Introduction
Glioma is the most prevalent and fatal primary tumor
of the central nervous system (CNS) in adults, accounting for 80%
of all primary CNS tumors, with an annual incidence of ~6
cases/100,000 people in the United States (1). The poor outcomes for glioma patients
primarily result from the malignant biological characteristics of
glioma cells, including highly aggressive proliferative and
invasive abilities (1,2). While persistent efforts to
investigate glioma molecular oncology have achieved accumulating
progress, the detailed mechanisms remain to be elucidated.
Developing effective treatment therapies against specific molecular
targets in human glioma remain a significant challenge (3–5).
The nucleotide-binding domain leucine-rich family
pyrin-containing 3 (NLRP3) inflammasome consists of the scaffold
protein NLRP3, the adaptor protein apoptosis-associated speck-like
protein containing a caspase-recruitment domain (ASC) and caspase-1
(6,7). Upon activation by endogenous
pathogen-and damage-associated molecular pattern molecules or
exogenous irritants, the NLRP3 inflammasome is assembled, and acts
as a platform for the maturation of the proinflammatory cytokines,
including interleukin (IL)-1β and IL-18 (7–9).
Subsequently this allows proinflammatory cytokine secretion, which
promotes inflammation and immunity (7–9). The
activation mechanism and function of the NLRP3 inflammasome during
immuno-inflammatory responses have been widely investigated.
However, accumulating studies have demonstrated that the NLRP3
inflammasome is associated with cancer progression (10–15).
Notably, certain studies reported that NLRP3 functions in an
unidentified inflammasome-independent manner, whereby NLRP3 may
serve independently of the production of bioactive
cleaved-caspase-1 and mature IL-1β (16,17).
However, the in depth role of NLRP3 in glioma has not been well
clarified.
In order to investigate the biological role of NLRP3
in glioma, the expression levels of NLRP3, as well as ASC,
caspase-1 and IL-1β protein in glioma tissues were analyzed using
immunohistochemical staining. Subsequently, an NLRP3-specific small
interfering RNA (si-NLRP3) was constructed to deplete NLRP3
expression levels, and a plasmid system [pIRES2-enhanced green
fluorescent protein (EGFP)-NLRP3] was produced for upregulating
NLRP3 protein in human glioma cell lines, SHG44 and A172. In order
to determine the effects of NLRP3 on malignant cellular behaviors,
in vitro biological function experiments and western blot
assays were performed to determine alterations to
epithelial-mesenchymal transition (EMT) components, and its
potential molecular mechanism in the phosphatase and tensin homolog
(PTEN)/AKT serine/threonine kinase (AKT) signaling pathway,
providing a novel insight into the contributions of NLRP3 to human
glioma malignancy.
Materials and methods
Human glioma tissue samples
A total of 39 human glioma tissues samples were
obtained from patients who underwent surgery at Nanfang Hospital,
Southern Medical University (Guangzhou, China) between October 2014
and October 2015. Written informed consent was obtained from all
enrolled patients. The present study was approved by the Ethics
Committee of Nanfang Hospital, Southern Medical University and was
performed in accordance with The Declaration of Helsinki. The age
of the patients (24 male and 15 female) ranged between 11 and 80
years, with a mean age of 42±11 years. All specimens had been
confirmed pathological diagnosis and classified according to the
2007 World Health Organization (WHO) classification criteria
(18).
Immunohistochemical staining
The glioma specimens were fixed with 10% formalin at
room temperature for 24 h and paraffin-embedded, and sectioned into
4-µm thick slices that were mounted onto microscope slides.
Then, the slides were deparaffinized with xylene three times and
rehydrated in a descending ethanol series (100, 95, 90, 80, 70%).
Antigen retrieval was performed by heating the samples for 15 min
at 95°C in citrate buffer (pH 6.0), samples were then cooled at
room temperature for 30 min. The endogenous peroxidase activity was
eliminated by 3% hydrogen peroxide in distilled water for 10 min at
room temperature, then the slides were blocked by incubation with
protein blocking solution (Dako; Agilent Technologies, Inc., Santa
Clara, CA, USA) at room temperature for 30 min. Subsequently,
primary antibodies against NLRP3 (cat. no. ABF23; Merck KGaA,
Darmstadt, Germany; 1:200; rabbit Ab), ASC (cat. no. ab128097;
Abcam, Cambridge, MA, USA; 1:100; mouse mAb), caspase-1 (cat. no.
2225; Cell Signaling Technology, Inc., Danvers, MA, USA; 1:100;
rabbit Ab), IL-1β (cat. no. 12242; Cell Signaling Technology, Inc.;
1:100; mouse mAb) were applied overnight at 4°C. Following washing
with PBS, Universal prediluted secondary antibody from the
Vecstain™ ABC kit (cat. no. PK-6200; Vector Laboratories, Inc.,
Burlingame, CA, USA) was applied for 30 min at room temperature
followed by washing with PBS. Then, the slides were incubated with
the Vecstain ABC reagent for 30 min at room temperature, washed
with PBS and then stained with 3,3′-diaminobenzidine using
immunoperoxidase staining procedure (cat. no. SK-4105; Vector
Laboratories, Inc.) according to the manufacturer's protocol. The
sections were counterstained with hematoxylin solution (cat. no.
C0107; Beyotime Institute of Technology, Haimen, China) according
to the manufacturer's protocol. Images of the sliders were captured
under a light microscope at ×100 magnification.
Cell culture and transient
transfection
Human glioma SHG44 and A172 cell lines, purchased
from the Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China), were cultured at 37°C with 5% CO2 in
Dulbecco's modified Eagle's medium (DMEM) (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (FBS; Invitrogen; Thermo Fisher Scientific, Inc.).
Glioma cells were divided into five groups: Untransfected,
si-negative control (NC), si-NLRP3, pIRES2-EGFP and
pIRES2-EGFP-NLRP3 groups. To reduce endogenous NLRP3 expression,
glioma cells of 70–80% confluence were transfected with 1
µg/well NLRP3-specific si-NLRP3 (cat. no. siB1261155342;
sequence, 5′-GGTGTTGGAATTAGACAAC-3′) or si-NC (cat. no.
siN05815122147; sequence, 5′-AAGCCUCGAAAUAUCUCCU-3′) (Guangzhou
RiboBio Co., Ltd., Guangzhou, China) dissolved in a transfection
medium mixture of Opti-MEM (Gibco; Thermo Fisher Scientific, Inc.)
and Lipofectamine 3000 (Invitrogen; Thermo Fisher Scientific, Inc.)
at a working concentration of 50 nM according to the manufacturer's
protocol. In parallel, for overexpression of NLRP3 in glioma cells,
transfection with plasmid vectors pIRES2-EGFP-NLRP3 or the negative
control pIRES2-EGFP [2 µg/well; both from Obio Technology
(Shanghai) Corporation, Ltd., Shanghai, China] was performed
according to the manufacturer's protocol. The untransfected group
was only cultured with a transfection medium mixture of Opti-MEM
and Lipofectamine 3000. After 6 h of transfection, the transfection
medium was replaced and cells were cultured for another 24 h in
DMEM with 10% FBS. The pIRES2-EGFP-NLRP3 groups were imaged at ×100
magnification using an inverted phase contrast fluorescent
microscope to assess GFP fluorescence. For confirmation of NLRP3
knockdown or overexpression efficiency, the cells were harvested
and analyzed for NLRP3 protein expression using western blotting.
The transfected glioma cells were subjected for in vitro
functional experiments or western blot analysis experiments
detecting molecular markers as described below.
Western blot analysis
Cells were extracted using ice-cold radio
immunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology). Protein concentration in the cell lysate was
determined using a bicinchoninic acid assay (Beyotime Institute of
Biotechnology). A total of 50 µg protein/lane were loaded
and separated using 10% SDS-PAGE, then transferred to
polyvinylidene fluoride membranes (Merck KGaA), and blocked in
PBS/Tween-20 containing 5% bovine serum albumin (EMD Millipore,
Billerica, MA, USA) for 2 h at room temperature. Subsequently, the
membranes were incubated overnight at 4°C with primary antibodies
against the following: NLRP3 (cat. no. ABF23; Sigma-Aldrich; Merck
KGaA; 1:2,000; rabbit Ab), ASC (cat. no. ab128097; Abcam; 1:500;
mouse mAb), caspase-1 (cat. no. 2225; 1:1,000; rabbit Ab), IL-1β
(cat. no. 12242; 1:1,000; mouse mAb), E-cadherin (cat. no. 3195),
zonula occluden-1 (ZO-1) (cat. no. 8193), claudin-1 (cat. no.
13255), vimentin (cat. no. 5741), N-cadherin (cat. no. 13116),
Snail-1 (cat. no. 3879), AKT (cat. no. 4691), phosphorylated
(p)-AKT (Ser473) (cat. no. 4060), p-AKT (Thr308) (cat. no. 13038),
glycogen synthase kinase-3β (GSK-3β) (cat. no. 12456), p-GSK-3β
(Ser9) (cat. no. 5558), p-PTEN (Ser380) (cat. no. 9551), PTEN (cat.
no. 9188) and β-actin (cat. no. 8457) (all from Cell Signaling
Technology, Inc.; 1:1,000; rabbit Ab). The membranes were then
incubated with the goat anti-rabbit or goat anti-mouse
IgG/horseradish peroxidase-conjugated secondary antibody
(anti-rabbit cat. no. bs-0295G-hrp; anti-mouse cat. no.
bs-0296G-hrp; both 1:5,000; BIOSS, Beijing, China) for 1 h at room
temperature. To visualize protein bands, ECL system (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) was used. The intensity of
protein bands was analyzed using ImageJ version 1.6.0 software
(National Institutes of Health, Bethesda, MD, USA) and normalized
by β-actin.
Colony formation assay
Glioma cells were seeded in 6-well plates at 100
cells/well. Two weeks later, cell colonies were fixed with 4%
paraformaldehyde for 30 min at room temperature and stained with
0.1% crystal violet for 30 min at room temperature. The numbers of
colonies with >50 cells were counted under a light microscope
and the images were captured using a digital camera.
Cell counting kit (CCK)-8 assay
Cell viability was detected using a Cell Counting
kit-8 assay (Dojindo Molecular Technologies, Inc., Kumamoto,
Japan). The glioma cells were seeded into 96-well plates
(2×103 cells/200 µl/well) and cultured at 37°C,
and 10 µl CCK-8 solution was added at 8, 24, 48 and 72 h.
Following incubation for 2 h, the absorbance at 450 nm was
measured.
5-ethynyl-2′-deoxyuridine (EdU) cell
proliferation assay
Cell proliferation rates were measured using an EdU
cell proliferation assay kit (cat. no. C10310; Guangzhou RiboBio
Co., Ltd.) according to the manufacturer's protocol. Briefly,
glioma cells (2×104 cells/well) were cultured in 24-well
plates for 24 h. Then, 10 µM of EdU and Apollo 643 staining
for red fluorescence was added to each well, and the cells were
cultured for an additional 2 h. Next, the cells were fixed with 4%
formaldehyde for 30 min at room temperature. Subsequently, the
cells were stained with Hoechst 33342 for 30 min at room
temperature, and visualized in blue fluorescence using a
fluorescent microscope at ×100 and ×200 magnifications. The
proliferating cells (EdU-positive cells in red) and total cells
(Hoechst 33342-positive cells in blue) were counted using Image-Pro
Plus 6.0 software (Media Cybernetics, Inc., Bethesda, MD, USA). The
ratio of EdU positive cells to total Hoechst 33342 positive cells
was used as cell proliferation rate.
Quantification of cell apoptosis using
flow cytometric assays
Cells were harvested and stained using the Annexin
V-APC/7-AAD kit (BD Biosciences, San Jose, CA, USA) for
quantification of cell apoptosis according to the manufacturer's
protocol. Cell detection and data analysis were performed on a
fluorescent activated cell sorting caliber flow cytometer (FACScan;
BD Biosciences) with CellQuest Pro software (version 5.1; BD
Biosciences).
Wound healing assay
For the wound healing assays, 2×105
glioma cells were seeded on 6-well plates and cultured to 90%
confluence. The confluent cell monolayer was scratched with a
sterile pipette tip. The suspended cells were washed with non-serum
DMEM. The rate of wound closure was monitored by measuring the
ratio of the distance of the wound at 0 h and 24 h under an
inverted microscope with a digital camera in five fields of view at
×100 magnification and quantified using Adobe Photoshop (version
7.0; Adobe Systems, Inc., San Jose, CA, USA). Each experiment was
performed in triplicate.
Cell migration and invasion assays
Cell migration and invasion assays were performed
using 24-well Transwell chambers with 8-µm pore size
polycarbonate membrane (Corning, Cambridge, MA, USA). The
transfected cells were cultured for 48 h and then 5×104
cells were seeded on the top side of the membrane without Matrigel
for the cell migration assay or pre-coated with Matrigel (BD
Biosciences) for the cell invasion assay. A total of 500 µl
DMEM containing 20% FBS was added to the lower chambers. Following
incubation in 12 h for the migration assay or 48 h for the invasion
assay, cells inside the upper chamber were removed with cottons
swabs. Migrated and invaded glioma cells attached to the lower
membrane surface were fixed with 75% methanol for 30 min at room
temperature, and then stained for 30 min with crystal violet at
room temperature. Five random fields of view at ×100 magnification
were counted in each well under a light microscope and expressed as
the mean number of migrated or invaded cells. Three independent
assays were performed.
Statistical analysis
Data are presented as the mean ± standard deviation.
Statistical analyses were performed with SPSS software version 13.0
(SPSS, Inc., Chicago, IL, USA). Spearman's correlation coefficient
was used to assess the correlation between protein expression
levels and glioma WHO grades. Student's t-tests were used to
compare data variances between two groups, and one-way analysis of
variance with the Dunnett's post hoc test was used to compare
difference among multiple groups. P<0.05 (two-tailed) was
considered to indicate a statistically significant difference.
Results
High expression levels of NLRP3, ASC,
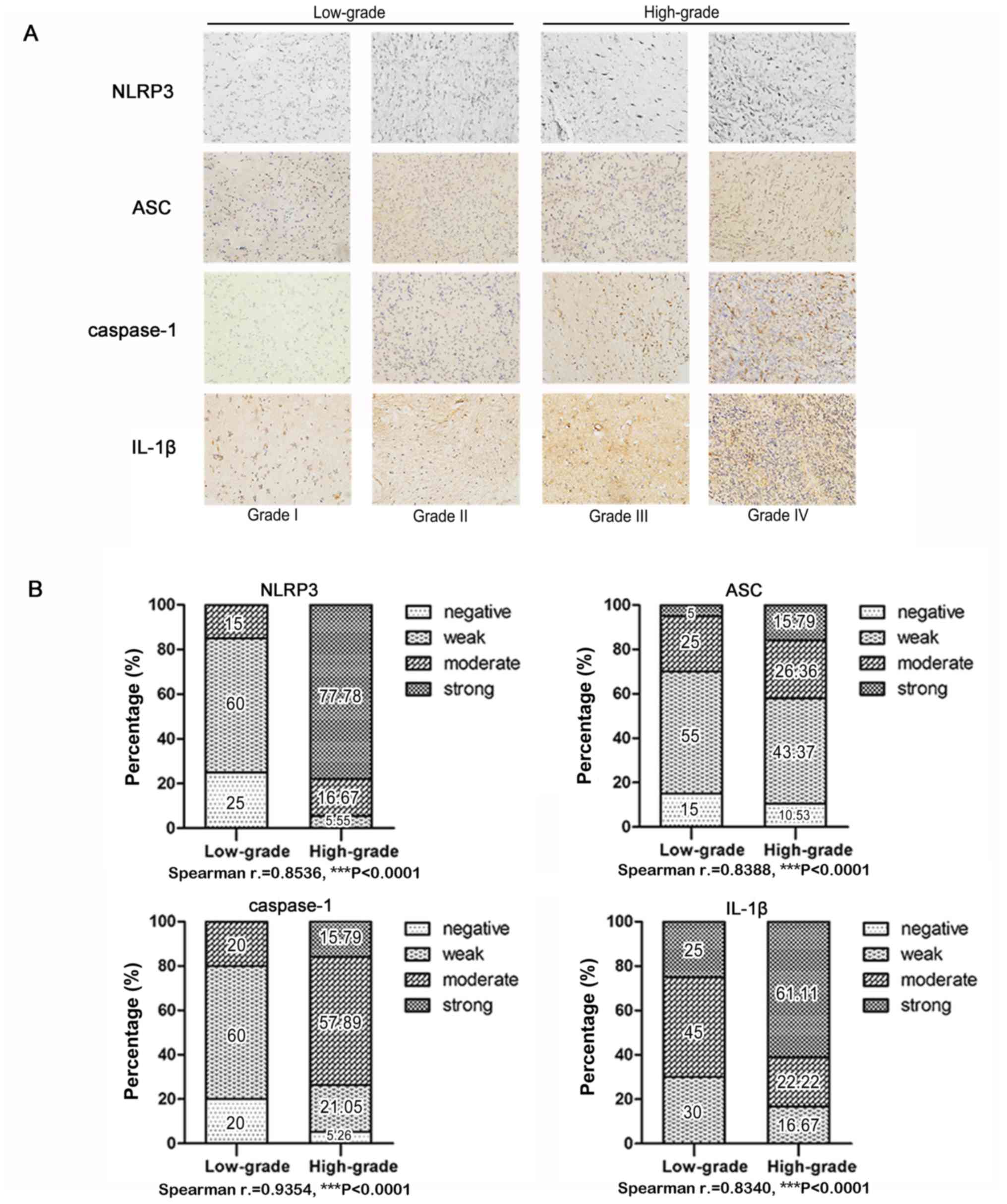
caspase-1 and IL-1β are correlated with glioma WHO grade
NLRP3, ASC, caspase-1 and IL-1β protein compose the
NLRP3 inflammasome. Immunohistochemical detection of human glioma
tissue sections revealed that NLRP3, ASC, caspase-1 and IL-1β
protein expression levels were all upregulated in high-grade glioma
(WHO III and IV) tissue samples compared with in low-grade glioma
(WHO I and II) tissue, as presented in Fig. 1A. Statistical analysis confirmed
significantly positive correlations between NLRP3, ASC, caspase-1
and IL-1β protein expression levels, and the histological grades of
human glioma (Fig. 1B), indicating
an important role for NLRP3 in glioma malignancy.
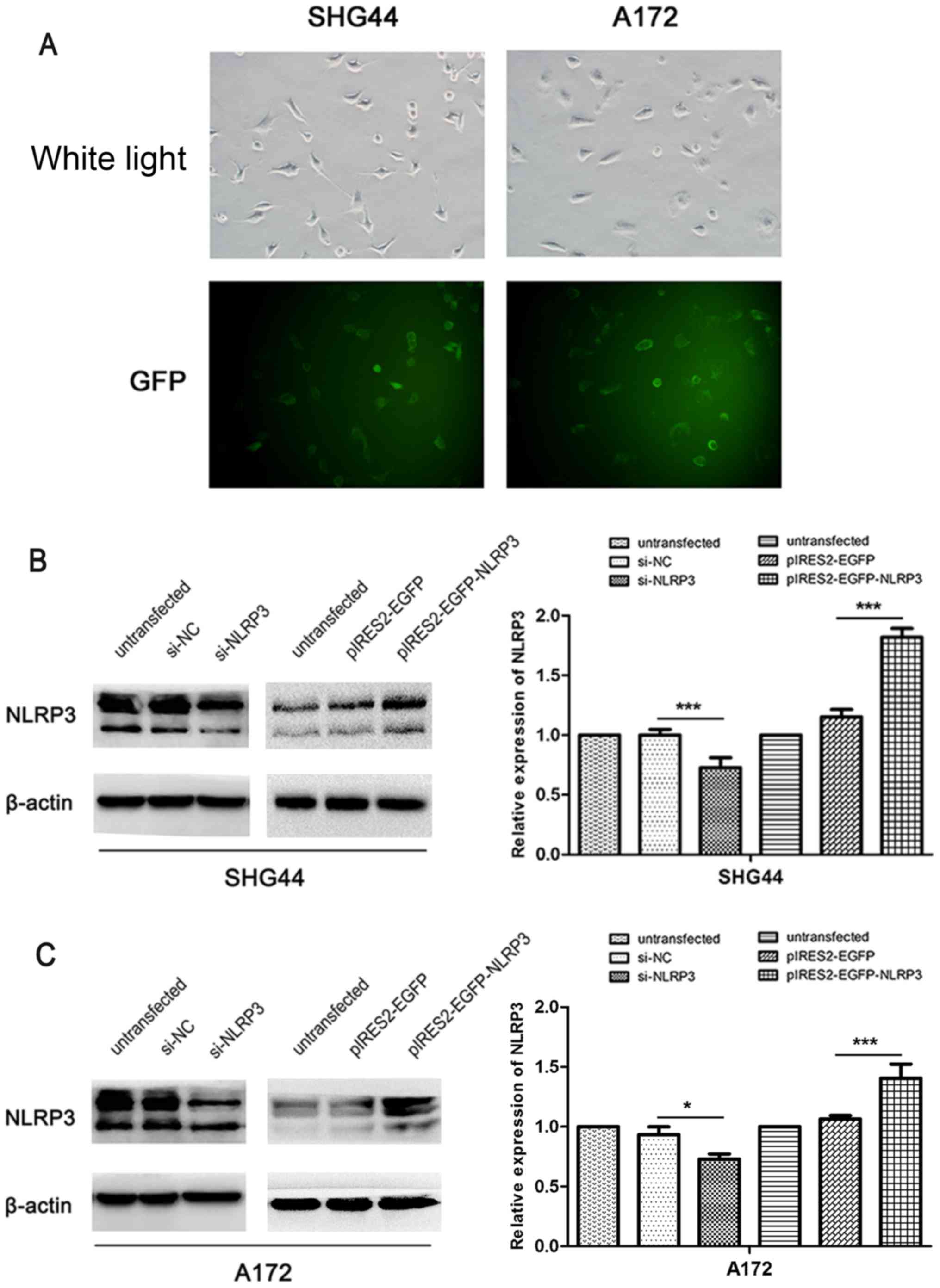
Verification of NLRP3 downregulation and
overexpression efficiencies in glioma cells
To evaluate the role of NLRP3 in the malignant
biological properties of glioma, siRNA-NLRP3 and the plasmid vector
pIRES2-EGFP-NLRP3 were constructed to downregulate and upregulate
NLRP3 expression, respectively, in glioma cell lines SHG44 and
A172. The plasmid vector pIRES2-EGFP-NLRP3 exhibited a transfection
efficiency of >80% in SHG44 and A172 glioma cells as indicated
by GFP fluorescence (Fig. 2A).
Western blot analysis verified a significant reduction in NLRP3
protein levels following the transfection of si-NLRP3 compared with
si-NC (Fig. 2B and C).
Furthermore, pIRES2-EGFP-NLRP3 transfection resulted in a
significant increase in NLRP3 expression compared with the
corresponding untransfected and pIRES2-EGFP control groups. These
results confirmed the efficiency of si-NLRP3 and pIRES2-EGFP-NLRP3
for use in the NLRP3 downregulation or overexpression in
vitro functional experiments in glioma cells.
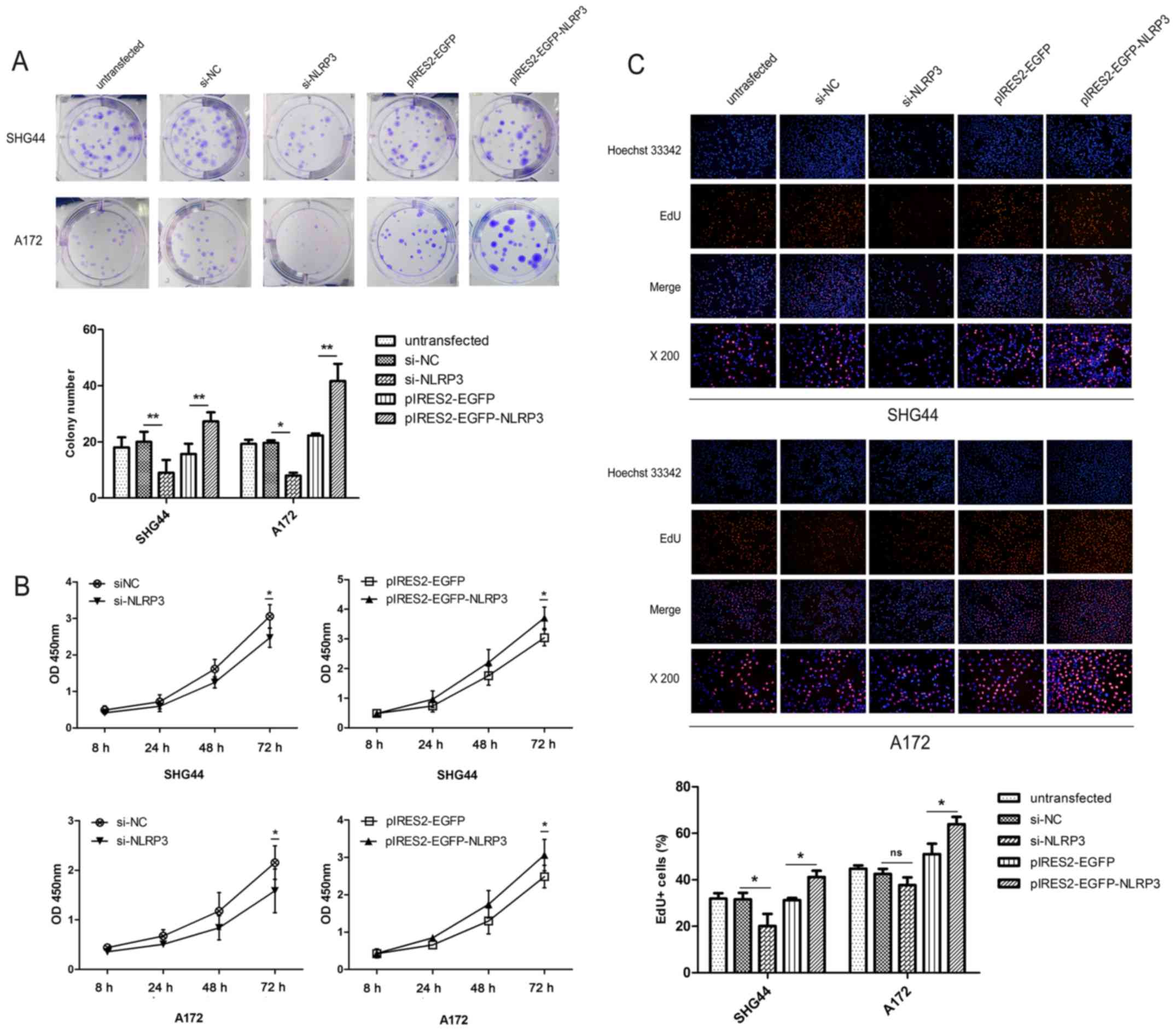
Facilitative effects of NLRP3 on the
proliferation of glioma cells
The effect of NLRP3 on cell proliferation was
assessed using colony formation, CCK-8 and EdU assays. In glioma
SHG44 and A172 cell lines, downregulation of NLRP3 significantly
suppressed colony formation, whereas overexpression of NLRP3 using
pIRES2-EGFP-NLRP3 significantly promoted colony formation compared
with the corresponding untransfected and negative control groups
(Fig. 3A). Then, the growth
effects of NLRP3 on human glioma cell lines were evaluated using
CCK-8 assays. The si-NLRP3 groups demonstrated a significant
reduction in cell viability compared with si-NC group, while NLRP3
overexpression significantly increased the viabilities of human
glioma cell lines compared with the pIRES2-EGFP control groups in a
time-dependent manner (Fig. 3B).
Furthermore, similar effects on cell proliferation were confirmed
using an EdU assay. The ratio of EdU-positive cells to total cell
nuclei was used to determine the cell proliferation rate. The
results revealed that the cell proliferation rate in the si-NLRP3
group was significantly reduced compared with the scramble si-NC
group (Fig. 3C). Furthermore,
pIRES2-EGFP-NLRP3 transfection significantly increased the cell
proliferation rate compared with pIRES2-EGFP group in SHG44 and
A172 glioma cells (Fig. 3C).
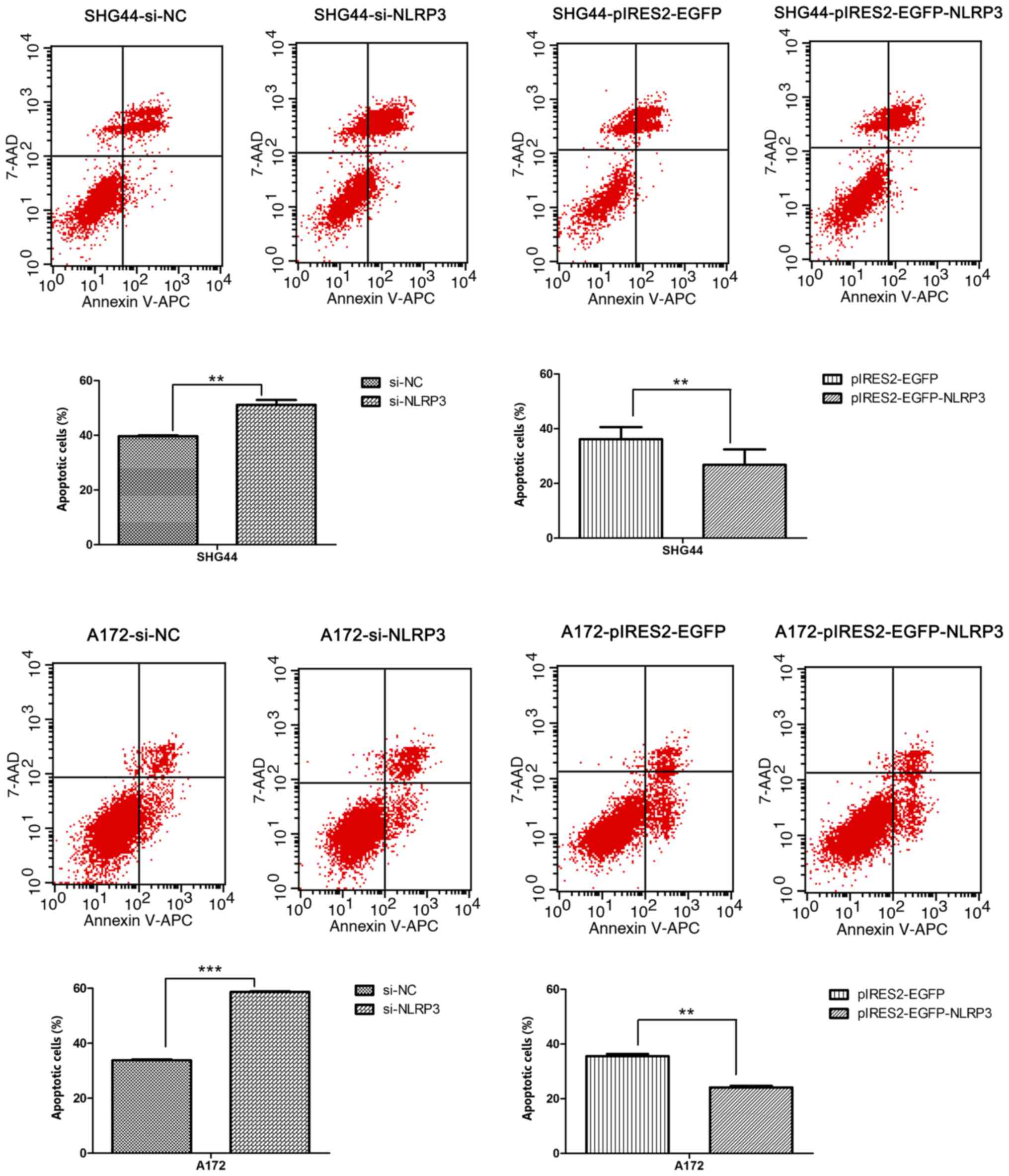
Inhibitory effects of NLRP3 on glioma
cell apoptosis
To further investigate the effect of NLRP3 on the
cell apoptosis, Annexin V-APC and 7-AAD staining of human glioma
cells was performed, and the apoptosis rate of each group was
measured by flow cytometry after 48 h of transfection.
Downregulation of NLRP3 by si-NLRP3 significantly induced apoptosis
of SHG44 and A172glioma cell lines compared with the si-NC group
(Fig. 4). Following overexpression
of NLRP3, the apoptosis rates of pIRES2-EGFP-NLRP3 groups were
significantly reduced compared with that of the pIRES2-EGFPgroupsin
SHG44 and A172 glioma cells (Fig.
4).
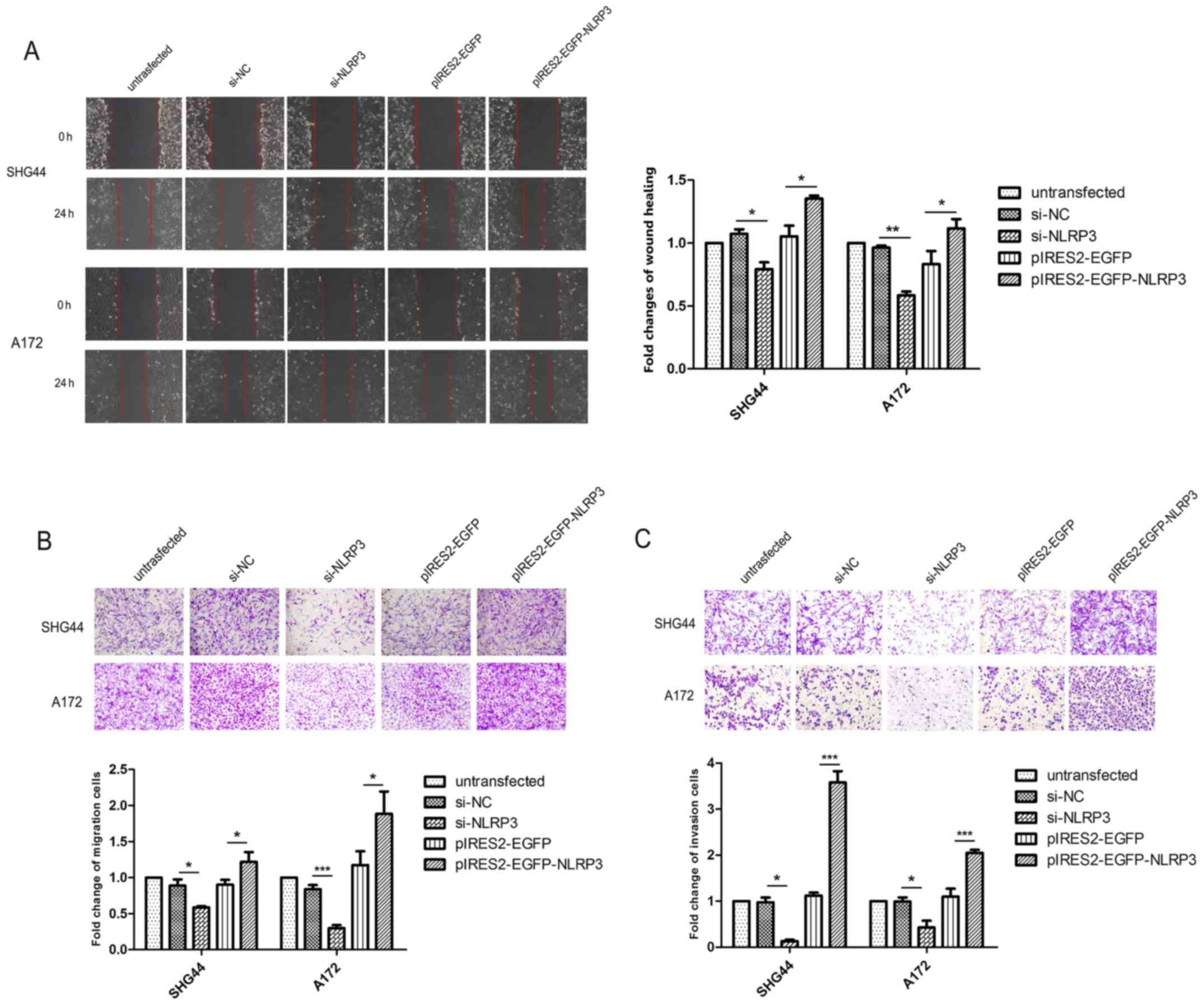
NLRP3 promotes glioma cell migration and
invasion
To explore the effect of NLRP3 on glioma cell
migration and invasion, wound healing assays, and Transwell
migration and invasion assays were performed. The wound healing
assays demonstrated that the migratory ability of glioma cells was
significantly suppressed following transfection of si-NLRP3 and
pIRES2-EGFP-NLRP3 significantly accelerated the wound closure
compared with the corresponding negative control groups (Fig. 5A). In the Transwell migration
assays, the number of migrated glioma cells in thesi-NLRP3 group
was significantly decreased compared with untransfected group and
si-NC groups (Fig. 5B).
Accordingly, pIRES2-EGFP-NLRP3 transfection significantly increased
cell migratory abilities in SHG44 and A172 glioma cell lines
(Fig. 5B). The results of the
Transwell invasion assays revealed that, compared with the
untransfected and si-NC groups, si-NLRP3 significantly reduced the
number of glioma cells that invaded through the Matrigel-coated
membrane at 48 h, whereas pIRES2-EGFP-NLRP3 significantly promoted
the invasiveness of glioma cells (Fig.
5C).
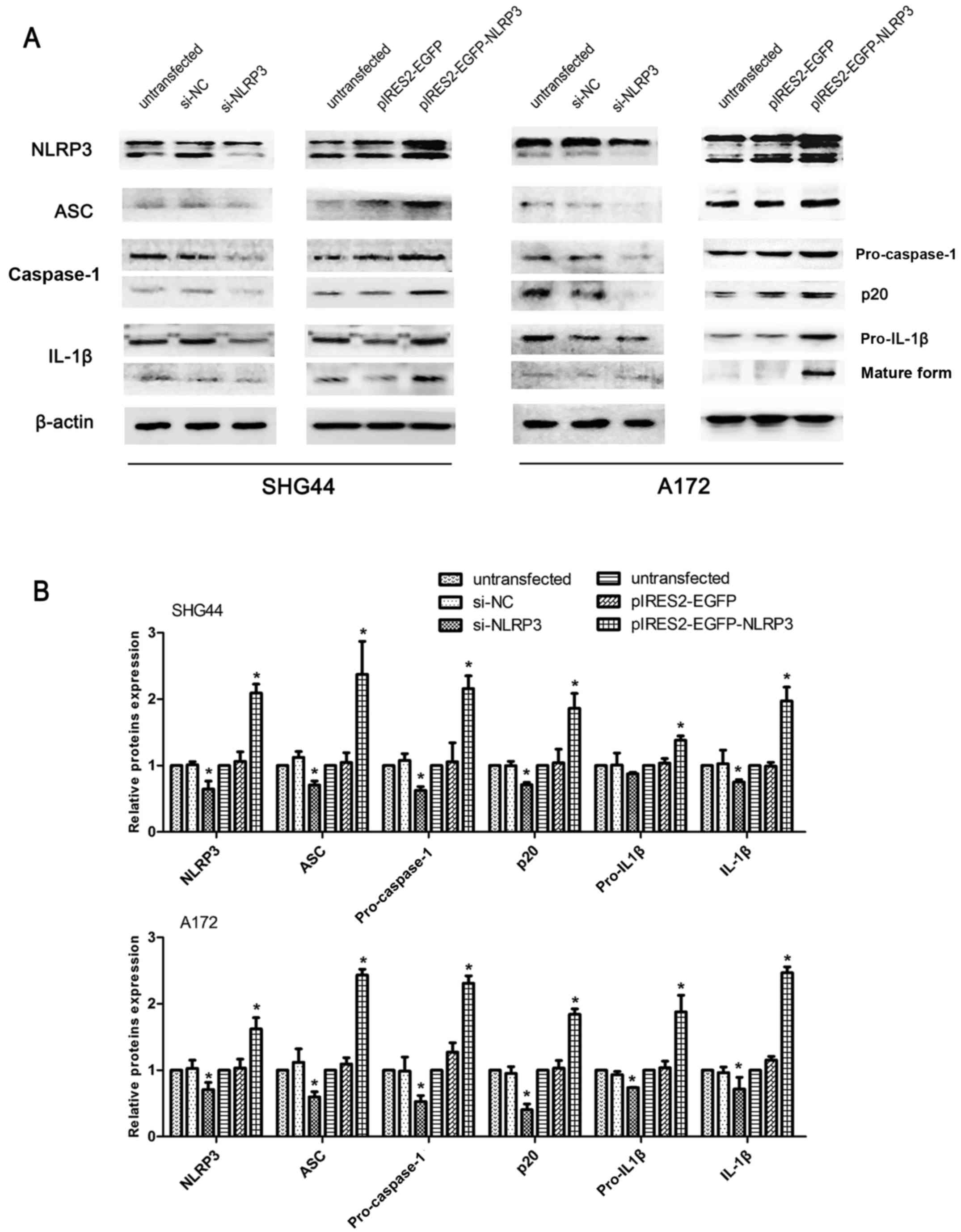
Regulatory effects of NLRP3 on
intracellular inflammasome activation, EMT and the AKT pathway in
glioma cells
Western blot analysis was performed on molecules of
the NLRP3 inflammasome complex, ASC, pro-caspase-1 and pro-IL-1β,
and molecules that indicate inflammasome activation,
cleaved-caspase-1 (p20) and mature IL-1β (17 kDa) in SHG44 and A172
glioma cell lines. The protein bands of pro-caspase-1 and
cleaved-caspase-1 (p20) were visualized using the same primary
antibodies against caspase-1 at 50 and 25 kDa, respectively.
Similarly, pro-IL-1β and mature IL-1β were visualized using the
same primary antibodies against IL-1β at 35 and 17 kDa,
respectively. Compared with the untransfected and si-NC groups,
si-NLRP3 transfection significantly decreased the protein
expression of all molecules of the NLRP3 inflammasome complex, and
cleaved-caspase-1 and mature IL-1β. Furthermore, NLRP3
overexpression significantly increased the expression levels of all
6 inflammasome complex-associated markers compared with the
untransfected and pIRES2-EGFP negative control groups (Fig. 6).
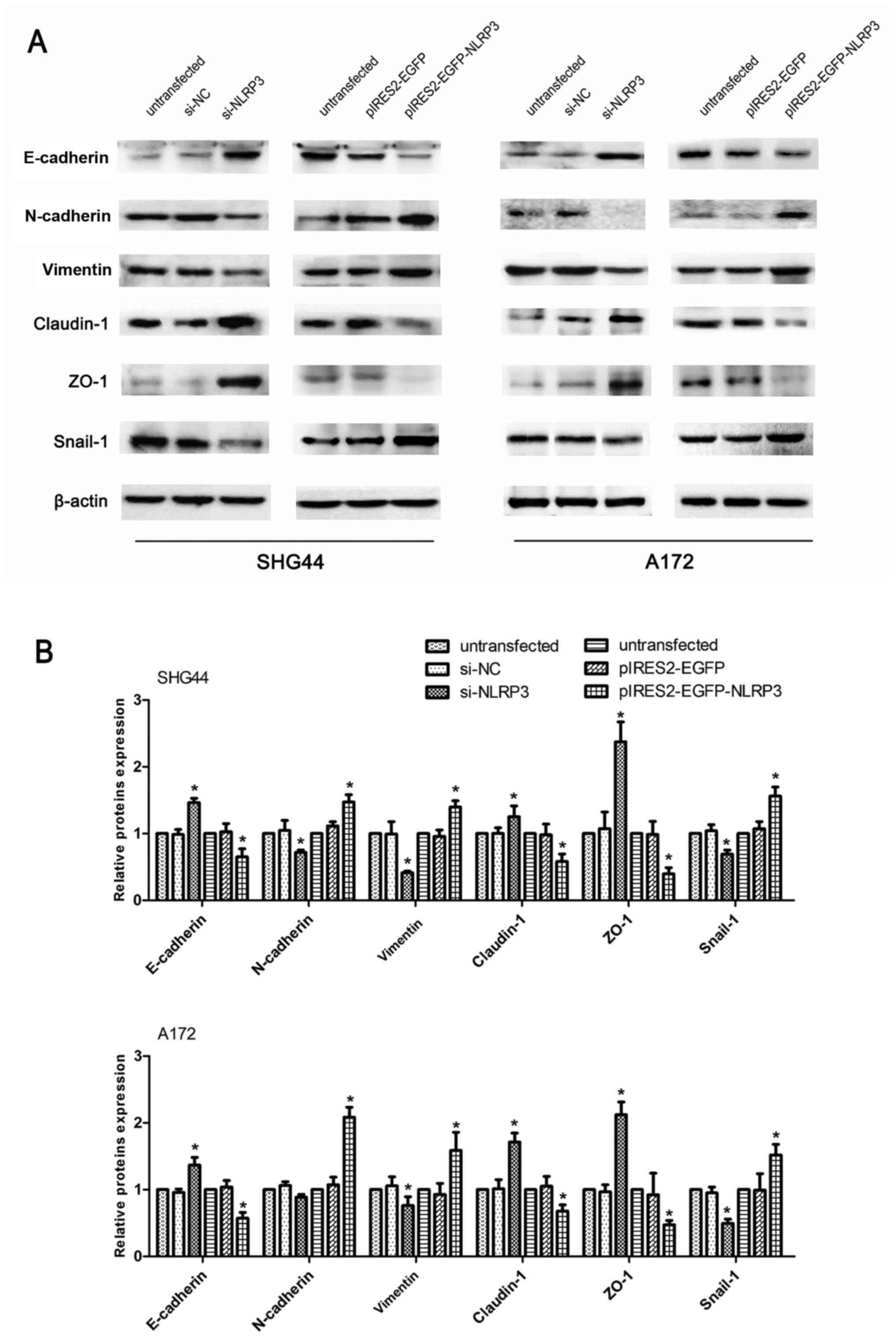
As indicated by the aforementioned results, NLRP3
demonstrated an intimate association with tumor malignances
involving apoptosis, proliferation, migration and invasion in human
glioma cells. EMT is widely regarded as a major contributor to
increased tumor metastasis, resulting in a higher capacity of
cellular migration and invasion (19,20).
Furthermore, cellular junctions serve an essential role in
structural connection, which may be involved in cancer metastasis
(21). The major components of
tight junctions include ZO-1) and claudin-1. To further explore the
facilitative effect of NLRP3 on the migration and invasion of
glioma cells, western blot analysis assays were performed on
molecules associated with EMT and cellular tight junctions. The
results revealed that si-NLRP3 significantly downregulated the
expression levels of mesenchymal proteins (N-cadherin and vimentin)
and EMT-associated transcription factor Snail-1, while it
upregulated the expression of epithelial markers and cellular tight
junctions proteins (E-cadherin, ZO-1 and claudin-1) compared with
the untransfected and si-NC groups. Compared with the untransfected
and control vector groups, the pIRES2-EGFP-NLRP3 group
significantly reduced E-cadherin, ZO-1 and claudin-1 protein
expression, and increased the expression of N-cadherin, vimentin
and snail-1 (Fig. 7).
The AKT signaling pathway has been associated with
the development of glioma malignances via its inhibitory effect on
apoptosis, as well as its promoter roles on proliferation,
migration and invasion (22,23).
As indicated in Fig. 8, p-AKT
(Ser473), p-AKT (Thr308) and p-GSK3β (Ser9) expression levels were
significantly reduced in the si-NLRP3 group, and significantly
increased in NLRP3-overexpressing glioma cells compared with
untransfected groups and negative control groups. Notably, no
marked differences in total AKT and GSK3β protein levels were noted
in the knockdown and overexpression experiments. Furthermore, the
si-NLRP3 groups reduced p-PTEN levels, while overexpression of
NLRP3 significantly attenuated the phosphorylation of PTEN with no
marked differences observed on total PTEN protein expression in
glioma cell lines (Fig. 8).
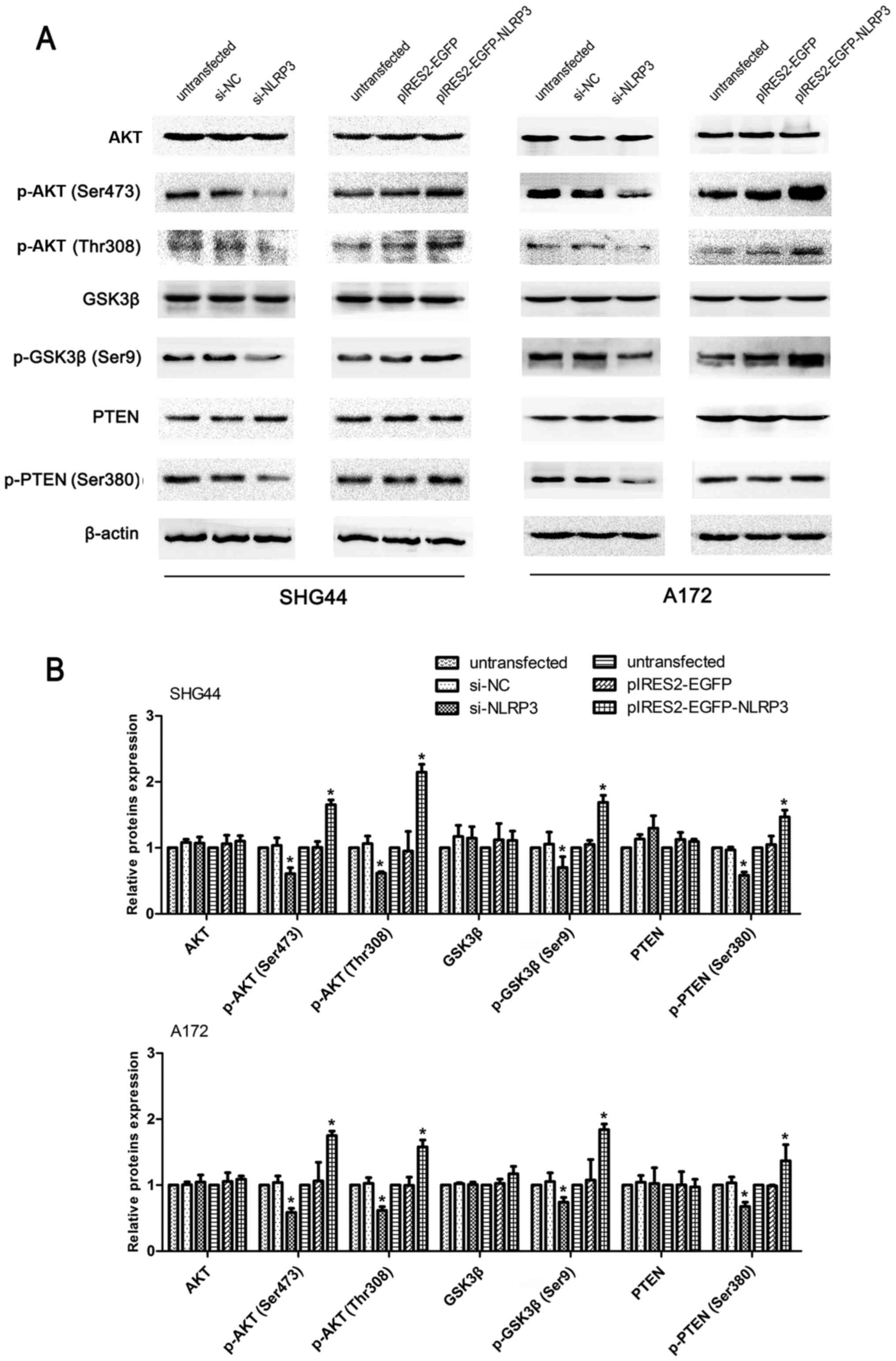 | Figure 8Regulatory effects of NLRP3 on the
PTEN/AKT signaling pathway in glioma cells. (A) Western blotting
was used to determine the level of proteins associated with
PTEN/AKT signaling pathway, including total AKT, p-AKT (Ser473),
p-AKT (Thr308), GSK3β, p-GSK3β (Ser9), and its regulators p-PTEN
(Ser380) and total PTEN. (B) The intensity of protein bands was
measured and the values are presented as the mean ± standard
deviation (n=3). *P<0.05. NLRP3, NLR family pyrin
domain containing 3; NC, negative control; EGFP, enhanced green
fluorescent protein; AKT serine/threonine kinase; GSK3β, glycogen
synthase kinase-3β; PTEN, phosphatase and tensin homolog. |
Discussion
The expression and function of the NLRP3 has been
previously investigated in inflammatory and autoimmune diseases
(6,7). Notably, there is an increasing amount
of evidence indicating an important role of NLRP3 in the
carcinogenesis of various tumors; however, the results appear to be
inconsistent among different types of cancer (10–12,15,24–26).
NLRP3 tissue expression and its function in glioma remain
unclear.
In the present study, upregulated protein expression
of NLRP3, ASC, caspase-1 and IL-1β in human glioma tissue was
significantly associated with higher WHO grades as confirmed using
immunohistochemical assays. These results provided a foundation for
further NLRP3 functional studies and suggesting a promising role
for NLRP3 in glioma progression. Furthermore, western blot assays
demonstrated that NLRP3 significantly affected the expression of
other constructive molecules of the inflammasome complex, including
ASC, caspase-1 and pro-IL-1β, as well as inflammasome-activated
status markers, cleaved-caspase1 and mature IL-1β. These finding
implied that the overexpression of NLRP3 in human glioma may
elevate the expression of other inflammasome complex proteins and
the cellular inflammasome-activated status. Subsequently, the in
vitro biological function experiments and western blot assays
identified that NLRP3 participated in the regulation of glioma
cellular proliferation, apoptosis, migration, invasion, EMT and the
PTEN/AKT signaling pathway.
A previous study reported that the assembly and
activation of the NLRP3 inflammasome complex, constructed of NLRP3,
ASC, caspase-1 and pro-IL-1β proteins, functions as a negative
regulator in the carcinogenesis of colitis-associated cancer
(27). In hepatocellular carcinoma
(HCC), the NLRP3 inflammasome complex was identified to be
downregulated and its upregulation induced by the
17β-estradiol/estrogen receptor β/mitogen-activated protein kinase
signaling pathway attributes to inhibition of HCC cell malignancy
(14,24). However, Wang et al (17) reported that NLRP3 expression, but
not inflammasome activation, is required for the EMT process in
colorectal cancer cells through regulation of Snail-1. It was also
suggested that NLRP3 contributes to renal fibrosis by promoting
transforming growth factor-β signaling and R-Smad activation,
independent of inflammasome activation and cleaved-caspase-1
production in renal tubular epithelial cells (16). In lung cancer, NLRP3 mediates the
release of IL-1β and IL-18 through a caspase-1-dependent or
independent pathway (28). NLRP3
also enhances the proliferation and migration of A549 cells, the
activation of phosphorylation of AKT and the expression of Snail-1,
while downregulating the expression of E-cadherin (28). The contradiction in the
aforementioned studies suggests that NLRP3 may affect tumorigenesis
and progression independent of its involvement in the inflammasome
complex.
As one of the most important effectors of the NLRP3
inflammasome upon activation, IL-1β is an essential cytokine in the
glioma inflammatory microenvironment (29), and considered a potent enhancer of
glioma proliferation and metastasis (30–32).
Tarassishin et al (33)
reported that human glioma cells produce IL-1β upon highly
sensitive NLRP3 inflammasome activation; however, in human normal
astrocytes, NLRP3 inflammasome activation and mature IL-1β protein
production are suppressed almost entirely, indicating that the
ability of glioma cells to activate the NLRP3 inflammasome and
produce IL-1β may lead to the development of an aggressively
oncogenetic phenotype (33).
Numerous signaling pathways are involved in
regulating the malignant anti-apoptotic and proliferative
characteristics of glioma, of these molecular mechanisms the AKT
signaling pathway is particularly important (34–36).
The mutation/deletion of the tumor suppressor PTEN is an inhibitive
regulator of the AKT signaling pathway (37,38).
Furthermore, EMT is considered one of the most important processes
that contribute to glioma progression and metastasis (19,20).
Consistent with the aforementioned studies, the observations in the
present study identified that NLRP3 functioned as a promoter of
glioma cell migration and invasion by increasing Snail-1
expression, subsequently inducing EMT. Additionally, NLRP3
alleviated cellular apoptosis, promoted cell proliferation upon
activation of the AKT signaling pathway and resulted in a reduction
in PTEN phosphorylation.
A previous study suggested that the induction of
p-PTEN induces a loss of PTEN activity, resulting in the activation
of the AKT signaling pathway (39). Mizushina et al (40) demonstrated that NLRP3−/−
mice exhibited diminished signal transducer and activator of
transcription 3 (STAT3) expression in an inflammasome-independent
way. STAT3 is a transcription factor that regulates various genes,
including polo like kinase 1 (PLK1), a phosphatase of PTEN that
leads to its inactivation and blockade of AKT signaling pathway
(41,42). In addition, the upregulation of
STAT3 enhances Snail expression (43), and increased Snail-1 expression was
observed in the NLRP3 overexpression model used in the present
study. Thus, we hypothesized that the NLRP3/STAT3/PLK1 signaling
axis is involved in the regulation of NLRP3 on the PTEN/AKT
signaling pathway. In parallel, reduced Smad2 phosphorylation
combined with less Smad2/3 translocation have been reported in
NLRP3-deficient B6lpr mice (44).
Furthermore, reduced Smad2/3 phosphorylation results in decreased
binding with Smad4 and impaired translocation of the Smad2/3/4
complex into the nucleus (45,46).
The Smad complex has been confirmed as a direct binding
site-targeted PTEN genetic promoter, which subsequently regulates
PTEN transcription (45,46). There appears to be multiple
pathways and complex crosstalk connecting NLRP3 to PTEN/AKT. The
results of the present study indicate that NLRP3 regulates glioma
malignancy through enhanced inflammasome activation, and undefined
inflammasome-independent mechanisms that target EMT and the AKT
signaling pathway by inducing PTEN phosphorylation. The in depth
regulatory mechanism between NLRP3 and PTEN remains to be explored,
and will be the focus of our future investigations.
To the best of our knowledge, this is the first
study to investigate the association between NLRP3 protein
expression and the WHO grades of human glioma. The findings of the
present study suggest that NLRP3 may function as a tumor promoter,
and that the aberrant activation of the AKT signaling pathway and
EMT process caused by NLRP3 upregulation may provide a novel
insight into the underlying mechanism of NLRP3 in glioma
malignancy.
Acknowledgments
The authors would like to thank Professor Songtao
Qi, Professor Yawei Liu and Dr Guozhong Yi (Department of
Neurosurgery, Nanfang Hospital, Southern Medical University) for
providing experimental suggestion and technical aid in
immunohistochemical analysis of human glioma tissue samples.
References
|
1
|
Wen PY and Reardon DA: Neuro-oncology in
2015: Progress in glioma diagnosis, classification and treatment.
Nat Rev Neurol. 12:69–70. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mao K, Lei D, Zhang H and You C:
MicroRNA-485 inhibits malignant biological behaviour of
glioblastoma cells by directly targeting PAK4. Int J Oncol.
51:1521–1532. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Omuro A and DeAngelis LM: Glioblastoma and
other malignant gliomas: A clinical review. JAMA. 310:1842–1850.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Olar A and Sulman EP: Molecular markers in
low-grade glioma-toward tumor reclassification. Semin Radiat Oncol.
25:155–163. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Christofides A, Kosmopoulos M and Piperi
C: Pathophysiological mechanisms regulated by cytokines in gliomas.
Cytokine. 71:377–384. 2015. View Article : Google Scholar
|
|
6
|
Strowig T, Henao-Mejia J, Elinav E and
Flavell R: Inflammasomes in health and disease. Nature.
481:278–286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lamkanfi M and Dixit VM: Mechanisms and
functions of inflammasomes. Cell. 157:1013–1022. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Haneklaus M, O'Neill LA and Coll RC:
Modulatory mechanisms controlling the NLRP3 inflammasome in
inflammation: Recent developments. Curr Opin Immunol. 25:40–45.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
He Y, Hara H and Núñez G: Mechanism and
regulation of NLRP3 inflammasome activation. Trends Biochem Sci.
41:1012–1021. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kent A and Blander JM: Nod-like receptors:
Key molecular switches in the conundrum of cancer. Front Immunol.
5:1852014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Terlizzi M, Casolaro V, Pinto A and
Sorrentino R: Inflammasome: Cancer's friend or foe. Pharmacol Ther.
143:24–33. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Paugh SW, Bonten EJ, Savic D, Ramsey LB,
Thierfelder WE, Gurung P, Malireddi RK, Actis M, Mayasundari A, Min
J, et al: NALP3 inflammasome upregulation and CASP1 cleavage of the
glucocorticoid receptor cause glucocorticoid resistance in leukemia
cells. Nat Genet. 47:607–614. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schneider SL, Ross AL and Grichnik JM: Do
inflammatory pathways drive melanomagenesis. Exp Dermatol.
24:86–90. 2015. View Article : Google Scholar
|
|
14
|
Wei Q, Guo P, Mu K, Zhang Y, Zhao W, Huai
W, Qiu Y, Li T, Ma X, Liu Y, et al: Estrogen suppresses
hepatocellular carcinoma cells through ERβ-mediated upregulation of
the NLRP3 inflammasome. Lab Invest. 95:804–816. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sharma N and Jha S: NLR-regulated pathways
in cancer: Opportunities and obstacles for therapeutic
interventions. Cell Mol Life Sci. 73:1741–1764. 2016. View Article : Google Scholar
|
|
16
|
Wang W, Wang X, Chun J, Vilaysane A, Clark
S, French G, Bracey NA, Trpkov K, Bonni S, Duff HJ, et al:
Inflammasome-independent NLRP3 augments TGF-β signaling in kidney
epithelium. J Immunol. 190:1239–1249. 2013. View Article : Google Scholar
|
|
17
|
Wang H, Wang Y, Du Q, Lu P, Fan H, Lu J
and Hu R: Inflammasome-independent NLRP3 is required for
epithelial-mesenchymal transition in colon cancer cells. Exp Cell
Res. 342:184–192. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kahlert UD, Nikkhah G and Maciaczyk J:
Epithelial-to-mesenchymal(-like) transition as a relevant molecular
event in malignant gliomas. Cancer Lett. 331:131–138. 2013.
View Article : Google Scholar
|
|
20
|
Ling G, Ji Q, Ye W, Ma D and Wang Y:
Epithelial-mesenchymal transition regulated by p38/MAPK signaling
pathways participates in vasculogenic mimicry formation in SHG44
cells transfected with TGF-β cDNA loaded lentivirus in vitro and in
vivo. Int J Oncol. 49:2387–2398. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhuang Y, Peng H, Mastej V and Chen W:
MicroRNA regulation of endothelial junction proteins and clinical
consequence: MicroRNA regulation of endothelial junction proteins
and clinical consequence. Mediators Inflamm. 2016:50786272016.
View Article : Google Scholar
|
|
22
|
Tang M, Zhao Y, Liu N, Chen E, Quan Z, Wu
X and Luo C: Overexpression of HepaCAM inhibits bladder cancer cell
proliferation and viability through the AKT/FoxO pathway. J Cancer
Res Clin Oncol. 143:793–805. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lim SY, Menzies AM and Rizos H: Mechanisms
and strategies to overcome resistance to molecularly targeted
therapy for melanoma. Cancer. 123(S11): 2118–2129. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wei Q, Mu K, Li T, Zhang Y, Yang Z, Jia X,
Zhao W, Huai W, Guo P and Han L: Deregulation of the NLRP3
inflammasome in hepatic parenchymal cells during liver cancer
progression. Lab Invest. 94:52–62. 2014. View Article : Google Scholar
|
|
25
|
Kong H, Wang Y, Zeng X, Wang Z, Wang H and
Xie W: Differential expression of inflammasomes in lung cancer cell
lines and tissues. Tumour Biol. 36:7501–7513. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li Y, Li J, Li S, Li Y, Wang X, Liu B, Fu
Q and Ma S: Curcumin attenuates glutamate neurotoxicity in the
hippocampus by suppression of ER stress-associated TXNIP/NLRP3
inflammasome activation in a manner dependent on AMPK. Toxicol Appl
Pharmacol. 286:53–63. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Allen IC, TeKippe EM, Woodford RM, Uronis
JM, Holl EK, Rogers AB, Herfarth HH, Jobin C and Ting JP: The NLRP3
inflammasome functions as a negative regulator of tumorigenesis
during colitis-associated cancer. J Exp Med. 207:1045–1056. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang Y, Kong H, Zeng X, Liu W, Wang Z, Yan
X, Wang H and Xie W: Activation of NLRP3 inflammasome enhances the
proliferation and migration of A549 lung cancer cells. Oncol Rep.
35:2053–2064. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Munoz L, Yeung YT and Grewal T: Oncogenic
Ras modulates p38 MAPK-mediated inflammatory cytokine production in
glioblastoma cells. Cancer Biol Ther. 17:355–363. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fathima Hurmath K, Ramaswamy P and
Nandakumar DN: IL-1β microenvironment promotes proliferation,
migration, and invasion of human glioma cells. Cell Biol Int.
38:1415–1422. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sun W, Depping R and Jelkmann W:
Interleukin-1β promotes hypoxia-induced apoptosis of glioblastoma
cells by inhibiting hypoxia-inducible factor-1 mediated
adrenomedullin production. Cell Death Dis. 5:e10202014. View Article : Google Scholar
|
|
32
|
Tarassishin L, Lim J, Weatherly DB,
Angeletti RH and Lee SC: Interleukin-1-induced changes in the
glioblastoma secretome suggest its role in tumor progression. J
Proteomics. 99:152–168. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tarassishin L, Casper D and Lee SC:
Aberrant expression of interleukin-1β and inflammasome activation
in human malignant gliomas. PLoS One. 9:e1034322014. View Article : Google Scholar
|
|
34
|
Zheng X, Jiang F, Katakowski M, Zhang ZG,
Lu QE and Chopp M: ADAM17 promotes breast cancer cell malignant
phenotype through EGFR-PI3K-AKT activation. Cancer Biol Ther.
8:1045–1054. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Song H, Zhang Y, Liu N, Zhao S, Kong Y and
Yuan L: miR-92a-3p exerts various effects in glioma and glioma
stem-like cells specifically targeting CDH1/β-catenin and
Notch-1/Akt signaling pathways. Int J Mol Sci. 17:E17992016.
View Article : Google Scholar
|
|
36
|
Li X, Wu C, Chen N, Gu H, Yen A, Cao L,
Wang E and Wang L: PI3K/Akt/mTOR signaling pathway and targeted
therapy for glioblastoma. Oncotarget. 7:33440–33450.
2016.PubMed/NCBI
|
|
37
|
Mueller S, Phillips J, Onar-Thomas A,
Romero E, Zheng S, Wiencke JK, McBride SM, Cowdrey C, Prados MD,
Weiss WA, et al: PTEN promoter methylation and activation of the
PI3K/Akt/mTOR pathway in pediatric gliomas and influence on
clinical outcome. Neuro-oncol. 14:1146–1152. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Moon SH, Kim DK, Cha Y, Jeon I, Song J and
Park KS: PI3K/Akt and Stat3 signaling regulated by PTEN control of
the cancer stem cell population, proliferation and senescence in a
glioblastoma cell line. Int J Oncol. 42:921–928. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang F, Wang Y, Sun P, Wang ZQ, Wang DS,
Zhang DS, Wang FH, Fu JH, Xu RH and Li YH: Fibrinogen promotes
malignant biological tumor behavior involving
epithelial-mesenchymal transition via the p-AKT/p-mTOR pathway in
esophageal squamous cell carcinoma. J Cancer Res Clin Oncol.
143:2413–2424. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mizushina Y, Shirasuna K, Usui F, Karasawa
T, Kawashima A, Kimura H, Kobayashi M, Komada T, Inoue Y, Mato N,
et al: NLRP3 protein deficiency exacerbates hyperoxia-induced
lethality through Stat3 protein signaling independent of
interleukin-1β. J Biol Chem. 290:5065–5077. 2015. View Article : Google Scholar
|
|
41
|
Morris EJ, Kawamura E, Gillespie JA, Balgi
A, Kannan N, Muller WJ, Roberge M and Dedhar S: Stat3 regulates
centrosome clustering in cancer cells via Stathmin/PLK1. Nat
Commun. 8:152892017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cao YY, Yu J, Liu TT, Yang KX, Yang LY,
Chen Q, Shi F, Hao JJ, Cai Y, Wang MR, et al: Plumbagin inhibits
the proliferation and survival of esophageal cancer cells by
blocking STAT3-PLK1-AKT signaling. Cell Death Dis. 9:172018.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Saitoh M, Endo K, Furuya S, Minami M,
Fukasawa A, Imamura T and Miyazawa K: STAT3 integrates cooperative
Ras and TGF-β signals that induce Snail expression. Oncogene.
35:1049–1057. 2016. View Article : Google Scholar
|
|
44
|
Lech M, Lorenz G, Kulkarni OP, Grosser MO,
Stigrot N, Darisipudi MN, Günthner R, Wintergerst MW, Anz D,
Susanti HE, et al: NLRP3 and ASC suppress lupus-like autoimmunity
by driving the immunosuppressive effects of TGF-β receptor
signalling. Ann Rheum Dis. 74:2224–2235. 2015. View Article : Google Scholar
|
|
45
|
Xiong S, Cheng JC, Klausen C, Zhao J and
Leung PC: TGF-β1 stimulates migration of type II endometrial cancer
cells by downregulating PTEN via activation of SMAD and ERK1/2
signaling pathways. Oncotarget. 7:61262–61272. 2016.PubMed/NCBI
|
|
46
|
Eritja N, Felip I, Dosil MA, Vigezzi L,
Mirantes C, Yeramian A, Navaridas R, Santacana M, Llobet-Navas D,
Yoshimura A, et al: Smad3-PTEN regulatory loop controls
proliferation and apoptotic responses to TGF-beta in mouse
endometrium. Cell Death Differ. 24:1443–1458. 2017. View Article : Google Scholar : PubMed/NCBI
|