Introduction
Worldwide, colorectal cancer (CRC) is the fourth
most common cancer and the fourth leading cause of cancer-related
mortality, with 1.4 million estimated cases and 700,000 estimated
deaths in 2012 (1,2). As in many other solid tumours,
metastasis is the primary cause of mortality in CRC, and defines
stage IV CRC, which is characterized by a relatively short overall
survival. One of the main issues encountered in CRC treatment is
drug resistance, which is responsible for relapses in a number of
patients and the failure of medical treatments for metastatic
disease.
The normal gastrointestinal epithelium is organized
along a crypt-villus axis, with a pool of colon stem cells and
progenitor cells, which are the most undifferentiated cell types
which can undergo self-renewal and exhibit pluripotency, residing
at the bottom of the crypts. These cells move along the
crypt-villus axis while differentiating into all the epithelial
colon lineages, such as Paneth cells, goblet cells, enterocytes and
enteroendocrine cells. In approximately 14 days, they arrive at the
top of the villus, and undergo apoptosis (3-5).
It has been suggested that an oncogenic hit in a
stem cell generates cancer stem cells (CSCs), which are
tumour-initiating cells that represent the only cell type able to
promote cancer onset, progression and metastases. CSCs are probably
responsible for both tumour relapse and resistance to therapy
(6,7). The source from which CSCs arise
remains unclear. It remains to be determined whether
trans-amplificating (TA) cancer cells can de-differentiate,
transforming themselves into CSCs, or alternatively, whether
healthy stem cells are transformed into CSCs by accumulating
genetic hits in oncogenes or tumour suppressor genes (8,9). It
has been hypothesized that microenvironment-derived signals, such
as transforming growth factor (TGF)-β, epidermal growth factor
(EGF), Wnt and Notch, can activate epithelial-to-mesenchymal
transition (EMT) and stem-cell properties in differentiated cells
(10,11); thus, an oncogenic hit in this
population may give rise to CSCs that can initiate cancer.
During tumour progression, epithelial cancer cells
undergo EMT. Cells that undergo EMT acquire a mesenchymal
phenotype, losing their epithelial features and cell polarity and
acquiring motility, stem cell-like properties, self-renewal and
apoptosis resistance. These cells are also able to degrade the
basement membrane and extracellular matrix. Thus, it has been
suggested that EMT confers to cancer cells, including CRC cells,
many of the features required to develop metastases (12-14).
Chemo-radiotherapy often kills differentiated cancer cells, while
the mesenchymal, stem-like cells are resistant and can survive
after therapy, giving rise to therapy-resistant tumours.
EMT-associated transcription factors are also involved in apoptosis
resistance in cells that have undergone EMT. It has been
demonstrated that TWIST1 inhibits cell cycle arrest and apoptosis
by blocking the p53 target genes (15). Moreover, SNAIL suppresses
TGF-β-induced cell death and regulates the components of the early
to late G1 transition and the G1/S checkpoint, including the
repression of cyclin D2 transcription and the increase in p21
expression (16,17).
However, the only crucial cell feature conferring to
cancer cells the ability to complete the metastatic process, is
cellular plasticity. Cellular plasticity is defined as the ability
of cells to switch from an epithelial to a mesenchymal phenotype
and vice versa. Indeed, epithelial adenocarcinoma cells
escape from the primary in situ carcinomas and spread to
distant organs through mesenchymal intermediates, while only
disseminated cancer cells capable of switching again to an
epithelial hyper-proliferative stem cell phenotype are able to
colonize distant organs and generate metastatic secondary lesions.
Cancer cells that undergo EMT, but lose this plasticity phenotype
are ineffective for seeding metastatic colonies (18-20).
Glycogen synthase kinase 3 β (GSK-3β) is a key
regulator of the crosstalk between different signalling pathways
involved in CRC development, such as WNT/β-catenin, Ras, PI3K/Akt,
cMET and vascular endothelial growth factor (VEGF). Data have also
been published regarding the role of GSK-3β in CRC cell drug
resistance (21,22).
Genomic instability facilitates the accumulation of
multiple mutations during CRC development; chromosomal instability
(CIN) is observed in 85% and microsatellite instability (MSI) is
detected in 15% of sporadic CRCs. The molecular mechanisms
underlying CRC progression remain poorly understood, particularly
as regards CRCs with MSI (23). We
previously isolated two primary colon cancer cell cultures, one
exhibiting a CIN phenotype (T93) and the other exhibiting an MSI
phenotype (T88). They both exhibited mesenchymal and epithelial
features and a high level expression of EMT-associated
transcription factors and stemness markers. Thus, we hypothesized
that they were epithelial adenocarcinoma cells that had undergone
EMT. These cells were also able to grow in conditioned medium as
non-adherent tumourspheres. Finally, we demonstrated in
vitro that LiCl-induced mesenchymal-to-epithelial transition
(MET), cellular differentiation and the downregulation of the
EMT-associated transcription factors, Twist1 and Snail, in these
primary CRC cell cultures (24).
Herein, we investigated the expression and
localisation of key markers of EMT and stemness in CRC cells
exhibiting both CIN and MIN by establishing a system of adherent
primary mesenchymal colon cancer cells and paired tumourspheres.
These cells exhibited plasticity. We also observed an atypical
nuclear localisation of N-cadherin, CD133 and the v6 splice form of
CD44 glycoprotein (CD44v6) in the majority of the mesenchymal
cells, suggesting a change in localisation from the plasma membrane
to the nucleus, which could allow cell plasticity in CRC
progression. Finally, we demonstrated that GSK-3β inhibition
reduced cell migration and cell plasticity in our experimental cell
model, thus suggesting that GSK-3β may be a target for CRC
therapy.
Materials and methods
Sample collection
CRC tissues and normal colorectal mucosa were
obtained from patients with sporadic CRC, who were operated at the
AOU Federico II and Istituto Nazionale dei Tumori (Naples, Italy)
and primary cell cultures were established from these tissues. Data
regarding tumour stage were recovered from the medical records of
each patient, in accordance with the TNM classifications and tumour
budding grades.
Samples from all subjects who participated in this
study were collected after obtaining authorisation from the
Comitato etico per le attività Biomediche - Carlo Romano of the
University of Naples Federico II (protocol no. 432/17).
Authorisation was granted only once the study had received ethical
approval and written informed consent had been obtained from all
participants. All methods were performed in accordance with the
relevant guidelines and regulations.
Cell culture
The T88 and T93 cells were previously isolated and
stabilized in vitro (24).
The HM110 colon cells were isolated and stabilized during this
study from the healthy colon mucosa (HM) of a patient with sporadic
colon cancer, as previously described (24). Briefly, samples were washed
overnight at 4°C in PBS containing antibiotics, finely minced and
digested in collagenase II in DMEM/FBS-10% for 1 h at 37°C, 5%
CO2. The obtained cell suspension was then collected by
centrifugation at 1,000 × g, at room temperature, washed twice and
subsequently cultured in DMEM/F12-10% FBS medium (1:1), 100 U/ml
penicillin, 100 μg/ml streptomycin, and 2.5 μg/ml
amphotericin B. The HT29 and RKO cells were obtained from ATCC
(Manassas, VA, USA). To inhibit GSK-3β activity, cells were
incubated in medium containing 30 mM LiCl for 10 days. The T88 and
T93 primary colon cancer cells were then cultured as spheres in
serum-free stem cell medium on low-adhesion plates as previously
described (24). Cancer spheroids
were also obtained by the hanging drop assay. Briefly, the cells
were diluted to a final concentration of 3.7×104
cells/ml in DMEM/F12 (1:1) containing 2% foetal bovine serum (FBS),
100 U/ml penicillin, 100 μg/ml streptomycin, 2.5
μg/ml amphotericin B, 10 μg/ml basic fibroblast
growth factor (bFGF) and 20 μg/ml epidermal growth factor
(EGF), with or without 35 mM LiCl, (all reagents were purchased
from Sigma-Aldrich Merk KGaA, Darmstadt, Germany). The drops (27
μl) were seeded on the cover of a plate and incubated at
37°C at 5% CO2 for 72 h. Under these conditions, the
cells aggregated to form 3D spheroids. Cancer spheroids obtained in
presence or absence of LiCl were finally disaggregated into single
cells, which were again grown in adhesion.
Migration assays
Cell migration was evaluated using in vitro
wound healing assays and the Boyden chamber assay. In vitro
wound healing assays were performed as previously described by
Liang et al (25). Briefly,
the cells were seeded at 1×104 cells/well in 24-well
plates. After the cells formed a monolayer, a scratch wound was
made with the tip of a 1,000-μl pipette tip, and the scratch
was photographed under a light microscope (Leica DM IL LED inverted
microscope, type 11090137002, serial no. 11521258/335209; Meyer
Instruments, Inc. Houston, TX, USA) at ×20 magnification.
Subsequently, cells were incubated with or without (untreated
control) 35 mM LiCl. After 24 h, cells were rinsed, and a second
set of images were acquired.
Boyden chamber assays were performed using the
Corning® Transwell® polycarbonate membrane cell culture inserts
(pore size, 8.0 μm; Sigma-Aldrich, Merk KGaA). A 300
μl of aliquot of a 10,000 cells/ml suspension was
resuspended in serum-free medium and seeded into the upper chamber
of the polycarbonate membrane. Subsequently, 500 μl of
medium containing 10% FBS (chemoattractant) were added to the lower
well of the migration plate. Finally, the cells were removed from
the top of the membrane and the migrated cells were stained with
crystal violet solution for 15 min at room temperature
(Sigma-Aldrich, Merk KGaA). Alternatively, 24-well Transwell plates
(pore size: 8.0 μm; Corning Inc., Corning, New York, NY,
USA) were used. Each membrane was released from the apparatus using
a scalpel. The permeable membranes were then washed twice with
PBS/1% FBS at room temperature. The cells on the membranes were
fixed, permeabilized and stained with DAPI (Sigma-Aldrich, Merk
KGaA) for 15 min at room temperature. Finally, the membranes were
mounted on glass slides, covered with coverslips, and fluorescence
was visualized and imaged under a fluorescence microscope (Leica DM
IL LED inverted microscope, type 11090137002, no. 11521258/335209;
Meyer Instruments, Inc.).
Reverse transcription-quantitative
(real-time) PCR (RT-qPCR)
Total RNA was extracted from the cells using QIAzol
reagent (Qiagen, Hilden, Germany) according to the manufacturer's
instructions. Following DNase incubation, cDNA was synthesized from
1 μg total RNA, 500 ng random hexamers, and 1 μl
Superscript III reverse transcriptase in the presence of 4
μl 5X RT buffer, 1 μl DTT (0.1 M) and 1 mM dNTPs
(Invitrogen/Thermo Fisher Scientific, Waltham, MA, USA) as
previously described (26).
Quantitative (real-time) PCR (qPCR) was performed on 0.5 μl
cDNA, Snail and Twist1 primer pairs as previously described
(24,27). Relative expression was calculated
with the 2−ΔΔCq method (28) and normalized against glucuronidase
(GUS) mRNA. qPCR was performed on a Bio-Rad iCycler iQ Real-Time
PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA). A
non-template control was run for each assay, all determinations
were performed at least in duplicate to ensure reproducibility and
each experiment was performed two times. The synthesis of the
expected PCR product was confirmed by melting curve analysis.
Western blot analysis
Fractionated nuclear and cytosolic protein extracts
were isolated from the T88 and T93 cells using the Qproteome
Nuclear Protein kit (Qiagen Hilden, Germany), and quantified by
using Traditional Bradford kit (Bio-Rad Laboratories Srl, Segrate,
Italy), adopting bovine serum albumin standards. A total of 50
μg of proteins were separated by 10% SDS-polyacrylamide gel
electrophoresis. Blots were prepared on Amersham Hybond-ECL
nitrocellulose membranes (Amersham Pharmacia Biotech, Cologno
Monzese, Italy), and proteins were blocked in non-fat dried milk
diluted in TBST to reduce the background, as previously described
(29,30). Primary antibodies against
N-cadherin (rabbit monoclonal anti-human; #4061; 1:1,000) and CD133
(rabbit monoclonal anti-human; #5860; 1:1,000) were obtained from
Cell Signaling Technology (Danvers, MA, USA). The primary antibody
against CD44v6 (mouse monoclonal anti-human; #5640; 1:1,000) was
from R&D Systems (Minneapolis, MN, USA), the anti-actin (goat
polyclonal anti-human; sc-1615; 1:8,000) and anti-Histone H1
(monoclonal anti-mouse; sc-393358; 1:1,000) antibodies were from
Santa Cruz Biotechnology (Santa Cruz, CA, USA). The membranes were
probed with peroxidase-conjugated secondary antibodies against
rabbit (#7074), mouse (#7076; both from Cell Signaling Technology
or goat IgG (#ab97110; from Abcam, Cambridge, UK), all used at
1:3,000 dilution, and immunoreactive bands were detected using the
enhanced chemiluminescence HRP Substrate Immobilon Western
(Millipore, Billerica, MA, USA).
Immunofluorescence
Primary CRC cells were seeded and grown in 12-well
cultivation chambers with removable microscopy glass slides (ibidi,
Martinsried, Germany); cancer spheroids obtained by the hanging
drop assay were also sedi-mented in the same chamber.
Immunofluorescence analyses were performed as previously described
by Di Maio et al (31).
Briefly, following fixation in 4% paraformaldehyde in PBS for 10
min, the cells were permeabilized in 0.1% Triton X-100 in PBS for
30-120 min, and then blocked in 10% bovine serum albumin for 30
min. The cells were incubated with primary antibodies (Table I) overnight, and then with
secondary antibodies (Alexa Fluor 546 donkey anti-rabbit, A10040;
Alexa Fluor 488 donkey anti-mouse, A21202; Thermo Fisher
Scientific) for 1 h, and then with DAPI (Sigma-Aldrich) for 30 min
at room temperature to label the nuclei. Negative controls without
primary antibodies were also included, and these exhibited no
staining. Following the indicated treatments, coverslips were
mounted on glass slides and examined under a fluorescence confocal
microscope (Zeiss LSM 700, Carl Zeiss, Oberkochen, Germany).
 | Table IAntibodies and dilutions used for
immunofluorescence staining. |
Table I
Antibodies and dilutions used for
immunofluorescence staining.
| Antibody: Dilution
(Cat. no., provider) | Antibody: Dilution
(Cat. no., provider) |
|---|
| Pan-cytokeratin
(CK): 1:50 (MA5-13203, Invitrogen, Thermo Fisher Scientific) | E-cadherin: 1:50
(ab76055, Abcam) |
| Nanog: 1:50 (3580,
Cell Signaling Technology) | N-cadherin: 1:50
(ab76057, Abcam) |
| Sox2: 1:50 (4900,
Cell Signaling Technology) | Snail: 1:150
(ab180714, Abcam) |
| Oct4: 1:50 (2840,
Cell Signaling Technology) | LGR5: 1:50
(sc135238, Santa Cruz Biotechnology) |
| Vimentin: 1:200
(5741, Cell Signaling Technology) | ALDH1: 1:50
(ab24343, Abcam) |
| CD44: 1:500 (5640,
Cell Signaling Technology) | CD133: 1:50
(ab16518, Abcam) |
| β-catenin: 1:100
(9581, Cell Signaling Technology) | CD44v6: 1:50
(BBA13, R&D Systems) |
Results
CRC cells that have undergone EMT exhibit
nuclear N-cadherin, CD133 and Cd44v6 localisation
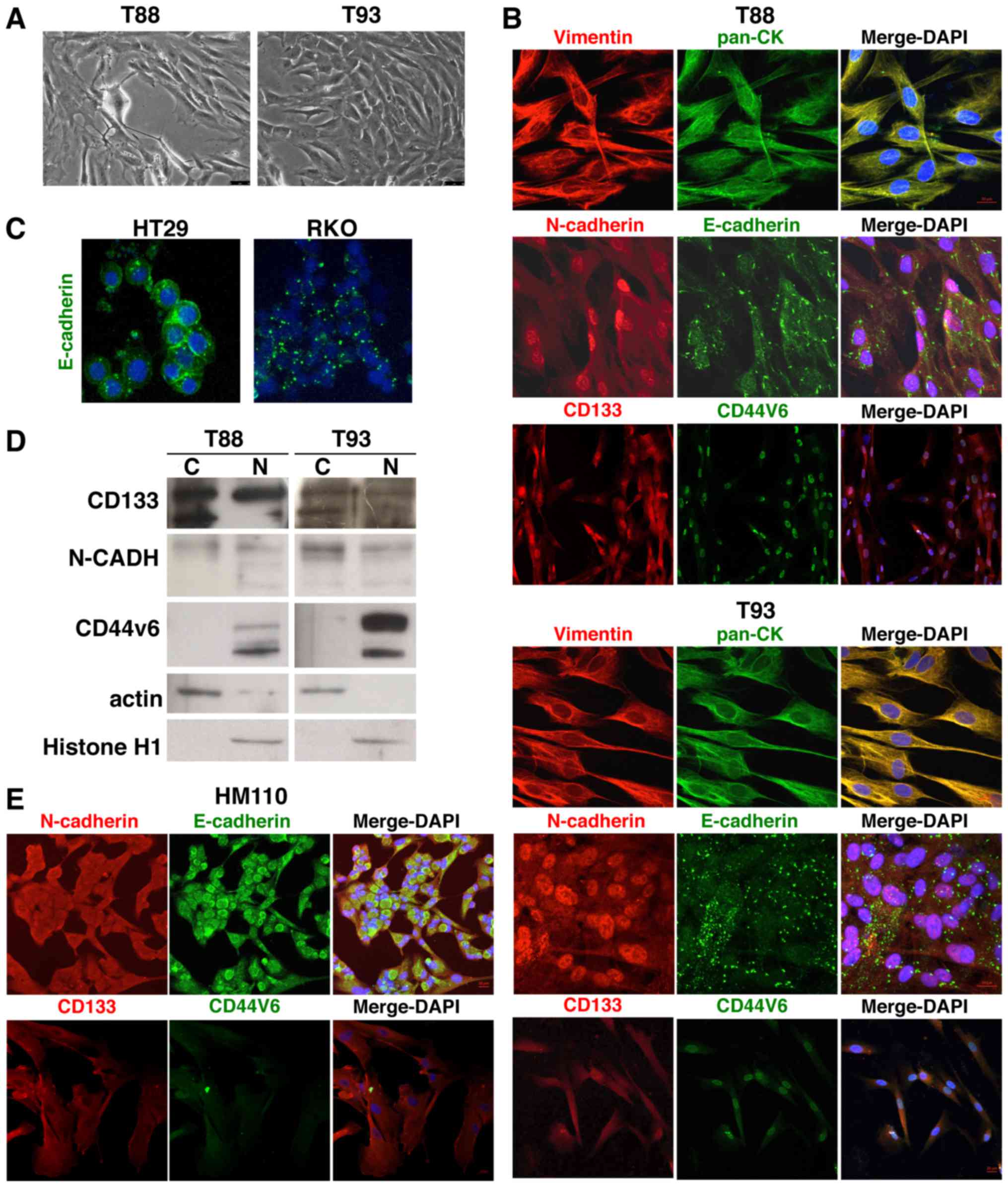
We first confirmed, by immunofluorescence analyses,
that both the T88 and T93 cells (Fig.
1A) expressed epithelial and mesenchymal proteins, in
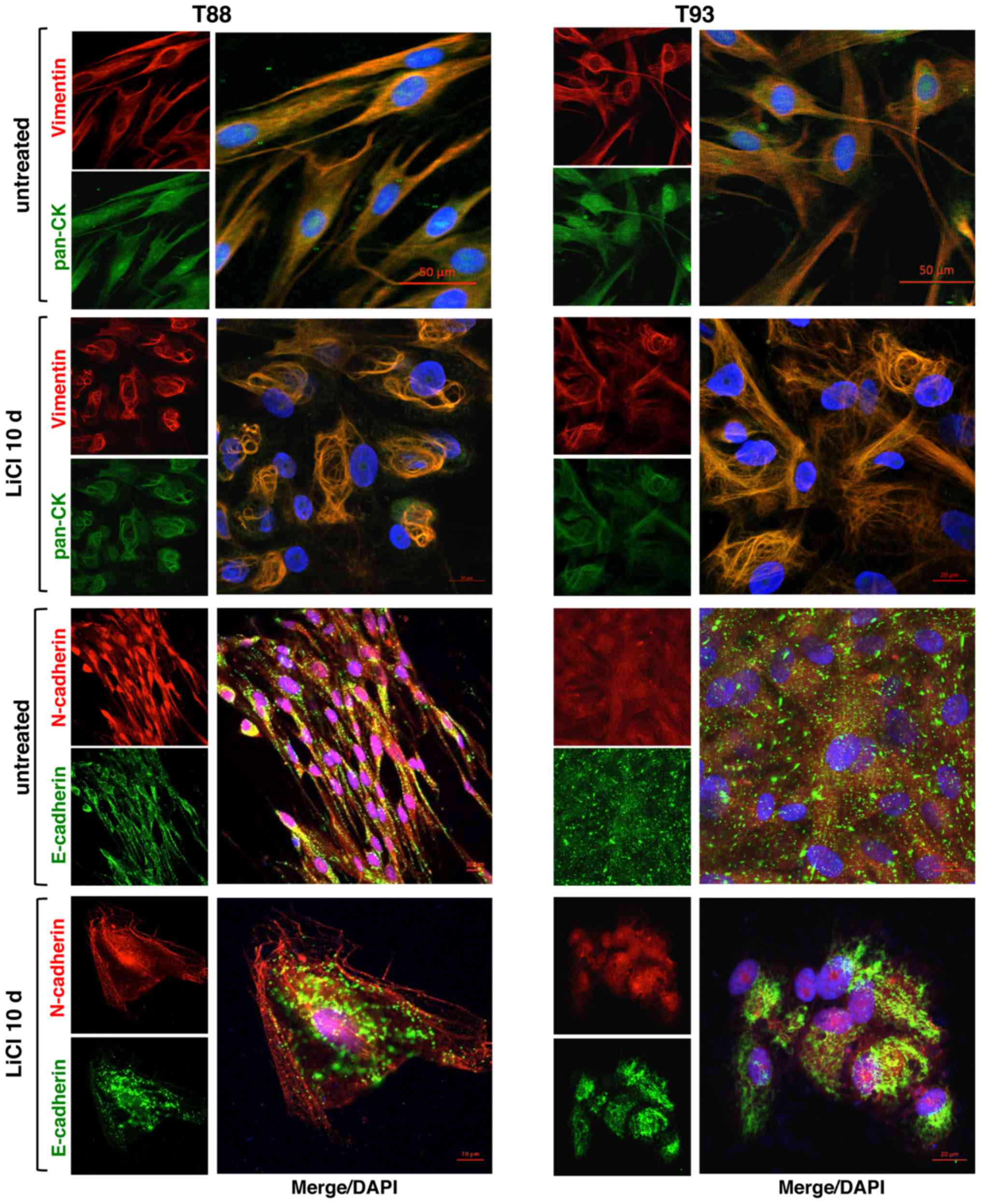
accordance with our previous data (24). As shown in Fig. 1B, Vimentin and Cytokeratin
co-localized at the cytoskeleton level in both cell lines.
E-cadherin did not exhibit the classic membrane staining of
epithelial cells, such as in commercial HT29 CRC cells (Fig. 1C), but rather a diffuse punctate
signal, as observed in the commercial RKO CRC cells, was found
(Fig. 1C). Commercially available
CRC cell cultures are largely characterised; thus, it is well known
that HT29 cells have a more epithelial phenotype than RKO cells,
which primarily exhibit mesenchymal features (32). N-cadherin exhibited an unexpected
nuclear localisation (Fig. 1B). We
also analysed two cell surface glycoproteins involved in stemness
in several types of cancer cells, CD133 and CD44v6. Of note,
similar to N-cadherin, these markers were mainly localised to the
nucleus. As shown in Fig. 1D,
these findings were confirmed by western blot analysis of the
nuclear and cytosolic protein fractions of the T88 and T93
cells.
Furthermore, to evaluate whether the observed
nuclear localisation phenotypes were tumour-specific, we isolated,
cultured and analysed cells from the healthy mucosae of a sporadic
CRC patient (HM110). Through immunofluorescence staining, we
demonstrated that these cultures also expressed both epithelial and
mesenchymal proteins (N- and E-cadherin, respectively), but found
that they did not exhibit the same nuclear localisation of
N-cadherin, CD133 and CD44v6, as was observed in the tumour cells
(Fig. 1E). This finding strongly
suggests a tumour-specific nature of this feature.
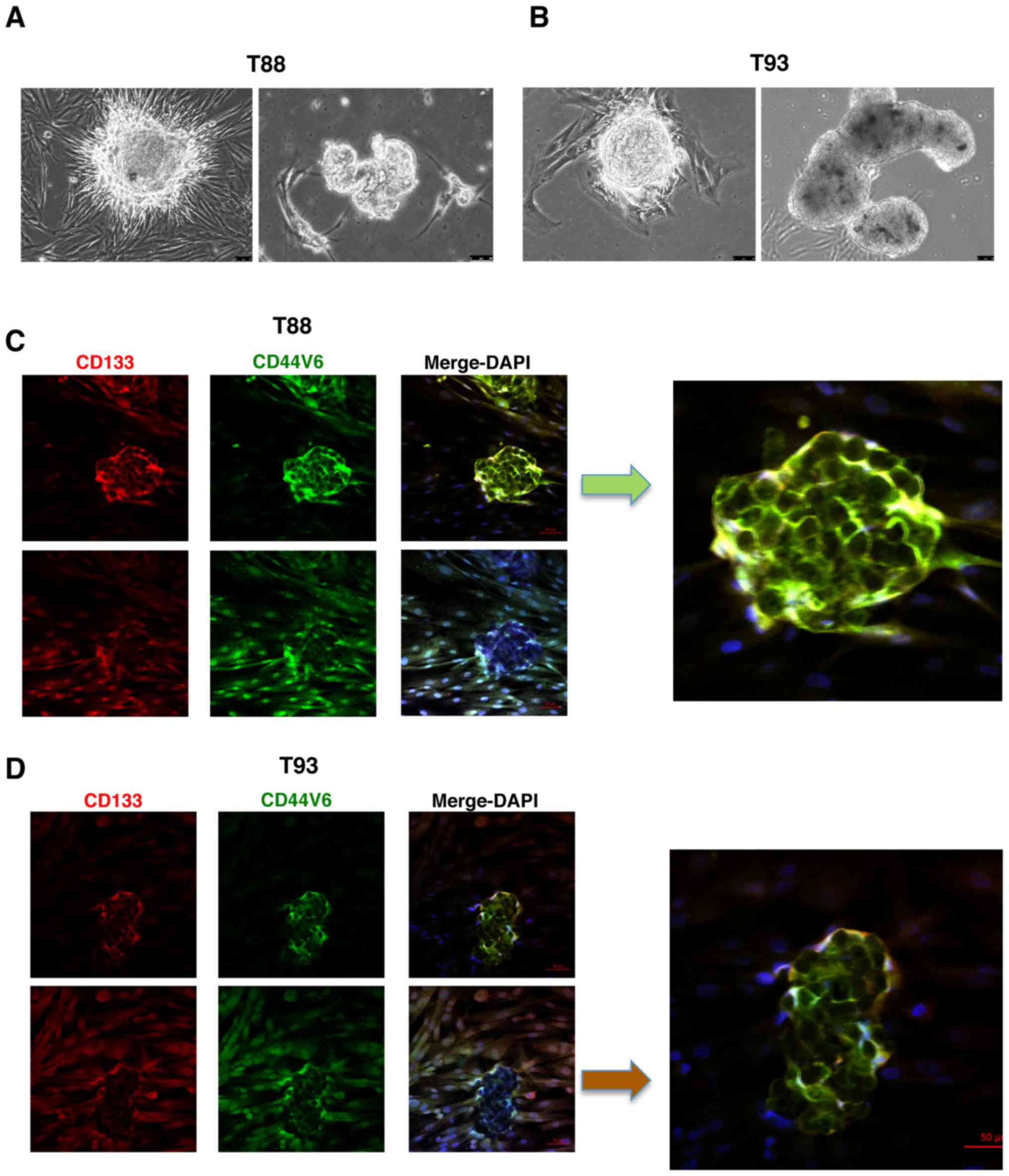
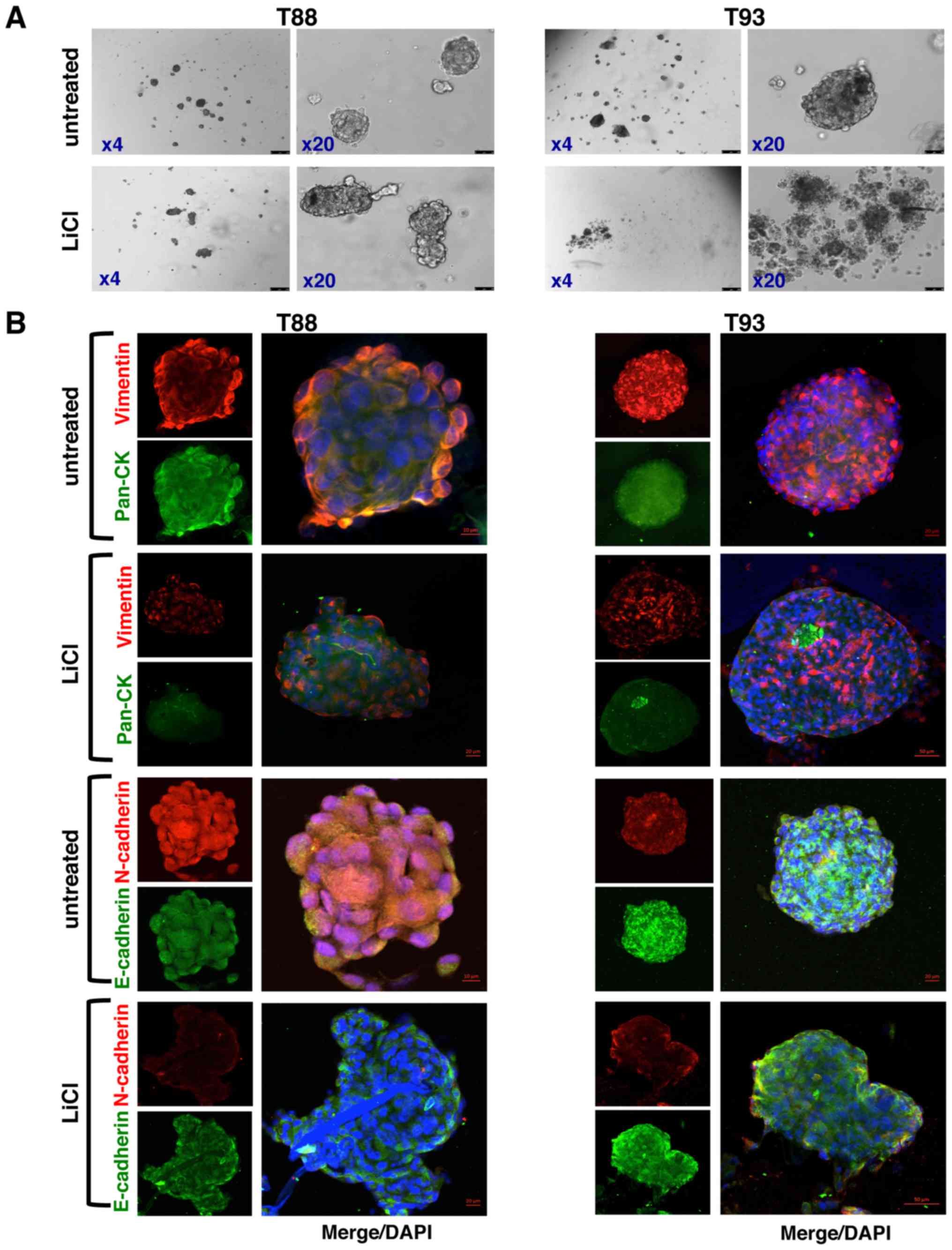
It is known that primary colon cultures are able to
generate in vitro organoids, which are propagated by intestinal
stem cells, and thus, represent an organotypic culture system.
Furthermore, it has been demonstrated that it is possible to
generate intestinal organoids starting from a single sorted
leucine-rich repeat-containing G-protein coupled receptor 5
(LGR5)-positive intestinal stem cell from dissociated intestinal
crypts (33,34). As shown in Fig. 2A and B, the T88 and T93 cells, all
LGR5-positive (please also see Fig.
6), generated organoid-like in vitro three-dimensional
bodies. Immunofluorescence staining revealed that cells of these
three-dimensional bodies expressed both CD133 and CD44v6 mainly at
the plasma membrane (Fig. 2C and
D; images on top panels), contrary to their mesenchymal
counterparts in monolayer, which retained nuclear localisation
(Fig. 2C and D; images on bottom
panels).
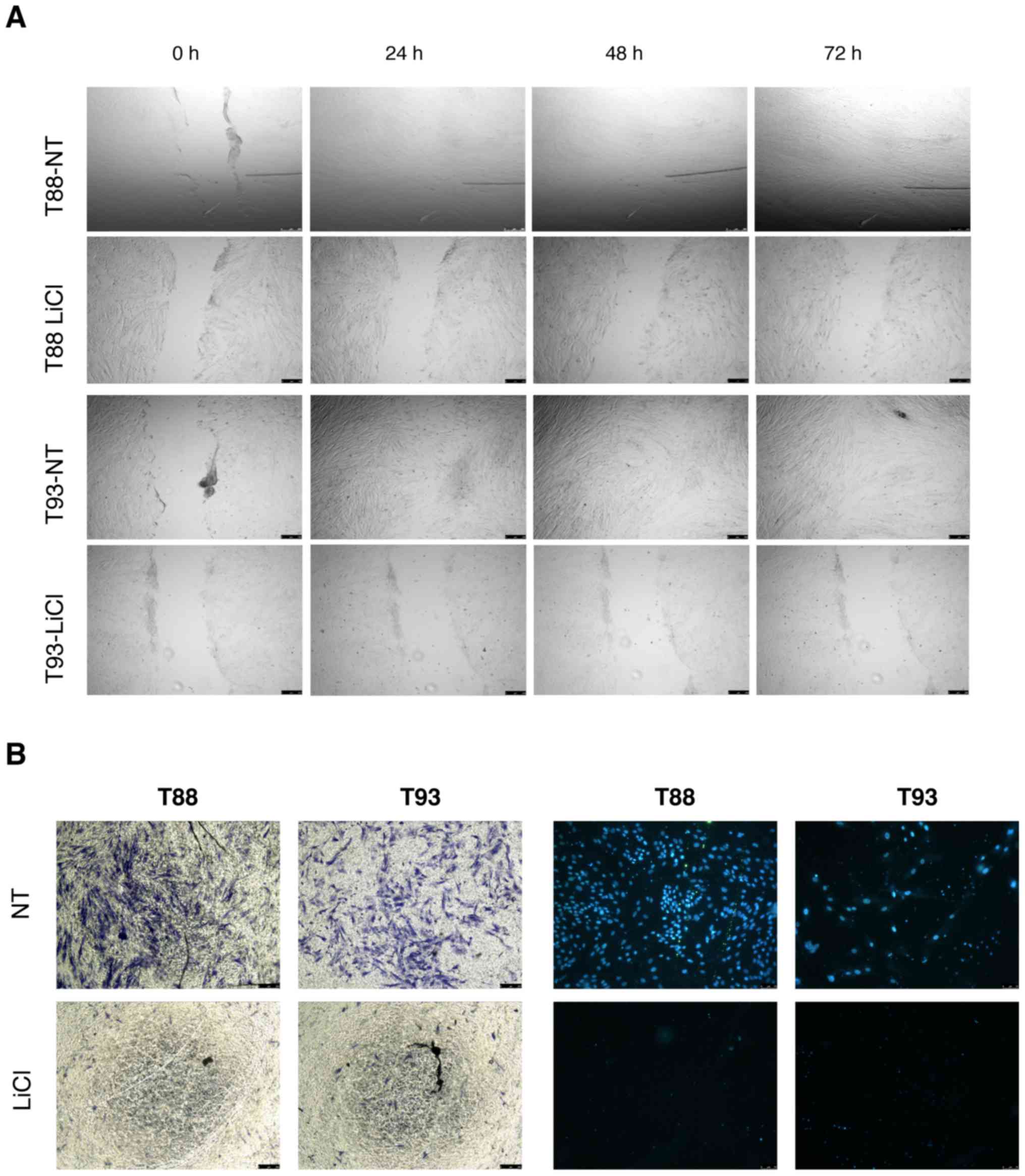
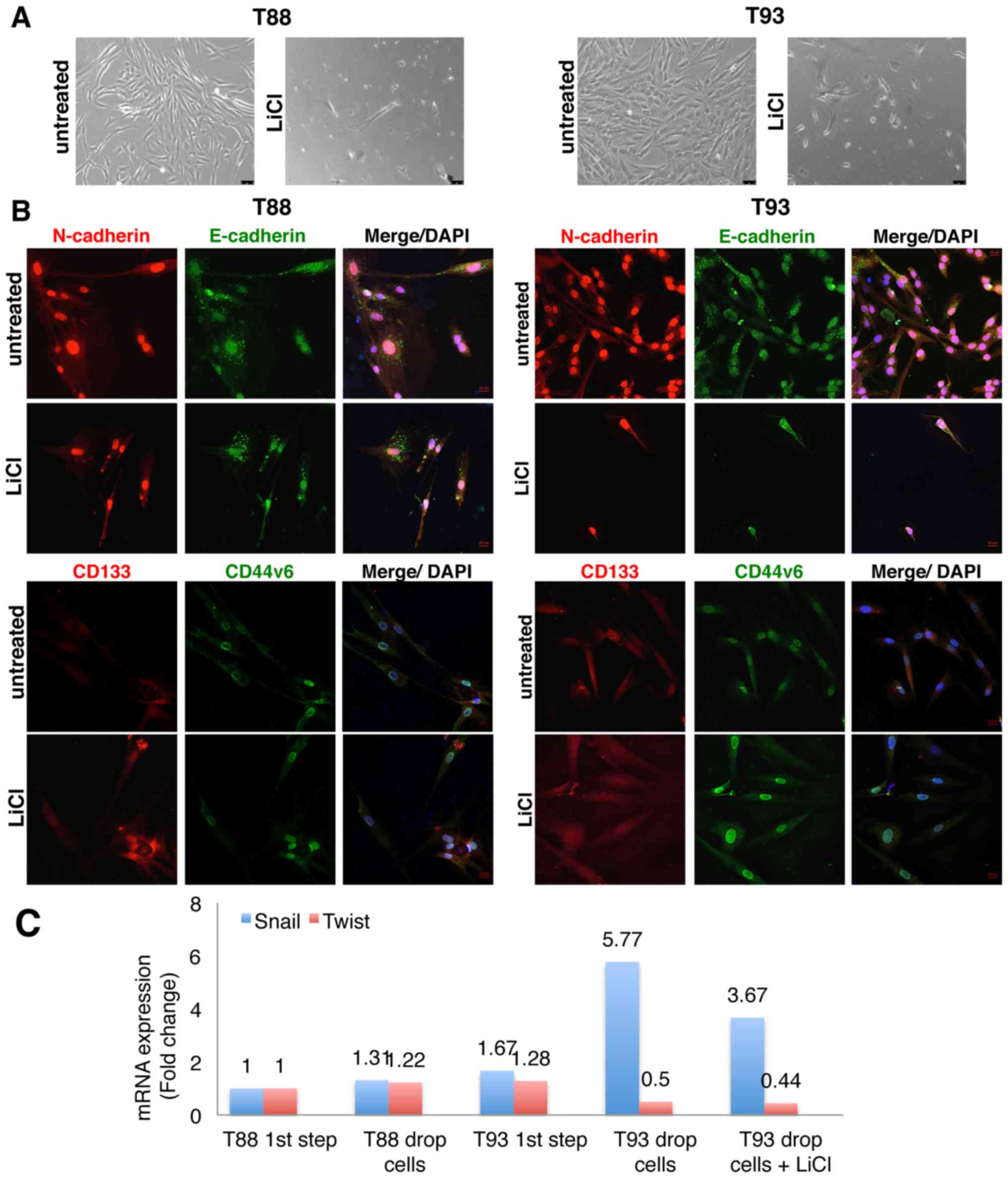
Incubation with LiCl reduces T88 and T93
cell migration
As GSK-3β is overexpressed and functions as an
oncogene in CRC, we examined the effects of GSK-3β inhibition on
tumour cells using LiCl, a specific inhibitor. We found that LiCl
treatment inhibited cell migration using wound healing and
Transwell migration assays. As shown in Fig. 3A, the wounds were almost completely
healed after 24 h in the untreated cells, while the cells incubated
with LiCl had wound sizes similar to time 0 after 72 h. We
confirmed this observation using Transwell migration assays; no
cell migration was detected when the cells were incubated with LiCl
using crystal violet or DAPI staining (Fig. 3B).
LiCl incubation affects the stemness
features of CRC cells that have undergone EMT
We confirmed our previous finding showing that LiCl
induces MET in T88 and T93 cell cultures (24). Indeed, by confocal microscopy, we
confirmed the finding of E-cadherin upregulation described in our
previous study (24). We assessed
the expression and localization of the epithelial markers,
E-cadherin and pan-cytokeratin (Ck), and the expression of the
mesenchymal markers, Vimentin and N-cadherin, in untreated and
LiCl-treated cells. As shown in Fig.
4, Vimentin and Cytokeratin co-localised in both untreated and
treated cells, the latter exhibiting a different cytoskeletal
organization. Following LiCl treatment, E-cadherin maintained a
discontinuous punctate pattern, although the treated cells
exhibited a differentiated, more polarized shape than the untreated
cells. Furthermore, following LiCl incubation, a nucleolar
localization of N-cadherin appeared visible, whereas it was not
detected in the untreated cells.
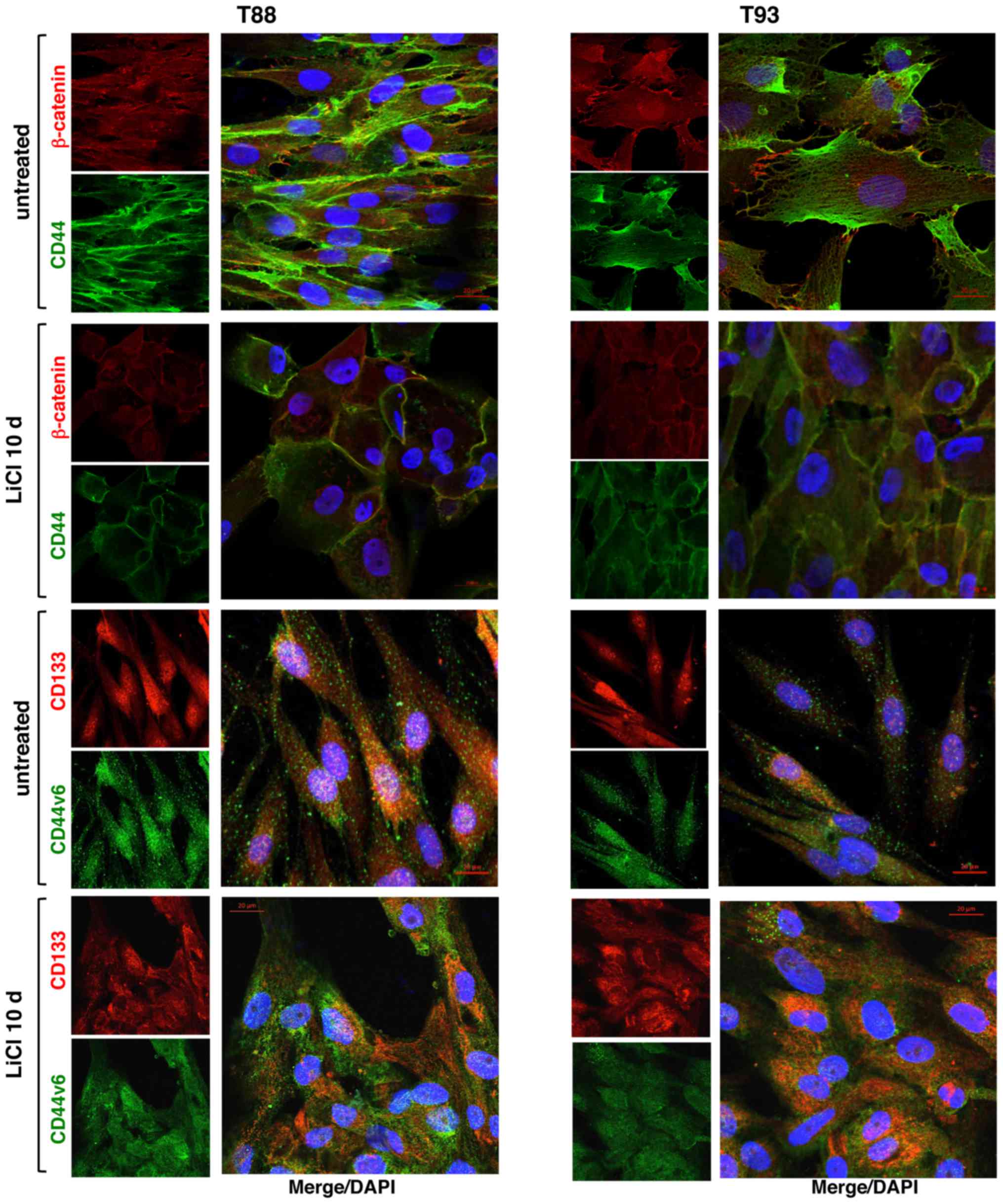
Thereafter, we evaluated in both the untreated cells
and cells incubated with LiCl, the expression of CD44, its splice
isoform CD44v6, CD133 and β-catenin, all proteins playing pivotal
roles in CRC development and progression. In accordance with our
previous data (24), following 10
days of LiCl treatment, CD44 and β-catenin expression decreased and
they both localised mainly at the plasma membrane, while CD44v6 and
CD133 proteins both exhibited cytosolic localisation (Fig. 5).
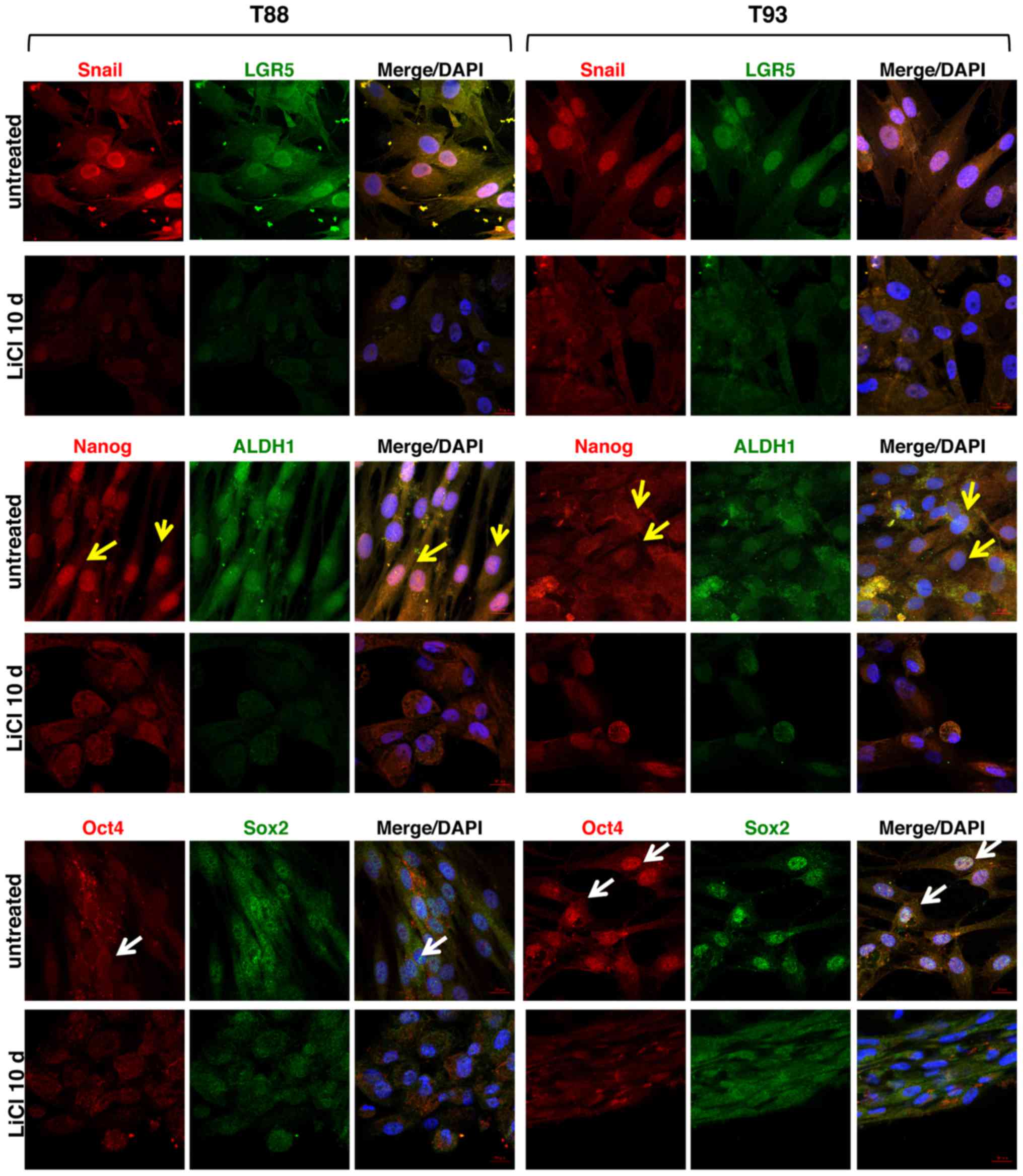
We also analysed the expression of the
EMT-associated transcription factor, Snail, and the expression of
several stem cell-specific markers, including LGR5, Nanog, aldehyde
dehydrogenase 1 (ALDH1), octamer-binding transcription factor 4
(Oct4) and sex determining region Y-box 2 (Sox2), in addition to
the aforementioned CD133, CD44 and CD44v6, following LiCl
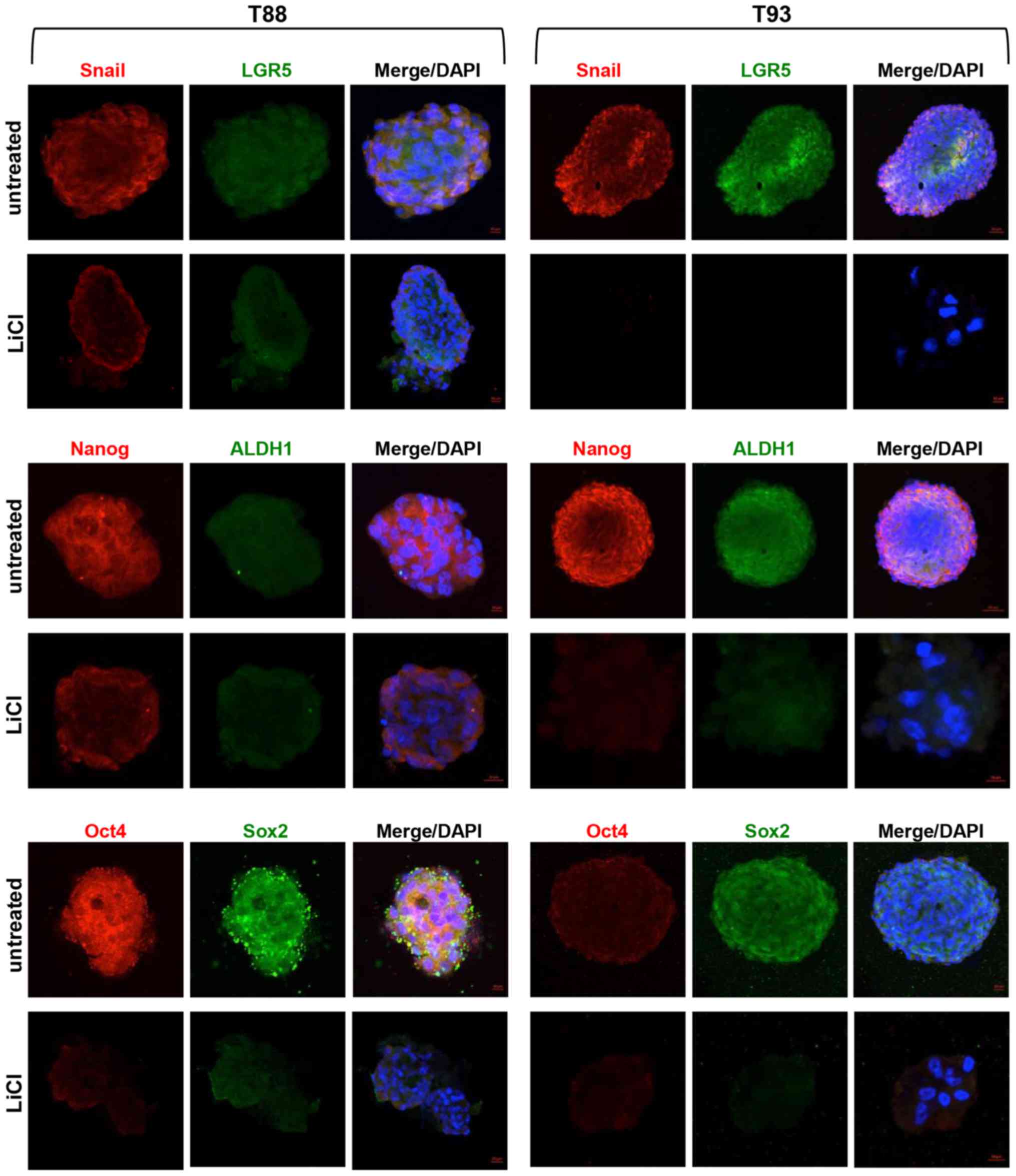
treatment. As shown in Fig. 6, we
observed that LiCl induced a downregulation in the expression of
the EMT-associated transcription factor, Snail, and all stemness
markers (LGR5, ALDH1, Nanog, Oct4 and Sox2), which in the untreated
cells exhibited nuclear localisation. Furthermore, only in the
untreated cells, Nanog exhibited a higher nuclear expression in the
T88 cells than in the T93 cells (yellow arrows), while Oct4
exhibited an opposite trend (white arrows).
Furthermore, we generated tumourspheres from the T88
and T93 cells by the hanging drop assay, thus establishing a system
of adherent primary mesenchymal colon cancer cells and paired
tumourspheres, which are useful for studying stemness features,
cell plasticity and drug responses. It has been reported that only
stem cells and/or stem cell-like cells are able to survive and grow
in suspension (35), as the loss
of adhesion induces death through anoikis in non-malignant and
cancer differentiated cells. Under these culture conditions,
undifferentiated tumour cells proliferate and grow as floating
clusters termed tumourspheres. As shown in Fig. 7A, the T88 and T93 cells formed
spheroid aggregates in hanging drop assays, indicating the
acquisition of a dedifferentiated state. However, following 72 h of
incubation with LiCl, the T88 cells lost their spherical shape,
consistent with a differentiated state, and the T93 cells
completely lost their ability to form spheres, generating only
cellular aggregates, in agreement with the more epithelial
phenotype of T93 cells compared with the T88 cells (24).
When analysed by confocal microscopy, both the
epithelial (Cks and E-cadherin) and mesenchymal (Vimentin and
N-cadherin) markers were expressed in the untreated tumourspheres
(Fig. 7B). However, these spheres
exhibited an upregulation of epithelial markers and a
downregulation of mesenchymal ones, together with the retention of
the expression of many stemness markers, thus suggesting a
switching from a mesenchymal stem-cell-like phenotype to a more
epithelial stem-cell-like phenotype, compared with the adherent T88
and T93 cells (Fig. 1). Moreover,
in the untreated tumourspheres, N-cadherin did not localise to the
nucleus (Fig. 7B), as observed in
the adherent cells (Fig. 1),
exhibiting a prevalent cytoplasmic localization. In addition,
N-cadherin was downregulated in the untreated T93 tumourspheres,
while E-cadherin was upregulated in both the untreated and treated
T88 and T93 tumourspheres (Fig.
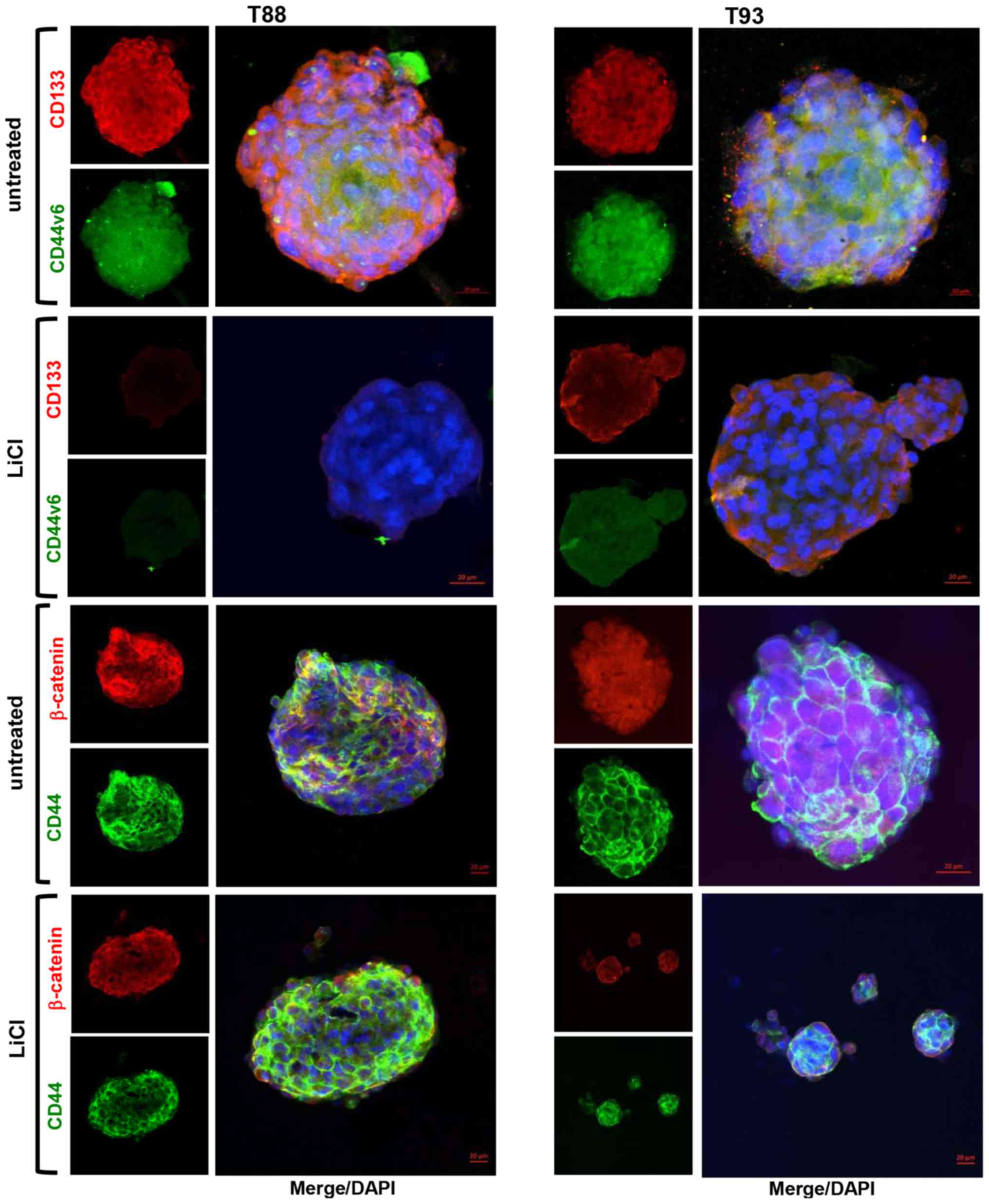
7B) compared with the adherent cells (Fig. 1). The untreated tumourspheres also
exhibited CD133, CD44v6, CD44 and β-catenin expression; the last
two were mainly expressed at the plasma membrane (Fig. 8). Furthermore, as shown in Fig. 9, the cells in the untreated cancer
spheroids retained the expression of mesen-chymal and stemness
biomarkers, exhibiting a lower nuclear Snail expression compared
with the adherent T88 and T93 cells (Fig. 6). ALDH1 expression was very low in
the T88 cancer spheroids (Fig. 9)
compared with both its paired adherent cells (Fig. 6) and T93 tumourspheres (Fig. 9). The T93 tumourspheres, exhibited
a very low Oct4 expression (Fig.
9) compared with both the paired adherent cells (Fig. 6) and T88 tumourspheres (Fig. 9).
We further investigated the molecular basis of the
GSK-3β-mediated inhibition of the ability of T88 and T93 cells to
generate tumourspheres, by the immunofluorescence analysis of
epithelial, mesenchymal and stemness markers, on spheroids obtained
in medium containing LiCl. In accordance with above-mentioned
findings on adherent cells, we observed a drastic downregulation of
all stemness and mesenchymal markers analysed. As shown in Fig. 7, LiCl induced a down-regulation of
Vimentin and N-cadherin. Moreover, the pan-Ck signal was lower in
the LiCl-treated tumourspheres compared with the untreated
tumourspheres, while E-cadherin (Fig.
7), CD44 and β-catenin (Fig.
8) exhibited a similar expression in the T88 and T93 spheroids
obtained in medium containing or not containing LiCl. In accordance
with the effect of LiCl on adherent cells, the expression of Snail,
CD133, CD44v6 proteins (Fig. 8)
and of all other stem-cell markers analysed (Fig. 9) strongly decreased in the
tumourspheres following incubation with LiCl.
Incubation with LiCl alters T88 and T93
cell plasticity
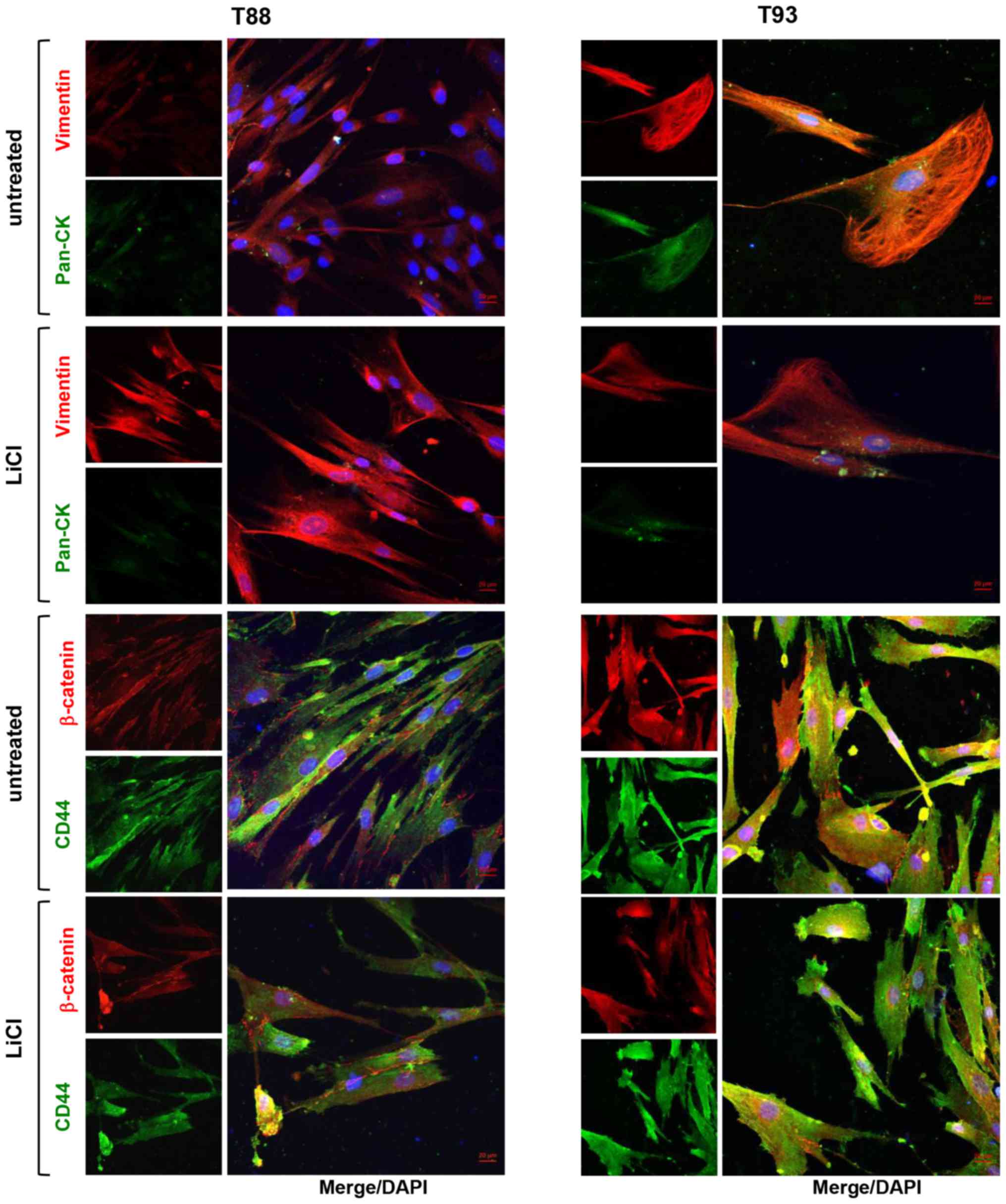
To further investigate the plasticity of the T88 and
T93 cells, we disaggregated spheroids obtained by hanging drop
assays, grown in medium with or without LiCl, (hereinafter referred
to as treated and untreated tumourspheres) and cultivated them in
adhesion, to examine the ability of the tumourspheres to re-adhere
to a matrix and grow in adherent culture. As shown in Fig. 10A, these cells were able to adhere
to the cell culture dishes and assume a mesenchymal phenotype.
However, the numbers of surviving cells were significantly lower
when they were derived from the treated spheroids compared to the
cells derived from the untreated spheroids. Furthermore, the T93
cells, but not the T88 cells, derived from the treated spheroids,
were able to proliferate and grow in adhesion (data not shown). By
confocal microscopy, we observed that all cells obtained by
disaggregating tumourspheres exhibited a similar phenotype with
regards to the expression of mesenchymal and stemness markers
(Figs. 10B and 11). However, cells derived from the
treated tumourspheres exhibited a major nuclear localization of
stem cells and mesenchymal markers (Fig. 10B) and an upregulation of the
Snail transcription factor was observed only in the T93 cells, when
analysed by RT-qPCR (Fig.
10C).
In both cell cultures, and under both experimental
conditions, we observed the nuclear localisation of E-cadherin,
N-cadherin, CD133 and CD44v6 (Fig.
10B), but not that of the canonical isoform of CD44 or
β-catenin (Fig. 11). When
analysed by RT-qPCR, Twist1 mRNA expression was downregulated in
both cell cultures, while Snail mRNA was upregulated, mainly in the
T93 cells, and even in cells disaggregated from the treated
tumourspheres (Fig. 10C). Indeed,
both cell cultures again expressed the mesenchymal protein,
Vimentin, which was upregulated in the T88 cells derived from the
treated tumourspheres (Fig. 11),
in accordance with our previous observation showing that following
10 days of incubation with LiCl, Vimentin mRNA and protein
expression levels strongly decreased in the T93 cells, while they
increased in the T88 cells (24).
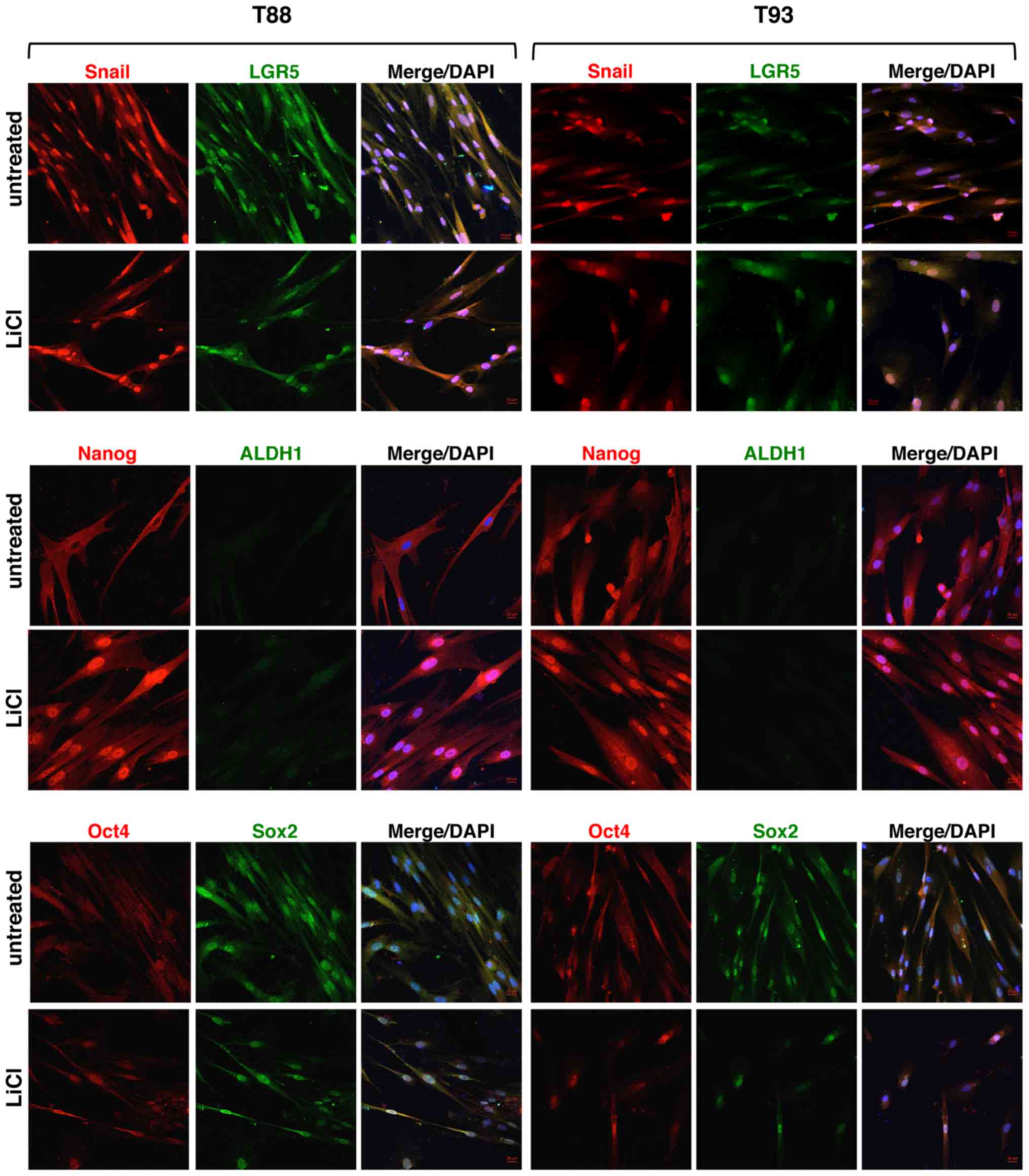
Nuclear localisation was also observed for Snail, LGR5 and Sox2
(Fig. 12). Nanog exhibited
cytoplasmic localisation in the cells derived from the untreated
tumourspheres, particularly in the T88 cells, while a clear nuclear
localisation in the cells derived from the treated tumourspheres
was observed (Fig. 12). Oct4 also
exhibited cytosolic localisation in the T88 cells derived from the
untreated tumourspheres, but moved into the nucleus when the
tumourspheres were treated with LiCl (Fig. 12). By contrast, Oct4 exhibited a
nuclear expression in the T93 cells under both experimental
conditions (Fig. 12). ALDH1 was
undetectable under all the conditions analysed (Fig. 12).
Discussion
EMT and its reverting process, MET, are
physiological processes occurring during embryonic development and
tissue remodelling that confer plasticity to cancer cells. It has
been suggested that EMT and cell plasticity may be responsible for
the acquisition of chemotherapeutic resistance and metastasis
development in several tumours, including CRCs (12).
We previously isolated and characterized at a
molecular level two primary CRC cell cultures from the tumour
tissues of patients 88 and 93 of our bio-bank (the T88 and T93
cultures). As previously described, the T93 cells exhibited a CIN
phenotype, while the T88 cells exhibited a MIN one, with high MSI.
We demonstrated that the T88 and T93 cells were mesenchymal colon
cancer cells that had undergone EMT from epithelial adenocarcinoma
cells and simultaneously expressed epithelial (Cks and E-cadherin)
and mesenchymal (Vimentin and N-cadherin) markers. High levels of
EMT-associated transcription factors (Twist and Snail) and several
stemness markers were also found (24). These finding were in accordance
with previous data indicating that EMT induces the expression of
stem cell-specific genes, and may represent a source of cancer
stem-like cells (36). We also
demonstrated that incubation with LiCl, a specific GSK-3β
inhibitor, induced MET (24).
In this study, we characterised our experimental
system of adherent primary mesenchymal colon cancer cells and their
paired tumourspheres more in depth, by analysing the localisation
and expression of a larger panel of markers, including E- and
N-cadherin, CD133, CD144v6, ALDH1 and LGR5. Furthermore, we
explored the effects of LiCl on cell motility and cell plasticity
of CRC cell cultures.
Thus, we confirmed the epithelial/mesenchymal
features of these cells and demonstrated that they were
characterised by the nuclear localisation of several stemness
markers, including Nanog, Oct4, Sox2, LGR5, ALDH1, CD133 and
CD44v6.
Of note, we observed atypical nuclear N-cadherin,
CD133 and Cd44v6 localisation in mesenchymal CRC cells. N-cadherin
is a crucial protein during EMT, cancer progression and invasion,
and an elevated N-cadherin expression is often associated with a
poor prognosis (37,38); however, its role in cell nuclei
requires further investigation. CD133 is a transmembrane
glycoprotein often expressed on adult stem cells, where it
functions in maintaining stem cell properties by suppressing
differentiation. CD133 expression is also associated with several
types of cancer. It was one of the first molecular biomarkers
associated with cancer stemness, although it later became clear
that it is expressed both in differentiated and undifferentiated
CRC cells (39,40).
Data regarding nuclear CD133 localisation are rarely
described. To the best of our knowledge, there are only three
studies that have reported nuclear CD133 localisation, one in
triple-negative breast cancer (41), one in a small subpopulation of
rhabdomyosarcoma cell lines (ranged from 3.4-7.5% of the total
population) (42), and one in
non-small cell lung cancer (NSCLC). In the latter study, the
authors found the nuclear and cytoplasmic CD133 localisation in 239
NSCLC cases using tissue immunohistochemistry, demonstrating that a
high nuclear and cytoplasmic CD133 expression were associated with
poor prognoses in these carcinomas (43).
CD44 is a plasma membrane glycoprotein that is a
receptor for hyaluronan and many other extracellular matrix
components. Thus, it transduces signals to membrane-associated
cytoskeletal proteins or to the nucleus, to regulate the expression
of genes related to cell-matrix adhesion, cell migration,
proliferation, differentiation and survival. Previous data have
indicated that CD44, particularly CD44v isoforms, are specific CSC
markers. CD44 proteins integrate environmental and cellular
signalling to regulate cancer stemness, they are also involved in
EMT regulation (44) and in the
regulation of reactive oxygen species metabolism in CSCs (45).
Several cell surface proteins are known to migrate
into the nucleus as intact polypeptides or proteolytic fragments,
where they function as transcription factors. It was previously
described that the canonical isoform of CD44 is imported into the
nucleus through the nuclear pore complex, thus promoting cell
proliferation (46). In this
study, we observed, to the best of our knowledge, for the first
time, that the CD44v6 isoform exhibited a mainly nuclear
localisation pattern in mesenchymal CRC cells, while the canonical
form retained a primarily membrane and cytosolic pattern. Of note,
cells of the organoid-like bodies, exhibited mainly a plasma
membrane and cytosolic localisation of CD133 and CD44v6, suggesting
that changes in localisation between the membrane and nucleus of
these specific proteins could allow the identification of cell
plasticity in CRCs, a crucial feature of cancer cells that will
develop into metastatic disease.
GSK-3β is a multitasking serine-threonine kinase
that can function as a pro-apoptotic or anti-apoptotic factor
(47). Usually, GSK-3β
phosphorylates β-catenin, inducing its proteasomal degradation.
When GSK-3β is inactivated, it cannot phosphorylate β-catenin,
which is then stabilized, and translocates into the nucleus, where
it activates target genes that drive cell proliferation. In colon
and pancreatic cancer cells, GSK-3β, which is constitutively
overexpressed, activates a proliferative signal (48,49),
thus acting as an oncogene. In this study, we examined the effects
of LiCl, a specific GSK-3β inhibitor, on our cellular model,
demonstrating that LiCl blocks the migration of T88 and T93 cells,
as it has also been reported for commercially available glioma,
glioblastoma, retinoblastoma and SW480 cell lines (50-53).
We have also observed that LiCl affects stemness
features, abolishing the expression of all mesenchymal and stemness
markers, thus altering the dynamics of tumoursphere formation and
cell plasticity. As previously described (24), we found that following incubation
with LiCl, cells differentiated, lost their symmetrical
(mesenchymal) shape and restored their polarity. In these cells,
β-catenin did not exhibit nuclear accumulation, but rather
co-localised with CD44 canonical protein at the plasma membrane.
Conversely, E-cadherin maintained a discontinuous punctate pattern
at the plasma membrane in cell contact areas, denoting that cells
continued to establish minimal contacts with their neighbours.
Taken together, these observations suggested an incomplete
epithelial differentiation of the LiCl-treated cells. Furthermore,
following incubation with LiCl, nucleolar N-cadherin localisation
was observed, whereas this was not observed in the untreated cells.
This intriguing finding supports the fascinating hypothesis of a
transcriptional inhibition mechanism mediated by the nucleolar
localisation of proteins (54);
nevertheless, the role of nucleolar localization of several protein
in cancer needs to be further elucidated.
Cellular plasticity plays an important role in
cancer development and progression; only cancer cells that are able
to switch from epithelial to mesenchymal phenotypes and vice
versa have metastasis-initiating potential, while disseminated
cancer cells that have undergone EMT, but lose their plasticity are
ineffective at seeding metastatic colonies (19). In light of our observations, we
suggest that during cell plasticity, a mesenchymal cell phenotype
could be characterised by the nuclear localisation of several
protein, some of which are usually expressed at the membrane in the
epithelial phenotype. Thus, changes in localisation between
membrane and nucleus of such proteins (CD133 and CD44v6), could
allow and characterize cell plasticity in colorectal cancer
progression. By analysing the molecular features of cells able to
generate tumourspheres, we identified a panel of biomarkers
(including E- and N-cadherin, CD133, CD44v6, Snail, Oct4, Sox2,
Nanog and LGR5) expressed by the most resistant subpopulation of
cancer cells with a mainly nuclear localisation. We suggest that
these proteins could represent a prognostic and/or predictive
biomarker panel that needs to be validated for supporting CRC
care.
Finally, we observed that when cells were
disaggregated from tumourspheres performed in LiCl, only the T93
cells, which exhibited a microsatellite stable (MSS) and CIN
phenotype, were able to re-grow, while the T88 cells, which
exhibited an MSI-high phenotype, appeared quiescent. This
observation is in agreement with findings in the literature, which
indicates a less aggressive phenotype of MSI CRC compared with MSS
subtypes (1,23). Since the T93 cells exhibited the
upregulation of Snail, even if tumourspheres were performed
following incubation with LiCl, a pivotal role for this protein in
the aggressiveness and drug resistance of CRC cells is suggested.
Although this hypothesis warrants further investigation, first of
all enlarging the number of cultures analysed and supporting the
finding with a statistical analysis, it has been demonstrated that
Snail knockdown results in an upregulation of the Raf kinase
inhibitor protein (RKIP), increased apoptosis, MET and reduction of
CSC markers, all of which contribute to decreased chemoresistance
(55).
In conclusion, in this study, we established a
system of adherent primary mesenchymal colon cancer cells and
paired tumourspheres, which are useful for studying the mechanisms
underlying CRC progression and drug response. In light of our
finding, we suggest that LiCl, a specific GSK-3β inhibitor, could
represent a drug candidate and that GSK-3β inhibition may be a
promising direction for future cancer therapy that needs to be
better elucidated. As recently demonstrated for other cancer types,
such as glioma (51), LiCl, a drug
already used in clinical practice for the treatment of bipolar
disorders, could represent an alternative therapy in colon cancer
care and/or able to sensitize cancer cells to chemo-radio-therapy.
Our findings indicate that it could act through the downregulation
of EMT and stem cell biomarkers, thus inhibiting crucial cancer
cell features, such as motility and plasticity. We also
demonstrated that the observed atypical nuclear N-cadherin, CD133
and Cd44v6 localisation in mesenchymal CRC cells is a specific
cancer feature, which may play a role during tumour progression. In
our humble opinion, this finding may open new perspectives to
clarify the molecular basis of EMT in cancer cells.
Abbreviations:
|
EMT
|
epithelial-to-mesenchymal
transition
|
|
MET
|
mesenchymal-to-epithelial
transition
|
|
CRC
|
colorectal cancer
|
|
CIN
|
chromosomal instability
|
|
MIN
|
microsatellite instability
|
|
HM
|
healthy colon mucosa
|
Acknowledgments
The authors would like to thank Dr James P. Mahaffey
from Edanz Group (www.edanzediting.com/ac) for editing a draft of this
manuscript.
Funding
This study was supported by: Regione Campania,
LR5/2002-2014; 'Fondo Straordinario di Ateneo-2017-Università di
Napoli Federico II'.
Availability of data and materials
All data generated or analysed during this study are
included in this published article. No datasets were generated or
analysed during the current study.
Authors' contributions
MDR and PI designed the study; MDR, MT, VC and AC,
performed research; PD, DR, UP, CAD and MM provided sample
collection and clinical support; MDR, PI and MT contributed to data
interpretation. MDR and MT wrote the manuscript, and FD and RL
critically revised the manuscript and participated in the analysis
and interpretation of the data. All authors reviewed, edited and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Samples from all subjects who participated in this
study were collected after obtaining authorisation from the
Comitato etico per le attività Biomediche - Carlo Romano of the
University of Naples Federico II (protocol no. 432/17).
Authorisation was granted only once the study had received ethical
approval and written informed consent had been obtained from all
participants. All methods were performed in accordance with the
relevant guidelines and regulations.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
De Rosa M, Rega D, Costabile V, Duraturo
F, Niglio A, Izzo P, Pace U and Delrio P: The biological complexity
of colorectal cancer: Insights into biomarkers for early detection
and personalized care. Therap Adv Gastroenterol. 9:861–886. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
3
|
Peifer M: Developmental biology: Colon
construction. Nature. 420:274–275. 2772002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kosinski C, Li VS, Chan AS, Zhang J, Ho C,
Tsui WY, Chan TL, Mifflin RC, Powell DW, Yuen ST, et al: Gene
expression patterns of human colon tops and basal crypts and BMP
antagonists as intestinal stem cell niche factors. Proc Natl Acad
Sci USA. 104:15418–15423. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
De Rosa M, Pace U, Rega D, Costabile V,
Duraturo F, Izzo P and Delrio P: Genetics, diagnosis and management
of colorectal cancer (Review). Oncol Rep. 34:1087–1096. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gangemi R, Paleari L, Orengo AM, Cesario
A, Chessa L, Ferrini S and Russo P: Cancer stem cells: A new
paradigm for understanding tumor growth and progression and drug
resistance. Curr Med Chem. 16:1688–1703. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fanali C, Lucchetti D, Farina M, Corbi M,
Cufino V, Cittadini A and Sgambato A: Cancer stem cells in
colorectal cancer from pathogenesis to therapy: Controversies and
perspectives. World J Gastroenterol. 20:923–942. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
O'Brien CA, Pollett A, Gallinger S and
Dick JE: A human colon cancer cell capable of initiating tumour
growth in immunodeficient mice. Nature. 445:106–110. 2007.
View Article : Google Scholar
|
|
9
|
Greaves M and Maley CC: Clonal evolution
in cancer. Nature. 481:306–313. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cano A, Pérez-Moreno MA, Rodrigo I,
Locascio A, Blanco MJ, del Barrio MG, Portillo F and Nieto MA: The
transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li X, Pei D and Zheng H: Transitions
between epithelial and mesenchymal states during cell fate
conversions. Protein Cell. 5:580–591. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Loboda A, Nebozhyn MV, Watters JW, Buser
CA, Shaw PM, Huang PS, Van't Veer L, Tollenaar RA, Jackson DB,
Agrawal D, et al: EMT is the dominant program in human colon
cancer. BMC Med Genomics. 4:92011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dallas NA, Xia L, Fan F, Gray MJ, Gaur P,
van Buren G II, Samuel S, Kim MP, Lim SJ and Ellis LM:
Chemoresistant colorectal cancer cells, the cancer stem cell
phenotype, and increased sensitivity to insulin-like growth
factor-I receptor inhibition. Cancer Res. 69:1951–1957. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fan F, Samuel S, Evans KW, Lu J, Xia L,
Zhou Y, Sceusi E, Tozzi F, Ye XC, Mani SA, et al: Overexpression of
snail induces epithelial-mesenchymal transition and a cancer stem
cell-like phenotype in human colorectal cancer cells. Cancer Med.
1:5–16. 2012. View
Article : Google Scholar
|
|
15
|
Vichalkovski A, Gresko E, Hess D,
Restuccia DF and Hemmings BA: PKB/AKT phosphorylation of the
transcription factor Twist-1 at Ser42 inhibits p53 activity in
response to DNA damage. Oncogene. 29:3554–3565. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang Y, Pan X, Lei W, Wang J and Song J:
Transforming growth factor-beta1 induces epithelial-to-mesenchymal
transition and apoptosis via a cell cycle-dependent mechanism.
Oncogene. 25:7235–7244. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vega S, Morales AV, Ocaña OH, Valdés F,
Fabregat I and Nieto MA: Snail blocks the cell cycle and confers
resistance to cell death. Genes Dev. 18:1131–1143. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ong BA, Vega KJ and Houchen CW: Intestinal
stem cells and the colorectal cancer microenvironment. World J
Gastroenterol. 20:1898–1909. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Doherty MR, Smigiel JM, Junk DJ and
Jackson MW: Cancer stem cell plasticity drives therapeutic
resistance. Cancers (Basel). 8. pp. E82016, View Article : Google Scholar
|
|
20
|
Chaffer CL, San Juan BP, Lim E and
Weinberg RA: EMT, cell plasticity and metastasis. Cancer Metastasis
Rev. 35:645–654. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Grassilli E, Narloch R, Federzoni E,
Ianzano L, Pisano F, Giovannoni R, Romano G, Masiero L, Leone BE,
Bonin S, et al: Inhibition of GSK3B bypass drug resistance of
p53-null colon carcinomas by enabling necroptosis in response to
chemotherapy. Clin Cancer Res. 19:3820–3831. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
McCubrey JA, Steelman LS, Bertrand FE,
Davis NM, Sokolosky M, Abrams SL, Montalto G, D'Assoro AB, Libra M,
Nicoletti F, et al: GSK-3 as potential target for therapeutic
intervention in cancer. Oncotarget. 5:2881–2911. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liccardo R, De Rosa M, Izzo P and Duraturo
F: Novel Implications in Molecular Diagnosis of Lynch Syndrome.
Gastroenterol Res Pract. 2017:25950982017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Costabile V, Duraturo F, Delrio P, Rega D,
Pace U, Liccardo R, Rossi GB, Genesio R, Nitsch L, Izzo P, et al:
Lithium chloride induces mesenchymal-to-epithelial reverting
transition in primary colon cancer cell cultures. Int J Oncol.
46:1913–1923. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liang CC, Park AY and Guan JL: In vitro
scratch assay: A convenient and inexpensive method for analysis of
cell migration in vitro. Nat Protoc. 2:329–333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Paparo L, Rossi GB, Delrio P, Rega D,
Duraturo F, Liccardo R, Debellis M, Izzo P and De Rosa M:
Differential expression of PTEN gene correlates with phenotypic
heterogeneity in three cases of patients showing clinical
manifestations of PTEN hamartoma tumour syndrome. Hered Cancer Clin
Pract. 11:82013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Duraturo F, Liccardo R, Cavallo A, De Rosa
M, Rossi GB and Izzo P: Multivariate analysis as a method for
evaluating the pathogenicity of novel genetic MLH1 variants in
patients with colorectal cancer and microsatellite instability. Int
J Mol Med. 511–517. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Δ Δ C(T)) Method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
29
|
Galatola M, Paparo L, Duraturo F, Turano
M, Rossi GB, Izzo P and De Rosa M: Beta catenin and cytokine
pathway dysregu-lation in patients with manifestations of the 'PTEN
hamartoma tumor syndrome'. BMC Med Genet. 13:282012. View Article : Google Scholar
|
|
30
|
Angrisani A, Turano M, Paparo L, Di Mauro
C and Furia M: A new human dyskerin isoform with cytoplasmic
localization. Biochim Biophys Acta. 1810:1361–1368. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Di Maio N, Vicidomini R, Angrisani A,
Belli V, Furia M and Turano M: A new role for human dyskerin in
vesicular trafficking. FEBS Open Bio. 7:1453–1468. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Feng B, Dong TT, Wang LL, Zhou HM, Zhao
HC, Dong F and Zheng MH: Colorectal cancer migration and invasion
initiated by microRNA-106a. PLoS One. 7:e434522012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sato T, Stange DE, Ferrante M, Vries RG,
Van Es JH, Van den Brink S, Van Houdt WJ, Pronk A, Van Gorp J,
Siersema PD, et al: Long-term expansion of epithelial organoids
from human colon, adenoma, adenocarcinoma, and Barrett's
epithelium. Gastroenterology. 141:1762–1772. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sato T, Vries RG, Snippert HJ, van de
Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters
PJ, et al: Single Lgr5 stem cells build crypt-villus structures in
vitro without a mesenchymal niche. Nature. 459:262–265. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Weiswald LB, Bellet D and Dangles-Marie V:
Spherical cancer models in tumor biology. Neoplasia. 17:1–15. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Reya T, Morrison SJ, Clarke MF and
Weissman IL: Stem cells, cancer, and cancer stem cells. Nature.
414:105–111. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yilmaz M and Christofori G: Mechanisms of
motility in metastasizing cells. Mol Cancer Res. 8:629–642. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
DI Domenico M, Pierantoni GM, Feola A,
Esposito F, Laino L, DE Rosa A, Rullo R, Mazzotta M, Martano M,
Sanguedolce F, et al: Prognostic significance of N-Cadherin
expression in oral squamous cell carcinoma. Anticancer Res.
31:4211–4218. 2011.PubMed/NCBI
|
|
39
|
LaBarge MA and Bissell MJ: Is CD133 a
marker of metastatic colon cancer stem cells? J Clin Invest.
118:2021–2024. 2008.PubMed/NCBI
|
|
40
|
Shmelkov SV, Butler JM, Hooper AT, Hormigo
A, Kushner J, Milde T, St Clair R, Baljevic M, White I, Jin DK, et
al: CD133 expression is not restricted to stem cells, and both
CD133+ and CD133- metastatic colon cancer cells initiate
tumors. J Clin Invest. 118:2111–2120. 2008.PubMed/NCBI
|
|
41
|
Cantile M, Collina F, D'Aiuto M, Rinaldo
M, Pirozzi G, Borsellino C, Franco R, Botti G and Di Bonito M:
Nuclear localization of cancer stem cell marker CD133 in
triple-negative breast cancer: A case report. Tumori. 99:e245–e250.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nunukova A, Neradil J, Skoda J, Jaros J,
Hampl A, Sterba J and Veselska R: Atypical nuclear localization of
CD133 plasma membrane glycoprotein in rhabdomyosarcoma cell lines.
Int J Mol Med. 36:65–72. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Huang M, Zhu H, Feng J, Ni S and Huang J:
High CD133 expression in the nucleus and cytoplasm predicts poor
prognosis in non-small cell lung cancer. Dis Markers.
2015:9860952015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bessède E, Staedel C, Acuña Amador LA,
Nguyen PH, Chambonnier L, Hatakeyama M, Belleannée G, Mégraud F and
Varon C: Helicobacter pylori generates cells with cancer stem cell
properties via epithelial-mesenchymal transition-like changes.
Oncogene. 33:4123–4131. 2014. View Article : Google Scholar
|
|
45
|
Yan Y, Zuo X and Wei D: Concise Review:
Emerging role of CD44 in cancer stem cells: A promising biomarker
and therapeutic target. Stem. Cells Transl Med. 4:1033–1043. 2015.
View Article : Google Scholar
|
|
46
|
Lee JL, Wang MJ and Chen JY: Acetylation
and activation of STAT3 mediated by nuclear translocation of CD44.
J Cell Biol. 185:949–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Beurel E and Jope RS: The paradoxical pro-
and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic
apoptosis signaling pathways. Prog Neurobiol. 79:173–189. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shakoori A, Ougolkov A, Yu ZW, Zhang B,
Modarressi MH, Billadeau DD, Mai M, Takahashi Y and Minamoto T:
Deregulated GSK3beta activity in colorectal cancer: Its association
with tumor cell survival and proliferation. Biochem Biophys Res
Commu. 334:1365–1373. 2005. View Article : Google Scholar
|
|
49
|
Shakoori A, Mai W, Miyashita K, Yasumoto
K, Takahashi Y, Ooi A, Kawakami K and Minamoto T: Inhibition of
GSK-3 beta activity attenuates proliferation of human colon cancer
cells in rodents. Cancer Sci. 98:1388–1393. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Fu Y, Jiao Y, Zheng S, Liang A and Hu F:
Combination of lithium chloride and pEGFP-N1-BmK CT effectively
decreases proliferation and migration of C6 glioma cells.
Cytotechnology. 68:197–202. 2016. View Article : Google Scholar :
|
|
51
|
Cockle JV, Picton S, Levesley J, Ilett E,
Carcaboso AM, Short S, Steel LP, Melcher A, Lawler SE and
Brüning-Richardson A: Cell migration in paediatric glioma;
characterisation and potential therapeutic targeting. Br J Cancer.
112:693–703. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Silva AK, Yi H, Hayes SH, Seigel GM and
Hackam AS: Lithium chloride regulates the proliferation of
stem-like cells in retinoblastoma cell lines: A potential role for
the canonical Wnt signaling pathway. Mol Vis. 16:36–45.
2010.PubMed/NCBI
|
|
53
|
Li H, Huang K, Liu X, Liu J, Lu X, Tao K,
Wang G and Wang J: Lithium chloride suppresses colorectal cancer
cell survival and proliferation through ROS/GSK-3β/NF-κB signaling
pathway. Oxid Med Cell Longev. 2014:2418642014. View Article : Google Scholar
|
|
54
|
Lee TY, Liu CL, Chang YC, Nieh S, Lin YS,
Jao SW, Chen SF and Liu TY: Increased chemoresistance via Snail-Raf
kinase inhibitor protein signaling in colorectal cancer in response
to a nicotine derivative. Oncotarget. 7:23512–23520.
2016.PubMed/NCBI
|
|
55
|
Sirri V, Urcuqui-Inchima S, Roussel P and
Hernandez-Verdun D: Nucleolus: The fascinating nuclear body.
Histochem Cell Biol. 129:13–31. 2008. View Article : Google Scholar
|