Introduction
Glioblastoma multiforme (GBM) is the most common
primary malignant brain tumor in adults. Despite treatment
optimization and improved standard of care, the median survival of
patients with GBM is only 14.5-16.6 months (1). Thus, the development of novel
therapeutic targets is crucial for the future treatment of GBM.
Over the last decade, substantial efforts have been
focused on elucidating the molecular characteristics of GBM, in
order to identify molecular subsets as targets for specific
targeted therapies, potentially improving the outcome (2). The most common genetic
characteristics of GBM are activated oncogenic receptor tyrosine
kinase (RTK) signaling pathways via epidermal growth factor
receptor (EGFR) or platelet-derived growth factor receptor (PDGFR),
and inactivated tumor suppressor pathways regulated by
retinoblastoma protein and tumor protein p53 (3). Among these, PDGFR signaling plays a
key role in the pathogenesis of GBM and represents a target for
tyrosine kinase inhibitors (4),
whereas PDGFR inhibitors to date have failed to elicit significant
responses in patients with GBM. Due to the key role of PDGFR
activation in GBM progression and the yet obscure mechanisms
underlying the poor clinical outcomes with PDGFR inhibitors, the
understanding of the endogenous regulators of PDGFR signaling has
been an area of intensive research.
The leucine-rich repeats and immunoglobulin-like
domains (LRIG) gene family is composed of three paralogs, namely
LRIG1, LRIG2 and LRIG3, which encode a family of integral membrane
proteins, with a signal peptide, an extracellular part consisting
of 15 leucine-rich repeats (LRR) and three immunoglobulin-like
domains, followed by a transmembrane domain and a cytoplasmic tail
(5). LRIG proteins have attracted
attention as key regulators of growth factor receptors, including
receptor tyrosine and serine/threonine kinases (6). LRIG1, the most extensively
investigated LRIG family member, is considered to be a tumor
suppressor by negatively regulating the signaling pathways mediated
by ErbB (7), MET (8) and RET (9) receptor tyrosine kinases. LRIG1 is
downregulated and associated with a favorable prognosis in various
types of cancer (6,10-15),
including glioma (16,17); however, our knowledge on the
biological and molecular functions of mammalian LRIG2 in tumors is
limited. It has been reported that LRIG2 expression is associated
with poor survival in oligodendroglioma (18) and uterine cervical carcinoma
(19), and wild-type mice
developed PDGFB-induced gliomas at a higher frequency and of higher
malignancy compared with LRIG2-deficient mice (20). Over the past decade, our research
team has focused on the functions of LRIGs in gliomagenesis, and
demonstrated that LRIG2 promotes the growth of GBM by positively
modulating EGFR-mediated signaling (21,22),
which adds to the evidence supporting the hypothesis that LRIG2 may
play an oncogenic role in the progression of glioma, possibly
contrary to the role of LRIG1 (23,24);
however, more compelling evidence is required to support the
concept of LRIG2 as a tumor promoter in GBM and to elucidate the
mechanistic insights.
In the present study, the role of LRIG2 in the
progression of GBM and the possible underlying mechanisms by which
LRIG2 modulates PDGFRβ signaling were investigated. First, the
association between the expression levels of LRIG2 and PDGFRβ in
human GBM was evaluated. Furthermore, stable GBM cells with LRIG2
overexpression or knockdown were established and the effects of
LRIG2 on PDGF-BB-induced proliferation and cell cycle progression
were investigated in vitro and in vivo. Finally, the
mechanism underlying these effects was explored. To the best of our
knowledge, the present study is the first to investigate the effect
of LRIG2 on PDGFRβ signaling pathways, and thereby on the
proliferation and cell cycle progression of human GBM.
Materials and methods
Cell culture
The U87 human GBM cell line was purchased from
American Type Culture Collection (ATCC; Manassas, VA, USA). U87
cells were cultured in Dulbecco's modified Eagle's medium (DMEM)
containing 10% fetal bovine serum (FBS) in a humidified incubator
with 5% CO2 at 37°C. According to Allen et al
(25), the fact that U87 from ATCC
originated from an unknown patient and is not the original U87
established at the University of Uppsala does not affect the
authenticity of U87 as a human GBM cell line. Thus, the use of U87
from ATCC in the present study is considered appropriate and the
results from the use of U87 as a GBM cell line are not
affected.
shRNA-mediated gene knockdown
To knock down LRIG2 expression, a vector-based short
hairpin RNA (shRNA) expression system was used. A total of two
nucleotide sequences, targeting LRIG2 (NM_014813) nucleotides
451-471 (shRNA1) and 1379-1399 (shRNA2), and one non-silencing
scrambled shRNA (scr) were designed and synthesized (Table I). The shRNA inserts were digested
with EcoRI and AgeI prior to ligation into the
pLKO.1-TRC cloning vector (Addgene Inc., Cambridge, MA, USA)
according to the manufacturer's protocol. The constructed plasmid
contained the ampicillin resistance gene for selection of
ampicillin-resistant colonies in bacteria. All the inserted
sequences were verified by DNA sequencing.
 | Table IOligonucleotide sequences of
LRIG2-specific siRNA. |
Table I
Oligonucleotide sequences of
LRIG2-specific siRNA.
| Name | siRNA sequences
(5′→3′) | Target sequence on
LRIG2 cDNA |
|---|
| shRNA1 |
AATCGGTTGTCTAACTGGAAC | 451-471 |
| shRNA2 |
AAGGAAACCAGATTAAGTCAA | 1379-1399 |
| Scramble |
CAACAAGATGAAGAGCACCAA | |
Stable cell transduction
The stable LRIG2-overexpressing U87 cells
(U87-LRIG2) and the corresponding control cells (U87-Con) had been
established and used previously (22). U87 cells were stably infected with
lentivirus expressing pLKO.1-TRC-scr or pLKO.1-TRC-LRIG2shRNA using
Lenti-X lentiviral Expression Systems (Clontech Laboratories, Inc.,
Mountainview, CA, USA) according to the manufacturer's instructions
as described previously (22). In
brief, 1 day before the transfection, 4×106 293T cells
(ATCC) were seeded in a 100-mm plate and cultured in DMEM
containing 10% Tc-free FBS (Clontech Laboratories, Inc.). A total
of 3 μg of each plasmid DNA, including the constructed
pLKO.1-TRC-scr and pLKO.1-TRC-LRIG2sh, was mixed with Lenti-X HT
Packaging Mix (Clontech Laboratories, Inc.) and added to the 293T
cell culture medium. After 48 h of incubation, the
lentivirus-containing supernatants were harvested and filtered
through a 0.45-μm filter (EMD Millipore, Billerica, MA,
USA). The Lenti-X qRT-PCR Titration Kit (Clontech Laboratories,
Inc.) was used to determine the viral titer. U87 cells were seeded
into 6-well plates at a density of 3×105 cells/well and
incubated overnight prior to transduction. The viral supernatant
was mixed with culture medium containing 4 μg/ml polybrene
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), added to the cells
and transduced for 24 h. Then, the transduced cells were selected
with culture medium containing 1 μg/ml puromycin
(Sigma-Aldrich; Merck KGaA) for 2 weeks and the puro-resistant
clones that represented possible stably transduced cells were
expanded for further experiments.
Patients and tissue samples
A total of 40 GBM samples were obtained by surgical
resection from Tongji Hospital, Tongji Medical College, Huazhong
University of Science and Technology (Wuhan, China), between 2012
and 2013. The subset of patients with GBM included 24 men and 16
women aged 38-76 years (median age, 58 years). All the patients
provided written informed consent for the use of their tissues, and
the study was approved by the Institutional Review Board of Tongji
Hospital (ID: 20121202). Following surgical resection, fresh tumors
were immediately fixed with formalin and embedded in paraffin for
routine histopathological evaluation and immunohistochemical
staining.
Immunohistochemical staining
The immunohistochemical staining procedures were
performed as previously described (22). Briefly, GBM samples from humans and
nude mice were fixed in a phosphate-buffered 4% formaldehyde
solution and embedded into paraffin blocks using standard methods.
Tissue sections (4-μm) were labeled with the following
primary antibodies at the indicated concentrations: Anti-LRIG2
rabbit polyclonal antibody 1:100 (cat. no. ab157452; Abcam,
Cambridge, UK), anti-PDGFRβ rabbit monoclonal antibody 1:100 (cat.
no. 3169), anti-pPDGFRβ rabbit monoclonal antibody 1:50 (cat. no.
4549), anti-pAkt rabbit monoclonal antibody 1:100 (cat. no. 4058),
anti-pStat3 rabbit monoclonal antibody 1:100 (cat. no. 9145) (all
from Cell Signaling Technology, Inc., Danvers, MA, USA),
anti-cyclin B1 rabbit polyclonal antibody 1:100 (cat. no. BA0766),
anti-Cyclin D1 rabbit polyclonal antibody 1:100 (cat. no. BA0770)
(both from Boster Biological Technology), anti-Ki67 rabbit
monoclonal antibody 1:100 (cat. no. ab16667), anti-PCNA rabbit
monoclonal antibody 1:100 (cat. no. ab92552;) (both from Abcam).
The evaluation of the immunostaining was performed by two
independent observers in a blinded manner. In brief, positive
immunohistochemical staining in tumor cells was evaluated with
respect to the intensity of positive staining and the percentage of
positive tumor cells. From these scoring data, a four-grade
semiquantitative score was defined as follows: 0, no or very faint
immunoreactivity (IR) or immunopositive cells <5%; 1, weak IR or
immunopositive cells 5-20%; 2, moderate IR or immunopositive cells
26-50%; 3, strong IR or immunopositive cells >50%. A total score
based on low intensity and fraction (scored as 0 or 1) vs. high
intensity and fraction (scored as 2 or 3) was created for each
patient and statistically analyzed.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde for 1 h at
4°C, permeabilized with 0.25% Triton X-100 for 5 min, blocked with
10% bovine serum albumin (Boster Biological Technology) for 1 h and
labeled with primary anti-LRIG2 1:50 (cat. no. ab157452; Abcam),
anti-PDGFRβ 1:100 (cat. no. 3169; Cell Signaling Technology, Inc.),
and anti-Flag 1:100 (cat. no. F1804; Sigma-Aldrich; Merck KGaA)
antibodies overnight at 4°C, as previously described (22). Following washing with
phosphate-buffered saline (PBS), cells were incubated with
fluorescein isothiocyanate or cyanine-labeled secondary antibodies
(1:100; ProteinTech Group, Inc., Chicago, IL, USA) and subjected to
immunofluorescence microscopy using appropriate filters.
Cell proliferation assay
In order to examine cellular proliferation, cell
counting kit-8 (CCK8) assays and cell count analysis were
performed. For CCK8 assays, 5×103 cells in 200 μl
suspension per well were seeded in triplicate in 96-well
flat-bottomed plates and allowed to attach overnight at 37°C.
Subsequently, the cells were treated with indicated concentrations
of PDGF-BB for indicated times. At the indicated time points, CCK8
(CK04; Dojindo Molecular Technologies, Inc., Kumamoto, Japan) was
added to the wells prior to incubation for 2 h at 37°C. The optical
density was measured with a microplate reader (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) using a wavelength of 450 nm.
For cell count determination, U87 GBM cells with LRIG2
overexpression or downregulation and the corresponding control
cells were seeded in triplicate at a density of 10×104
cells/well in 6-well plates. Similar to the CCK8 assay, cells were
allowed to attach, followed by serum starvation with DMEM for 24 h.
Synchronized cells were then treated with or without STI571 (cat.
no. 13139; Cayman Chemical Company, Ann Arbor, MI, USA) 10
μM for 2 h prior to stimulation with PDGF-BB (cat. no.
100-14B; PeproTech, Rocky Hill, NJ, USA) 50 ng/ml for 24 h. At the
end of the experiment, the cells were enzymatically detached, and
the cell numbers were counted in triplicate for each group.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Synchronized cells were cultured with DMEM
containing 10% FBS for 48 h. Total RNA was isolated and
quantitative RT-qPCR was performed as previously described
(24). The DNA primer sequences of
LRIG2 were designed as follows: Sense, 5′-CAGTGCATAGCTGGAGGGAGTC-3′
and antisense, 5′-TACAATGATGAGAAGCTGATTGGCTGCA-3′. The primer
sequences of the control GAPDH were designed as follows: Sense,
5′-CACCAGGGCTGCTTTTAACTCTGGTA-3′ and antisense,
5′-CCTTGACGGTGCCATGGAATTTGC-3′.
Flow cytometric analysis of the cell
cycle
The cells were synchronized to the G0/G1 stage by 24
h of serum deprivation and then cultured in DMEM with 0.5% FBS with
or without PDGF-BB for an additional 24 h. Cell cycle analysis was
performed by propidium iodide (PI) staining and flow cytometry. In
brief, cells were collected and washed twice with cold PBS and
resuspended in pre-cooled 70% ethanol at 4°C overnight.
Subsequently, fixed cells were collected by centrifugation and
incubated with RNase (25 μg/ml) and PI (50 μg/ml) at
37°C for 30 min in the dark. The number of cells in the G0/G1, S
and G2/M phases was analyzed by flow cytometry using a FACSCalibur
Flow Cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Soft agar colony formation assay
Anchorage-independent cell proliferation was
determined by soft agar colony formation assay as previously
described (22). Briefly, 6-well
plates were first covered with a bottom layer of 0.6% agar made in
10% FBS DMEM and 4×104 cells per well were embedded in
triplicate into 0.3% top agar gel containing DMEM supplemented with
10% FBS. Cells were fed with 0.5% FBS DMEM with or without PDGF-BB
(50 ng/ml) every third day and allowed to grow for 2 weeks. For
each well, 30 randomly selected fields were photographed under a
microscope (ECLIPSE TS100; Nikon, Tokyo, Japan) with a ×10
microscopic lens and a ×6 optical lens (magnification, ×60). The
colonies were then counted and the mean number of colonies per
field was calculated. Each experiment was performed in
triplicate.
In vivo tumor formation in nude mice
All animal procedures were conducted in accordance
with the guidelines for animal experimentation, and the animal
protocol used in this study was approved by the Institutional
Review Board of Tongji Hospital, Tongji Medical College, Huazhong
University of Science and Technology (IRB ID: 2011A01). A total of
10 immune-deficient female nude mice (BALB/c), aged 6 weeks and
weighing 20-22 g, were purchased from the Wuhan Laboratory Animal
Center, bred at the facility of laboratory animals at Tongji
Medical College, and randomly assigned to two groups (n=5 per
group): U87-scr and U87-LRIG2sh. For subcutaneous inoculation, a
total of 2×106 cells in 100 μl PBS were injected
subcutaneously into the right flank of each mouse as previously
described (22). Tumor volume was
measured with calipers every 5 days and calculated using the
formula: Volu me = length × width2 × (π/6) (length,
larger diameter in mm; width, smaller diameter in mm). At the end
of the experiment, the mice were euthanized and the tumors were
surgically harvested, measured, weighed, fixed in 4%
paraformaldehyde overnight and analyzed by
immunohistochemistry.
Western blotting and
co-immunoprecipitation
Western blotting and co-immunoprecipitation were
performed as previously described (22,24).
The primary antibodies used in western blotting at the indicated
concentrations were as follows: Anti-pPDGFRβ rabbit monoclonal
antibody 1:500 (cat. no. 4549), anti-PDGFRβ rabbit monoclonal
antibody 1:500 (cat. no. 3169), anti-pAkt rabbit monoclonal
antibody 1:1,000 (cat. no. 4058), anti-Akt rabbit polyclonal
antibody (cat. no. 9273), anti-pStat3 rabbit monoclonal antibody
1:1,000 (cat. no. 9145), anti-Stat3 rabbit monoclonal antibody
1:1,000 (cat. no. 4904) (all from Cell Signaling Technology, Inc.),
anti-cyclin B1 rabbit polyclonal antibody 1:500 (cat. no. BA0766),
anti-cyclin D1 rabbit polyclonal antibody 1:500 (cat. no. BA0770),
β-actin mouse monoclonal antibody 1:1,000 (cat. no. BM0627) (all
from Boster Biological Technology). Co-immunoprecipitation of LRIG2
and PDGFRβ were performed using flag-tagged LRIG2-overexpressing
U87 cells. Briefly, cells were washed twice with ice-cold PBS and
lysed with IP lysis buffer (cat. no. 87787; Thermo Fisher
Scientific, Inc.). Lysates were pre-cleared by adding 20 μl
of Protein A/G Agarose (cat. no. A10001; Abmart, Berkeley Heights,
NJ, USA), followed by overnight incubation with anti-Flag (cat. no.
F1804; Sigma-Aldrich; Merck KGaA) or anti-PDGFRβ (cat. no. 3169;
Cell Signaling Technology, Inc.) on a rocker at 4°C.
Immunocomplexes were captured by incubating with Protein A/G
Agarose for 2 h at 4°C, followed by washing with ice-cold lysis
buffer to eliminate non-specific interactions. The agarose-bound
immunocomplexes were then eluted by boiling the beads in 2X SDS
loading buffer (cat. no. P0015B; Beyotime Institute of
Biotechnology, Shanghai, China) for 10 min at 100°C. The whole-cell
lysates were used as positive controls. In negative controls,
primary antibodies used for co-IP were replaced with corresponding
isotype IgG controls. Eluted proteins and the input (whole lysates)
were subjected to western blotting.
Statistical analysis
Statistical analyses were performed using SPSS 16.0
software for Windows (SPSS Inc., Chicago, IL, USA). The
significance of the correlation between LRIG2 and PDGFRβ in the
immunohistochemical staining analysis was determined using
Pearson's χ2 test. Multiple comparisons were performed
using one-way analysis of variance followed by Bonferroni as a
post-hoc test. Statistical differences between the two groups were
analyzed with the Student's t-test. Values are expressed as the
mean ± standard deviation of at least three independent
experiments. P<0.05 was considered to indicate a statistically
significant difference.
Results
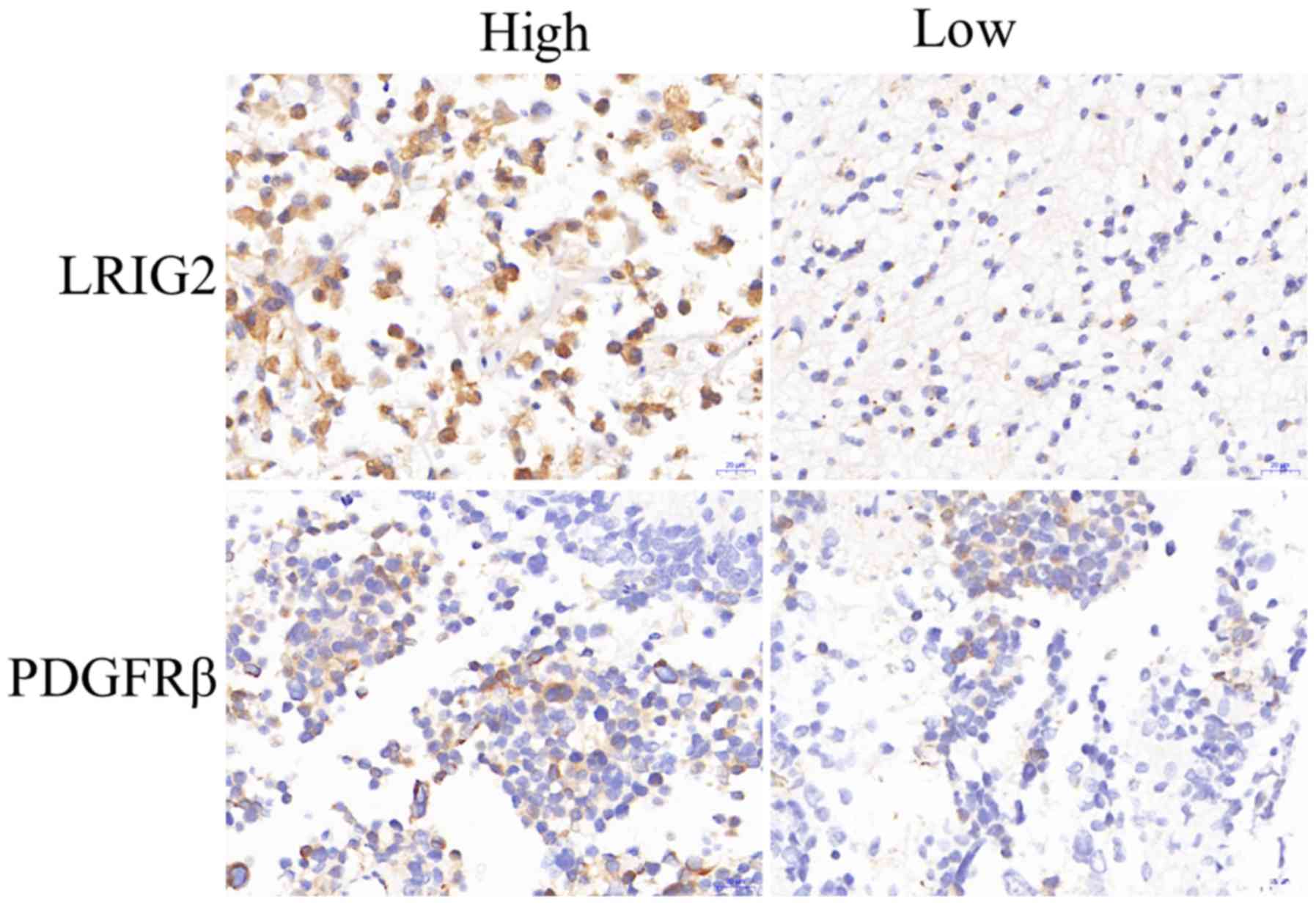
Protein expression level of LRIG2 is
positively correlated with the level of PDGFRβ in human GBM
To investigate the correlation between the
expression levels of LRIG2 and PDGFRβ in human GBM, the protein
expression levels of LRIG2 and PDGFRβ were determined in 40
patient-derived biopsies of GBM, obtained from surgical resections
using immunohistochemical staining. According to the total score
based on the relative intensity of LRIG2 expression, the patients
were categorized into two groups: LRIG2 high expression (scored as
2 or 3) and LRIG2 low expression (scored as 0 or 1). Of the 20
cases in the LRIG2 high expression group, the PDGFRβ IR of 7 cases
was defined as low expression and of 13 cases as high expression.
Of the 20 cases in the LIRG2 low expression group, the PDGFRβ IR of
14 cases was scored as low expression and of 6 cases as high
expression. The difference in PDGFRβ IR was significant between the
LRIG2 high expression and low expression groups (P<0.05;
Fig. 1 and Table II), which indicated that the
protein expression level of PDGFRβ was higher in GBMs with high
LRIG2 expression, compared with GBMs with low LRIG2 expression.
| Table IIProtein expression levels of LRIG2
and PDGFRβ in human glioblastoma tissues. |
Table II
Protein expression levels of LRIG2
and PDGFRβ in human glioblastoma tissues.
| LRIG2
expression | PDGFRβ expression
|
|---|
| High | Low |
|---|
| High | 13 | 7 |
| Low | 6 | 14 |
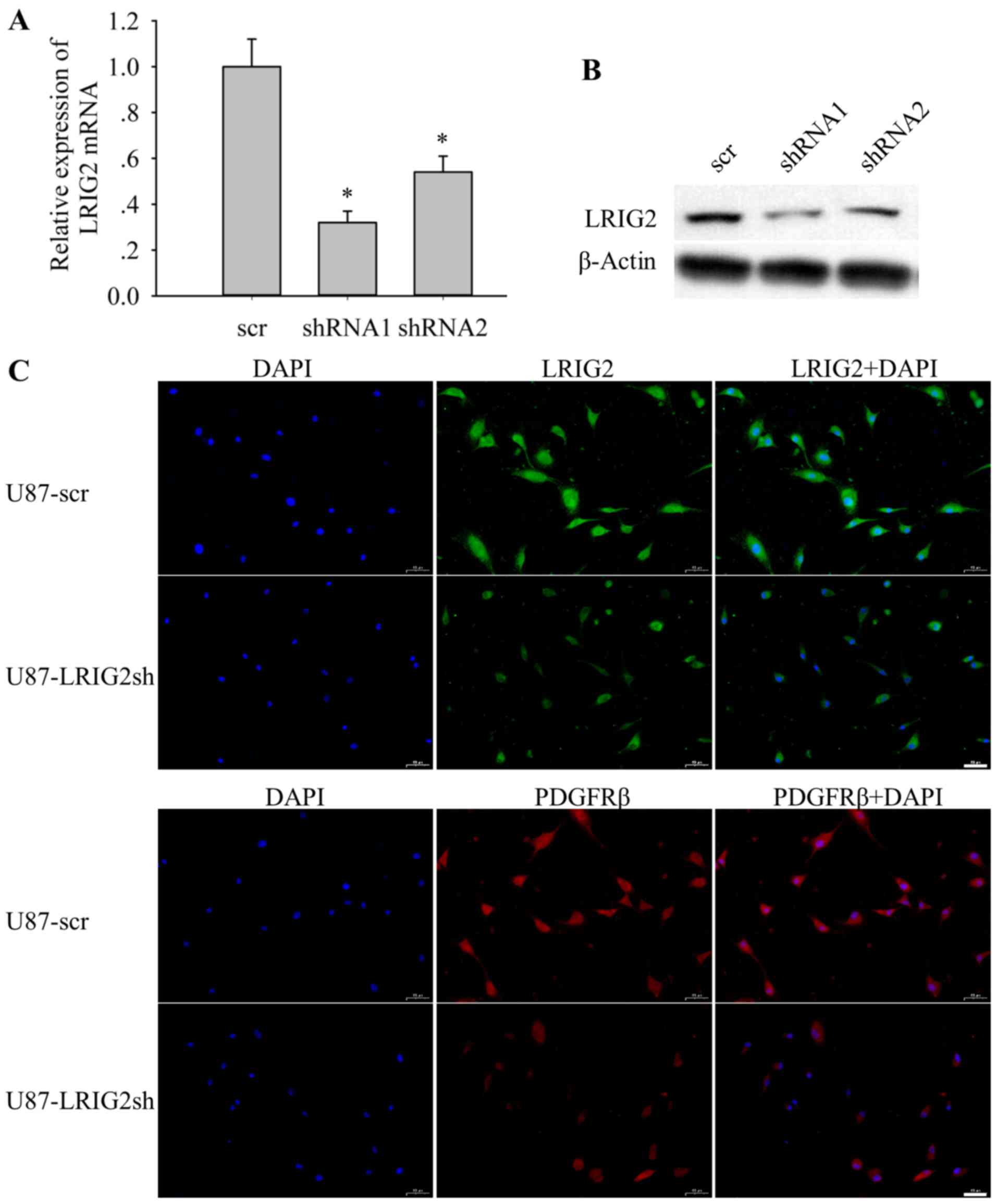
Establishment of the stable LRIG2
knockdown GBM cell line
In order to evaluate the role of LRIG2 in GBM, the
expression of LRIG2 was downregulated via shRNA mediated-knockdown
in U87 cells. A total of three plasmids expressing two nucleotide
sequences targeting LRIG2 (shRNA1 and shRNA2) and one non-silencing
scramble shRNA (scr) (Table I)
were constructed and stably transduced into U87 GBM cells. RT-qPCR
and western blotting were used to verify the mRNA and protein
expression levels of LRIG2, respectively, in stably transduced
cells. Compared with the scramble control cells, LRIG2 transcripts
were markedly reduced in the shRNA1- and shRNA2-transduced cells
(Fig. 2A). In line with the
RT-qPCR results, LRIG2 protein levels were also significantly
decreased in the shRNA1- and shRNA2-transduced cells (Fig. 2B). These results indicated that the
expression levels of LRIG2 were notably downregulated by LRIG2
shRNAs, and the transduced cells with shRNA1, which exhibited
higher knockdown efficiency, were selected for the subsequent
experiments. Furthermore, fluorescence microscopy was used to
investigate the expression of LRIG2 and PDGFRβ in U87 cells with
LRIG2 knockdown by shRNA1. The results demonstrated that the
fluorescent intensity of LRIG2 was markedly attenuated in the
U87-LRIG2sh cells (Fig. 2C).
Notably, the fluorescent intensity of PDGFRβ was also significantly
decreased (Fig. 2C), similar to
the result of LRIG2 expression being positively correlated with the
expression of PDGFRβ in human GBM samples.
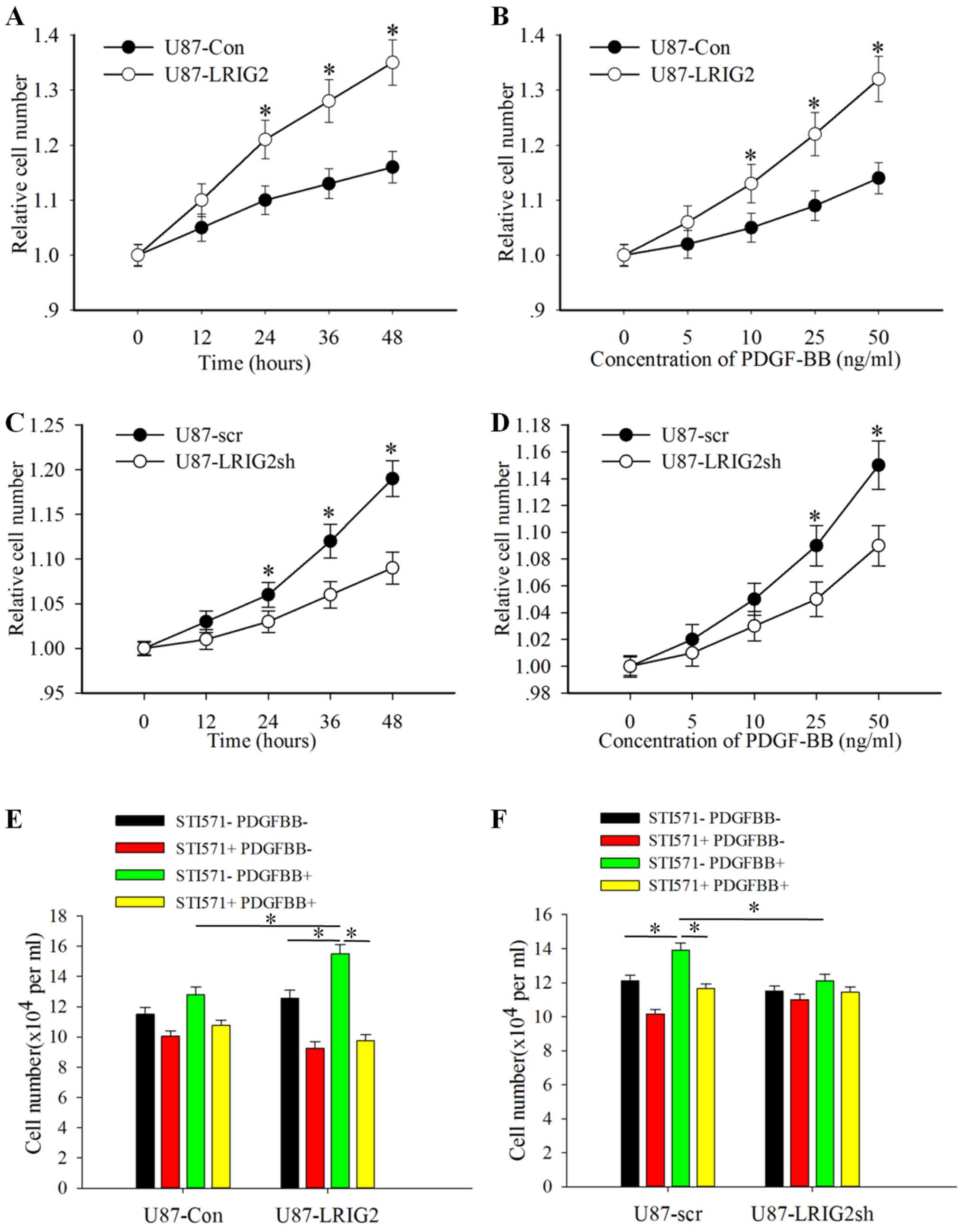
LRIG2 promotes PDGF-BB-stimulated
proliferation of human GBM cells in vitro
To determine the potential effects of the LRIG2
protein on PDGF-BB-induced cell proliferation of GBM cells, U87
cells were first treated with different concentrations of PDGF-BB
for various durations. The CCK8 assay was used to evaluate cell
proliferation. Overexpression of LRIG2 was found to markedly
promote the PDGF-BB-induced proliferation of U87 GBM cells in a
concentration- and time-dependent manner (Fig. 3A and B). Congruently,
downregulation of LRIG2 markedly inhibited the PDGF-BB-induced
proliferation of U87 cells in a concentration- and time-dependent
manner (Fig. 3C and D).
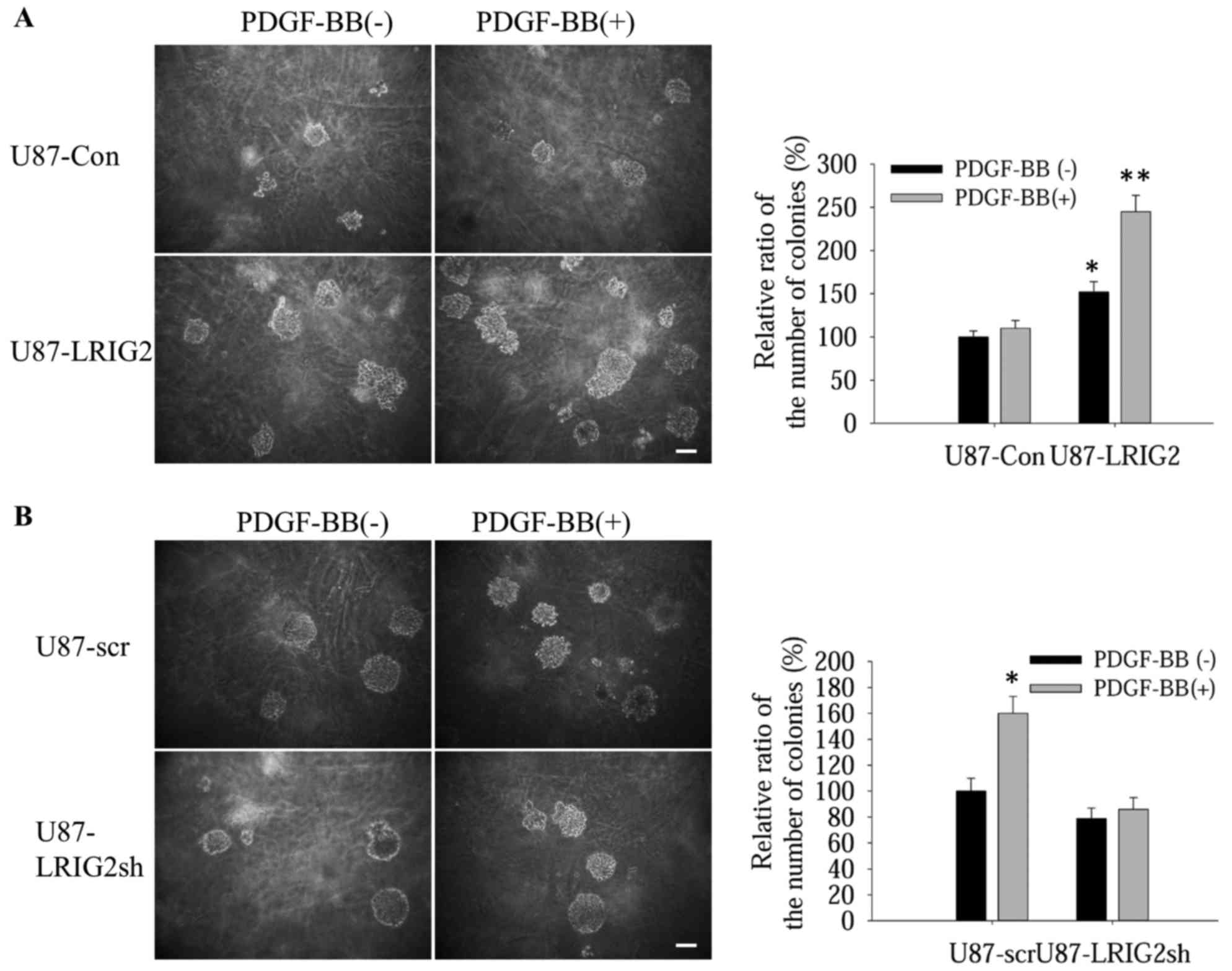
Subsequently, we investigated the effects of LRIG2 on the
PDGF-BB-induced anchorage-independent growth of U87 cells, a
property that mimics tumorigenesis in vivo. Using soft agar
colony formation assays, it was observed that, when stimulated with
PDGF-BB, LRIG2-overexpressing GBM cells formed more colonies
compared with the control cells (Fig.
4A); however, when LRIG2 was downregulated in U87 cells, the
effect of LRIG2 on enhancement of PDGF-BB-induced
anchorage-independent cell growth was abrogated (Fig. 4B), indicating that the LRIG2
protein enhances the ability of PDGF-BB-stimulated
anchorage-independent proliferation in GBM cells.
Furthermore, STI571, the most promising PDGFRβ
inhibitor, was used to investigate whether the impact of LRIG2 on
the proliferation of GBM cells relies on PDGFRβ RTK regulation. U87
GBM cells with LRIG2 overexpression or downregulation and the
corresponding control cells were pretreated with or without STI571
to block PDGFRβ signaling, and then stimulated with or without
PDGF-BB for 24 h. As shown in Fig. 3E
and F, pretreatment of LRIG2-overexpressing U87 cells with
STI571 resulted in marked attenuation of the increased
proliferation induced by PDGF-BB; however, when LRIG2 was
downregulated, the PDGF-BB-induced proliferation of GBM cells was
markedly decreased, and pretreatment with STI571 exerted no
significant effect on PDGF-BB-induced proliferation. Taken
together, these findings confirmed that the LRIG2 protein promotes
PDGF-BB-induced proliferation of human GBM cells in vitro by
regulating the activation of PDGFRβ.
Effects of LRIG2 on the
PDGF-BB-stimulated cell cycle distribution of GBM cells
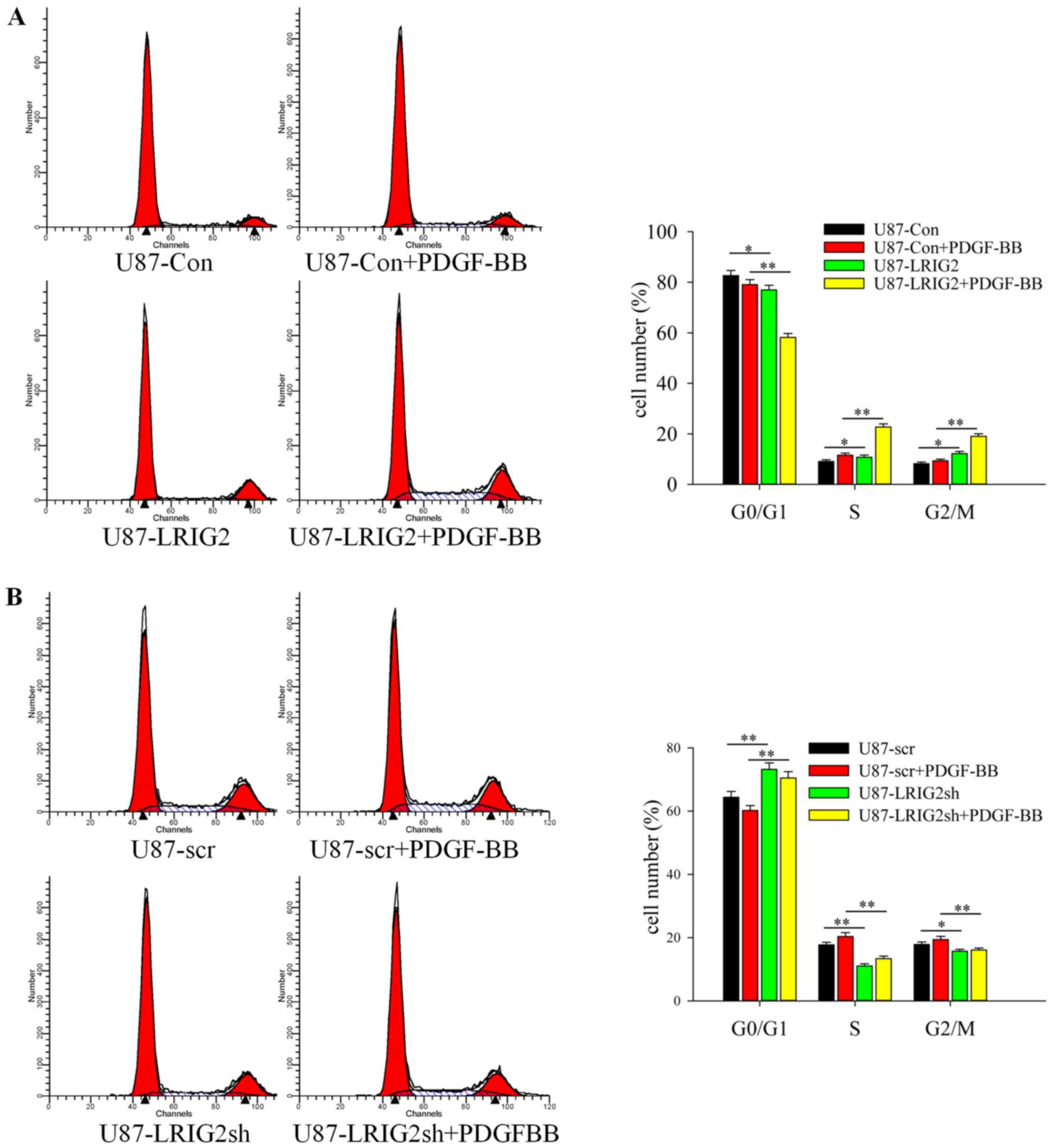
To investigate the mechanism underlying LRIG2
promoting the proliferation of PDGF-BB-induced GBM cells, an
experiment was performed to assess the effects of LRIG2 on U87 cell
cycle progression stimulated by PDGF-BB. The synchronized cells
were harvested, cultured in DMEM with 0.5% FBS with or without
PDGF-BB for 24 h, and the cell cycle distribution was analyzed by
flow cytometry. The results revealed that the percentage of cells
in the G0/G1 phase was markedly decreased and the percentage of
cells in the S or G2/M phase was markedly increased in the
PDGF-BB-induced LRIG2-overexpressing U87 cells compared with the
control cells (Fig. 5A).
Concordantly, down-regulation of LRIG2 caused increased
accumulation of cells in the G0/G1 phase and a significantly
decreased percentage of cells in the S or G2/M phase (Fig. 5B), which was in line with the
results reported previously (21).
More importantly, when stimulated with PDGF-BB, LRIG2-knockdown GBM
cells exhibited markedly increased accumulation in the G0/G1 phase
and a strikingly decreased percentage of cells in the S or G2/M
phase compared with the scramble control cells (Fig. 5B). Taken together, these results
demonstrated that the LRIG2 protein promoted PDGF-BB-induced DNA
synthesis and the G0/G1 to S phase cell cycle transition in GBM
cells, resulting in a higher number of cells entering the G2/M
phase.
LRIG2 promotes the growth of U87 tumor
xenograft through regulating the PDGFRβ signaling pathway in
vivo
The aforementioned data confirmed the role of LRIG2
in promoting the proliferation of human GBM cells in vitro;
therefore, the present study was further extended in vivo to
investigate the effects of LRIG2 knockdown on U87 GBM xenografts,
and to explore the possible underlying mechanisms. As mentioned
above, stably transduced U87 GBM cells expressing LRIG2 shRNA or
scramble RNA were inoculated via subcutaneous injection to the
right flank of mice with severe combined immunodeficiency (SCID).
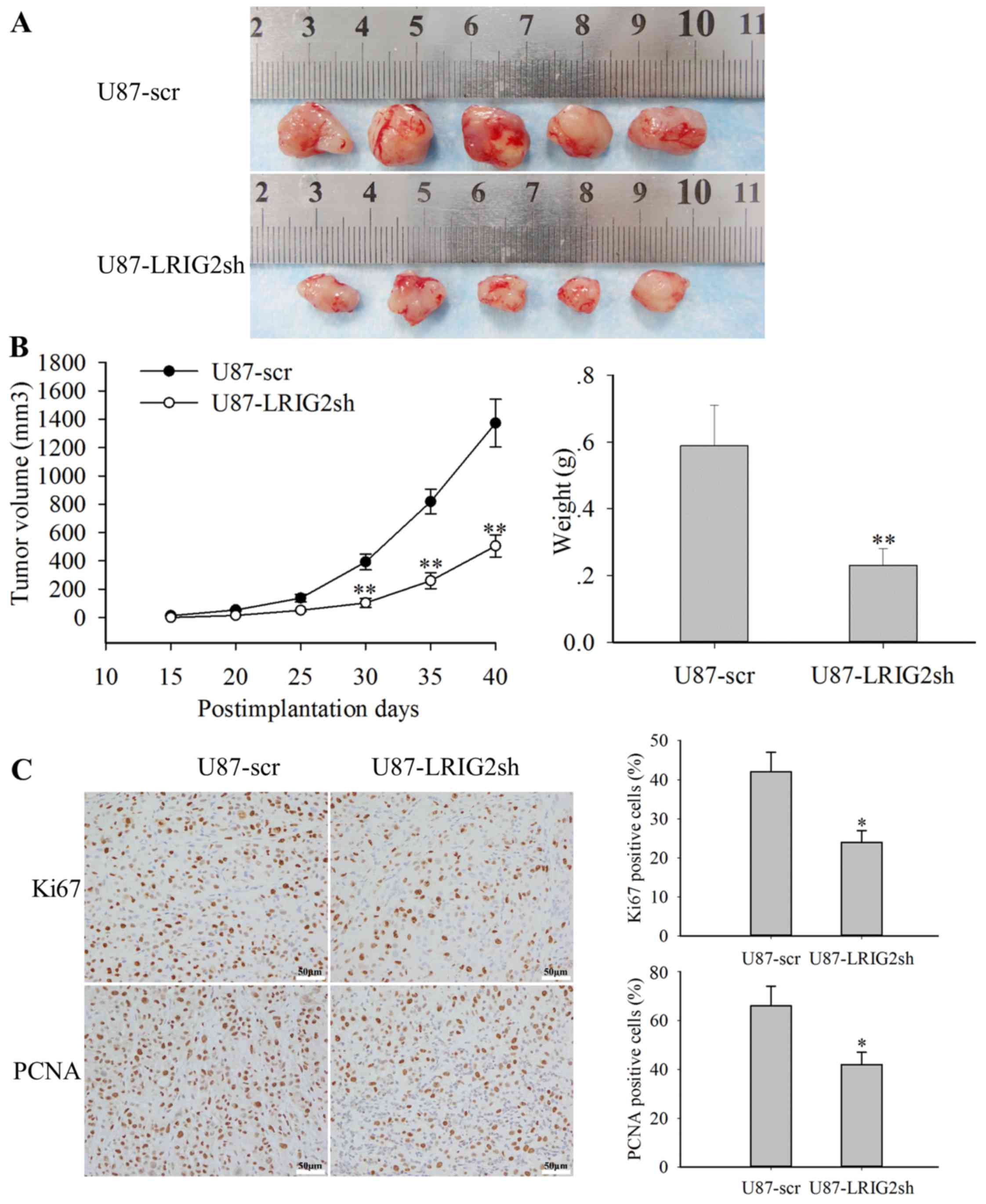
Tumor volume was recorded every 5 days from day 15
post-implantation, and the tumor growth curve results demonstrated
that tumor growth was markedly inhibited in mice injected with
LRIG2-knockdown U87 cells compared with the scramble group
(Fig. 6A and B). At the end of the
experimental period (day 40), the xenografts were surgically
removed, weighed and subjected to immunohistochemical staining for
Ki-67 and proliferating cell nuclear antigen (PCNA), two classical
markers of cell proliferation. Consistently, the results revealed
that tumor weight was also notably lower in tumors with LRIG2
downregulation (Fig. 6B), and the
expression levels of Ki-67 and PCNA were both significantly
decreased in LRIG2-shRNA-expressing U87 xenograft tumor tissues
(Fig. 6C). Combining the effects
of LRIG2 knockdown on tumor growth of in vivo, which were in
line with the in vitro data, with the previously reported
results that overexpression of LRIG2 promoted the growth of GBM
xenograft in vivo (22),
the evidence supporting a role of LRIG2 in promoting the
proliferation of human GBM cells in vivo is compelling.
Based on the aforementioned in vitro results
that LRIG2 promoted the PDGF-BB-induced proliferation of GBM cells
via regulating the cell cycle distribution, the possible PDGFRβ
signaling pathways mediated by LRIG2 was further explored in
vivo. Fresh LRIG2-overexpressing and LRIG2-silenced tumor
xenografts were subjected to immunohistochemical staining for
LRIG2, PDGFRβ and its downstream pathways and effectors. Notably,
the expression levels of phosphorylated and total PDGFRβ, as well
as its downstream targets, phosphorylated Akt (pAkt) and
phosphorylated Stat3 (pStat3), were significantly upregulated in
LRIG2-overexpressing GBM xenografts compared with the control
xenografts (Fig. 7), whereas the
levels of those proteins were markedly decreased in
LRIG2-downregulated tumor xenografts (Fig. 7). Furthermore, the expression
levels of PDGFRβ downstream effectors cyclin D1 and cyclin B1, a
key regulator of the G1/S phase transition and a G2/M regulator,
respectively, were positively correlated with the expression levels
of the LRIG2 protein in GBM xenografts (Fig. 7), providing a convincing
explanation for the effects of LRIG2 on the PDGF-BB-stimulated cell
cycle distribution of GBM cells in vitro. Collectively,
these results demonstrated that LRIG2 played a pivotal role in
promoting the growth of GBM xenografts through regulating PDGFRβ
signaling and its downstream effectors in vivo.
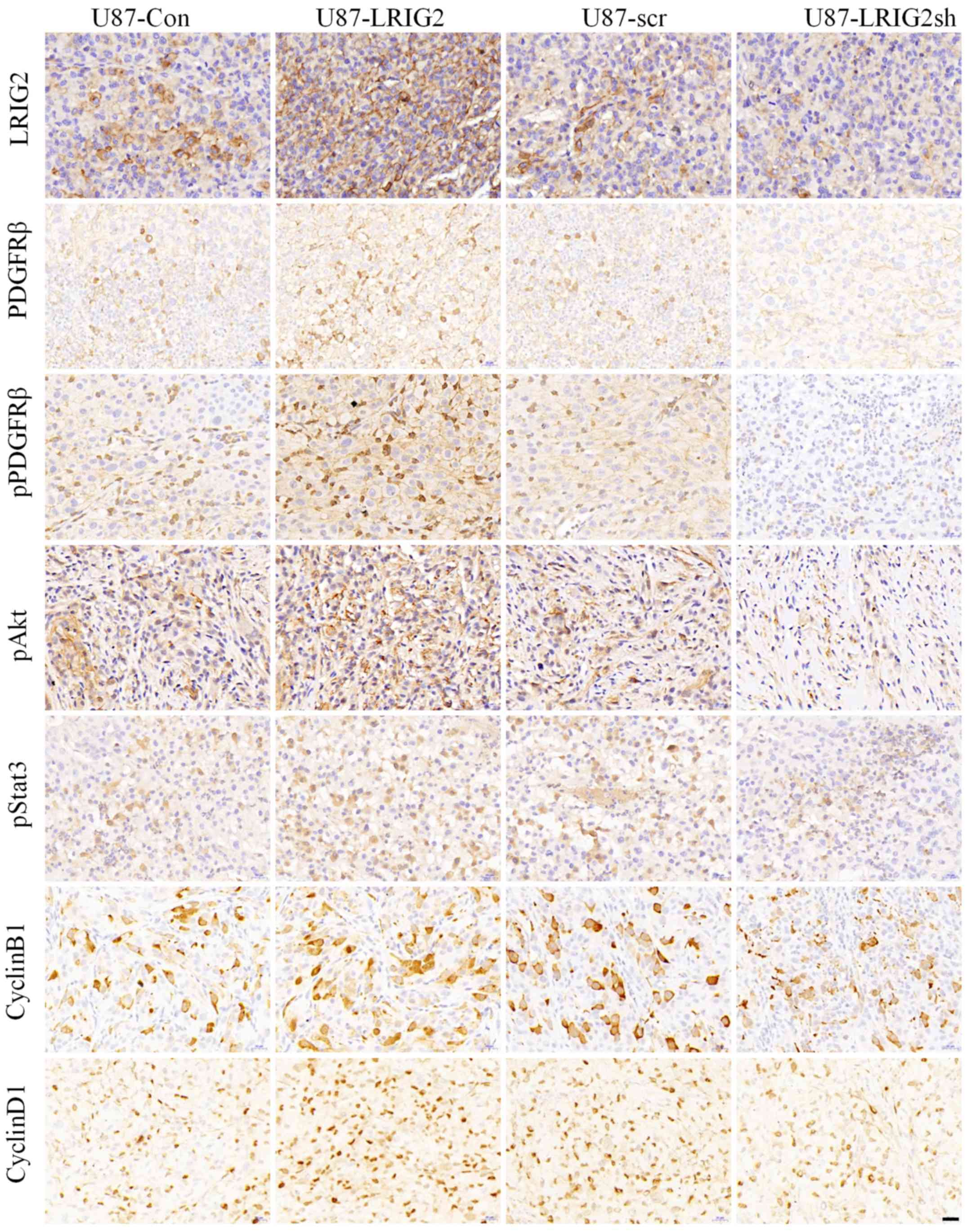 | Figure 7LRIG2 enhances the activation of
PDGFRβ and regulates its downstream pathways and effectors in
vivo. A total of four groups of indicated stable U87
glioblastoma cells (U87-LRIG2, LRIG2-overexpressing; U87-Con,
control U87 cells; U87-LRIG2sh, LRIG2-knockdown cells; and U87-scr,
scramble U87 cells) were injected subcutaneously into the flanks of
mice with severe combined immunodeficiency. At the end of the
animal experiments, tumor xenografts were isolated and
immunohistochemistry was performed to evaluate the expression of
LRIG2, PDGFRβ and its downstream pathways, as indicated.
Representative images are depicted (scale bars, 20 μm).
LRIG2, leucine-rich repeats and immunoglobulin-like domain 2;
PDGFRβ, platelet-derived growth factor receptor β. |
LRIG2 physically interacts with PDGFRβ
RTK in GBM cells
Having demonstrated that LRIG2 promotes the growth
of GBM and the possible role of LRIG2 in the enhancement of PDGFRβ
signaling activation, in order to gain insight into the molecular
mechanisms underlying the activation of PDGFRβ by LRIG2, confocal
microscopy was performed to evaluate the expression pattern of the
two fluorescent proteins in U87 cells. In line with previous
studies regarding fluorescent LRIG2 subcellular distribution
(17,26), the present study demonstrated via
immunofluorescence staining that Flag-tagged LRIG2 was primarily
localized in the cytoplasm, and it is noteworthy that the
distribution of PDGFRβ fluorescent signals mirrored the LRIG2
distribution (Fig. 8A), indicating
a colocalization of LRIG2 and PDGFRβ expression in GBM cells. Based
on the co-expression pattern and the similar protein structure of
LRIG2 and PDGFRβ, both containing extracellular immunoglobulin
loops and an intracellular tail (27), it is reasonable to hypothesize that
LRIG2 may physically interact with PDGFRβ in GBM cells. To confirm
this hypothesis, the Flag-tagged LRIG2 was precipitated from the
cell lysates of LRIG2-overexpressing cells using an anti-Flag
antibody. Coprecipitation of PDGFRβ was observed by probing the
resulting blot with anti-PDGFRβ, and when PDGFRβ was
immunoprecipitated from the cell lysates, coprecipitation of the
Flag-LRIG2 was also detected by western blotting with the anti-Flag
antibody (Fig. 8B), indicating
that LRIG2 physically associates with PDGFRβ. In conclusion, the
results presented in Fig. 8
indicate that LRIG2 physically interacts with the PDGFRβ RTK in GBM
cells, which may result in the enhancement of PDGFRβ signaling
activation.
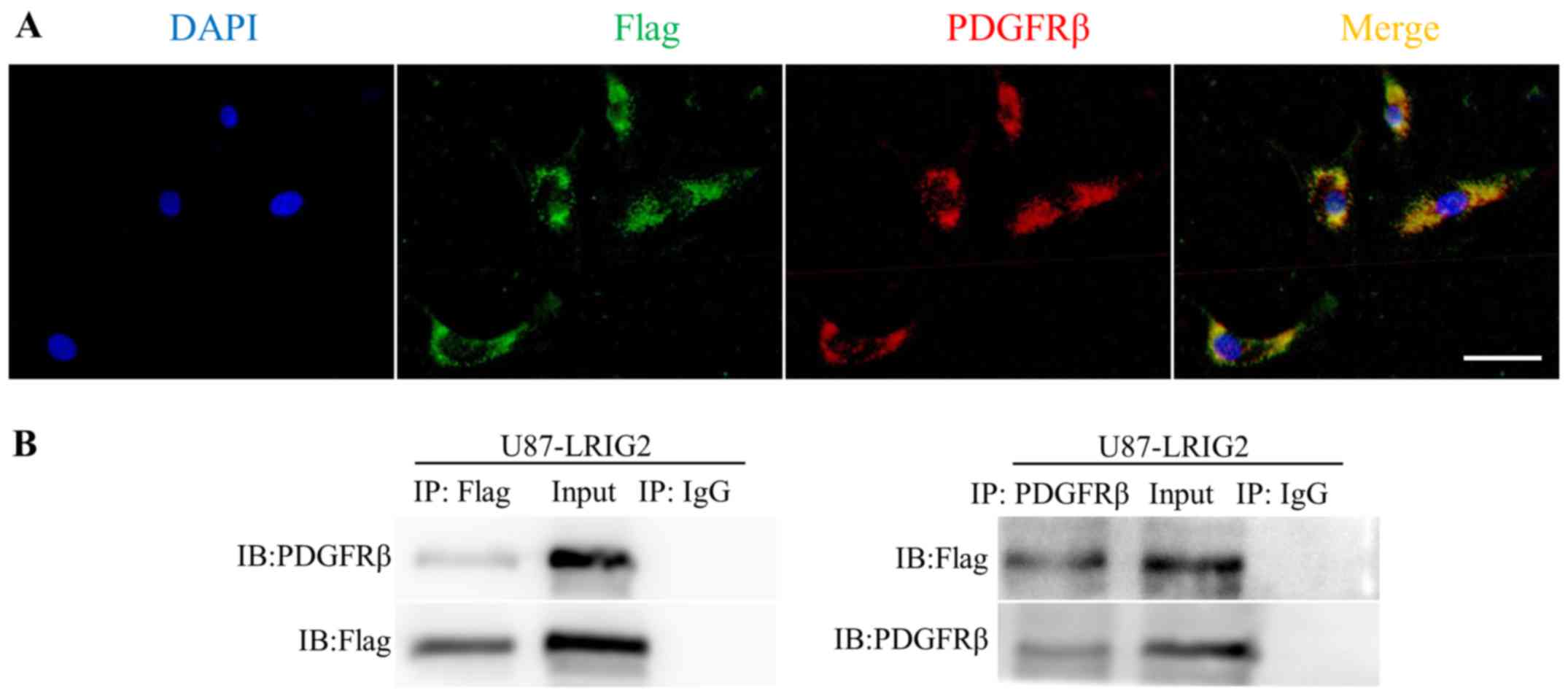 | Figure 8LRIG2 physically interacts with
PDGFRβ in glioblastoma cells. (A) Colocalization of LRIG2 and
PDGFRβ in LRIG2-overexpressing U87 glioblastoma cells. Confocal
micrographs reflecting the distribution of fluorescent Flag-LRIG2
(green fluorescence) and PDGFRβ (red fluorescence) are depicted,
along with a merge graph depicting both signals (yellow
fluorescence), indicating regions of colocalization (scale bar, 50
μm). (B) Lysates from cells overexpressing LRIG2 were
immunoprecipitated with indicated antibodies or corresponding
control IgG, and the immunoprecipitates were western blotted with
indicated antibodies. Representative western blotting images are
shown. The labels of the lanes are indicated as follows: IP,
immunoprecipitated with; IB, immunoblotted with; Input, whole cell
lysates used as positive controls; IgG, the isotype IgG used as
negative control. LRIG2, leucine-rich repeats and
immunoglobulin-like domain 2; PDGFRβ, platelet-derived growth
factor receptor β. |
LRIG2 promotes PDGF-BB-induced activation
of PDGFRβ RTK and the downstream Akt and Stat3 signaling path-ways
and effectors
Subsequently, the physical association of LRIG2 and
PDGFRβ prompted an investigation into whether LRIG2 can modulate
the ligand-dependent activation of the PDGFRβ receptor and its
downstream signaling. Stably transduced U87 cells with LRIG2
overexpression or LRIG2 knockdown were synchronized for 24 h and
then treated with or without various concentrations of PDGF-BB for
a further 24 h. As depicted in Fig.
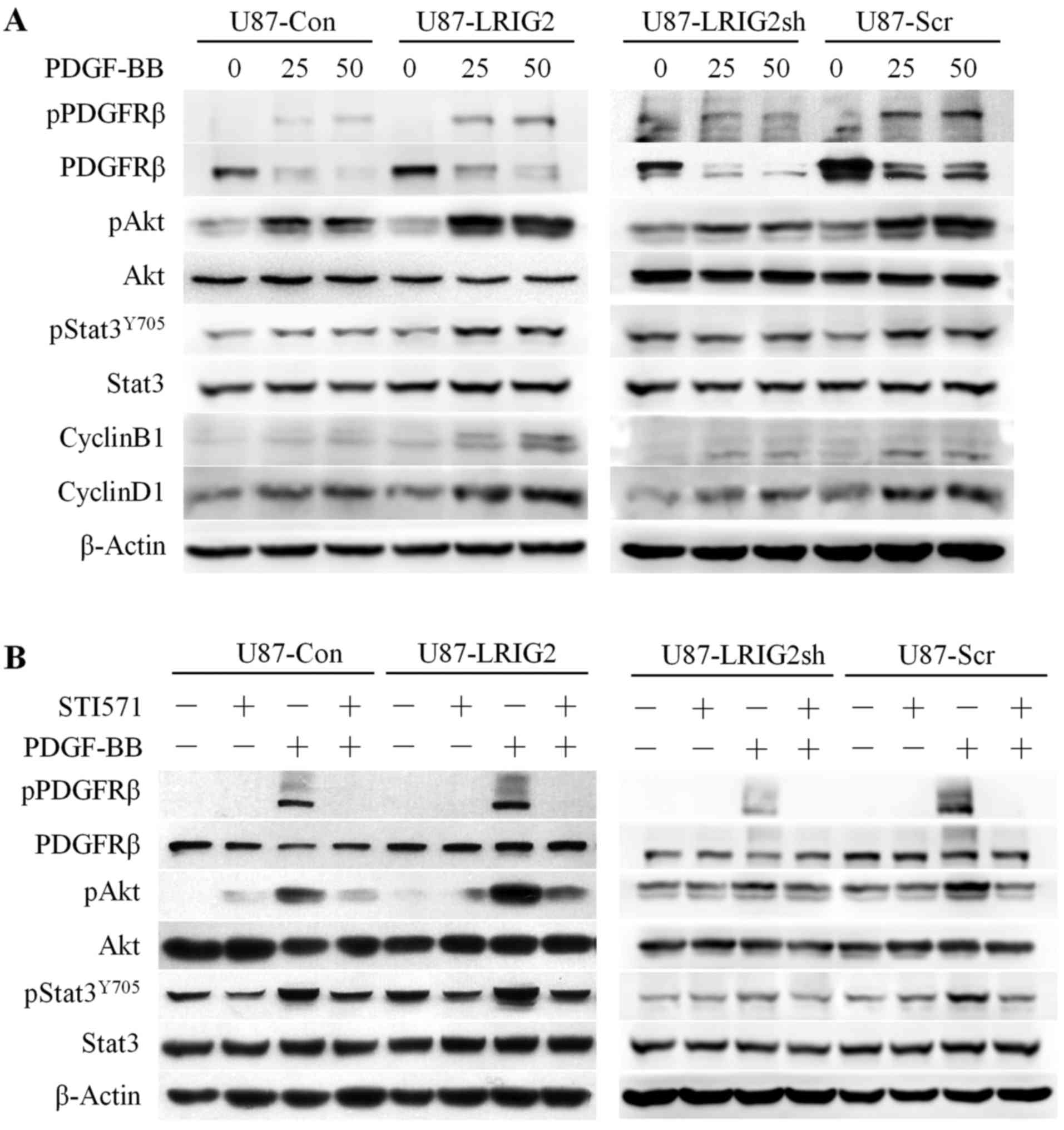
9A, the western blotting results demonstrated that the total
expression level and the phosphorylation of PDGFRβ following
stimulation with PDGF-BB (the primary activator of PDGFRβ) were
significantly increased in LRIG2-overexpressing cells and
drastically reduced in LRIG2-knockdown cells, as compared with the
corresponding control cells. Consistently, the levels of
PDGF-BB-induced phosphorylation of Akt and Stat3, two key
downstream signaling pathways of PDGFRβ, were markedly increased in
LRIG2-overexpressing cells, resulting in substantial accumulation
of PDGF-BB-induced downstream effectors of cyclin B1 and cyclin D1,
which are key regulators of cell cycle progression (Fig. 9A). Notably, when LRIG2 was
downregulated in GBM cells, the PDGF-BB-induced activation of Akt,
Stat3 and the downstream effectors of cyclin B1 and cyclin D1 was
markedly attenuated (Fig. 9A). To
further substantiate the role of LRIG2 in the regulation of PDGFRβ
signaling pathways, STI571, the most promising PDGFRβ inhibitor,
was used to inhibit the phosphorylation of PDGFRβ prior to
detecting the aforementioned downstream pathways. Cells were
serum-starved for 24 h and then treated with STI571 for 2 h prior
to a 10-min exposure to PDGF-BB, and the total cell lysates were
subjected to western blotting. As expected, the expression level of
total PDGFRβ was increased and the activation of PDGFRβ, as well as
its downstream targets Akt and Stat3, was significantly enhanced
following a 10-min stimulation with PDGF-BB in LRIG2-overexpressing
cells, whereas the PDGFRβ expression level was decreased and the
activation of PDGFRβ, Akt and Stat3 was attenuated in cells with
LRIG2 knockdown (Fig. 9B).
Notably, the effects of LRIG2 overexpression or knockdown on
PDGF-BB-induced enhanced or attenuated activation of Akt and Stat3
were significantly abrogated following the inhibition of PDGFRβ
phosphorylation by STI571 (Fig.
9B), which demonstrated that LRIG2 played a pivotal role in the
regulation of PDGF-BB-induced PDGFRβ activation and its downstream
pathways. Taken together, the aforementioned results provide
compelling evidence supporting that LRIG2 physically interacts with
PDGFRβ, leading to stabilization and activation of PDGFRβ and
enhancing the downstream Akt and Stat3 pathways, ultimately
resulting in accumulation of the downstream pro-proliferative
effectors of cell cycle proteins to promote the proliferation of
GBM cells.
Discussion
In the present study, it was demonstrated that
LRIG2, the least investigated and documented member of the LRIG
gene family, is positively correlated with the expression of PDGFRβ
RTK in human GBM, and markedly promoted the PDGF-BB-induced growth
of GBM cells in vitro and in vivo through modulating
PDGFRβ activation and its downstream signaling pathways and
effectors of cell cycle progression. These data indicated that
LRIG2 exerts its pro-tumor effects on GBM cells by positively
regulating PDGFRβ signaling, another important aberrant oncogenic
RTK signaling modulated by LRIG2 in GBM, similar to our previously
reported EGFR signaling (22).
GBM, the most common and lethal type of malignant
primary brain tumor (1), is a
devastating and intractable disease with a poor outcome. A hallmark
of malignant glioma is activation of aberrant RTK signaling
pathways, most commonly caused by EGFR amplification/mutation or
PDGFR amplification/overexpression (28,29),
the genomic alterations of which ultimately lead to enhanced tumor
cell proliferation, increased transition through the cell cycle and
resistance to treatment. PDGFRs are well-characterized RTKs in GBM
that have been found to be overexpressed in human gliomas of all
grades, are most highly expressed in GBM, and are closely
associated with GBM initiation and progression (20,28).
Similar to previous studies (30),
we observed that overexpression of PDGFRβ was a frequent event in
human GBM. To the best of our knowledge, this is the first study to
demonstrate that the expression levels of PDGFRβ vary and are
positively correlated with the expression levels of LRIG2 in human
GBM, indicating that the inter-individual variation in PDGFRβ
expression may be determined by the expression pattern of LRIG2,
and PDGFRβ expression may be positively regulated by LRIG2 in human
GBM, which was demonstrated in vitro and in vivo in
the present study. Congruently, Rondahl et al (20) used animal models of PDGFB-induced
glioma to demonstrate that Lrig2E12−/− mice, generated
by the ablation of Lrig2 exon 12, developed lower-grade tumors
(77%) or had no detectable tumors (23%), and Lrig2E12+/+
mice developed lower-grade tumors (82%) or high-grade GBM-like
tumors (18%). The Lrig2E12+/+ mice developed
PDGFB-driven gliomas at a higher frequency and of higher malignancy
compared with Lrig2E12−/− mice (20), suggesting a key promoting role of
Lrig2 in the regulation of PDGF signaling and in the development
and/or progression of glioma, particularly in mice, consistently
with the findings of the present study, which demonstrated that
LRIG2 positively regulates PDGFRβ signaling in human GBM.
Furthermore, the present results, which indicated that LRIG2 is
positively correlated with PDGFRβ in human GBM, are also in line
with those of a previous study reporting that LRIG2 expression is
associated with poor survival of patients with oligodendroglioma,
in which dysregulated PDGFR signaling is a common feature (18). To the best of our knowledge, this
is the first study demonstrating a positive association between
LRIG2 and PDGFR signaling in human GBM, providing compelling
evidence in support of the hypothesis that LRIG2 serves as a tumor
promoter in human glioma, which is distinct from the functions of
LRIG1 (23,24) and LRIG3 in glioma (6).
Furthermore, in order to provide further evidence to
support the concept that LRIG2 acts as a tumor promoter by
positively regulating PDGFRβ signaling in GBM, PDGF-BB-induced
biological effects and signaling events were analyzed in GBM cells
with LRIG2 overexpression or downregulation. The pathogenesis of
GBM is complex, due to a highly deregulated tumor genome with a
network of interconnected signaling pathways of three steps: i) An
input step in which membrane receptors are triggered from the
signals outside the cell; ii) a core system in which protein
kinases transmit the signal to the nucleus; and iii) an output step
in which transcription factors regulate the genes that affect
various cellular functions (29).
Congruently, LRIG2 initially plays a critical role in the first
input step by promoting the PDGF-BB-induced activation of PDGFRβ,
followed by enhancing the activation of Akt kinases and the Stat3
transcription factor, the core system of the second step,
ultimately resulting in the output step, in which the
pro-proliferative cell cycle proteins are accumulated and the
uncontrolled proliferation of GBM cells is promoted. Due to the
modulation of PDGFRβ signaling by LRIG2 upstream and the importance
of abnormal PDGFRβ signaling in gliomagenesis, it is reasonable to
propose that LRIG2 fits the role of a single critical oncogene, the
antagonism of which should abrogate the important downstream
deregulated signaling cascades and interrupt cell proliferation. It
is well documented that deregulation of the phosphoinositide
3-kinase/Akt pathway is an obligate event frequently observed in
gliomagenesis (2), with Akt
activation reported in ~80% of human GBMs (31). Stat3 is also aberrantly activated
in human GBM tissues and serves as a 'molecular hub' connecting
extracellular signals to the transcriptional control of critical
cellular events in gliomagenesis, including cell cycle progression,
proliferation, angiogenesis and immune evasion (32). Of note, our findings demonstrated
that LRIG2 positively regulated Akt and Stat3 pathways to promote
cell cycle progression and proliferation of GBM cells by modulating
PDGFRβ activation. However, Rondahl et al (20) reported that Lrig2 exerted no
effects on PDGFR protein levels or the phosphorylation events of
PDGFR and Akt in mouse embryonic fibroblasts (MEFs), which was
distinct from the results of the present study. Due to the fact
that the experiments by Rondahl et al were carried out in
MEFs, rather than in tumor cells or GBM cells, we hypothesized that
the positive regulation of LRIG2 on PDGFRβ and its downstream
pathways may be prominent in GBM cells or other tumor cells, but
subtle in non-tumor cells, highlighting that the role of LRIG2 in
the modulation of PDGFR signaling is oncogenic and
tumor-specific.
In addition to the pivotal role of LRIG2 in the
positive regulation of PDGFR signaling in GBM, we previously
demonstrated that LRIG2 also positively regulated EGFR signaling,
another common dysregulated RTK signaling pathway in GBM, to exert
its pro-tumor effects on GBM (21,22),
indicating that LRIG2 may play a key role in the genesis and
progression of GBM by positively regulating EGFR and PDGFR
signaling, the most commonly dysregulated RTKs in GBM. Over the
past decade, GBM research has primarily focused on determining and
investigating specific inhibitors targeting key RTK signaling
pathways, in order to develop novel and effective therapeutic
strategies to improve the prognosis of GBM. However, accumulating
clinical trial data to date, evaluating RTK inhibitors, including
EGFR tyrosine kinase inhibitors (TKIs) and PDGFR TKIs, support the
conclusion that RTK inhibitors are not beneficial for patients with
GBM and the underlying mechanisms have not yet been fully
elucidated (33). It has been
demonstrated that multiple RTKs may be activated simultaneously in
GBM cells (34,35), which is a plausible explanation for
the disappointing effects of RTK inhibitors, in that targeting
specific RTK signaling may result in compensatory activation of
alternative signaling mediators. Our determination of the positive
regulation of LRIG2 on EGFR and PDGFR, the most common dysregulated
RTKs in GBM, not only improved our understanding of the endogenous
regulatory mechanisms of EGFR and PDGFR, but also provided one
potential explanation for the limited clinical responses to
anti-EGFR or anti-PDGFR therapies using corresponding inhibitors,
which is that targeting specific RTK signaling may confer
activation of alternative RTK signaling through LRIG2, the key
mediator of multiple RTK signals. Our data provide the rationale
for targeting LRIG2 as a strategy to achieve the dual inhibition of
EGFR and PDGFR signaling in GBM, highlighting the possibility of
targeting LRIG2 as a promising therapeutic strategy for the future
treatment of GBM.
To the best of our knowledge, this is the first
study to demonstrate that LRIG2 positively regulates multiple RTKs,
including PDGFRβ, EGFR (22) and
other RTKs in GBM (data not published). The role of LRIG2 in the
positive regulation of RTKs in glioma is contrary to the role of
LRIG1, which has been well documented to negatively regulate
multiple RTKs and is a proposed tumor suppressor in a wide spectrum
of human cancer types, including glioma (6,23,24).
LRIG1 has been demonstrated to restrict RTK signaling through
different mechanisms, such as enhancing the degradation of EGFR in
a ligand-induced cbl-mediated ubiquitylation process (7,36),
destabilizing the MET receptor in a lysosome-dependent and
cbl-independent manner (8), and
inhibiting glial cell-derived neurotrophic factor binding with RET
(9). Little is known regarding the
mechanisms through which LRIG2 positively regulates RTKs. In the
present study, it was demonstrated that LRIG2 could physically
interact with PDGFRβ in the cytoplasm and increase the
PDGF-BB-induced total expression level and phosphorylation of
PDGFRβ, which was in line with the effects of LRIG2 on EGFR
(22). We hypothesized that the
physical interaction of LRIG2 with RTKs may stabilize the
expression level of RTKs in GBM cells; however, the critical
questions of how LRIG2 stabilizes RTKs, and why the function of
LRIG2 as a tumor promoter is largely distinct from that of LRIG1 in
glioma, remain unanswered and require further investigation.
Collectively, it was demonstrated that LRIG2
promoted cell cycle progression and proliferation of GBM cells
in vitro and in vivo by positively regulating the
PDGFRβ signaling pathways, a critical aberrant oncogenic RTK
signaling modulated by LRIG2 in GBM, similar to the previously
reported effects on EGFR signaling. Targeting LRIG2, the key
oncogenic regulator of multiple RTKs signals, appears to be
promising for developing novel therapeutic strategies for the
future treatment of GBM.
Abbreviations:
|
GMB
|
glioblastoma multiforme
|
|
LRIG
|
leucine-rich repeats and
immunoglobulin-like domains
|
|
RTK
|
receptor tyrosine kinase
|
|
PDGFR
|
platelet-derived growth factor
receptor
|
|
Stat3
|
signal transducer and activator of
transcription 3
|
Acknowledgments
The authors would like to thank Anding Liu for
providing technical assistance.
References
|
1
|
Wen PY and Reardon DA: Neuro-oncology in
2015: Progress in glioma diagnosis, classification and treatment.
Nat Rev Neurol. 12:69–70. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Patel M, Vogelbaum MA, Barnett GH, Jalali
R and Ahluwalia MS: Molecular targeted therapy in recurrent
glioblastoma: Current challenges and future directions. Expert Opin
Investig Drugs. 21:1247–1266. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cancer Genome Atlas Research Network:
Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Paulsson J, Lindh MB, Jarvius M, Puputti
M, Nistér M, Nupponen NN, Paulus W, Söderberg O, Dresemann G, von
Deimling A, et al: Prognostic but not predictive role of
platelet-derived growth factor receptors in patients with recurrent
glioblastoma. Int J Cancer. 128:1981–1988. 2011. View Article : Google Scholar
|
|
5
|
Guo D, Holmlund C, Henriksson R and Hedman
H: The LRIG gene family has three vertebrate paralogs widely
expressed in human and mouse tissues and a homolog in Ascidiacea.
Genomics. 84:157–165. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Simion C, Cedano-Prieto ME and Sweeney C:
The LRIG family: Enigmatic regulators of growth factor receptor
signaling. Endocr Relat Cancer. 21:R431–R443. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gur G, Rubin C, Katz M, Amit I, Citri A,
Nilsson J, Amariglio N, Henriksson R, Rechavi G, Hedman H, et al:
LRIG1 restricts growth factor signaling by enhancing receptor
ubiquitylation and degradation. EMBO J. 23:3270–3281. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shattuck DL, Miller JK, Laederich M, Funes
M, Petersen H, Carraway KL III and Sweeney C: LRIG1 is a novel
negative regulator of the Met receptor and opposes Met and Her2
synergy. Mol Cell Biol. 27:1934–1946. 2007. View Article : Google Scholar :
|
|
9
|
Ledda F, Bieraugel O, Fard SS, Vilar M and
Paratcha G: Lrig1 is an endogenous inhibitor of Ret receptor
tyrosine kinase activation, downstream signaling, and biological
responses to GDNF. J Neurosci. 28:39–49. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lindström AK, Ekman K, Stendahl U, Tot T,
Henriksson R, Hedman H and Hellberg D: LRIG1 and squamous
epithelial uterine cervical cancer: Correlation to prognosis, other
tumor markers, sex steroid hormones, and smoking. Int J Gynecol
Cancer. 18:312–317. 2008. View Article : Google Scholar
|
|
11
|
Hellberg D, Tot T and Stendahl U: Pitfalls
in immunohistochemical validation of tumor marker expression -
exemplified in invasive cancer of the uterine cervix. Gynecol
Oncol. 112:235–240. 2009. View Article : Google Scholar
|
|
12
|
Krig SR, Frietze S, Simion C, Miller JK,
Fry WH, Rafidi H, Kotelawala L, Qi L, Griffith OL, Gray JW, et al:
Lrig1 is an estrogen-regulated growth suppressor and correlates
with longer relapse-free survival in ERalpha-positive breast
cancer. Mol Cancer Res. 9:1406–1417. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Y, Poulin EJ and Coffey RJ: LRIG1 is
a triple threat: ERBB negative regulator, intestinal stem cell
marker and tumour suppressor. Br J Cancer. 108:1765–1770. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sheu JJ, Lee CC, Hua CH, Li CI, Lai MT,
Lee SC, Cheng J, Chen CM, Chan C, Chao SC, et al: LRIG1 modulates
aggressiveness of head and neck cancers by regulating
EGFR-MAPK-SPHK1 signaling and extracellular matrix remodeling.
Oncogene. 33:1375–1384. 2014. View Article : Google Scholar
|
|
15
|
Ranhem C, Lillsunde Larsson G, Hedman H,
Lindquist D, Karlsson MG, Hellström AC, Östensson E, Sorbe B,
Hellman K and Andersson S: Expression of LRIG proteins as possible
prognostic factors in primary vaginal carcinoma. PLoS One.
12:e01838162017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rouam S, Moreau T and Broët P: Identifying
common prognostic factors in genomic cancer studies: A novel index
for censored outcomes. BMC Bioinformatics. 11:1502010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guo D, Nilsson J, Haapasalo H, Raheem O,
Bergenheim T, Hedman H and Henriksson R: Perinuclear leucine-rich
repeats and immunoglobulin-like domain proteins (LRIG1-3) as
prognostic indicators in astrocytic tumors. Acta Neuropathol.
111:238–246. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Holmlund C, Haapasalo H, Yi W, Raheem O,
Brännström T, Bragge H, Henriksson R and Hedman H: Cytoplasmic
LRIG2 expression is associated with poor oligodendroglioma patient
survival. Neuropathology. 29:242–247. 2009. View Article : Google Scholar
|
|
19
|
Hedman H, Lindström AK, Tot T, Stendahl U,
Henriksson R and Hellberg D: LRIG2 in contrast to LRIG1 predicts
poor survival in early-stage squamous cell carcinoma of the uterine
cervix. Acta Oncol. 49:812–815. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rondahl V, Holmlund C, Karlsson T, Wang B,
Faraz M, Henriksson R and Hedman H: Lrig2-deficient mice are
protected against PDGFB-induced glioma. PLoS One. 8:e736352013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang B, Han L, Chen R, Cai M, Han F, Lei T
and Guo D: Downregulation of LRIG2 expression by RNA interference
inhibits glioblastoma cell (GL15) growth, causes cell cycle
redistribution, increases cell apoptosis and enhances cell adhesion
and invasion in vitro. Cancer Biol Ther. 8:1018–1023. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xiao Q, Tan Y, Guo Y, Yang H, Mao F, Xie
R, Wang B, Lei T and Guo D: Soluble LRIG2 ectodomain is released
from glioblastoma cells and promotes the proliferation and inhibits
the apoptosis of glioblastoma cells in vitro and in vivo in a
similar manner to the full-length LRIG2. PLoS One. 9:e1114192014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mao F, Wang B, Xiao Q, Xi G, Sun W, Zhang
H, Ye F, Wan F, Guo D, Lei T, et al: A role for LRIG1 in the
regulation of malignant glioma aggressiveness. Int J Oncol.
42:1081–1087. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xie R, Yang H, Xiao Q, Mao F, Zhang S, Ye
F, Wan F, Wang B, Lei T and Guo D: Downregulation of LRIG1
expression by RNA interference promotes the aggressive properties
of glioma cells via EGFR/Akt/c-Myc activation. Oncol Rep.
29:177–184. 2013. View Article : Google Scholar
|
|
25
|
Allen M, Bjerke M, Edlund H, Nelander S
and Westermark B: Origin of the U87MG glioma cell line: Good news
and bad news. Sci Transl Med. 8:354re32016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Holmlund C, Nilsson J, Guo D, Starefeldt
A, Golovleva I, Henriksson R and Hedman H: Characterization and
tissue-specific expression of human LRIG2. Gene. 332:35–43. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Andrae J, Gallini R and Betsholtz C: Role
of platelet-derived growth factors in physiology and medicine.
Genes Dev. 22:1276–1312. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nazarenko I, Hede SM, He X, Hedrén A,
Thompson J, Lindström MS and Nistér M: PDGF and PDGF receptors in
glioma. Ups J Med Sci. 117:99–112. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nakada M, Kita D, Watanabe T, Hayashi Y,
Teng L, Pyko IV and Hamada J: Aberrant signaling pathways in
glioma. Cancers (Basel). 3:3242–3278. 2011. View Article : Google Scholar
|
|
30
|
Paradowski M, Bilinska M and Bar J:
Characteristics of the expression of KAI1/CD82 and PDGFRβ and their
impact on glioma progression. Folia Neuropathol. 54:241–248. 2016.
View Article : Google Scholar
|
|
31
|
Holland EC, Celestino J, Dai C, Schaefer
L, Sawaya RE and Fuller GN: Combined activation of Ras and Akt in
neural progenitors induces glioblastoma formation in mice. Nat
Genet. 25:55–57. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Brantley EC and Benveniste EN: Signal
transducer and activator of transcription-3: A molecular hub for
signaling pathways in gliomas. Mol Cancer Res. 6:675–684. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Reardon DA, Wen PY and Mellinghoff IK:
Targeted molecular therapies against epidermal growth factor
receptor: past experiences and challenges. Neuro Oncol. 16(Suppl
8): viii7–13. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Van Meir EG, Hadjipanayis CG, Norden AD,
Shu HK, Wen PY and Olson JJ: Exciting new advances in
neuro-oncology: The avenue to a cure for malignant glioma. CA
Cancer J Clin. 60:166–193. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Snuderl M, Fazlollahi L, Le LP, Nitta M,
Zhelyazkova BH, Davidson CJ, Akhavanfard S, Cahill DP, Aldape KD,
Betensky RA, et al: Mosaic amplification of multiple receptor
tyrosine kinase genes in glioblastoma. Cancer Cell. 20:810–817.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Laederich MB, Funes-Duran M, Yen L,
Ingalla E, Wu X, Carraway KL III and Sweeney C: The leucine-rich
repeat protein LRIG1 is a negative regulator of ErbB family
receptor tyrosine kinases. J Biol Chem. 279:47050–47056. 2004.
View Article : Google Scholar : PubMed/NCBI
|