Introduction
Each year, >273,000 new cases of malignant
nephrotic tumors are reported worldwide, accounting for ~2% of all
cases of cancer (1), among which
>85% of cases are renal cell carcinomas (RCCs). RCC is
considered the most life-threatening type of common urinary cancer
and, despite significant progress in RCC treatment in previous
years, 25–30% of patients continue to present with metastases.
Patients with metastatic RCC have a poor prognosis, the median
survival rate of which can be <1 year (2). Surgical resection remains the primary
treatment for RCC, however, 30–40% of patients continue to present
with recurrence or disease progression following surgery (3). The majority of RCC treatment failures
are due to radiation not being an ideal treatment option and
traditional chemotherapy not always being effective. Therefore, it
is critical to identify novel therapeutic strategies and more
effective agents to treat RCC, and to improve patient survival
rates and quality of life.
Parthenolide (PTL) is a sesquiterpene lactone
compound obtained from Tanacetum parthenium (feverfew).
Owing to the anti-inflammatory activity of PTL, it has been used
worldwide for the treatment of migraine and rheumatoid arthritis
for many years (4). Several
studies have shown that PTL can inhibit the activity of nuclear
factor (NF)-κB, and it has been shown in cell and animal
experiments to inhibit the expression of proinflammatory cytokines
(5,6). One study reported on the PTL-based
treatment of RCC by inhibiting the activity of NF-κB (7). On investigating the inhibition of
NF-κB activity, it was observed that PTL compounds and their
derivatives are promising therapeutic agents for the treatment of
different inflammatory-related diseases. It has also been reported
that PTL can inhibit cell proliferation, promote apoptosis and
enhance the anticancer effect of certain drugs in various types of
human cancer cell in vitro, including non-small cell lung
cancer, colon cancer, pancreatic cancer, acute myelogenous leukemia
and cervical cancer (8–12). Owing to the instability of PTL in
acidic and alkaline conditions (13), a structurally-related compound,
epoxymicheliolide (EMCL), has been developed; however, whether EMCL
can treat cancer, and its anticancer mechanism, remain to be fully
elucidated.
NF-κB is an important class of transcription factor,
which has a wide range of biological effects through downstream
target genes, including proliferation, metastasis and apoptosis
(7,14). There is increasing evidence that
the NF-κB signaling pathway is the central coordinator for cancer
(15). In tumors, mainly through
internal or external factors, an elevated NF-κB activity is
established, thereby enhancing tumor proliferation, migration and
invasion (16). The NF-κB
signaling pathway has been investigated in several malignancies,
including RCC (17). In addition,
it has been shown that RCC can be treated by inhibiting activation
of the NF-κB signaling pathway (18).
The inflammatory antitumor response is important in
inhibiting tumor growth in human malignancies (19). Cyclooxygenase-2 (COX-2) is a
rate-limiting enzyme for the synthesis of prostaglandins from
arachidonic acid, which is regulated by NF-κB (20). COX-2 regulates the formation of
carcinogens, assists in tumor progression, and inhibits apoptosis,
angiogenesis and the metastatic process (21). COX-2 is commonly unregulated in
various types of human cancer, including RCC (22–24).
In vitro, in vivo, observational and clinical data
have demonstrated that selective COX-2 inhibitors are effective in
preventing proliferation, angiogenesis and inflammation, and
inducing apoptosis in human cancer cells (5,14,25).
These findings indicate that COX-2 may be a useful target for the
treatment of RCC in chemotherapeutic strategies.
In the present study, the mechanism of EMCL in
inhibiting the expression of COX-2 was investigated, and it was
revealed that EMCL inhibited the NF-κB/COX-2 signaling pathways by
targeting leucine 21 and leucine 25 in inhibitor of NF-κB (IκB)
kinase β (IKKβ) in RCC cells.
Materials and methods
Chemicals and reagents
EMCL and pyromellitic acid (PMA) were purchased from
Nanjing Spring & Autumn Biological Engineering Co., Ltd.
(Nanjing, China). Ammonium pyrrolidine dithiocarbamate (PDTC) and
celecoxib were purchased from Sigma-Aldrich; EMD Millipore
(Billerica, MA, USA). All reagents were dissolved in dimethyl
sulfoxide (DMSO) as the initial concentrate and diluted with medium
prior to use; the final concentration of DMSO was <0.1%. The
pGL3-NF-κB plasmid, which contained four NF-κB binding motifs
(GGGACTTTCC), pGL3-Basic plasmid, and pRL-TK plasmid were purchased
from GenePharma (Suzhou, China). Anti-cyclin B1 (cat. no. 4138),
anti-cyclin E1 (cat. no. 4129), anti-cyclin-dependent kinase 2
(CDK2) (cat. no. 2546), anti-cyclin D1 (cat. no. 2922),
anti-E-cadherin (cat. no. 3195), anti-vimentin (cat. no. 12826),
anti-N-cadherin (cat. no. 4061), anti-B-cell lymphoma 2 (Bcl-2)
(cat. no. 15071), anti-Bcl-2-associated X protein (Bax) (cat. no.
2774), anti-cleaved caspase-3 (cat. no. 9664), anti-cleaved
caspase-9 (cat. no. 7237), anti-cleaved poly (ADP-ribose)
polymerase (PARP) (cat. no. 5625), anti-COX-2 (cat. no. 12282),
anti-phosphorylated (p)-IKKβ (cat. no. 2697), anti-IKKβ (cat. no.
2684), anti-p-IκBα (cat. no. 2859), anti-IκBα (cat. no. 9242),
anti-p65 (cat. no. 8242), anti-p-p65 (cat. no. 3033), histone H3
(cat. no. 4499) and anti-β-actin (cat. no. 3700) antibodies were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Anti-cytochrome c (cat. no. sc-13561), and anti-p50 (cat.
no. sc-81710) antibodies were purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). Anti-matrix
metalloproteinase (MMP)-2 (cat. no. ab86607), anti-MMP-9 (cat. no.
ab76003) and anti-tissue inhibitor of metalloproteinase (TIMP)-2
(cat. no.ab180630) were purchased from Abcam (Cambridge, MA,
USA).
Cell culture
The 786-0, Caki-1 and A498 human RCC cell lines were
obtained from the American Type Culture Collection (Manassas, VA,
USA). The 786-0, Caki-1 and A498 cells were cultured in RPMI-1640
medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
McCoy's 5a modified medium (Gibco; Thermo Fisher Scientific, Inc.),
and Dulbecco's modified Eagle's medium (Gibco; Thermo Fisher
Scientific, Inc.), respectively, with 10% fetal bovine serum (FBS;
Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin and 100
ng/ml streptomycin. The cells were cultured at 37°C in a humidified
atmosphere with 5% CO2. The authenticity of all cell
lines was verified through the genomic short tandem repeat profile
by Shanghai ZhongQiao Xin Zhou Biotechnology Co., Ltd. (Shanghai,
China) and the cell lines were confirmed to be free of mycoplasma
using a Mycoplasma Detection Kit-Quick Test (Biotool, Houston, TX,
USA).
Cell Counting Kit-8 (CCK-8) assay
Cell viability was measured using a CCK-8 assay
(DojindoMolecular Laboratories, Inc., Kumamoto, Japan). Briefly,
3×103 cells were counted and seeded into 96-well
flat-bottomed plates in 100 µl of complete medium, allowed
to adhere overnight, and then treated with various concentrations
of EMCL (0, 5, 10, 15, 20, 30, 50, 100 and 200 µM) for 24
and 48 h at 37°C. CCK-8 (10 µl) was added to each well, and
the absorbance was measured following incubation for 2 h at 37°C.
Each experiment was repeated at least three times.
Colony formation assay
The human RCC cells were treated with various
concentrations of EMCL (0, 5, 10 and 20 µM) for 24 h, and
then trypsinized to form single-cell suspensions, and seeded into
6-well plates (800 cells/well). Following incubation at 37°C in 5%
CO2 for 14 days until colonies were large enough to be
visualized, the cells were washed with phosphate-buffered saline
(PBS) and fixed in methanol/glacial acetic/ddH2O (1:1:8)
for 10 min and stained with 0.1% crystal violet for 30 min. The
colonies were then photographed using a Canon camcera (PowerShot
SX620 HS).
Wound-healing assay
Wound-healing assays (scratch assays) were performed
to detect cell migration. Briefly, the 786-0 and Caki-1 cells were
grown to full confluence in 6-well culture plates. Following 6 h of
serum starvation, the confluent cell monolayer was scraped with a
sterile 10-µl pipette tip and treated with the indicated
doses of EMCL. After 24 h, the wound gap was observed and images
were captured.
Cell invasion assay
The human RCC cells were treated with various
concentrations of EMCL (0, 5, 10 and 20 µM) for 24 h.
According to the manufacturer's protocol, Transwell filters were
precoated with 70 µl of 1.1 mg/ml Matrigel (BD Biosciences,
Franklin Lakes, NJ, USA), following inoculation of 105
cells per pore in 200 µl cell sap. Culture medium (600
µl) with 10% FBS was added into the lower chamber and
cultured in a CO2 incubator. After 24 h, the lower
chamber cells were fixed in methanol/glacial
acetic/ddH2O (1:1:8) for 10 min and stained with 0.1%
crystal violet for 30 min. Three independent experiments were
performed in triplicate.
Fabrication of the microfluidic chip and
in vitro cell migration/invasion assay
The microfluidic system was fabricated on a
micro-patterned polydimethylsiloxane (PDMS; Sylgard 184; Dow
Chemical, Midland, MI, USA) chip using the standard soft
lithographic method, as described in our previous study (26). As shown in Fig. 3D, the microfluidic system consisted
of a glass coverslip, a central gel micro-channel (9,000×1,000×40
µm), and two lateral media micro-channels (6,000×1,000×40
µm), which were separated by two arrays of micro-columns
(200×50×40 µm). The gap between each micro-column was
50×20×40 µm. To quantify the invasive capability of the
cancer cells, the gel micro-channels were precoated with Matrigel
(BD Biosciences). The human RCC cells (5×106/ml;3
µl) pre-treated with varying concentrations of EMCL (0, 5,
10 and 20 µM) for 24 h containing 10% FBS medium were seeded
into the cell micro-channel, and medium containing 20% FBS was
added as a chemoattractant in the medium micro-channel. The chip
was cultured in a CO2 incubator for 24 h, following
which images were captured using a Leica DM14000B microscope fitted
with digital camera.
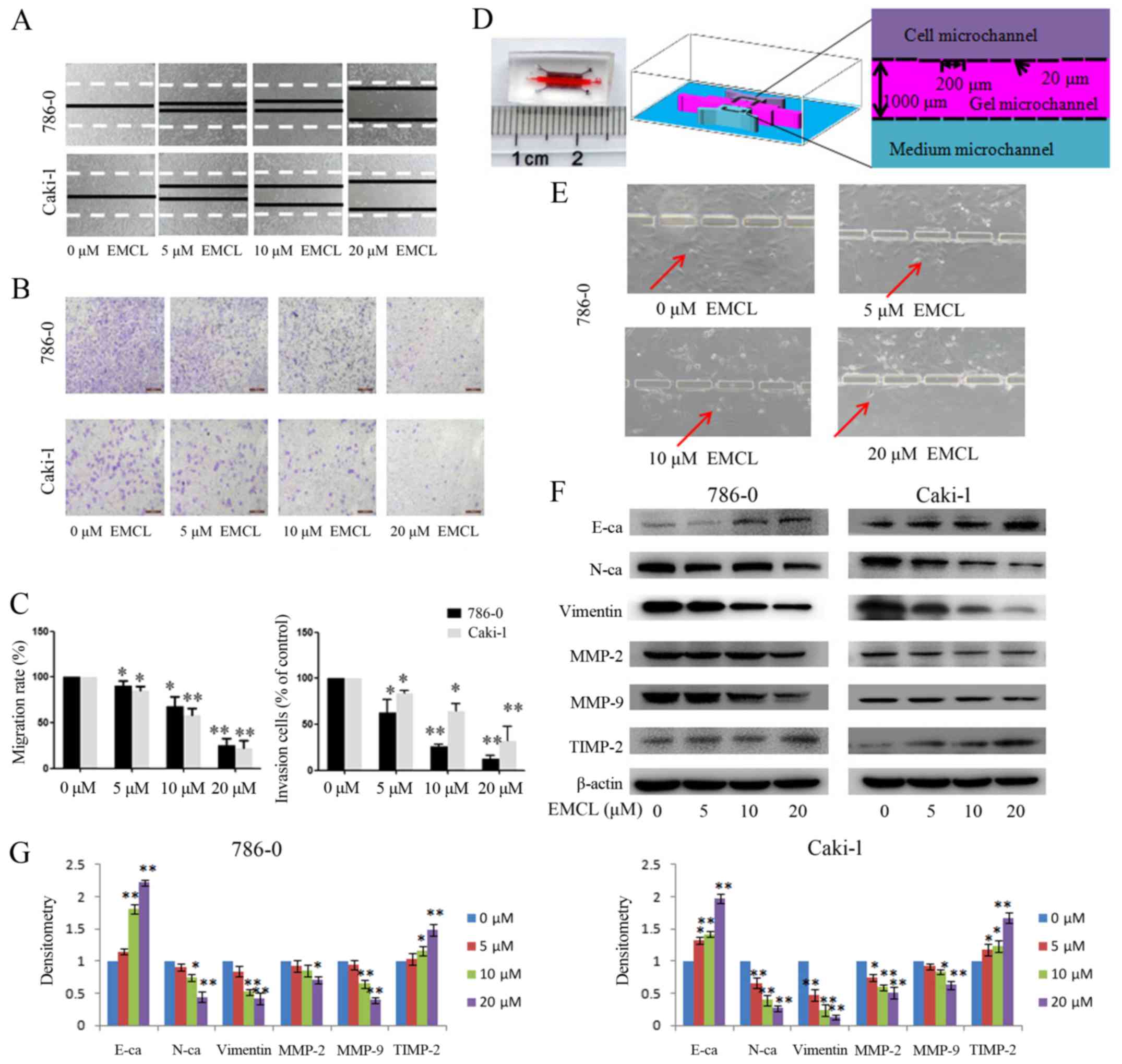 | Figure 3Effects of treatment with EMCL on
cell migration and invasion. (A) Migration of 786-0 and Caki-1
cells was analyzed using a scratch assay. Following24 h of
treatment with EMCL at the indicated doses, the wound gap was
observed and images were captured. (B) Cell invasion was analyzed
in 786-0 and Caki-1 cells treated with EMCL at the indicated doses
for 24 h. Cell invasion was observed and images were captured. (C)
Percentages of migrated cells were calculated relative to the
original gap and the percentage of invaded cells was calculated.
(D) Image and illustration of the homemade microfluidic chip. The
chip comprised two-sided micro-channels sandwiching a gel
micro-channel with two rows of micro-gaps. The gel micro-channel
was filled with Matrigel (pink), and the cell micro-channel and
medium micro-channel were used for plating the cells (purple) and
the chemoattractant (20% FBS; green), respectively. (E) Invasion of
786-0 cells to the gel micro-channel following 24 h of treatment
with EMCL at the indicated doses. Red arrows indicate invasion of
786-0 cells. (F) Protein expression levels of MMP-2/9, TIMP-2,
E-cadherin, N-cadherin and vimentin were analyzed by western blot
analysis. (G) Quantitative analysis of proteins. Data are presented
as the mean ± standard deviation of three independent experiments.
*P<0.05 and **P<0.01. vs. dimethyl
sulfoxide-treated group. EMCL, epoxymicheliolide; E-ca, E-cadherin;
N-ca, N-cadherin; MMP, matrix metalloproteinase; TIMP-2, tissue
inhibitor of metalloproteinase 2. |
Confocal immunofluorescence analysis
The 786-0 cells were seeded on coverslips and
treated with varying concentrations of EMCL (0, 5, 10 and 20
µM) for 24 h. Subsequently, the cells were fixed with 4%
paraformaldehyde at room temperature, permeabilized with 0.2%
Triton X-100, and blocked in PBS containing 5% bovine serum albumin
(Sigma-Aldrich, St. Louis, MO, USA). The cells were washed with
PBS, incubated with 3% FBS for 30 min at room temperature, and
incubated overnight at 4°C with antibodies specific to NF-κB p50
(1:100 dilution) and NF-κB p65 (1:500 dilution). Following this,
the cells were incubated with fluorescein isothiocyanate- or
rhodamine isothiocyanate-conjugated secondary antibodies (cat. no.
4408 or 4413, 1:1,000 dilution, Cell Signaling Technology) for 60
min at room temperature in a darkroom. Finally, DAPI was added to
each sample for nuclear counterstaining, and fluorescent images
were examined using a Leica DM 14000B confocal microscope (Leica
Microsystems, Inc., Buffalo Grove, IL, USA).
Western blot analysis
Cell lysate preparation and western blot analysis
were performed as previously described (25). Briefly, cells were lysed in lysis
buffer for 30 min. The lysates were then centrifuged at 12,000 × g
for 15 min at 4°C, and the total protein concentration was
determined using a BCA protein assay kit. Equivalent quantities of
protein (40 µg) from each cell group were separated on a 12%
SDS-PAGE gel and transferred electrophoretically onto
polyvinylidene difluoride membranes (EMD Millipore). The membranes
were blocked with 5% non-fat dry-milk for 1 h and then incubated
overnight at 4°C with antibodies specific to COX-2 (1:1,000
dilution), NF-κB p65 (1:1,000 dilution), IKKβ (1:1,000 dilution),
IκBα (1:800 dilution), p-NF-κB p65 (1:500 dilution), p-IKKβ (1:500
dilution), p-IκBα (1:500 dilution), cyclin B1 (1:1,000 dilution),
cyclin E1 (1:1,000 dilution), CDK2 (1:1,000 dilution), cyclin D1
(1:1,000 dilution), E-cadherin (1:1,000 dilution), vimentin
(1:1,000 dilution), N-cadherin (1:1,000 dilution), Bcl-2 (1:1,000
dilution), Bax (1:1,000 dilution), cleaved caspase-3 (1:1,000
dilution), cleaved caspase-9 (1:1,000 dilution), cleaved
caspase-PARP (1:1,000 dilution), NF-κB p50 (1:200 dilution), MMP-2
(1:500 dilution), MMP-9 (1:500 dilution), TIMP-2 (1:500 dilution),
GAPDH (1:2,500 dilution), histone H3 (1:2,000 dilution) and β-actin
(1:2,000 dilution). The membrane was then washed three times with
1XTris-buffered saline with Tween and incubated with the
anti-rabbit IgG (1:5,000 dilution) (cat. no. 7074) or anti-mouse
IgG (1:5,000 dilution) (cat. no. 7076) secondary antibodies for 2 h
at room temperature. The protein bands were visualized by enhanced
chemiluminescence, and the integrated optical density of bands was
quantified using ImageQuant TL 7.0 (GE Healthcare Life Sciences,
Chalfont, UK).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from the cells using the
RNAprep pure Cell kit (Tiangen Biotech Co., Ltd., Beijing, China).
The cDNA was synthesized from 1 µg of total RNA as the
template in a 20-µl reaction volume using the FastQuant RT
kit (with gDNase; Tiangen Biotech Co., Ltd.) at 42°C for 15 min and
95°C for 3 min according to the manufacturer's instructions. The
mRNA level of genes was detected with SuperRealPreMix Plus
(SYBR-Green) from Tiangen Biotech Co., Ltd. The relative
quantification for the mRNA level of the genes was performed by
RT-qPCR using the Agilent Mx3005P qPCR system (Agilent
Technologies, Inc., Santa Clara, CA, USA) in triplicate for each of
the independently prepared RNAs. The primer sequences used were as
follows: COX2 (PTGS2) forward, 5′-CAG CCA TAC AGC AAA TCC TTG-3′
and reverse, 5′-CAA ATG TGA TCT GGA TGT CAA C-3′; β-actin forward,
5′-CAC CAG GGC GTG ATG GT-3′ and reverse, 5′-CTC AAA CAT GAT CTG
GGT CAT-3′. qPCR was performed at 95°C for 15 min, 95°C for 10 sec
and 60°C for 20 sec for 40 cycles. The relative levels of COX-2
transcripts were normalized to the expression of β-actin and
calculated based on the 2−ΔΔCq method (27).
Flow cytometric analysis
To determine the distribution of cells in the cell
cycle and the proportion of apoptotic cells, flow cytometric
analysis was performed using a flow cytometer (BD FACS Accuri C6;
BD Biosciences). Briefly, the 786-0 cells were treated with
different concentrations of EMCL (0, 5, 10, and 20 µM) for
24 h, following which the treated cells were collected and fixed
with ice-cold 70% ethanol at 4°C for 4 h, and then stained with
propidium iodide (PI) staining buffer (0.2% Triton X-100, 100
µg/ml DNase-free RNase A, and 50 µg/ml PI in PBS) in
the dark for 30 min. For apoptosis examination, the treated cells
were stained with the Annexin V-FITC Apoptosis Detection kit in the
dark at room temperature for 15 min. The cell cycle distribution
and the fraction of apoptotic cells were determined using the FACS
analysis system.
Dual-luciferase reporter assay
In the 786-0 cells, the pRL-TK plasmid was
co-transfected with pGL3-NFκB or pGL3-basic (negative control) in a
96-well plate using Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific). Following treatment with different concentrations of
EMCL (0, 5, 10, and 20 µM) for 24 h, the dual-luciferase
reporter assay system (Promega, Madison, WI, USA) was used to
measure the changes in luciferase activity. Firefly luciferase
activity was normalized to Renilla luciferase activity. All
values are shown as the mean ± standard deviation of triplicate
samples.
Electrophoretic mobility shift assay
(EMSA)
The 786-0 cells were treated with different
concentrations of EMCL (0, 5, 10, and 20 µM) for 24 h. The
nuclear proteins of the human RCC cells were then isolated using
the Nuclear and Cytoplasmic Protein Extraction kit (Beyotime
Institute of Biotechnology, Shanghai, China) according to the
manufacturer's protocol. The protein concentrations were measured
using the bicinchoninic acid method. Double-stranded
oligonucleotides encoding the NF-κB consensus sequence were 5′-AGT
TGA GGG GAC TTT CCC AGG C-3′ and 3′-TCA ACT CCC CTG AAA GGG TCC
G-5′, which were end-labeled with biotin (Beyotime Institute of
Biotechnology). Nuclear extracts (5 µg) were added to 20
µl of binding reactions and incubated for 20 min at room
temperature. The EMSA reactions were performed according to the
manufacturer's protocol (Light Shift Chemiluminescent EMSA kit;
Pierce; Thermo Fisher Scientific, Inc.). The same unlabeled probe
was used as a competitor in the assay. Three independent
experiments were performed in triplicate.
Molecular modeling
Molecular docking was used to investigate whether
EMCL specifically binds to IKKβ protein complexes. EMCL was
optimized using the semi-empirical PM3 method with the
Polak-Ribière-Polyak conjugate gradient algorithm, with an RMS
gradient of 0.01 kcal mol−1 Å−1 as
convergence criterion. The optimized structure of EMCL was docked
into the active site of IKKβ with ligand K-252A (PDB code: 4KIK).
The crystallographic ligand was extracted from the active site, and
the residues within a 6.5 Å radius around the IKKβ molecule were
defined as the active pocket. The Surflex-Dock of sybyl-x 1.10
program package (Tripos, St. Louis, MO, USA) was used for the
docking calculations with default parameters.
Statistical analysis
All data are expressed as the mean ± standard
deviation of at least three independent experiments. Statistical
analysis was performed using SPSS 19.0 software (IBM SPSS, Armonk,
NY, USA). statistical significance for each variable was estimated
by a one-way analysis of variance followed by Tukey's test for post
hoc analysis. P<0.05 was considered to indicate a statistically
significant difference.
Results
EMCL inhibits RCC proliferation and
changes cell morphology
In order to investigate the functional role of EMCL
in human RCC cells, the present study quantitatively examined the
effect of EMCL on cell morphology and cell proliferation in several
human RCC cell lines. First, the effect of EMCL on cell
proliferation was quantitatively examined in 786-0, Caki-1and A498
cells using a CCK-8 assay. As shown in Fig. 1A, following treatment of the three
RCC cell lines with different concentrations of EMCL for 24 and 48
h, the survival rates of the three cell lines decreased in a dose-
and time-dependent manner. The IC50 values of EMCL
against the 786-0, Caki-1 and A498 cells were then calculated
(Table I). As shown in Fig. 1B, EMCL reduced cell-to-cell contact
and filopodia formation in the 786-0 and Caki-1 cells, compared
with the cells in the DMSO vehicle control groups.
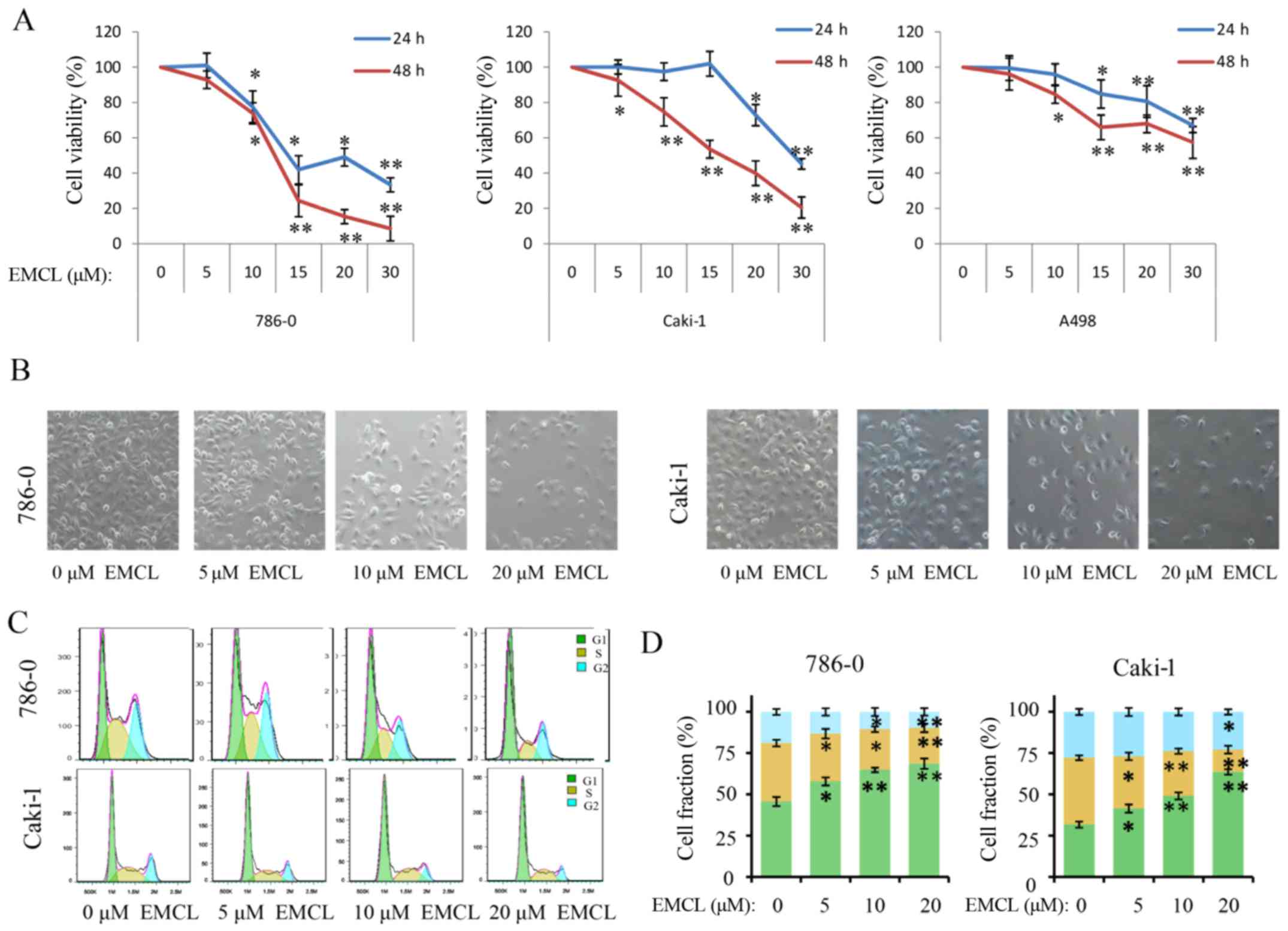 | Figure 1EMCL inhibits cell viability, changes
morphology, and induces cell cycle arrest. (A) 786-0, Caki-1, and
A498 cells were treated with EMCL at the indicated doses. Following
treatment for 24 and 48 h, cell viability was determined using a
Cell Counting Kit-8 assay. Cells treated with vehicle control
(DMSO) were used as the reference group (viability set at 100%).
(B) Changes in cell morphology and spreading of 786-0 and Caki-1
cells treated with EMCL at the indicated doses for 24 h were
observed. Images were captured using a microscope fitted with a
digital camera (magnification, ×100). (C) Cell cycle analyses of
786-0 and Caki-1 cells were performed following 24 h of treatment
with EMCL, using a BD Accuri C6 flow cytometer. (D) Cell fractions
were calculated. Data are presented as the mean ± standard
deviation of three independent experiments. *P<0.05
and **P<0.01, vs. DMSO-treated group. EMCL,
epoxymicheliolide; DMSO, dimethyl sulfoxide. |
 | Table ITwenty-four and 48 h IC50
values of EMCL for inhibition of cell viability. |
Table I
Twenty-four and 48 h IC50
values of EMCL for inhibition of cell viability.
| Cell line | IC50
(µM)
|
|---|
| 24 h | 48 h |
|---|
| 786-0 | 18.82±2.24 | 12.98±1.35 |
| Caki 1 | 25.38±1.61 | 15.16±0.90 |
| A498 | 37.58±1.58 | 28.95±1.46 |
EMCL induces cell cycle arrest of RCC
cell lines
As changes in cell proliferation are usually
accompanied by changes in the cell cycle (28), the present study next evaluated the
effect of EMCL on the cell cycle. As shown in Fig. 1C and D, induction of cell cycle
arrest was observed in the G0/G1 phase. Compared with the control
group, the percentages of cells in the G0/G1 phase increased from
43.2 to 67.3% and from 30.6 to 59.1% following EMCL treatment in
the 786-0 and Caki-1 cells, respectively; whereas, the percentages
of cells in the S phase decreased from 33.24 to 18.05% and from
29.70 to 7.67%, respectively.
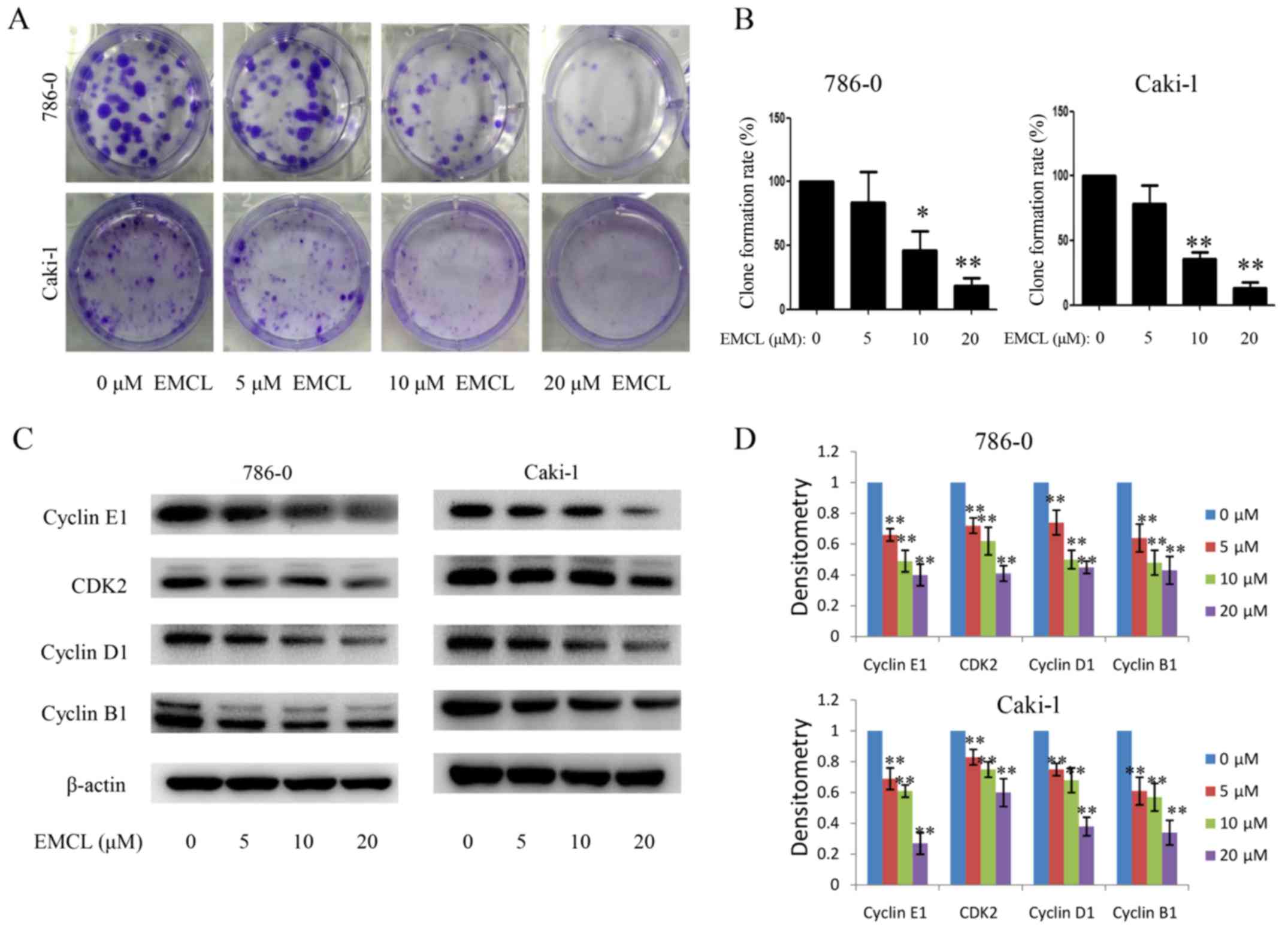
Subsequently, the present study investigated the
influence of EMCL on colony formation in 786-0 and Caki-1 cells by
using the colony formation assay. As shown in Fig. 2A and B, treatment with EMCL
resulted in a significant reduction of RCC cell colony formation
and colony formation rate. These results showed that EMCL may be a
potent inhibitor of RCC cell proliferation.
To determine the detailed mechanisms underlying the
above, the protein expression of several core cell cycle regulators
were examined by western blot analysis in human RCC cells following
EMCL treatment, including cyclin B1, cyclin E1, cyclin-dependent
kinase 2 (CDK2) and cyclin D1 (Fig. 2C
and D). It is likely that cell cycle arrest may have caused the
resulting reduced expression of these proteins. These data
indicated that EMCL promoted G1 cell cycle arrest and delayed entry
into the S phase.
EMCL suppresses migration and invasion of
RCC
To verify that EMCL inhibited cell motility, cell
migration and invasion were evaluated using wound-healing and
Transwell assays. As shown in Fig.
3A–C, the migratory ability of the 786-0 and Caki-1 cells was
inhibited by EMCL treatment in a dose-dependent manner. The same
result was obtained when the microfluidic chip was used to detect
the invasive capability of cancer cells (Fig. 3D and E). These results demonstrated
that EMCL inhibited cell migration and invasion. To ascertain the
detailed underlying mechanisms of the effect of EMCL on cell
migration and invasion, key protein markers were analyzed in the
786-0 and Caki-1 cells by western blot analysis. As shown in
Fig. 3F and G, treatment with EMCL
significantly upregulated the expression of E-cadherin (epithelial
marker), whereas the expression of mesenchymal markers (vimentin
and N-cadherin) were downregulated. Consistently, EMCL also
inhibited the protein expression of MMP-2 and MMP-9 and upregulated
the expression of TIMP-2 in a dose-dependent manner. These results
indicated that EMCL treatment inhibited cell migration and invasion
via the regulation of EMT markers and the expression of MMPs.
EMCL induces apoptosis via modulating
cytochrome c and caspase signaling
The present study also determined whether EMCL
inhibited cell proliferation associated with activation of the RCC
apoptotic pathway. As shown in Fig. 4A
and B, treatment with EMCL at three effective concentrations
for 24 h induced 7, 10.4 and 12.2% of the 786-0 cells to become
apoptotic, and 6.4, 9.2, and 15.7% of the Caki-1 cells to become
apoptotic. The levels of apoptosis-related proteins, including
cleaved caspase-3, cleaved caspase-9, cleaved PARP, B-cell lymphoma
2 (Bcl-2) and Bcl-2-associated X protein (Bax) were also detected
in the 24 h-treated cells by western blot analysis. As shown in
Fig. 4C–E, treatment with EMCL
resulted in increased levels of cleaved caspase-3/9, cleaved PARP,
and the ratio of Bax/Bcl-2.
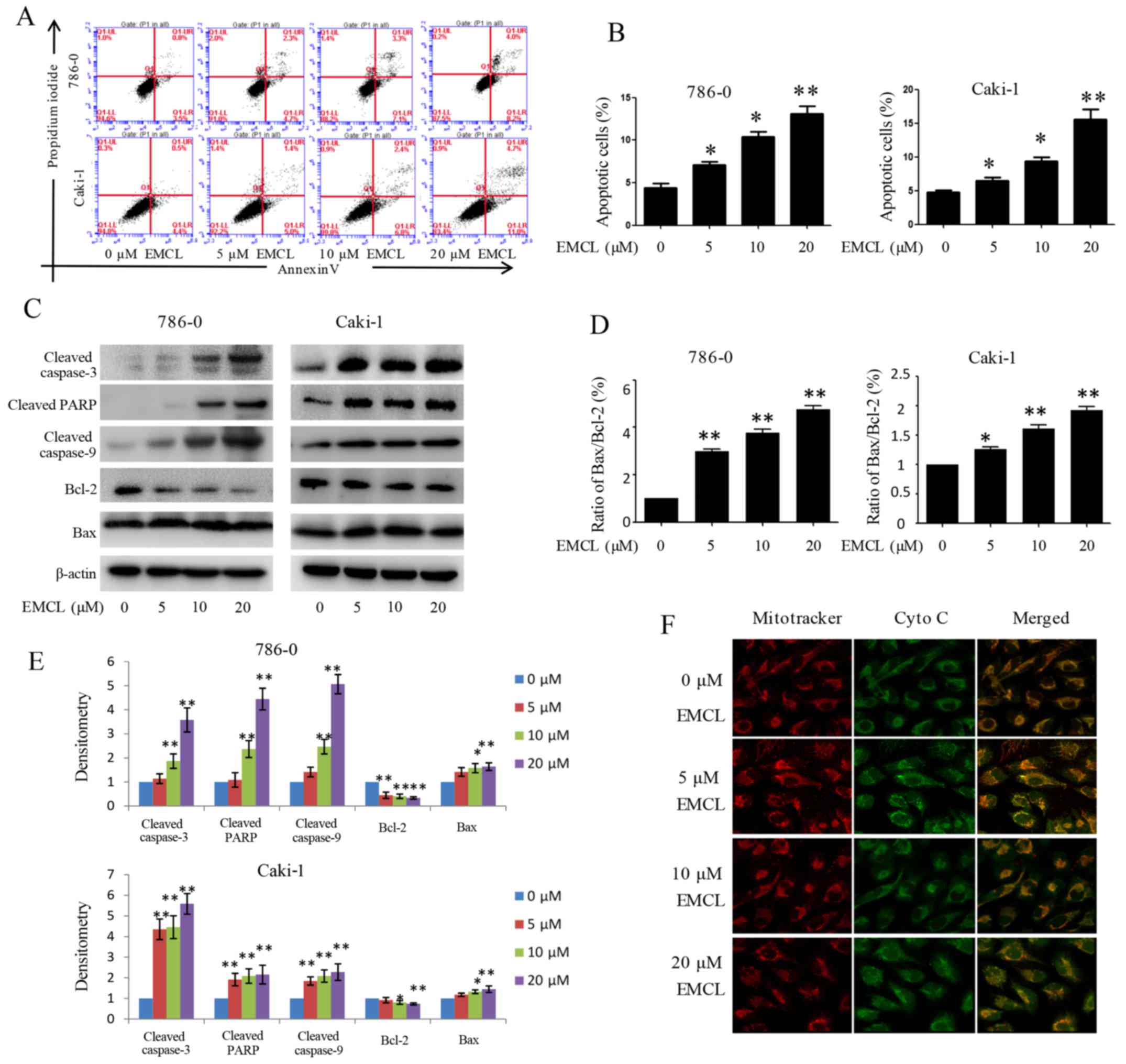 | Figure 4Effects of treatment with EMCL on
caspase-dependent apoptosis. 786-0 and Caki-1 cells were treated
with EMCL at the indicated doses. (A) Following treatment for 24 h,
levels of apoptosis were determined by FACS analysis, (B) and the
percentage of apoptotic cells was calculated. (C) Protein
expression levels of cleaved caspase-3, cleaved caspase-9, cleaved
caspase-PARP, and BCL-2/BAX in 786-0 and Caki-1 cells were analyzed
by western blot analysis. (D) Ratio of Bax/Bcl-2. (E) Quantitative
analysis of proteins. (F) Release of cytochrome c from the
mitochondria to the cytoplasm was observed by immunofluorescence
imaging analysis in 786-0 and Caki-1 cells (magnification, ×630).
Data are presented as the mean ± standard deviation of three
independent experiments. *P<0.05 and
**P<0.01, vs. dimethyl sulfoxide-treated group. EMCL,
epoxymicheliolide; PARP, poly (ADP-ribose) polymerase ; Bcl-2,
B-lymphoma 2; Bax, Bcl-2-assocated X protein; Cyto C, cytochrome
c. |
Previous studies have shown that mitochondrial
cytochrome c released into the cytoplasm can induce
apoptosis (14,29). The present study performed
immunofluorescence imaging analysis to determine whether EMCL can
induce the release of cytochrome c in RCC cells. The results
showed that treatment with EMCL effectively induced the release of
cytochrome c from the inter-mitochondrial space into the
cytosol of the 786-0 cells (Fig.
4F). These results showed that EMCL promoted the induction of
cell apoptosis by triggering the release of cytochrome c and
facilitating caspase activation in the cytosol.
EMCL suppresses the expression of COX-2
in RCC
Multiple lines of evidence and clinical data have
confirmed that selective COX-2 inhibitors can suppress
inflammation, angiogenesis and cell proliferation, and induce
apoptosis in human cancer cells (5,16).
The present study evaluated the activities of EMCL on the
expression of COX-2 in human RCC cells at the protein and mRNA
levels by western blot and RT-qPCR analyses, respectively. As shown
in Fig. 5A and B, EMCL effectively
inhibited the expression of COX-2 at the protein and mRNA levels in
786-0 cells and Caki-1 cells in a dose-dependent manner. The 786-0
and Caki-1 cells were then pretreated with the COX-2-selective
inhibitor (celecoxib; CB; 50 µM) for 8 h, followed by EMCL
treatment. Following incubation for 24 h, cell viability was
analyzed using the CCK-8 assay. As shown in Fig. 5C, treatment with either CB or EMCL
significantly inhibited cell proliferation compared with that in
the controls, whereas CB pretreatment followed by EMCL treatment
did not significantly alter the inhibition of cell viability,
compared with EMCL treatment alone. Furthermore, the 786-0 and
Caki-1 cells were incubated with EMCL following pretreatment with
the COX-2 inducer PMA (200 nM) for 8 h, following which cell death
was evaluated. The results showed that inducing the exogenous
overexpression of COX-2 by PMA restored the cell viability of the
EMCL-treated cells (Fig. 5D).
These results indicated that the inhibition of proliferation by
EMCL treatment of RCC cells was partially mediated by the
inactivation of COX-2 signaling.
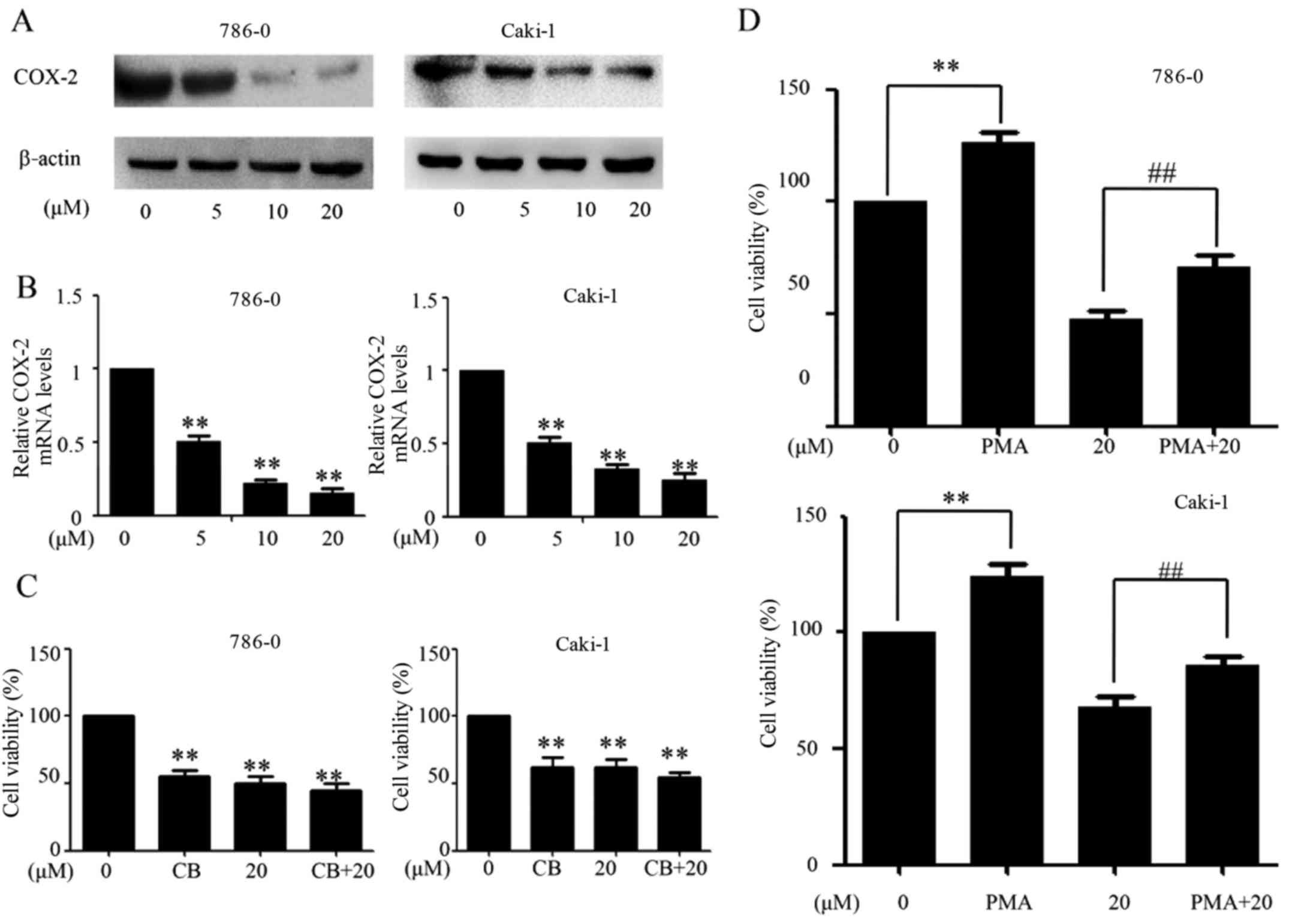 | Figure 5EMCL suppresses the expression of
COX-2. 786-0 and Caki-1 cells were treated with EMCL at the
indicated doses. After 24 h, expression levels of COX-2 protein and
gene expression were analyzed by (A) western blot and (B) reverse
transcription-quantitative polymerase chain reaction analyses in
the 786-0 and Caki-1 cells, respectively. (C) 786-0 and Caki-1
cells were treated with EMCL (20 µM) for 24 h following
pretreatment with the COX-2 selective inhibitor CB (50 µM)
for 8 h, and the cell viability was determined by CCK-8 analysis.
(D) 786-0 and Caki-1 cells were treated with EMCL (20 µM)
for 24 h following pretreatment with the COX-2 selective inducer
PMA (200 nM) for 8 h, and the cell viability was determined by
CCK-8 analysis. Data are presented as the mean ± standard deviation
of three independent experiments.**P<0.01, vs.
DMSO-treated group; ##P<0.01 between EMCL treatment
group and combination treatment group. EMCL, epoxymicheliolide;
COX-8, cyclooxygenase-2; CB, celecoxib; PMA, pyromellitic acid.
CCK-8, Cell Counting Kit-8. |
EMCL inhibits NF-κB and its binding
activity
The above-mentioned results showed that EMCL
inhibited the expression of COX-2; however, several transcription
factors and transcriptional coactivators affect the expression of
COX-2 by regulating the promoter region of COX-2, including NF-κB
(30). In the present study, the
role of the NF-κB signaling pathway and their binging activity in
response to EMCL challenge was examined. The EMCL concentration of
20 µM was selected as the highest treatment concentration,
as it inhibited cell proliferation with only marginal cytotoxicity.
In addition, selecting a concentration close to the IC50
can make the experimental results more obvious, therefore, in order
to examine the role of NF-κB/COX-2 signaling pathway in
EMCL-treated tumor cells, this concentration was selected. Firstly,
the 786-0 and Caki-1 cells were pretreated with the NF-κB inhibitor
PDTC (30 µM) for 6 h, followed by EMCL treatment. Following
incubation for 24 h, cell viability was analyzed by the CCK-8
assay. As shown in Fig. 6A,
treatment with either PDTC or EMCL significantly inhibited cell
proliferation compared with that in the controls; whereas PDTC
pretreatment followed by EMCL treatment did not significantly alter
the inhibition of cell viability compared with EMCL treatment
alone. Furthermore, exposure of the 786-0 cells to EMCL was
detected by EMSA, which indicated that EMCL treatment significantly
decreased NF-κB DNA-binding activity in a dose-dependent manner
(Fig. 6B). Simultaneously, the
786-0 cells were transfected with luciferase reporter plasmids
containing NF-κB binding sites and treated with EMCL to investigate
changes in their gene expression levels. As shown in Fig. 6C, EMCL treatment significantly
suppressed gene expression by 1.2-, 1.4-, and 2.2-fold in the
dual-luciferase reporter assay at the three effective
concentrations of 5, 10, and 20 µM, respectively. These
results indicated that inhibition of the expression of COX-2 by
EMCL treatment of RCC cells was mediated by inactivation of the
NF-κB signaling pathway.
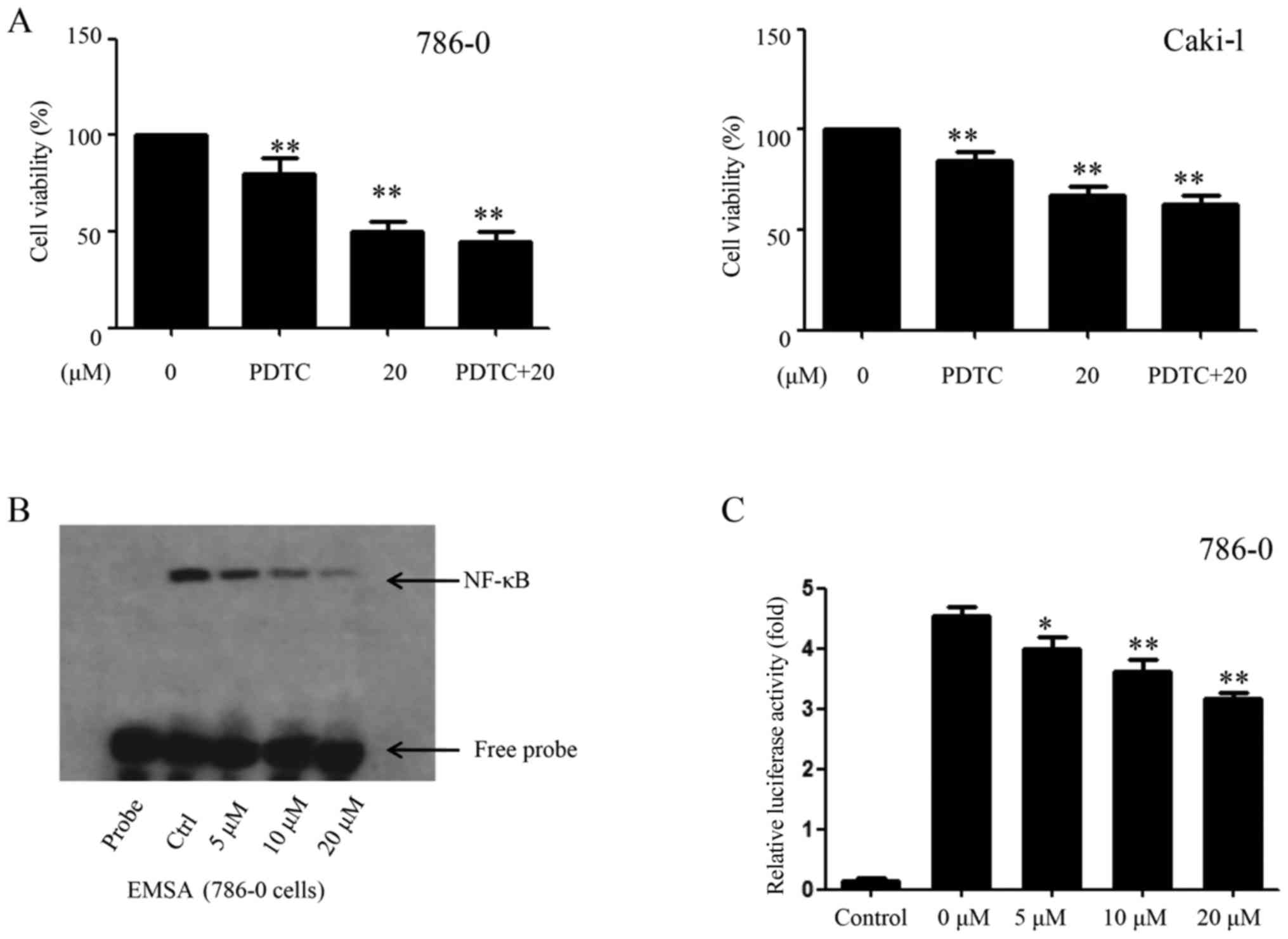 | Figure 6EMCL inhibits NF-κB and its binding
activity. (A) 786-0 and Caki-1 cells were treated with EMCL (20
µM) for 24 h following pretreatment with PDTC, an inhibitor
of NF-κB (30 µM), for 6 h, and the cell viability was
determined by Cell Counting Kit-8 analysis. (B) 786-0 was either
untreated (lane 2, control cultures) or treated with the indicated
doses of EMCL (lanes 3-5) for 24 h. Nuclear extracts were prepared
and analyzed by an electrophoretic mobility shift assay. (C)
NF-κB-dependent gene expression in the 786-0 cell line. Cells were
transfected with Bwt-luc reporter plasmid, stimulated with EMCL (5,
10, and 20 µM). These stimuli were administered 24 h
following transfection, and the cell lysates were prepared
following an additional 24 h incubation for the determination of
luciferase activity. As an internal control, pRL-TK, expressing
Renilla luciferase under the control of TK promoter, was
co-transfected. All luciferase activities were corrected by the
internal control activity of Renilla luciferase. Data are
presented as the mean ± standard deviation of three independent
experiments. *P<0.05 and **P<0.01, vs.
dimethyl sulfoxide-treated group. NF-κB, nuclear factor-κB; EMCL,
epoxymicheliolide; PDTC, ammonium pyrrolidine dithiocarbamate;
Ctrl, control. |
EMCL inhibits NF-κB activity by targeting
IKKβ
NF-κB leads to IκBα phosphorylation mainly through
the activation of IKKβ kinase activity. The p-IκBα is then degraded
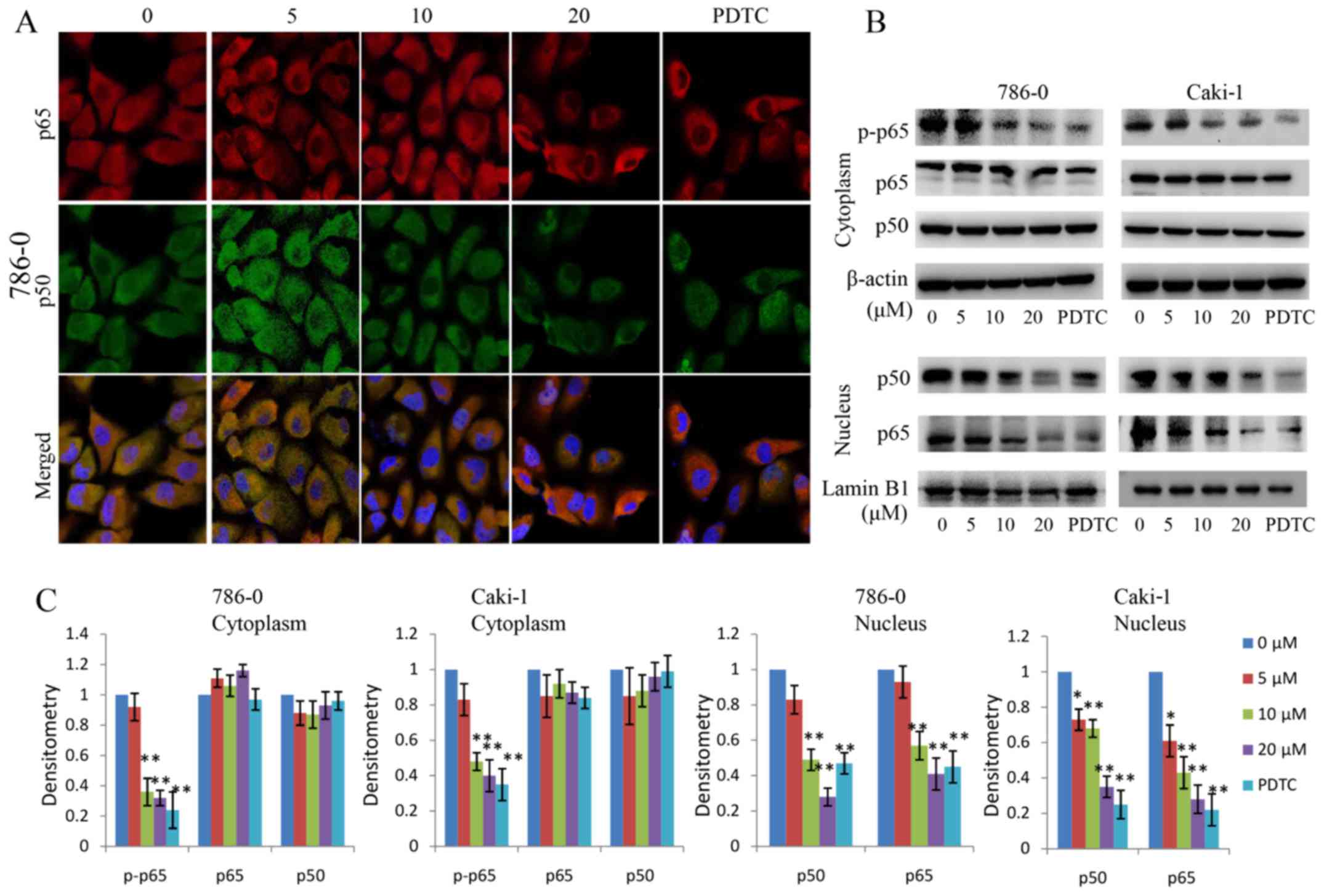
by the proteasome, releasing the p50/p65 NF-κB dimers and
activating NF-κB signaling. In the present study, the effect of
EMCL on NF-κB signaling in RCC was examined. The protein changes of
NF-κBp50/p65 subunits in the cytoplasm and nuclear lysates were
detected in the EMCL-treated RCC cells. As shown in Fig. 7A–C, the protein levels of p65/p50
in nuclear lysates decreased significantly following treatment with
EMCL or PDTC (an inhibitor of NF-κB, used as a positive control).
In addition, the protein levels of p-p65 in the cytoplasm decreased
significantly following treatment with EMCL or PDTC. These data
indicated that EMCL effectively inhibited activation of the NF-κB
pathway in human RCC cells.
In order to determine the molecular targets of EMCL
on the NF-κB pathway, the 786-0 and Caki-1 cells were treated with
EMCL or PDTC, and the molecular markers of the NF-κB pathway were
examined. The effects of EMCL on IκBα protein degradation and IKKβ
activity were also examined. As shown in Fig. 8A and B, EMCL significantly
inhibited the phosphorylation of IKKβ and IκBα, but did not affect
the total IKKβ content. Subsequently, the protein levels of NF-κB
p50/p65 subunits were detected in the nuclear lysates of 786-0 and
Caki-1 cells treated with EMCL (20 µM) for an indicated
time. The results showed that EMCL inhibited the NF-κB pathway in
the 786-0 and Caki-1 cells in a time-dependent manner (Fig. 8C and D). Furthermore, the 786-0 and
Caki-1 cells were treated with EMCL and NF-κB inhibitor (PDTC)
alone or together, and the expression of COX-2 was detected at the
mRNA and protein levels. The results showed that treatment with
EMCL or PDTC alone significantly inhibited the expression of COX-2,
whereas the combination of EMCL and PDTC did not significantly
alter the inhibition of COX-2 at the protein or mRNA levels
(Fig. 8E and F). These results
indicated that the EMCL-induced inhibition of the NF-κB signaling
pathway may partly involve the repression of COX-2.
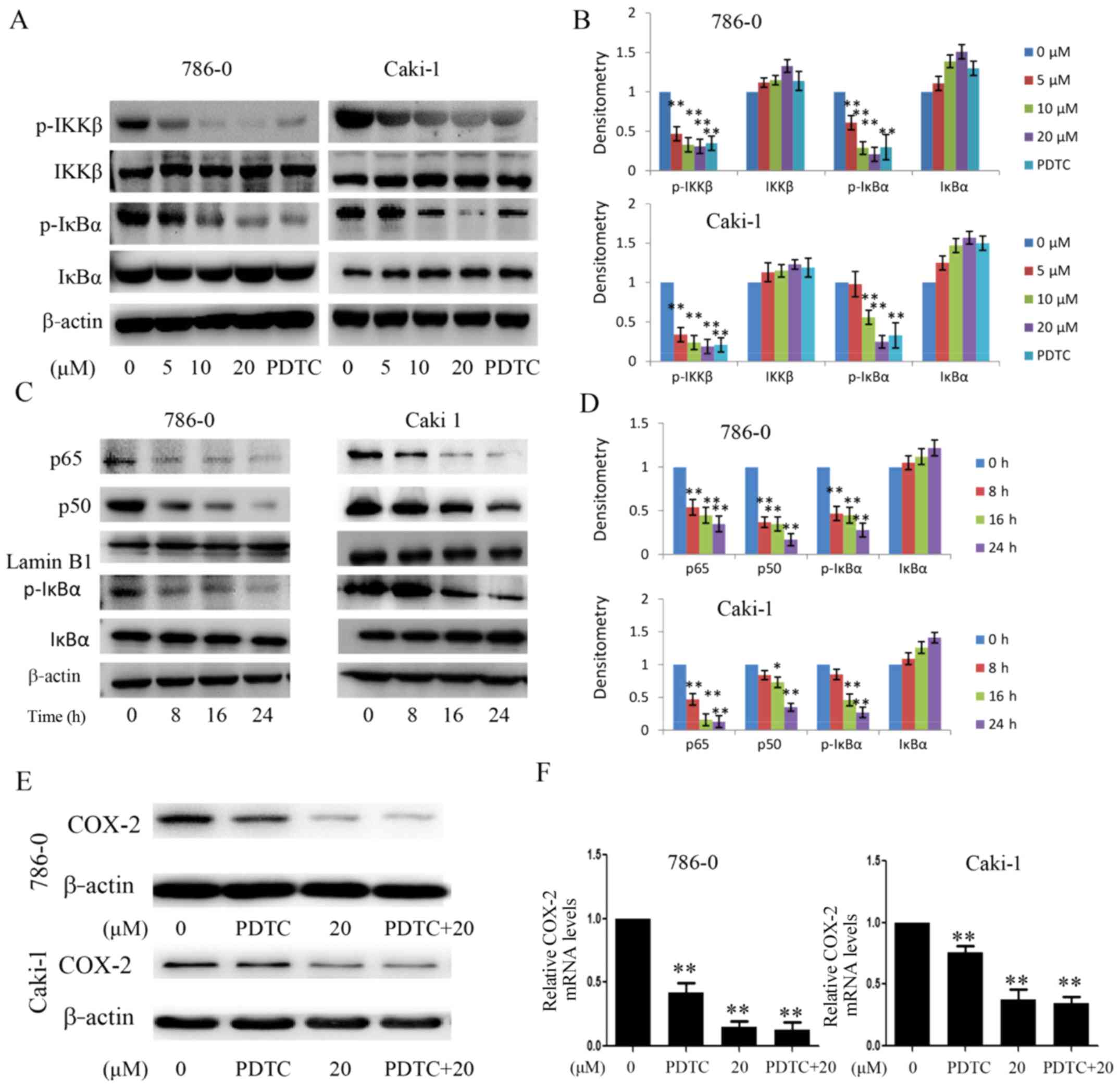 | Figure 8Effect of EMCL on IKKβ/NF-κB
signaling and the expression of COX-2. (A) Following treatment with
EMCL at the indicated doses and PDTC (30 µM) for 24 h, the
protein expression levels of IKKβ, p-IKKβ, IκBα, and p-IκBα were
analyzed by western blot analysis. (B) Quantitative analysis of the
proteins. (C) Following treatment with EMCL (20 µM) at the
indicated time, cytoplasmic and nuclear extracts were prepared for
the western blot analysis of p65, p50, IκBα, and p-IκBα. (D)
Quantitative analysis of the proteins. Following treatment with
EMCL (20 µM) or PDTC (30 µM) alone or together for 24
h, protein and gene expression levels of COX-2 were analyzed by (E)
western blot analysis and (F) reverse transcription-quantitative
polymerase chain reaction analyses were performed in 786-0 and
Caki-1 cells, respectively. Data are presented as means ± SD of
three independent experiments. *P<0.05 and
**P<0.01. vs. dimethyl sulfoxide-treated group. EMCL,
epoxymicheliolide; PDTC, ammonium pyrrolidine dithiocarbamate;
IκBα, inhibitor of nuclear factor-κB; IKK, IκB kinase; p-,
phosphorylated; COX-2, cyclooxygenase-2. |
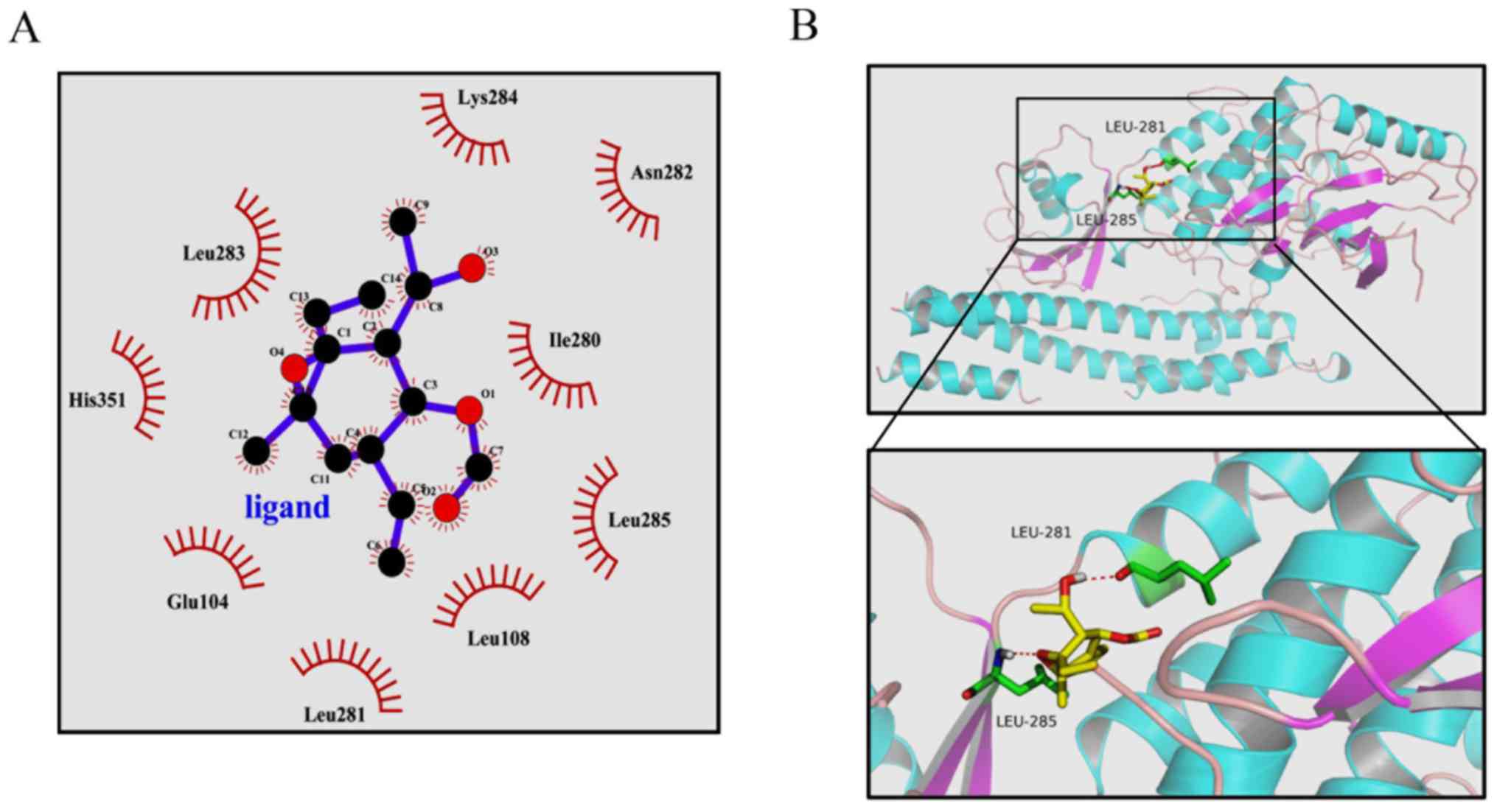
Previous studies have clearly shown that the
activity of IKKβ is essential for activation of the canonical NF-κB
signaling pathway (14,29). From the above results, it was
hypothesized that EMCL may bind to IKKβ and subsequently inhibit
its kinase activity. To test this hypothesis, a computer molecular
modeling assay was performed to model the interaction between EMCL
and IKKβ. Molecular docking experiments predicted that EMCL was
able to bind to the ATP binding site of IKKβ. As shown in Fig. 9A and B, EMCL formed two hydrogen
bonds with the ATP binding pocket of the IKKβ kinase domain. The
COC motif at the 9,10-epoxide moiety of EMCL formed a hydrogen bond
with the backbone NH of Leu285. The OH group at the C-3 position
formed an additional hydrogen bond to the carbonyl oxygen of
Leu281. These computational docking results indicated that EMCL
occupied the deep hydrophobic pocket in the ATP-binding site of
IKKβ to inhibit its kinase activity, and then attenuated the
activation of the NF-κB signaling pathway.
All of the above results support the hypothesis that
EMCL promotes the suppression of NF-κB/COX-2 signaling through the
inhibition of IKKβ kinase activity.
Discussion
PTL, which is a major sesquiterpene lactone
responsible for the bioactivity of feverfew, is a traditional drug
that has been used for the treatment of fever, migraine, and
arthritis (31). PTL exhibits
significant antitumor activities, including inhibition of cell
proliferation, induction of apoptosis, and enhancement of the role
of anticancer drugs (8-12). As PTL is unstable under acidic and
alkaline conditions (13), the
structurally related compound, EMCL, was developed; however, the
biological effects of EMCL in vivo and in vitro
remain to be fully elucidated. In particular, to the best of our
knowledge, there has been no report on the investigation of EMCL in
the treatment of RCC.
In the present study, it was found that EMCL
effectively inhibited the growth of human RCC cells and enhanced
the induction of apoptosis in a dose- and time-dependent manner.
Furthermore, it was shown that EMCL suppressed growth and apoptosis
of human RCC cells by inhibiting the NF-κB/COX-2 signaling pathway,
activating the cytochrome c/caspase-dependent apoptotic
pathway, and promoting G1 cell cycle arrest. The experiments showed
that EMCL inhibited the expression of COX-2through suppressing the
phosphorylation of IKKβ, preventing IκBα degradation, and resulting
in the NF-κB p65/p50 protein-sustained cytoplasmic retention,
thereby inhibiting its nuclear translocation and transcriptional
activity. In this regard, to the best of our best knowledge, this
may be the first report of the effect of EMCL on the expression of
COX-2 and its potential mechanisms in vitro.
The inflammatory antitumor response is important in
inhibiting tumor growth in human malignancies (19). COX-2, a rate-limiting enzyme for
the synthesis of prostaglandins from arachidonic acid, is important
in the inflammatory process. Inflammatory mediators, cytokines,
growth factors and tumor promoters can rapidly induce the
expression of COX-2 (30,32,33).
One of the key aspects of the inflammatory process
is the association of COX-2 with the formation of carcinogens,
tumor progression and inhibition of apoptosis, angiogenesis, and
metastatic processes (21). COX-2
is commonly upregulated in various types of human cancer, including
RCC (22-24). In order to elucidate the mechanism
of EMCL as an anticancer agent, the present study investigated
whether COX-2 is important in the biological activity of EMCL and
found that EMCL inhibited the expression of COX-2, and inhibited
the viability, migration and colony formation of human RCC
cells.
The transcription factor NF-κB is key in regulating
the expression of COX-2 at the transcriptional level (34). It appears likely that the mechanism
by which EMCL inhibits the expression of COX-2 is associated with
the inhibition of NF-κB in human RCC cells. In agreement with this
hypothesis, the experiments in the present study showed that EMCL
inhibited NF-κB signaling in human RCC cells by inhibiting
phosphorylation of the IKK complex, thereby preventing IκBα
degradation and resulting in sustained cytoplasmic retention of
NF-κB. In the 786-0 cells, treatment with EMCL at two effective
concentrations for 24 h suppressed gene expression from the
reporter plasmid containing NF-κB binding sites by 1.4- and
2.2-fold, respectively. Simultaneously, exposure of the 786-0 cells
to EMCL at three effective concentrations, as detected by EMSA,
indicated that EMCL treatment significantly decreased NF-κB
DNA-binding activity in a dose-dependent manner. Therefore,
inhibition of the NF-κB pathway may be a potential mechanism
involved in the suppression of human RCC cells by EMCL.
Chronic inflammation is a major activator of the
metastatic cascade (35). This
tumor-associated inflammation is important in regulating EMT, which
contributes to cancer invasion and metastasis. The results of the
present study indicated that EMCL inhibited the EMT process of RCC
cells by upregulating levels of E-cadherin and downregulating
levels of N-cadherin and vimentin. Activation of the MMP proteins
leads to cell migration and penetration into the basement membrane,
which is important in the EMT process (36). The results indicated that EMCL
inhibited the protein expression and enzyme activity ofMMP-9/-2 in
RCC cells. TIMPs are key endogenous regulators of MMP activity in
the tissue, specifically inhibiting MMPs and maintaining matrix
integrity (37). The levels of
TIMP-2 were significantly increased in the EMCL-treated human RCC
cells in a dose-dependent manner. These findings indicated that
EMCL inhibited EMT and the synthesis of MMPs and increased TIMP-2,
which is a novel finding.
In conclusion, the present study showed that EMCL is
a potent suppressor of the biological characteristics of RCC with
respect to cell proliferation, migration, invasion, apoptosis, and
cell cycle. Furthermore, its anticancer properties were mediated,
at least in part, through inhibition of the NF-κB/COX-2 signaling
pathway by targeting IKKβ. These results provide evidence
supporting EMCL as a novel anticancer drug for the treatment of
RCC.
Acknowledgments
Not applicable.
References
|
1
|
Zhou S, Wang J and Zhang Z: An emerging
understanding of long noncoding RNAs in kidney cancer. J Cancer Res
Clin Oncol. 140:1989–1995. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Smaletz O: Current management and future
directions in the treatment of advanced renal cell carcinoma - a
latin american perspective: 10 years in review. Int Braz J Urol.
41:835–843. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lu J, Wei JH, Feng ZH, Chen ZH, Wang YQ,
Huang Y, Fang Y, Liang YP, Cen JJ, Pan YH, et al: miR-106b-5p
promotes renal cell carcinoma aggressiveness and stem-cell-like
phenotype by activating Wnt/β-catenin signalling. Oncotarget.
8:21461–21471. 2017.PubMed/NCBI
|
|
4
|
Heinrich M, Robles M, West JE, Ortiz de
Montellano BR and Rodriguez E: Ethnopharmacology of Mexican
asteraceae (Compositae). Annu Rev Pharmacol Toxicol. 38:539–565.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liao K, Xia B, Zhuang QY, Hou MJ, Zhang
YJ, Luo B, Qiu Y, Gao YF, Li XJ, Chen HF, et al: Parthenolide
inhibits cancer stem-like side population of nasopharyngeal
carcinoma cells via suppression of the NF-κB/COX-2 pathway.
Theranostics. 5:302–321. 2015. View Article : Google Scholar :
|
|
6
|
Saadane A, Masters S, DiDonato J, Li J and
Berger M: Parthenolide inhibits IkappaB kinase, NF-kappaB
activation, and inflammatory response in cystic fibrosis cells and
mice. Am J Respir Cell Mol Biol. 36:728–736. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Oka D, Nishimura K, Shiba M, Nakai Y, Arai
Y, Nakayama M, Takayama H, Inoue H, Okuyama A and Nonomura N:
Sesquiterpene lactone parthenolide suppresses tumor growth in a
xenograft model of renal cell carcinoma by inhibiting the
activation of NF-kappaB. Int J Cancer. 120:2576–2581. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lin M, Bi H, Yan Y, Huang W, Zhang G,
Zhang G, Tang S, Liu Y, Zhang L, Ma J, et al: Parthenolide
suppresses non-small cell lung cancer GLC-82 cells growth via
B-Raf/MAPK/Erk pathway. Oncotarget. 8:23436–23447. 2017.PubMed/NCBI
|
|
9
|
Kim SL, Kim SH, Park YR, Liu YC, Kim EM,
Jeong HJ, Kim YN, Seo SY, Kim IH, Lee SO, et al: Combined
parthenolide and balsalazide have enhanced antitumor efficacy
through blockade of NF-κB activation. Mol Cancer Res. 15:141–151.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu W, Wang X, Sun J, Yang Y, Li W and
Song J: Parthenolide suppresses pancreatic cell growth by
autophagy-mediated apoptosis. OncoTargets Ther. 10:453–461. 2017.
View Article : Google Scholar
|
|
11
|
Pei S, Minhajuddin M, D'Alessandro A,
Nemkov T, Stevens BM, Adane B, Khan N, Hagen FK, Yadav VK, De S, et
al: Rational design of a parthenolide-based drug regimen that
selectively eradicates acute myelogenous leukemia stem cells. J
Biol Chem. 291:21984–22000. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jeyamohan S, Moorthy RK, Kannan MK and
Arockiam AJ: Parthenolide induces apoptosis and autophagy through
the suppression of PI3K/Akt signaling pathway in cervical cancer.
Biotechnol Lett. 38:1251–1260. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jin P, Madieh S and Augsburger LL: The
solution and solid state stability and excipient compatibility of
parthenolide in feverfew. AAPS PharmSciTech. 8:E1052007. View Article : Google Scholar
|
|
14
|
Yu Z, Guo W, Ma X, Zhang B, Dong P, Huang
L, Wang X, Wang C, Huo X, Yu W, et al: Gamabufotalin, a
bufadienolide compound from toad venom, suppresses COX-2 expression
through targeting IKKβ/NF-κB signaling pathway in lung cancer
cells. Mol Cancer. 13:2032014. View Article : Google Scholar
|
|
15
|
Karin M: Nuclear factor-kappaB in cancer
development and progression. Nature. 441:431–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mantovani A, Allavena P, Sica A and
Balkwill F: Cancer-related inflammation. Nature. 454:436–444. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kankaya D, Kiremitci S, Tulunay O and
Baltaci S: Gelsolin, NF-κB, and p53 expression in clear cell renal
cell carcinoma: Impact on outcome. Pathol Res Pract. 211:505–512.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Peri S, Devarajan K, Yang DH, Knudson AG
and Balachandran S: Meta-analysis identifies NF-κB as a therapeutic
target in renal cancer. PLoS One. 8:e767462013. View Article : Google Scholar
|
|
19
|
Ohshima H, Tazawa H, Sylla BS and Sawa T:
Prevention of human cancer by modulation of chronic inflammatory
processes. Mutat Res. 591:110–122. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Surh YJ, Chun KS, Cha HH, Han SS, Keum YS,
Park KK and Lee SS: Molecular mechanisms underlying chemopreventive
activities of anti-inflammatory phytochemicals: Down-regulation of
COX-2 and iNOS through suppression of NF-kappa B activation. Mutat
Res. 480–481:243–268. 2001. View Article : Google Scholar
|
|
21
|
Dannenberg AJ, Altorki NK, Boyle JO, Dang
C, Howe LR, Weksler BB and Subbaramaiah K: Cyclo-oxygenase 2: A
pharmacological target for the prevention of cancer. Lancet Oncol.
2:544–551. 2001. View Article : Google Scholar
|
|
22
|
Williams CS, Mann M and DuBois RN: The
role of cyclooxygenases in inflammation, cancer, and development.
Oncogene. 18:7908–7916. 1999. View Article : Google Scholar
|
|
23
|
Erdem H, Aydin HR, Bahadir A, Gündogdu B,
Balta H, Sener E, Kayikci MA, Albayrak A and Erdogan F:
Relationship of CD95 and COX-2 in renal cell carcinomas with
survival and other prognostic parameters: A tissue microarray
study. J Pak Med Assoc. 65:597–601. 2015.PubMed/NCBI
|
|
24
|
Yang S, Gao Q and Jiang W: Relationship
between tumour angiogenesis and expression of cyclo-oxygenase-2 and
vascular endothelial growth factor-A in human renal cell carcinoma.
J Int Med Res. 43:110–117. 2015. View Article : Google Scholar
|
|
25
|
Shrestha S, Zhu J, Wang Q, Du X, Liu F,
Jiang J, Song J, Xing J, Sun D, Hou Q, et al: Melatonin potentiates
the antitumor effect of curcumin by inhibiting IKKβ/NF-κB/COX-2
signaling pathway. Int J Oncol. 51:1249–1260. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao H, Wang J, Kong X, Li E, Liu Y, Du X,
Kang Z, Tang Y, Kuang Y, Yang Z, et al: CD47 promotes tumor
invasion and metastasis in non-small cell lung cancer. Sci Rep.
6:297192016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
28
|
Hu Q, Hou YC, Huang J, Fang JY and Xiong
H: Itraconazole induces apoptosis and cell cycle arrest via
inhibiting Hedgehog signaling in gastric cancer cells. J Exp Clin
Cancer Res. 36:502017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang X, Yu Z, Wang C, Cheng W, Tian X, Huo
X, Wang Y, Sun C, Feng L, Xing J, et al: Alantolactone, a natural
sesquiterpene lactone, has potent antitumor activity against
glioblastoma by targeting IKKβ kinase activity and interrupting
NF-κB/COX-2-mediated signaling cascades. J Exp Clin Cancer Res.
36:932017. View Article : Google Scholar
|
|
30
|
Zhang C, Su ZY, Wang L, Shu L, Yang Y, Guo
Y, Pung D, Bountra C and Kong AN: Epigenetic blockade of neoplastic
transformation by bromodomain and extra-terminal (BET) domain
protein inhibitor JQ-1. Biochem Pharmacol. 117:35–45. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Knight DW: Feverfew: Chemistry and
biological activity. Nat Prod Rep. 12:271–276. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Boolbol SK, Dannenberg AJ, Chadburn A,
Martucci C, Guo XJ, Ramonetti JT, Abreu-Goris M, Newmark HL, Lipkin
ML, DeCosse JJ, et al: Cyclooxygenase-2 overexpression and tumor
formation are blocked by sulindac in a murine model of familial
adenomatous polyposis. Cancer Res. 56:2556–2560. 1996.PubMed/NCBI
|
|
33
|
Zimmermann KC, Sarbia M, Weber AA,
Borchard F, Gabbert HE and Schrör K: Cyclooxygenase-2 expression in
human esophageal carcinoma. Cancer Res. 59:198–204. 1999.PubMed/NCBI
|
|
34
|
Heiss E, Herhaus C, Klimo K, Bartsch H and
Gerhäuser C: Nuclear factor kappa B is a molecular target for
sulforaphane-mediated anti-inflammatory mechanisms. J Biol Chem.
276:32008–32015. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee DF and Hung MC: Advances in targeting
IKK and IKK-related kinases for cancer therapy. Clin Cancer Res.
14:5656–5662. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Horejs CM: Basement membrane fragments in
the context of the epithelial-to-mesenchymal transition. Eur J Cell
Biol. 95:427–440. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gomez DE, Alonso DF, Yoshiji H and
Thorgeirsson UP: Tissue inhibitors of metalloproteinases:
Structure, regulation and biological functions. Eur J Cell Biol.
74:111–122. 1997.PubMed/NCBI
|