Introduction
MicroRNAs (miRNAs or miRs) are small,
single-stranded non-coding RNAs consisting of 21–23 nucleotides
that regulate protein expression by pairing with the 3′
untranslated region (3′UTR) of target mRNAs (1). The role of miRNAs, which are
associated with multiple facets of cellular activities, is one of
the most important and exciting emerging paradigms in cell biology.
Accumulating genetic evidence indicates that miRNAs also play an
essential part in stress responses, such as hypoxia, hydrogen
peroxide (H2O2) and ionizing radiation (IR)
(2–5). For example, it has been reported that
several miRNAs are regulated by IR treatment (6,7). The
miRNAs, let-7, miR-34 and miR-16, have been identified to be
upregulated in response to irradiation (8–10).
Of note, the majority of these IR-induced miRNAs are also
frequently dysregulated in cancer cells, and specific miRNAs are
known to regulate both cell cycle progression and apoptosis
(11,12). These miRNAs may be predictive
markers for clinical outcomes in human cancers or may be exploited
to enhance sensitivity to radiotherapy, impede DNA double-strand
break repair and induce chromosomal instability in γ-irradiated
cells (13–15).
Radiotherapy, as the one of the three primary cancer
treatment modalities, is used in approximately 50% of all cancer
patients. It can disrupt cancer growth primarily by modulating
several signal transduction pathways, causing DNA damage and
enhancing tumor reaction to other therapies (16). However, the mechanisms underlying
IR-induced miRNA expression and its possible effects are not yet
well understood.
In our previous study, miRNA expression profiles
were analyzed in HeLa cells at 6 h following treatment with 8 Gy
IR. Using reverse transcription-quantitative PCR (RT-qPCR), the
expression levels of 21 miRNAs, including the let-7 family, miR-34a
and miR-320a were found to be significantly altered (17).
miR-320a is an important miRNA and can regulate the
expression of various target genes. For example, it has reported
that survivin and c-Myc are suppressed by miR-320a (18,19).
Bronisz et al demonstrated that miR-320 functions as a
critical component of the PTEN tumour-suppressor axis and curtailed
tumour progression (20). We
previously found that the expression of miR-320a was altered in
HeLa cells treated with 8 Gy IR (17). However, the mechanisms through
which miR-320a is regulated in response to IR and its target genes
in this process remain unclear.
In the present study, we identified the promoter
region of miR-320a and verified three transcription factors,
activating transcription factor 2 (ATF2), ETS transcription factor
(ELK1) and YY1 transcription factor (YY1), to be responsible for
inducing miR-320a expression under IR conditions. We provided
evidence identifying the novel miR-320a target gene, XIAP,
and suggest that miR-320a enhances the radiosensitivity of cancer
cells. We aim to further investigate whether such a regulation of
miR-320a by IR occurs in vivo, and the possibility of
whether miR-320a can be used as a radiation sensitizer in future
studies.
Materials and methods
Cell culture conditions and
treatments
Human uterine cervical cancer cells (HeLa) were
cultured in RPMI-1640 (Thermo Fisher Scientific, Waltham, MA, USA)
and human colon cancer cells (HCT116) were cultured in Dulbecco's
modified Eagle's medium (DMEM) (Thermo Fisher Scientific)
supplemented with 10% fetal bovine serum, penicillin and
streptomycin. The cells were maintained at 37°C in the presence of
5% CO2. The cells were plated in T-25 flasks and treated
with IR at various doses (0, 1, 3, 5, 8 and 12 Gy) following
incubation at 37°C for 24 h to achieve logarithmic growth.
miRNAs, siRNAs and transfection
Synthetic miRNA mimics and siRNAs were obtained from
GenePharma (Shanghai, China) (Table
I). The HeLa and HCT116 cells were grown to 50% confluence in
6-well plates. The transfection of siRNAs and miRNAs was performed
with jetPRIME (Polyplus, Illkirch, France) according to the
manufacturer's instructions. The transfection medium was replaced
with fresh medium after 6 h of incubation at 37°C. Total RNA and
protein were prepared at 48 h following transfection and used for
RT-qPCR detection or western blot analysis. The p38 and JNK
agonist, anisomycin (Ani), the p38 inhibitor, SB203580 (SB), and
the JNK inhibitor, SP600125 (SP), were purchased from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany) and dissolved in
dimethyl sulfoxide (DMSO). The cells were respectively treated with
Ani (20 µM), SB (10 µM), and SP (20 µM) for 1
h prior to IR and harvested for western blot analysis after 24 h of
incubation at 37°C.
 | Table IList of siRNAs, primers and miRNAs
utilized in the experiments and their sequences. |
Table I
List of siRNAs, primers and miRNAs
utilized in the experiments and their sequences.
| Primer name | Application | Sequence |
|---|
| Negative
control-F | siRNA |
UGAAUUAGAUGGCGAUGUUTT |
| Negative
control-R | siRNA |
AACAUCGCCAUCUAAUUCATT |
| ATF2-F | siRNA |
GUUGGCGAGUCCAUUUGAGTT |
| ATF2-R | siRNA |
CUCAAAUGGACUCGCCAACTT |
| ELK1-F | siRNA |
GGUGAGCGGCCAGAAGUUCTT |
| ELK1-R | siRNA |
GAACUUCUGGCCGCUCACCTT |
| YY1-F | siRNA |
GGGAGCAGAAGCAGGUGCAGAUTT |
| YY1-R | siRNA |
AUCUGCACCUGCUUCUGCUCCCTT |
| XIAP-F | siRNA |
GGACAUAGUUUGAAGGUGATT |
| XIAP-R | siRNA |
UCACCUUCAAACUAUGUCCTT |
| miR-320a-guide | mimics |
AAAAGCUGGGUUGAGAGGGCGAA |
|
miR-320a-passenger | mimics |
CGCCCUCUCAACCCAGCUUUUUU |
| miR-320a
inhibitor | 2′ Ome |
CCUCUCAACCCAGCUUUU |
| microRNA inhibitor
N.C | 2′ Ome |
CAGUACUUUUGUGUAGUACAA |
| miRNA RT
primer | RT |
GCGAGCACAGAATTAATACGACTCACTATAGG(T)18VN |
| mRNA RT primer | RT |
TTTTTTTTTTTTTTTTTT |
| Universal
primer | Real-time PCR |
GCGAGCACAGAATTAATACGAC |
| miRNA-320a | Real-time PCR |
AAAAGCTGGGTTGAGAGGGCGA |
| U6 snRNA-F | Real-time PCR |
CTCGCTTCGGCAGCACA |
| U6 snRNA-R | Real-time PCR |
AACGCTTCACGAATTTGCGT |
| GAPDH-F | Real-time PCR |
TCAGTGGTGGACCTGACCTG |
| GAPDH-R | Real-time PCR |
TGCTGTAGCCAAATTCGTTG |
| pri-miR-320a-F | Real-time PCR |
TGAGGTGACTGGTCTGGGCTAC |
| pri-miR-320a-R | Real-time PCR |
TCACATTGCGGCGTGGTG |
| XIAP-F | Real-time PCR |
GGGGTTCAGTTTCAAGGACA |
| XIAP-R | Real-time PCR |
CGCCTTAGCTGCTCTTCAGT |
| ATF2-F | Real-time PCR |
CTCCAGCTCACACAACTCCA |
| ATF2-R | Real-time PCR |
TGTTTCAGCTGTGCCACTTC |
| ELK1-F | Real-time PCR |
CCACCTTCACCATCCAGTCT |
| ELK1-R | Real-time PCR |
TCTTCCGATTTCAGGTTTGG |
| YY1-F | Real-time PCR |
GGAGGAATACCTGGCATTGA |
| YY1-R | Real-time PCR |
TTCTGCACAGACGTGGACTC |
|
ChIP-miR-320a-F | Real-time PCR |
ACCAGCCGCCAGCCTTCGGTCTCC |
|
ChIP-miR-320a-R | Real-time PCR |
GCCCAGTCCCGCCCCCCTAACGTC |
| ChIP-GAPDH-F | Real-time PCR |
TGATGACATCAAGAAGGTGGTGAAG |
| ChIP-GAPDH-R | Real-time PCR |
TCCTTGGAGGCCATGTGGGCCAT |
| 5′ RACE RT
Primer | 5′ RACE |
TCACATTGCGGCGTGGTG |
| 5′ RACE Primer | 5′ RACE |
CGACTGGAGCACGAGGACACTGA |
| 5′ RACE Nested
Primer | 5′ RACE |
GGACACTGACATGGACTGAAGGAGTA |
| XIAP-1-UTR-F | Cloning | agttctagaTTGAATGTGTGATGTGAACTG |
| XIAP-1-UTR-R | Cloning | agtcatatgAACTAGCAAGGATTAAGGATG |
| XIAP-1-UTR-M-F | Cloning |
GCAAAACTACTTATCACTCTGGTATAGTTTTTCTAATCCAAGAAG |
| XIAP-1-UTR-M-R | Cloning |
CTTCTTGGATTAGAAAAACTATACCAGAGTGATAAGTAGTTTTGC |
| XIAP-2-UTR-F | Cloning | agttctagaTTTTTGGTGCCAATGTGAAA |
| XIAP-2-UTR-R | Cloning | agtcatatgAAGGCGAGTGGATCTCTTGA |
| XIAP-2-UTR-M-F | Cloning |
CACTGCTTGTAGTTATAGTGACTGGTATCCATGTTGAGATTCTCATATC |
| XIAP-2-UTR-M-R | Cloning |
GATATGAGAATCTCAACATGGATACCAGTCACTATAACTACAAGCAGTG |
| XIAP-3-UTR-F | Cloning | agttctagaTGCTGTTGCTTCAGGTTCTTA |
| XIAP-3-UTR-R | Cloning | agtcatatgAGCCCCATTTTATTTCATTTC |
| XIAP-3-UTR-M-F | Cloning |
CCAGTTAATTAATTAATACCATAATATTGCCTTTCCTGCTAC |
| XIAP-3-UTR-M-R | Cloning |
GTAGCAGGAAAGGCAATATTATGGTATTAATTAATTAACTGG |
| pCMV6-ATF2-F | Cloning | agtggcgcgccaATGAAATTCAAGTTACATGTG |
| pCMV6-ATF2-R | Cloning | agtctcgagAGGTTTTTAATCAACTTCCT |
| pCMV6-ELK1-F | Cloning | agtggcgcgccaATGGACCCATCTGTGACGCTG |
| pCMV6-ELK1-R | Cloning | agtctcgagGGTGGTGGTGGTGGTGGTAGTA |
| pCMV6-YY1-F | Cloning | agtggcgcgccaATGGCCTCGGGCGACACCCT |
| pCMV6-YY1-R | Cloning | agtctcgagTCACTGGTTGTTTTTGGCCTTAGCATGT |
| 1,000 bp
Fragment-F | Cloning | agtctcgagGGAGAGGCCACAGAGCCTCG |
| 1,000 bp
Fragment-R | Cloning | agtaagcttGCCGCCTGATAAATACTGTGG |
| 500 bp
Fragment-F | Cloning | agtctcgagGCCGGCCGCCGCCCGATGAG |
| 500 bp
Fragment-R | Cloning | agtaagcttGCCGCCTGATAAATACTGTGG |
| 200 bp
Fragment-F | Cloning | agtctcgagCAGGAACCAGACAGGGACG |
| 200 bp
Fragment-R | Cloning | agtaagcttGCCGCCTGATAAATACTGTGG |
| Δ200
Fragment-F | Cloning | agtctcgagGGAGAGGCCACAGAGCCTCG |
| Δ200
Fragment-R | Cloning | agtaagcttGGCGGGGTTCCTGGAGAGG |
| mut-200-ATF2-F | Cloning |
CTTGGCGCGGGGCGGAAGAAGCGTTAGGGGGGCGGGAC |
| mut-200-ATF2-R | Cloning |
GTCCCGCCCCCCTAACGCTTCTTCCGCCCCGCGCCAAG |
| mut-200-ELK1-F | Cloning |
TCGGTCTCCGCGCCGGGCTTCAGTCTGCGTGGCAGGGCC |
| mut-200-ELK1-R | Cloning |
GGCCCTGCCACGCAGACTGAAGCCCGGCGCGGAGACCGA |
| mut-200-YY1-F | Cloning |
GGCAGGGCCTGGGCGCCGTTCTCTTGGCGCGGGGCGGAA |
| mut-200-YY1-F | Cloning |
TTCCGCCCCGCGCCAAGAGAACGGCGCCCAGGCCCTGCC |
Plasmid constructs
The 1,000, 800, 500 and 200 bp fragment of the
genomic sequence immediately upstream of the miR-320a stem-loop
structure were amplified by PCR from HeLa cell cDNA using Phusion
DNA polymerase (Thermo Fisher Scientific). A pair of primers,
including the XhoI and HindIII restriction sites were
designed to amplify the miR-320a promoter inserts. The PCR products
were cloned into the pGL3-enhancer vector (Promega, Madison, WI,
USA).
Firstly, we searched the putative target sites of
3′UTR of XIAP by bioinformatics analysis using TargetScan
(http://www.targetscan.org/) according to
the instructions provided (http://www.targetscan.org/faqs.Release_7.html). The
3′UTR of XIAP was then cloned into a modified pGL3-control
plasmid (21) following the PCR
amplification of genomic DNA. The coding sequences of ATF2,
ELK1 and YY1 were amplified from HeLa cell cDNA using
a pair of primers, including the AscI and XhoI
restriction sites and were ligated into pCMV6-AN-3DDK. The
pCMV6-3DDK-XIAP vector was purchased from Origene (Rockville, MD,
USA). The primer sequences are presented in Table I. The sequences of all constructs
were confirmed by DNA sequencing.
Quantification of miRNA and
pri-miRNA
Total RNA was extracted from the cells using TRIzol
reagent (Sigma-Aldrich; Merck KGaA) according to the manufacturer's
instructions. miRNA quantification was performed using a Quantitect
SYBR-Green PCR kit (Qiagen, Hilden, Germany). Reverse transcription
followed by RT-qPCR was carried out using a high capacity cDNA
reverse transcription kit (Thermo Fisher Scientific) as previously
described by Fu et al (22). Specific primer sequences were
designed according to miRBase 22.0 (http://www.mirbase.org/). For pri-miRNA
quantification, RT-qPCR was performed according to mRNA
quantitation method (23). GAPDH
was used as an endogenous control. All qPCR experiments were
carried out using the Mx3000p detection system (Agilent
Technologies, Palo Alto, CA, USA). The PCR thermal cycle began at
95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C
for 1 min. The primer sequences are presented in Table I. GenEX software version 5.0 was
employed to analyze the qPCR data. One-way ANOVA with Dunnett's
post hoc tests were performed to determine the difference in miRNA
expression levels observed in the cells treated with IR at various
doses (0, 1, 3, 5, 8 and 12 Gy) and treatment times (0, 3, 6, 12,
24, 48 and 72 h).
Chromatin immunoprecipitation (ChIP)
ChIP assays were carried out using a commercial kit
from Cell Signaling Technology (CST, Danvers, MA, USA). Briefly,
the cells were first fixed with 1% formaldehyde, and chromatin DNA
was isolated with 1 µg anti-ATF2 (sc-187X), 1 µg
anti-ELK1 (sc-355X), 1 µg anti-YY1 (sc-1703X), or 1
µg IgG (sc-2027) antibodies from Santa Cruz Biotechnology
(Santa Cruz, CA, USA) the indicated antibody and bound protein was
digested with proteinase K. qPCR was performed using SYBR Premix Ex
Taq (Takara, Dalian, China). The sequences of the primers used to
amplify the ChIP samples are provided in Table I.
Western blot analysis
Total cell protein lysates were prepared using a
modified radio-immunoprecipitation assay (RIPA) buffer (50 mM
Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate
and 0.5% sodium dodecyl sulfate) in the presence of a proteinase
inhibitor cocktail (Roche, Basel, Switzerland). Protein
concentrations were determined using BCA protein assay reagent
(Beyotime, Shanghai, China). Protein samples (30 µg per
lane) were separated by 10% sodium dodecyl sulfate-polyacrylamide
gel electrophoresis (SDS-PAGE) and transferred onto Immobilon
Hybond-C membranes (Amersham Biosciences, Piscataway, NJ, USA).
After blocking with 5% non-fat dried milk for 3 h at room
temperature, the membranes were incubated overnight at 4°C with
primary antibodies. The antibodies used were as follows: ATF2
(1:1,000; ab32160), ELK1 (1:1,000; ab32106), YY1 (1:1,000;
ab109237) (all from Abcam), XIAP (1:1,000; 2045), p-ATF2 (1:1,000;
9225), JNK (1:1,000; 9258), p-ELK1 (1:1,000; 9181), p-JNK (1:1,000;
4668), p38 (1:1,000; 9212), p-p38 (1:1,000; 4511), ERK (1:1,000;
4695), p-ERK (1:1,000; 4370) (all from Cell Signaling Technology)
and β-actin (1:1,000; sc-47778; Santa Cruz Biotechnology). The
blots were then incubated with horseradish peroxidase-conjugated
secondary antibodies (1:3,000; ZB2301; Zhongshan Jinqiao, Beijing,
China). Signals were detected with the ECL western blot detection
reagent (Thermo Fisher Scientific).
Cell proliferation assays
The HeLa cells were seeded in 60-mm flasks at
1×105 cells, simultaneously transfected with a serial of
RNA duplex and incubated at 37°C for a further 24 h. The cells were
then treated with 8 Gy IR and incubated at 37°C for 24 h. The
number of living cells was directly counted using the trypan blue
exclusion method during the period of 5 days. Briefly, the cells
were harvested and resuspended in 1 ml PBS. Subsequently, 1 part
cell susupension was mixed with 1 part of 0.4% trypan blue
(Beyotime). The mixture was incubated 3 min at room temperature and
counted using a hemacytometer.
Luciferase reporter assays
The HeLa cells were co-transfected with 100 ng.
Firefly luciferase reporter vector containing plasmid constructs
and 1 ng of a control vector containing Renilla luciferase
pRL-TK (Promega) in a final volume of 0.5 ml RPMI-1640 in 24-well
plates using jetPRIME. The Firefly and Renilla luciferase
activities were measured consecutively using a dual-luciferase
reporter assay system (Promega) at 48 h following transfection.
Annexin V apoptosis assays
Annexin V apoptosis assays were performed using an
Annexin V-EGFP kit (Biolegend, San Diego, CA, USA) according to the
manufacturer's instructions. The stained cells were immediately
analyzed on a FACSCalibur flow cytometer (BD Biosciences, Franklin
Lakes, NJ, USA). Each measurement was carried out at least in
triplicate. The data were displayed as the percentage of apoptotic
cells.
Cell proliferation assays and colony
forming unit assays
The cell proliferation rate was determined using
cell growth curve analysis. Cells were seeded in 60 mm flasks with
1×105 cells, simultaneously transfected with a serial of
RNA duplex and incubated for a further 24 h. The cells were then
treated with 8 Gy IR and incubated at 37°C for 24 h. The number of
living cells was directly counted using the trypan blue exclusion
method during the period of 5 days.
For colony forming unit (CFU) assays, the cells were
transfected with miR-320a mimics, and 900 cells were seeded in each
well of a fresh 6-well plate for 24 h prior to transfection. The
CFU assay was performed 7 days after transfection, and the scoring
criteria for colonies required at least 50 cells per colony.
Statistical analysis of the data
Unless otherwise indicated, all the values are
reported as the means ± standard deviation. Data analyses were
estimated using SPSS software version 18.0 (IBM SPSS, Armonk, NY,
USA). When the datasets contained multiple groups, statistical
differences were evaluated by a one-way ANOVA with Dunnett's post
hoc test. When only 2 groups were compared once in the datasets,
statistical differences were evaluated using a Student's t-test. A
value of P<0.05 was considered to indicate a statistically
significant difference.
Results
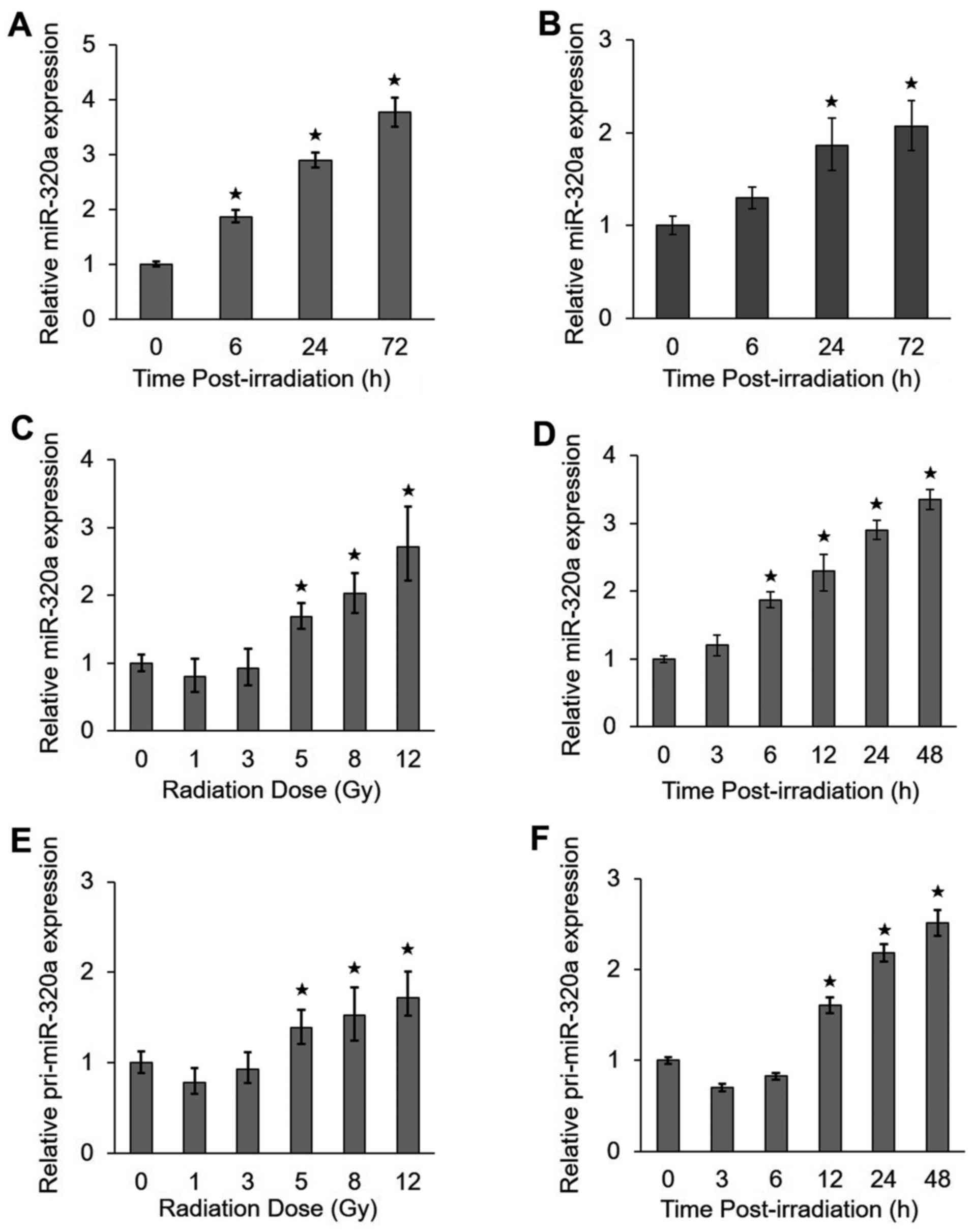
IR upregulates miR-320a expression
To identify which miRNAs are regulated by IR, we
performed a miRNA microarray analysis in HeLa cells treated with 8
Gy IR. We previously found that the upregulation of miR-320a
expression by IR was significant using a strict cut-off value of
P<0.01 (>2.7-fold) (17). To
confirm the microarray results, we analyzed miR-320a expression by
RT-qPCR in the HeLa and HCT-116 cells under IR conditions. We found
that the expression of miR-320a increased with time from 0 to 72 h
in the HeLa cells (Fig. 1A) and
HCT116 cells (Fig. 1B) treated
with 8 Gy IR. In addition, the expression of miR-320a also
increased with radiation doses ranging from 1 to 12 Gy at 24 h
following exposure to IR (Fig. 1C)
and with the radiation time ranging from 0 to 48 h following
exposure to 8 Gy IR (Fig. 1D) in
the HeLa cells. To determine whether miR-320a accumulation is
mediated by transcriptional regulation, we also detected the
expression of the primary precursor of miR-320a (pri-miR-320a). Of
note, we found that pri-miR-320a exhibited similar expression
profiles as miR-320a (Fig. 1E and
F). Taken together, these data suggested that miR-320a was
highly responsive to treatment with IR.
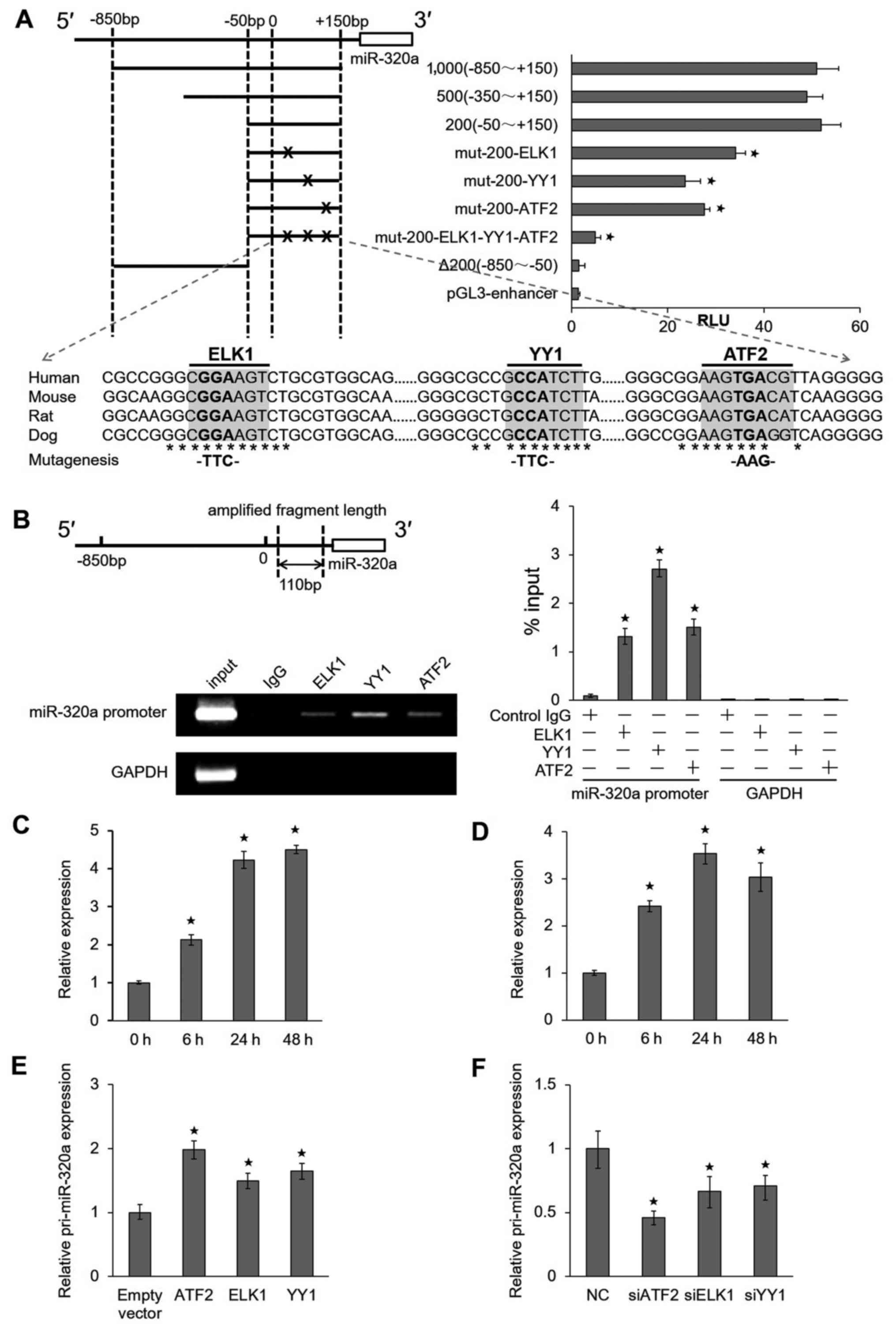
ATF2, ELK1 and YY1 interact directly with
miR-320a promoter
To elucidate the mechanisms through which miR-320a
is upregulated by IR, we examined the precise location of the
miR-320a promoter region. Four serial deletion luciferase reporter
gene vectors (1,000, 800, 500 and 200 bp fragments) were
constructed (Fig. 2A, top left
panel), and their relative luciferase reporter units were measured.
We observed that a 200 bp fragment was responsible for the robust
induction of the miR-320a promoter. The analysis of the 200 bp
miR-320a core-promoter sequence using the TFSEARCH program
(http://diyhpl.us/~bryan/irc/protocol-online/protocol-cache/TFSEARCH.html)
suggested that there were three potential transcription factor
binding sites for ATF2, ELK1 and YY1. The mutation of any of these
three binding sites could attenuate luciferase activities, and the
simultaneous mutation of all three sites completely abolished the
responsiveness of the promoter to these transcription factors
(Fig. 2A, top right panel).
Moreover, these three sites were highly conserved in the human,
mouse, rat and dog miR-320a genomic sequences (Fig. 2A, bottom panel), implying that
these elements were functionally important.
To determine whether these transcription factors
directly bind the miR-320a promoter, we performed ChIP assays using
anti-ATF2, anti-ELK1 and anti-YY1 antibodies. The results revealed
that the miR-320a promoter sequences were enriched in these samples
compared to the samples precipitated with an isotype-matched normal
IgG. Moreover, an irrelevant DNA sequence (GAPDH coding
region) was not specifically precipitated by these antibodies
(Fig. 2B, top left panel). The
ChIP results were also confirmed by RT-qPCR (Fig. 2B, bottom left and right panel).
Moreover, we performed the ChIP assay using anti-ATF2 with a
radiation time ranging from 0 to 48 h in HeLa cells (Fig. 2C) and HCT116 cells (Fig. 2D) treated with 8 Gy IR. The results
indicated that the miR-320a promoter sequences were enriched by
ATF2. Furthermore, the overexpression of ATF2, ELK1 and YY1 in HeLa
cells could lead to the upregulation of pri-miR-320a expression
(Fig. 2E). By contrast, the
knockdown of ATF2, ELK1 and YY1 significantly suppressed the
expression of pri-miR-320a (Fig.
2F). We obtained similar results in the HCT116 cells (data not
shown). Taken together, these results suggest that ATF2,
ELK1 and YY1 can bind the miR-320a promoter and
promote the transcription of miR-320a under IR conditions.
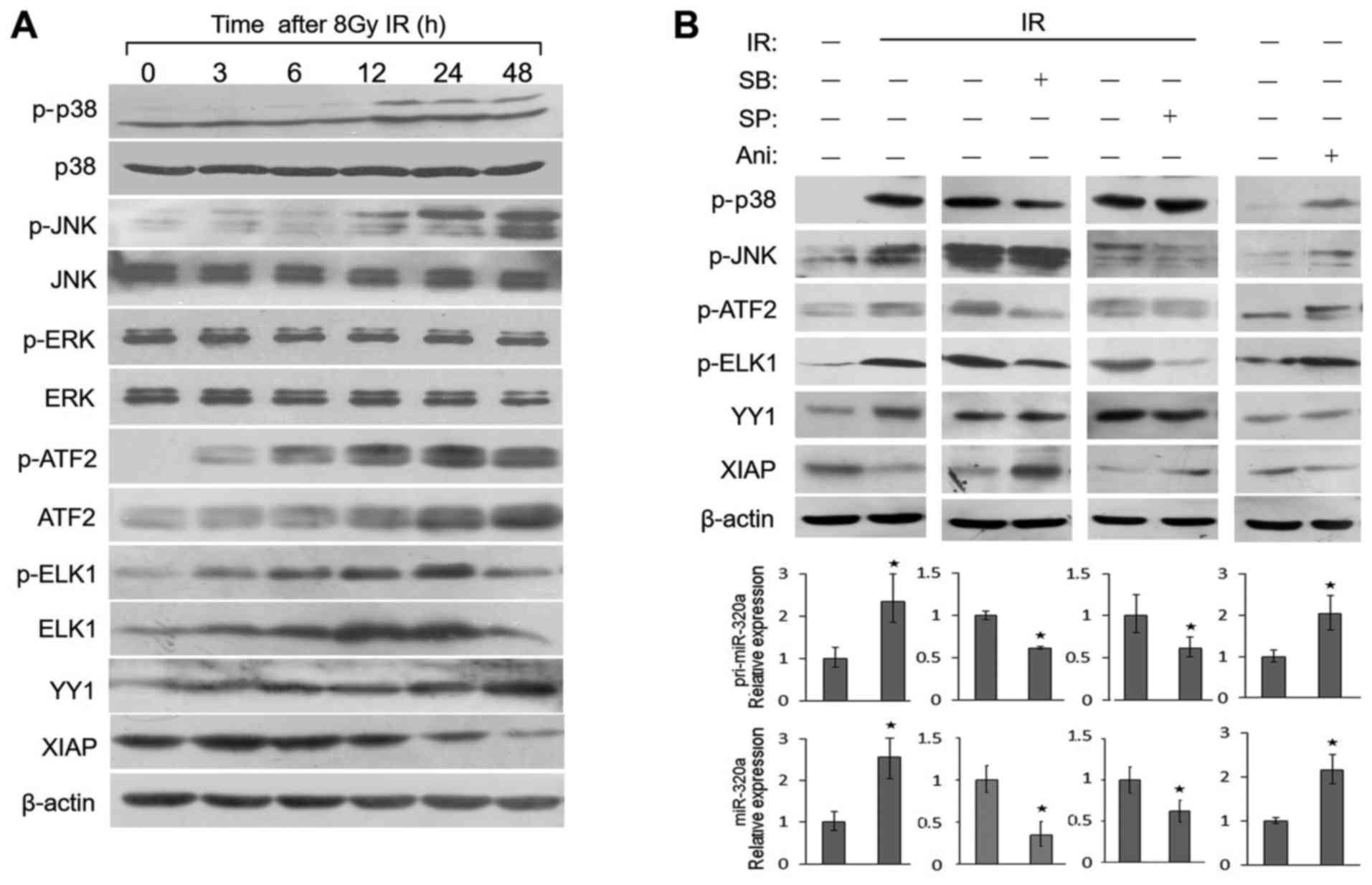
p38 MAPK and JNK activate transcription
factors to regulate miR-320a expression under IR conditions
Both the p38 MAPK and JNK signaling pathways play a
vital role in cellular response to IR, UV light and oxygen stress
(24,25). ATF2 and ELK1 can be both
phosphorylated by p38 and JNK. In order to identify whether these
signal transduction pathways stimulate miR-320a expression under IR
conditions, we first examined the phosphorylation and activity of
these two signal transduction pathways in response to IR in HeLa
cells. As shown in Fig. 3A, the
levels of phosphorylated p38 (p-p38) and phosphorylated JNK (p-JNK)
were found to gradually increase from 3 to 48 h following treatment
with 8 Gy IR, with a peak expression at 48 h; the total protein
levels of p38 and JNK were not altered. By contrast, IR did not
alter the phosphorylation or expression levels of endogenous ERK
protein. As targets of p38 and JNK, the phosphorylation or
expression levels of ATF2 and ELK1 were also activated (Fig. 3A); similar results were obtained
with the HCT116 cells (data not shown). Moreover, these expression
profiles were consistent with those of miR-320a expression
(Fig. 1D).
To investigate whether the regulation of miR-320a
expression by ATF2, YY1 and ELK1 is mediated by p38 and JNK, we
detected their expression levels in HeLa cells treated with two
specific chemical inhibitors of p38 (SB203580) and JNK (SP600125)
or an agonist of p38 and JNK (anisomycin). The results revealed
that either anisomycin or IR directly increased the protein
expression or phosphorylation of a series of proteins, including
p38, JNK, ATF2, ELK1 and YY1 (Fig.
3B, top panels). Moreover, the transcription of pri-miR-320a
and miR-320a expression also increased (Fig. 3B, bottom panels). SB203580 or
SP600125 inhibited the phosphorylation of ATF2 and ELK1, but not
that of YY1 (Fig. 3B, top panels),
resulting in the decreased expression of pri-miR-320a and miR-320a
(Fig. 3B, bottom panels). The
results suggested that IR induces the expression of YY1 and the
phosphorylation of ATF2 and ELK to induce miR-320a expression
through the p38 MAPK/JNK pathway.
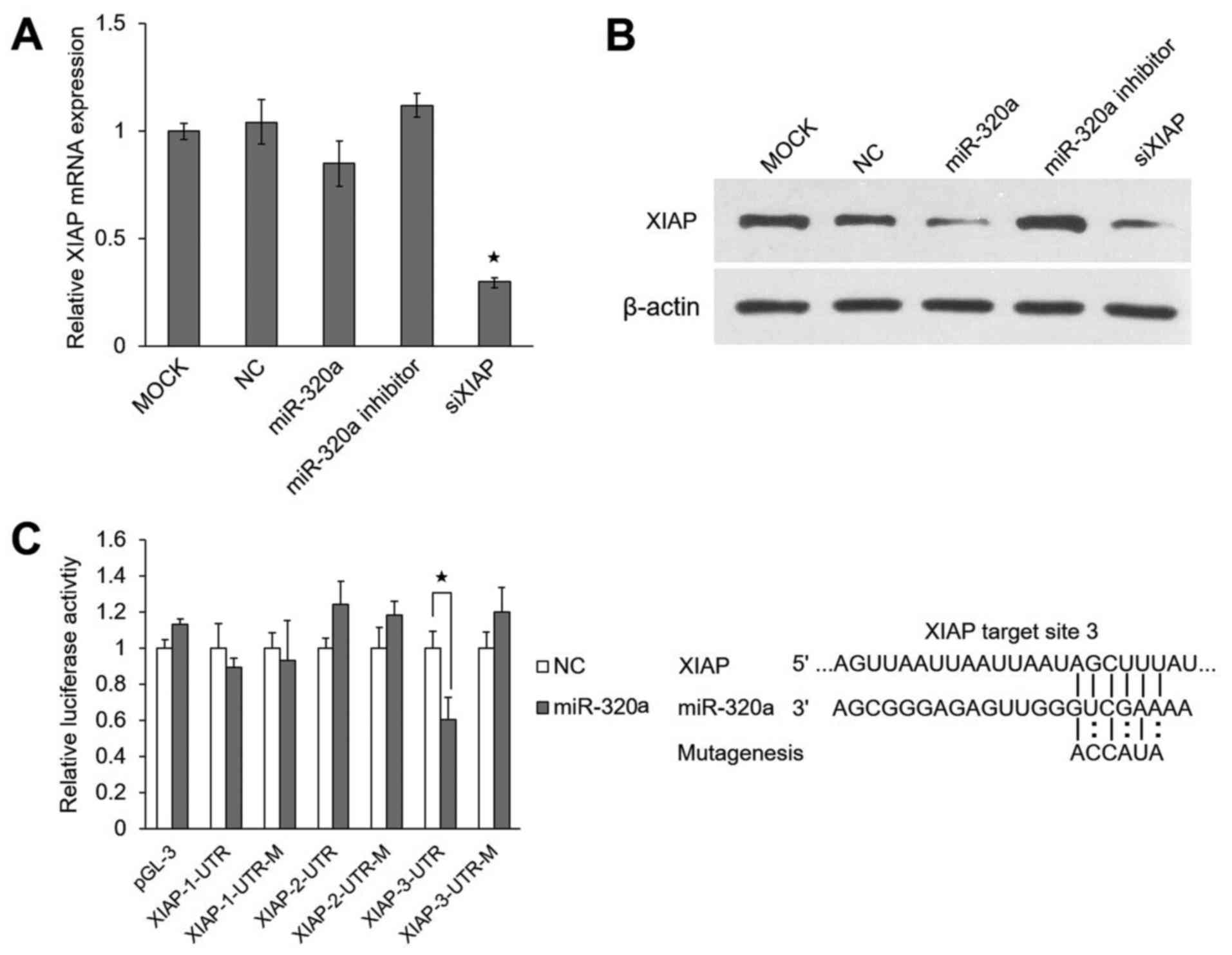
miR-320a suppresses XIAP expression by
targeting its 3′UTR
miRNAs can regulate their target genes by inhibiting
protein translation and/or degrading mRNAs. In this study, we found
that the 3′UTR of XIAP had 3 predicted target sites, through
TargetScan (http://www.targetscan.org/) (26). XIAP protein is necessary for
anti-apoptotic function and is usually overexpressed in cancer
cells (27,28).
In this study, we found that the overexpression or
the inhibition of miR-320a had little effect on the mRNA level of
XIAP in HeLa cells (Fig.
4A), but markedly affected its protein level (Fig. 4B). These results support the
hypothesis that miR-320a regulates the expression of XIAP
directly at the translational level.
To determine whether miR-320a targets the 3′UTR of
the XIAP gene directly, luciferase reporter assays were
performed using the HeLa cells. The results revealed that miR-320a
significantly decreased the relative luciferase activity (~42%) of
XIAP-3-3′UTR as compared to the negative control. However, the
reporter with mutated effective target was not suppressed by
miR-320a (Fig. 4C). These results
indicated that XIAP was indeed a direct target of
miR-320a.
miR-320a enhances cellular
radiosensitivity by inducing apoptosis and suppressing
proliferation
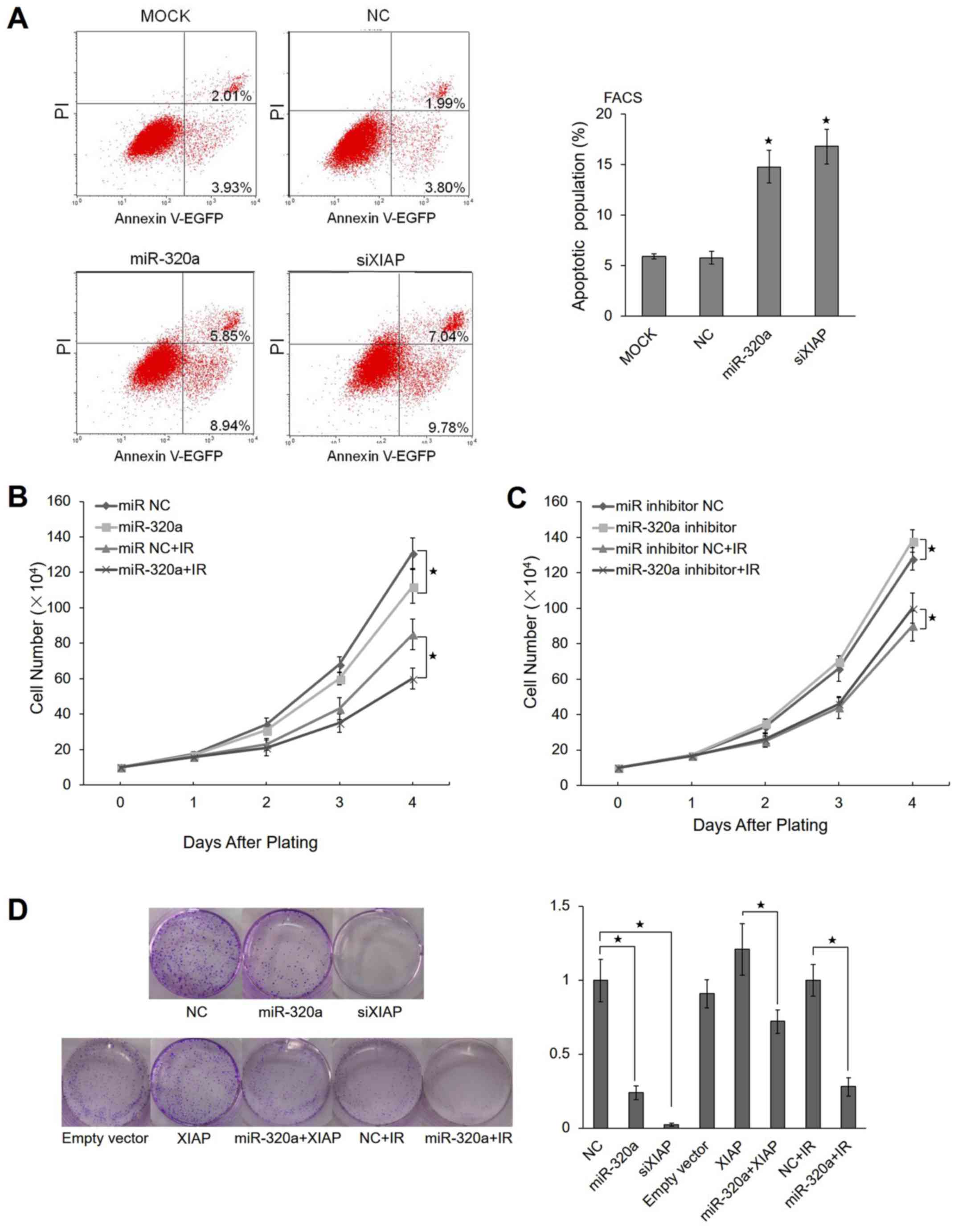
To validate whether miR-320a induces apoptosis, we
performed an Annexin V apoptosis assay using the HeLa cells. The
results revealed that approximately 14.79% of HeLa cells
transfected with miR-320a underwent apoptosis compared with 5.94%
of untreated (mock) cells and 5.79% of the control cells.
Similarly, 16.82% of the cells transfected with XIAP siRNA
underwent apoptosis, suggesting that miR-320a promotes apoptosis by
regulating XIAP expression (Fig.
5A).
IR can cause multiple types of cellular responses,
such as DNA strand breaks, cell cycle arrest and apoptosis
(29). Radiotherapy is an
important treatment for cancer patients, and miR-320a expression
can be induced by IR. This led us to examine whether miR-320a can
affect the proliferation of tumor cells. We used the trypan blue
exclusion method to evaluate the proliferation of HeLa cells
overexpressing miR-320a. We observed that miR-320a inhibited cell
proliferation compared with the negative control (Fig. 5B). In IR-treated HeLa cells,
miR-320a also inhibited cell proliferation (Fig. 5B). When the miR-320a inhibitor was
transfected into HeLa cells simultaneously, the HeLa cells
exhibited a partial reversal of IR-induced apoptosis (Fig. 5C).
To determine the potential role of miR-320a in
tumori-genesis, the colony formation capacity of the HeLa cells was
evaluated following transfection with miR-320a. Notably, the
miR-320a-overexpressing group displayed evidently fewer and smaller
colonies compared with the control group (Fig. 5D). These data indicated that
miR-320a enhanced radiosensitivity by inducing apoptosis and
suppressing proliferation under IR conditions.
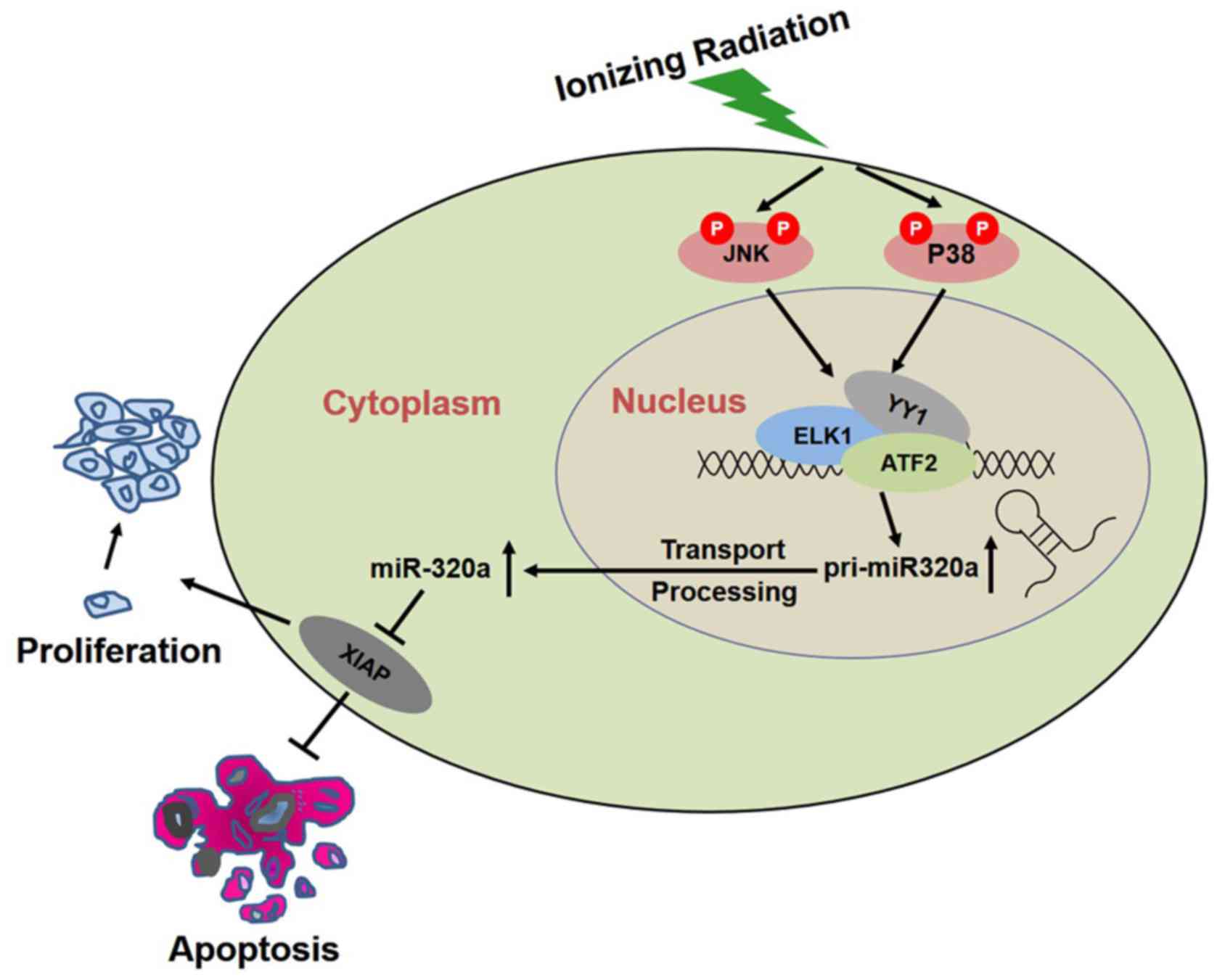
Overall, the results of this study suggested that IR
induced the phosphorylation of p38 and JNK, and activated their
downstream targets-ATF2, ELK1 and YY1, resulting in the
transcription of miR-320a, thus increasing the expression of
miR-320a to inhibit XIAP, and consequently inducing apoptosis and
inhibiting proliferation to realize its biological function
(Fig. 6).
Discussion
Although it has reported that miRNA expression was
induced by IR in cancer cells (4,30,31),
the expression of the majority of these IR-induced miRNAs was
irregular with radiation dose or time. In this study, we observed
that miR-320a expression increased in a dose- and time-dependent
manner under IR conditions, particularly with high doses or longer
treatment periods. It would be of interest to elucidate the
mechanisms through which miR-320a is regulated and to examine its
target gene in response to IR in cancer cells.
In this study, miR-320a expression, as a potential
tumor suppressor, increased linearly with radiation doses or
treatment duration under IR conditions. However, the mechanisms
underlying the response of miR-320a to IR are unclear. We analyzed
the precise location of the miR-320a promoter region and
transcriptional start site (TSS). We identified that ATF2, ELK1 and
YY1 were activated by the p38 MAPK and JNK pathways under IR
conditions and regulated miR-320a expression by binding to its
promoter.
ATF2 is a member of the bZIP transcription factor
family, which has been implicated in the transcriptional regulation
of cytokines, cell cycle control proteins and apoptotic factors
(32–34). ATF2 is regulated by p38 MAPK/JNK in
response to stress and plays a role in the DNA damage response and
ATM phosphorylates ATF2 on Ser490 and Ser498 following exposure to
IR (35,36). ELK1 is a component of the ternary
complex that binds the serum response element (SRE) in response to
serum growth factors (37). ELK1
is phosphorylated by MAPKs [extracellular signal-related kinase
(ERK)1 and ERK2], and the Ser383 residue of ELK1 is also a target
of several stress-activated kinases (SAPK/JNK1, JNK2 and p38). The
expression of ELK1 phosphorylated on Ser383 in the rat jejunum and
transverse colon has been shown to be dependent on doses and time
following whole-body γ-irradiation (38). YY1 is a 414-amino-acid
Krüppel-related zinc finger transcription factor and enhancer of
many cellular and viral genes. YY1 attenuates p53-dependent
transcription in a subset of p53-target genes. Notably, YY1 is
associated with cellular proliferation and transformation (39).
In this study, the overexpression or inhibition of
ATF2 observably influenced pri-miR-320a expression, suggesting that
ATF2 may be the key transcription factor regulating miR-320a.
Whether other miRNAs are controlled by ATF2 may be worth studying
in the future. Our results suggested that the p38 MAPK and JNK
pathways induced miR-320a expression, leading to cell apoptosis
under IR conditions. It would be of interest to investigate whether
they also regulate miR-320a expression during oncogenesis in the
future.
The data of this study identified that XIAP
was a novel target gene of miR-320a. XIAP is a member of the
inhibitors of apoptosis (IAP) family, which are endogenous
inhibitors of caspase-3, -7 and -9 (40). XIAP has been shown to be critical
for regulating sensitivity to apoptosis induced by cellular stress
and DNA damage (41). The present
study revealed that miR-320a enhanced radiosensitivity at least
partly by inhibiting XIAP expression. Furthermore, we aimed to
examine the association between XIAP and miR-320a in these radiated
samples using Spearman's rank correlation analysis. The results
revealed that there was not a significant negative correlation
(r=0.278, P=0.066) (data not shown) between them in the cell
samples at 24 h after 8 Gy IR. The possible reason leading to this
result may be as follows: Small cell sample sizes, the complexity
of IR and the miR-320a regulatory network.
In this study, our data suggested that the
overexpression of miR-320a enhanced the radiosensitivity of HeLa
cells by inducing cell apoptosis, and inhibiting proliferation or
colony formation. We hypothesized that miR-320a was upregulated
under IR conditions by the activated MAPK pathway and then
regulated its target genes, including XIAP, resulting in cell
apoptosis, and reduced proliferation or colony formation (Fig. 6). As shown in Fig. 5A, 14.79% of HeLa cells transfected
with miR-320a underwent apoptosis compared with 5.94% of untreated
cells and 5.79% of control cells. In general, the apoptotic
population is in the range from 10 to 30% in HeLa cells treated
with miRNAs (42–44). In addition, in this study, we
observed that miR-320a inhibited the proliferation of either
IR-untreated or -treated HeLa cells (Fig. 5B), while miR-320a inhibitor
exhibited a partial reversal of IR-induced apoptosis (Fig. 5C), suggesting that miR-320a indeed
enhanced radiosensitivity in response to IR. As for that miR-320a
inhibitor had a weak effect, this was probably due to the low
expression of miR-320a in HeLa cells (data not shown).
In conclusion, this study provides evidence that
miR-320a expression increases in a dose- or time-dependent manner
under IR conditions and that its transcription is directly
regulated by ATF2, YY1 and ELK1. The findings of this study also
demonstrated that miR-320a enhanced radiosensitivity by inhibiting
its target gene, XIAP. XIAP served as a specific tumor
therapeutic target due to its important roles in tumori-genesis.
Since miR-320a can regulate multiple target genes to inhibit cell
growth, it is possible that the exogenous expression of miR-320a
may be a more effective tumor therapy than the depletion of a
single oncogene. In this study, we only investigated the role of
miR-320a in response to IR in vitro. It would be interesting
to reveal whether such a regulation of miR-320a occurs in
vivo. In the future, the re-introduction of miR-320a in
vivo may be useful as a radiation sensitizer, once miRNA
delivery to tumor becomes feasible.
Acknowledgments
The authors would like to express their sincere
gratitude to Dr Lingqiang Zhang, from the Beijing Institute of
Radiation Medicine for reading the manuscript and providing and
helpful suggestions.
Abbreviations:
|
miRNA or miR
|
microRNA
|
|
IR
|
ionizing radiation
|
|
ATF2
|
activating transcription factor 2
|
|
ELK1
|
ETS transcription factor
|
|
YY1
|
YY1 transcription factor
|
|
MAPK
|
mitogen-activated protein kinase
|
|
JNK
|
mitogen-activated protein kinase 8
|
|
XIAP
|
X-linked inhibitor of apoptosis
|
|
3′UTR
|
3′ untranslated region
|
|
RT-qPCR
|
reverse transcription-quantitative
PCR
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
ChIP
|
chromatin immunoprecipitation
|
|
RIPA
|
radio-immunoprecipitation assay
|
|
SDS-PAGE
|
sodium dodecyl sulfate-polyacrylamide
gel electrophoresis
|
|
ERK
|
p44/42 MAPK
|
|
TSS
|
transcriptional start site
|
|
SRE
|
serum response element
|
|
IAP
|
inhibitor of apoptosis
|
Funding
This study was supported by Chinese National Natural
Science Foundation projects (31370760, 91540202, 31470782 and
81702925) and by the Natural Science Foundation of Heilongjiang
Province (H201403 and H2018002).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HF and XZ conceived the project and contributed to
the study design; ZH, YT, GL and JZ performed data collection; ZH,
YT and HF analyzed and interpreted the data; ZH, YT and HF wrote
the manuscript; all authors HAVE read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Huang X, Ding L, Bennewith KL, Tong RT,
Welford SM, Ang KK, Story M, Le QT and Giaccia AJ:
Hypoxia-inducible mir-210 regulates normoxic gene expression
involved in tumor initiation. Mol Cell. 35:856–867. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Leung AK and Sharp PA: MicroRNA functions
in stress responses. Mol Cell. 40:205–215. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Simone NL, Soule BP, Ly D, Saleh AD,
Savage JE, Degraff W, Cook J, Harris CC, Gius D and Mitchell JB:
Ionizing radiation-induced oxidative stress alters miRNA
expression. PLoS One. 4:e63772009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wilke CM, Hess J, Klymenko SV, Chumak VV,
Zakhartseva LM, Bakhanova EV, Feuchtinger A, Walch AK,
Selmansberger M, Braselmann H, et al: Expression of miRNA-26b-5p
and its target TRPS1 is associated with radiation exposure in
post-Chernobyl breast cancer. Int J Cancer. 142:573–583. 2018.
View Article : Google Scholar
|
|
6
|
Niemoeller OM, Niyazi M, Corradini S,
Zehentmayr F, Li M, Lauber K and Belka C: MicroRNA expression
profiles in human cancer cells after ionizing radiation. Radiat
Oncol. 6:292011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yano H, Hamanaka R, Nakamura-Ota M, Zhang
JJ, Matsuo N and Yoshioka H: Regulation of type I collagen
expression by microRNA-29 following ionizing radiation. Radiat
Environ Biophys. 57:41–54. 2018. View Article : Google Scholar
|
|
8
|
Chaudhry MA, Sachdeva H and Omaruddin RA:
Radiation-induced micro-RNA modulation in glioblastoma cells
differing in DNA-repair pathways. DNA Cell Biol. 29:553–561. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
He L, He X, Lim LP, de Stanchina E, Xuan
Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, et al: A microRNA
component of the p53 tumour suppressor network. Nature.
447:1130–1134. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kovalchuk O, Zemp FJ, Filkowski JN,
Altamirano AM, Dickey JS, Jenkins-Baker G, Marino SA, Brenner DJ,
Bonner WM and Sedelnikova OA: microRNAome changes in bystander
three-dimensional human tissue models suggest priming of apoptotic
pathways. Carcinogenesis. 31:1882–1888. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cha HJ, Seong KM, Bae S, Jung JH, Kim CS,
Yang KH, Jin YW and An S: Identification of specific microRNAs
responding to low and high dose gamma-irradiation in the human
lymphoblast line IM9. Oncol Rep. 22:863–868. 2009.PubMed/NCBI
|
|
12
|
Sokolov M and Neumann R: Global gene
expression alterations as a crucial constituent of human cell
response to low doses of ionizing radiation exposure. Int J Mol
Sci. 17:172015. View Article : Google Scholar
|
|
13
|
Begg AC, Stewart FA and Vens C: Strategies
to improve radiotherapy with targeted drugs. Nat Rev Cancer.
11:239–253. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lal A, Pan Y, Navarro F, Dykxhoorn DM,
Moreau L, Meire E, Bentwich Z, Lieberman J and Chowdhury D:
miR-24-mediated downregulation of H2AX suppresses DNA repair in
terminally differentiated blood cells. Nat Struct Mol Biol.
16:492–498. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mateescu B, Batista L, Cardon M, Gruosso
T, de Feraudy Y, Mariani O, Nicolas A, Meyniel JP, Cottu P,
Sastre-Garau X, et al: miR-141 and miR-200a act on ovarian
tumorigenesis by controlling oxidative stress response. Nat Med.
17:1627–1635. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Adusumilli PS, Chan MK, Hezel M, Yu Z,
Stiles BM, Chou TC, Rusch VW and Fong Y: Radiation-induced cellular
DNA damage repair response enhances viral gene therapy efficacy in
the treatment of malignant pleural mesothelioma. Ann Surg Oncol.
14:258–269. 2007. View Article : Google Scholar
|
|
17
|
Hu Z, Tie Y, Lv G, et al: Correlation of
microRNAs responding to high dose γ-irradiation with predicted
target mRNAs in HeLa cells using microarray analyses. Chin Sci
Bull. 58:4622–4629. 2013. View Article : Google Scholar
|
|
18
|
Diakos C, Zhong S, Xiao Y, Zhou M,
Vasconcelos GM, Krapf G, Yeh RF, Zheng S, Kang M, Wiencke JK, et
al: TEL-AML1 regulation of survivin and apoptosis via miRNA-494 and
miRNA-320a. Blood. 116:4885–4893. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xie F, Yuan Y, Xie L, Ran P, Xiang X,
Huang Q, Qi G, Guo X, Xiao C and Zheng S: miRNA-320a inhibits tumor
proliferation and invasion by targeting c-Myc in human
hepatocellular carcinoma. OncoTargets Ther. 10:885–894. 2017.
View Article : Google Scholar
|
|
20
|
Bronisz A, Godlewski J, Wallace JA,
Merchant AS, Nowicki MO, Mathsyaraja H, Srinivasan R, Trimboli AJ,
Martin CK, Li F, et al: Reprogramming of the tumour
microenvironment by stromal PTEN-regulated miR-320. Nat Cell Biol.
14:159–167. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu Q, Fu H, Sun F, Zhang H, Tie Y, Zhu J,
Xing R, Sun Z and Zheng X: miR-16 family induces cell cycle arrest
by regulating multiple cell cycle genes. Nucleic Acids Res.
36:5391–5404. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fu HJ, Zhu J, Yang M, Zhang ZY, Tie Y,
Jiang H, Sun ZX and Zheng XF: A novel method to monitor the
expression of microRNAs. Mol Biotechnol. 32:197–204. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jiang J, Lee EJ, Gusev Y and Schmittgen
TD: Real-time expression profiling of microRNA precursors in human
cancer cell lines. Nucleic Acids Res. 33:5394–5403. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ho SY, Wu WJ, Chiu HW, Chen YA, Ho YS, Guo
HR and Wang YJ: Arsenic trioxide and radiation enhance apoptotic
effects in HL-60 cells through increased ROS generation and
regulation of JNK and p38 MAPK signaling pathways. Chem Biol
Interact. 193:162–171. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang J and Bowden GT: Activation of p38
MAP kinase and JNK pathways by UVA irradiation. Photochem Photobiol
Sci. 11:54–61. 2012. View Article : Google Scholar
|
|
26
|
Shin C, Nam JW, Farh KK, Chiang HR,
Shkumatava A and Bartel DP: Expanding the microRNA targeting code:
Functional sites with centered pairing. Mol Cell. 38:789–802. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kashkar H: X-linked inhibitor of
apoptosis: A chemoresistance factor or a hollow promise. Clin
Cancer Res. 16:4496–4502. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Owens TW, Foster FM, Valentijn A, Gilmore
AP and Streuli CH: Role for X-linked Inhibitor of apoptosis protein
upstream of mitochondrial permeabilization. J Biol Chem.
285:1081–1088. 2010. View Article : Google Scholar :
|
|
29
|
Ward JF: DNA damage produced by ionizing
radiation in mammalian cells: Identities, mechanisms of formation,
and reparability. Prog Nucleic Acid Res Mol Biol. 35:95–125. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kraemer A, Anastasov N, Angermeier M,
Winkler K, Atkinson MJ and Moertl S: MicroRNA-mediated processes
are essential for the cellular radiation response. Radiat Res.
176:575–586. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li B, Shi XB, Nori D, et al:
Down-regulation of microRNA 106b is involved in p21-mediated cell
cycle arrest in response to radiation in prostate cancer cells.
Prostate. 71:567–574. 2011. View Article : Google Scholar
|
|
32
|
Bhoumik A, Lopez-Bergami P and Ronai Z:
ATF2 on the double - activating transcription factor and DNA damage
response protein. Pigment Cell Res. 20:498–506. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Breitwieser W, Lyons S, Flenniken AM,
Ashton G, Bruder G, Willington M, Lacaud G, Kouskoff V and Jones N:
Feedback regulation of p38 activity via ATF2 is essential for
survival of embryonic liver cells. Genes Dev. 21:2069–2082. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hai T and Hartman MG: The molecular
biology and nomenclature of the activating transcription
factor/cAMP responsive element binding family of transcription
factors: Activating transcription factor proteins and homeostasis.
Gene. 273:1–11. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kanzawa T, Iwado E, Aoki H, Iwamaru A,
Hollingsworth EF, Sawaya R, Kondo S and Kondo Y: Ionizing radiation
induces apoptosis and inhibits neuronal differentiation in rat
neural stem cells via the c-Jun NH2-terminal kinase (JNK) pathway.
Oncogene. 25:3638–3648. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kumar P, Miller AI and Polverini PJ: p38
MAPK mediates gamma-irradiation-induced endothelial cell apoptosis,
and vascular endothelial growth factor protects endothelial cells
through the phosphoinositide 3-kinase-Akt-Bcl-2 pathway. J Biol
Chem. 279:43352–43360. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Marais R, Wynne J and Treisman R: The SRF
accessory protein Elk-1 contains a growth factor-regulated
transcriptional activation domain. Cell. 73:381–393. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Driák D, Osterreicher J, Reháková Z,
Vilasová Z and Vávrová J: Expression of phospho-Elk-1 in rat gut
after the whole body gamma irradiation. Physiol Res. 57:753–759.
2008.
|
|
39
|
Gordon S, Akopyan G, Garban H and Bonavida
B: Transcription factor YY1: Structure, function, and therapeutic
implications in cancer biology. Oncogene. 25:1125–1142. 2006.
View Article : Google Scholar
|
|
40
|
Eckelman BP, Salvesen GS and Scott FL:
Human inhibitor of apoptosis proteins: Why XIAP is the black sheep
of the family. EMBO Rep. 7:988–994. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Datta R, Oki E, Endo K, Biedermann V, Ren
J and Kufe D: XIAP regulates DNA damage-induced apoptosis
downstream of caspase-9 cleavage. J Biol Chem. 275:31733–31738.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Park SY, Lee JH, Ha M, Nam JW and Kim VN:
miR-29 miRNAs activate p53 by targeting p85 alpha and CDC42. Nat
Struct Mol Biol. 16:23–29. 2009. View Article : Google Scholar
|
|
43
|
Cui F, Li X, Zhu X, Huang L, Huang Y, Mao
C, Yan Q, Zhu J, Zhao W and Shi H: MiR-125b inhibits tumor growth
and promotes apoptosis of cervical cancer cells by targeting
phosphoinositide 3-kinase catalytic subunit delta. Cell Physiol
Biochem. 30:1310–1318. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu L, Yu X, Guo X, Tian Z, Su M, Long Y,
Huang C, Zhou F, Liu M, Wu X, et al: miR-143 is downregulated in
cervical cancer and promotes apoptosis and inhibits tumor formation
by targeting Bcl-2. Mol Med Rep. 5:753–760. 2012.
|