Introduction
In the last few years, significant progress has been
made in the field of cancer immunotherapy (1). These advances have resulted in the
approval of agents which target components of the immune system
including cytotoxic T-lymphocyte-associated protein 4 (CTLA-4),
programmed cell death protein-1 (PD-1) and programmed cell death
protein ligand 1 (PD-L1). Although these agents have yielded
encouraging results in a subset of patients, the majority of
patients fail to respond to these therapies. Emerging data have
indicated that the status of the tumor microenvironment (TME) is
critical to the therapeutic outcome. Treatment is most effective in
tumors that have an immunogenic environment (hot tumors) with a
number of neoantigens and a high mutation load. A number of
approaches are being pursued to modulate the TME such as talimogene
laherparepvec, an oncolytic modified herpes simplex virus, which
has been approved for the treatment of melanoma (2,3).
Toll-like receptor (TLR) agonists provide another mechanism for
modulating TME without the inherent infectious risks that are
associated with the use of oncolytic viruses.
It was recognized as early as 1893 that bacterial
extracts injected into human tumors led to a decrease in tumor
burden and occasionally cures (4).
Later, the intratumoral (i.t.) injection of live Bacillus
Calmette-Guérin (BCG) (5) was also
identified to be therapeutically effective (6-10).
In certain cases, the regression of distant (uninjected) metastatic
cancer was also observed in patients following i.t. treatment of
the primary tumor (7,11,12).
This notable therapeutic activity of BCG was identified to reside
mainly in its nucleic acid fraction (13). Later, it was determined that
bacterial nucleic acids contain unmethylated CpG dinucleotides,
which activate immune responses through TLR9.
In total, 10 human TLR subtypes have been identified
to date including TLR9, which is located almost exclusively on the
endosomes of naïve B and dendritic cells. TLR9 is potently
stimulated by DNA (or DNA mimetics) containing unmethylated CpG
synthetic nucleotide sequences assembled to mimic those found in
bacterial DNA (oligodeoxynucleotides or ODNs). Synthetic nucleic
acid agonists of TLR9 signal the clonal expansion and maturation of
plasmacytoid dendritic cells (14–17).
As part of their activation response, plasmacytoid dendritic cells
are able to secrete high levels of interferon (IFN)α (18), which promotes a Th1-type immune
response favoring the activation of cluster of differentiation
(CD)8+ killer T-cells that are crucial for an antitumor
immune response.
Extensive structure-activity association studies of
CpG ODNs have been performed to create optimized TLR9 agonists
(15,19–29).
One such TLR9 agonist, IMO-2125, consists of a phosphorothioate ODN
sequence with three immunostimulatory dinucleotide motifs which
consist of cytosine-7-deazaguanosine (CpG*) (30). Activation of TLR9 by IMO-2125 leads
to the production of Th1-type cytokines and chemokines, including
high levels of IFNα in human peripheral blood mononuclear cell and
plasmacytoid dendritic cell cultures (15). IFNα is a member of the type I class
of interferons that produce the first line of defense against viral
or bacterial infections. We have previously demonstrated the safety
of subcutaneously administered IMO-2125, along with evidence for
clinical activity in a phase I clinical trial in patients with
hepatitis C. IMO-2125 elicited a dose-dependent increase in
circulating IFNα levels (31). In
the present study, a dually implanted syngeneic CT26 colon
carcinoma model was used to demonstrate that the injection of
IMO-2125 directly into one tumor leads to potent tumor regression
of the injected and the uninjected distant tumors by a
CD8+ T-cell directed Th1 response with a long-term
tumor-specific memory.
Materials and methods
Synthesis and purification of
compounds
IMO-2125 is a potent synthetic ODN TLR9 agonist
containing a phosphorothioate backbone with the sequence
5′-TCG*AACG*TTCG*-X-G*CT TG*CAAG*CT-5′ where G* represents
2′-deoxy-7-deaza-guanosine and X is a glycerol linker. A control
oligonucleotide, 'IMO control', is a phosphorothioate ODN,
5′-CTATCTGUC†G*TCCTTCTGU-3′, where G* is
2′-deoxy-7-deaza-guanosine, C† is 2′-deoxy-5-methylcytidine, and
G and U are
2′-O-methylribonucleotides. The two compounds were synthe-sized,
purified and evaluated as described previously (15,30,32).
The synthetic peptides β-galactosidase (β-gal), TPHPARIGL
(representing the naturally processed H-2Ld restricted
epitope spanning amino acids 876-884 of β-gal) and AH1, SPSYVYHQF
(containing a CTL determinant from CT26) were synthesized by New
England Peptide, Inc. (Gardner, MA, USA), to a purity of >99% as
determined using high-performance liquid chromatography (33,34).
Animals and cell lines
A total of 128 female BALB/c mice (6-8-week-old;
17–19 g) were purchased from Jackson Laboratory (Bar Harbor, ME,
USA). All animal studies were approved by the Institutional Animal
Care and Use Committee of Idera Pharmaceuticals, Inc. Mice were
maintained under standard conditions (room temperature, 22±2°C;
relative humidity, 55±10%) on a 12-h light/12-h dark schedule
(lights on at 6:00 a.m.). Animals had ad libitum access to
food and water. All cell lines were obtained from the American Type
Culture Collection (Manassas, VA, USA). CT26 is a
carcinogen-induced undifferentiated syngeneic colon carcinoma
(33). CT26.CL25 is a subclone of
CT26 that has been transduced with the β-gal gene from
Escherichia coli (33). A20
is a murine B-cell lymphoma (35).
Tumor cells were cultured at 37°C in 5% CO2 in RPMI-1640
medium supplemented with 10% heat-inactivated fetal bovine serum
(HyClone; GE Healthcare, Logan, UT, USA), 2 mM L-glutamine, 100
μg/ml streptomycin and 100 U/ml penicillin (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA). CT26. CL25 was
maintained in the culture medium with 400 μg/ml G418 sulfate
(Invitrogen; Thermo Fisher Scientific, Inc.).
Comparative antitumor activity of i.t.
and subcutaneous (s.c.) IMO-2125
BALB/c mice (n=8) were injected subcutaneously with
5×106 A20 lymphoma cells into the right flank. Treatment
was initiated on day 8 when the tumor volume reached ~200
mm3. PBS or IMO-2125 (50 μg in 100 μl PBS,
2.5 mg/kg) was administered to tumor-bearing mice directly into the
implanted tumor (i.t.) or subcutaneously (s.c.) at a site distal to
the tumor graft. Treatment was given on days 8, 11, 13 and 15.
Tumor growth was determined by measuring the long and short
diameters of the tumor using calipers. Tumor volume was calculated
using the formula 0.5 × length × width2.
Antitumor activity and dose response of
IMO-2125
BALB/c mice (n=8) were injected subcutaneously with
2×106 CT26 cells into the right flank and
2×106 CT26.CL25 cells into the left flank. Once the
tumor volume reached between 50 and 150 mm3, treatment
was initiated. IMO control (100 μg) or IMO-2125 (10, 50 or
100 μg) were injected into the CT26 tumor (i.t.) once every
3 days for four doses.
Tumor re-challenge in previously treated
mice
Mice (n=6) from the 100 μg IMO-2125 treatment
group that exhibited complete tumor regression were re-challenged
with a second subcutaneous inoculation of 1×106 CT26
tumor cells into the right and left flanks on day 33. In total, 5
mice survived due to rejection of the CT26 tumor and these mice
were then subcutaneously inoculated with 1×106 A20 cells
on day 73 in the upper back area. Age-matched naïve BALB/c mice
inoculated with 1×106 A20 (n=5) served as naïve
inoculation controls.
Depletion of CD4+ and/or
CD8+ cells
Naïve BALB/c mice (two groups, n=8 each group) or
T-cell-depleted BALB/c mice (three groups, n=5 each group,
CD4+-depleted, CD8+-depleted or
CD4+/CD8+-co-depleted) were injected
subcutaneously with 2×106 CT26 cells into the right
flank and 2×106 CT26. CL25 cells into the left flank, on
study day 0. CD4+ and/or CD8+ T-cells were
depleted by intraperitoneal injection of 25 mg/kg (500 μg)
anti-mouse CD4 monoclonal antibody (mAb) (clone GK1.5; cat. no.
BE0003-1), anti-mouse CD8 mAb (clone YTS 169.4; cat. no. BE0117)
(both from BioXcell, West Lebanon, NH, USA), or anti-CD4 and
anti-CD8 mAb together on days −1, 5 and 12. Antibodies were diluted
to 2.5 mg/ml in PBS and 200 μl was administered per mouse.
The depletion conditions were validated by flow cytometry of
peripheral blood exhibiting complete depletion of CD4+
and CD8+ T-cells. Treatment with IMO-2125 or IMO control
was initiated on day 6 when the tumor volume ranged between 100 and
250 mm3. IMO-2125 or IMO control (50 μg) was
injected i.t into the right-flank CT26 tumor on days 6, 10 and
13.
Flow cytometry
Fresh peripheral blood cells and single cell
suspensions of splenocytes were preblocked by mouse Fc blocker
(anti-mouse CD16/CD32 antibody, 1:50 dilution; cat. no. 553142; BD
Biosciences, San Jose, CA, USA) then stained with fluorescently
labeled anti-CD4 (1:200 dilution; cat. no. 553049) and/or anti-CD8
antibody (1:200 dilution, cat. no. 553035) (both from BD
Biosciences). The stained peripheral blood cells and splenocytes
were fixed/hemolysed and extensively washed prior to being analyzed
using a BD Accuri flow cytometer. The flow cytometry data analysis
was performed using FlowJo software (version 10; FlowJo LLC,
Ashland, OR, USA).
T-cell responses by enzyme-linked
immunospot (ELISPOT) assay
Splenocytes from individual tumor-bearing mouse were
prepared on day 28 from 50 μg IMO-2125 or IMO control groups
(n=3). T-cells were purified using T-cell enrichment columns
(R&D Systems, Inc., Minneapolis, MN, USA). Purified T-cells
(2.5×105) were stimulated with 2.5×105
mitomycin C (50 μg/ml)-treated syngeneic spleen cells pulsed
with medium, 100 μg/ml AH1 peptide (SPSYVYHQF) or β-gal
peptide (TPHPARIGL) for 24 h. The frequencies of T-cells
specifically responsive to AH1 epitope in CT26 and to AH1 and β-gal
epitopes in CT26.CL25 were determined in duplicate using an IFNγ
ELISPOT assay (R&D Systems, Inc.), according to the
manufacturer's protocol. Spots were enumerated with an automated
ELISpot reader system (Zellnet, Port Lee, NJ, USA).
Immunohistochemistry of
tumor-infiltrating CD3+ T-cells
Injected and uninjected contralateral tumor samples
were collected from mice on day 28. Formalin-fixed
paraffin-embedded sections from injected and distant tumors were
immunohistochemically stained for mouse T-cells by the Mass
Histology Service (Worcester, MA, USA). Rabbit monoclonal
anti-mouse CD3 antibody (clone SP7; cat. no. ab16669; Abcam,
Cambridge, MA, USA) was used as primary antibody (1:200 dilution)
and a horseradish peroxidase system was used to detect the signal.
Sections from mouse spleen were used as positive controls.
CD3+ cells in the tumor tissues were examined under a
microscope, and counted in high-power fields (HPF; magnification,
×400). A minimum of 10 HPF was counted to generate a mean value per
animal.
Gene expression analysis
BALB/c mice (four groups, n=5/group) were
subcutaneously inoculated on the flanks with 3×106 CT26
cells. On day 8, a single i.t. injection on the right flank of PBS,
250 μg IMO control, 50 μg IMO-2125 or 250 μg
IMO-2125 was delivered. Tumor nodules from injected and distant
sites were collected on day 10 and were evaluated for the
expression of indoleamine 2,3-dioxygenase (IDO)-1, IDO-2,
programmed cell death protein ligand 1 (PD-L1), programmed cell
death protein-1 (PD-1), carcinoembryonic antigen-related cell
adhesion molecule-1 (CEACAM-1), tumor necrosis factor receptor
superfamily member 4 (OX40), B- and T-lymphocyte attenuator, T-cell
immunoglobulin and mucin-domain-containing-3 protein (TIM-3),
lymphocyte-activation gene 3 (LAG-3), cytotoxic
T-lymphocyte-associated protein 4 (CTLA-4) and OX40 ligand (OX40L).
Total RNA was extracted using TRIzol® reagent (Thermo
Fisher Scientific, Inc.), according to the manufacturer's protocol.
Equal amounts of RNA were reverse-transcribed using a High-Capacity
cDNA Reverse Transcription kit (Applied Biosystems; Thermo Fisher
Scientific, Inc.). cDNAs were then amplified using the quantitative
polymerase chain reaction (qPCR) using TaqMan® gene
expression assays (Applied Biosystems; Thermo Fisher Scientific,
Inc.). Relative gene expression was analyzed using the
2−ΔΔCq method (36).
The amplification cycles were performed using a QuantStudio™ 12K
Flex Real-Time PCR system (Applied Biosystems; Thermo Fisher
Scientific, Inc.) as follows: 95°C for 20 sec, followed by 40
cycles of 95°C for 1 sec and 60°C for 20 sec. The housekeeping gene
peptidyl-prolyl isomerase (Ppib; probe Mm00478295_m1 Ppib
VIC®; Thermo Fisher Scientific, Inc.) was used as an
internal control for normalization of each sample.
Statistical analysis
Statistical analysis was performed using unpaired
two-tailed or one-tailed (checkpoint gene expression) Student's
t-test for comparisons between two groups or a one-way analysis of
variance (ANOVA) followed by either Dunnett's or Tukey's multiple
comparison test was used in studies comparing more than two groups.
The survival data were compared using a log-rank (Mantel-Cox) test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Comparison of i.t. and s.c. IMO-2125
treatment of syngeneic A20 lymphoma
The i.t. and s.c. routes of delivery were compared
in the A20 syngeneic tumor model bearing single tumor grafts on
their right flank. Tumor dimensions were measured over time and the
resulting tumor volumes are presented in Fig. 1A (i.t. PBS control), Fig. 1B (s.c. IMO-2125) and Fig. 1C (i.t. IMO-2125). On day 18, the
mean tumor volumes were identified to be significantly different
from each other using ANOVA (P<0.0001) followed by Tukey's
multiple comparison test: PBS vs. i.t. IMO-2125 (P<0.0001); PBS
vs. s.c. IMO-2125 (P<0.016) and s.c. IMO-2125 vs. i.t. IMO-2125
(P=0.002). s.c. IMO-2125 (50 μg) exhibited a minimal
antitumor effect (2/8-treated mice) over time compared with PBS
controls. However, tumor growth inhibition was observed over time
in all 8 animals following i.t. treatment with an equal 50
μg dose of IMO-2125. The number of tumor-infiltrating
lymphocytes (TILs) was assessed using CD3 immunohistochemistry and
representative micro-graphs from PBS controls (Fig. 1D), s.c. IMO-2125-treated mice
(Fig. 1E) and i.t. IMO-2125
treated mice (Fig. 1F). The
delivery of IMO-2125 by the i.t. route resulted in an increased
number of CD3+ TILs into the tumor compared with the
s.c. route or i.t. PBS control (Fig.
1G).
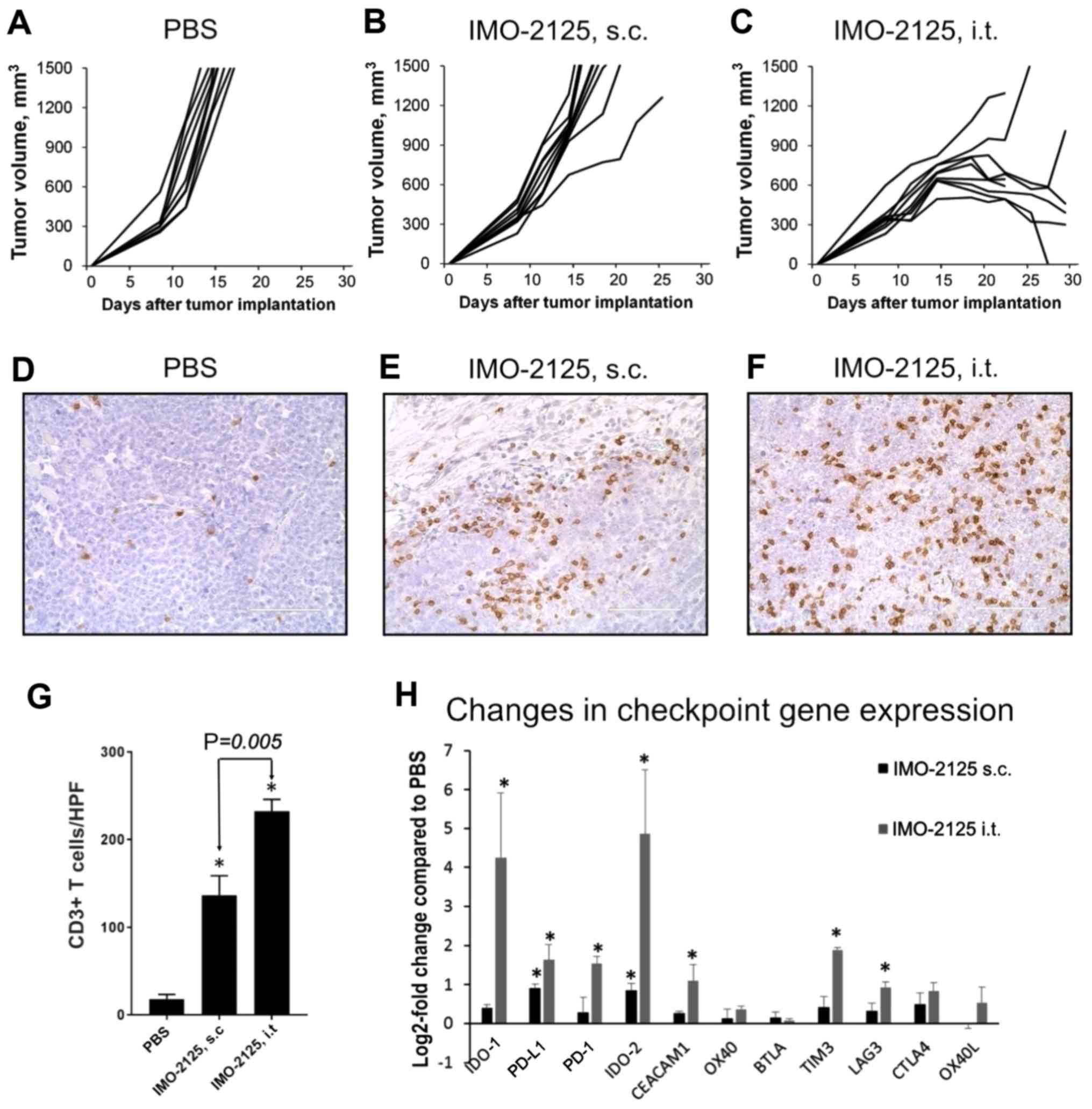 | Figure 1Comparative antitumor activity of
i.t. and s.c. IMO-2125 administration. Three groups of BALB/c mice
(n=9) bearing A20 lymphoma grafts (~200 mm3) on their
right flank were injected with (A) i.t. PBS, (B) 50 μg s.c.
IMO-2125 or (C) 50 μg i.t. IMO-2125 on days 8, 11, 13 and
15, and tumor volumes were determined to day 30. On day 18, the
mean tumor volumes were significantly different from each other by
analysis of variance (P<0.0001) followed by Tukey's multiple
comparison test: PBS vs. i.t. IMO-2125 (P<0.0001); PBS vs. s.c.
IMO-2125 (P<0.016) and s.c. IMO-2125 vs. i.t. IMO-2125
(P=0.002). Tumors were processed, sectioned and stained to detect
the CD3+ T-cells. T-cells staining positively for CD3
antigen are indicated by punctate brown staining in representative
A20 tumor sections from mice injected with (D) i.t. PBS, (E) 50
μg s.c. IMO-2125 or (F) 50 μg i.t. IMO-2125. Scale
bar, 100 μm. (G) CD3+ T-cell staining.
*P<0.05 vs. PBS group. (H) Alterations in checkpoint
gene expression. *P<0.05 vs. s.c. IMO-2125. i.t.,
intratumoral; s.c., subcutaneous; CD, cluster of differentiation;
HPF, high-power field; IDO, indoleamine 2,3-dioxygenase; PD-L1,
programmed cell death protein ligand 1; PD-1, programmed cell death
protein-1; CEACAM1, carcinoembryonic antigen-related cell adhesion
molecule 1; OX40, tumor necrosis factor receptor superfamily member
4; BTLA, B- and T-lymphocyte attenuator; TIM3, T-cell
immunoglobulin and mucin-domain-containing 3 protein; LAG3,
lymphocyte-activation gene 3; CTLA, cytotoxic
T-lymphocyte-associated protein 4; OX40L, OX40 ligand. |
Comparison of changes in checkpoint gene
expression in i.t or s.c. injected A20 lymphoma
Compared with systemic s.c. administration, i.t.
IMO-2125 resulted in significantly increased modulation of
checkpoint gene expression (Fig.
1H). The fold increase in gene expression in A20 tumors (50
μg group) from i.t. treated mice compared with s.c. treated
mice was 3.8 for IDO-1, 3.1 for IDO-2, 1.4 for PD-L1, 2.0 for PD-1,
1.6 for CEACAM-1, 1.2 for OX40, 2.0 for TIM-3, 1.5 for LAG-3, 1.2
for CTLA-4 and 1.6 for OX40L.
i.t. IMO-2125 treatment leads to abscopal
effects
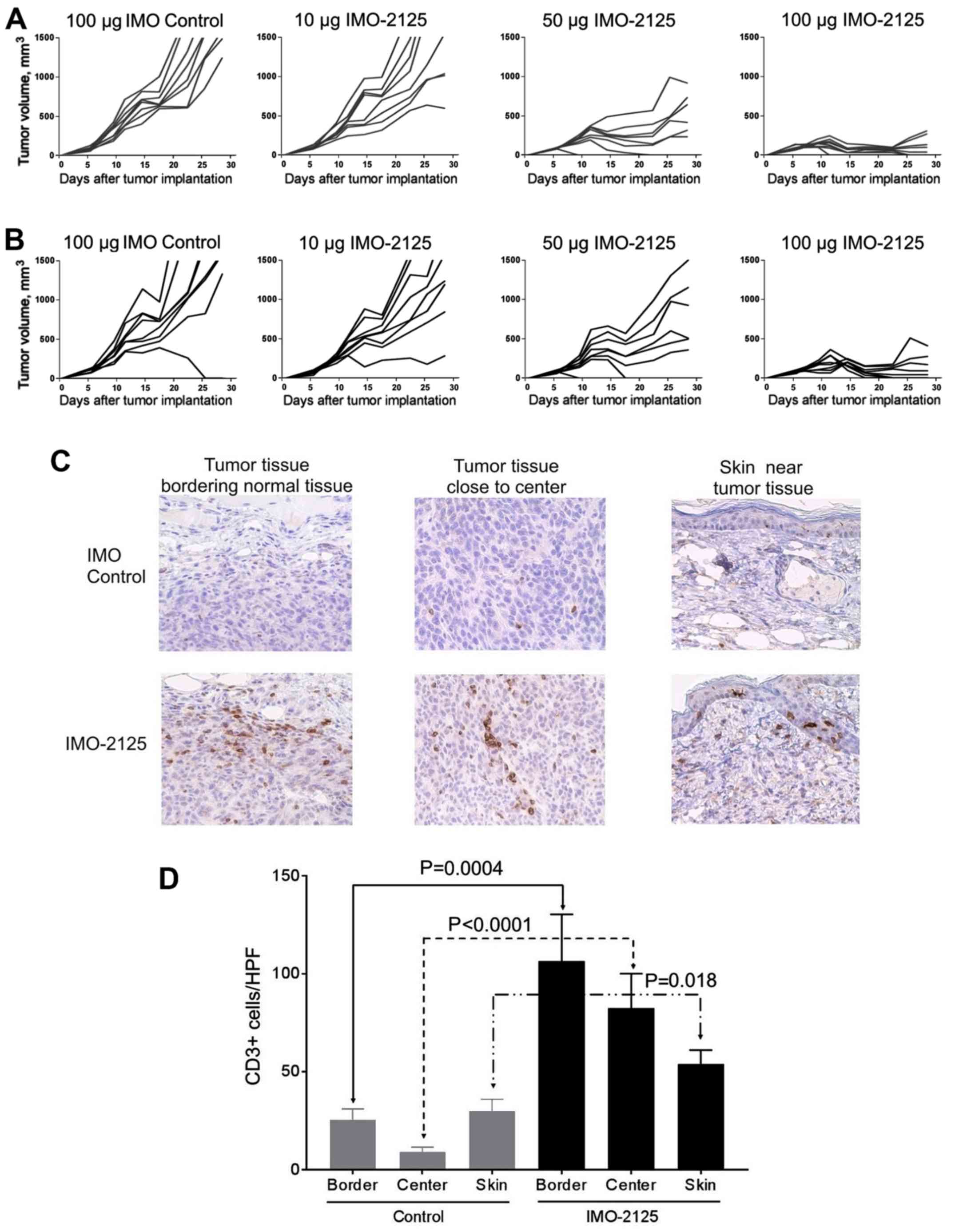
IMO-2125 injected directly (i.t.) into right flank
CT26 syngeneic colon tumor grafts of mice also bearing uninjected
left flank CT26. CL25 tumor grafts on days 5, 8, 11 and 14 resulted
in a significant growth suppression of the two tumors in a
dose-dependent manner (Fig. 2A and
B). A one-way ANOVA conducted to compare the mean tumor volumes
on day 28 in the treatment groups with the IMO control group
demonstrated a significant difference between groups. Tumor growth
of the injected CT26 tumor was inhibited 15.4, 79.6 and 94.7% at
respective doses of 10 (0.5 mg/kg), 50 (2.5 mg/kg) and 100 (5
mg/kg) μg IMO-2125 compared with IMO control on day 28. The
growth of uninjected CT26.CL25 tumors, were similarly inhibited 18,
65.6 and 93.2% at doses of 10, 50 and 100 μg, respectively,
compared with control group on day 28. Treated and distant tumors
injected with 50 and 100 μg IMO-2125 exhibited a significant
decrease in tumor growth (P<0.01) compared with the tumors in
IMO control group. The degree of tumor growth inhibition was
dose-dependent, and complete ablation (<100 mm3
consisting mostly of scar tissue) of the injected and uninjected
tumors was observed in none of the 8 mice in the IMO control group,
none of the 8 mice in the 10-μg dose-group, 2 of 8 mice in
the 50-μg dose-group and 5 of 8 mice in the 100-μg
dose-group on day 28. Tumor volumes for the injected (Fig. 2A) and uninjected contralateral
(Fig. 2B) tumors of each
individual animal are presented. The five mice with ablated tumors
in the 100-μg dose group were used in subsequent
re-challenge experiments to determine tumor-specific immune memory.
All other mice were sacrificed on day 28 and selected tumors from
each group were used for CD3 immunohistochemical staining
studies.
i.t. IMO-2125 leads to increased
CD3+ T-cell infiltration
The presence of TILs was determined by
immunohistochemical staining of CD3+ T-cells in tumors
treated with IMO control and 50 μg i.t. IMO-2125. In total,
three samples were taken from each tumor: The edge of the tumor
bordering normal muscle tissue, central tumor tissue and tumor
tissue near the skin. A limited number of CD3+ cells was
observed from samples taken from the i.t. IMO control group
(Fig. 2C, upper panels). In
contrast, mice treated i.t with IMO-2125 exhibited a marked
increase in the number of CD3+ T-cells in the tumor
tissue from all three regions (Fig.
2D), indicating that the anti-tumor activity of IMO-2125 was
associated with the increased infiltration of TILs. Representative
micrographs of uninjected CT26.CL25 tumor samples are presented in
the lower panels of Fig. 2C.
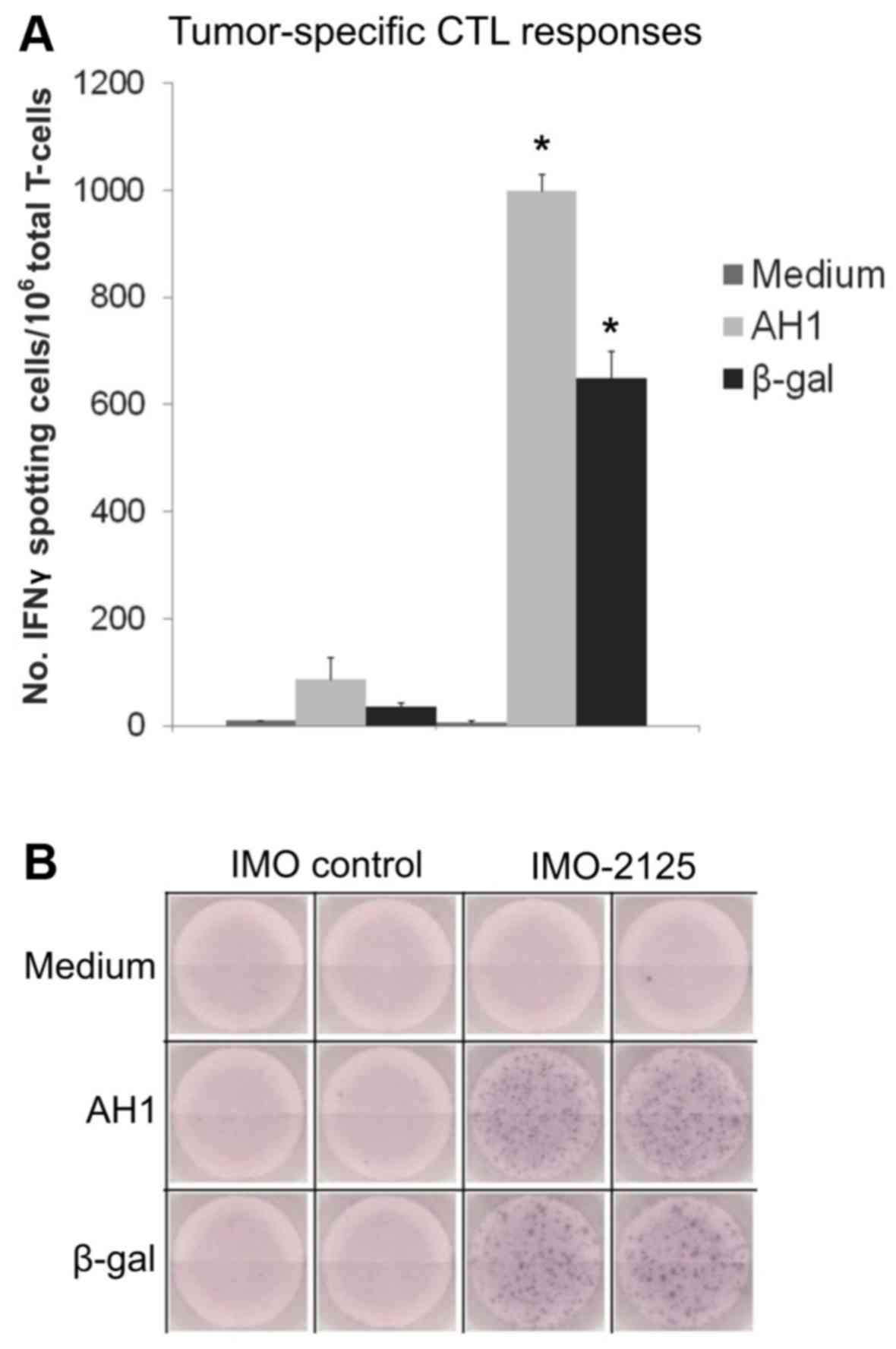
i.t. IMO-2125 induces cytotoxic T-cell
responses against treated and distant tumors
The splenic T-cell responses to tumor antigens (AH1
and β-gal) were determined in mice from the IMO control or 50
μg IMO-2125 treatment groups on day 28. The tumor internal
antigen (AH1, present in CT26 and CT26.CL25) and β-gal (present
only in CT26.CL25) were used as challenge peptide antigens in the
ELISPOT assay. In total, 3 mice in each of IMO control or 50
μg IMO-2125 treatment groups were sacrificed on day 28.
T-cells isolated from splenocytes were stimulated with mitomycin
C-treated syngeneic spleen cells pulsed with medium, AH1 or β-gal
peptide for 24 h. The number of T-cells responding to antigen
challenge in the IMO-2125-treated and IMO control mice are
quantified in Fig. 3A, and
representative images of ELISPOT plates indicating IFNγ-positive
spots are presented in Fig. 3B.
i.t. IMO-2125 in the single CT26 tumor induced significant
increases in tumor antigen-specific IFNγ-secreting effector cell
responses against the CT26 endogenous antigen, AH1, and also
against the β-gal antigen that existed in the distant CT26.CL25
tumor. In contrast, IMO control-treated mice displayed negligible
T-cell responses to either of the peptide challenges
(P<0.001).
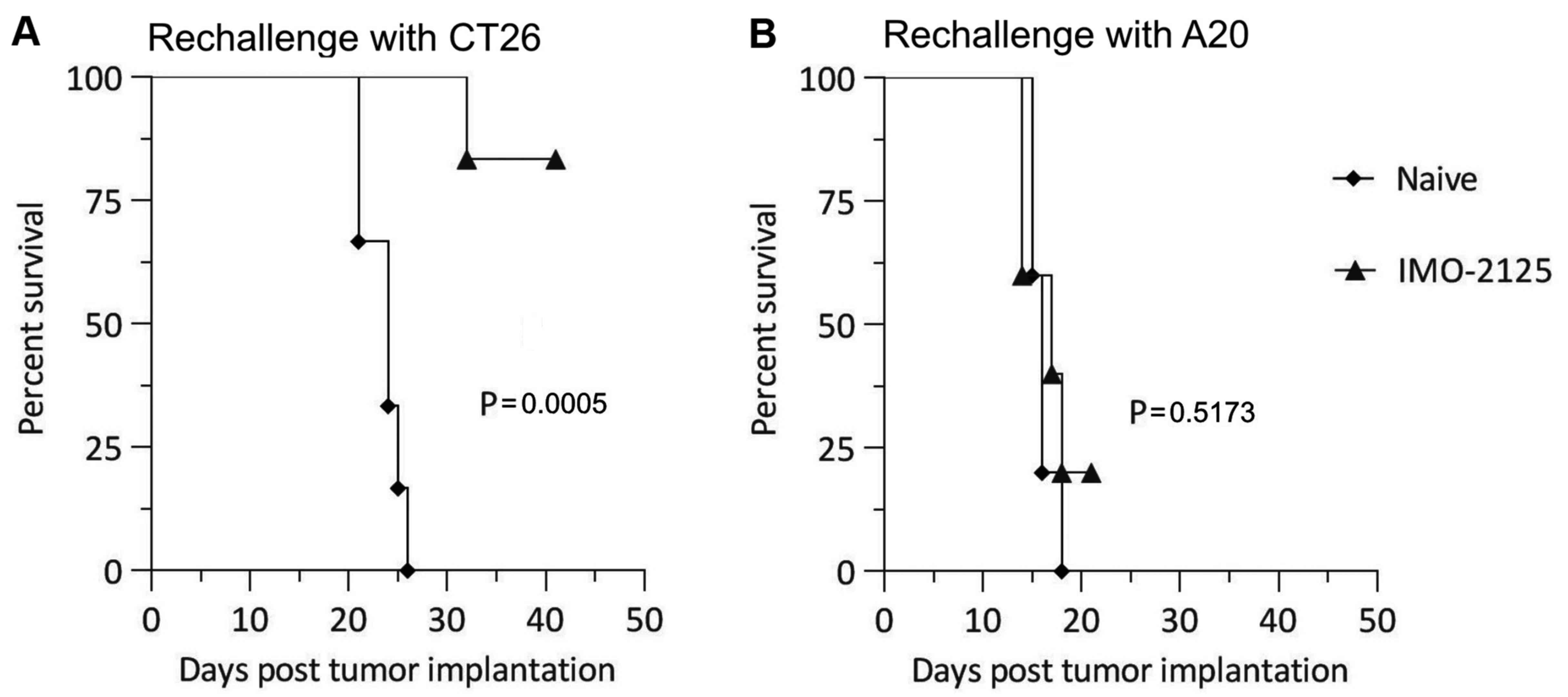
Treatment with IMO-2125 induces a durable
tumor-specific response
Animals from the high-dose group that exhibited
complete tumor regression following 100 μg i.t. IMO-2125
treatment (n=5) were re-implanted with 1×106 CT26 tumor
cells on day 33 to determine the duration of T-cell memory.
Age-matched naïve BALB/c mice inoculated with 1×106 CT26
cells (n=5) were used as controls. IMO-2125-treated tumor-free mice
developed tumor-specific protections and rejected certain tumor
(CT26) re-challenge (Fig. 4A).
However, such immune memory was tumor-specific and the same mice
that rejected CT26 implantation were not protected from syngeneic
non-organ-associated A20 lymphoma challenge (Fig. 4B).
Antitumor activity of IMO-2125 is
dependent on CD8+ T-cells
The association between IMO-2125-mediated anti-tumor
activity and the requirement of CD4+ and CD8+
T-cells was investigated in groups of mice depleted of
CD4+ and/or CD8+ T-cells by intraperitoneal
injection of anti-mouse CD4 mAb or anti-mouse CD8 mAb.
Fluorescence-activated cell sorting analysis of spleen cells was
used to demonstrate the effective depletion of CD4+
lymphocytes and CD8+ lymphocytes (Fig. 5A). All mice exhibited CT26 tumors
on their right flank and CT26.CL25 tumors on their left flank. In
non-T-cell-depleted mice, IMO-2125 administered i.t. into the right
flank CT26 tumor resulted in a robust inhibition of tumor growth in
CT26 and CT26.CL25 tumors compared with IMO control. No inhibition
of tumor growth was observed by i.t. IMO-2125 treatment in mice
depleted of CD8+ T-cells in either of their tumor
grafts, suggesting that CD8+ T-cells are required for
the antitumor activity of IMO-2125. Interestingly, depletion of
CD4+ T-cells resulted in a more robust antitumor
response to IMO-2125 treatment compared with IMO-2125 treatment in
naïve mice (Fig. 5B), possibly due
to the removal of regulatory T-cells that are inhibitory to
antitumor immune response.
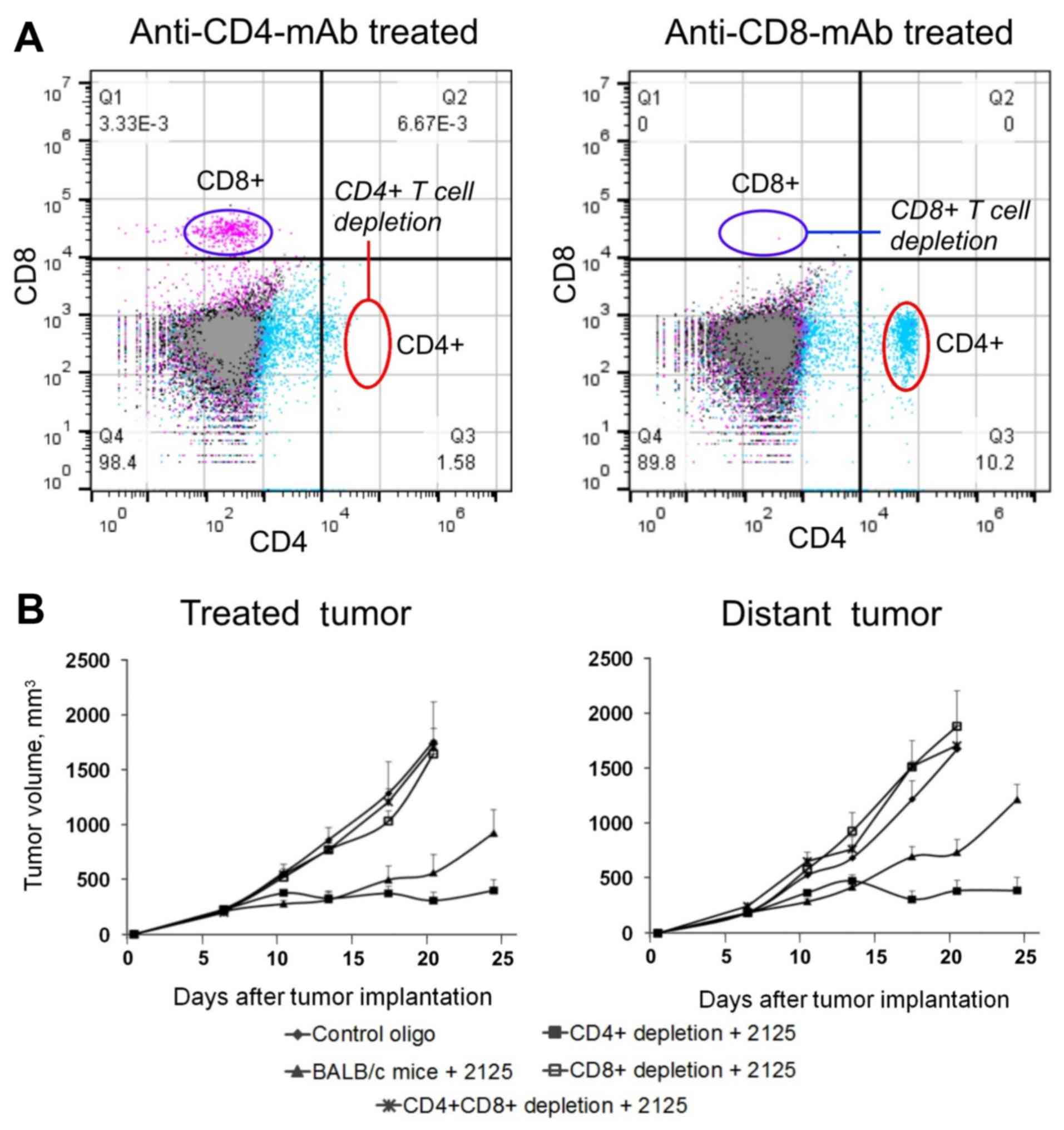 | Figure 5Antitumor activity of i.t. IMO-2125
is dependent on CD8+ T-cells. Mice with bilateral CT26
tumor grafts were injected i.t. into the right flank tumor with 50
μg IMO control or IMO-2125 on days 6, 10 and 13.
CD4+ or CD8+, or CD4+ and
CD8+ T lymphocytes were depleted by the intraperitoneal
administration of 25 mg/kg anti-CD4 or anti-CD8 antibodies. (A)
Depletion by anti-CD4 (left) or anti-CD8 (right) monoclonal
antibodies was validated by flow cytometry. (B) Tumor volumes (mean
± standard error of the mean) from mice treated with IMO control
(diamonds, n=8), IMO-2125 (triangles, n=8), CD4 depletion and
IMO-2125 (filled squares, n=5), CD8 depletion and IMO-2125 (open
squares, n=5), or CD4 and CD8 depletion, and IMO-2125 (crosses,
n=5) in injected (left) and uninjected contralateral tumors
(right). i.t., intratumoral; CD, cluster of differentiation; oligo,
oligonucleotide. |
IMO-2125 treatment leads to modulation of
checkpoint gene expression
Immune checkpoint gene expression levels were
evaluated using qPCR in mice bearing subcutaneous CT26 tumors
following a single i.t. injection of either 50 μg or 250
μg IMO-2125, or 250 μg IMO control 8 days after tumor
inoculation. Mice treated with IMO-2125 exhibited slower tumor
growth in the injected and uninjected contralateral CT26 tumors
that was evident even 2 days after IMO-2125 injection (on day 10)
(Fig. 6A). Mice were sacrificed
and the tumors were removed for immune checkpoint gene expression
studies on day 10 (Fig. 6A). The
checkpoint gene expression of IDO-1, PD-L1, CEACAM-1 and OX40 in
cDNA preparations from excised tumors was determined by qPCR as
presented in Fig. 6B (n=3). A
dose-dependent increase in IDO-1, PD-L1 and CEACAM1 gene expression
was observed, although it was not statistically significant. OX40
was not altered 48 h after a single dose. The lack of statistical
significance is possibly due to tumor tissue sampling.
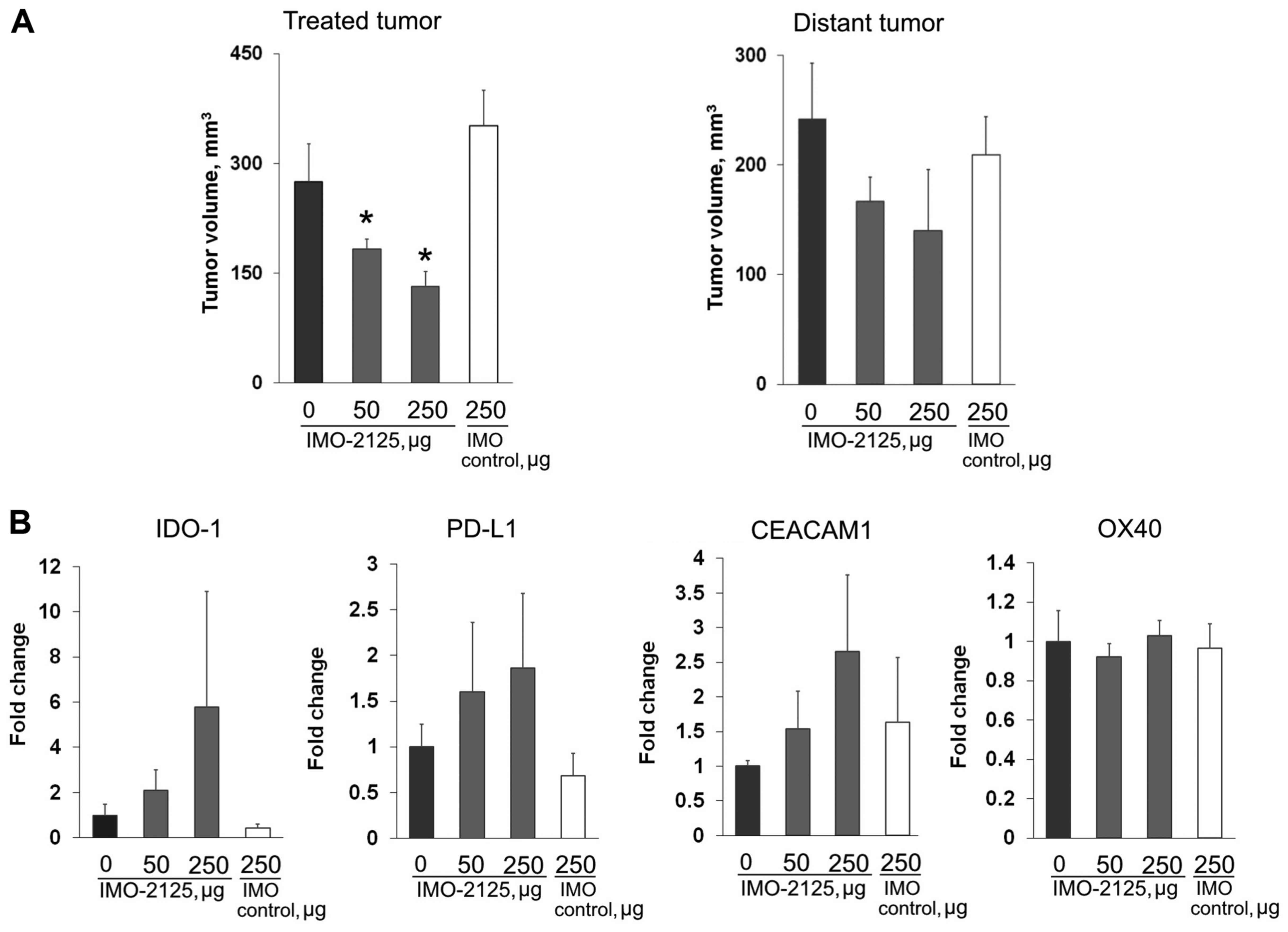 | Figure 6Treatment with i.t. IMO-2125 leads to
modulation of immune checkpoint gene expression. (A) Tumor volumes
on day 10 of bilaterally implanted mice bearing CT26 tumor grafts
following a single i.t. injection on day 8 into the right flank
tumor with PBS, 50 μg IMO-2125 (2.5 mg/kg), 250 μg
IMO-2125 (12.5 mg/kg) or 250 μg IMO control in the injected
tumor on the right flank (left) or the uninjected tumor on the left
flank (right). Tumor volumes are presented as the mean ± SEM (n=5,
each group). *P<0.05 vs. PBS. (B) Alterations in
checkpoint gene expression from uninjected tumors are depicted as
the mean ± SEM fold change over IMO control levels for IDO-1,
PD-L1, CEACAM-1 and OX40. i.t., intratumoral; SEM, standard error
of the mean; IDO, indoleamine 2,3-dioxygenase; PD-L1, programmed
cell death protein ligand 1; CEACAM1, carcinoembryonic
antigen-related cell adhesion molecule 1; OX40, tumor necrosis
factor receptor superfamily member 4. |
Discussion
IMO-2125 is a potent synthetic oligonucleotide
designed to activate TLR9 by initiating a strong Th1-polarized
immune response. In the present study, it was identified that the
direct i.t. administration of IMO-2125 into murine A20 lymphoma
tumor grafts led to increased antitumor activity compared with
equal doses administered subcutaneously. This observation is
consistent with the hypothesis that immunostimulatory drugs target
immune cells in the TME and prime the immune system to recognize
cancer cells as foreign. TLR9 is located on the endosomes of
plasmacytoid dendritic cells and B-cells that respond to IMO-2125
stimulation through the induction of Th1 cytokines including
interleukin-12, IFNα and IFNγ (15,30,31).
Mature dendritic cells located in the TME act as sentries to
recognize bacterial and viral DNA and may be key cells that
initiate the receptor mediated immune response to i.t. injection of
IMO-2125. Clinical trials are currently being conducted using i.t.
IMO-2125 in patients with cancer (clinicaltrials.gov identifiers NCT03052205,
NCT03445533 and NCT02644967).
IMO-2125 is composed of two 11-mer phosphorothioate
oligonucleotides, covalently linked via their 3′-ends, which each
contain three appropriately placed CpG* motifs (G* representing
7-deazaguanosine). The resulting palindromic sequence form
double-stranded secondary structures with 5′ overhangs on each end.
This unique structure facilitates the dimerization of the compound
and thereby potentiates the activation of the Th1 pathway
associated with increased IFNα induction (15,30,31).
The i.t. delivery of IMO-2125 to a single tumor site
led to systemic antitumor responses at uninjected distal tumors.
Thus, targeting IMO-2125 directly to the TME by i.t injection
resulted in systemic efficacy. This delivery route allows IMO-2125
to act as a danger signal within a local tumor compartment,
triggering antigen recognition by antigen-presenting cells, and
causing the host immune system to recognize and attack the injected
tumor. In the present study, a syngeneic colon carcinoma model with
the CT26 colon graft on one flank and a CT26.CL25 subclone
engineered to express an additional antigenic fragment of β-gal on
the opposite flank was used. The i.t. administration of IMO-2125 to
the CT26 graft on one flank resulted in potent and equivalent
antitumor activity at the injected CT26 graft as well as the
uninjected CT26.CL25 distal tumor in a dose-dependent manner.
Immunohistochemical staining of CD3+, a
marker of infiltrating T lymphocytes, exhibited increased densities
of CD3+ T-cells in the central and peripheral regions of
injected CT26 and uninjected CT26.CL25 tumor grafts compared with
IMO controls. Depletion of CD8+ T lymphocytes with
CD8-specific antibodies abrogated the antitumor response, whereas
depletion of CD4+ lymphocytes with CD4-specific
antibodies did not, suggesting that IMO-2125 primarily elicited a
CD8+ T-cell-mediated response indicative of Th1
polarization. Interestingly, the treatment of
CD4+-depleted mice with IMO-2125 led to an antitumor
response that was more marked compared with that observed in
IMO-2125-treated mice with no T lymphocyte depletion, suggesting
that CD4+ regulatory T lymphocytes may negatively
modulate the immunotherapeutic response of IMO-2125.
ELISPOT assays indicated that T-cells purified from
the spleens of dually implanted CT26/CT26.CL25 mice responded
equally well to AH1 or β-gal antigens, even though the β-gal
antigen was only expressed in the uninjected CT26. CL25 subclone.
Thus, a clonal expansion of T-cells responsive to the two
tumor-specific antigens (AH1 expressed in the two tumors and β-gal
expressed only in the CT26.CL25 subclone) occurred in response to
i.t. IMO-2125 administration. These results further suggested that
the i.t. injection of IMO-2125 was able to initiate antigen
spreading by cells present in the injected and the uninjected
tumors. Activated splenic T-cells were quantified in the ELISPOT
assay by their release of IFNγ upon stimulation with AH1 or β-gal.
IFNγ is also an important cytokine in the Th1 response that is
reported to lead to the M1 polarization of immunostimulatory
macrophages rather than the M2 immunosuppressive macrophages
(37), so it is possible that
IMO-2125 supports M1 macrophage polarization directed against tumor
antigens.
IMO-2125 has been identified to induce surface
marker expression and the activation of human dendritic cells and
B-cells, as well as increased levels of IFNα in vitro and
in vivo (15,30). In clinical trials, IMO-2125
elicited a potent dose-dependent increase in systemic IFNα and
other Th1-type cytokines in patients with hepatitis C at doses that
were well-tolerated following systemic administration (31). Interferons are known to interact
with specific cellular receptors, which promote the production of
second messengers leading to expression of immune modulatory genes.
Type I interferons activate the majority of immune cells including
macrophages, dendritic cells, B-cells and T-cells to suppress
cancer. They are also able to upregulate the expression of tumor
antigens (38,39), stimulate the presentation of
antigens by dendritic cells (40,41),
and promote optimal effector function of both CD8+
T-cells and natural killer cells (42,43).
Immune checkpoints are ligand-receptor pairs that
feedback signals to properly balance the immune response to rid the
body of abnormal cells while protecting normal cells from an
autoimmune attack. However, neoplastic progression and
transformation often involve the epigenetic modification of genes
that allow cancer cells to escape immune surveillance (44,45).
The therapeutic approach to cancer treatment has expanded to
include drugs that reverse the immune tolerance that cancer cells
develop. In the last decade, immunotherapy has been demonstrated to
improve the survival of a subset of patients with advanced cancers.
The US Food and Drug Administration-approved ipilimumab
(CTLA-4-blocking antibody), nivolumab and pembrolizumab
(PD-1-blocking antibodies), and atezolizumab and durvalumab
(PD-L1-blocking antibodies) have led to favorable outcomes in a
subset of patients (46–51). It has become increasingly evident
that the TME must be in a receptive state for cancer immunotherapy
to be successful (52). IMO-2125
may provide the stimulus to create a more receptive TME and thereby
improve the efficacy of immune checkpoint inhibitors currently
approved and in clinical trials (53,54).
Immune checkpoints protect against excessive
activation though a feedback loop and are therefore upregulated
during an inflammatory response. For example, the checkpoint
inhibitors CTLA-4, PD-1, PD-L1 and IDO-1 negatively modulate the
immune system's ability to attack cancerous tumors allowing for
their continuous growth and potential metastasis (55). Previous studies have identified
increased expression levels of CTLA-4 and PD-1 in TILs of patients
with metastatic melanoma compared with those of healthy donors
(56). Other studies have also
demonstrated that activation of TLR9 by CpG ODN agonists leads to
the expression of IDO-1 in dendritic cells (57,58).
In the present study, it was identified that i.t. IMO-2125
treatment increased the levels of mRNA for various immune
checkpoint inhibitors including PD-1, PD-L1 and CEACAM-1 above IMO
control-treated levels in A20 and CT26 tumors. In addition,
increased CTLA-4 gene expression levels in the A20 tumor 20 days
after initiation of IMO-2125 treatment was also identified. The
upregulation of checkpoint gene expression levels following i.t.
IMO-2125 treatment suggests that IMO-2125 co-administered with
checkpoint inhibitors may lead to additive or synergistic antitumor
activity compared with monotherapies. Clinical trials are currently
in progress evaluating i.t. IMO-2125 monotherapy and IMO-2125 in
combination with ipilimumab, an anti-CTLA-4 antibody, or in
combination with pembrolizumab, an anti-PD-1 antibody, in patients
previously treated for metastatic melanoma (clinicaltrials.gov identifiers NCT03052205,
NCT03445533 and NCT02644967).
In summary, these studies suggest that IMO-2125 is a
potent synthetic TLR9 agonist that activates TLR9 on B-cells and
dendritic cells in the TME to initiate and potentiate a
Th1-polarized local and systemic immune response when administered
by i.t. injection. The i.t. injection of IMO-2125 to a single tumor
site led to systemic and equivalent antitumor responses at
uninjected contralateral tumors suggesting that treatment with
IMO-2125 may effectively treat primary and metastatic disease when
injected into one tumor lesion. Indeed, in an ongoing trial of i.t.
IMO-2125 in combination with ipilimumab, certain patients exhibited
responses in injected and metastatic lesions (59). The stimulation of TLR9 by IMO-2125
by the i.t. route of administration led to a tumor-specific immune
response in the injected and uninjected tumors requiring
CD8+, but not CD4+, T lymphocytes. The
immunostimulatory modulation of IMO-2125 was accompanied by an
upregulation of immune checkpoint genes including IDO-1, PD-L1,
CEACAM-1 and CTLA-4, suggesting that feedback inhibition by these
checkpoints may negatively modulate the immunostimulatory effects
of IMO-2125. Thus, although IMO-2125 exhibited potent antitumor
activity as a monotherapy, the co-administration of approved immune
checkpoint inhibitors with IMO-2125 may provide additive or
synergistic antitumor efficacy.
A phase I/II clinical trial of i.t. IMO-2125 in
combination with ipilimumab or pembrolizumab in patients with
anti-PD-1 refractory melanoma is ongoing (NCT02644967), and
encouraging evidence of changes in immune markers and clinical
activity has been observed for the ipilimumab combination (59,60).
A phase III trial is in progress (NCT03445533). In addition, a
phase I trial of i.t. IMO-2125 monotherapy in refractory solid
tumors (NCT03052205) is also ongoing.
Acknowledgments
The authors thank Jillian DiMuzio (Idera
Pharmaceuticals, Inc., Cambridge, MA, USA) and Evren K. Argon
(Idera Pharmaceuticals, Inc.) for assistance with qPCR analysis,
Katherine Worsham (Idera Pharmaceuticals, Inc.) for editing and
administrative assistance, and Carol A. Nelson (Translational
Pharmacology Consulting, Westford, MA, USA) for assistance with the
manuscript preparation.
References
|
1
|
Alexander W: The Checkpoint Immunotherapy
Revolution: What started as a trickle has become a flood, despite
some daunting adverse effects; New drugs, indications, and
combinations continue to emerge. PT. 41:185–191. 2016.
|
|
2
|
Fountzilas C, Patel S and Mahalingam D:
Review: Oncolytic virotherapy, updates and future directions.
Oncotarget. 8:102617–102639. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yin J, Markert JM and Leavenworth JW:
Modulation of the intratumoral immune landscape by oncolytic herpes
simplex virus virotherapy. Front Oncol. 7:1362017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Coley WB: The treatment of malignant
tumors by repeated inoculations of erysipelas. With a report of ten
original cases 1893 Clin Orthop Relat Res. 262:3–11. 1991.
|
|
5
|
Zbar B and Tanaka T: Immunotherapy of
cancer: Regression of tumors after intralesional injection of
living Mycobacterium bovis. Science. 172:271–273. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cohen MH, Jessup JM, Felix EL, Weese JL
and Herberman RB: Intralesional treatment of recurrent metastatic
cutaneous malignant melanoma: A randomized prospective study of
intralesional Bacillus Calmette-Guerin versus intralesional
dinitrochloro-benzene. Cancer. 41:2456–2463. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Morton DL, Eilber FR, Holmes EC, Hunt JS,
Ketcham AS, Silverstein MJ and Sparks FC: BCG immunotherapy of
malignant melanoma: Summary of a seven-year experience. Ann Surg.
180:635–643. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Silverstein MJ, DeKernion J and Morton DL:
Malignant melanoma metastatic to the bladder. Regression following
intratumor injection of BCG vaccine. JAMA. 229:6881974. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Krown SE, Hilal EY, Pinsky CM, Hirshaut Y,
Wanebo HJ, Hansen JA, Huvos AG and Oettgen HF: Intralesional
injection of the methanol extraction residue of Bacillus
Calmette-Guerin (MER) into cutaneous metastases of malignant
melanoma. Cancer. 42:2648–2660. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bier J, Kleinschuster S, Bier H and Rapp
H: Intratumor immunotherapy with BCG cell wall preparations:
Development of a new therapy approach for head-neck tumors. Arch
Otorhinolaryngol. 236:245–255. 1982.In German. View Article : Google Scholar
|
|
11
|
Bast RC Jr, Zbar B, Borsos T and Rapp HJ:
BCG and cancer (first of two parts). N Engl J Med. 290:1413–1420.
1974. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bast RC Jr, Zbar B, Borsos T and Rapp HJ:
BCG and cancer. N Engl J Med. 290:1458–1469. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shimada S, Yano O, Inoue H, Kuramoto E,
Fukuda T, Yamamoto H, Kataoka T and Tokunaga T: Antitumor activity
of the DNA fraction from Mycobacterium bovis BCG. II. Effects on
various syngeneic mouse tumors. J Natl Cancer Inst. 74:681–688.
1985.PubMed/NCBI
|
|
14
|
Yamamoto S, Yamamoto T, Nojima Y, Umemori
K, Phalen S, McMurray DN, Kuramoto E, Iho S, Takauji R, Sato Y, et
al: Discovery of immunostimulatory CpG-DNA and its application to
tuberculosis vaccine development. Jpn J Infect Dis. 55:37–44.
2002.PubMed/NCBI
|
|
15
|
Yu D, Kandimalla ER, Bhagat L, Tang JY,
Cong Y, Tang J and Agrawal S: 'Immunomers' - novel 3′-3′-linked CpG
oligodeoxyribonucleotides as potent immunomodulatory agents.
Nucleic Acids Res. 30:4460–4469. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hemmi H, Takeuchi O, Kawai T, Kaisho T,
Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, et al:
A Toll-like receptor recognizes bacterial DNA. Nature. 408:740–745.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Krieg AM, Yi AK, Matson S, Waldschmidt TJ,
Bishop GA, Teasdale R, Koretzky GA and Klinman DM: CpG motifs in
bacterial DNA trigger direct B-cell activation. Nature.
374:546–549. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mandl JN, Barry AP, Vanderford TH, Kozyr
N, Chavan R, Klucking S, Barrat FJ, Coffman RL, Staprans SI and
Feinberg MB: Divergent TLR7 and TLR9 signaling and type I
interferon production distinguish pathogenic and nonpathogenic AIDS
virus infections. Nat Med. 14:1077–1087. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao Q, Temsamani J, Iadarola PL, Jiang Z
and Agrawal S: Effect of different chemically modified
oligodeoxynucleotides on immune stimulation. Biochem Pharmacol.
51:173–182. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kandimalla ER, Yu D, Zhao Q and Agrawal S:
Effect of chemical modifications of cytosine and guanine in a
CpG-motif of oligonucleotides: Structure-immunostimulatory activity
relationships. Bioorg Med Chem. 9:807–813. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu D, Zhao Q, Kandimalla ER and Agrawal S:
Accessible 5′-end of CpG-containing phosphorothioate
oligodeoxynucleotides is essential for immunostimulatory activity.
Bioorg Med Chem Lett. 10:2585–2588. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kandimalla ER, Bhagat L, Yu D, Cong Y,
Tang J and Agrawal S: Conjugation of ligands at the 5′-end of CpG
DNA affects immunostimulatory activity. Bioconjug Chem. 13:966–974.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kandimalla ER, Bhagat L, Wang D, Yu D, Zhu
FG, Tang J, Wang H, Huang P, Zhang R and Agrawal S: Divergent
synthetic nucleotide motif recognition pattern: Design and
development of potent immunomodulatory oligodeoxyribonucleotide
agents with distinct cytokine induction profiles. Nucleic Acids
Res. 31:2393–2400. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu D, Kandimalla ER, Zhao Q, Bhagat L,
Cong Y and Agrawal S: Requirement of nucleobase proximal to CpG
dinucleotide for immunostimulatory activity of synthetic CpG DNA.
Bioorg Med Chem. 11:459–464. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang D, Li Y, Yu D, Song SS, Kandimalla ER
and Agrawal S: Immunopharmacological and antitumor effects of
second-generation immunomodulatory oligonucleotides containing
synthetic CpR motifs. Int J Oncol. 24:901–908. 2004.PubMed/NCBI
|
|
26
|
Damiano V, Caputo R, Bianco R, D'Armiento
FP, Leonardi A, De Placido S, Bianco AR, Agrawal S, Ciardiello F
and Tortora G: Novel toll-like receptor 9 agonist induces epidermal
growth factor receptor (EGFR) inhibition and synergistic antitumor
activity with EGFR inhibitors. Clin Cancer Res. 12:577–583. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Damiano V, Caputo R, Garofalo S, Bianco R,
Rosa R, Merola G, Gelardi T, Racioppi L, Fontanini G, De Placido S,
et al: TLR9 agonist acts by different mechanisms synergizing with
bevacizumab in sensitive and cetuximab-resistant colon cancer
xenografts. Proc Natl Acad Sci USA. 104:12468–12473. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Damiano V, Garofalo S, Rosa R, Bianco R,
Caputo R, Gelardi T, Merola G, Racioppi L, Garbi C, Kandimalla ER,
et al: A novel toll-like receptor 9 agonist cooperates with
trastuzumab in trastuzumab-resistant breast tumors through multiple
mechanisms of action. Clin Cancer Res. 15:6921–6930. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rosa R, Melisi D, Damiano V, Bianco R,
Garofalo S, Gelardi T, Agrawal S, Di Nicolantonio F, Scarpa A,
Bardelli A, et al: Toll-like receptor 9 agonist IMO cooperates with
cetuximab in K-ras mutant colorectal and pancreatic cancers. Clin
Cancer Res. 17:6531–6541. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yu D, Putta MR, Bhagat L, Dai M, Wang D,
Trombino AF, Sullivan T, Kandimalla ER and Agrawal S: Impact of
secondary structure of toll-like receptor 9 agonists on interferon
alpha induction. Antimicrob Agents Chemother. 52:4320–4325. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rodriguez-Torres M, Ghalib RH, Gordon SC
and McHutchison JG: IMO-2125, a TLR9 agonist, induces immune
responses which correlate with reductions in viral load in null
responder HCV patients. Hepatology. 52:336A2010.
|
|
32
|
Kandimalla ER, Bhagat L, Wang D, Yu D,
Sullivan T, La Monica N and Agrawal S: Design, synthesis and
biological evaluation of novel antagonist compounds of Toll-like
receptors 7, 8 and 9. Nucleic Acids Res. 41:3947–3961. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang M, Bronte V, Chen PW, Gritz L,
Panicali D, Rosenberg SA and Restifo NP: Active immunotherapy of
cancer with a nonreplicating recombinant fowlpox virus encoding a
model tumor-associated antigen. J Immunol. 154:4685–4692.
1995.PubMed/NCBI
|
|
34
|
Huang AY, Gulden PH, Woods AS, Thomas MC,
Tong CD, Wang W, Engelhard VH, Pasternack G, Cotter R, Hunt D, et
al: The immunodominant major histocompatibility complex class
I-restricted antigen of a murine colon tumor derives from an
endogenous retroviral gene product. Proc Natl Acad Sci USA.
93:9730–9735. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Touitou V, Daussy C, Bodaghi B, Camelo S,
de Kozak Y, Lehoang P, Naud MC, Varin A, Thillaye-Goldenberg B,
Merle-Béral H, et al: Impaired th1/tc1 cytokine production of
tumor-infiltrating lymphocytes in a model of primary intraocular
B-cell lymphoma. Invest Ophthalmol Vis Sci. 48:3223–3229. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−ΔΔC(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
37
|
Duluc D, Corvaisier M, Blanchard S, Catala
L, Descamps P, Gamelin E, Ponsoda S, Delneste Y, Hebbar M and
Jeannin P: Interferon-gamma reverses the immunosuppressive and
protumoral properties and prevents the generation of human
tumor-associated macrophages. Int J Cancer. 125:367–373. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Greiner JW, Hand PH, Noguchi P, Fisher PB,
Pestka S and Schlom J: Enhanced expression of surface
tumor-associated antigens on human breast and colon tumor cells
after recombinant human leukocyte alpha-interferon treatment.
Cancer Res. 44:3208–3214. 1984.PubMed/NCBI
|
|
39
|
Boyer CM, Dawson DV, Neal SE, Winchell LF,
Leslie DS, Ring D and Bast RC Jr: Differential induction by
interferons of major histocompatibility complex-encoded and
non-major histo-compatibility complex-encoded antigens in human
breast and ovarian carcinoma cell lines. Cancer Res. 49:2928–2934.
1989.PubMed/NCBI
|
|
40
|
Schiavoni G, Mattei F and Gabriele L: Type
I interferons as stimulators of DC-mediated cross-priming: Impact
on anti-tumor response. Front Immunol. 4:4832013. View Article : Google Scholar
|
|
41
|
Joffre OP, Segura E, Savina A and
Amigorena S: Cross-presentation by dendritic cells. Nat Rev
Immunol. 12:557–569. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nguyen KB, Salazar-Mather TP, Dalod MY,
Van Deusen JB, Wei XQ, Liew FY, Caligiuri MA, Durbin JE and Biron
CA: Coordinated and distinct roles for IFN-alpha beta, IL-12, and
IL-15 regulation of NK cell responses to viral infection. J
Immunol. 169:4279–4287. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Herberman RB, Ortaldo JR, Rubinstein M and
Pestka S: Augmentation of natural and antibody-dependent
cell-mediated cytotoxicity by pure human leukocyte interferon. J
Clin Immunol. 1:149–153. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu M, Zhou J, Chen Z and Cheng AS:
Understanding the epigenetic regulation of tumours and their
microenvironments: Opportunities and problems for epigenetic
therapy. J Pathol. 241:10–24. 2017. View Article : Google Scholar
|
|
45
|
Dunn J and Rao S: Epigenetics and
immunotherapy: The current state of play. Mol Immunol. 87:227–239.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ott PA, Hodi FS and Robert C: CTLA-4 and
PD-1/PD-L1 blockade: New immunotherapeutic modalities with durable
clinical benefit in melanoma patients. Clin Cancer Res.
19:5300–5309. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mahoney KM, Freeman GJ and McDermott DF:
The next immune-checkpoint inhibitors: PD-1/PD-L1 blockade in
melanoma. Clin Ther. 37:764–782. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Postow MA, Callahan MK and Wolchok JD:
Immune checkpoint blockade in cancer therapy. J Clin Oncol.
33:1974–1982. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Khalil DN, Smith EL, Brentjens RJ and
Wolchok JD: The future of cancer treatment: Immunomodulation, CARs
and combination immunotherapy. Nat Rev Clin Oncol. 13:273–290.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Serra-Bellver P, Valpione S and Lorigan P:
Sequential immunotherapy regimens-expect the unexpected. Lancet
Oncol. 17:854–855. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Weber JS, Gibney G, Sullivan RJ, Sosman
JA, Slingluff CL Jr, Lawrence DP, Logan TF, Schuchter LM, Nair S,
Fecher L, et al: Sequential administration of nivolumab and
ipilimumab with a planned switch in patients with advanced melanoma
(CheckMate 064): An open-label, randomised, phase 2 trial. Lancet
Oncol. 17:943–955. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Devaud C, John LB, Westwood JA, Darcy PK
and Kershaw MH: Immune modulation of the tumor microenvironment for
enhancing cancer immunotherapy. OncoImmunology. 2:e259612013.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Arpaia N and Barton GM: Toll-like
receptors: Key players in antiviral immunity. Curr Opin Virol.
1:447–454. 2011. View Article : Google Scholar
|
|
54
|
Iwasaki A and Medzhitov R: Toll-like
receptor control of the adaptive immune responses. Nat Immunol.
5:987–995. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
55
|
Pardoll DM: The blockade of immune
checkpoints in cancer immunotherapy. Nat Rev Cancer. 12:252–264.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ahmadzadeh M, Johnson LA, Heemskerk B,
Wunderlich JR, Dudley ME, White DE and Rosenberg SA: Tumor
antigen-specific CD8 T cells infiltrating the tumor express high
levels of PD-1 and are functionally impaired. Blood. 114:1537–1544.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Mellor AL, Baban B, Chandler PR, Manlapat
A, Kahler DJ and Munn DH: Cutting edge: CpG oligonucleotides induce
splenic CD19+ dendritic cells to acquire potent
indoleamine 2,3-dioxygenase-dependent T cell regulatory functions
via IFN Type 1 signaling. J Immunol. 175:5601–5605. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Fallarino F and Puccetti P: Toll-like
receptor 9-mediated induction of the immunosuppressive pathway of
tryptophan catabolism. Eur J Immunol. 36:8–11. 2006. View Article : Google Scholar
|
|
59
|
Diab A, Haymaker C, Uemura M, Murthy R,
James M, Geib J, Cornfeld M, Swann S, Yee C, Wargo J, et al: A
phase 1/2 trial of intratumoral (i.t.) IMO-2125 (IMO) in
combination with checkpoint inhibitors (CPI) in PD-(L)1-refractory
melanoma. Ann Oncol. 28(Suppl 5): 1187P2017. View Article : Google Scholar
|
|
60
|
Haymaker C, Uemura M, Murthy R, James M,
Wang D, Brevard J, Swann S, Geib J, Cornfeld M, Chunduru S, et al:
Translational evidence of reactivated innate and adaptive immunity
with intratumoral IMO-2125 in combination with systemic checkpoint
inhibitors from a phase I/II study in patients with anti-PD-1
refractory metastatic melanoma. Cancer Res. 77(Suppl 13): 56522017.
View Article : Google Scholar
|