Introduction
Malignant pleural mesothelioma (MPM) is a relatively
rare form of cancer of the chest cavity lining, which is difficult
to control (1). There have been an
increasing number of new cases of MPM globally since the 1960s,
with ~2,000 new cases diagnosed per year in the United States
(2). This progressive increase has
been attributed to the increased use of asbestos, and exposure to
its fibers, which can escape from the lung into the pleural cavity
following inhalation, causing chronic and malignant transformation
(3). The prognosis for patients
with MPM remains poor, despite advances in medical technology and
treatment modalities. Depending on the histological subtype
(epithelioid, sarcomatoid or biphasic), the median life expectancy
is only 8-18 months from diagnosis (4,5).
Current treatments are limited to either a multimodal strategy
involving surgery plus chemotherapy and/or radiotherapy, or
palliative chemotherapy (6). For
eligible patients with resectable disease, surgery improves
survival; either extrapleural pneumonectomy (EPP) or lung-sparing
extended pleurectomy/decortication (EPD) are used to remove all
macroscopic nodules, alongside chemotherapy or radiotherapy to
target residual microscopic disease (7). However, even with the use of
aggressive multimodal treatment, the majority of patients have
local recurrence, yet little is currently known about the precise
pattern of relapse (8-11). Therefore, other means to eliminate
microscopic disease following surgery are urgently required.
Several groups are exploring novel intrapleural
therapies, including immunotherapy, gene therapy and
cisplatin-based chemotherapy (12,13).
In line with these novel modalities, intraoperative photodynamic
therapy (PDT) has recently been reported as a potential therapeutic
strategy for this purpose (14,15).
Effective PDT requires a light-absorbing compound
(photosensitizer), light at a specific wavelength to activate the
photosensitizer and, for most photosensitizers, oxygen (16). The activated photosensitizer
generates reactive singlet oxygen species as the primary cytotoxic
agent to initiate cell death, often via apoptosis (17). At present, a few photosensitizers
have been clinically approved for cancer therapy, including
porfimer sodium (Photofrin®) in the USA and
m-tetrahydroxyphenylchlorin (Foscan®) in Europe.
Photofrin, which is a multi-component derivative of
hematoporphyrin, was the first photosensitizer to be evaluated for
intrapleural PDT following surgery (18); it involves systemic (intravenous,
i.v.) administration followed by red light delivery to irradiate
the pleural cavity. Although the feasibility of this approach has
been demonstrated, fatal toxicities have been noted in previous
studies (18,19). Similarly, Foscan-PDT may cause
severe toxicity and even mortality (20,21).
Compiled data from 10 trials exhibited an overall mortality rate of
4.9% with Photofrin and 13.3% for Foscan, with morbidities of 38
and 70%, respectively (22).
Neither of these otherwise potent photosensitizers possesses marked
tumor selectivity; therefore, treatment is limited by normal-tissue
phototoxicity.
Porphysomes are liposome-mimicking nanoparticles
(~100 nm diameter) that self-assemble from porphyrin-phospholipid
conjugates (23). They have an
extremely high porphyrin density (>80,000 per nanoparticle), so
that PDT efficacy and associated fluorescence are highly quenched
in solution (24). However, these
are restored upon disassembly of the nanoparticles following
cellular internalization. In our previous study, folic acid was
integrated into the porphysome formulation at 1 mole% to enable
targeting of folate receptors (folate-porphysomes, FPs), in order
to actively target tumor cells that had overexpressed folate
receptors (25). FPs initially
accumulate in the tumor interstitial space through the enhanced
permeation and retention (EPR) effect and are then taken up by
tumor cells through folate receptor 1 (FOLR1)-mediated active
transport. Both fluorescence and the resulting PDT efficacy were
demonstrated in a mouse subcutaneous tumor model (25). High expression levels of FOLR1 have
been clinically reported in 39-72% of MPM cases (26,27).
It was hypothesized that a nanostructure-based photosensitizer may
be advantageous to obtain significant tumor-specific accumulation
in MPM, and to minimize toxicity to surrounding normal tissues in
the pleural cavity (28). PDT,
enabled by folate receptor targeting of porphysomes, has recently
been evaluated by the University Health Network (Toronto, ON,
Canada) as a promising novel strategy to treat various types of
cancer (23,24). In the present study, the targeting
efficiency of FPs towards MPM cells was determined in vitro
and in vivo, based on fluorescence imaging, and the FP-PDT
efficacy was also measured in vitro and in vivo in a
mouse subcutaneous MPM tumor model.
Another aspect of PDT is that it has been reported
to stimulate survival pathways via the nuclear accumulation of
activated epidermal growth factor receptor (EGFR) (29). Notably, growing tumors stimulate
neovascularization through the secretion of various proangiogenic
growth factors, including vascular endothelial growth factor and
EGFR (30). Therefore,
deactivating these pathways using EGFR-tyrosine kinase inhibitors
(EGFR-TKIs) may potentially enhance PDT efficacy (29). Combinations of PDT with small
molecules or anti-EGFR antibodies have been reported to increase
the cytotoxicity of PDT in non-small cell lung cancer,
nasopharyngeal carcinoma and bladder carcinoma (31-34).
Furthermore, EGFR has been reported, using immunohistochemistry, to
be overexpressed in 44-97% of patients with MPM, with high
variability across studies (35-39).
Therefore, the present study initially evaluated the expression
levels of FOLR1 and EGFR in MPM clinical samples, as confirmation
of their overexpression, using histological subset analysis. It was
further hypothesized that blocking EGFR pathways by EGFR-TKI
alongside FP-PDT could achieve a greater response in MPM with fewer
side effects; therefore, the present study evaluated the expression
levels of EGFR, and the efficacy of combined EGFR-TKIs and FP-PDT
in vivo.
Materials and methods
MPM clinical tissue samples
A total of 156 MPM tissue samples (EPP, n=22;
pleural biopsy, n=26; recurrent tumor resection, n=3; radical
pleurectomy, n=1) were obtained from 52 patients undergoing surgery
at Hokkaido University Hospital (Sapporo, Japan) and affiliated
hospitals (Sapporo Minami-Sanjyo Hospital, Sapporo, Japan and
Kinikyo-Chuo Hospital, Sapporo, Japan) between February 1990 and
April 2012. All specimens were fixed in 10% formalin for 3-5 days
at room temperature and embedded in paraffin. Three tissue cores (1
mm diameter) taken from each tumor block were placed into recipient
paraffin blocks using a tissue microarrayer (JF-4; Sakura Finetek
Japan, Tokyo, Japan). Tissue areas for sampling were selected based
on visual alignment of paraffin-embedded blocks with the
corresponding hematoxylin and eosin (H&E)-stained sections on
slides. H&E staining was performed using an automated slide
stainer and coverslipper (Tissue-Tek Prisma Plus; Sakura Finetek
Japan). All tumors were histologically reviewed by two experienced
pathologists (HK and KCH). Clinical information of the patients was
obtained from the medical records. The protocol was approved by the
Independent Clinical Research Review Board of Hokkaido University
Hospital [approval no. 012-0136]. All patients provided written
informed consent prior to surgery. The median age at the time of
diagnosis was 65.6 years (range, 35-80 years) and 92.3% of the
patients were men. Among the 52 MPM cases, 33 were of the
epithelioid type (63.5%), 13 were biphasic (25.0%), five were
sarcomatoid (9.6%) and one was desmoplastic (1.9%) (Table I). Histological classification of
tumors and stage were performed according to the Union for
International Cancer Control pathological tumor/node/metastasis
classification criteria (40).
 | Table IPatients and tumor characteristics
(n=52). |
Table I
Patients and tumor characteristics
(n=52).
| Variables | Values |
|---|
| Sex, male/female
(%) | 48/4
(92.3/7.7) |
| Mean age, years
(range) | 65.6 (35-80) |
| Histology | |
| Epithelioid, n
(%) | 33 (63.5) |
| Biphasic, n
(%) | 13 (25.0) |
| Sarcomatoid, n
(%) | 5 (9.6%) |
| Desmoplastic, n
(%) | 1 (1.9%) |
| Surgical
procedure | |
| Extrapleural
pneumonectomy, n (%) | 22 (42.3) |
| Pleural biopsy, n
(%) | 26 (50.0) |
| Recurrent tumor
resection, n (%) | 3 (5.8) |
| Radical
pleurectomy, n (%) | 1 (1.9) |
| Total | 52 |
MPM cell lines
Murine (AE17, AE17-sOVA, AK7, AB12 and RN5) and
human (H28, H226, H2052 and H2452) MPM cells lines were used, as
well as a control human adult normal mesothelial cell line (MES-F)
(41). A lung large cell carcinoma
cell line (H460) that overexpresses folate receptor was used as a
positive control. Mixed cancer type KB cell line (42) was also used as a FOLR1-positive
control, as in our previous study (25). The human MPM cell lines (H28, H226,
H2052 and H2452) were purchased from American Type Culture
Collection (Manassas, VA, USA). AB12 was donated by Dr. Jay Kolls
(University of Pittsburgh, Pittsburgh, PA, USA). AE17 cells were
obtained from the European Collection of Authenticated Cell
Cultures (Salisbury, UK). AE17-sOVA cells were developed by stably
transfecting the AE17 parental cell line with secretory ovalbumin
(sOVA) (43,44). The AK7 cell line (45) was kindly provided by Dr Steven
Albelda (University of Pennsylvania, Philadelphia, PA, USA) and Dr
Delia Nelson (University of Western Australia, Crawley, WA,
Australia). The RN5 cell line was developed in collaboration with
the University of Fribourg (Fribourg, Switzerland) (46). MES-F was purchased from Zen-Bio,
Inc. (Research Triangle Park, NC, USA) and was grown in mesothelial
cell growth medium (Zen-Bio, Inc.). KB and H460 cells were provided
by Dr Ming-Sound Tsao (University of Toronto, Toronto, ON, Canada).
All cells, with the exception of the MES-F cell line, were grown as
monolayers in RPMI-1640 medium (Sigma- Aldrich; Merck KGaA,
Darmstadt, Germany) supplemented with 10% fetal bovine serum (FBS,
Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA). All cells
were maintained at 37°C in a humidified atmosphere containing 5%
CO2.
Western blotting for in vitro FOLR1
expression
After cells were grown to >80% confluence, they
were lysed with radioimmunoprecipitation assay buffer (pH 7.5; 50
mmol/l Tris-HCl, 1 mmol/l EDTA, 100 mmol/l NaCl, 1% Triton X-100)
containing protease inhibitors (20 µmol/l leupeptin, 0.8
µmol/l aprotinin, 10 µmol/l pepstatin and 1.25 mmol/l
phenylmethylsulfonyl fluoride). Cell lysates were maintained at 4°C
for 15 min and were then centrifuged at 14,000 x g for 15 min at
4°C. The total protein concentration in the supernatant was
quantified using a bicinchoninic acid protein assay reagent kit
(Bio-Rad Laboratories, Inc., Mississauga, ON, Canada). Protein (10
µg/lane) was loaded for each cell line and was separated by
10% SDS-PAGE at 100 V for 90 min. Proteins were then transferred
onto a nitrocellulose membrane using a Miniprotein III
electro-blotter (Bio-Rad Laboratories, Inc.). The membranes were
blocked in 5% skimmed milk-Tris-buffered saline (TBS) containing
0.1% Tween-20 (TBST) for 2 h at 4°C, after which, the immunoblots
were washed in TBST and probed overnight at 4°C with anti- FOLR1
primary antibody [EPR4708(2)]
(cat. no. ab125030; Abcam, Cambridge, MA, USA) at a 1:1,000
dilution. Membranes were washed and were then incubated for 1 h at
room temperature with an anti-rabbit horseradish peroxidase
(HRP)-conjugated secondary antibody at a 1:2,000 dilution (cat. no.
A00098; GenScript Biotech Corporation, Pascataway, NJ, USA). Bound
antibodies were detected using a Gel Logic 2200 Imaging system
(Kodak, Rochester, NY, USA) following treatment with Clarity
Western enhanced chemiluminescence reagent (Bio-Rad Laboratories,
Inc.). The membranes were then stripped and immunoblotted with a
mouse monoclonal antibody against β-actin (cat. no. A5441, 1:5,000;
Sigma- Aldrich; Merck KGaA) and anti-mouse HRP-conjugated secondary
antibody (cat. no. A00160; GenScript Biotech Corporation) at a
1:2,000 dilution.
Porphysome synthesis
Non-targeted (regular) porphysomes and FPs were
synthesized according to a previously described protocol (23,25).
The lipid film for non-targeted porphysomes consisted of 55 mole%
pyropheophorbide-lipid, 40 mole% cholesterol (Avanti Polar Lipids,
Avanti, AL, USA) and 5 mole%
distearoyl-sn-glycero-3-phosphoethanolamine- N-methoxy (polyethene
glycol) (PEG2000-DSPE; Avanti Polar Lipids). For FPs, 1 mole% 1,
2-distearoyl-sn-glycero- 3-phosphoethanolamine-folate (polyethylene
glycol) (Folate-PEG2000-DSPE; Avanti Polar Lipids) was added to the
formulation, together with 4 mole% PEG2000-DSPE, 55 mole%
porphyrinlipid (pyropheophorbide-lipid) and 40 mole% cholesterol
(Avanti Polar Lipids). The lipid films were dried under a gentle
stream of nitrogen gas, followed by 1 h under a vacuum to remove
the remaining solvent. The dried lipid films were stored at 20°C
under argon gas until synthesis. To obtain fresh porphysomes for
each experiment, the lipid films were rehydrated with 1.0 ml PBS
(150×10−3 M, pH 7.5) and extruded 10 times under high
N2 pressure through a poly- carbonate membrane (pore
size, 100 nm), after which, they were kept sterile at 4°C. The
nanoparticle sizes were measured using dynamic light scattering
(ZS90 Nanosizer; Malvern Instruments, Malvern, UK) and the
porphyrin concentration was determined by UV-Vis absorption
spectrometry (Varian Medical Systems, Inc., Palo Alto, CA,
USA).
Cellular uptake and fluorescence
activation of porphysomes
Confocal fluorescence microscopy was used to
determine the cellular internalization of non-targeted porphysomes
and FPs. Upon reaching >80% confluence, 105 cells
were seeded in a 2-well chamber slide (Lab-Tek™; Sigma-Aldrich;
Merck KGaA) and were incubated for 24 h at 37°C. The medium was
then replaced with fresh medium containing non-targeted porphysomes
(5×10−6 M), FPs (5×10−6 M) or FPs
(5×10−6 M) plus free folic acid (1×10−3 M, MW
441.40, CAS no. 59-30-3; Sigma-Aldrich; Merck KGaA), and the cells
were incubated for 3 h at 37°C. The cells were then washed gently
three times with PBS. Fresh medium was added and the cells were
allowed to grow at 37°C for a further 21 h, at which time they were
fixed in 4% paraformaldehyde for 15 min at 4°C, nuclei were stained
with 300 nM DAPI stain solution (Thermo Fisher Scientific, Inc.)
for 5 min at room temperature, and mounted using Dako
fluorescence-mounting medium (Dako; Agilent Technologies, Inc.,
Santa Clara, CA, USA). Images of cells were captured using a
confocal laser-scanning microscope (FV1000; Olympus Corporation,
Tokyo, Japan). The cells in each well were detected by open field
imaging under a 60× oil-immersion lens, and the area of relatively
high cell density in each well was selected for subsequent
fluorescence microscopy. The cell number was 20-100 in the selected
300×300 µm area; this variance arose from the different size
and growth rates of the various cell lines. Excitation wavelengths
of 405 and 633 nm were used to visualize DAPI and porphysome
fluorescence, respectively.
In vitro dark toxicity and PDT
efficacy
To evaluate the dark toxicity of non-targeted
porphysomes and FPs, H2052 and AE17-sOVA cells were seeded at a
concentration of 5×103 cells/well in 96-well
black-walled plates (cat. no. 655946; CELLCOAT®; Greiner
Bio-One GmbH, Frickenhausen, Germany) 24 h prior to porphysome
incubation. The cells were then grouped into four incubation
conditions: Normal medium only, medium containing non-targeted
porphysomes (5×10−6 M), medium containing FPs
(5×10−6 M), and medium containing FPs (5×10−6
M) and free folic acid (1×10−3 M). Cells were incubated
for 3 h at 37°C to allow folate receptor-mediated internalization,
and each well was then gently rinsed with PBS three times and
incubated for a further 21 h in fresh medium without porphysomes.
Cell viability was measured using a colorimetric proliferation
assay (CellTiter96® AQueous One Solution Cell Assay;
Promega Corporation, Madison, WI, USA), according to the
manufacturer’s protocol. Each experiment was performed in
quadruplicate. The light absorbance was measured at 630 nm using a
µQuant microplate reader (BioTek Instruments, Inc.,
Winooski, VT, USA), with absorbance at 490 nm serving as
background, in order to determine the relative cell viability
normalized to the no-porphysome control group.
To investigate the PDT efficacy of FP, in
vitro treatment was conducted. The same growth and incubation
conditions were used as for the aforementioned dark toxicity
studies, with eight replicates of each condition. The cells were
then irradiated using a 671 nm continuous-wave diode laser beam
expanded to 6.4 mm diameter to fully cover each well at a power
density of 50 mW/cm2. Four wells in each treatment group
received a light dose of 5 J/cm2 and four received 10
J/cm2. Immediately after the light treatment, the
incubation medium was replaced with fresh medium without
porphysomes. The cell viability was measured after 24 h using the
CellTiter96® assay as aforementioned.
In vivo mesothelioma models
The animal study was approved by the ethics
committee of the University Health Network. An orthotopic
mesothelioma model was established in Nu/nu mice (female; age, 6-8
weeks; weight, 20-25 g; Taconic Biosciences, Inc. (Rensselaer, NY,
USA) by first inducing general anesthesia with 2% isoflurane.
Xylazine (20 mg/kg)/ketamine (100 mg/kg) was then injected
intraperitoneally (0.1 ml/10 g body weight) and the mouse was
placed in the left lateral position. After sterilization of the
chest wall with 70% isopropyl alcohol, a bolus of 106
AE17-sOVA cells (cultured in RPMI-1640 media supplemented with 10%
FBS) in 100 µl normal saline was injected into the left
pleural cavity through the intercostal space using a 27G needle.
After stable spontaneous respiration was confirmed, the animals
were returned to their cages. Tumor growth in the pleural cavity
was confirmed by micro chest computed tomography (CT)-scanning at 1
week. A subcutaneous mesothelioma model was induced using the same
cell line inoculated in the thigh (2×106 cells in 50
µl saline). The tumor growth was monitored using calipers
every 2 days for ~10 days, at which time the tumor diameter was 6-7
mm.
In vivo porphysome accumulation in
tumors
Fluorescence is a useful indicator of porphysome
accumulation and cellularization in vivo, as porphyrin
fluorescence is highly quenched until cellular uptake. Qualitative
studies were performed in ex vivo tissues from the
orthotopic tumor model using a commercial fluorescence imaging
system (Maestro® EX 2.10; PerkinElmer, Inc., Waltham,
MA, USA) (25). Briefly,
porphysomes were injected via the tail vein at 10 mg/kg, based on
porphyrin content. The mice were sacrificed by CO2
followed by cervical dislocation at 6 h post-injection, after
which, the chest was opened to expose the lung and the tumors were
imaged (575-605 nm excitation, 647 nm emission). The tumors were
then resected under fluorescence image guidance, together with
other organs (diaphragm, chest wall, heart, lung, liver, kidney,
spleen, adrenal gland, small intestine and large intestine), and
placed on a 24-well dish. To further compare the non-targeted
porphysomes vs. FPs, tumor and normal lung tissues (n=3) were snap
frozen in OCT gel, cryosectioned (5 µm), mounted and stained
with VECTASHIELD Antifade Mounting Medium with DAPI (cat. no.
H-1200; Vector Laboratories, Inc., Burlingame, CA, USA) at room
temperature for ≥15 min and imaged by confocal microscopy (x60
magnification; 633 nm excitation for porphyrin, 408 nm excitation
for DAPI). To quantify porphysome uptake, freshly resected tissues
were weighed and homogenized in 1 ml PBS. The tissue suspension was
then lysed at room temperature by adding Triton X-100 (final
concentration, 1%) and centrifuging at 10,000 x g for 10 min at
room temperature (5415D Benchtop Microcentrifuge; Eppendorf North
America, Hauppauge, NY, USA); Triton X-100 was used to unquench the
porphysomes. The supernatant was then measured by
spectrofluorimetry (420 nm excitation, 600-800 nm emission), and
the percent- injected dose per gram tissue (%ID/g) was calculated
using a pyro-lipid concentration standard curve.
PDT efficacy in vivo
The PDT efficacy of FPs was investigated in the
subcutaneous tumor model using three groups (n=6/ group), as
follows: No treatment, light only and FP-PDT at 24 h after i.v.
injection of 10 mg/kg FPs, with the mice kept under dim ambient
lighting after injection. Treatment was administered under general
anesthesia (2% isoflurane), using a 671 nm diode laser (DPSS;
LaserGlow Technologies, Toronto, ON, Canada) at an incident power
density of 100 mW/cm2 over a 9 mm spot size for total
light dose of 100 J/cm2. The tumor size was then
measured with calipers every 2 days to estimate the volume.
Survival was measured as a function of time after treatment and the
mice were euthanized when the maximum diameter of the tumor reached
15 mm. In a separate group, the same PDT treatment was given, but
the mice were sacrificed at 24 h to evaluate the treatment response
by histology, for which tumors were harvested, fixed in 10%
formaldehyde, sectioned (8 µm) and stained with H&E.
H&E staining was carried out according to standard methods at
the Pathology Research Program Laboratory at University Health
Network. Digital images of the stained slides were obtained under
x20 magnification using a whole slide scanner (ScanScope CS, Leica
Microsystems GmbH, Wetzlar, Germany) and Aperio ImageScope software
(version 12.1.0.5029; Leica Microsystems GmbH). Alternatively,
sectioned tumors underwent immunohistochemistry (IHC) to detect the
expression of cleaved caspase-3 (apoptosis) and Ki-67 (tumor cell
proliferation).
Effects of an EGFR inhibitor on FP-PDT
responses
The potential combined effect of molecular targeted
therapy using an EGFR-TKI (erlotinib; Selleck Chemicals, Houston,
TX, USA) and FP-PDT was evaluated in vitro and in
vivo. The expression levels of EGFR in the MPM clinical samples
were initially evaluated by histological subset analysis, comparing
FOLR1 and EGFR. For the in vitro studies, AE17-sOVA and
H2052 cells were incubated under three conditions for 3 h at 37°C,
as follows (n=16/each condition): Normal medium without
porphysomes; medium containing non-targeted porphysomes
(5×10−6 M); and medium containing FPs (5×10−6
M). Each well was then rinsed gently three times with PBS. For each
condition, half of the 16 wells were then incubated for 21 h with 5
µM erlotinib dissolved in dimethyl sulfoxide (DMSO), while
the other wells were treated with DMSO only. The cells were then
treated with PDT at 50 mW/cm2 for doses of 5 or 10
J/cm2 (n=4 each); after 24 h, cell viability was
measured using the CellTiter96® assay (Promega
Corporation).
The in vivo efficacy of pretreatment with
EGFR-TKIs on the FP-PDT response was investigated in the AE17-sOVA
subcutaneous tumor model. Erlotinib (50 mg/kg) was administered
daily by oral gavage using CAPTISOL® (Ligand, San Diego,
CA, USA) 2 days, 1 day and 3 h before PDT. This dose has exhibited
antitumor efficacy without toxicity in a previous study (33). Five subgroups were used: No
treatment; light only; FP-PDT; erlotinib only; and erlotinib +
FP-PDT. In the PDT groups, 10 mg/kg FP was injected i.v. 24 h
before light irradiation (100 J/cm2 at 100
mW/cm2). After 24 h, the tumors were excised for
histological analyses, including H&E and immunohistochemical
staining, which was conducted at UHN and the efficacy was evaluated
using Aperio ScanScope system (Leica Microsystems GmbH).
IHC
FOLR1 immunostaining was performed using the
catalyzed signal amplification (CSA) system, according to the
manufacturer’s protocol (CSA II Biotin-free Tyramide Signal
Amplification system; cat. no. K1497; Dako; Agilent Technologies,
Inc.). Briefly, specimens were incubated with 3% hydrogen peroxide
for 5 min to quench endogenous peroxidase activity and were then
rinsed with distilled water and washed with TBST (0.05 M Tris-HCl,
pH 7.6, containing 0.3 M NaCl and 0.1% Tween-20) for 5 min. The
specimens were then incubated with a protein block (serum-free
protein in PBS with 0.015 M sodium azide) for 5 min to suppress
non-specific binding. Subsequently, they were incubated overnight
at 4°C with a primary folate receptor α antibody (Novocastra™;
product code, NCL-L-FRalpha; Leica Microsystems GmbH) at a 1:30
dilution with mixed antibody diluent (S2022 Antibody Diluent; Dako;
Agilent Technologies, Inc.) and washed three times in TBST for 5
min. Subsequently, the specimens were sequentially incubated for 15
min each with Anti-Mouse Immunoglobulin-HRP, Amplification Reagent,
and Anti-Fluorescein-HRP. Each of these sequential incubations
included three washing steps with TBST (5 min/wash). Colorimetric
signals were localized after incubation in Liquid DAB
Substrate-Chromogen for 5 min. Aperio ScanScope system (Leica
Microsystems GmbH) was used for analyses.
For cleaved caspase-3, Ki-67 and EGFR staining,
heat- induced epitope retrieval was conducted by microwaving the
tissue sections in 10 mM citrate buffer (pH 6.0). Endogenous
peroxidase was blocked with 3% hydrogen peroxide for 5 min at room
temperature, and 10% normal horse serum blocking solution (cat. no.
S-2000; Vector Laboratories, Inc.) was then applied to block
non-specific binding for 10 min (for EGFR staining only). Sections
were then drained and incubated at room temperature with the
appropriate primary antibodies, under previously optimized
conditions: Cleaved caspase-3 (cat. no. 9661; Cell Signaling
Technology, Inc., Danvers, MA, USA) at 1:600 dilution overnight;
Ki-67 (cat. no. NB110-90592; Novus Biologicals, LLC) at 1:700
dilution for 1 h; EGFR (cat. no. 280005 (31G7); Invitrogen; Thermo
Fisher Scientific, Inc.) at 1:100 dilution for 1 h. Subsequently,
sections were incubated with biotin-labeled anti-mouse/rabbit
secondary antibodies (cat. no. BA-1400, 1:50; Vector Laboratories,
Inc.) for 30 min at room temperature and horseradish
peroxidase-conjugated ultrastreptavidin labeling reagent (Empire
Genomics, LLC, Buffalo, NY, USA) for 30 min. After washing well in
TBS, color development was conducted with freshly prepared DAB. The
slides were then dehydrated and cover-slipped. Aperio ScanScope
(Leica Microsystems GmbH) was used for analyses.
For immunohistochemical analysis of Ki-67, the
percentage of positively stained nuclei was calculated using
commercial software (Aperio Nuclear v9; Leica Microsystems GmbH),
with the manufacturer’s default settings. Cleaved caspase-3 and
FOLR1 expression were calculated (Aperio Positive Pixel Count v9;
Leica Microsystems GmbH) with the default settings ‘Positive
Cell/Total Cell’. FOLR1 expression was quantified by
immunohistochemical scoring, the percentage area stained at each
intensity level was multiplied by the weighted intensity, as
reported in other studies (47,48).
Initially, the weighted intensity was graded as follows: Grade 0
(Negative), 1+ (weak positive: Intensity threshold weak; upper
limit 240, lower limit 220), 2+ (moderate positive: Intensity
threshold medium; upper limit 220, lower limit 180), and 3+ (strong
positive: Intensity threshold strong; upper limit 180, lower limit
0). FOLR1 expression was then divided into four groups: Negative
(IHC score <0.50), weak (IHC score 0.50-0.99), moderate (IHC
score 1.00-1.49) and strong (IHC score ≥1.50). FOLR1 expression was
finally judged as positive (weak, moderate and strong) or negative.
For EGFR, each core was scored semi-quantitatively in tumor cells
under x200 magnification; staining intensity was as follows: Grade
0 (negative), 1+ (weak positive), 2+ (moderate positive) and 3+
(strong positive).
Statistical analysis
Statistical analyses were performed with StatFlex
version 6.0 for Windows (Artech Co., Ltd., Osaka, Japan). In
vitro experiments were repeated at least four times. Multiple
comparison analyses were used to determine statistical
significance. One-factor analysis of variance, followed by Newman
Keuls or Dunnett post hoc tests, was conducted when comparing the
difference between treatment groups. The survival rate was
calculated by the Kaplan-Meier method and the differences between
groups were compared by the log-rank test. P<0.05 was considered
to indicate a statistically significant difference.
Results
FOLR1 and EGFR expression
Positive FOLR1 staining on tissue microarrays (TMAs)
generally exhibited a membranous and cytoplasmic pattern (Fig. 1A) and was observed in 41 of 52 MPM
cases (79%) (Table II), 27 of
which were epithelioid (82% of 33 cases), 12 were biphasic (92% of
13 cases) and 2 were sarcomatoid (40% of 5 cases). Conversely, the
benign pleuritis portion exhibited just faint FOLR1 expression. In
addition, chronic pleuritis revealed almost no staining of FOLR1.
EGFR was overexpressed in 89% of cases (91% of epithelioid, 92% of
biphasic, 60% of sarcomatoid and 100% of desmoplastic cases)
(Table III and Fig. 1A). Notably, 71% of MPM cases
expressed both FOLR1 and EGFR (76% of epithelioid and 85% of
biphasic cases) (Table IV). FOLR1
was also highly expressed in the majority of the MPM cell lines
(Fig. 1B). H28, H2052, AB12 and
AE17-sOVA cell lines were selected as FOLR1-positive cell lines,
and RN5 cells were selected as a FOLR1-negative cell line for
subsequent porphysome uptake studies. RN5 cells are derived from
sarcomatoid MPM; these findings were consistent with the TMA
results, which detected low FOLR expression in sarcomatoid cases
(Table II).
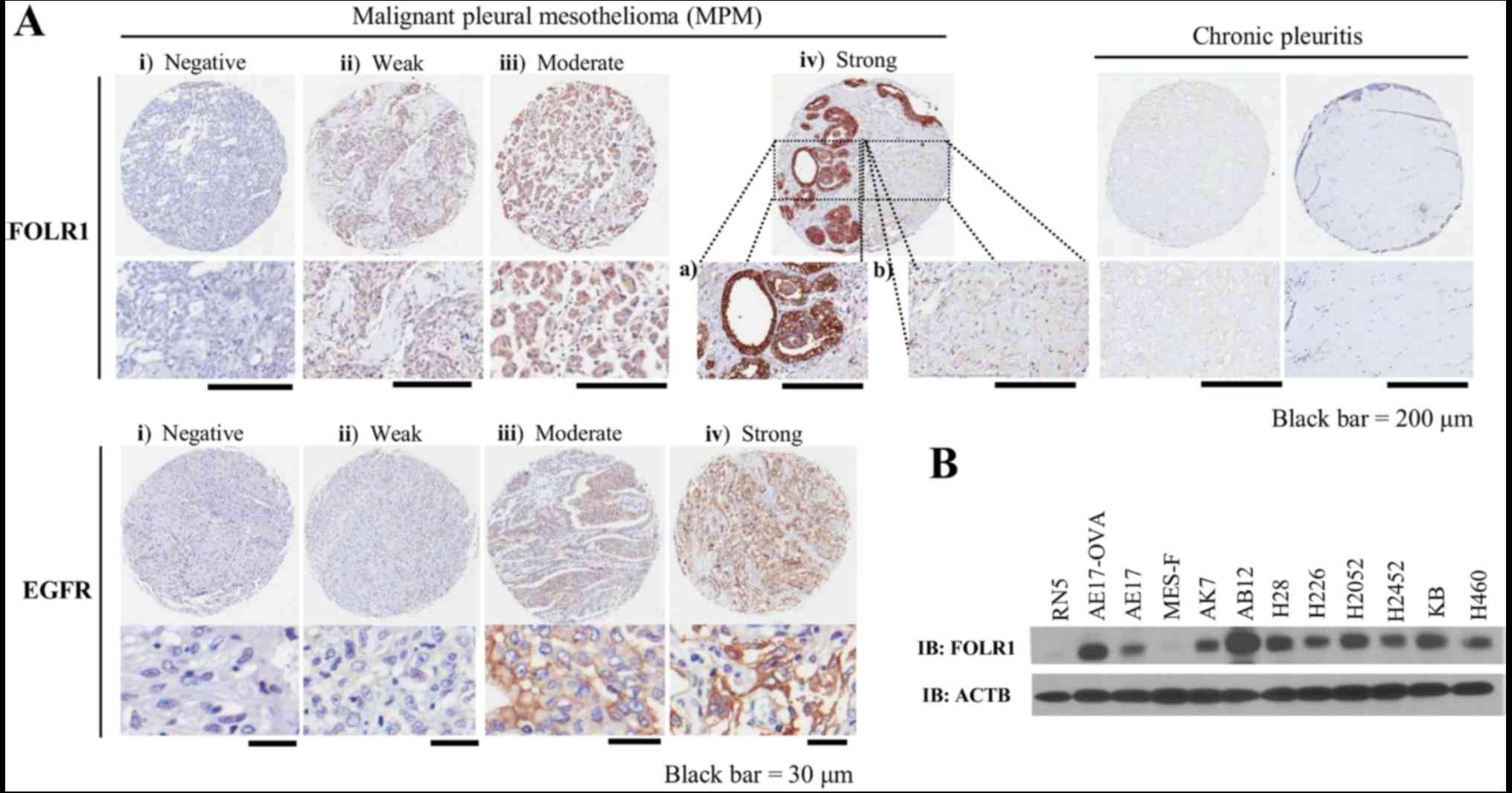 | Figure 1Expression of FOLR1 and EGFR in MPM.
(A) Representative examples of FOLR1 (upper panel) and EGFR (lower
panel) staining in MPM tissues on a tissue microarray. (i)
Negative, (ii) weak, (iii) moderate and (iv_a) strong staining is
presented. Adjacent benign pleuritis samples exhibited faint FOLR1
expression (iv_b); chronic pleuritis cases revealed almost no
staining of FOLR1. (B) Western blotting of mesothelioma cell lines
for the expression of FOLR1, including mouse MPM cell lines (AE17,
AE17-sOVA, AK7, AB12 and RN5), human MPM cell lines (H28, H226,
H2052 and H2452) and a human adult normal mesothelial cell line
(MES-F). A lung large cell carcinoma (H460) and a mixed cancer type
(KB) cell line were both used as positive controls, which are known
to overexpress FOLR1. EGFR, epidermal growth factor receptor;
FOLR1, folate receptor 1; MPM, malignant pleural mesothelioma;
sOVA, secretory ovalbumin. |
| Table IIImmunopositivity of FOLR1 in
malignant pleural mesothelioma (n=52). |
Table II
Immunopositivity of FOLR1 in
malignant pleural mesothelioma (n=52).
| Tissue type | FOLR1 expression
(n=52)
| Total (n) | Percentage of
FOLR1- positive case |
|---|
| Negative (IHC
score: 0.00-0.49) | Weak (IHC score:
0.50-0.99) | Moderate (IHC
score: 1.00-1.49) | Strong (IHC score:
≥1.50) |
|---|
| Epithelioid | 6 (18.2%) | 8 (24.2%) | 11 (33.3%) | 8 (24.2%) | 33 | 81.8 (27/33) |
| Biphasic | 1 (7.7%) | 1 (7.7%) | 10 (76.9%) | 1 (7.7%) | 13 | 92.3 (12/13) |
| Sarcomatoid | 3 (60.0%) | 0 (0%) | 1 (20.0%) | 1 (20.0%) | 5 | 40.0 (2/5) |
| Desmoplastic | 1 (100.0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 | 0.0 (0/1) |
| Total | 11 (21.2%) | 9 (17.3%) | 22 (42.3%) | 10 (19.2%) | 52 | 78.8 (41/52) |
| Table IIIImmunopositivity of EGFR in malignant
pleural mesothelioma (n=52). |
Table III
Immunopositivity of EGFR in malignant
pleural mesothelioma (n=52).
| Tissue type | EGFR expression
(n=52)
| Total (n) | Percentage of EGFR-
positive case |
|---|
| Negative (−) | Weak (+) | Moderate (++) | Strong (+++) |
|---|
| Epithelioid | 3 (9.1%) | 8 (24.2%) | 10 (30.3%) | 12 (36.4%) | 33 | 90.9 (30/33) |
| Biphasic | 1 (7.7%) | 4 (30.8%) | 3 (23.1%) | 5 (38.5%) | 13 | 92.3 (12/13) |
| Sarcomatoid | 2 (40.0%) | 2 (40.0%) | 1 (20.0%) | 0 (0.0%) | 5 | 60.0 (3/5) |
| Desmoplastic | 0 (100.0%) | 0 (0%) | 1 (0%) | 0 (0%) | 1 | 100.0 (1/1) |
| Total | 6 (11.5%) | 14 (26.9%) | 15 (28.8%) | 17 (32.7%) | 52 | 88.5 (46/52) |
| Table IVCoexpression of EGFR and FOLR1 in
malignant pleural mesothelioma (n=52). |
Table IV
Coexpression of EGFR and FOLR1 in
malignant pleural mesothelioma (n=52).
| Tissue type | FOLR1/EGFR
expression (n=52)
| Percentage of FOLR1
and EGFR coexpression |
|---|
| −/− | −/+ | +/− | +/+ | Total (n) |
|---|
| Epithelioid | 1 (3.0%) | 5 (15.2%) | 2 (6.0%) | 25 (75.8%) | 33 | 75.8 (25/33) |
| Biphasic | 0 (0%) | 1 (7.7%) | 1 (7.7%) | 11 (84.6%) | 13 | 84.6 (11/13) |
| Sarcomatoid | 1 (20.0%) | 2 (40.0%) | 1 (20.0%) | 1 (20.0%) | 5 | 20.0 (1/5) |
| Desmoplastic | 0 (0%) | 1 (100%) | 0 (0%) | 0 (0%) | 1 | 0.0 (0/1) |
| Total | 2 (3.8%) | 9 (17.3%) | 4 (7.7%) | 37 (71.2%) | 52 | 71.2 (37/52) |
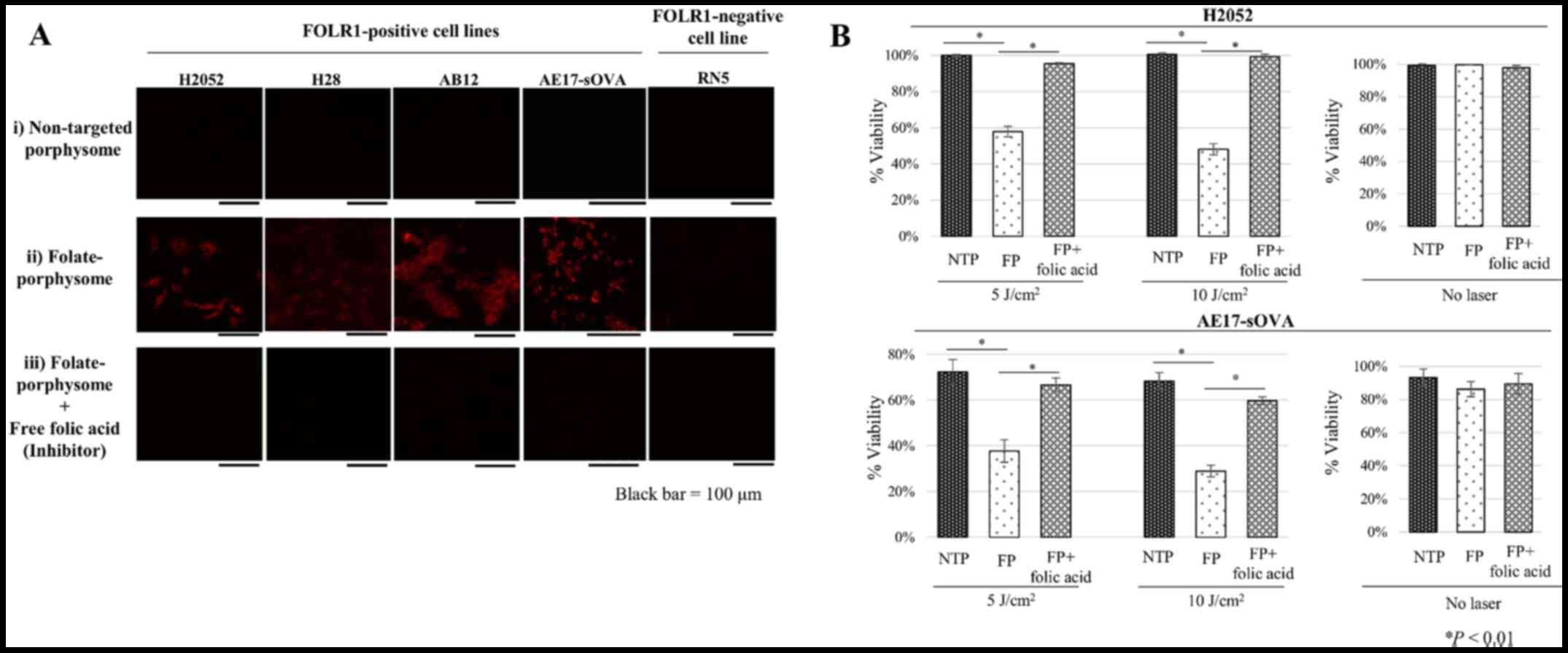
Cellular uptake and FP-PDT efficacy in
vitro
The porphysome fluorescence is unquenched upon
internalization into cells and therefore serves as a direct
indicator of uptake. None of the cells lines exhibited significant
fluorescence after incubation with non-targeting porphysomes
(Fig. 2A). The FOLR1-positive
H2052, H28, AB12 and AE17-sOVA cells exhibited high intra- cellular
florescence following incubation with FPs, which was efficiently
inhibited by excess free folic acid (Fig. 2A). No fluorescence signal was
observed in the FOLR1-negative RNS cells (Fig. 2A). Together, these data
demonstrated the specificity of cellular targeting of FPs mediated
by FOLR1. The pattern of enhanced intracellular uptake with FOLR1
targeting was similar to what we previously detected in other
FOLR1-positive cell lines, including KB cells (25). Fluorescence was only detected in
the cytoplasm, particularly in the lysosomes (23), with no significant nuclear
concentration.
Cell viability of FOLR1-positive H2052 and AE17-sOVA
cells was measured with or without light irradiation, in order to
verify the in vitro FP-PDT efficacy (Fig. 2B). H2052 and AE17-sOVA cells
treated with non-targeted porphysomes were used as a negative
control. Neither non-targeted porphysomes nor FP had measurable
dark toxicity at the maximum porphyrin concentration of
5×10−6 M in RPMI-1640 medium (Fig. 2B). However, 5 and 10 J
cm−2 FP incubation plus light irradiation (after 24 h
reduced cell viability to 57.9±2.9 and 48.1±3.1% in H2052 cells and
37.7±4.9 and 28.9±2.6% in AE17- sOVA cells, respectively (Fig. 2B). Conversely, the cytotoxicity was
completely inhibited by free folic acid, and there was no
measurable phototoxicity with non-targeted porphysomes.
Tumor uptake and FP-PDT efficacy in
vivo
In vivo fluorescence imaging in the pleural
dissemination model was conducted after chest CT confirmation of
tumor development. Both non-targeted porphysomes and FPs exhibited
fluorescence activation in the tumor at 6 h post i.v. injection
(Fig. 3A); FP-treated mice
exhibited clear fluorescence of small nodules on the surface of the
lungs, allowing surgical resection under image guidance (data not
shown). Since porphysome fluorescence was only partially
unquenched, fluorescence imaging yielded only the relative uptake
between the targeted and untargeted nanoparticles. Both showed high
uptake in tumor (1.22±0.47 vs. 0.82±0.20 %ID/g), and the difference
was not statistically significant (P=0.11). Conversely, there was a
significant increase in FP uptake in the liver (4.6±0.5 vs.
0.63±0.23 %ID/g) and the spleen (2.11±0.59 vs. 0.92±0.29 %ID/g) at
the same time-point (Fig. 3B).
Porphysome fluorescence was clearly visible under confocal
microscopy of tumor sections in the tumor cell cytoplasm (Fig. 3C).
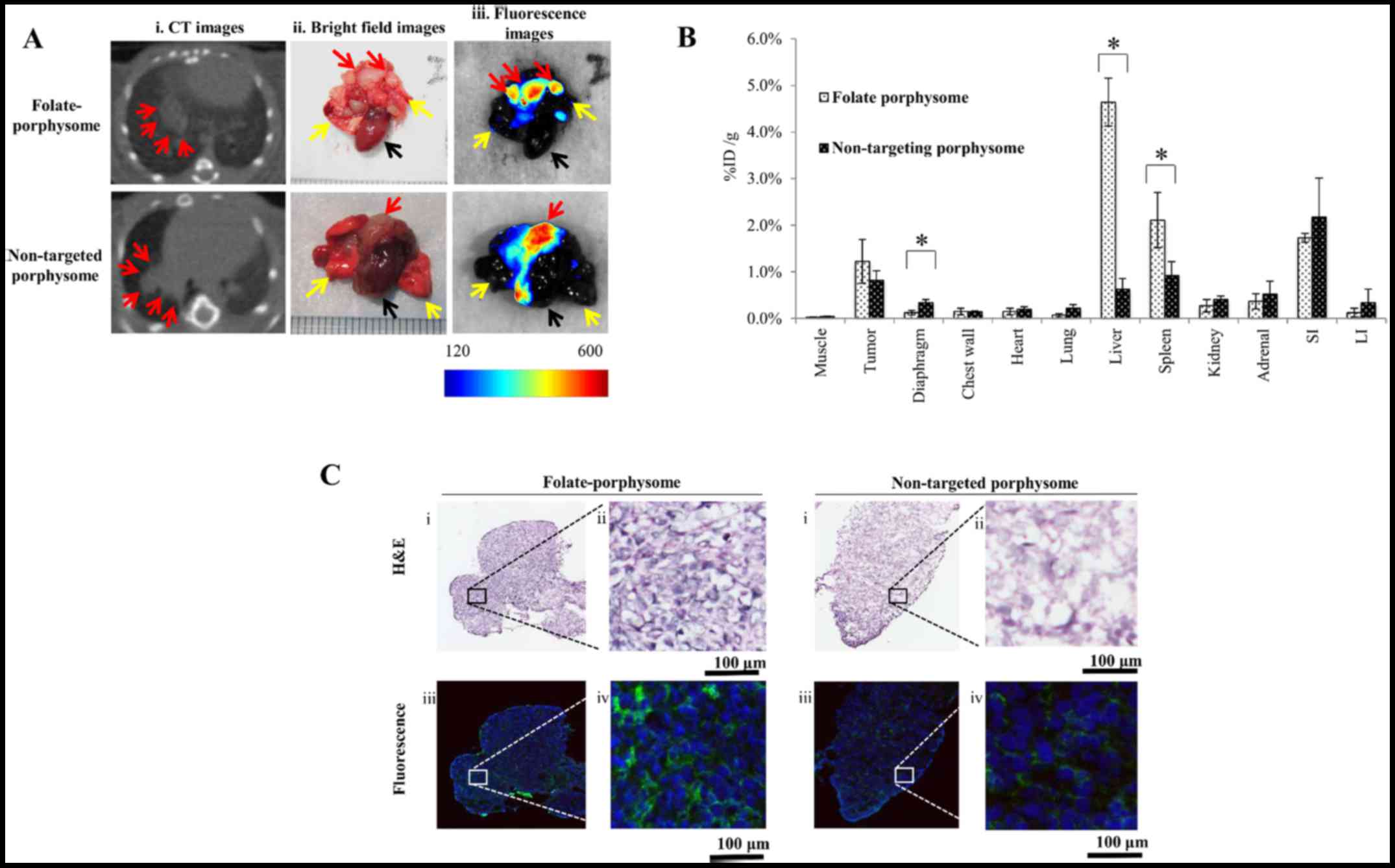 | Figure 3In vivo fluorescence
activation and biodistribution of porphysomes in nude mice with
disseminated intrapleural AE17-sOVA tumors. (A) (i) Representative
chest CT, and (ii) bright field and (iii) Maestro®
fluorescence images of the heart (black arrow), lungs (yellow
arrows) and tumor nodules (red arrows) 6 h post-injection of FP or
non-targeted porphysome (10 mg/kg). (B) Accumulation of FP and
non-targeted porphysome in organs harvested 6 h after
administration in mice with disseminated pleural mesothelioma.
*P<0.05. (C) Histological analysis and confocal
fluorescence microscopy images of AE17-sOVA cells: (i and ii)
H&E staining, and (iii and iv) fluorescence images showing the
cytoplasmic distribution (blue, DAPI at 408 nm excitation; green,
pyro signal at 635 nm excitation). Scale bar represents 100
µm. CT, computed tomography; FP, folate-porphysome; H&E,
hematoxylin and eosin; ID/g, injected dose per gram of tissue;
sOVA, secretory ovalbumin. |
The in vivo PDT response was assessed
in AE17-sOVA subcutaneous tumors, compared with the growth of
untreated tumors (Fig. 4A and B),
which reached the defined endpoint of 15 mm diameter after 5 days.
All mice were sacrificed by day 10 post-treatment (Fig. 4C). In a second control group that
received laser treatment only, the same growth trend was observed
as in the untreated controls, reaching the endpoint on or before
day 7 (Fig. 4A-C). Conversely,
FP-PDT caused typical initial tumor swelling at day 1
post-treatment (49,50), which resolved by day 2, with
subsequent inhibition of tumor growth (Fig. 4A and B). However, tumor regrowth
was then observed at 7 days post-treatment for all mice in this
group (Fig. 4B), with one mouse
reaching the endpoint at day 14 and the other five on day 21
(Fig. 4C). There were no
behavioral changes or local toxicities, such as skin necrosis.
Therefore, FP-PDT at these photosensitizer and light doses did not
completely eliminate the tumor, but significantly suppressed tumor
growth and prolonged survival up to 3 weeks post-treatment.
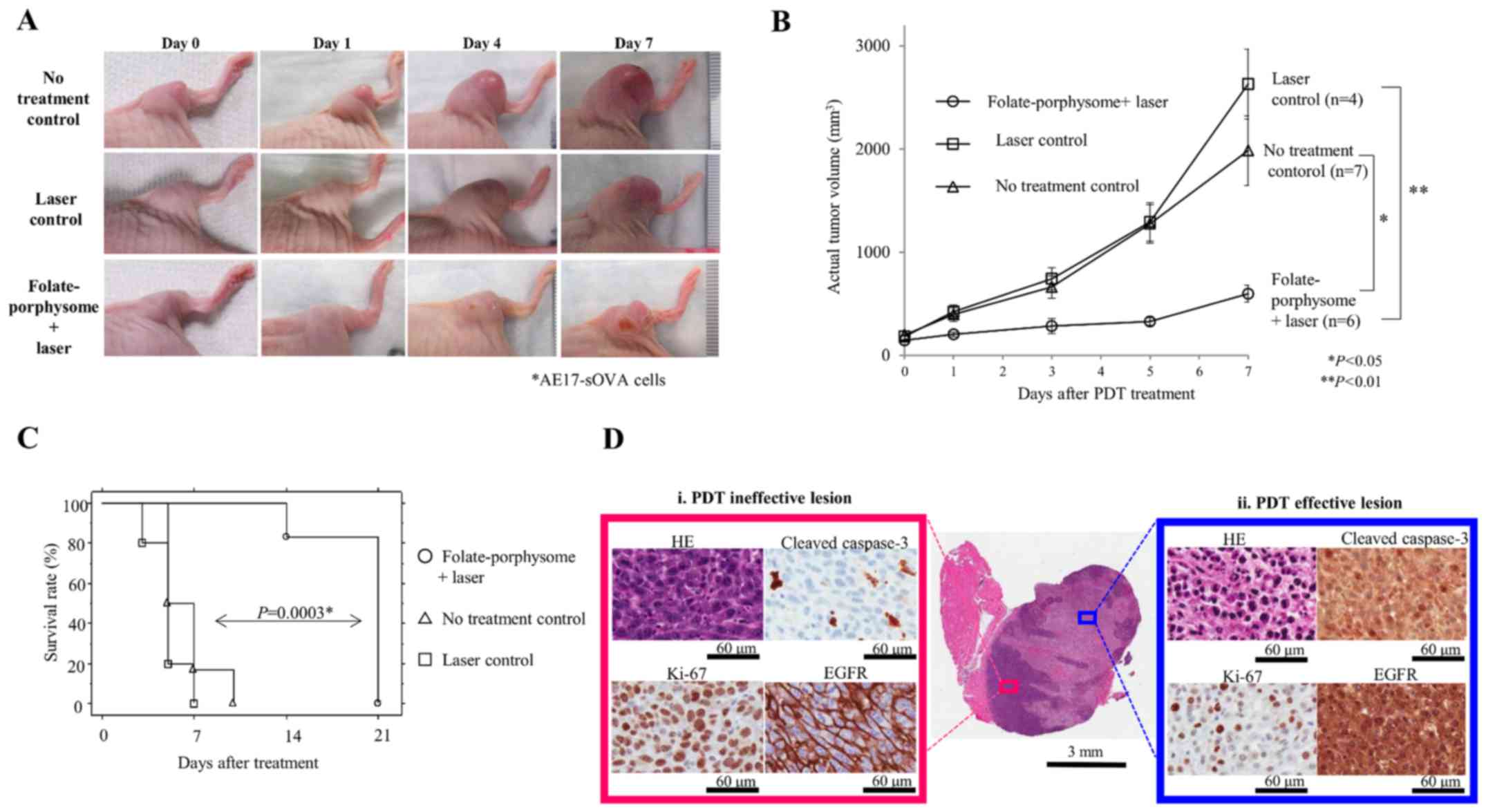 | Figure 4Post-PDT response in AE17-sOVA
tumor-bearing mice. (A) Representative tumor images on days 0, 1, 4
and 7 post-PDT treatment. (B) Tumor growth curve following
treatment for each group. Tumor volume in the FP-PDT group at each
time-point was separately compared to each of the control groups.
Statistical comparison at each time-point between groups was made
by one-factor repeated measures analysis of variance and
Newman-Keuls post hoc test was used to evaluate the significance of
the differences between two groups on day 7. *P<0.05,
**P<0.01. (C) Survival rate of animals in each group:
Laser control (n=5), no treatment control (n=6), and FP + laser
(n=6). Survival rate of the FP-PDT group was compared with the no
treatment control and the laser control groups; rates were
calculated by the Kaplan-Meier method and differences between the
no treatment control group and the FP-PDT group was compared using
the log-rank test (P=0.0003). (D) Representative
immunohistochemical staining images of tumors treated with
FP-enabled PDT, showing (i) a PDT ineffective lesion (red box) and
(ii) a PDT effective lesion (blue box). HE, cleaved caspase-3,
Ki-67 and EGFR staining are presented. ANOVA, analysis of variance;
EGFR, epidermal growth factor receptor; FP, folate-porphysome;
H&E, hematoxylin and eosin; PDT, photodynamic therapy; sOVA,
secretory ovalbumin. |
In a previous study, FP-PDT was revealed to achieve
complete tumor elimination using optimized dosing in subcutaneous
KB tumors (25); therefore,
optimizing the doses in the MPM model is likely to further enhance
the efficacy. However, the sub-optimized FP-PDT doses allowed for
investigation of the potential combined effect of EGFR inhibitor
plus PDT.
Combined FP-PDT plus EGFR inhibitor
One contributor to tumor regrowth/recurrence
following PDT (in general) is induced activation of cell cycle
progression pathways, such as EGFR (51). Benzoporphyrin derivative-mediated
PDT stimulates EGFR tyrosine phosphorylation and nuclear
translocation from the cellular membrane, whereas EGFR inhibition
by erlotinib results in a reduction of PDT-mediated EGFR activation
(29,52). In the MPM in vivo model, IHC
demonstrated that EGFR was markedly activated in the region of
effective FP-PDT (Fig. 4D, blue
box), with nuclear translocation of EGFR from the cell membrane
detected alongside a decrease in Ki-67 positivity and an increase
in the cleaved caspase-3 apoptotic index. In addition, these
effects were intratumorally heterogeneous, with some regions
showing viable tumor cells, higher Ki-67 positivity, and reduced
apoptotic index and membranous EGFR staining; these regions
appeared to be located where PDT was ineffective (Fig. 4D, red box).
In order to investigate whether inhibiting
PDT-dependent EGFR signaling may increase direct cell cytotoxicity,
cells in vitro were pretreated with an EGFR-TKI (erlotinib, 5
µM) and cell viability was measured following FP-PDT (5 or
10 J/cm2). The combination decreased the viability of
AE17-sOVA cells (from 55.2±2.4 and 31.2±2.9% with FP-PDT only to
36.1±0.9 and 21.7±2.4 with FP-PDT plus EGFR-TKI for 5 and 10
J/cm2, P<0.01 and P<0.05, respectively) and of
H2052 cells (from 36.5±5.1 to 16.4±0.8% with FP-PDT only for 5
J/cm2, P<0.05). In addition, there was a slight
decrease in the viability of H2052 cells treated with FP-PDT (10
J/cm2) and EGFR-TKI, as compared with cells treated with
FP-PDT only, even though there was no statistical significance
(from 19.0±1.1 to 12.1±0.7%; Fig.
5A).
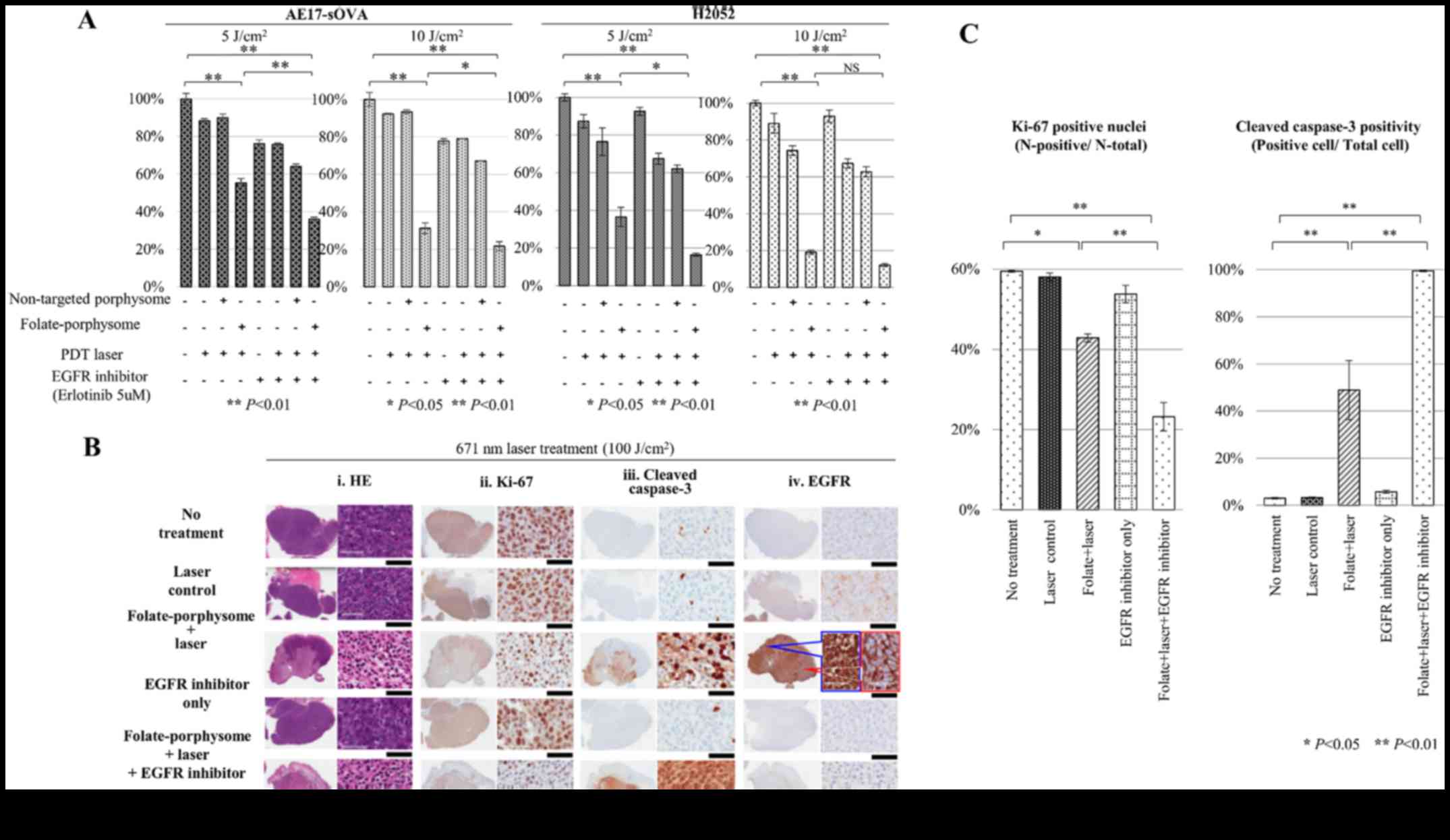 | Figure 5In vitro and in vivo
evaluation of the combined therapeutic efficacy of EGFR inhibitor
plus FP-enabled PDT in malignant pleural mesothelioma. (A) In
vitro cytotoxicity induced by eight treatments, tested on
AE17-sOVA and H2052 cell lines (both EGFR-positive and
FOLR1-positive); two PDT doses (5 and 10 J/cm2) were
compared. One-factor analysis of variance and a Newman-Keuls test
was conducted to evaluate the significance of differences between
the groups. (B) Histopathological analysis of tumors 24 h
post-treatment, including: i) H&E; ii) Ki-67; iii) cleaved
caspase-3; and iv) EGFR staining. Black scale bar, 60 µm.
(C) Quantitative evaluation of Ki-67 and cleaved caspase-3
staining, showing the percentage of positively stained cells in
tumors from the different treatment groups. Each treatment group
was compared with the control group (no treatment) using one-factor
analysis of variance and Dunnett post hoc test.
*P<0.05, **P<0.01. EGFR, epidermal
growth factor receptor; FP, folate-porphysome; H&E, hematoxylin
and eosin; PDT, photodynamic therapy; sOVA, secretory
ovalbumin. |
For corresponding in vivo studies, histology
was used to evaluate therapeutic efficacy. Erlotinib was
administered orally at a dose of 50 mg/kg to mice bearing AE17-sOVA
subcutaneous tumors 2 days, 1 day and 3 h before PDT treatment.
Mice were sacrificed after 24 h for H&E and immunohistochemical
analyses (Fig. 5B). EGFR
activation was completely inhibited by erlotinib (Fig. 5B). As expected, the control groups
(no treatment, laser only and erlotinib only groups) exhibited
59.5±0.2, 58.0±1.0 and 53.8±2.2% positive Ki-67 staining in the
viable tumor fractions, and 3.0±0.3, 3.3±0.2 and 5.7±0.6% damage,
as indicated by positive cleaved caspase-3 staining (Fig. 5B and C). These findings indicated
that the tumors in the control groups were minimally affected.
Conversely, in the FP-PDT group, a decrease in cell viability
(42.9±1.0 %) was detected by Ki-67 staining (P<0.05 compared
with the control groups; Fig. 5B
and C) and significant apoptosis was observed, as determined by
48.9±12.6% positive staining of cleaved caspase-3 (P<0.01
compared with the control groups; Fig.
5B and C). However, tumors treated with FP-PDT still exhibited
high EGFR activation and partial nuclear translocation (Fig. 5B), compared with the control
groups. Since nuclear translocation of EGFR is thought to mediate
anti-apoptotic signaling (29),
these results suggested that FP-PDT initiated nuclear signaling of
EGFR, which resulted in reduced PDT cytotoxicity. However, the
combination of pretreatment with an EGFR inhibitor and FP-PDT
further decreased Ki-67 positivity compared with in the FP-PDT
groups (23.2±3.6%, P<0.01), and much higher cleaved caspase-3
positivity, and thus apoptosis, was also detected (99.5±0.2%,
P<0.01).
Discussion
Mesothelioma is associated with a very poor
prognosis, due to the late onset of clinical symptoms that delay
diagnosis and limit therapeutic options (53). Surgical treatment is most efficient
when the tumor is of epithelioid subtype and essentially confined
to the hemithorax without lymph node metastasis at the time of
diagnosis (54). The addition of
localized PDT has been reported to limit the rate of tumor growth
after recurrence following EPD surgery (14,15)
and technical advances have improved the uniformity of light
delivery, including the use of a light-scattering medium to fill
the pleural cavity (18,55). We aim to focus on the use of an
optimal light-scattering medium within the pleural cavity in the
future. The relatively poor tumor specificity of current approved
photosensitizers remains a major challenge to achieve safe and
effective PDT treatment. Therefore, the present study introduced a
novel tumor- specific nanostructure-based photosensitizer to
enhance PDT efficacy and tumor selectivity, and evaluated the
effects of pretreatment with an EGFR inhibitor. Firstly, IHC
staining of TMA samples and western blot analysis of cell lysates
demonstrated that FOLR1 was highly expressed in MPM, which is in
line with a previous study, in which 72% of MPM cases had a 2- to
4-fold increase in FOLR1 mRNA expression compared with in normal
tissues; in addition, IHC detected FOLR1 expression on the cellular
membrane in 13 of 17 frozen samples (26). Likely as a consequence of this, FPs
exhibited markedly higher cellular uptake compared with
non-targeted porphysomes in vitro, and possessed higher
photocytotoxicity in FOLR1-positive MPM cell lines. FPs have a
shorter circulation half-life than non-targeted porphysome in
vivo (3.5 vs. 10.4 h); therefore, a 6 h FP-light interval was
used as the optimal interval in a previous FP-PDT study in
FOLR1-positive KB tumors (25).
The present study also compared the biodistribution of FPs and
non-targeted porphysomes at this time-point. As expected,
significantly more FPs, compared with non-targeted porphysomes,
accumulated in the liver and spleen, which was probably due to the
extended retention of FPs in these two organs caused by receptor-
mediated endocytosis, whereas non-targeting porphysomes were
cleared out more quickly by the reticulo-endothelial system
(56). However, the uptake in
FOLR1-positive tumors was not statistically different, likely since
both are mediated by the EPR effect due to their nano-scale size
(57). Notably, however, only FPs
were internalized into the FOLR1- positive tumor cells and became
reactivated for PDT upon disruption of the nanostructure, whereas
non-targeted porphysomes remained in the interstitial space and
most of them remained photodynamically quenched (25). The therapeutic efficacy and
mechanism of FPs were investigated previously (25). FPs are unquenched inside targeted
cells, and porphyrins, as PDT agents, are activated to generate
singlet oxygen for Type II PDT reactions upon irradiation of the
PDT laser. Consequently, significant FP-PDT efficacy has been
observed. This MPM tumor-specific PDT efficacy may translate into a
wider therapeutic window for treating disseminated MPM in the
pleural cavity and may avoid the dose-limiting normal tissue
phototoxicity of conventional photosensitizers. Since FOLR1 is
overexpressed in several tumors, including adenocarcinoma of the
ovary, uterus and the pituitary gland, testicular choriocarcinoma
and ependymal brain tumor (26,58-63),
the technology should be more widely applicable.
Although tumor growth in vivo was
significantly suppressed by FP-PDT, the tumor recurred and all mice
eventually reached the endpoint by 21 days after treatment. This
may be caused in part by heterogeneous FP distribution in the tumor
(64) and, as indicated by the
heterogeneous distribution of residual viable tumor cells in
regions of EGFR overexpression, by activation of survival pathways
and nuclear translocation. Since it has been reported that
inhibiting EGFR signaling increases PDT cytotoxicity through a
mechanism that involves increased apoptotic cell death (29), the present study investigated this
as a strategy to improve FP-PDT. The human-TMA analysis revealed
~89% EGFR positivity and, more importantly, ~70% coexpression of
FOLR1 and EGFR in MPM. In the present study, erlotinib pretreatment
synergistically enhanced the efficacy of FP-PDT, which was
associated with reduced EGFR activation, decreased expression of
the cell proliferation marker Ki-67 and increased apoptosis, as
determined by cleaved caspase-3 positivity. Numerous mechanisms may
be involved here, including erlotinib-induced increase of
photosensitizer accumulation and/or sensitization of the tumor
vasculature (33). While this
enhanced response was measured only by histological analysis, these
findings suggested that further preclinical and clinical
development of this strategy may be beneficial.
Finally, the formulation of porphysomes can be
modified easily due to their liposomal structure (23). Therefore, other targeting motifs,
such as prostate specific membrane antigen and EGFR, could be
integrated into the porphysome formulation to target other tumor
types. Porphyrins are also natural metal chelators and we
previously developed 64Cu folate-porphysomes as a
radioactive tracer for positron emission tomography imaging
(65), in order to detect tumors
at different stages, assist in pretreatment planning, provide
real-time guidance for surgical navigation, or to monitor post-
treatment tumor responses. FP-PDT could also be combined with other
therapies, for example by loading the porphysome core or lipid
bilayer with chemotherapeutic drugs, such as doxorubicin, for more
targeted tumor delivery (23).
In conclusion, the present study demonstrated the
efficacy of FP-enabled PDT for the treatment of FOLR1- positive MPM
in preclinical models in vitro and in vivo, as well
as the combined therapeutic effect of pretreatment with EGFR-TKI.
If clinically translatable, this should improve the efficacy and
safety of intrapleural PDT as an adjuvant treatment that could
improve the prognosis of this highly fatal disease.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study
are included in this published article.
Authors’ contributions
TK and CSJ performed the experimental design, most
of the experiments and analysis, drafted the manuscript, and were
involved in the conception and design of the study. TK was also
involved in collecting clinical tissue samples and accessing
clinical databases. DL conducted experiments, analyzed the data,
performed the statistical analysis and contributed to the writing
of the manuscript. HU, KF, HPH, HW and LW conducted some supporting
in vivo experiments. HK, YH, and KCH contributed to the
preparation and the review of the tissue microarray. RAW performed
some supporting laser experiments. JC, KK, YM, MDP, BCW, GZ and KY
supervised the study, were involved in the conception and design of
the study, and proofread the manuscript. YM participated in the
planning/design of the tissue microarray experiments and supervised
the pathological review. All authors have read and approved the
final manuscript.
Ethics approval and consent to
participate
The protocol was approved by the appropriate
institutional review board of Hokkaido University [approval no.
012-0136]. Each patient provided written informed consent at the
time of surgery. The animal study was approved by the ethics
committee of University Health Network.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Mr. Takatomo
Funayama (Morphotechnology Co., Ltd., Sapporo, Japan) who conducted
FOLR1 and EGFR immunohistochemical studies; Ms. Melanie Peralta
(Pathology Research Program Laboratory at University Health
Network, Toronto, ON, Canada) who performed Ki-67, cleaved
caspase-3 and EGFR immunohistochemical studies; and Ms. Judy
McConnell, Ms. Alexandria Grindlay and Ms. Kimberley Hudson
(Toronto General Hospital, Toronto, ON, Canada) for laboratory
management.
References
|
1
|
Yang H, Testa JR and Carbone M:
Mesothelioma epidemiology, carcinogenesis, and pathogenesis. Curr
Treat Options Oncol. 9:147–157. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kotova S, Wong RM and Cameron RB: New and
emerging therapeutic options for malignant pleural mesothelioma:
Review of early clinical trials. Cancer Manag Res. 7:51–63.
2015.PubMed/NCBI
|
|
3
|
Robinson BM: Malignant pleural
mesothelioma: An epidemiological perspective. Ann Cardiothorac
Surg. 1:491–496. 2012.
|
|
4
|
Robinson BW and Lake RA: Advances in
malignant mesothelioma. N Engl J Med. 353:1591–1603. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Becklake MR, Bagatin E and Neder JA:
Asbestos-related diseases of the lungs and pleura: Uses, trends and
management over the last century. Int J Tuberc Lung Dis.
11:356–369. 2007.PubMed/NCBI
|
|
6
|
Scherpereel A, Astoul P, Baas P, Berghmans
T, Clayson H, de Vuyst P, Dienemann H, Galateau-Salle F, Hennequin
C, Hillerdal G, et al: European Respiratory Society/European
Society of Thoracic Surgeons Task Force: Guidelines of the European
Respiratory Society and the European Society of Thoracic Surgeons
for the management of malignant pleural mesothelioma. Eur Respir J.
35:479–495. 2010. View Article : Google Scholar
|
|
7
|
Flores RM, Pass HI, Seshan VE, Dycoco J,
Zakowski M, Carbone M, Bains MS and Rusch VW: Extrapleural
pneumonectomy versus pleurectomy/decortication in the surgical
management of malignant pleural mesothelioma: results in 663
patients. J Thorac Cardiovasc Surg. 135:620–626. 626e621–623. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kostron A, Friess M, Crameri O, Inci I,
Schneiter D, Hillinger S, Stahel R, Weder W and Opitz I: Relapse
pattern and secondline treatment following multimodality treatment
for malignant pleural mesothelioma. Eur J Cardiothorac Surg.
49:1516–1523. 2016. View Article : Google Scholar
|
|
9
|
Baldini EH, Richards WG, Gill RR, Goodman
BM, Winfrey OK, Eisen HM, Mak RH, Chen AB, Kozono DE, Bueno R, et
al: Updated patterns of failure after multimodality therapy for
malignant pleural mesothelioma. J Thorac Cardiovasc Surg.
149:1374–1381. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gomez DR, Hong DS, Allen PK, Welsh JS,
Mehran RJ, Tsao AS, Liao Z, Bilton SD, Komaki R and Rice DC:
Patterns of failure, toxicity, and survival after extrapleural
pneumonectomy and hemithoracic intensity-modulated radiation
therapy for malignant pleural mesothelioma. J Thorac Oncol.
8:238–245. 2013. View Article : Google Scholar
|
|
11
|
Gupta V, Krug LM, Laser B, Hudka K, Flores
R, Rusch VW and Rosenzweig KE: Patterns of local and nodal failure
in malignant pleural mesothelioma after extrapleural pneumonectomy
and photon-electron radiotherapy. J Thorac Oncol. 4:746–750. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tsao AS, Mehran R and Roth JA: Neoadjuvant
and intrapleural therapies for malignant pleural mesothelioma. Clin
Lung Cancer. 10:36–41. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sterman DH, Alley E, Stevenson JP,
Friedberg J, Metzger S, Recio A, Moon EK, Haas AR, Vachani A, Katz
SI, et al: Pilot and feasibility trial evaluating immuno-gene
therapy of malignant mesothelioma using intrapleural delivery of
adenovirus-IFNα combined with chemotherapy. Clin Cancer Res.
22:3791–3800. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Friedberg JS, Culligan MJ, Mick R,
Stevenson J, Hahn SM, Sterman D, Punekar S, Glatstein E and Cengel
K: Radical pleurectomy and intraoperative photodynamic therapy for
malignant pleural mesothelioma. Ann Thorac Surg. 93:1658–65;
discussion 1665-7. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Friedberg JS: Photodynamic therapy for
malignant pleural mesothelioma. J Natl Compr Canc Netw. 10(Suppl
2): S75–S79. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vrouenraets MB, Visser GW, Snow GB and van
Dongen GA: Basic principles, applications in oncology and improved
selectivity of photodynamic therapy. Anticancer Res. 23B:505–522.
2003.
|
|
17
|
Dolmans DE, Fukumura D and Jain RK:
Photodynamic therapy for cancer. Nat Rev Cancer. 3:380–387. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pass HI, DeLaney TF, Tochner Z, Smith PE,
Temeck BK, Pogrebniak HW, Kranda KC, Russo A, Friauf WS, Cole JW,
et al: Intrapleural photodynamic therapy: Results of a phase I
trial. Ann Surg Oncol. 1:28–37. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Takita H, Mang TS, Loewen GM, Antkowiak
JG, Raghavan D, Grajek JR and Dougherty TJ: Operation and
intracavitary photodynamic therapy for malignant pleural
mesothelioma: A phase II study. Ann Thorac Surg. 58:995–998. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schouwink H, Rutgers ET, van der Sijp J,
Oppelaar H, van Zandwijk N, van Veen R, Burgers S, Stewart FA,
Zoetmulder F and Baas P: Intraoperative photodynamic therapy after
pleuropneumonectomy in patients with malignant pleural
mesothelioma: Dose finding and toxicity results. Chest.
120:1167–1174. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Friedberg JS, Mick R, Stevenson J, Metz J,
Zhu T, Buyske J, Sterman DH, Pass HI, Glatstein E and Hahn SM: A
phase I study of Foscan-mediated photodynamic therapy and surgery
in patients with mesothelioma. Ann Thorac Surg. 75:952–959. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Moghissi K and Dixon K: Photodynamic
therapy in the management of malignant pleural mesothelioma: A
review. Photodiagn Photodyn Ther. 2:135–147. 2005. View Article : Google Scholar
|
|
23
|
Lovell JF, Jin CS, Huynh E, Jin H, Kim C,
Rubinstein JL, Chan WC, Cao W, Wang LV and Zheng G: Porphysome
nanovesicles generated by porphyrin bilayers for use as multimodal
biophotonic contrast agents. Nat Mater. 10:324–332. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jin CS, Lovell JF, Chen J and Zheng G:
Ablation of hypoxic tumors with dose-equivalent photothermal, but
not photodynamic, therapy using a nanostructured porphyrin
assembly. ACS Nano. 7:2541–2550. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jin CS, Cui L, Wang F, Chen J and Zheng G:
Targeting-triggered porphysome nanostructure disruption for
activatable photodynamic therapy. Adv Healthc Mater. 3:1240–1249.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bueno R, Appasani K, Mercer H, Lester S
and Sugarbaker D: The alpha folate receptor is highly activated in
malignant pleural mesothelioma. J Thorac Cardiovasc Surg.
121:225–233. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nutt JE, Razak AR, O’Toole K, Black F,
Quinn AE, Calvert AH, Plummer ER and Lunec J: The role of folate
receptor alpha (FRalpha) in the response of malignant pleural
mesothelioma to pemetrexed-containing chemotherapy. Br J Cancer.
102:553–560. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lovell JF, Liu TWB, Chen J and Zheng G:
Activatable photo-sensitizers for imaging and therapy. Chem Rev.
110:2839–2857. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Edmonds C, Hagan S, Gallagher-Colombo SM,
Busch TM and Cengel KA: Photodynamic therapy activated signaling
from epidermal growth factor receptor and STAT3: Targeting survival
pathways to increase PDT efficacy in ovarian and lung cancer.
Cancer Biol Ther. 13:1463–1470. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bhuvaneswari R, Yuen GY, Chee SK and Olivo
M: Antiangiogenesis agents avastin and erbitux enhance the efficacy
of photodynamic therapy in a murine bladder tumor model. Lasers
Surg Med. 43:651–662. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Weyergang A, Selbo PK and Berg K:
Sustained ERK [corrected] inhibition by EGFR targeting therapies is
a predictive factor for synergistic cytotoxicity with PDT as
neoadjuvant therapy. Biochim Biophys Acta. 1830.2659–2670.
2013.
|
|
32
|
Bhuvaneswari R, Gan YY, Soo KC and Olivo
M: Targeting EGFR with photodynamic therapy in combination with
Erbitux enhances in vivo bladder tumor response. Mol Cancer.
8:942009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gallagher-Colombo SM, Miller J, Cengel KA,
Putt ME, Vinogradov SA and Busch TM: Erlotinib pretreatment
improves photodynamic therapy of non-small cell lung carcinoma
xenografts via multiple mechanisms. Cancer Res. 75:3118–3126. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Postiglione I, Chiaviello A, Aloj SM and
Palumbo G: 5-aminolaevulinic acid/photo-dynamic therapy and
gefitinib in non-small cell lung cancer cell lines: A potential
strategy to improve gefitinib therapeutic efficacy. Cell Prolif.
46:382–395. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kurai J, Chikumi H, Hashimoto K, Takata M,
Sako T, Yamaguchi K, Kinoshita N, Watanabe M, Touge H, Makino H, et
al: Therapeutic antitumor efficacy of anti-epidermal growth factor
receptor antibody, cetuximab, against malignant pleural
mesothelioma. Int J Oncol. 41:1610–1618. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Destro A, Ceresoli GL, Falleni M, Zucali
PA, Morenghi E, Bianchi P, Pellegrini C, Cordani N, Vaira V,
Alloisio M, et al: EGFR overexpression in malignant pleural
mesothelioma. An immunohistochemical and molecular study with
clinico-pathological correlations. Lung Cancer. 51:207–215. 2006.
View Article : Google Scholar
|
|
37
|
Garland LL, Rankin C, Gandara DR, Rivkin
SE, Scott KM, Nagle RB, Klein-Szanto AJ, Testa JR, Altomare DA and
Borden EC: Phase II study of erlotinib in patients with malignant
pleural mesothelioma: A Southwest Oncology Group Study. J Clin
Oncol. 25:2406–2413. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Agarwal V, Lind MJ and Cawkwell L:
Targeted epidermal growth factor receptor therapy in malignant
pleural mesothelioma: Where do we stand? . Cancer Treat Rev.
37:533–542. 2011. View Article : Google Scholar
|
|
39
|
Okuda K, Sasaki H, Kawano O, Yukiue H,
Yokoyama T, Yano M and Fujii Y: Epidermal growth factor receptor
gene mutation, amplification and protein expression in malignant
pleural mesothelioma. J Cancer Res Clin Oncol. 134:1105–1111. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Union for International Cancer Control
(UICC). TNM Classification of Malignant Tumours. 7.
Wiley-Blackwell; Hoboken: 2009
|
|
41
|
ZenBio I: Human mesothelial cells.
http://www.zen-bio.com/products/cells/mesothelial.php,
Accessed July 18, 2014.
|
|
42
|
ExPASy: the SIB Bioinformatics Resource
Portal. https://web.expasy.org/cellosaurus/CVCL_0372.
|
|
43
|
Jackaman C, Bundell CS, Kinnear BF, Smith
AM, Filion P, van Hagen D, Robinson BW and Nelson DJ: IL-2
intratumoral immunotherapy enhances CD8+ T cells that
mediate destruction of tumor cells and tumor-associated
vasculature: A novel mechanism for IL-2. J Immunol. 171:5051–5063.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
ExPASy: the SIB Bioinformatics Resource
Portal_AE17-sOVA. https://web.expasy.org/cellosaurus/CVCL_LJ85.
|
|
45
|
Cordier Kellerman L, Valeyrie L, Fernandez
N, Opolon P, Sabourin JC, Maubec E, Le Roy P, Kane A, Legrand A,
Abina MA, et al: Regression of AK7 malignant mesothelioma
established in immunocompetent mice following intratumoral gene
transfer of interferon gamma. Cancer Gene Ther. 10:481–490. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Blum W, Pecze L, Felley-Bosco E,
Worthmüller-Rodriguez J, Wu L, Vrugt B, de Perrot M and Schwaller
B: Establishment of immortalized murine mesothelial cells and a
novel mesothelioma cell line. In Vitro Cell Dev Biol Anim.
51:714–721. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kato T, Wada H, Patel P, Hu HP, Lee D,
Ujiie H, Hirohashi K, Nakajima T, Sato M, Kaji M, et al:
Overexpression of KIF23 predicts clinical outcome in primary lung
cancer patients. Lung Cancer. 92:53–61. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kato T, Lee D, Wu L, Patel P, Young AJ,
Wada H, Hu HP, Ujiie H, Kaji M, Kano S, et al: Kinesin family
members KIF11 and KIF23 as potential therapeutic targets in
malignant pleural mesothelioma. Int J Oncol. 49:448–456. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Castano AP, Demidova TN and Hamblin MR:
Mechanisms in photodynamic therapy: Part two-cellular signaling,
cell metabolism and modes of cell death. Photodiagn Photodyn Ther.
2:1–23. 2005. View Article : Google Scholar
|
|
50
|
Moor AC: Signaling pathways in cell death
and survival after photodynamic therapy. J Photochem Photobiol B.
57:1–13. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Pugh CW and Ratcliffe PJ: Regulation of
angiogenesis by hypoxia: Role of the HIF system. Nat Med.
9:677–684. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang SC and Hung MC: Nuclear translocation
of the epidermal growth factor receptor family membrane tyrosine
kinase receptors. Clin Cancer Res. 15:6484–6489. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Boffetta P, Donaldson K, Moolgavkar S and
Mandel JS: A systematic review of occupational exposure to
synthetic vitreous fibers and mesothelioma. Crit Rev Toxicol.
44:436–449. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Friedberg JS, Simone CB II, Culligan MJ,
Barsky AR, Doucette A, McNulty S, Hahn SM, Alley E, Sterman DH,
Glatstein E, et al: Extended pleurectomy-decortication-based
treatment for advanced stage epithelial mesothelioma yielding a
median survival of nearly three years. Ann Thorac Surg.
103:912–919. 2017. View Article : Google Scholar
|
|
55
|
Rodriguez E, Baas P and Friedberg JS:
Innovative therapies: Photodynamic therapy. Thorac Surg Clin.
14:557–566. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lovell JF, Jin CS, Huynh E, MacDonald TD,
Cao W and Zheng G: Enzymatic regioselection for the synthesis and
biodegradation of porphysome nanovesicles. Angew Chem Int Ed Engl.
51:2429–2433. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Albanese A, Tang PS and Chan WC: The
effect of nanoparticle size, shape, and surface chemistry on
biological systems. Annu Rev Biomed Eng. 14:1–16. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Weitman SD, Weinberg AG, Coney LR,
Zurawski VR, Jennings DS and Kamen BA: Cellular localization of the
folate receptor: Potential role in drug toxicity and folate
homeostasis. Cancer Res. 52:6708–6711. 1992.PubMed/NCBI
|
|
59
|
Chancy CD, Kekuda R, Huang W, Prasad PD,
Kuhnel JM, Sirotnak FM, Roon P, Ganapathy V and Smith SB:
Expression and differential polarization of the reduced-folate
transporter-1 and the folate receptor alpha in mammalian retinal
pigment epithelium. J Biol Chem. 275:20676–20684. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Toffoli G, Cernigoi C, Russo A, Gallo A,
Bagnoli M and Boiocchi M: Overexpression of folate binding protein
in ovarian cancers. Int J Cancer. 74:193–198. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ross JF, Chaudhuri PK and Ratnam M:
Differential regulation of folate receptor isoforms in normal and
malignant tissues in vivo and in established cell lines.
Physiologic and clinical implications. Cancer. 73:2432–2443. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Evans CO, Young AN, Brown MR, Brat DJ,
Parks JS, Neish AS and Oyesiku NM: Novel patterns of gene
expression in pituitary adenomas identified by complementary
deoxyribonucleic acid microarrays and quantitative reverse
transcription-polymerase chain reaction. J Clin Endocrinol Metab.
86:3097–3107. 2001.PubMed/NCBI
|
|
63
|
Weitman SD, Frazier KM and Kamen BA: The
folate receptor in central nervous system malignancies of
childhood. J Neurooncol. 21:107–112. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Jain RK and Stylianopoulos T: Delivering
nanomedicine to solid tumors. Nat Rev. Clin Oncol. 7:653–664.
2010.
|
|
65
|
Ni NC, Jin CS, Cui L, Shao Z, Wu J, Li SH,
Weisel RD, Zheng G and Li RK: Non-invasive macrophage tracking
using novel porphysome nanoparticles in the post-myocardial
Infarction murine heart. Mol Imaging Biol. 18:557–568. 2016.
View Article : Google Scholar : PubMed/NCBI
|