagated by iPSCs. The
correctness and accuracy of the CRISPR system was validated from
the fact that hemophiliac mice were rescued upon the engraftment of
corrected iPSCs (70).
In the frame of genetic diseases, gene correction
seems to be very beneficial in hemoglobinopathies, such as sickle
cell disease (SCD) and β-thalassemia. In the context of SCD, the
causal origin of the disease is a replacement of valine with
glutamate due to a homozygous missense point mutation in the
hemoglobin subunit beta (HBB) gene, which results in the
accumulation of hemoglobin and the circulation of red blood cells
with conformational changes. The correction of mutated sickle cell
gene expression was accomplished with the use of the CRISPR system
in iPSCs derived from patients, followed by their differentiation
into functional erythrocytes (71,72).
It was shown that Cas9 nuclease was superior to ZNF in modulating
the expression of the sickle gene (73). The benefit of the CRISPR system on
SCD has also been demonstrated in a clinical trial
(NCT03167450).
In the case of β-thalassemia, iPSCs have been shown
to differentiate into specific lineages, thereby presenting as a
possible therapeutic means (74).
Similarly, the hemoglobin subunit beta (HBB) gene has been
effectively engineered by the CRISPR approach in tripronuclear
zygotes, at a 15% rate, considering that the mutations in the human
HBB gene are believed to be responsible for the disease.
However, the low effectiveness of the CRISPR approach highlighted
the need for precise optimization in a clinical setting.
In the field of gene editing, a potential obstacle
is the immune rejection of 'ex vivo' modified cells that
carry newly expressed or corrected proteins. The infusion of
modified cells harboring new proteins can evoke the stimulation of
the immune system, recruiting cytotoxic T lymphocytes in exhausting
the potential 'enemy'. In another case, it has been shown that
neutralizing antibodies can arise against replacement blood
clotting factors in patients with hemophilia (75). However, the possibility of inducing
an immune reaction has been avoided with the CRISPR system,
particularly if one considers that the gene therapy of SCD has been
achieved without adverse effects. Specifically, autologous
hematopoietic stem cells (CD34+ cells) have been
transduced with lentiviruses expressing the modified globin gene,
leading to the rescue of the phenotype at the patient level. In
other words, by exploiting the CRISPR gene engineering tool, it was
possible to alleviate the symptoms of the disease.
As regards Huntington's disease (HD), previous RNA
interference-based method reduced the expression of the Huntingtin
gene (HTT), but did not manage to completely attenuate gene
expression, as anticipated (76).
By contrast, the CRISPR system proved to be invaluable in
eradicating the expression of HTT, if one considers that the
origin of HD is ascribed to CAG repeat expansion at the 1st exon of
HTT. Therefore, using the CRISPR system, researchers have
managed to disrupt the sequence of the mutant HTT allele
(77), while at the same time
another research team highlighted the ablation of HTT gene
expression both in patients and in a transgenic mouse model bearing
the human HTT, using the CRISPR system (78). In the latter case, they
characteristically created small deletions and produced single
nucleotide polymorphisms (SNPs), which altered the determinant PAM
sequence ('NGG'), increasing the effectiveness of Cas9 nuclease. In
both cases, the researchers used the CRISPR strategy to cure HD. In
parallel, iPSCs derived from patients with HD were repaired with
the aid of the CRISPR and the piggyBac transposon-based approach
(79). In addition, in the case of
neurogenerative diseases, such as Parkinson's disease, iPSCs have
been employed to differentiate into neurons and have been used as a
substrate of the CRISPR screen for the identification of activators
of toxic protein a-Synuclein (aSyn) (80). In the frame of inheritable genetic
disorders, the CRISPR approach has also been shown to mediate the
repair of dystrophin gene mutations in zygotes of mdx mice
(C57BL/10ScSn-Dmdmdx/J) at 2% efficiency (81).
Of note, the CRISPR approach has been used as an
exciting therapeutic technological tool for the treatment of cystic
fibrosis. Cystic fibrosis essentially constitutes a disease that is
the outcome of many genetic alterations encountered at the CF
transmembrane conductor receptor (CFTR). Organoid buds from
iPSCs derived from patients with cystic fibrosis have been
successfully modulated by the CRISPR system, holding great promise
for organ transplantation and gene therapy (82). The correction of the CFTR
locus was performed in a separate case, generating iPSCs with the
correct allele of CFTR, as demonstrated by Firth et
al (83). In parallel, the
efficiency of the CRISPR genome engineering tool has been verified
in organoids derived from patient intestinal stem cells, where the
mutation (Phe 508) of the CFTR was repaired (82). It should be mentioned that the
elimination of the side-effects caused by the CFTR mutation
was verified via normal secretory functions (82). Apart from the repair of the
CFTR locus in stem cells of various origins, studies have
supported the CFTR gene editing in one step, upon influx of
the CRISPR system in zygotes. In such cases, an oligonucleotide of
wild-type CFTR sequence is inserted as a donor template and
used by the Cas9 nuclease via homologous recombination (84). The results were profoundly
spectacular due to germ-line transmission of the repaired
CFTR locus allele, even though off-target mutagenesis
remained a possibility. Nonetheless, the danger of insertional
mutagenesis in murine germ-line cells was abrogated by engineering
the mutant loci at spermatogonial stem cells (SSCs). Wu et
al reported that the mutational repair of the EGFP
transgene or endogenous Crypc gene via the CRISPR approach
in SSCs that were competent to form male gametes after injection
into testes, resulted in the formation of round spermatids
following fusion with mature oocytes (85). The major advantage of genetic
manipulation via the CRISPR approach in SSCs, as compared to gene
editing in zygotes, has been the lack of side-effects and the
generation of anticipated descendants at 100% efficiency (85). Therefore, the CRISPR method has
proven to be a promising approach for the treatment of genetic
diseases.
In addition to the above-mentioned findings, Xie
et al (86) demonstrated
that the epigenetic dormancy of the FMR1 gene was reversed
using the revolutionary genome editing tool, CRISPR in iPSCs
derived from patients or in somatic hybrid cell lines. The effect
of Cas9 was directed to the removal of the CGG repeats that are
encountered in the FMR1 gene. Specifically, the researchers
cleaved the FMR1 gene repeats with high efficiency, inducing
DSBs and using homologous recombination (86). The same genetic disorder was also
reported to be repaired via NHEJ, albeit less efficiently (86). A landmark study by Horii et
al successfully demonstrated the genetic correction of a
mutation at the DNMT3B locus in iPSCs with the use of the
CRISPR system, thus repairing a rare abnormality [termed
immunodeficiency centromeric region instability facial anomalies
syndrome (ICF)] (87). In the
prospect of curing human severe immunodeficiency, researchers
observed that iPSCs deficient for JAK3 perished during the initial
stages of the disease due to a low BCL2 expression pattern;
nonetheless, the correction of the JAK3 mutation with the
CRISPR approach caused iPSCs to differentiate into fully functional
T cells, including the full spectrum of T cell receptors (TCRs)
(88). Consequently, the
combination of the CRISPR approach and iPSCs has been reported to
be particularly beneficial in chronic granulomatous disease (CGD),
where patients suffer from the accumulation of oxidative molecules
that are used as a phagocytic weapons against fungi and bacteria
(89).
Furthermore, patient-derived pluripotent stem cells
have been exploited by the CRISPR method for therapeutic
intervention in other diseases, such as Fanconi anemia (90), dominant dystrophic epidermolysis
bullosa (91), retinitis
pigmentosa (92) and severe cases
of retinal dystrophy [Leber congenital amaurosis-10 (LCA10)]
(93).
Finally, the CRISPR system has been highlighted as
a unique system with profound impact on repairing gene mutations in
adult tissues 'in vivo'. Yin et al (39) rescued the phenotype generated by
Fah gene deficiency via repairing of the Fah mutation
in hepatocytes using the CRISPR system. Notably, Cas9 nuclease
mediated its beneficial action by restoring the function of
hepatocytes at very low (0.4%) efficiency in mouse models harboring
hereditary tyrosinemia (39).
Despite the initial effectiveness of the genome engineering tool
being very low, it was culminated over time, repairing 33% of
deficient hepatocytes. In another experimental setting, the
disruption of Pcsk9 gene expression in the liver resulted in
an attenuation of the concentration of LDL-C, which would otherwise
be very harmful for the heart (58).
As a general note, it has been claimed that the
Cas9 nuclease constitutes a therapeutic model for the treatment of
several genetic disorders. In the case of monogenic recessive
disorders, such as cystic fibrosis, sickle-cell anemia, hereditary
tyrosinemia or Duchenne muscular dystrophy, the target mutation can
be repaired with the aid of Cas9. In this manner, the protein
derived from the corrected gene can be developed in native
conditions. In the case of dominant-negative disorders where the
target gene is represented by one allele (haploin-sufficiency
phenomenon), the CRISPR system seems to be the most advantageous
method in inactivating the mutated allele. Alternatively, the use
of inactivated Cas9 fused to a transcriptional repression domain
can be used to rescue the phenotype (94). In addition, the elimination of
duplicated regions could be accomplished through Cas9 nuclease and
NHEJ-mediated repair, whereas therapeutic benefit has also been
observed by introducing protection mutations in mitotic tissues in
complex diseases. Finally, the CRISPR system has been employed in
the modification of T cells, particularly with chimeric antigen
receptor (CAR) or artificial TCRs, prior to introducing them into
the body of cancer patients (95)
(Fig. 3).
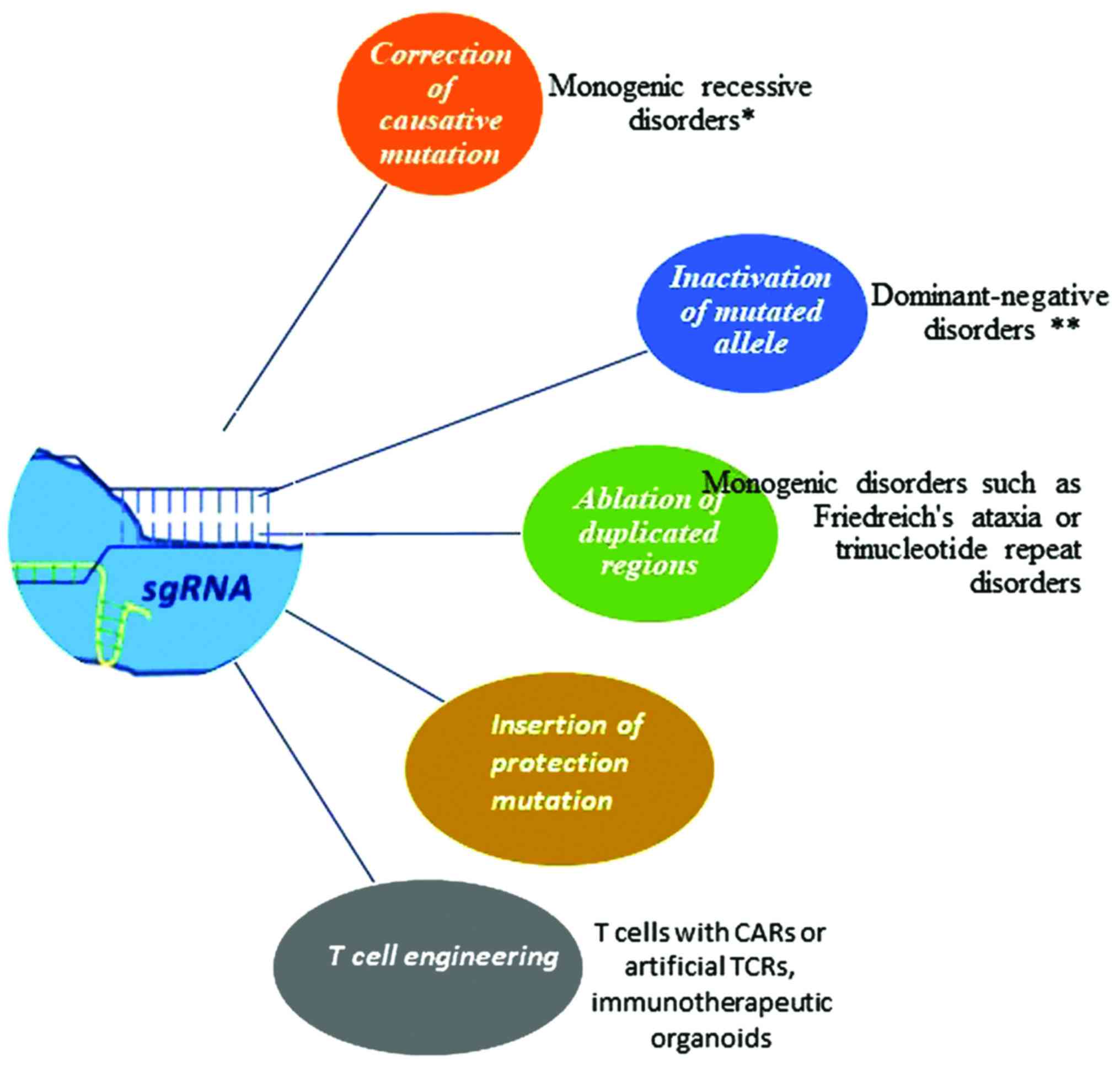 | Figure 3Cas9 nuclease as a therapeutic model
for the treatment of genetic disorders. In the case of monogenic
recessive disorders such as cystic fibrosis, sickle cell anemia,
hereditary tyrosinemia or Duchenne muscular dystrophy, the target
mutation is repaired with the aid of Cas9. In this manner, the
protein derived from corrected gene can be developed in native
conditions. In the case of dominant-negative disorders in which the
target gene is represented by one allele (haploinsufficiency
phenomenon), the CRISPR system seems to be the most advantageous
method in inactivating the mutated allele. In other instances, the
elimination of duplicated regions could be accomplished through
Cas9 nuclease and NHEJ mediated repair, whereas therapeutic benefit
has also been observed by introducing protection mutations in
mitotic tissues in complex diseases. Finally, CRISPR system has
been employed for the modification of T cells, especially with CAR
or artificial TCRs, with the aim to introduce modified cells into
the body of cancer patients. CRISPR, clustered regularly
interspaced short palindromic repeats; Cas9, CRISPR-associated
protein 9; NHEJ, non-homologous end joining; CAR, chimeric antigen
receptor; TCR, T-cell receptor. The single asterisk (*) indicates
cystic fibrosis, sickle cell anemia or Duchenne muscular dystrophy.
The double asterisks (**) indicate transthyretin-related hereditary
amyloidosis or dominant forms of retinitis pigmentosum. |
5. Organoids: Smart weapons against complex
genetic diseases
A significant part of the research community has
focused on the generation of the reliable biological models (animal
and cell lines) that will be able to mimic all the characteristic
mechanisms of cancer cells with high fidelity. In many cases, the
complete understanding of the genetic perturbations involved in
cancer outgrowth has been accomplished; however, researchers have
been unable to directly modify genes in the human body, thus
resulting in a highly anticipated therapeutic strategy (96).
Despite the successful creation of GEMMs, there is
significant shortage of therapeutic applications, mainly due to
difficulties in the isolation of specific neoplastic cells from the
multitude of extended stromal compartments in animal models. The
scarcity of therapies can also be explained by the fact that animal
models are time-consuming and costly. Cancer cell lines, on the
other hand, are known to harbor genetic profiles that are not
identical to the initial tumor mass and are cultured in
two-dimensional directions. Furthermore, cancer cell lines are
uncoupled to the amount of non-neoplastic cells, which are usually
located in the tumor microenvironment (97). As a consequence of the above,
organoids have been postulated as a novel facile tool that holds
great promise in the field of cancer research. Tumor-derived
organoids can mimic all the typical characteristics of the initial
tumor mass, the three-dimensional (3D) structural framework and the
property for uncontrolled growth. It has therefore been shown that
organoids can not only recapitulate the genetic modifications that
arise in cancer cells with high fidelity (98), but can also provide unique
opportunities for the generation of fully characterized models at
an unprecedented rate.
Hans Clevers and colleagues were the pioneer
investigators in the field of organoids, as they managed to create
intestinal epithelial organoids with distribution of all cell types
(such as stem, goblet and villus cells), maintaining the procedures
of cell division and differentiation in physiological conditions
[Sato et al (99)]. Hans
Clevers supported that organoids are able to efficiently simulate
the tissue microenvironment, as represented by their structural and
functional hallmarks (100).
Lancaster and Knoblich, on the other hand, defined an organoid as a
3D structure in which progenitor cells are self-organized in order
to commit to specific cell lineages, in a manner which is
consistent with the 'in vivo' conditions (101). Characteristically, the cells of
organoids have internal gates of self-assembling and
self-regulation, even though their manipulation is not restricted
to exogenous signals.
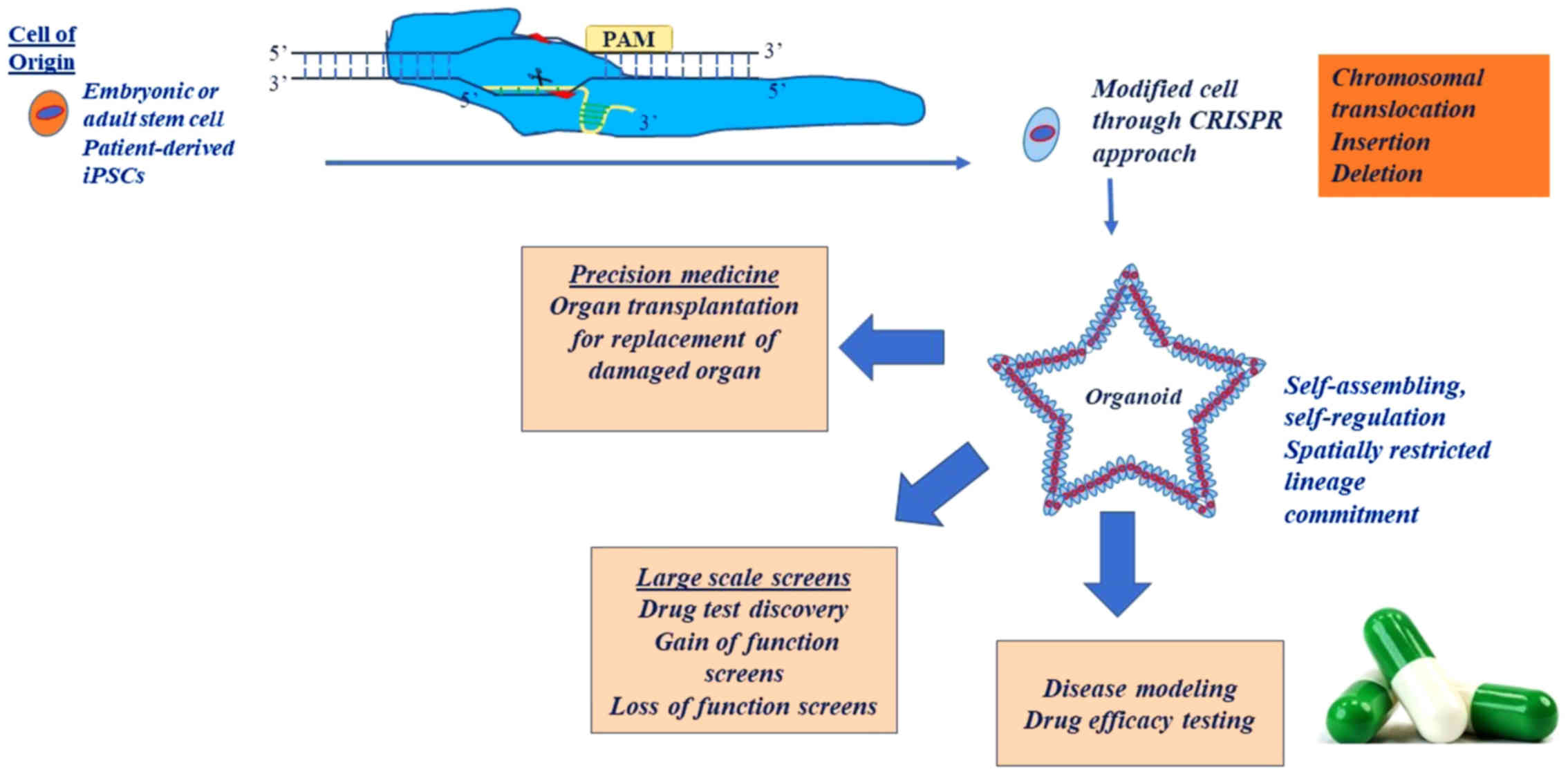
The origin of organoids can be either embryonic or
adult stem cells or patient-derived stem cells (100). On the one hand, organoids derived
from stem cells can be crucial to the study of organ development or
organ pathologies (101). While
pluripotent stem cell-based organoids exploit developmental
processes, adult stem cells can be coerced to form organoids by
creating conditions that mimic the stem cell environment during
physiological tissue self-renewal or during DNA damage/repair
(102). On the other hand,
patient-derived organoids can be used to gain drug-response
feedback in patients. Tumor organoids display a differential
mutational landscape indicative of each parental tumor. The
feasibility of culturing solid tumors directly from the patient in
the form of organoids holds great promise (102).
From a functional aspect, the generation of human
organ-oids is regarded highly important, as it has the potential to
enable the study of human pathogenesis 'in vitro'. For
example, murine neoplastic organoids have been generated from
Kras+/LSL-G12D; Pdx1-Cre mice ('KC mice')
in order to recapitulate the phenotype of human preinvasive
pancreatic intraepithelial neoplasms (PanINs) and
Kras+/LSL-G12D;
Trp53+/LSL-R172H; Pdx1-Cre mice ('KPC
mice') (103). It was
demonstrated that many candidate driver genes of the neoplastic
procedure are represented in the proteomic and transcriptional
genetic profile of murine pancreatic ductal organoids (103). The functional significance of
organoids was demonstrated when human tumor organoids were
engrafted into immunocompromised (Nu/Nu) mice, thus highlighting
the presence of the stromal compartment and validating the accuracy
of pancreatic tumorigenesis through organoids (103).
Nonetheless, the use of organoids is not restricted
to the study of molecular mechanisms that drive developmental
processes (104,105), but may also be used for the
modeling of diseases (Fig. 4). At
the same time, the generation of patient-derived organoids can
serve as an 'ex vivo' rational platform that has the
potential to predict the patient response to specific drug
administration, thereby helping towards deciding on the appropriate
patient treatment individually, particularly if one considers that
the majority of organoids are amenable to pharmacological studies
(Fig. 4). For example, van de
Wetering et al used patient-derived organoids as a potential
model to simulate the networks that characterize intestinal
diseases, submitted them in high-throughput drug screens, thereby
investigating the interactions among target genes (106). Intestinal cancer organoids
derived from 20 sequential colorectal carcinoma patients were
employed to create a living colorectal biobank (106), given that conventional cancer
cell lines do not represent the tissue structure and genetic
characteristics of original neoplasms. Remarkably, the differential
expression of parental intestinal neoplasms was well detected in
colorectal organoids (106) and
was shown that colorectal tumor organoids retained the molecular
and histopathological features of original tumors, as well as their
transcriptional profile. Importantly, van de Wetering et al
demonstrated the existence of an association between drugs and the
genomic profile of tumor organoids. Furthermore, p53
gene-deficient tumor organoids appeared to be prone to treatment
with the MDM2-inhibitor, Nutlin-3a, whereas organoids with the
elimination of RAS mutations were respectively sensitive to
therapy with specific antibodies against epidermal growth factor
receptor (EGFR) (106).
Consequently, the pharmacodynamic profile of primary cancers, as
well as of infectious and developmental diseases, has the potential
to be recapitulated from a colorectal biobank derived from
organoids that have originated from patient biopsy samples, thus
aiding in the conduction of personalized therapy that is pertinent
to genes of drug sensitivity or resistance.
The potential use of organoids in the field of
transplantation, as an alternative to iPSCs, holds immense promise.
Organoids have been transplanted in an autologous manner without
immunogenicity reactions, without the risk of teratomas and with
assured stability (107).
Notably, researchers have found the culture conditions for gastric
(108), pancreatic (109), hepatic (110), prostatic (111,112) and intestinal organoids (113). The ultimate goal is to replace
damaged organs inpatients with organoids that do not harbor the
usual genetic perturbations, given that the colonization of cancer
organoids in the human body can contribute to metastasis (60,114,115). A feasible personalized approach
can be performed through the correction of genetic modifications by
Cas9 nuclease in patient-derived organoids, and subsequently, the
transplantation of these modified organoids into the bodies of
patients. Nonetheless, the engraftment of organoids and the
delivery of Cas9 nuclease and its components need to be further
optimized, as thus far, the engraftment of organoids has been
accomplished at only 1% efficiency, whereas a 10% efficiency is
usually required for the replacement of determined protein
elimination, as shown in the case of liver organoids (110).
From a therapeutic point of view, the use of
organoids and their genetic manipulation through the CRISPR system
is regarded extremely important in precision medicine. Recently, a
colorectal tumor organoid library (CTOL) was constructed,
encompassing 55 colorectal tumor organoids and 41 respective normal
colorectal organoids (116),
using the CRISPR approach. Fujii et al (116) demonstrated that the transition of
carcinoma to more progressive stages was not associated with niche
signals; at the same time, it was suggested that the mutations in
oncogenic pathways are responsible for conferring the selective
advantage of neoplastic growth, thus paving the way for the
development of patient-specific therapies. Personalized therapy
based on organoids seems feasible if one considers the reported
mutations of the main five signaling pathways: WNT,
RAS/mitogen-activated protein kinase (MAPK), phosphoinositide
3-kinase (PI3K), transforming growth factor (TGF) and p53 (60). The study by Fujii et al
provided compelling evidence that tumor organoids and the CRISPR
approach can be effectively used in xenotransplantation assays
'in vivo', thus bridging the gap between genetic profile and
personalized medicine. Specifically, organoids were injected in
splenic compartments or into renal cell types of NOG mice,
demonstrating that the drivers of metastatic process are not
related to mutations or niche requirements (116). Additionally, Fujii et al
managed to modify genes of interest in organoids derived from
patient colon tissue using the CRISPR approach, in an attempt to
elucidate the functionality of intestinal cell types. Last but not
least, the powerfulness of the CRISPR approach was confirmed by
Drost et al (117), via
sequential editing of genetic loci Apc, p53,
KRAS and SMAD4 and converting normal colon organoids
derived from human intestinal crypt stem cells to tumor organoids,
without the need for stem-cell niche factors. Characteristically,
it was shown that APC and p53 deficiency can induce
chromosomal instability and aneuploidy, i.e., characteristics of
cancer. The efficiency of tumor organoids was mostly highlighted by
the fact that they sustained their tumor heterogeneity upon
engraftment into immunodeficient mice (117). The innovative results were
attributed to the plasticity of stem cell organoids, which
undoubtedly expand our understanding of the underlying molecular
mechanisms responsible for colorectal carcinogenesis.
Overall, it is considered that organoids can
function as smart weapons against many cancer types, by the release
of soluble proteins with immunomodulatory activity or recombinant
antibodies lacking Fc, with modified properties, thus circumventing
the cytokine storm induced by the cross-reaction of cells bearing
Fc receptors (118,119). Thus, cells are precisely
committed to continuously providing immunotherapeutic molecules
against cancer. The rationale is based on allowing cells to be
developed in a constrained and controlled environment, secreting
therapeutic molecules when the cancer cells are present. For
example, organoids constructed by mesenchymal stem cells have been
mentioned to secrete interleukins (IL-2, IL-12), thus preventing
the onset of melanoma (120,121).
Even though tumor CRISPR-modified organoids hold
great promise for gene therapy in a number of diseases, their use
may be hindered by certain disadvantages, as shown in the case of
intestinal tumor organoids. For example, drug side-effects cannot
be properly evaluated as intestinal organoids lack immune, nervous
and vascular system (122,123).
In addition, heterogeneity is usually observed between human and
murine organoids, which can be ascribed to the existence of
secreted factors in the intestinal microenvironment, as for example
epigenetic factors, hormones, etc. Finally, in certain cases, the
composition of organoids is not consistent with the structure of
cells naturally occurring in an organism (124).
6. Applying bioengineering approaches and
creating the appropriate niche to improve organoid-based therapies
'in vivo'
Organoids, despite attempting to simulate the
proxies of 'in vivo' tissues, they do not recapitulate the
complexity of an organism. The whole procedure of generating
organoids requires optimization at many levels. First of all, a
specific extracellular matrix (ECM) is a prerequisite for various
cell types, considering that the ECM is the driver of signaling and
responsible for the structural landscape (125). Matrigel is usually used for the
engraftment of stem cell-derived organoids; however, it is not
sufficient in meeting the requirements in a cell type-dependent
manner (126). Several approaches
have been used for the deposition of specific ECM in various cell
types, such as nanolithography, soft lithography, electron-beam,
nano-imprint lithography, etc. (127), which mimic the basement membrane
fibers. Another method for constructing natural organoids is to
design surfaces that ensure the engagement of adhesion molecules.
However, the regulation of signaling cues in a spatial-dependent
manner poses a significant challenge when it comes to natural
engraftment of organoids into organisms. The delivery of soluble
growth factors with the use of nanoparticles or bioresponsive
materials can confer the control of signal orchestration in a
spatial-dependent manner (125).
Finally, it should be taken into consideration that other cell
types, such as immune and mesenchymal cells, can play a fundamental
role in the successful engraftment of organoids, as it has
previously been shown by Lindemans et al (128). In the same context, luminal cells
have been shown to play a vital role in conducting the necessary
interactions between microbial flora and epithelial cells, as well
as cells of the epithelial layer (129).
7. Towards personalized therapy
The comprehensive understanding of the molecular
alterations that have a profound impact on gene expression is
essential in order to achieve personalized care for patients with
neoplasia. Undoubtedly, personalized medicine has already been used
by clinicians (130). The
national scheme of clinical trials will enable clinicians to
determine the appropriate drug administration for each patient
individually and the CRISPR approach has the capacity to confer
additional therapeutic benefit.
A recent study demonstrated that patient-specific
therapy can bypass the drug resistance that is usually associated
with lung cancer and is caused by tyrosine kinase inhibitors (TKIs)
targeting against EGFR. Genomic mutations in EGFR
(E19del, T790M at exon20 and L858R) have been highly associated
with the drug resistance of lung tumors following the
administration of TKIs, particularly the T790M mutation, which is
found in >50% of patients. In this context, Cas9 nuclease
emerges as a molecular scalpel that can modify the genome in such a
manner that the outcome of the disease is improved in a
personalized and permanent manner. The proposed proof of concept is
the repair of the mutated EGFR gene, using Cas9 nickase.
Specifically, Cas9 nickase can either induce single-strand breaks
and repair the mutated gene via homologous recombination (HDR), or
decay the mutated EGFR. It has been postulated that CRISPR
component assembly can be achieved in plasmids and delivered
intratracheally or intravascularly in some cases (131).
8. The contribution of functional
genome-wide pooled sgRNA screens
The rarity of experimental results derived from
animal models or immortalized cell lines has led researchers to
conduct observational studies, such as genome-wide association
studies (GWAS), in order to identify genes are strongly associated
with disease onset. Data from numerous studies [The Cancer Genome
Atlas (CGA) (132), the Cancer
Cell Line Encyclopedia (CCLE) (133) and the Encyclopedia of DNA
Elements (ENCODE) (134)] were
previously collected to provide deeper insight into the association
of genes with disease predisposition (132–134); however, they were proven to be
insufficient. Respectively, genome-wide loss-of-function screens
employed RNAi approaches, but they did not prove to be particularly
beneficial due to the partial knockdown of predetermined genes,
random side-effects and their propagation in protein-coding genes
(135). Therefore, despite the
accumulation and reliability of the existing results, the
researchers were unable to discriminate which genetic variants are
implicated in particular disease phenotypes. The recent generation
of unbiased genome-wide functional CRISPR screens has identified
the functional role of thousands of genomic elements in parallel,
irrespective of their position in the coding or non-coding
compartment. In addition, CRISPR screens have been used for both
the positive and negative selection of genes that are usually
implicated in tumorigenesis.
As regards the construction of sgRNA libraries
mediated by the CRISPR system, it has been noted that the
principles of constructing large scale screens are as follows: i)
The use of cloning tools for the pooled synthesis of sgRNAs
(135–137); ii) the design of three up to ten
sgRNAs that mark a specific gene (135); and iii) the consistency of Cas9
nuclease and its relevant sgRNAs. In brief, libraries are produced
as DNA and are incorporated into plasmids via cloning to generate
lentiviruses (138). The pool of
sgRNAs (represented by oligonucleotides) often targets 104 to 105
different genes and multiple sgRNAs are designed to augment the
accuracy at specific target genes. In addition, lentiviruses
expressing Cas9 nuclease and sgRNA are delivered at a low
multiplicity of infection (MOI), enabling a single sgRNA to be
introduced into the cell (138).
At the end of the procedure, the lentiviral library of sgRNAs is
amenable to phenotypical tests and high-throughput sequencing in
order to identify and classify the gene targets that are enriched
or diminished in various conditions (139). Despite the advances in designing
pooled libraries, certain biases have not been circumvented, such
as library synthesis and defaults during experimental procedures,
such as cloning.
When it comes to the classification of CRISPR
screens, they can be subdivided into CRISPR nuclease screens
(CRISPRn screens), CRISPR interference screens (CRISPRi screens)
and CRISPR activation screens (CRISPRa screens). The main
differences between the CRISPRn and CRISPRi screens are as follows:
In CRISPRn screens, Cas9 nuclease targets any gene surrounded by a
PAM sequence, causing its elimination. For example, CRISPRn screens
have been used in the identification of significant developmental
genes (135), as well as genes
implicated in cancer growth (140). However, the results have not been
particularly encouraging, due to the restricted number of cells
(141) and the phenotypes
following inactivation of the anticipated gene, possibly resulting
from in-frame insertion/deletions (INDELs) and hypomorphic alleles
(142). These pitfalls have led
scientists to the revelation that CRISPR screens can be modified to
included Cas9 using CRISPR interference (CRISPRi) (143). Thus, CRISPRi screens, which
conjugated dCas9 with different transcriptional repression domains,
accomplished highly efficient transcriptional silencing (144,145) in any given cell type, including
iPSCs (146). In general, the
effects mediated by CRISPRi are more efficient, rapid, specific and
homogenous as opposed to those caused by Cas9 nuclease. The only
difficulty associated with CRISPRi screens is that dCas9-KRAB
inhibits gene expression only when sgRNA is targeted to the
transcription start site (TSS) of a gene (145). Overall, the efficacy of CRISPR
interference has been shown to be significantly affected by certain
parameters, such as the length of sgRNA, the sequence
complementarity, the distance of target gene from transcriptional
start site (TSS) and the chromatin state constitute the factors
that influence the power of CRISPRi. The minimal length of sgRNA
for efficient silencing should be 12 nucleotides, whereas the
minimum length of PAM should be two nucleotides (143). Theoretically, the genomic
sequence that can be targeted by Cas9 for efficient silencing
should be 268 Mb (414). The specificity is dictated by
two PAM nucleotides and 12 nucleotides between the sgRNA and DNA
stretch, indicating that the alteration, on average, of one PAM
nucleotide is efficient to abolish CRISPR interference (143). Notably, CRISPRi is the most
effective when the sgRNAs targeted a region of 150 bp downstream or
upstream of the TSS (145).
dCas9, coupled with multiple copies of the
activator effector domain, have also been used, thereby
accelerating the transcriptional enrichment of the gene of interest
and inducing the generation of CRISPRa screens (145). In this manner, CRISPRa screens
have the potential to elucidate phenotypes based on the
overexpression of certain genes (145), thus offering advantage over the
previously used cDNA screens that included elaborate design of
cDNAs. An additional advantage of CRISPRa screens over cDNA screens
is that they induce transcriptional expression even from secondary
transcriptional start sites, particularly if one considers that the
design principle of sgRNAs is based on targeting any sequence with
a transcriptional start site that is surrounded by a PAM sequence.
For example, novel CRISPRa screens have been designed, conjugating
dCas9 with distinct transcriptional activators in order to search
for gain-of-function phenotypes (147,148). Nonetheless, CRISPRa screens
cannot be used to stimulate the expression of highly repressed
genes. Another method includes fusing dCas9 to different domains of
epigenetic modifiers, as an attempt to elucidate the effects of
epigenetic modifications on chromatin states. Using truncated
sgRNAs or building redundancy with several sgRNAs targeting each
locus, constitute important design principles for filtering out
false positive signals and improving the interpret-ability of
screening data.
sgRNA libraries were first employed for the
identification of novel target gene high-confidence biomarkers,
thus aiding in the design of innovative therapeutic options. At a
genome-wide level, CRISPR knock-out libraries were constructed to
show sensitivity or resistance to classical therapeutic inhibitors
(6-thioguanine and vemurafenib), thus revealing novel drug
resistant genes (135,136). For example, deficient libraries
(termed Cas9 knockout-GeCKO libraries) were constructed using
sgRNAs with the aim of identifying driver genes, whose loss confers
resistance to the classical therapy of melanoma, using the BRAF
inhibitor, vemurafenib (135). In
the same context, a loss of function CRISPR library was generated
at the genome-wide level in order to identify genes that were
involved in uncontrolled cell growth and pluripotency (135). Following this, the candidate
genes were engrafted into mice and generated tumor formation,
demonstrating their functional significance in metastasis. In the
same context, another CRISPR-mediated screen eliminated candidate
genes that were downregulated in Ara-C resistant AML cell lines and
highlighted the functional significance of Dck as the primary
contributor conferring resistance to the chemotherapeutic Ara-C
(149). Tzelepis et al
provided convincing evidence regarding the functional sensitivities
and potential therapeutic targets in five cell lines that mirrored
the transcriptional landscape of AML: MOLM-13,
MV4-11, HL-60, OCI-1ML2 and OCI-AML-3.
As a result, the KAT2A molecule (histone lysine
acetylatransferase-SAGA member) was identified as a therapeutic
target of utmost importance. The therapeutic potency of
KAT2A was confirmed in an ex vivo leukemia mouse model
(Rosa26; Flt3ITD−/+ with the retroviral
infection of MML-AF9 or MLLAF4), further demonstrating its harmless
nature in normal hematopoietic cells (150). Respectively, the sensitivity of
certain genes to ATR inhibition (151) and p53 expression status
(152) on a genome-wide and
high-throughput level using CRISPR screens was also
demonstrated.
The benefits of CRISPR-based screens are not
restricted to the identification of genes that are implicated in
drug resistance and tumorigenesis; their use also expands to the
study of the genomic alterations that confer resistance to classic
therapeutic options. When it comes to elucidating the genomic
alterations that cause tumor resistance, researchers have used the
CRISPR approach to identify nucleotide alterations (including
kinesin-5 A133P mutation or exportin-1 cysteine 528 residue) that
confer loss of sensitivity to classic clinical drugs, such as
Ispinesib (an inhibitor of kinesin-5) in osteosarcoma therapy
(153) and Selinexor (inhibitor
of exportin-1) in multiple myeloma (154). Last but not least, Steinhart
et al used CRISPR screens in RNF43-mutant pancreatic ductal
adenocarcinoma (PDAC) cells, identifying novel therapeutic targets
and opening up new avenues for antibody generation (155).
Zhu et al modified CRISPR screens in a
manner that paired guide RNAs were used to ablate regions of long
non-coding RNAs of 700 bp in length, as the indel mediated by one
sgRNA was insufficient to cause loss-of-function phenotype. Using
this particular method, 700 lncRNAs in the human genome were
screened, taking into consideration that the careful design of
guide RNAs is a prerequisite for avoiding overlap with other
functional elements (156).
Undoubtedly, the optimization of the procedure is essential for the
identification of the underlying mechanisms of action of non-coding
RNAs, as the modified method could be amenable to other regulatory
elements, such as microRNAs. In other words, unbiased genome-wide
functional screens can be used to dissect new enhancers and other
regulatory elements that have profound impact on the protein
level.
Jaitin et al reported a method in which
single-cell RNA sequencing (RNA-seq) was combined with
CRISPR-pooled screens in an attempt to decipher the regulatory
circuits of myeloid development. Specifically, they constructed
lenti-viral backbones, each of which was composed of a sgRNA
expression cassette, transcribed UGI (unique guide index) and the
fluorescent selection marker. The concept was based on tracing each
sgRNA located in CRISPR-pooled screens, taking into consideration
the expression pattern of unique guide index (UGI) and the data
acquired by single-cell RNA-seq. The presence of fluorescent marker
aided in the selection of cells. In other words, the researchers
implemented the method such that they managed to compare
fluorescent densities with transcription modules, as represented by
UGI read counts, thus highlighting the factors involved in
developing the distinct immune subpopulations and delineating the
genotype-to-associations in single cells. The results supported
that Cebpb was the determinant factor controlling the
differentiation of myeloid cells to either dendritic or monocyte
cells. In the case of Cebpb deficiency, Irf8 played a vital
role in the dendritic cell differentiation route (157).
9. The combination of immune and CRISPR
system against complex diseases
Immunotherapy has arisen as a novel method in the
treatment of cancer. Tumor-specific T cells can be modulated and
infused into the body of patients suffering from synovial cell
sarcoma, lymphoma or melanoma or leukemia (158), with significant implications in
personalized therapy.
A recent immunotherapy breakthrough was based on
the use of CARs, which bind to specific antigens in cancer cells
(159). These receptors have
attracted much attention as they can be very easily engrafted in
any patient, circumventing any possibility of immunogenicity.
Evidently, the concept is based on the complete matching of CAR on
T cells with the antigen of cancer cells, with the aim of
eliminating cancer cells. In the past, genetically edited CAR-T
cells produced by ZFNs, TALEN nucleases did not yield the desired
results. Nowadays, T cells can be reprogrammed using the CRISPR
gene-manipulating system and can be used to eradicate leukemia
cells or to treat patients with relapsed B-cell malignancies in
remission. For example, Eyquem et al demonstrated that
Cas9-modified effector T cells displayed CD19-specific CAR
receptor, recognizing the specific sequence of T cell receptor
alpha constant (TRAC) and successfully treating acute lymphoblastic
leukemia in a mouse model (160).
In this manner, outpaced modified effector-T cells proved to be
very efficient in recognizing antigen following repeated exposures,
and prevented the expansion of cancer cells as well as exhaustion
of T cells. In parallel, certain pharmaceutical companies,
including Novartis, have used the CRISPR technology in CAR-T cells
in order to decipher the most suitable therapeutic application in
each cancer type. Following approval from the Recombinant DNA
Advisory Committee at the US National Institutes of Health,
clinical trials have been conducted on castration-resistant
prostate cancer, muscle-invasive bladder cancer, lung cancer and
metastatic renal cell carcinoma, using CRISPR technology (161).
A cornerstone strategy in the field of cancer
immunotherapy is the abrogation of 'immune checkpoints', such as
PD-1 and CTLA-4. The principle is based on PD-L1 binding expressed
by cancer cells at the PD-1 receptor in chronically activated or
exhausted T cells, leading to an exacerbation of cancer cell growth
and in turn compromising the anti-tumor immune effect of activated
T cells against cancer cells, through engagement of PD-1 receptor
to the PDL-1 molecule (162). For
this reason, in an attempt to couple the 'immune check-point' and
the CRISPR system, researchers have produced RNP complexes composed
of sgRNA and Cas9 (Cas9 RNPs), as well as single-stranded DNA
oligonucleotides as a template for homology-directed repair to
replace the expression of PD-1 in human primary T cells, rendering
them more effective (40). The
same research team employed Cas9-RNA RNPs to introduce knock-in
modifications in the CXCR4 coreceptor of human primary T cells in
the face against HIV infection (40). Undoubtedly, the particular research
study became a landmark in T cell genome engineering, presentinga
fast, efficient and very safe method of modulating primary immune
cells, as Cas9 RNP complex delivery was reduced to 24 h, with
significant implications in the amelioration of side-effects. Cas9
RNP technology needs to be improved due to the low rate of the
resulting modifications (20%), following the enrichment of
FAC-sorted cells with low protein expression. In addition, one
should take into consideration that the efficiency of T cell
editing can be affected by the dynamics of their chromatin state,
their activation potential or other exogenous signals.
Yang et al identified the molecular changes
that inhibited BCR-dependent NF-κB stimulation in the activated B
cell-like (ABC) type of diffuse large B cell lymphoma (DLBCL),
using SMAC mimetics. The researchers used the CRISPR system to
demonstrate the specificity of SMAC mimetics, particularly in
lymphoma subtypes and to show that SMAC mimetics prevent the
recruitment of LUBAC ligase or BCL10 to the CBM complex
(CARD11-MALT1-BCL10) and the existing redundancy between cIAPs
(163).
On the other hand, the CRISPR approach seems to be
a beneficial therapeutic tool in cancer types originating from
viruses. In particular, it has been shown that the CRISPR system
can combat oncolytic viruses by removing viral oncogenes or
targeting genes responsible for viral genome maintenance and
replication. For example, the CRISPR genome engineering tool has
emerged as a great therapeutic option in patients infected with
hepatitis B virus (HBV), which has life-threatening consequences,
such as cirrhosis and most likely, hepatocellular carcinoma
(164). The attack against the
HBV virus was demonstrated through the suppression of HBV antigen
expression in mice deficient for cccDNA, via the CRISPR system
(165). Recently, another
application of the CRISPR method against the HBV virus was shown to
eliminate the recombinant form of cccDNA that causes viral
persistence (166). In the
context of hepatic defects caused by infections, nuclease-deficient
FnCas9 has been reported to hinder the synthesis of HCV protein
upon engagement of sgRNAs in the genome (167).
The Epstein-Barr Virus (EBV), which is related to
an increased incidence of lymphomas, such as Burkitt lymphoma,
Hodgkin's lymphoma, non-Hodgkin's lymphoma, as well as
nasopharyngeal carcinoma, constitutes another example of a virus
causing severe infections in humans. The modification of Burkitt
lymphoma cells mediated by Cas9 nuclease has resulted in the
cessation of viral replication and cell proliferation (168). Notably, the CRISPR system has
also been shown to disrupt the HPV virus genes in both 'in
vitro' and 'in vivo' xenograft models (169).
10. The CRISPR system against HIV
infection
Antiretroviral therapy (ART) has thus far been used
to eliminate infections caused by HIV, but without the anticipated
outcomes. The disadvantages that render ART ineffective include
administration for a prolonged period of time, the appearance of
transient results in patients and the incidence of several
side-effects. Nonetheless, adoptive T cell therapy has been applied
as a therapeutic option against cytomegalovirus (CMV) and EBV
(158,170,171). When it comes to therapeutic
strategies against human immunodeficiency virus (HIV), three
promising therapies have been postulated thus far: i) Remodeling of
the immune response through the enhancement of HIV-infected cell
awareness and not via the recruitment of a large number of T cells,
so that T cells can boost the response of other immunological
populations; ii) increasing the action and endurance of T cells;
and iii) triggering the immune system to develop memory cells,
since HIV escapes immune mechanisms through viral quasispecies
(vQS) or via mutagenesis at specific epitopes (Fig. 5). In principle, the memory T cell
population can be triggered to create viral particles upon
interruption of antiviral therapy, in the sense that HIV can be
managed through an edited reservoir of memory T cells.
The remodeling of the immune response can be
achieved by three different methods, all of which are aimed at
expanding the compartment of HIV-specific T cells. These
methodologies also attempt to refine the immune response against
HIV by providing either mono-specific T cells or poly-specific T
cells or modified T cells. In this context, HIV-directed T cells,
activated by cytokines, have been used as a shield against HIV
(171). However, mono-specific T
cell expansion has proven to be insufficient against HIV, as the
virus can circumvent the host immune system. Clinical studies have
demonstrated that CD8+ T cells can be directed towards
HLA A2 epitopes, thus eliciting responses against gp120, p17, p24
and Nef (172) or CD8+
T cells specific for Gag, in conditions that included OKT3 and IL-2
(173), without causing any
differences in the viral load. At the same time, another clinical
study reported no significant changes in the viral load using T
cells specific for HIV: Gag p17-8 SLYNTVATL and Pol VIYQYMDDL
(174). It was also postulated
that the cure of HIV patients was unresolved due to the transient
nature and the narrowed efficiency of T cells. Notably, the
importance of increasing the persistence and efficacy of the immune
response against HIV was highlighted.
An infusion of 'poly-specific T cells' that
recognize different HIV antigens has also been employed to
eliminate the HIV virus. Specifically, 'polyclonal HIV-specific T
cells were stimulated following exposure to multiple HIV antigens,
even upon non-immunodominant epitopes. Additionally, the
poly-specific T cells were concentrated without any requirement of
class presentation, contributing to a broader range of therapeutic
implementation. In the clinical setting, broadly-specific cytotoxic
T cells (HXTCs) were constructed to exhibit immune reactions with
Gag, Nef and Pol epitopes and resulted in eradication of HIV
replication (175). The
beneficial effect of HXTCs was also validated in HIV-positive
individuals with hematological perturbations (176). The results of the clinical trials
have been registered (https://clinicaltrials.gov/). The implantation of
multi-antigen T cell clones against HIV is currently being studied
in a clinical trial (NCT02208167) (177).
Another promising approach is altering the genetic
properties of T cells, i.e., equipping them with artificial TCRs or
CARs that are composed of an extracellular region responsible for
recognition of HIV antigens and an activation-domain (178,179). Both types of receptors are
modified in such a manner that they bind epitopes, apart from the
individual HLA type of each patient, thus enhancing the therapeutic
potential of the approach. The distinguishable characteristic of
artificial TCRs is their crosstalk with a broad range of epitopes,
thus aiding in the design of specific immune responses against
certain epitopes and their interaction with crude viral proteins,
irrespective of the patient's HLA type. Of note, a clinical study
conducted in 2008 which included modified T cells loaded with
specific SL9 TCRs, demonstrated the attenuation of HIV levels in
immunodeficient mice (NOD/scid/IL-2Rγcnul)
(180). It should be noted,
however, that the modified T cells were loaded with TCR receptors
recognizing HLA-A*02 restricted P17 epitope SLYNTVATL (A2-SL9),
which are related to depletion of HIV in chronic infection
(180), thereby helping to
overcome the immune escape exhibited by some HIV variants.
Respectively, the hallmarks of CARs that distinguish them from TCRs
are their ability to provide long-term protection without
side-effects and their low immunogenicity. T cells were constructed
to include a CAR receptor comprised from a CD4 extracellular domain
that recognizes the Env glycoprotein of HIV and the intracellular
signaling domain, termed 'CD4zeta-modified T cells'. When such
modified T cells were used in a clinical trial (NCT01013415) in
distinct phases, they produced very exciting results with regards
to specified immunity and persistence of T cells against HIV, in
combination with a reduction of the HIV burden (181,182).
As a second immunological approach, it was observed
that CD4+ T cells are recruited at the site of
infection, supporting the presence and persistence of cytotoxic T
cells against HIV (183).
Specifically, the complex assembled between CD4 and MHCII
determined the response of T helper cells to antigen and
orchestrated a complete immune response mediated by T
CD8+ cells. In the same context, gene editing strategies
have been employed to abolish the entry of HIV in an organism, via
eradication of its receptors. For example, it was previously
demonstrated that a patient who received a transplant with mutated
CCR5 receptor at delta 32 position was cured, acquiring natural HIV
resistance (184). Initially, the
elimination of HIV coreceptors was achieved using ZNFs that
combined Fok1 endonuclease with the DNA-binding zinc finger domain
(185). At the preclinical level,
ZNFs have been employed for the disruption of HIV coreceptors
(CXCR4 or CCR5) (186) at a
successful rate, allowing the implementation of ZNF nucleases
against HIV in the clinical setting (187). However, the use of ZNFs against
HIV infection was hindered by the potential cross-reactivity and
side-effects (188). Compared to
ZNFs, the CRISPR system was shown to be more beneficial due to its
multiplex capacity to disrupt the function of both HIV coreceptors
with amenable side-effects (189). In addition, Kaminski et al
demonstrated another convincing method through which persistent
expression of Cas9 can impede HIV-1 replication in infected
CD4+ T cells and protect HIV-1-infected T cells against
new infection. Specifically, it was shown that the excision of the
genomic regions of the HIV-1 promoter that are located at the 5′
long terminal repeat (LTR) in 2D10 CD4+ T cells, was
responsible for HIV eradication (190).
In an attempt to increase the persistence of T
cells, the latter method employed enrichment of T cells with the
memory phenotype so as to combat every new infection more rapidly
and efficiently. Apart from the infusion of T memory cells in the
cancer setting (191), the T cell
memory reservoir has been employed against HIV in the clinical
setting (192). To sum up, T cell
engineering holds great promise in the therapeutic management of
HIV in the absence of ART.
11. Challenges associated with the
CRISPR-based gene method and alternative methods
For the successful implementation of a therapeutic
approach based on the CRIPSR method, the existing challenges
include increasing the specificity of CRISPR system, as well as
delivering Cas9 nuclease and its sgRNAs without side-effects. In
this session, we analyze proposed methods with which to circumvent
these difficulties.
Challenge 1: Delivery
First, the 'ex vivo' delivery of Cas9
nuclease and its sgRNAs has been reported to be achieved with the
use of plasmids or classical viral methods (adenovirus, lentivirus,
adeno-associated virus or retrovirus). In parallel, Cas9 nuclease
has been shown to be delivered through non-viral methods, including
cationic lipids or nanoparticle encapsulation or electroporation
(193) or microinjection
(194,195). In these cases, Cas9 nuclease and
its gRNA are delivered in the form of RNPs, resulting in a
long-term response.
In the case of plasmids, the transferred components
are inserted into the nuclear area, thus ensuring that delivery is
restricted to dividing cells and the danger of insertional
mutagenesis usually associated with viruses is eliminated (196). Regarding the 'ex vivo'
delivery of the CRISPR system constituents, the iTOP technique and
mechanical cell derangement are the current methods. In the case of
mechanical cell deformation, membranes are temporally disturbed,
following the collection of cell samples in microfluidic devices,
allowing the passive diffusion of substances in non-nuclear
regions. Single-strand DNA and plasmids have been reported to
employ the mechanism of delivery (197). In the iTOP method, macromolecules
are delivered into cells via micropinocytosis and using
propanebetaine with minimal toxicity (198). In addition, Ha et al have
presented an innovative way of delivery through Poly-sgRNA/siRNA
nanoparticles, in order to disrupt gene expression (199). The principle in using these
nanoparticles is based on the assembly of siRNA (substrate of
Dicer) and sgRNAs, which are cleaved through the action of Dicer
following rolling circle transcription (RCT). The advantages are as
follows: i) Minimal toxicity; ii) nanoparticle strength and
stability; and iii) single administration of components to ensure
the permanent disarrangement of target gene expression (199). Other delivery methods have been
reported to be involved in the 'in vivo' transfer of
constituents of the CRISPR system. The first includes the
encapsulation of Cas9 nuclease and the appropriate sgRNAs in DNA
nanoclews, which constitute DNA nanoparticles constructed by
rolling circle amplification (200). DNA nanoclews have been postulated
as a suitable choice for delivery as they offer a high intrusion of
DNA nanoparticle delivery into cells (200). A different rationale has been
suggested that fuses the C-terminal end of Cas9 nuclease with a
'nona-arginine' based cell penetrating peptide (CPP), thereby
recruiting the appropriates gRNA (201). In both cases, the positive charge
of particles is responsible for the direct entry of the CRISPR
system in cells.
Typically, Cas9 nuclease and sgRNA can be delivered
either as DNA or RNA, and both can be packaged into viral vectors.
The advantage of viral vectors in transferring the CRISPR system is
attributed to their ability to regulate gene expression in a
spatial-temporal manner, given that a viral vector has the selected
promoter to guide transcription of Cas9 and its sgRNA, even though
insertional mutagenesis and immunogenic responses can still occur
(202). Adenoviruses have been
proposed as the safest viral method in transferring the CRISPR
components, due to their serotype specificity and low
immunogenicity, despite their low packaging capacity. For example,
recent data have highlighted the AAV9 vector as the ideal method in
transferring the CRISPR components in the Duchenne dystrophin gene
(81,203,204). In another study, Gomez et
al engineered adeno-associated (AAV) virus exposing phytochrome
interacting factor 6 motifs (PIF6), to interact with
phytochrome B (PhyB) factor in response to red light, thus
attaining high nuclear translocation of virus in cells and implying
a proposal of transferring CRISPR system through adeno-associated
virus (205). Further on, Ran
et al used an alternative version of Cas9 (SaCas9), which
was 1 kb shorter in length than Cas9 nuclease, and was packaged
into the AAV serotype 8 vector. The deficiency of ApoB and PSCK9
was determined to be at a rate of 40% in hepatocytes (206).
In order to avoid the danger of insertional
mutagenesis, cationic lipid nanoparticles (LNPs) have been
suggested as an extraordinary non-immunogenic selection for the
intracellular delivery of anionic molecules that can be
encapsulated (207,208). The LNPs can specifically transfer
their load in a cell-dependent manner for a long period, taking
advantage of the charge, the hydrophobicity/hydrophilicity of the
load and biocompatibility/clearance (207). For example, LNPs have been
devised to contain the lipid-like material C12–200 for the
successful introduction of modifications in hepatocytes, thus
highlighting the CRISPR constructs and the homology-directed repair
mechanism. The efficacy of gene repair with this method appears to
be higher than that accomplished with the use of viruses (39).
Future efforts should focus on delivering the
CRISPR components in cell types other than hepatocytes, including
non-dividing cells and stem cells, in an attempt to increase intake
of the CRISPR components and to pave the road for new therapeutic
interventions. In conjunction with studies on materials science,
other delivery methods could be implemented in order to boost the
efficiency of the CRISPR system and to circumvent the side-effects
induced by the classical means of delivery.
Challenge 2: Methods for enhancing Cas9
nuclease activity
Even though most efforts are currently focused on
exploring the molecular mechanisms underlying the fundamental
biological processes or on improving the therapeutic outcome of a
number of diseases, research teams explore several options for the
validation or enhancement of Cas9 nuclease activity. The precision
of the CRISPR system can be increased by modifying Cas9
nuclease.
To validate Cas9 nuclease activity, several
sequencing methods, in conjunction with in silico prediction
programs, have been extensively used, for the detection of the
off-target effects in a genome-wide high sensitive manner.
Specifically, the efficacy of Cas9 has been confirmed by
integrase-defective lentiviral vector capture (IDLV) (37,209), linear amplification-mediated PCR
(LAM-PCR), high-throughput genome-wide translocation sequencing
(HTGTS) (210), genome-wide
unbiased identification of DSBs enabled by sequencing (GUIDE-seq)
(211,212), next-generation sequencing (BLESS)
(213) using in situ
adapter ligation and digested genome sequencing (Digenome-seq)
(214). The above-mentioned tools
have been generated with the notion in mind that potential
mismatches can be created between sgRNA and complementary DNA
sequence. These techniques have enriched our knowledge in bypassing
unintended effects mediated by CRISPR system and in determining the
implementation of the advanced engineered toolbox. In other words,
the challenging nature of the CRISPR system has led to enhancement
of Cas9 nuclease precision.
12. Ways to circumvent the off-target
effects of the CRISPR system
To date, two methods have been reported to
eliminate the off-target cleavage effects generated by Cas9
nuclease, either by augmenting the specificity or by decreasing the
duration of Cas9 nuclease action.
To enhance the specificity, a first approach is
based on truncating the guide RNA from 20 nucleotides to 17 or 18
nucleotides in the genomic region where RNA creates complementary
bonds, away from the PAM sequence, while at the same time, Cas9
nuclease retains its activity at desired genomic sequences
(215,216). The application of the
aforementioned approach has demonstrated the reduction of random
effects (18,37,215,217,218). The validity of the method (using
truncated sgRNA) has been confirmed by GUIDE-seq or targeted deep
sequencing in human cells (216).
One potential explanation for the above-mentioned property of the
CRISPR system lies on reducing the excess energy that is released
by RNA-DNA complementarity, thereby rendering Cas9 nuclease to be
very specific on cleavage events. Another method for elevating Cas9
specificity is based on using two additional guanine nucleotides in
the 5′ end of the guide sequence (36,217). The underlying mechanism of action
remains elusive, but one can envisage that disorganization of the
association network between Cas9 nuclease and the 5′ end of the
sgRNA is responsible for reducing the off-target effects mediated
by Cas9. In an attempt to boost the Cas9 precision and to minimize
the random non-specific cleavage effects, paired Cas9 nickases have
been reportedly used. The nickases introduce a single-strand cut
(nick) with the same specificity as a regular Cas9 nuclease and a
pair of nickases are used to initiate double nicks in each DNA
strand, using two separate sgRNAs. The major difference is when two
Cas9 nickases are used, long overhangs are produced on each of the
cleaved ends, instead of blunt ends. Cas9 nickase is also
distinguishable from Cas9 nuclease because the former has an
inactivated catalytic domain (either in RuvC or HNH part) among its
six domains: REC I, REC II, Bridge Helix, PAM Interacting, HNH and
RuvC (219). Specifically, a D10A
replacement at RuvC domain or H840A substitution at HNH takes
place, producing a highly specific DNA-binding complex. A
characteristic example includes the assembly of one sgRNA with Cas9
nickase, resulting in the reduction of off-target effects in human
cells (220). Nevertheless, Cas9
nickase has not been reported as particularly effective in
introducing DSBs, as compared to Cas9 nuclease, since Cas9 nickase
has been shown to insert point mutations in anticipated target
sites (220,221).
Another approach for minimizing side-effects is to
use dimerized factors. Dimerized proteins, in particular, use
synergistic energy to achieve the desired efficiency. For example,
dCas9 was combined with a dimerization-dependent FokI nuclease
domain, resulting in a complex named RNA-guided FokI-dCas9 nuclease
(RFN) (222). In the same
context, RNA-guided FokI-dCas9 nuclease (RFN) has been used with
truncated sgRNA at the 5′ end, attaining the best specificity of
Cas9 nuclease (223). Finally, it
has been shown that Cas9 nuclease precision can be improved by
modulating Cas9 with alanine replacements at four residues
(25). Of note, the
GUIDE-sequencing method (GUIDE-seq) has provided convincing
evidence that the Cas9 version containing the alanine substitutions
is far more specific than the unmodified Cas9 nuclease (224), preventing non-targeted cleavage
events at the DNA sequence.
Hou et al isolated a different form of Cas9
nuclease from Neisseria meningitidis (NmCas9), which exerts its
effects very efficiently and with great specificity (225). In-depth analyses have reported
that NmCas9 binds a 24 nt crRNA, providing superior specificity
over the previous 20 nt crRNA used by Cas9 derived from
Streptococcus pyogenes. Notably, the PAM sequence of NmCas9
comprises an either 5′-NNNNGATT-3′ or 5′-NNNNGCTT-3′ genomic
sequence, through which it accomplishes efficient DNA cleavage. As
would be expected, the long PAM sequence employed by NmCas9 confers
high specificity. The efficiency offered by NmCas9 is higher (60%)
in hESCs and iPSCs compared to that offered by other nucleases and
in parallel its use can be expanded to trace cell fate of rare
cells (226).
13. Conclusion
The rationale based on which the prokaryotic viral
defense mechanism is converted to the eminent engineering approach
underscores the significance of basic science research. We conclude
that the CRISPR system is unique and capable of providing
therapeutic applications using stem cells or organoids of different
origins. The utility of the CRISPR system seems to be beneficial in
the genetic screening for cancer gene validation/discovery. The
multilayered characterization of new targets will give
comprehensive insights into the therapeutic potential of the
particular system, thus guiding its appropriate implementation
directly into patients (Fig.
6).
Abbreviations:
|
ABC
|
activated B cell-like type
|
|
AML
|
acute myeloid leukemia
|
|
Apc
|
adenomatous polyposis coli
|
|
ART
|
antiretroviral therapy
|
|
APOBEC1
|
apolipoprotein B mRNA editing enzyme
catalytic polypeptide-like family protein 1
|
|
aSyn
|
A-synuclein
|
|
HXTCs
|
broadly-specific cytotoxic T
cells
|
|
CCLE
|
Cancer Cell Line Encyclopedia
|
|
CGA
|
Cancer Genome Atlas
|
|
Ctnnb1
|
cateninb1 gene
|
|
CPP
|
cell penetrating peptide
|
|
CFTR
|
CF transmembrane conductor
receptor
|
|
CARs
|
chimeric antigen receptors
|
|
CGD
|
chronic granulomatous disease
|
|
CRISPR
|
clustered regularly interspaced short
palindromic repeats
|
|
CTOL
|
colorectal tumor organoid library
|
|
CRISPRa screens
|
CRISPR activation screens
|
|
Cas9
|
CRISPR-associated protein 9
|
|
CRISPRi
|
CRISPR interference
|
|
CRISPRn screens
|
CRISPR nuclease screens
|
|
crRNA
|
CRISPR RNA
|
|
CMV
|
cytomegalovirus
|
|
dCas9
|
deactivated version of Cas9
nuclease
|
|
DOCK8
|
dedicator of cytokinesis 8
|
|
DLBCL
|
Diffuse large B cell lymphoma
|
|
Digenome-seq
|
digested genome sequencing
|
|
DSBs
|
double-strand breaks
|
|
Eml4-Alk fusion protein
|
echinoderm microtubule associated
protein like 4-anaplastic lymphoma kinase
|
|
ENCODE
|
Encyclopedia of DNA Elements
|
|
EGFR
|
epidermal growth factor receptor
|
|
EBV
|
Epstein-Barr virus
|
|
Ecm
|
extracellular matrix
|
|
Flt3-ITD
|
Flt3 internal tandem
duplication
|
|
GEMMs
|
genetically engineered mouse
models
|
|
GWAS
|
genome-wide association studies
|
|
HSPCs
|
hematopoietic stem and progenitor
cells
|
|
HBV
|
hepatitis B virus
|
|
HDR
|
homology directed repair
|
|
HBB
|
human hemoglobin beta gene
|
|
HITI
|
homology-independent targeted
integration
|
|
HIV
|
human immunodeficiency virus
|
|
ICF
|
immunodeficiency centromeric region
instability facial anomalies syndrome
|
|
iPSCs
|
induced pluripotent stem cells
|
|
IDLV
|
integrase-defective lentiviral
vector
|
|
LAM-PCR
|
linear amplification-mediated PCR
|
|
LNPs
|
lipid nanoparticles
|
|
LTR
|
long terminal repeats
|
|
HTGTS
|
high-throughput genome-wide
translocation sequencing
|
|
HD
|
Huntington's disease
|
|
HBB
|
hemoglobin subunit beta
|
|
HTT
|
Huntingtin gene
|
|
GUIDE-seq
|
genome-wide unbiased identification
of DSBs enabled by sequencing
|
|
LCA10
|
Leber congenital amaurosis 10
|
|
MAPK
|
mitogen-activated protein kinase
|
|
Mll3
|
mixed lineage leukemia 3
|
|
MGEs
|
mobile genetic elements
|
|
MOI
|
multiplicity of infection
|
|
NHEJ
|
non-homologous end joining
|
|
NSCLC
|
non-small cell lung cancer
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
Pten
|
phosphatase and tensin homolog
|
|
PanINs
|
pre-invasive pancreatic
intraepithelial neoplasms
|
|
PAM
|
protospacer adjacent motif
|
|
RFLP
|
restriction fragment length
polymorphism
|
|
RNPs
|
ribonucleoproteins
|
|
RFN
|
RNA-guided FokI-dCas9 nuclease
|
|
RNA-seq
|
RNA sequencing
|
|
RCT
|
rolling circle transcription
|
|
SCD
|
sickle cell disease
|
|
sgRNA
|
single-guide RNA
|
|
SNPs
|
single nucleotide polymorphisms
|
|
ssRNA
|
single-strand RNA
|
|
SSCs
|
spermatogonial stem cells
|
|
TCRs
|
T cell receptors
|
|
TRAC
|
T cell receptor alpha constant
|
|
3D
|
three-dimensional
|
|
tracrRNA
|
trans-activating RNA
|
|
TALEN
|
transcription activator-like effector
nuclease
|
|
TGF
|
transforming growth factor
|
|
TSS
|
transcriptional start site
|
|
TKIs
|
tyrosine kinase inhibitors
|
|
UGI
|
unique guide index
|
|
vQS
|
viral quasispecies
|
|
ZNFs
|
Zinc finger nucleases
|
Acknowledgments
Not applicable.
Funding
No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
All authors were involved in the conception of the
study, and have revised and approved the final manuscript. All
authors have taken the responsibility for publishing this review
paper. SB performed the literature search, has written the
manuscript, has critically analyzed the existing knowledge and has
designed the pictures; SB, MA and IC were involved in the
acquisition and analysis of 'in vitro' data. MA
significantly contributed to editing the manuscript; SB and AMK
were involved in the analysis of data in the clinical setting. SB
and DAS were involved in analysis of data in the 'HIV setting'. SB,
MP and VZ were involved in analysis of 'in vivo' data. SB,
DAS, MP and VZ were significantly involved in the drafting of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
DAS is the Editor-in-Chief for the journal, but had
no personal involvement in the reviewing process, or any influence
in terms of adjudicating on the final decision, for this
article.
References
|
1
|
Bedell VM, Wang Y, Campbell JM, Poshusta
TL, Starker CG, Krug RG II, Tan W, Penheiter SG, Ma AC and Leung
AY: In vivo genome editing using a high-efficiency TALEN system.
Nature. 491:114–118. 2012. View Article : Google Scholar
|
|
2
|
Urnov FD, Miller JC, Lee YL, Beausejour
CM, Rock JM, Augustus S, Jamieson AC, Porteus MH, Gregory PD and
Holmes MC: Highly efficient endogenous human gene correction using
designed zinc-finger nucleases. Nature. 435:646–651. 2005.
View Article : Google Scholar
|
|
3
|
Kim EJ, Kang KH and Ju JH: CRISPR-Cas9: A
promising tool for gene editing on induced pluripotent stem cells.
Korean J Intern Med (Korean Assoc Intern Med). 32:42–61. 2017.
|
|
4
|
Cong L, Ran FA, Cox D, Lin S, Barretto R,
Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al: Multiplex
genome engineering using CRISPR/Cas systems. Science. 339:819–823.
2013. View Article : Google Scholar
|
|
5
|
Wu X, Scott DA, Kriz AJ, Chiu AC, Hsu PD,
Dadon DB, Cheng AW, Trevino AE, Konermann S, Chen S, et al:
Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian
cells. Nat Biotechnol. 32:670–676. 2014. View Article : Google Scholar
|
|
6
|
Doudna JA and Charpentier E: Genome
editing. The new frontier of genome engineering with CRISPR-Cas9.
Science. 346:12580962014. View Article : Google Scholar
|
|
7
|
Wiedenheft B, Sternberg SH and Doudna JA:
RNA-guided genetic silencing systems in bacteria and archaea.
Nature. 482:331–338. 2012. View Article : Google Scholar
|
|
8
|
Mali P, Yang L, Esvelt KM, Aach J, Guell
M, DiCarlo JE, Norville JE and Church GM: RNA-guided human genome
engineering via Cas9. Science. 339:823–826. 2013. View Article : Google Scholar
|
|
9
|
Wright AV, Nuñez JK and Doudna JA: Biology
and applications of CRISPR systems: Harnessing nature' s toolbox
for genome engineering. Cell. 164:29–44. 2016. View Article : Google Scholar
|
|
10
|
van der Oost J, Westra ER, Jackson RN and
Wiedenheft B: Unravelling the structural and mechanistic basis of
CRISPR-Cas systems. Nat Rev Microbiol. 12:479–492. 2014. View Article : Google Scholar
|
|
11
|
Mougiakos I, Bosma EF, de Vos WM, van
Kranenburg R and van der Oost J: Next generation prokaryotic
engineering: The CRISPR-Cas Toolkit. Trends Biotechnol. 34:575–587.
2016. View Article : Google Scholar
|
|
12
|
Bolotin A, Quinquis B, Sorokin A and
Ehrlich SD: Clustered regularly interspaced short palindrome
repeats (CRISPRs) have spacers of extrachromosomal origin.
Microbiology. 151:2551–2561. 2005. View Article : Google Scholar
|
|
13
|
Westra ER, Semenova E, Datsenko KA,
Jackson RN, Wiedenheft B, Severinov K and Brouns SJ: Type I-E
CRISPR-cas systems discriminate target from non-target DNA through
base pairing-independent PAM recognition. PLoS Genet.
9:e10037422013. View Article : Google Scholar
|
|
14
|
Jinek M, Chylinski K, Fonfara I, Hauer M,
Doudna JA and Charpentier E: A programmable dual-RNA-guided DNA
endonuclease in adaptive bacterial immunity. Science. 337:816–821.
2012. View Article : Google Scholar
|
|
15
|
Larson MH, Gilbert LA, Wang X, Lim WA,
Weissman JS and Qi LS: CRISPR interference (CRISPRi) for
sequence-specific control of gene expression. Nat Protoc.
8:2180–2196. 2013. View Article : Google Scholar
|
|
16
|
Mojica FJ, Díez-Villaseñor C,
García-Martínez J and Almendros C: Short motif sequences determine
the targets of the prokaryotic CRISPR defence system. Microbiology.
155:733–740. 2009. View Article : Google Scholar
|
|
17
|
Mei Y, Wang Y, Chen H, Sun ZS and Ju XD:
Recent progress in CRISPR/Cas9 technology. J Genet Genomics.
43:63–75. 2016. View Article : Google Scholar
|
|
18
|
Ran FA, Hsu PD, Wright J, Agarwala V,
Scott DA and Zhang F: Genome engineering using the CRISPR-Cas9
system. Nat Protoc. 8:2281–2308. 2013. View Article : Google Scholar
|
|
19
|
Drost J and Clevers H: Who is in the
Driver' s Seat: Tracing cancer genes using CRISPR-barcoding. Mol
Cell. 63:352–354. 2016. View Article : Google Scholar
|
|
20
|
Lin S, Staahl BT, Alla RK and Doudna JA:
Enhanced homology-directed human genome engineering by controlled
timing of CRISPR/Cas9 delivery. eLife. 3:e047662014. View Article : Google Scholar
|
|
21
|
Ghezraoui H, Piganeau M, Renouf B, Renaud
JB, Sallmyr A, Ruis B, Oh S, Tomkinson AE, Hendrickson EA,
Giovannangeli C, et al: Chromosomal translocations in human cells
are generated by canonical nonhomologous end-joining. Mol Cell.
55:829–842. 2014. View Article : Google Scholar
|
|
22
|
Roukos V and Misteli T: The biogenesis of
chromosome translocations. Nat Cell Biol. 16:293–300. 2014.
View Article : Google Scholar
|
|
23
|
Komor AC, Kim YB, Packer MS, Zuris JA and
Liu DR: Programmable editing of a target base in genomic DNA
without double-stranded DNA cleavage. Nature. 533:420–424. 2016.
View Article : Google Scholar
|
|
24
|
Suzuki K, Tsunekawa Y, Hernandez-Benitez
R, Wu J, Zhu J, Kim EJ, Hatanaka F, Yamamoto M, Araoka T, Li Z, et
al: In vivo genome editing via CRISPR/Cas9 mediated
homology-independent targeted integration. Nature. 540:144–149.
2016. View Article : Google Scholar
|
|
25
|
Nishimasu H, Ran FA, Hsu PD, Konermann S,
Shehata SI, Dohmae N, Ishitani R, Zhang F and Nureki O: Crystal
structure of Cas9 in complex with guide RNA and target DNA. Cell.
156:935–949. 2014. View Article : Google Scholar
|
|
26
|
Christian M, Cermak T, Doyle EL, Schmidt
C, Zhang F, Hummel A, Bogdanove AJ and Voytas DF: Targeting DNA
double-strand breaks with TAL effector nucleases. Genetics.
186:757–761. 2010. View Article : Google Scholar
|
|
27
|
Kabadi AM, Ousterout DG, Hilton IB and
Gersbach CA: Multiplex CRISPR/Cas9-based genome engineering from a
single lentiviral vector. Nucleic Acids Res. 42:e1472014.
View Article : Google Scholar
|
|
28
|
Sadikovic B, Al-Romaih K, Squire JA and
Zielenska M: Cause and consequences of genetic and epigenetic
alterations in human cancer. Curr Genomics. 9:394–408. 2008.
View Article : Google Scholar
|
|
29
|
Baliou S, Adamaki M, Kyriakopoulos AM,
Spandidos DA, Panayiotidis M, Christodoulou I and Zoumpourlis V:
Role of the CRISPR system in controlling gene transcription and
monitoring cell fate (Review). Mol Med Rep. 17:1421–1427. 2018.
|
|
30
|
Swiech L, Heidenreich M, Banerjee A, Habib
N, Li Y, Trombetta J, Sur M and Zhang F: In vivo interrogation of
gene function in the mammalian brain using CRISPR-Cas9. Nat
Biotechnol. 33:102–106. 2015. View Article : Google Scholar
|
|
31
|
Yang H, Wang H, Shivalila CS, Cheng AW,
Shi L and Jaenisch R: One-step generation of mice carrying reporter
and conditional alleles by CRISPR/Cas-mediated genome engineering.
Cell. 154:1370–1379. 2013. View Article : Google Scholar
|
|
32
|
Torres R, Martin MC, Garcia A, Cigudosa
JC, Ramirez JC and Rodriguez-Perales S: Engineering human
tumour-associated chromosomal translocations with the RNA-guided
CRISPR-Cas9 system. Nat Commun. 5:39642014. View Article : Google Scholar
|
|
33
|
Canver MC, Bauer DE, Dass A, Yien YY,
Chung J, Masuda T, Maeda T, Paw BH and Orkin SH: Characterization
of genomic deletion efficiency mediated by clustered regularly
interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system
in mammalian cells. J Biol Chem. 292:25562017. View Article : Google Scholar
|
|
34
|
Lupiáñez DG, Kraft K, Heinrich V, Krawitz
P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R,
et al: Disruptions of topological chromatin domains cause
pathogenic rewiring of gene-enhancer interactions. Cell.
161:1012–1025. 2015. View Article : Google Scholar
|
|
35
|
Choi PS and Meyerson M: Targeted genomic
rearrangements using CRISPR/Cas technology. Nat Commun. 5:37282014.
View Article : Google Scholar
|
|
36
|
Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae
S and Kim JS: Analysis of off-target effects of CRISPR/Cas-derived
RNA-guided endonucleases and nickases. Genome Res. 24:132–141.
2014. View Article : Google Scholar
|
|
37
|
Wang H, Yang H, Shivalila CS, Dawlaty MM,
Cheng AW, Zhang F and Jaenisch R: One-step generation of mice
carrying mutations in multiple genes by CRISPR/Cas-mediated genome
engineering. Cell. 153:910–918. 2013. View Article : Google Scholar
|
|
38
|
Yang L, Guell M, Byrne S, Yang JL, De Los
Angeles A, Mali P, Aach J, Kim-Kiselak C, Briggs AW, Rios X, et al:
Optimization of scarless human stem cell genome editing. Nucleic
Acids Res. 41:9049–9061. 2013. View Article : Google Scholar
|
|
39
|
Yin H, Xue W, Chen S, Bogorad RL,
Benedetti E, Grompe M, Koteliansky V, Sharp PA, Jacks T and
Anderson DG: Genome editing with Cas9 in adult mice corrects a
disease mutation and phenotype. Nat Biotechnol. 32:551–553. 2014.
View Article : Google Scholar
|
|
40
|
Schumann K, Lin S, Boyer E, Simeonov DR,
Subramaniam M, Gate RE, Haliburton GE, Ye CJ, Bluestone JA, Doudna
JA, et al: Generation of knock-in primary human T cells using Cas9
ribonucleoproteins. Proc Natl Acad Sci USA. 112:10437–10442. 2015.
View Article : Google Scholar
|
|
41
|
Renaud JB, Boix C, Charpentier M, De Cian
A, Cochennec J, Duvernois-Berthet E, Perrouault L, Tesson L,
Edouard J, Thinard R, et al: Improved genome editing efficiency and
flexibility using modified oligonucleotides with TALEN and
CRISPR-Cas9 nucleases. Cell Rep. 14:2263–2272. 2016. View Article : Google Scholar
|
|
42
|
Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech
L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, et
al: CRISPR-Cas9 knockin mice for genome editing and cancer
modeling. Cell. 159:440–455. 2014. View Article : Google Scholar
|
|
43
|
Kaulich M, Lee YJ, Lönn P, Springer AD,
Meade BR and Dowdy SF: Efficient CRISPR-rAAV engineering of
endogenous genes to study protein function by allele-specific RNAi.
Nucleic Acids Res. 43:e452015. View Article : Google Scholar
|
|
44
|
Li Y, Park AI, Mou H, Colpan C, Bizhanova
A, Akama-Garren E, Joshi N, Hendrickson EA, Feldser D, Yin H, et
al: A versatile reporter system for CRISPR-mediated chromosomal
rearrangements. Genome Biol. 16:1112015. View Article : Google Scholar
|
|
45
|
Xiao A, Wang Z, Hu Y, Wu Y, Luo Z, Yang Z,
Zu Y, Li W, Huang P, Tong X, et al: Chromosomal deletions and
inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic
Acids Res. 41:e1412013. View Article : Google Scholar
|
|
46
|
Heyer J, Kwong LN, Lowe SW and Chin L:
Non-germline genetically engineered mouse models for translational
cancer research. Nat Rev Cancer. 10:470–480. 2010. View Article : Google Scholar
|
|
47
|
Xue W, Chen S, Yin H, Tammela T,
Papagiannakopoulos T, Joshi NS, Cai W, Yang G, Bronson R and
Crowley DG: et al CRISPR-mediated direct mutation of cancer genes
in the mouse liver. Nature. 514:380–384. 2014. View Article : Google Scholar
|
|
48
|
Zuckermann M, Hovestadt V, Knobbe-Thomsen
CB, Zapatka M, Northcott PA, Schramm K, Belic J, Jones DT, Tschida
B, Moriarity B, et al: Somatic CRISPR/Cas9-mediated tumour
suppressor disruption enables versatile brain tumour modelling. Nat
Commun. 6:73912015. View Article : Google Scholar
|
|
49
|
Doerks T, Copley RR, Schultz J, Ponting CP
and Bork P: Systematic identification of novel protein domain
families associated with nuclear functions. Genome Res. 12:47–56.
2002. View Article : Google Scholar
|
|
50
|
Guerra C, Mijimolle N, Dhawahir A, Dubus
P, Barradas M, Serrano M, Campuzano V and Barbacid M: Tumor
induction by an endogenous K-ras oncogene is highly dependent on
cellular context. Cancer Cell. 4:111–120. 2003. View Article : Google Scholar
|
|
51
|
Findlay GM, Boyle EA, Hause RJ, Klein JC
and Shendure J: Saturation editing of genomic regions by multiplex
homology-directed repair. Nature. 513:120–123. 2014. View Article : Google Scholar
|
|
52
|
Hacisuleyman E, Goff LA, Trapnell C,
Williams A, Henao-Mejia J, Sun L, McClanahan P, Hendrickson DG,
Sauvageau M, Kelley DR, et al: Topological organization of
multichromosomal regions by the long intergenic noncoding RNA
Firre. Nat Struct Mol Biol. 21:198–206. 2014. View Article : Google Scholar
|
|
53
|
Krishnaswamy JK, Singh A, Gowthaman U, Wu
R, Gorrepati P, Sales Nascimento M, Gallman A, Liu D, Rhebergen AM,
Calabro S, et al: Coincidental loss of DOCK8 function in
NLRP10-deficient and C3H/HeJ mice results in defective dendritic
cell migration. Proc Natl Acad Sci USA. 112:3056–3061. 2015.
View Article : Google Scholar
|
|
54
|
Billon P, Bryant EE, Joseph SA, Nambiar
TS, Hayward SB, Rothstein R and Ciccia A: CRISPR-mediated base
editing enables efficient disruption of eukaryotic genes through
induction of STOP codons. Moll Cell. 67:1068–79.e4. 2017.
View Article : Google Scholar
|
|
55
|
Blasco RB, Karaca E, Ambrogio C, Cheong
TC, Karayol E, Minero VG, Voena C and Chiarle R: Simple and rapid
in vivo generation of chromosomal rearrangements using CRISPR/Cas9
technology. Cell Rep. 9:1219–1227. 2014. View Article : Google Scholar
|
|
56
|
Maddalo D, Manchado E, Concepcion CP,
Bonetti C, Vidigal JA, Han YC, Ogrodowski P, Crippa A, Rekhtman N,
de Stanchina E, et al: In vivo engineering of oncogenic chromosomal
rearrangements with the CRISPR/Cas9 system. Nature. 516:423–427.
2014. View Article : Google Scholar
|
|
57
|
Nishio M, Kim DW, Wu YL, Nakagawa K,
Solomon BJ, Shaw AT, Hashigaki S, Ohki E, Usari T, Paolini J, et
al: Crizotinib versus chemotherapy in Asian patients with advanced
ALK-positive non-small cell lung cancer. Cancer Res Treat. Jul
6–2017.Epub ahead of print. View Article : Google Scholar
|
|
58
|
Ding Q, Strong A, Patel KM, Ng SL, Gosis
BS, Regan SN, Cowan CA, Rader DJ and Musunuru K: Permanent
alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ
Res. 115:488–492. 2014. View Article : Google Scholar
|
|
59
|
Yang DG, Chung YC, Lai YK, Lai CW, Liu HJ
and Hu YC: Corrigendum to 'Avian Influenza Virus Hemagglutinin
Display on Baculovirus Envelope: Cytoplasmic Domain Affects Virus
Properties and Vaccine Potential'. Mol Ther. 15:17362007.
View Article : Google Scholar
|
|
60
|
Cancer Genome Atlas N; Cancer Genome Atlas
Network: Comprehensive molecular characterization of human colon
and rectal cancer. Nature. 487:330–337. 2012. View Article : Google Scholar
|
|
61
|
Sánchez-Rivera FJ, Papagiannakopoulos T,
Romero R, Tammela T, Bauer MR, Bhutkar A, Joshi NS, Subbaraj L,
Bronson RT, Xue W, et al: Rapid modelling of cooperating genetic
events in cancer through somatic genome editing. Nature.
516:428–431. 2014. View Article : Google Scholar
|
|
62
|
Chiou SH, Winters IP, Wang J, Naranjo S,
Dudgeon C, Tamburini FB, Brady JJ, Yang D, Grüner BM, Chuang CH, et
al: Pancreatic cancer modeling using retrograde viral vector
delivery and in vivo CRISPR/Cas9-mediated somatic genome editing.
Genes Dev. 29:1576–1585. 2015. View Article : Google Scholar
|
|
63
|
Annunziato S, Kas SM, Nethe M, Yücel H,
Del Bravo J, Pritchard C, Bin Ali R, van Gerwen B, Siteur B, Drenth
AP, et al: Modeling invasive lobular breast carcinoma by
CRISPR/Cas9-mediated somatic genome editing of the mammary gland.
Genes Dev. 30:1470–1480. 2016. View Article : Google Scholar
|
|
64
|
Oh B, Hwang S, McLaughlin J, Solter D and
Knowles BB: Timely translation during the mouse oocyte-to-embryo
transition. Development. 127:3795–3803. 2000.
|
|
65
|
Flemr M and Bühler M: Single-step
generation of conditional knockout mouse embryonic stem cells. Cell
Rep. 12:709–716. 2015. View Article : Google Scholar
|
|
66
|
Malina A, Mills JR, Cencic R, Yan Y,
Fraser J, Schippers LM, Paquet M, Dostie J and Pelletier J:
Repurposing CRISPR/Cas9 for in situ functional assays. Genes Dev.
27:2602–2614. 2013. View Article : Google Scholar
|
|
67
|
Heckl D, Kowalczyk MS, Yudovich D,
Belizaire R, Puram RV, McConkey ME, Thielke A, Aster JC, Regev A
and Ebert BL: Generation of mouse models of myeloid malignancy with
combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat
Biotechnol. 32:941–946. 2014. View Article : Google Scholar
|
|
68
|
Chen C, Liu Y, Rappaport AR, Kitzing T,
Schultz N, Zhao Z, Shroff AS, Dickins RA, Vakoc CR, Bradner JE, et
al: MLL3 is a haploinsufficient 7q tumor suppressor in acute
myeloid leukemia. Cancer Cell. 25:652–665. 2014. View Article : Google Scholar
|
|
69
|
Zhong C, Yin Q, Xie Z, Bai M, Dong R, Tang
W, Xing YH, Zhang H, Yang S, Chen LL, et al: CRISPR-Cas9-mediated
genetic screening in mice with haploid embryonic stem cells
carrying a Guide RNA Library. Cell Stem Cell. 17:221–232. 2015.
View Article : Google Scholar
|
|
70
|
Park CY, Kim DH, Son JS, Sung JJ, Lee J,
Bae S, Kim JH, Kim DW and Kim JS: Functional correction of large
factor VIII gene chromosomal inversions in hemophilia a
patient-derived iPSCs using CRISPR-Cas9. Cell Stem Cell.
17:213–220. 2015. View Article : Google Scholar
|
|
71
|
Huang L, Holtzinger A, Jagan I, BeGora M,
Lohse I, Ngai N, Nostro C, Wang R, Muthuswamy LB, Crawford HC, et
al: Ductal pancreatic cancer modeling and drug screening using
human pluripotent stem cell- and patient-derived tumor organoids.
Nat Med. 21:1364–1371. 2015. View Article : Google Scholar
|
|
72
|
Wang J, Exline CM, DeClercq JJ, Llewellyn
GN, Hayward SB, Li PW, Shivak DA, Surosky RT, Gregory PD, Holmes
MC, et al: Homology-driven genome editing in hematopoietic stem and
progenitor cells using ZFN mRNA and AAV6 donors. Nat Biotechnol.
33:1256–1263. 2015. View Article : Google Scholar
|
|
73
|
Hoban MD, Cost GJ, Mendel MC, Romero Z,
Kaufman ML, Joglekar AV, Ho M, Lumaquin D, Gray D, Lill GR, et al:
Correction of the sickle cell disease mutation in human
hematopoietic stem/progenitor cells. Blood. 125:2597–2604. 2015.
View Article : Google Scholar
|
|
74
|
Xie F, Ye L, Chang JC, Beyer AI, Wang J,
Muench MO and Kan YW: Seamless gene correction of β-thalassemia
mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac.
Genome Res. 24:1526–1533. 2014. View Article : Google Scholar
|
|
75
|
Roberts SA, Dong B, Firrman JA, Moore AR,
Sang N and Xiao W: Engineering factor Viii for hemophilia gene
therapy. J Genet Syndr Gene Ther. 1:12011.
|
|
76
|
Stanek LM, Sardi SP, Mastis B, Richards
AR, Treleaven CM, Taksir T, Misra K, Cheng SH and Shihabuddin LS:
Silencing mutant huntingtin by adeno-associated virus-mediated RNA
interference ameliorates disease manifestations in the YAC128 mouse
model of Huntington' s disease. Hum Gene Ther. 25:461–474. 2014.
View Article : Google Scholar
|
|
77
|
Shin JW, Kim KH, Chao MJ, Atwal RS, Gillis
T, MacDonald ME, Gusella JF and Lee JM: Permanent inactivation of
Huntington's disease mutation by personalized allele-specific
CRISPR/Cas9. Hum Mol Genet. 25:4566–4576. 2016.
|
|
78
|
Monteys AM, Ebanks SA, Keiser MS and
Davidson BL: CRISPR/Cas9 editing of the mutant huntingtin allele in
vitro and in vivo. Mol Ther. 25:12–23. 2017. View Article : Google Scholar
|
|
79
|
Xu X, Tay Y, Sim B, Yoon SI, Huang Y, Ooi
J, Utami KH, Ziaei A, Ng B, Radulescu C, et al: Reversal of
Phenotypic Abnormalities by CRISPR/Cas9-Mediated Gene Correction in
Huntington Disease Patient-Derived Induced Pluripotent Stem Cells.
Stem Cell Reports. 8:619–633. 2017. View Article : Google Scholar
|
|
80
|
Chung CY, Khurana V, Auluck PK, Tardiff
DF, Mazzulli JR, Soldner F, Baru V, Lou Y, Freyzon Y, Cho S, et al:
Identification and rescue of α-synuclein toxicity in Parkinson
patient-derived neurons. Science. 342:983–987. 2013. View Article : Google Scholar
|
|
81
|
Long C, McAnally JR, Shelton JM, Mireault
AA, Bassel-Duby R and Olson EN: Prevention of muscular dystrophy in
mice by CRISPR/Cas9-mediated editing of germline DNA. Science.
345:1184–1188. 2014. View Article : Google Scholar
|
|
82
|
Schwank G, Koo BK, Sasselli V, Dekkers JF,
Heo I, Demircan T, Sasaki N, Boymans S, Cuppen E, van der Ent CK,
et al: Functional repair of CFTR by CRISPR/Cas9 in intestinal stem
cell organoids of cystic fibrosis patients. Cell Stem Cell.
13:653–658. 2013. View Article : Google Scholar
|
|
83
|
Firth AL, Menon T, Parker GS, Qualls SJ,
Lewis BM, Ke E, Dargitz CT, Wright R, Khanna A, Gage FH, et al:
Functional gene correction for cystic fibrosis in lung epithelial
cells generated from patient iPSCs. Cell Rep. 12:1385–1390. 2015.
View Article : Google Scholar
|
|
84
|
Wu Y, Liang D, Wang Y, Bai M, Tang W, Bao
S, Yan Z, Li D and Li J: Correction of a genetic disease in mouse
via use of CRISPR-Cas9. Cell Stem Cell. 13:659–662. 2013.
View Article : Google Scholar
|
|
85
|
Wu Y, Zhou H, Fan X, Zhang Y, Zhang M,
Wang Y, Xie Z, Bai M, Yin Q, Liang D, et al: Correction of a
genetic disease by CRISPR-Cas9-mediated gene editing in mouse
spermatogonial stem cells. Cell Res. 25:67–79. 2015. View Article : Google Scholar
|
|
86
|
Xie N, Gong H, Suhl JA, Chopra P, Wang T
and Warren ST: Reactivation of FMR1 by CRISPR/Cas9-mediated
deletion of the expanded CGG-repeat of the Fragile X chromosome.
PLoS One. 11:e01654992016. View Article : Google Scholar
|
|
87
|
Horii T, Tamura D, Morita S, Kimura M and
Hatada I: Generation of an ICF syndrome model by efficient genome
editing of human induced pluripotent stem cells using the CRISPR
system. Int J Mol Sci. 14:19774–19781. 2013. View Article : Google Scholar
|
|
88
|
Chang CW, Lai YS, Westin E,
Khodadadi-Jamayran A, Pawlik KM, Lamb LS Jr, Goldman FD and Townes
TM: Modeling human severe combined immunodeficiency and correction
by CRISPR/Cas9-enhanced gene targeting. Cell Rep. 12:1668–1677.
2015. View Article : Google Scholar
|
|
89
|
Flynn R, Grundmann A, Renz P, Hanseler W,
James WS, Cowley SA and Moore MD: CRISPR-mediated genotypic and
phenotypic correction of a chronic granulomatous disease mutation
in human iPS cells. Exp Hematol. 43:838–848.e3. 2015. View Article : Google Scholar
|
|
90
|
Osborn MJ, Belanto JJ, Tolar J and Voytas
DF: Gene editing and its application for hematological diseases.
Int J Hematol. 104:18–28. 2016. View Article : Google Scholar
|
|
91
|
Shinkuma S, Guo Z and Christiano AM:
Site-specific genome editing for correction of induced pluripotent
stem cells derived from dominant dystrophic epidermolysis bullosa.
Proc Natl Acad Sci USA. 113:5676–5681. 2016. View Article : Google Scholar
|
|
92
|
Bassuk AG, Zheng A, Li Y, Tsang SH and
Mahajan VB: Precision Medicine: Genetic Repair of Retinitis
Pigmentosa in Patient-Derived Stem Cells. Sci Rep. 6:199692016.
View Article : Google Scholar
|
|
93
|
Ruan GX, Barry E, Yu D, Lukason M, Cheng
SH and Scaria A: CRISPR/Cas9-mediated genome editing as a
therapeutic approach for leber congenital amaurosis 10. Mol Ther.
25:331–341. 2017. View Article : Google Scholar
|
|
94
|
Mandegar MA, Huebsch N, Frolov EB, Shin E,
Truong A, Olvera MP, Chan AH, Miyaoka Y, Holmes K, Spencer CI, et
al: CRISPR Interference Efficiently Induces Specific and Reversible
Gene Silencing in Human iPSCs. Cell Stem Cell. 18:541–553. 2016.
View Article : Google Scholar
|
|
95
|
Couzin-Frankel J: Breakthrough of the year
2013. Cancer immunotherapy Science. 342:1432–1433. 2013.
|
|
96
|
Guye P, Ebrahimkhani MR, Kipniss N,
Velazquez JJ, Schoenfeld E, Kiani S, Griffith LG and Weiss R:
Genetically engineering self-organization of human pluripotent stem
cells into a liver bud-like tissue using Gata6. Nat Commun.
7:102432016. View Article : Google Scholar
|
|
97
|
Sharma SV, Haber DA and Settleman J: Cell
line-based platforms to evaluate the therapeutic efficacy of
candidate anticancer agents. Nat Rev Cancer. 10:241–253. 2010.
View Article : Google Scholar
|
|
98
|
Lutsenko S: Introduction to the minireview
series on modern technologies for in-cell biochemistry. J Biol
Chem. 291:3757–3758. 2016. View Article : Google Scholar
|
|
99
|
Sato T, Vries RG, Snippert HJ, van de
Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters
PJ, et al: Single Lgr5 stem cells build crypt-villus structures in
vitro without a mesenchymal niche. Nature. 459:262–265. 2009.
View Article : Google Scholar
|
|
100
|
Clevers H: Modeling development and
disease with organoids. Cell. 165:1586–1597. 2016. View Article : Google Scholar
|
|
101
|
Lancaster MA and Knoblich JA: Generation
of cerebral organoids from human pluripotent stem cells. Nat
Protoc. 9:2329–2340. 2014. View Article : Google Scholar
|
|
102
|
Sayin VI and Papagiannakopoulos T:
Application of CRISPR-mediated genome engineering in cancer
research. Cancer Lett. 387:10–17. 2017. View Article : Google Scholar
|
|
103
|
Boj SF, Hwang CI, Baker LA, Chio II, Engle
DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, et al:
Organoid models of human and mouse ductal pancreatic cancer. Cell.
160:324–338. 2015. View Article : Google Scholar
|
|
104
|
Nantasanti S, Spee B, Kruitwagen HS, Chen
C, Geijsen N, Oosterhoff LA, van Wolferen ME, Pelaez N, Fieten H,
Wubbolts RW, et al: Disease modeling and gene therapy of copper
storage disease in canine hepatic organoids. Stem Cell Rep.
5:895–907. 2015. View Article : Google Scholar
|
|
105
|
Walsh AJ, Cook RS, Sanders ME, Arteaga CL
and Skala MC: Drug response in organoids generated from frozen
primary tumor tissues. Sci Rep. 6:188892016. View Article : Google Scholar
|
|
106
|
van de Wetering M, Francies HE, Francis
JM, Bounova G, Iorio F, Pronk A, van Houdt W, van Gorp J,
Taylor-Weiner A, Kester L, et al: Prospective derivation of a
living organoid biobank of colorectal cancer patients. Cell.
161:933–945. 2015. View Article : Google Scholar
|
|
107
|
Huch M, Gehart H, van Boxtel R, Hamer K,
Blokzijl F, Verstegen MM, Ellis E, van Wenum M, Fuchs SA, de Ligt
J, et al: Long-term culture of genome-stable bipotent stem cells
from adult human liver. Cell. 160:299–312. 2015. View Article : Google Scholar
|
|
108
|
Barker N, Huch M, Kujala P, van de
Wetering M, Snippert HJ, van Es JH, Sato T, Stange DE, Begthel H,
van den Born M, et al: Lgr5(+ve) stem cells drive self-renewal in
the stomach and build long-lived gastric units in vitro. Cell Stem
Cell. 6:25–36. 2010. View Article : Google Scholar
|
|
109
|
Huch M, Bonfanti P, Boj SF, Sato T,
Loomans CJ, van de Wetering M, Sojoodi M, Li VS, Schuijers J,
Gracanin A, et al: Unlimited in vitro expansion of adult bi-potent
pancreas progenitors through the Lgr5/R-spondin axis. EMBO J.
32:2708–2721. 2013. View Article : Google Scholar
|
|
110
|
Huch M, Dorrell C, Boj SF, van Es JH, Li
VS, van de Wetering M, Sato T, Hamer K, Sasaki N, Finegold MJ, et
al: In vitro expansion of single Lgr5+ liver stem cells induced by
Wnt-driven regeneration. Nature. 494:247–250. 2013. View Article : Google Scholar
|
|
111
|
Gao D, Vela I, Sboner A, Iaquinta PJ,
Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora
VK, et al: Organoid cultures derived from patients with advanced
prostate cancer. Cell. 159:176–187. 2014. View Article : Google Scholar
|
|
112
|
Karthaus WR, Iaquinta PJ, Drost J,
Gracanin A, van Boxtel R, Wongvipat J, Dowling CM, Gao D, Begthel
H, Sachs N, et al: Identification of multipotent luminal progenitor
cells in human prostate organoid cultures. Cell. 159:163–175. 2014.
View Article : Google Scholar
|
|
113
|
Heo I and Clevers H: Expanding intestinal
stem cells in culture. Cell Res. 25:995–996. 2015. View Article : Google Scholar
|
|
114
|
Cooks T, Pateras IS, Tarcic O, Solomon H,
Schetter AJ, Wilder S, Lozano G, Pikarsky E, Forshew T, Rosenfeld
N, et al: Mutant p53 prolongs NF-κB activation and promotes chronic
inflammation and inflammation-associated colorectal cancer. Cancer
Cell. 23:634–646. 2013. View Article : Google Scholar
|
|
115
|
Kazanjian A and Shroyer NF: NOTCH
signaling and ATOH1 in colorectal cancers. Curr Colorectal Cancer
Rep. 7:121–127. 2011. View Article : Google Scholar
|
|
116
|
Fujii M, Shimokawa M, Date S, Takano A,
Matano M, Nanki K, Ohta Y, Toshimitsu K, Nakazato Y, Kawasaki K, et
al: A colorectal tumor organoid library demonstrates progressive
loss of niche factor requirements during tumorigenesis. Cell Stem
Cell. 18:827–838. 2016. View Article : Google Scholar
|
|
117
|
Drost J, van Jaarsveld RH, Ponsioen B,
Zimberlin C, van Boxtel R, Buijs A, Sachs N, Overmeer RM, Offerhaus
GJ, Begthel H, et al: Sequential cancer mutations in cultured human
intestinal stem cells. Nature. 521:43–47. 2015. View Article : Google Scholar
|
|
118
|
Bargou R, Leo E, Zugmaier G, Klinger M,
Goebeler M, Knop S, Noppeney R, Viardot A, Hess G, Schuler M, et
al: Tumor regression in cancer patients by very low doses of a T
cell-engaging antibody. Science. 321:974–977. 2008. View Article : Google Scholar
|
|
119
|
Chames P and Baty D: Bispecific antibodies
for cancer therapy: The light at the end of the tunnel. MAbs.
1:539–547. 2009. View Article : Google Scholar
|
|
120
|
Stagg J, Lejeune L, Paquin A and Galipeau
J: Marrow stromal cells for interleukin-2 delivery in cancer
immunotherapy. Hum Gene Ther. 15:597–608. 2004. View Article : Google Scholar
|
|
121
|
Compte M, Nuñez-Prado N, Sanz L and
Alvarez-Vallina L: Immunotherapeutic organoids: A new approach to
cancer treatment. Biomatter. 3:e238972013. View Article : Google Scholar
|
|
122
|
Fuller MK, Faulk DM, Sundaram N, Shroyer
NF, Henning SJ and Helmrath MA: Intestinal crypts reproducibly
expand in culture. J Surg Res. 178:48–54. 2012. View Article : Google Scholar
|
|
123
|
Grün D, Lyubimova A, Kester L, Wiebrands
K, Basak O, Sasaki N, Clevers H and van Oudenaarden A: Single-cell
messenger RNA sequencing reveals rare intestinal cell types.
Nature. 525:251–255. 2015. View Article : Google Scholar
|
|
124
|
Onuma K, Ochiai M, Orihashi K, Takahashi
M, Imai T, Nakagama H and Hippo Y: Genetic reconstitution of
tumori-genesis in primary intestinal cells. Proc Natl Acad Sci USA.
110:11127–11132. 2013. View Article : Google Scholar
|
|
125
|
Vazin T and Schaffer DV: Engineering
strategies to emulate the stem cell niche. Trends Biotechnol.
28:117–124. 2010. View Article : Google Scholar
|
|
126
|
Lancaster MA, Renner M, Martin CA, Wenzel
D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP and
Knoblich JA: Cerebral organoids model human brain development and
microcephaly. Nature. 501:373–379. 2013. View Article : Google Scholar
|
|
127
|
Murphy SV and Atala A: 3D bioprinting of
tissues and organs. Nat Biotechnol. 32:773–785. 2014. View Article : Google Scholar
|
|
128
|
Lindemans CA, Calafiore M, Mertelsmann AM,
O'Connor MH, Dudakov JA, Jenq RR, Velardi E, Young LF, Smith OM,
Lawrence G, et al: Interleukin-22 promotes
intestinal-stem-cell-mediated epithelial regeneration. Nature.
528:560–564. 2015. View Article : Google Scholar
|
|
129
|
Wilson SS, Tocchi A, Holly MK, Parks WC
and Smith JG: A small intestinal organoid model of non-invasive
enteric pathogen-epithelial cell interactions. Mucosal Immunol.
8:352–361. 2015. View Article : Google Scholar
|
|
130
|
Wu K, House L, Liu W and Cho WC:
Personalized targeted therapy for lung cancer. Int J Mol Sci.
13:11471–11496. 2012. View Article : Google Scholar
|
|
131
|
Tang H and Shrager JB: CRISPR/Cas-mediated
genome editing to treat EGFR-mutant lung cancer: A personalized
molecular surgical therapy. EMBO Mol Med. 8:83–85. 2016. View Article : Google Scholar
|
|
132
|
Cancer Genome Atlas Research Network;
Electronic address edsc, Cancer Genome Atlas Research N:
Comprehensive and Integrated Genomic Characterization of Adult Soft
Tissue Sarcomas. Cell. 171:950–65.e28. 2017. View Article : Google Scholar
|
|
133
|
Barretina J, Caponigro G, Stransky N,
Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV,
Sonkin D, et al: The cancer cell line encyclopedia enables
predictive modelling of anticancer drug sensitivity. Nature.
483:603–607. 2012. View Article : Google Scholar
|
|
134
|
Consortium EP; ENCODE Project Consortium:
An integrated encyclopedia of DNA elements in the human genome.
Nature. 489:57–74. 2012. View Article : Google Scholar
|
|
135
|
Shalem O, Sanjana NE, Hartenian E, Shi X,
Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, et
al: Genome-scale CRISPR-Cas9 knockout screening in human cells.
Science. 343:84–87. 2014. View Article : Google Scholar
|
|
136
|
Wang T, Wei JJ, Sabatini DM and Lander ES:
Genetic screens in human cells using the CRISPR-Cas9 system.
Science. 343:80–84. 2014. View Article : Google Scholar
|
|
137
|
Koike-Yusa H, Li Y, Tan EP,
Velasco-Herrera MC and Yusa K: Genome-wide recessive genetic
screening in mammalian cells with a lentiviral CRISPR-guide RNA
library. Nat Biotechnol. 32:267–273. 2014. View Article : Google Scholar
|
|
138
|
Sanjana NE: Genome-scale CRISPR pooled
screens. Anal Biochem. 532:95–99. 2017. View Article : Google Scholar
|
|
139
|
Fellmann C, Gowen BG, Lin PC, Doudna JA
and Corn JE: Cornerstones of CRISPR-Cas in drug discovery and
therapy. Nat Rev Drug Discov. 16:89–100. 2017. View Article : Google Scholar
|
|
140
|
Wang T, Birsoy K, Hughes NW, Krupczak KM,
Post Y, Wei JJ, Lander ES and Sabatini DM: Identification and
characterization of essential genes in the human genome. Science.
350:1096–1101. 2015. View Article : Google Scholar
|
|
141
|
González F, Zhu Z, Shi ZD, Lelli K, Verma
N, Li QV and Huangfu D: An iCRISPR platform for rapid,
multiplexable, and inducible genome editing in human pluripotent
stem cells. Cell Stem Cell. 15:215–226. 2014. View Article : Google Scholar
|
|
142
|
Shi J, Wang E, Milazzo JP, Wang Z, Kinney
JB and Vakoc CR: Discovery of cancer drug targets by CRISPR-Cas9
screening of protein domains. Nat Biotechnol. 33:661–667. 2015.
View Article : Google Scholar
|
|
143
|
Qi LS, Larson MH, Gilbert LA, Doudna JA,
Weissman JS, Arkin AP and Lim WA: Repurposing CRISPR as an
RNA-guided platform for sequence-specific control of gene
expression. Cell. 152:1173–1183. 2013. View Article : Google Scholar
|
|
144
|
Gilbert LA, Larson MH, Morsut L, Liu Z,
Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH,
Doudna JA, et al: CRISPR-mediated modular RNA-guided regulation of
transcription in eukaryotes. Cell. 154:442–451. 2013. View Article : Google Scholar
|
|
145
|
Gilbert LA, Horlbeck MA, Adamson B,
Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh
HL, Bassik MC, et al: Genome-Scale CRISPR-Mediated Control of Gene
Repression and Activation. Cell. 159:647–661. 2014. View Article : Google Scholar
|
|
146
|
Takahashi K, Tanabe K, Ohnuki M, Narita M,
Ichisaka T, Tomoda K and Yamanaka S: Induction of pluripotent stem
cells from adult human fibroblasts by defined factors. Cell.
131:861–872. 2007. View Article : Google Scholar
|
|
147
|
Konermann S, Brigham MD, Trevino AE, Joung
J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS,
Nishimasu H, et al: Genome-scale transcriptional activation by an
engineered CRISPR-Cas9 complex. Nature. 517:583–588. 2015.
View Article : Google Scholar
|
|
148
|
Cheng AW, Wang H, Yang H, Shi L, Katz Y,
Theunissen TW, Rangarajan S, Shivalila CS, Dadon DB and Jaenisch R:
Multiplexed activation of endogenous genes by CRISPR-on, an
RNA-guided transcriptional activator system. Cell Res.
23:1163–1171. 2013. View Article : Google Scholar
|
|
149
|
Rathe SK, Moriarity BS, Stoltenberg CB,
Kurata M, Aumann NK, Rahrmann EP, Bailey NJ, Melrose EG, Beckmann
DA, Liska CR, et al: Using RNA-seq and targeted nucleases to
identify mechanisms of drug resistance in acute myeloid leukemia.
Sci Rep. 4:60482014. View Article : Google Scholar
|
|
150
|
Tzelepis K, Koike-Yusa H, De Braekeleer E,
Li Y, Metzakopian E, Dovey OM, Mupo A, Grinkevich V, Li M, Mazan M,
et al: A CRISPR Dropout Screen Identifies Genetic Vulnerabilities
and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep.
17:1193–1205. 2016. View Article : Google Scholar
|
|
151
|
Ruiz S, Mayor-Ruiz C, Lafarga V, Murga M,
Vega-Sendino M, Ortega S and Fernandez-Capetillo O: A Genome-wide
CRISPR Screen Identifies CDC25A as a Determinant of Sensitivity to
ATR Inhibitors. Mol Cell. 62:307–313. 2016. View Article : Google Scholar
|
|
152
|
Wanzel M, Vischedyk JB, Gittler MP, Gremke
N, Seiz JR, Hefter M, Noack M, Savai R, Mernberger M, Charles JP,
et al: CRISPR-Cas9-based target validation for p53-reactivating
model compounds. Nat Chem Biol. 12:22–28. 2016. View Article : Google Scholar
|
|
153
|
Rath O and Kozielski F: Kinesins and
cancer. Nat Rev Cancer. 12:527–539. 2012. View Article : Google Scholar
|
|
154
|
Chen C, Siegel D, Gutierrez M, Jacoby M
and Hofmeister CC: Safety and efficacy of selinexor in relapsed or
refractory multiple myeloma and Waldenstrom macroglobulinemia.
Blood. 131:855–863. 2018. View Article : Google Scholar
|
|
155
|
Steinhart Z, Pavlovic Z, Chandrashekhar M,
Hart T, Wang X, Zhang X, Robitaille M, Brown KR, Jaksani S,
Overmeer R, et al: Genome-wide CRISPR screens reveal a Wnt-FZD5
signaling circuit as a druggable vulnerability of RNF43-mutant
pancreatic tumors. Nat Med. 23:60–68. 2017. View Article : Google Scholar
|
|
156
|
Zhu S, Li W, Liu J, Chen CH, Liao Q, Xu P,
Xu H, Xiao T, Cao Z, Peng J, et al: Genome-scale deletion screening
of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9
library. Nat Biotechnol. 34:1279–1286. 2016. View Article : Google Scholar
|
|
157
|
Jaitin DA, Weiner A, Yofe I, Lara-Astiaso
D, Keren-Shaul H, David E, Salame TM, Tanay A, van Oudenaarden A
and Amit I: Dissecting immune circuits by linking CRISPR-pooled
screens with single-cell RNA-Seq. Cell. 167:1883–1896.e15. 2016.
View Article : Google Scholar
|
|
158
|
Hinrichs CS and Rosenberg SA: Exploiting
the curative potential of adoptive T-cell therapy for cancer.
Immunol Rev. 257:56–71. 2014. View Article : Google Scholar
|
|
159
|
Jensen MC and Riddell SR: Designing
chimeric antigen receptors to effectively and safely target tumors.
Curr Opin Immunol. 33:9–15. 2015. View Article : Google Scholar
|
|
160
|
Eyquem J, Mansilla-Soto J, Giavridis T,
van der Stegen SJ, Hamieh M, Cunanan KM, Odak A, Gönen M and
Sadelain M: Targeting a CAR to the TRAC locus with CRISPR/Cas9
enhances tumour rejection. Nature. 543:113–117. 2017. View Article : Google Scholar
|
|
161
|
Cyranoski D: Chinese scientists to pioneer
first human CRISPR trial. Nature. 535:476–477. 2016. View Article : Google Scholar
|
|
162
|
Topalian SL, Drake CG and Pardoll DM:
Immune checkpoint blockade: A common denominator approach to cancer
therapy. Cancer Cell. 27:450–461. 2015. View Article : Google Scholar
|
|
163
|
Yang Y, Kelly P, Shaffer AL III, Schmitz
R, Yoo HM, Liu X, Huang DW, Webster D, Young RM, Nakagawa M, et al:
Targeting non-proteolytic protein ubiquitination for the treatment
of diffuse large B cell lymphoma. Cancer Cell. 29:494–507. 2016.
View Article : Google Scholar
|
|
164
|
DeWitt MA, Magis W, Bray L, Wang T, Berman
JR, Urbinati F, Heo SJ, Mitros T, Muñoz DP, Boffelli D, et al:
Selection-free genome editing of the sickle mutation in human adult
hemato-poietic stem/progenitor cells. Sci Transl Med.
8:360ra1342016. View Article : Google Scholar
|
|
165
|
Zhen S, Hua L, Liu YH, Gao LC, Fu J, Wan
DY, Dong LH, Song HF and Gao X: Harnessing the clustered regularly
inter-spaced short palindromic repeat (CRISPR)/CRISPR-associated
Cas9 system to disrupt the hepatitis B virus. Gene Ther.
22:404–412. 2015. View Article : Google Scholar
|
|
166
|
Dong C, Qu L, Wang H, Wei L, Dong Y and
Xiong S: Targeting hepatitis B virus cccDNA by CRISPR/Cas9 nuclease
efficiently inhibits viral replication. Antiviral Res. 118:110–117.
2015. View Article : Google Scholar
|
|
167
|
Price AA, Sampson TR, Ratner HK, Grakoui A
and Weiss DS: Cas9-mediated targeting of viral RNA in eukaryotic
cells. Proc Natl Acad Sci USA. 112:6164–6169. 2015. View Article : Google Scholar
|
|
168
|
Wang J and Quake SR: RNA-guided
endonuclease provides a therapeutic strategy to cure latent
herpesviridae infection. Proc Natl Acad Sci USA. 111:13157–13162.
2014. View Article : Google Scholar
|
|
169
|
Zhen S, Hua L, Takahashi Y, Narita S, Liu
YH and Li Y: In vitro and in vivo growth suppression of human
papillomavirus 16-positive cervical cancer cells by CRISPR/Cas9.
Biochem Biophys Res Commun. 450:1422–1426. 2014. View Article : Google Scholar
|
|
170
|
Leen AM, Myers GD, Sili U, Huls MH, Weiss
H, Leung KS, Carrum G, Krance RA, Chang CC, Molldrem JJ, et al:
Monoculture-derived T lymphocytes specific for multiple viruses
expand and produce clinically relevant effects in immunocompromised
individuals. Nat Med. 12:1160–1166. 2006. View Article : Google Scholar
|
|
171
|
Saglio F, Hanley PJ and Bollard CM: The
time is now: Moving toward virus-specific T cells after allogeneic
hematopoietic stem cell transplantation as the standard of care.
Cytotherapy. 16:149–159. 2014. View Article : Google Scholar
|
|
172
|
Lieberman J, Skolnik PR, Parkerson GR III,
Fabry JA, Landry B, Bethel J and Kagan J: Safety of autologous, ex
vivo-expanded human immunodeficiency virus (HIV)-specific cytotoxic
T-lymphocyte infusion in HIV-infected patients. Blood.
90:2196–2206. 1997.
|
|
173
|
Brodie SJ, Lewinsohn DA, Patterson BK,
Jiyamapa D, Krieger J, Corey L, Greenberg PD and Riddell SR: In
vivo migration and function of transferred HIV-1-specific cytotoxic
T cells. Nat Med. 5:34–41. 1999. View
Article : Google Scholar
|
|
174
|
Tan R, Xu X, Ogg GS, Hansasuta P, Dong T,
Rostron T, Luzzi G, Conlon CP, Screaton GR, McMichael AJ, et al:
Rapid death of adoptively transferred T cells in acquired
immunodeficiency syndrome. Blood. 93:1506–1510. 1999.
|
|
175
|
Lam S, Sung J, Cruz C, Castillo-Caro P,
Ngo M, Garrido C, Kuruc J, Archin N, Rooney C, Margolis D, et al:
Broadly-specific cytotoxic T cells targeting multiple HIV antigens
are expanded from HIV+ patients: implications for immunotherapy.
Mol Ther. 23:387–395. 2015. View Article : Google Scholar
|
|
176
|
Patel S, Lam S, Cruz CR, Wright K, Cochran
C, Ambinder RF and Bollard CM: Functionally active HIV-specific T
cells that target Gag and Nef can be expanded from virus-naive
donors and target a range of viral epitopes: Implications for a
cure strategy after allogeneic hematopoietic stem cell
transplantation. Biol Blood Marrow Transplant. 22:536–541. 2016.
View Article : Google Scholar
|
|
177
|
Sung JA, Lam S, Garrido C, Archin N,
Rooney CM, Bollard CM and Margolis DM: Expanded cytotoxic T-cell
lymphocytes target the latent HIV reservoir. J Infect Dis.
212:258–263. 2015. View Article : Google Scholar
|
|
178
|
Colovos C, Villena-Vargas J and Adusumilli
PS: Safety and stability of retrovirally transduced chimeric
antigen receptor T cells. Immunotherapy. 4:899–902. 2012.
View Article : Google Scholar
|
|
179
|
Scholler J, Brady TL, Binder-Scholl G,
Hwang WT, Plesa G, Hege KM, Vogel AN, Kalos M, Riley JL, Deeks SG,
et al: Decade-long safety and function of retroviral-modified
chimeric antigen receptor T cells. Sci Transl Med. 4:132ra532012.
View Article : Google Scholar
|
|
180
|
Joseph A, Zheng JH, Follenzi A, Dilorenzo
T, Sango K, Hyman J, Chen K, Piechocka-Trocha A, Brander C,
Hooijberg E, et al: Lentiviral vectors encoding human
immunodeficiency virus type 1 (HIV-1)-specific T-cell receptor
genes efficiently convert peripheral blood CD8 T lymphocytes into
cytotoxic T lymphocytes with potent in vitro and in vivo
HIV-1-specific inhibitory activity. J Virol. 82:3078–3089. 2008.
View Article : Google Scholar
|
|
181
|
Mitsuyasu RT, Anton PA, Deeks SG, Scadden
DT, Connick E, Downs MT, Bakker A, Roberts MR, June CH, Jalali S,
et al: Prolonged survival and tissue trafficking following adoptive
transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T
cells in human immunodeficiency virus-infected subjects. Blood.
96:785–793. 2000.
|
|
182
|
Deeks SG, Wagner B, Anton PA, Mitsuyasu
RT, Scadden DT, Huang C, Macken C, Richman DD, Christopherson C,
June CH, et al: A phase II randomized study of HIV-specific T-cell
gene therapy in subjects with undetectable plasma viremia on
combination antiretroviral therapy. Mol Ther. 5:788–797. 2002.
View Article : Google Scholar
|
|
183
|
Koretzky GA: Multiple roles of CD4 and CD8
in T cell activation. J Immunol. 185:2643–2644. 2010. View Article : Google Scholar
|
|
184
|
Hütter G and Ganepola S: Eradication of
HIV by transplantation of CCR5-deficient hematopoietic stem cells.
Sci World J. 11:1068–1076. 2011. View Article : Google Scholar
|
|
185
|
Bitinaite J, Wah DA, Aggarwal AK and
Schildkraut I: FokI dimerization is required for DNA cleavage. Proc
Natl Acad Sci USA. 95:10570–10575. 1998. View Article : Google Scholar
|
|
186
|
Yuan J, Wang J, Crain K, Fearns C, Kim KA,
Hua KL, Gregory PD, Holmes MC and Torbett BE: Zinc-finger nuclease
editing of human cxcr4 promotes HIV-1 CD4(+) T cell resistance and
enrichment. Mol Ther. 20:849–859. 2002. View Article : Google Scholar
|
|
187
|
Tebas P, Stein D, Tang WW, Frank I, Wang
SQ, Lee G, Spratt SK, Surosky RT, Giedlin MA, Nichol G, et al: Gene
editing of CCR5 in autologous CD4 T cells of persons infected with
HIV. N Engl J Med. 370:901–910. 2014. View Article : Google Scholar
|
|
188
|
Pattanayak V, Ramirez CL, Joung JK and Liu
DR: Revealing off-target cleavage specificities of zinc-finger
nucleases by in vitro selection. Nat Methods. 8:765–770. 2011.
View Article : Google Scholar
|
|
189
|
Hou P, Chen S, Wang S, Yu X, Chen Y, Jiang
M, Zhuang K, Ho W, Hou W, Huang J, et al: Genome editing of CXCR4
by CRISPR/cas9 confers cells resistant to HIV-1 infection. Sci Rep.
5:155772015. View Article : Google Scholar
|
|
190
|
Kaminski R, Chen Y, Fischer T, Tedaldi E,
Napoli A, Zhang Y, Karn J, Hu W and Khalili K: Elimination of HIV-1
genomes from human T-lymphoid cells by CRISPR/Cas9 gene editing.
Sci Rep. 6:225552016. View Article : Google Scholar
|
|
191
|
Berger C, Jensen MC, Lansdorp PM, Gough M,
Elliott C and Riddell SR: Adoptive transfer of effector
CD8+ T cells derived from central memory cells
establishes persistent T cell memory in primates. J Clin Invest.
118:294–305. 2008. View Article : Google Scholar
|
|
192
|
Chapuis AG, Casper C, Kuntz S, Zhu J,
Tjernlund A, Diem K, Turtle CJ, Cigal ML, Velez R, Riddell S, et
al: HIV-specific CD8+ T cells from HIV+ individuals receiving HAART
can be expanded ex vivo to augment systemic and mucosal immunity in
vivo. Blood. 117:5391–5402. 2011. View Article : Google Scholar
|
|
193
|
Hashimoto M and Takemoto T:
Electroporation enables the efficient mRNA delivery into the mouse
zygotes and facilitates CRISPR/Cas9-based genome editing. Sci Rep.
5:113152015. View Article : Google Scholar
|
|
194
|
Chu VT, Weber T, Graf R, Sommermann T,
Petsch K, Sack U, Volchkov P, Rajewsky K and Kühn R: Efficient
generation of Rosa26 knock-in mice using CRISPR/Cas9 in C57BL/6
zygotes. BMC Biotechnol. 16:42016. View Article : Google Scholar
|
|
195
|
Cottle RN, Lee CM, Archer D and Bao G:
Controlled delivery of β-globin-targeting TALENs and CRISPR/Cas9
into mammalian cells for genome editing using microinjection. Sci
Rep. 5:160312015. View Article : Google Scholar
|
|
196
|
Oude Blenke E, Evers MJ, Mastrobattista E
and van der Oost J: CRISPR-Cas9 gene editing: Delivery aspects and
therapeutic potential. J Control Release. 244:139–148. 2016.
View Article : Google Scholar
|
|
197
|
Han X, Liu Z, Jo MC, Zhang K, Li Y, Zeng
Z, Li N, Zu Y and Qin L: CRISPR-Cas9 delivery to hard-to-transfect
cells via membrane deformation. Sci Adv. 1:e15004542015. View Article : Google Scholar
|
|
198
|
D'Astolfo DS, Pagliero RJ, Pras A,
Karthaus WR, Clevers H, Prasad V, Lebbink RJ, Rehmann H and Geijsen
N: Efficient intracellular delivery of native proteins. Cell.
161:674–690. 2015. View Article : Google Scholar
|
|
199
|
Ha JS, Lee JS, Jeong J, Kim H, Byun J, Kim
SA, Lee HJ, Chung HS, Lee JB and Ahn DR: Poly-sgRNA/siRNA
ribonucleoprotein nanoparticles for targeted gene disruption. J
Control Release. 250:27–35. 2017. View Article : Google Scholar
|
|
200
|
Sun W, Ji W, Hall JM, Hu Q, Wang C, Beisel
CL and Gu Z: Self-assembled DNA nanoclews for the efficient
delivery of CRISPR-Cas9 for genome editing. Angew Chem Int Ed Engl.
54:12029–12033. 2015. View Article : Google Scholar
|
|
201
|
Ramakrishna S, Kwaku Dad AB, Beloor J,
Gopalappa R, Lee SK and Kim H: Gene disruption by cell-penetrating
peptide-mediated delivery of Cas9 protein and guide RNA. Genome
Res. 24:1020–1027. 2014. View Article : Google Scholar
|
|
202
|
Chew WL, Tabebordbar M, Cheng JK, Mali P,
Wu EY, Ng AH, Zhu K, Wagers AJ and Church GM: A multifunctional
AAV-CRISPR-Cas9 and its host response. Nat Methods. 13:868–874.
2016. View Article : Google Scholar
|
|
203
|
Long C, Amoasii L, Mireault AA, McAnally
JR, Li H, Sanchez-Ortiz E, Bhattacharyya S, Shelton JM, Bassel-Duby
R and Olson EN: Postnatal genome editing partially restores
dystrophin expression in a mouse model of muscular dystrophy.
Science. 351:400–403. 2016. View Article : Google Scholar
|
|
204
|
Nelson CE, Hakim CH, Ousterout DG, Thakore
PI, Moreb EA, Castellanos Rivera RM, Madhavan S, Pan X, Ran FA, Yan
WX, et al: In vivo genome editing improves muscle function in a
mouse model of Duchenne muscular dystrophy. Science. 351:403–407.
2016. View Article : Google Scholar
|
|
205
|
Gomez EJ, Gerhardt K, Judd J, Tabor JJ and
Suh J: Light-Activated Nuclear translocation of adeno-associated
virus nanoparticles using phytochrome B for enhanced, tunable, and
spatially programmable gene delivery. ACS Nano. 10:225–237. 2016.
View Article : Google Scholar
|
|
206
|
Ran FA, Cong L, Yan WX, Scott DA,
Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, et
al: In vivo genome editing using Staphylococcus aureus Cas9.
Nature. 520:186–191. 2015. View Article : Google Scholar
|
|
207
|
Zuris JA, Thompson DB, Shu Y, Guilinger
JP, Bessen JL, Hu JH, Maeder ML, Joung JK, Chen ZY and Liu DR:
Cationic lipid-mediated delivery of proteins enables efficient
protein-based genome editing in vitro and in vivo. Nat Biotechnol.
33:73–80. 2015. View Article : Google Scholar
|
|
208
|
Wang M, Zuris JA, Meng F, Rees H, Sun S,
Deng P, Han Y, Gao X, Pouli D, Wu Q, et al: Efficient delivery of
genome-editing proteins using bioreducible lipid nanoparticles.
Proc Natl Acad Sci USA. 113:2868–2873. 2016. View Article : Google Scholar
|
|
209
|
Gabriel R, Lombardo A, Arens A, Miller JC,
Genovese P, Kaeppel C, Nowrouzi A, Bartholomae CC, Wang J, Friedman
G, et al: An unbiased genome-wide analysis of zinc-finger nuclease
specificity. Nat Biotechnol. 29:816–823. 2011. View Article : Google Scholar
|
|
210
|
Chiarle R, Zhang Y, Frock RL, Lewis SM,
Molinie B, Ho YJ, Myers DR, Choi VW, Compagno M, Malkin DJ, et al:
Genome-wide translocation sequencing reveals mechanisms of
chromosome breaks and rearrangements in B cells. Cell. 147:107–119.
2011. View Article : Google Scholar
|
|
211
|
Hsu PD, Scott DA, Weinstein JA, Ran FA,
Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al: DNA
targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol.
31:827–832. 2013. View Article : Google Scholar
|
|
212
|
Heigwer F, Kerr G and Boutros M: E-CRISP:
Fast CRISPR target site identification. Nat Methods. 11:122–123.
2014. View Article : Google Scholar
|
|
213
|
Crosetto N, Mitra A, Silva MJ, Bienko M,
Dojer N, Wang Q, Karaca E, Chiarle R, Skrzypczak M, Ginalski K, et
al: Nucleotide-resolution DNA double-strand break mapping by
next-generation sequencing. Nat Methods. 10:361–365. 2013.
View Article : Google Scholar
|
|
214
|
Kim D, Bae S, Park J, Kim E, Kim S, Yu HR,
Hwang J, Kim JI and Kim JS: Digenome-seq: Genome-wide profiling of
CRISPR-Cas9 off-target effects in human cells. Nat Methods.
12:237–243. 2015. View Article : Google Scholar
|
|
215
|
Tsai SQ, Zheng Z, Nguyen NT, Liebers M,
Topkar VV, Thapar V, Wyvekens N, Khayter C, Iafrate AJ, Le LP, et
al: GUIDE-seq enables genome-wide profiling of off-target cleavage
by CRISPR-Cas nucleases. Nat Biotechnol. 33:187–197. 2015.
View Article : Google Scholar
|
|
216
|
Fu Y, Sander JD, Reyon D, Cascio VM and
Joung JK: Improving CRISPR-Cas nuclease specificity using truncated
guide RNAs. Nat Biotechnol. 32:279–284. 2014. View Article : Google Scholar
|
|
217
|
Kim D, Kim S, Kim S, Park J and Kim JS:
Genome-wide target specificities of CRISPR-Cas9 nucleases revealed
by multiplex Digenome-seq. Genome Res. 26:406–415. 2016. View Article : Google Scholar
|
|
218
|
Frock RL, Hu J, Meyers RM, Ho YJ, Kii E
and Alt FW: Genome-wide detection of DNA double-stranded breaks
induced by engineered nucleases. Nat Biotechnol. 33:179–186. 2015.
View Article : Google Scholar
|
|
219
|
Jinek M, Jiang F, Taylor DW, Sternberg SH,
Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S, et al: Structures
of Cas9 endonucleases reveal RNA-mediated conformational
activation. Science. 343:12479972014. View Article : Google Scholar
|
|
220
|
Ran FA, Hsu PD, Lin C-Y, Gootenberg JS,
Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, et
al: Double nicking by RNA-guided CRISPR Cas9 for enhanced genome
editing specificity. Cell. 154:1380–1389. 2013. View Article : Google Scholar
|
|
221
|
McCaffrey J, Sibert J, Zhang B, Zhang Y,
Hu W, Riethman H and Xiao M: CRISPR-CAS9 D10A nickase
target-specific fluorescent labeling of double strand DNA for whole
genome mapping and structural variation analysis. Nucleic Acids
Res. 44. e112016. View Article : Google Scholar
|
|
222
|
Tsai SQ, Wyvekens N, Khayter C, Foden JA,
Thapar V, Reyon D, Goodwin MJ, Aryee MJ and Joung JK: Dimeric
CRISPR RNA-guided FokI nucleases for highly specific genome
editing. Nat Biotechnol. 32:569–576. 2014. View Article : Google Scholar
|
|
223
|
Wyvekens N, Topkar VV, Khayter C, Joung JK
and Tsai SQ: Dimeric CRISPR RNA-guided FokI-dCas9 nucleases
directed by truncated gRNAs for highly specific genome editing. Hum
Gene Ther. 26:425–431. 2015. View Article : Google Scholar
|
|
224
|
Kleinstiver BP, Pattanayak V, Prew MS,
Tsai SQ, Nguyen NT, Zheng Z and Joung JK: High-fidelity CRISPR-Cas9
nucleases with no detectable genome-wide off-target effects.
Nature. 529:490–495. 2016. View Article : Google Scholar
|
|
225
|
Hou Z, Zhang Y, Propson NE, Howden SE, Chu
LF, Sontheimer EJ and Thomson JA: Efficient genome engineering in
human pluripotent stem cells using Cas9 fro Neisseria meningitidis.
Proc Natl Acad Sci USA. 110:15644–15649. 2013. View Article : Google Scholar
|
|
226
|
Walsh RM and Hochedlinger K: A variant
CRISPR-Cas9 system adds versatility to genome engineering. Proc
Natl Acad Sci USA. 110:15514–15515. 2013. View Article : Google Scholar
|
|
227
|
Raza U, Saatci Ö, Uhlmann S, Ansari SA,
Eyüpoğlu E, Yurdusev E, Mutlu M, Ersan PG, Altundağ MK, Zhang JD,
et al: The miR-644a/CTBP1/p53 axis suppresses drug resistance by
simultaneous inhibition of cell survival and epithelial-mesenchymal
transition in breast cancer. Oncotarget. 7:49859–49877. 2016.
View Article : Google Scholar
|
|
228
|
Singh R, Gupta SC, Peng WX, Zhou N,
Pochampally R, Atfi A, Watabe K, Lu Z and Mo YY: Regulation of
alternative splicing of Bcl-x by BC200 contributes to breast cancer
pathogenesis. Cell Death Dis. 7:e22622016. View Article : Google Scholar
|
|
229
|
Engel BJ, Bowser JL, Broaddus RR and
Carson DD: MUC1 stimulates EGFR expression and function in
endometrial cancer. Oncotarget. 7:32796–32809. 2016. View Article : Google Scholar
|
|
230
|
Zhang X, Choi PS, Francis JM, Imielinski
M, Watanabe H, Cherniack AD and Meyerson M: Identification of
focally amplified lineage-specific super-enhancers in human
epithelial cancers. Nat Genet. 48:176–182. 2016. View Article : Google Scholar
|
|
231
|
Kavlashvili T, Jia Y, Dai D, Meng X, Thiel
KW, Leslie KK and Yang S: Inverse relationship between progesterone
receptor and Myc in endometrial cancer. PLoS One. 11:e01489122016.
View Article : Google Scholar
|
|
232
|
Kawamura N, Nimura K, Nagano H, Yamaguchi
S, Nonomura N and Kaneda Y: CRISPR/Cas9-mediated gene knockout of
NANOG and NANOGP8 decreases the malignant potential of prostate
cancer cells. Oncotarget. 6:22361–22374. 2015. View Article : Google Scholar
|
|
233
|
Mazur PK, Herner A, Mello SS, Wirth M,
Hausmann S, Sánchez-Rivera FJ, Lofgren SM, Kuschma T, Hahn SA,
Vangala D, et al: Combined inhibition of BET family proteins and
histone deacet-ylases as a potential epigenetics-based therapy for
pancreatic ductal adenocarcinoma. Nat Med. 21:1163–1171. 2015.
View Article : Google Scholar
|
|
234
|
Zhang Z, Christin JR, Wang C, Ge K, Oktay
MH and Guo W: Mammary-stem-cell-based somatic mouse models reveal
breast cancer drivers causing cell fate dysregulation. Cell Rep.
16:3146–3156. 2016. View Article : Google Scholar
|
|
235
|
Walton J, Blagih J, Ennis D, Leung E,
Dowson S, Farquharson M, Tookman LA, Orange C, Athineos D, Mason S,
et al: CRISPR/Cas9-mediated Trp53 and Brca2 knockout to generate
improved murine models of ovarian high-grade serous carcinoma.
Cancer Res. 76:6118–6129. 2016. View Article : Google Scholar
|
|
236
|
Liang P, Xu Y, Zhang X, Ding C, Huang R,
Zhang Z, Lv J, Xie X, Chen Y, Li Y, et al: CRISPR/Cas9-mediated
gene editing in human tripronuclear zygotes. Protein Cell.
6:363–372. 2015. View Article : Google Scholar
|