Introduction
Epidermal growth factor (EGF) was isolated from
mouse submaxillary glands and demonstrated to promote incisor
eruption and eyelid opening in newborn rodents (1). After EGF binds to its cell surface
receptor (EGFR), EGFR undergoes dimerization, which in turn induces
EGFR auto-phosphorylation (2).
This auto-phosphorylation elicits downstream signal transduction
cascades such as the phosphoinositide 3-kinase-pyruvate
dehydrogenase kinase-protein kinase B (Akt) and
RAF-mitogen-activated protein kinase kinase-extracellular
signal-regulated kinase pathways, which promote cell survival and
proliferation. Phosphorylated (p-)EGFR is translocated to the
cytoplasm and degraded in lysosomes, which causes suppression of
downstream signaling cascades in normal cells (2). However, in cancer cells, p-EGFR is
accumulated in the nucleus, increasing cancer cell activity
(3). EGFR is overexpressed in many
cancers, including head and neck, breast, lung, colon, stomach,
kidney, prostate and ovarian cancer, and is associated with worse
outcome (4-6). An anti-EGFR monoclonal antibody,
cetuximab, is the first molecular target drug for head and neck
squamous cell carcinoma (HNSCC) (7,8).
When combined with radiotherapy and chemotherapy, cetuximab has
been demonstrated to have significant survival benefits in patients
with locally advanced and recurrent/metastatic HNSCC, respectively
(7,8). However, resistance to cetuximab is
known to develop gradually (9),
and presents an important challenge for the future.
Tobacco smoking is associated with the
carcinogenesis and development of cancer (10). Tobacco smoking is carcinogenic to
the oral cavity, pharynx, larynx, esophagus, stomach, colon, liver,
pancreas, lung, uterine cervix, ovary, kidney, renal pelvis and
ureter, and bladder (10). The
organs directly exposed to tobacco smoke are at high risk. Tobacco
smoke is divided into a particle and gas phase (11). The particle phase includes tar and
nicotine, while the gas phase includes carbon monoxide. Tar
contains many carcinogenic chemicals, including benzopyrene and
nitrosamine. Nicotine is responsible for tobacco addiction, and
thus nicotine replacement therapy, which supplies nicotine in the
form of gum or a patch, is used to help individuals with tobacco
addiction quit smoking (12).
The nicotine in the tobacco smoke is absorbed across
the epithelium of the lung, and the mucosal epithelia in the head
and neck region, e.g., the oral cavity, nasal cavity, pharynx,
nasopharynx and larynx (13).
Nicotine exerts its cellular functions through nicotinic
acetylcholine receptors (nAChRs). nAChRs are homomeric or
heteromeric pentameric proteins consisting of α1-10, β1-4, γ, δ and
ε subunits, and are located in the central nervous system and
neuromuscular junctions (14). The
binding of nicotine to nAChRs in the ventral tegmental area of the
midbrain induces the release of dopamine to the nucleus accumbens,
which is involved in the rewarding effects of nicotine and nicotine
addiction. Although nAChR localization is thought to be limited in
the nervous system, nAChR is present in a wide variety of
non-neuronal cells, including bronchial epithelial, urothelial,
skin, endothelial and vascular smooth muscle cells (15). The mucosal epithelia of the head
and neck region are the primary sites of exposure to tobacco smoke,
and nAChRs have been observed in mucosal epithelial cells in these
regions (16,17). Previous studies have suggested that
nAChRs are also present in lung cancer, breast cancer, pancreatic
cancer, colon cancer and head and neck cancer cells (15,18-21).
In lung and breast cancer, nicotine has been demonstrated to
contribute to tumor growth and metastasis (19,20).
Nicotine also controls the expression and subcellular localization
of EGFR in breast cancer cells (22). Specifically, nicotine decreases the
expression of EGFR, and enhances the accumulation of p-EGFR in the
nucleus, thereby increasing proliferation of breast cancer cells
(22). Finally, in lung cancer
cells, nicotine serves a role in the resistance against EGFR
inhibitors (23,24).
These facts prompted us to explore whether HNSCC
cells could utilize nicotine-nAChR signaling to metastasize from
the primary tumor to the regional lymph nodes and acquire
resistance to cetuximab through EGFR activation. In the present
study it was demonstrated that nicotine increased proliferation,
migration, invasion, p-EGFR nuclear translocation and Akt
activation in HNSCC cells. It was also demonstrated that nicotine
restored cetuximab-inhibited proliferation, migration and invasion
of HNSCC cells. Finally, it was demonstrated that an nAChR
inhibitor suppressed lymph node metastasis in a mouse model of
lymph node metastasis using OSC-19 cells.
Materials and methods
Cell culture and reagents
The following human HNSCC cell lines were used in
the present experiments: HSC-2, a mouth floor SCC cell line derived
from a metastatic cervical lymph node; HSC-3, a tongue SCC cell
line derived from a metastatic cervical lymph node; OSC-19, a
tongue SCC cell line derived from the primary site; and OSC-20, a
tongue SCC cell line derived from a metastatic cervical lymph node.
HSC-2 and HSC-3 were obtained from the Cell Engineering Division of
the RIKEN BioResource Center (Ibaraki, Japan). OSC-19 and OSC-20
were obtained from the Health Science Research Resources Bank
(Osaka, Japan). Human umbilical vein endothelial cells (HUVECs)
were purchased from Takara Bio, Inc. (Otsu, Japan). All cancer
cells were cultured in Dulbecco’s modified Eagle’s medium/Ham’s
F-12 nutrient mixture (DMEM/F-12: Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum
(FBS: Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) at 37°C.
HUVECs were cultured in endothelial cell growth medium-2 (Takara
Bio, Inc.) at 37°C. Nicotine (Sigma-Aldrich; Merck KGaA; 0.5
µM) (19,20,22),
mecamylamine hydrochloride (MCA; Sigma-Aldrich; Merck KGaA; 50 nM)
(22), α-bungarotoxin (α-BTX;
Abcam, Cambridge, UK; 1 µM) (17), cetuximab (Merck KGaA; 0.5
µg/ml) (30), SCH772984
(Santa Cruz Biotechnology, Inc., Dallas, TX, USA; 1 µM)
(25), Akt inhibitor II (Merck
KGaA; 10 µM) (26) and
temsirolimus (Pfizer, Inc., New York, NY, USA; 10 nM) (27) were purchased. The concentration of
nicotine (0.5 µM) used was decided based on previously
published basic studies (19,20,22)
and clinical investigations in which the concentrations of nicotine
in the bloodstream of smokers exposed to nicotine at
pharmacological concentrations were reported to be 0.09-1 µM
(28,29).
Cell proliferation
Cells were seeded at a density of 1x105
cells in a 6-well plate. After becoming subconfluent at 37°C, the
cells were cultured for 24 h in DMEM/F-12 without FBS. They were
then cultured in the presence or absence of nicotine (0.5
µM) (19,20,22),
MCA (50 nM) (22), α-BTX (1
µM) (17) or cetuximab (0.5
µg/ml) (30) in DMEM/F-12
supplemented with 0.5% FBS. The number of cells was counted with
TC10TM automated cell counter (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) daily for 5 days.
Invasion and migration assays
Invasion and migration of cells were studied as
reported previously, by using Boyden chambers with or without
Matrigel®, respectively (BD Biosciences, Franklin Lakes,
NJ, USA) (31). Cells in the
logarithmic growth phase were detached by trypsin-EDTA, and
3x104 cells in serum-free DMEM/F12 were added to
polycarbonate membranes (pore size, 8.0 µm). Nicotine (0.5
µM) was added to the lower chamber, and the system was
incubated at 37°C for 24 h in 5% CO2. Following
incubation and fixation with 70% ethanol for 10 min at room
temperature, the non-invading or migrating cells were removed with
a cotton swab and the remaining cells were stained with 2% crystal
violet (Sigma-Aldrich; Merck KGaA) for 5 min at room temperature.
The number of stained cells on the lower side of the membrane in 4
light microscopic fields were counted, and the mean value of 3
wells was determined.
Immunoblot analysis
HSC-3 and OSC-19 cells were transferred to DMEM/F-12
without FBS, incubated for 24 h, and then treated with nicotine
(0.5 µM) with or without MCA (50 nM), α-BTX (1 µM),
cetuximab (0.5 µg/ml), SCH772984 (1 µM), Akt
inhibitor II (10 µM) or temsirolimus (10 nM) at 37°C for 1
h. Cells in monolayer cultures were rinsed with ice-cold PBS and
lysed in an ice-cold lysis buffer (50 mM Tris-HCl, pH 7.4,
containing 150 mM NaCl, 1% Triton X-100, 1% NP-40, 10 mM NaF, 100
mM leupeptin, 2 mg/ml aprotinin and 1 mM phenylmethyl sulfonyl
fluoride). Protein concentration was determined using a BCA Protein
Assay Kit (Pierce, Thermo Fisher Scientific, Inc.) The cell lysates
containing 10 µg total protein in the lysis buffer were
electrophoresed in 12% SDS-PAGE gels, and the proteins were then
transferred to nylon membranes (Immobilon-P; EMD Millipore,
Billerica, MA, USA). The membranes were blocked with 2% non-fat dry
milk in TBS overnight at 4°C and then incubated with a 1:1,000
dilution of the desired antibody at 4°C for 12 h. Primary rabbit
anti-human p-EGFR monoclonal immunoglobulin G (IgG; ab32578), mouse
anti-human β-actin monoclonal IgG (ab49900), mouse anti-human PCNA
monoclonal IgG (ab29; all Abcam), rabbit anti-human EGFR monoclonal
IgG (4267S), rabbit anti-human p-Akt monoclonal IgG (4058), rabbit
anti-human Akt monoclonal IgG (4685), rabbit anti-human p-mTOR
polyclonal IgG (2974) and rabbit anti-human mTOR polyclonal IgG
(2972; all Cell Signaling Technology, Danvers, MA, USA) were used
for immunoblot analyses. Horseradish peroxidase (HRP)-conjugated
goat anti-rabbit (RPN4301) or sheep anti-mouse IgG (NA931) were
used as the secondary antibody at a 1:1,000 dilution (GE Healthcare
Life Sciences, Little Chalfont, UK). Bands were visualized via
enhanced chemiluminescence (RPN2109; GE Healthcare Life Sciences).
Proteins in the nuclear and cytosolic fraction were obtained with
NE-PERTM Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher
Scientific, Inc.) using the experimental procedures specified by
the manufacturer. For immunoblot analysis of α7 nAChR, the cell
lysates containing 40 µg total protein in the lysis buffer
were electrophoresed in 12% SDS-PAGE gels, and then the proteins
were transferred to nylon membranes. The membranes were blocked
with 2% non-fat dry milk in TBS overnight at 4°C and then incubated
with a 1:400 dilution of rabbit anti-human α7 nAChR polyclonal IgG
(ab10096; Abcam) overnight at 4°C. HRP-conjugated goat anti-rabbit
IgG (RPN4301) was used as the secondary antibody at a 1:10,000
dilution (GE Healthcare Life Sciences). Band densities of
immunoblotting were quantified with ImageJ (version 1.51; National
Institutes of Health, Bethesda, MD, USA).
Immunofluorescence staining
Following growth on culture slides, cells were
washed with PBS, fixed at -20°C for 20 min with 100% methanol, and
permeabilized with 0.1% NP-40 in PBS. The slides were blocked with
3% FBS in PBS for 10 min at room temperature. Following washing 3
times with PBS, the cells were incubated for 1 h with 1:50 dilution
of anti-p-EGFR (rabbit IgG; ab32578; Abcam) in 3% BSA-PBS, washed 3
additional times with PBS, and reacted with 1:1,000 dilution of
Alexa Fluor 647 (Invitrogen; Thermo Fisher Scientific, Inc.) in 3%
BSA-PBS for 2 h at room temperature. Following a final wash, the
cells were sealed with a coverslip and viewed under a fluorescent
microscope (IX81; Olympus Corporation, Tokyo, Japan).
Lymph node metastasis model
All animal experiments were approved by the
Institutional Animal Care and Use Committee of Okayama University
(Okayama, Japan; OKU-2016046). The lymph node metastasis model was
prepared as described previously (32). A total of 50 male athymic mice
(nu/nu; age, 5 weeks; mean body weight, 19.5 g) were obtained from
CLEA Japan, Inc. (Tokyo, Japan). OSC-19 cells, 8x105 per
mouse, were inoculated into a hind footpad. Groups of 10 mice each
were peritoneally injected with either PBS, nicotine (30
µg/mouse) (20), MCA (20
µg/mouse) (33), nicotine
and MCA, cetuximab (1 mg/mouse) (34), or cetuximab and nicotine every day.
Tumor sizes and body weights were measured weekly, and the former
were recorded in mm3 (length x width2 / 2).
Mice were sacrificed at day 42, the footpad tumor tissues and
popliteal lymph nodes were isolated and slides were prepared as
described previously (32).
Immunohistochemistry for xenograft tumor
specimens
Paraffin blocks of specimens were cut at 4-µm
thickness. The sections were deparaffinized, and then autoclaved in
0.2% citrate buffer for 15 min for antigen retrieval. Sections were
incubated with 3% hydrogen peroxide for 30 min at room temperature
to block endogenous peroxidase activity. Immunohistochemistry was
performed using rabbit anti-human EGFR monoclonal IgG (Cell
Signaling Technology, Inc.) and rabbit anti-human p-EGFR monoclonal
IgG (Abcam), each at 1:100 dilution. The sections were incubated
with the primary antibodies at 4°C for 16 h, and then treated with
Envision System Labeled Polymer (Dako; Agilent Technologies, Inc.,
Santa Clara, CA, USA) for 60 min at a dilution of 1:100. The
immunoreaction was visualized using a 3,3′-diaminobenzidine
substrate-chromogen solution. Finally, the sections were immersed
in an ethanol and xylene bath and mounted for examination. The
extent of p-EGFR staining was evaluated according to the percentage
of cells with strongly stained nuclei in 3 visual fields under a
light microscope (x200). All immunohistochemistry studies were
evaluated by 2 experienced observers who were blind to the
conditions of the experiments as reported previously (35).
Statistical analysis
Data were analyzed by using unpaired Student’s
t-test for analyses between two groups, and one-way analysis of
variance with Bonferroni post hoc tests for the analysis of
multiple group comparisons using SPSS statistical software (version
22; IBM Corp., Armonk, NY, USA). Results were expressed as the mean
± standard deviation. To control for multiple testing for data in
the incidence of popliteal lymph node metastasis in mice, q-values
were calculated with the false discovery rate method controlled by
the Benjamini-Hochberg procedure. P<0.05 and q<0.05 were
considered to indicate statistically significant differences.
Results
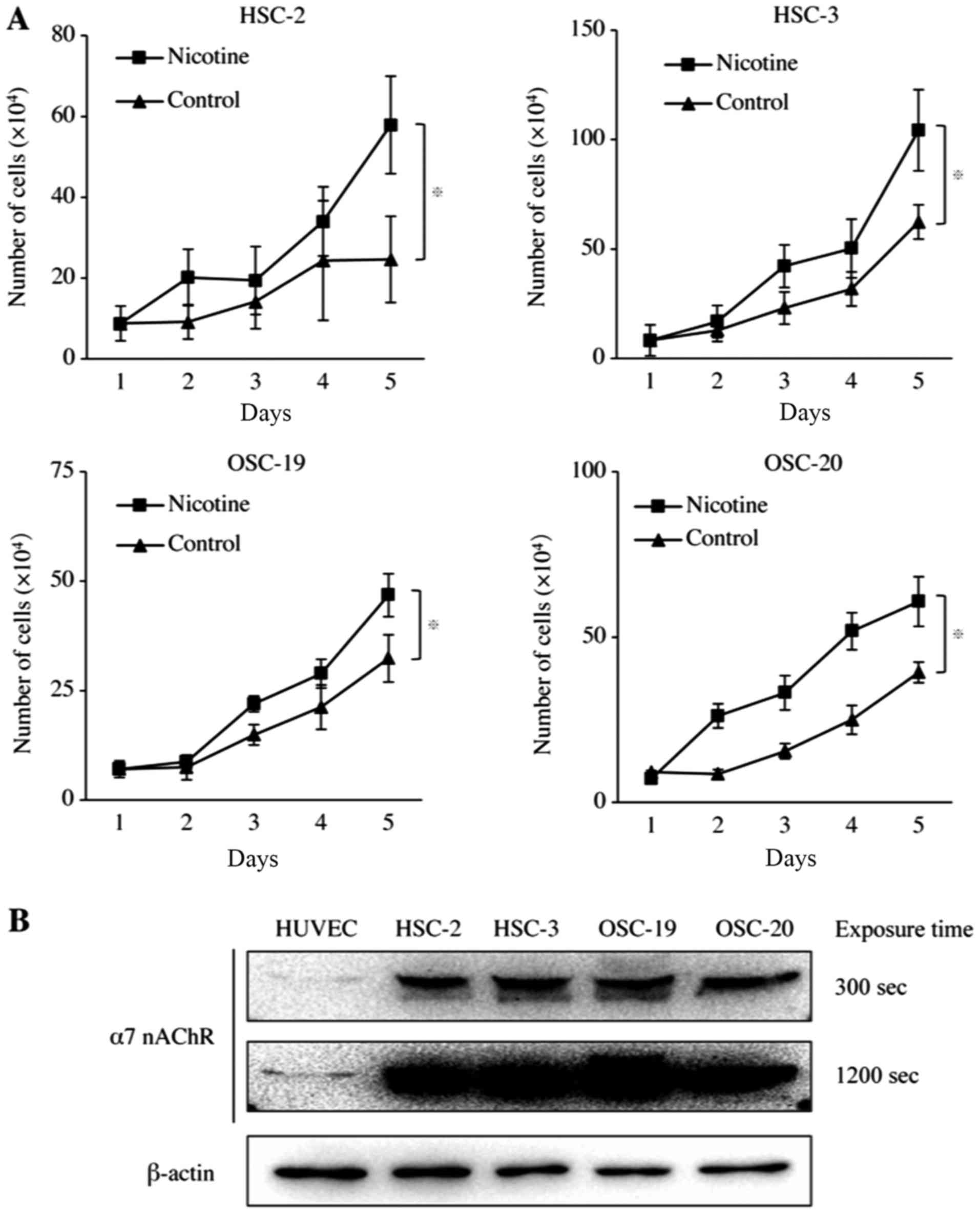
Nicotine upregulated HNSCC cell
proliferation
To examine the effect of nicotine in vitro,
HNSCC cells were treated with 0.5 µM nicotine for 5 days.
This treatment stimulated a 2.4-, 1.6-, 1.5- or 1.6-fold increase
in the proliferation of HSC-2, HSC-3, OSC-19 and OSC-20 cells at
day 5, respectively (Fig. 1A).
Having established that nicotine stimulated the proliferation of
HNSCC cells, whether nicotine receptors were expressed in HNSCC
cells was then examined. Among nicotinic acetylcholine receptors,
α7 nAChR has been reported as the primary receptor that mediates
the proliferative effects of nicotine in various cancer cells
(18). In the present study, it
was demonstrated that all four HNSCC cell lines exhibited stronger
α7 nAChR expression than that of the HUVEC used as the positive
control for α7 nAChR (Fig.
1B).
nAChR inhibitors suppressed
nicotine-upregulated cell activities in HNSCC cells
As nicotine increased the cell proliferation of
HNSCC cell lines, the effects of nAChR inhibitors on HNSCC cells
were evaluated. As HSC-3 and OSC-19 have been reported to
metastasize to the lymph nodes (36,37),
it was decided to use these cell lines in later experiments. HSC-3
and OSC-19 were treated with MCA (a non-selective nAChR inhibitor)
and α-BTX (an α7 nAChR inhibitor) in the presence of nicotine.
Nicotine increased cell viability 1.3-fold in both HSC-3 and OSC-19
cells. MCA and α-BTX inhibited nicotine-induced viability to the
same level as in the control group (Fig. 2A). In regard to cell migration,
nicotine increased cell migration 1.4- and 1.2-fold in HSC-3 and
OSC-19 cells, respectively, and MCA and α-BTX suppressed these
nicotine-induced effects (Fig.
2B). Finally, invasion of HSC-3 and OSC-19 cells was increased
1.4- and 1.3-fold by nicotine, respectively, and MCA and α-BTX
counteracted these effects as well (Fig. 2C).
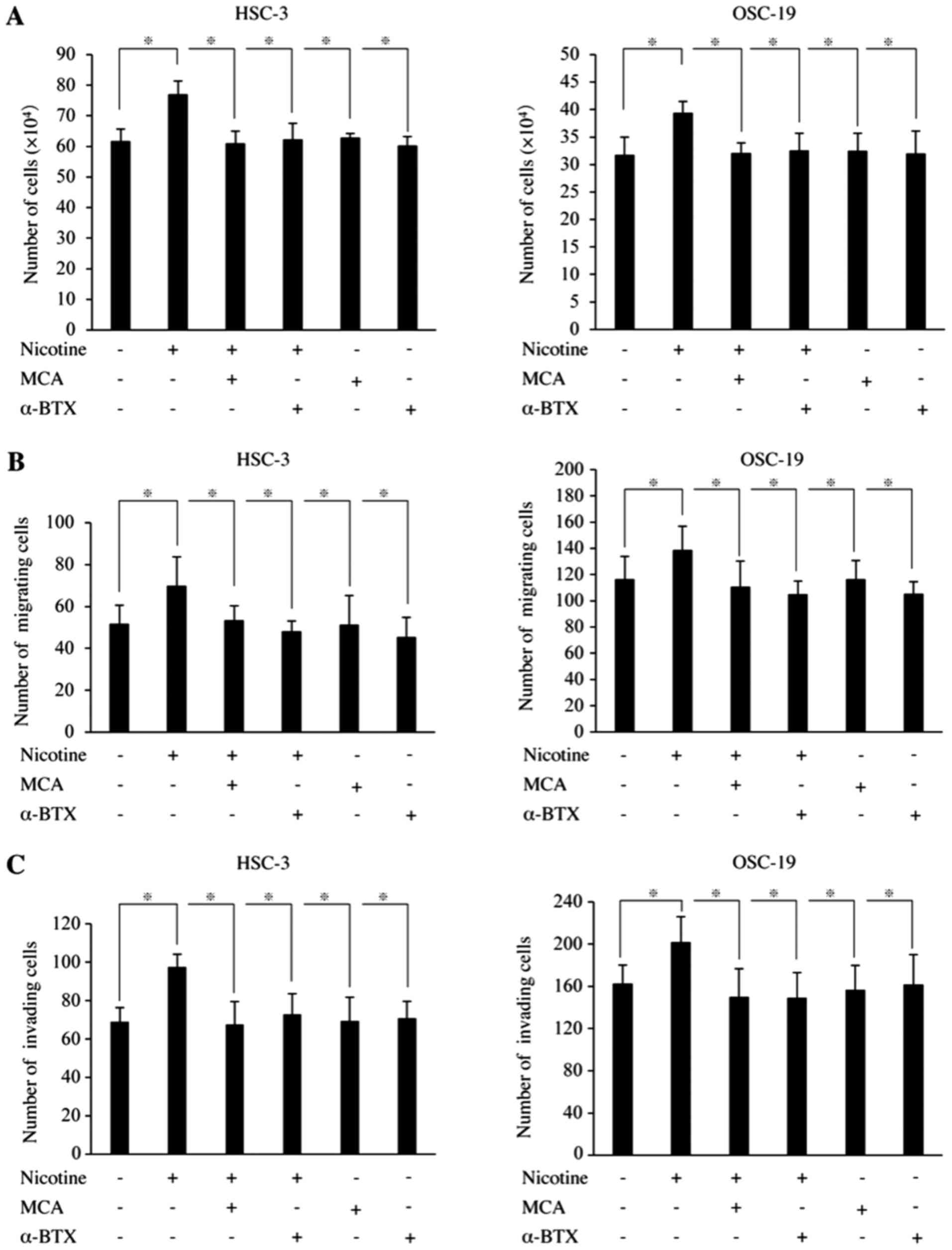 | Figure 2Effects of nAChR inhibitors on the
cell viability, migration and invasion of HNSCC cells. (A)
Viability. Cells were seeded at a density of 1x105 cells
in a 6-well plate. After becoming subconfluent, cells were cultured
for 24 h in DMEM/F-12 without FBS. They were then cultured in the
presence or absence of nicotine, MCA or α-BTX in DMEM/F-12
supplemented with 0.5% FBS. The cell number was measured at day 5.
(B) Migration. Migration was evaluated using Boyden chambers. Cells
were seeded at a density of 3x104 in medium with or
without MCA, α-BTX or cetuximab on polycarbonate membranes.
Nicotine was added to the lower chamber, and the system was
incubated for 24 h. Following incubation, the number of cells on
the lower side of the membrane was counted. (C) Invasion. Invasion
was evaluated using Boyden chambers with Matrigel®.
Incubation and cell counting were performed as in the migration
assay. All experiments were repeated 3 times. Data are presented as
the mean ± standard deviation of triplicates from a typical
experiment. *P<0.05. nAChR, nicotinic acetylcholine
receptor; HNSCC, head and neck squamous cell carcinoma; DMEM/F-12,
Dulbecco’s modified Eagle’s medium/Ham’s F-12 nutrient mixture;
FBS, fetal bovine serum; MCA, mecamylamine hydrochloride; α-BTX,
α-bungarotoxin. |
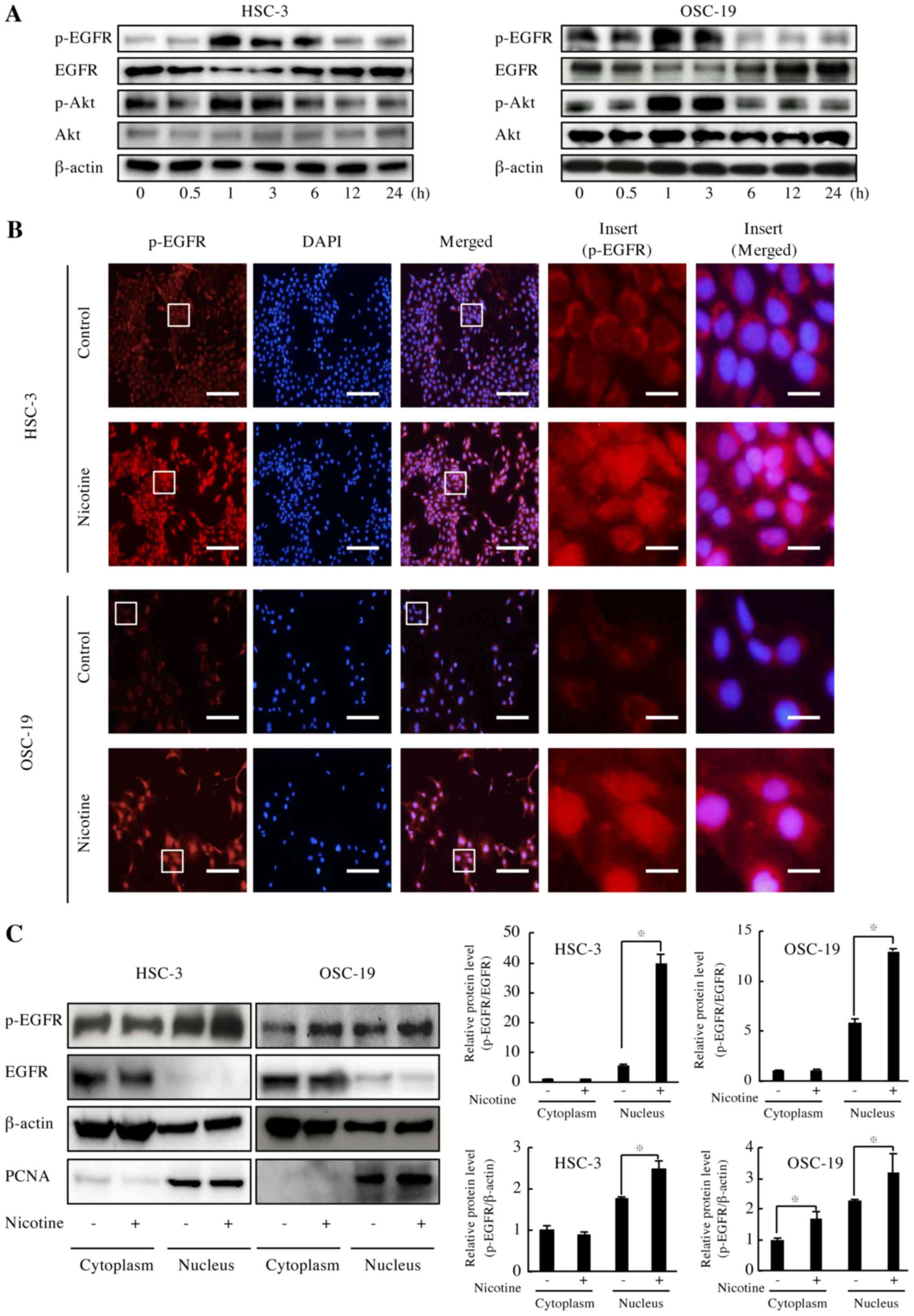
Nicotine induced phosphorylation and
nuclear translocation of EGFR
EGFR is known to be overexpressed and its signaling
pathway is one of the most important pathways in HNSCC (3). As nicotine upregulated the cell
activities of HNSCC in the present experiments, the effect of
nicotine on EGFR activation was evaluated. It was demonstrated that
both EGFR phosphorylation and Akt phosphorylation were increased 1
h following the addition of nicotine (Fig. 3A). Immunofluorescence analysis of
HNSCC cells demonstrated that nicotine induced p-EGFR nuclear
translocation (Fig. 3B). To obtain
quantitative data on this effect, p-EGFR distribution was evaluated
in the nuclear and cytosolic fractions. Immunoblot analysis
revealed an increase in nuclear p-EGFR in both HSC-3 (7.1-fold
p-EGFR/EGFR and 1.4-fold p-EGFR/ β-actin) and OSC-19 (2.2-fold
p-EGFR/EGFR and 1.6-fold p-EGFR/β-actin) cells exposed to nicotine,
respectively (Fig. 3C).
Nicotine induced phosphorylation of EGFR,
Akt and mTOR through nAChRs
To determine whether nAChRs served any role in the
activation of EGFR and Akt by nicotine, whether nAChR inhibitors
would suppress the effects of nicotine was evaluated. Treatment
with MCA and α-BTX reversed the nicotine-induced activation of EGFR
and Akt (Fig. 4A), suggesting that
nicotine activates EGFR and Akt via nAChRs. To examine whether Akt
and mTOR are involved in the region downstream of the
nicotine-nAChR-EGFR pathway, Akt inhibitor II (an Akt inhibitor)
and temsirolimus (an mTOR inhibitor) were used. Treatment with Akt
inhibitor II reversed the nicotine-dependent stimulation of Akt and
mTOR, whereas it did not change EGFR phosphorylation (Fig. 4B). In a similar manner,
temsirolimus also markedly counteracted the nicotine-dependent
stimulation of mTOR, whereas it did not change the levels of EGFR
or Akt phosphorylation (Fig. 4B).
SCH772984 (a MAPK inhibitor) did not change EGFR, Akt or mTOR
phosphorylation (Fig. 4B). Taken
together, our findings demonstrate that nicotine stimulated the
EGFR-Akt-mTOR signaling pathway via nAChRs in HNSCC cells.
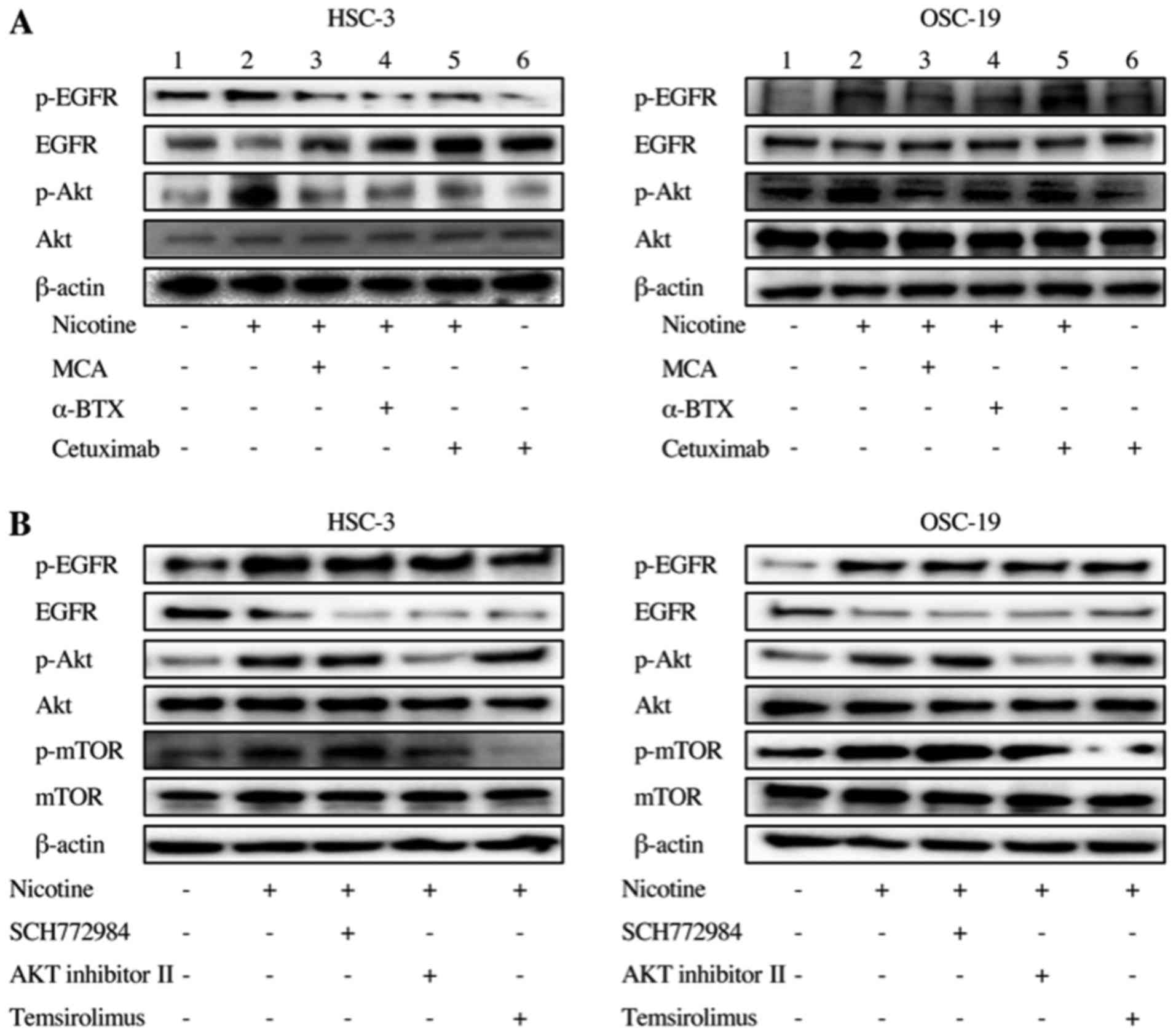 | Figure 4Effects of MCA, α-BTX, cetuximab,
SCH772984, Akt inhibitor II and temsirolimus on nicotine-dependent
activation of EGFR, Akt and mTOR in head and neck squamous cell
carcinoma cells. (A) Nicotinic acetylcholine receptor; downstream
signaling. HSC-3 and OSC-19 cells were transferred to DMEM/F-12
without FBS, incubated for 24 h, and then treated with 0.5
µM nicotine with or without MCA, α-BTX or cetuximab for 1 h.
Protein extracts were analyzed by immunoblotting with antibodies
recognizing p-EGFR and p-Akt. (B) EGFR downstream signaling. Cancer
cells were transferred to DMEM/F-12 without FBS, incubated for 24
h, and then treated with nicotine with or without SCH772984, Akt
inhibitor II or temsirolimus at 37°C for 1 h. Protein extracts were
analyzed by immunoblotting with antibodies recognizing p-EGFR,
p-Akt and p-mTOR. MCA, mecamylamine hydrochloride; α-BTX,
α-bungarotoxin; EGFR, epidermal growth factor receptor; Akt,
protein kinase B; mTOR, mechanistic target of rapamycin; DMEM/F-12,
Dulbecco’s modified Eagle’s medium/Ham’s F-12 nutrient mixture;
FBS, fetal bovine serum; p, phosphorylated. |
Notably, in lanes 5 and 6 of Fig. 4A, it was observed that nicotine
upregulated cetuximab-suppressed EGFR phosphorylation, suggesting
that nicotine may contribute to cetuximab resistance.
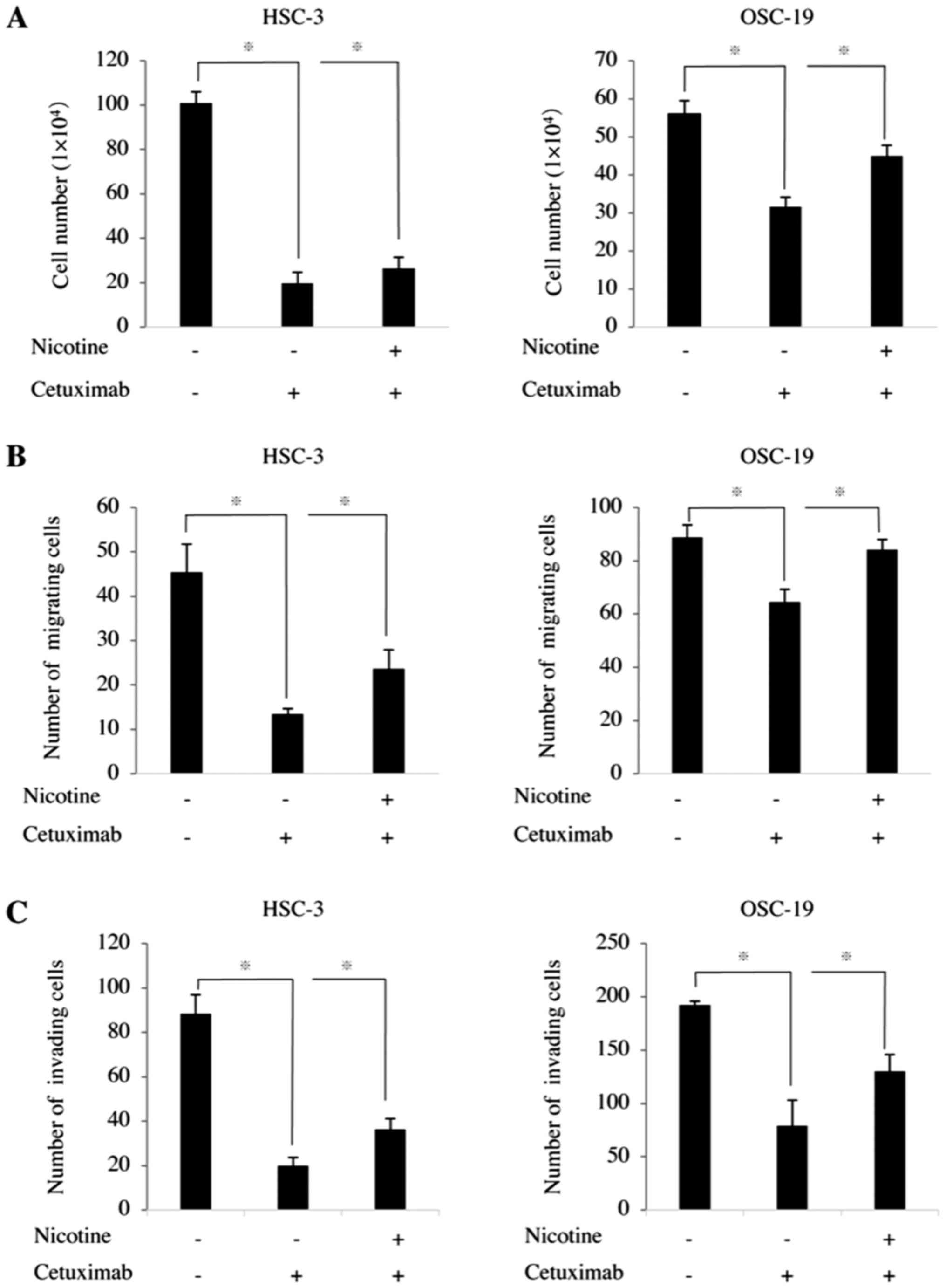
Nicotine counteracted the anti-tumor
effects of cetuximab in HNSCC cells
As nicotine upregulated cetuximab-suppressed EGFR
phosphorylation, it was speculated that nicotine counteracted
anti-EGFR therapy through EGFR activation. To test this hypothesis,
HNSCC cells were treated with cetuximab and nicotine, then the cell
activities were evaluated.
In the presence of cetuximab alone, the
proliferation of HSC-3 and OSC-19 cells was significantly
inhibited. When nicotine was added, however, the proliferation of
both HSC-3 and OSC-19 cells increased to 1.4-fold of that in the
group treated with cetuximab alone (Fig. 5A). Cetuximab also significantly
inhibited the cell migration of HNSCC cells. However, when nicotine
was added, the cell migrations of HSC-3 and OSC-19 cells were
increased to 1.7- and 1.3-fold of those in the cells treated with
cetuximab alone (Fig. 5B). In a
similar manner, the levels of invasion of HSC-3 and OSC-19 cells
treated with cetuximab and nicotine were 1.9- and 1.6-fold of those
in the cells treated with cetuximab alone, respectively.
An nAChR inhibitor suppressed the tumor
growth and lymph node metastasis of xenografted HNSCC in athymic
mice
To further investigate whether nicotine was
responsible for tumor growth and metastasis by HNSCC cells, an
animal model of lymph node metastasis was utilized (32). To inhibit nAChRs, only MCA was used
in animal experiments. MCA (38)
is well-known as an antihypertensive drug under the name
Inversine® (39),
whereas α-BTX is famous as a snake venom (40); therefore, MCA was speculated to be
more suitable for future clinical application.
In the animal experiments, nicotine increased the
growth rate of xenografted tumors compared with the rate in the
control group, and MCA decreased tumor growth compared with the
nicotine-treated group (Fig. 6A).
The tumor volumes at the end of the experiment (day 42) for the
control group, nicotine-treated group and MCA group were
615.2±65.3, 950.0±188.9 and 686.5±156.4 mm3,
respectively. These results indicated an ~54.5% increase in the
tumor growth rate for the nicotine-treated group. The findings also
suggested that MCA effectively inhibited the nicotine-induced
xenograft tumor growth of HNSCC cells in athymic mice to the same
level as in the control group (Fig.
6A). Furthermore, nicotine increased cetuximab-suppressed
xenografted tumor growth from 1.2±1.8 to 32.9±37.7 mm3,
suggesting that nicotine may contribute to local relapse following
anti-EGFR therapy.
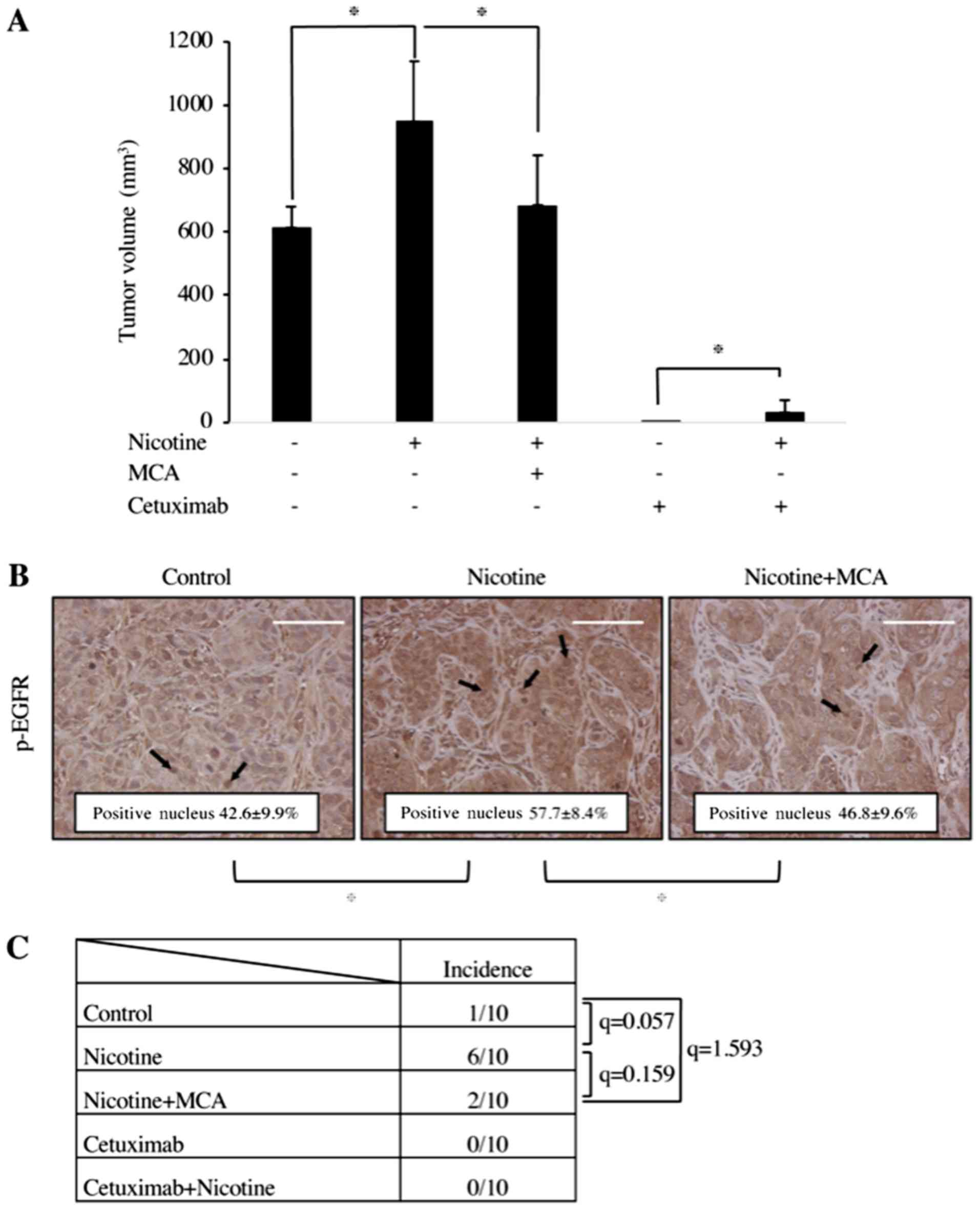 | Figure 6Effect of nicotine, MCA and cetuximab
on the growth of OSC-19 xenografts and lymph node metastasis in
athymic mice. (A) A total of 8x105 OSC-19 cells per
mouse (10 mice/group) were inoculated into the hind footpad of
athymic mice. Mice were peritoneally administered with PBS,
nicotine (30 µg/mouse), MCA (20 µg/mouse), nicotine
and MCA, cetuximab (1 mg/mouse), or cetuximab and nicotine every
day. Mice were sacrificed at day 42. In bar chart, the data are
presented as tumor volume in mm3 (length x
width2 / 2). Data are presented as the mean ± standard
deviation. *P<0.05. (B) IHC staining of OSC-19
xenograft tumor specimens from the hind footpad. IHC staining of
EGFR and p-EGFR in tumors from the OSC-19 treated with or without
nicotine or MCA is presented. Nicotine induced nuclear localization
of p-EGFR (arrows) whereas MCA suppressed nicotine-induced nuclear
localization of p-EGFR. Scale bar, 100 µm. (C) The rate of
popliteal lymph node metastasis is presented as the number of mice
with metastases/number of mice injected. Lymph node metastasis was
observed in the control group (10%), nicotine-treated group (60%)
and nicotine and MCA-treated group (20%). MCA, mecamylamine
hydrochloride; IHC, immunohistochemical; EGFR, epidermal growth
factor receptor; p, phosphorylated. |
To determine whether nicotine influences p-EGFR
localization in vivo, the percentage of p-EGFR-positive
nuclei in the tumor specimens was evaluated. As presented in
Fig. 6B, the percentage of
p-EGFR-positive nuclei was increased in the nicotine-treated group
(57.7±8.4%) compared with the control group (42.6±9.9%), which was
consistent with the in vitro analysis. Conversely, MCA
decreased the percentage of p-EGFR-positive nuclei to the level of
the control group (46.8±9.6%). Nicotine also increased the rate of
popliteal lymph node metastasis from 10 to 60%. MCA decreased the
rate of metastasis to 20%. Although there were no significant
differences between the any two groups, there was a trend toward
higher metastasis rate with nicotine compared with control.
(control vs. nicotine, q=0.057; nicotine vs. nicotine+MCA, q=0.159;
nicotine+MCA vs. control, q=1.593; Fig. 6C). There was no lymph node
metastasis in either the cetuximab-treated group or the cetuximab
and nicotine-treated group.
Taken together, the in vivo experiments
revealed that nicotine increased the tumor growth and lymph node
metastasis of HNSCC, whereas MCA suppressed them. It was also
demonstrated that nicotine restored the cetuximab-inhibited tumor
growth of HNSCC. The activation and nuclear localization of EGFR
may contribute to these tumor-promoting effects of nicotine.
Discussion
Nicotine, one of the crucial components in the
addictiveness of tobacco, also has important roles in the invasion
and metastasis of various cancers, including lung cancer, breast
cancer, glioma, bladder cancer, pheochromocytoma and colorectal
cancer (15,18). In lung cancer cells, nicotine
exposure induces epithelial-mesenchymal transition (41). In breast cancer cells, nicotine
promotes cell motility via PKC and cdc42, leading to lung
metastasis (20). However, its
effect in HNSCC cells is unknown. To determine whether nicotine
influences HNSCC in vivo, a mouse model of lymphatic
metastasis was prepared (32). In
this model, nicotine increased the tumor volume and incidence of
metastasis in regional lymph nodes compared with the control
groups. Consistent with the in vivo experiments, in in
vitro studies, cell proliferation, migration and invasion were
significantly elevated in the nicotine-treated groups than the
control groups in all four HNSCC cell lines. In the animal model,
nicotine failed to induce lymphangio-genesis in tumor tissues (data
not shown). This suggests that nicotine-induced lymphatic
metastasis was attributable to nicotine-driven invasive cell
activities. Conversely, MCA and α-BTX, two nAChR inhibitors,
reversed the nicotine-driven invasive cell activities, suggesting
that nicotine stimulation of cell activities occurs through nAChRs.
The head and neck mucous epithelia express α3, α5, α7, α9, β2 and
β4 nAChRs (16,17). Among these nAChR subunits, α7 has
been implicated as the most powerful nAChR subunit in terms of
mediating the proliferative effects of nicotine in various cancer
cells (18). Furthermore, nicotine
activates α7 nAChR of keratinocytes in the head and neck region
(17,42,43).
These findings indicate that nicotine accelerates cell activity of
HNSCC cells via α7 nAChR. This is consistent with the present in
vitro finding that α-BTX inhibited nicotine-increased cell
proliferation, migration and invasion to the level of the control
groups.
Cetuximab, a chimeric human mouse anti-EGFR
monoclonal antibody, was the first molecular target drug used in
head and neck cancer therapy (7,8). In
our experiments, nicotine induced phosphorylation and nuclear
translocation of EGFR. Nuclear translocation of EGFR is correlated
with worse prognosis of HNSCC (3).
Cetuximab is known to inhibit EGFR translocation to the nucleus,
while accumulation of intranuclear EGFR is known to have some
relevance to cetuximab resistance (3). Nicotine may suppress the anti-tumor
effect of cetuximab by promoting EGFR nuclear translocation. In the
present experiments, nicotine restored the cetuximab-inhibited cell
proliferation, migration and invasion of HNSCC cells. However, we
note here that cetuximab resistance might also arise through
another mechanism, since nicotine failed to restore the
cetuximab-inhibited cell activities to the level seen in the
untreated control groups.
In the in vitro experiments, cetuximab
inhibited EGFR phosphorylation in HSC-3 cells. Conversely,
cetuximab increased EGFR phosphorylation in OSC-19 cells, although
Akt phosphorylation was still inhibited by cetuximab in this cell
line. Recently, it was demonstrated that HNSCC secreted cetuximab
with EGFR-containing extracellular vesicles (44,45).
OSC-19 cells may incorporate cetuximab-bound EGFR in the cytoplasm
following EGFR phosphorylation, and the cetuximab-bound EGFR may be
excluded by this novel mechanism.
To date, no study has examined the direct effect of
nicotine on cetuximab sensitivity, to the best of our knowledge.
However, a few studies have demonstrated the effect of nicotine on
anti-EGFR cancer therapy. In non-small cell lung cancer, nicotine
induces resistance to erlotinib and gefitinib, which are EGFR
tyrosine kinase inhibitors (23,46).
In colorectal cancer, cigarette smoking during anticancer treatment
with a cetuximab-based regimen reduces the therapeutic benefit
(47). These findings support the
present hypothesis that nicotine serves a role in cetuximab
resistance. In addition, nicotine suppresses cisplatin-induced
apoptosis in HNSCC cells through Akt signaling, which is a
mandatory signaling pathway for cell survival and regulation of
apoptosis (48). Akt regulates the
expression of survivin, which inhibits apoptosis (48). In the present experiments, nicotine
treatment induced phosphorylation of Akt and its downstream
molecule mTOR. This suggests that MCA and α-BTX inhibited Akt
phosphorylation and that nicotine induced Akt phosphorylation
through nAChRs. Furthermore. Akt inhibitor II and temsirolimus
induced no changes in the expression or phosphorylation of EGFR,
suggesting that the nicotine-driven Akt phosphorylation resides
downstream of EGFR (Fig. 7).
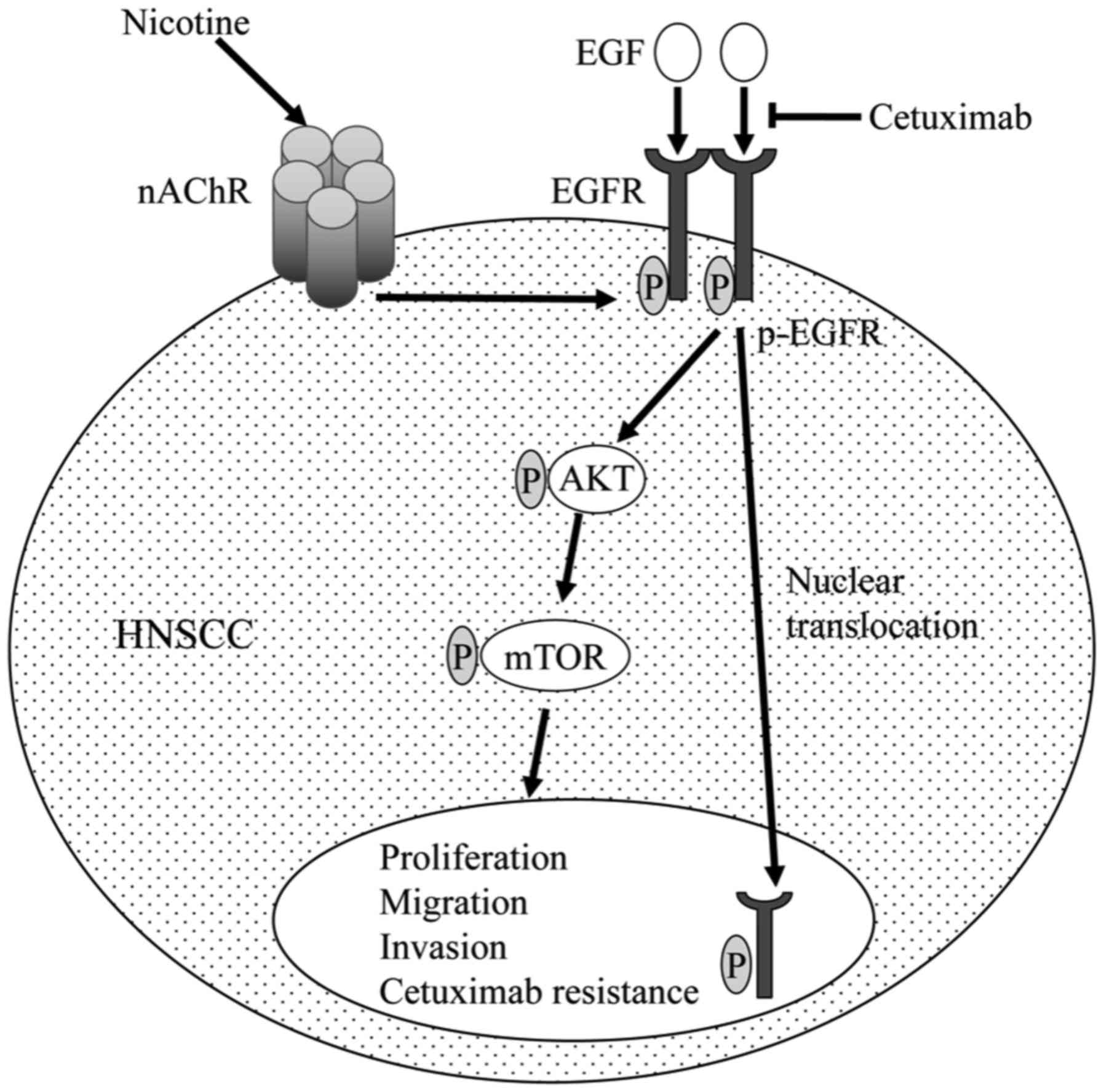 | Figure 7Function of nicotine in EGFR
signaling in HNSCC. Nicotine binds to nAChR and enhances EGFR
phosphorylation in HNSCC cells. EGFR phosphorylation elicits the
Akt-mTOR pathway, a downstream signal transduction cascade, and
p-EGFR itself is translocated to the nucleus, leading to cell
proliferation, migration, invasion and cetuximab resistance. EGFR,
epidermal growth factor receptor; HNSCC, head and neck squamous
cell carcinoma cells; nAChR, nicotinic acetylcholine receptor; Akt,
protein kinase B; mTOR, mechanistic target of rapamycin; EGF,
epidermal growth factor; P, phosphate group. |
In an in vivo experiment, cetuximab inhibited
metastasis. However, nicotine failed to restore the
cetuximab-inhibited metastasis. Therefore, the effect of nicotine
in metastatic HNSCC treated with cetuximab remains unclear, and
further investigation of this effect is required. In human
papillo-mavirus-positive oropharyngeal cancer, increased distant
metastases rates were noted in active smokers vs. never/former
smokers (22 vs. 5%), and cetuximab-based bio-radiotherapy (BRT) vs.
cisplatin-based chemoradiotherapy (CRT) (23 vs. 5%) (49). Although there was no difference in
nodal disease extent between CRT and BRT groups in that previous
study, nicotine and cetuximab may have synergistic negative effects
in certain types of HNSCC.
In conclusion, nicotine induced the phosphorylation
of EGFR through nAChR in the present experiments. Phosphorylated
EGFR was translocated from the cell surface to the nucleus, and
activated Akt and mTOR. These signaling pathways elevated the cell
activities of HNSCC cells, causing lymph node metastasis and
serving a role in cetuximab resistance (Fig. 7). Blockade of nicotine-nAChR
signaling may be of clinical benefit in cases of advanced HNSCC,
especially HNSCC in the oral cavity, which is the primary site of
exposure to nicotine, by inhibiting lymph node metastasis and
releasing cetuximab resistance.
Funding
The present study was supported by a Grant-in-Aid
for Scientific Research (B) (JSPS KAKENHI grant no. JP 17H04405) to
AS, a Grant-in-Aid for Scientific Research (C) (JSPS KAKENHI grant
no. JP 26463004, 17K11836) from the Ministry of Education, Culture,
Sports, Science, and Technology of Japan to SI and a Wesco
Scientific Promotion Foundation Grant to SI.
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors’ contributions
SI and AS conceptualized and designed the study. AS,
SI, TE, HN, KOk, RS, DK, SK and TN prepared resources and devised
methodology. RS, DK, SK, TN, TO KOb, KT, HK, KOn and KOk carried
out the experimentation. SI, TE, RS, DK, SK and TN interpreted
data. SI wrote the manuscript. AS, HN, KOk, TE and SI revised and
edited the manuscript. All authors reviewed the manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the
Institutional Animal Care and Use Committee of Okayama University
(Okayama, Japan; OKU-2016046).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors are grateful to Dr Takanori Tsuji
(Department of Radiation Oncology, Beth Israel Deaconess Medical
Center, Harvard Medical School, Boston, MA, USA) for suggesting the
topic treated in this paper. The authors also thank Ms. Kazuko
Funakoshi (Department of Oral Pathology and Medicine, Okayama
University Graduate School of Medicine, Dentistry, and
Pharmaceutical Sciences, Okayama, Japan) for the expert technical
assistance in histological preparations.
References
|
1
|
Cohen S: Isolation of a mouse submaxillary
gland protein accelerating incisor eruption and eyelid opening in
the new-born animal. J Biol Chem. 237:1555–1562. 1962.PubMed/NCBI
|
|
2
|
Avraham R and Yarden Y: Feedback
regulation of EGFR signalling: Decision making by early and delayed
loops. Nat Rev Mol Cell Biol. 12:104–117. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Burtness B, Bauman JE and Galloway T:
Novel targets in HPV-negative head and neck cancer: Overcoming
resistance to EGFR inhibition. Lancet Oncol. 14:e302-e3092013.
View Article : Google Scholar
|
|
4
|
Nicholson RI, Gee JM and Harper ME: EGFR
and cancer prognosis. Eur J Cancer. 37(Suppl 4): S9–S15. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Selvaggi G, Novello S, Torri V, Leonardo
E, De Giuli P, Borasio P, Mossetti C, Ardissone F, Lausi P and
Scagliotti GV: Epidermal growth factor receptor overexpression
correlates with a poor prognosis in completely resected
non-small-cell lung cancer. Ann Oncol. 15:28–32. 2004. View Article : Google Scholar
|
|
6
|
Hirsch FR, Varella-Garcia M, Bunn PA Jr,
Di Maria MV, Veve R, Bremmes RM, Barón AE, Zeng C and Franklin WA:
Epidermal growth factor receptor in non-small-cell lung carcinomas:
Correlation between gene copy number and protein expression and
impact on prognosis. J Clin Oncol. 21:3798–3807. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bonner JA, Harari PM, Giralt J, Azarnia N,
Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J, et al:
Radiotherapy plus cetuximab for squamous-cell carcinoma of the head
and neck. N Engl J Med. 354:567–578. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vermorken JB, Mesia R, Rivera F, Remenar
E, Kawecki A, Rottey S, Erfan J, Zabolotnyy D, Kienzer HR, Cupissol
D, et al: Platinum-based chemotherapy plus cetuximab in head and
neck cancer. N Engl J Med. 359:1116–1127. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wheeler DL, Dunn EF and Harari PM:
Understanding resistance to EGFR inhibitors-impact on future
treatment strategies. Nat Rev Clin Oncol. 7:493–507. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
International Agency for Research on
Cancer (IARC): IARC Working Group on the Evaluation of Carcinogenic
Risks to Humans. Tobacco smoke and involuntary smoking. IARC Work
Gr Eval Carcinog Risks Hum. 83:1–1438. 2004.
|
|
11
|
Valavanidis A, Vlachogianni T and Fiotakis
K: Tobacco smoke: Involvement of reactive oxygen species and stable
free radicals in mechanisms of oxidative damage, carcinogenesis and
synergistic effects with other respirable particles. Int J Environ
Res Public Health. 6:445–462. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Benowitz NL: Neurobiology of nicotine
addiction: Implications for smoking cessation treatment. Am J Med.
121(Suppl 1): S3–S10. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sanner T and Grimsrud TK: Nicotine:
Carcinogenicity and effects on response to cancer treatment - A
review. Front Oncol. 5:1962015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Davis R, Rizwani W, Banerjee S, Kovacs M,
Haura E, Coppola D and Chellappan S: Nicotine promotes tumor growth
and metastasis in mouse models of lung cancer. PLoS One.
4:e75242009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Egleton RD, Brown KC and Dasgupta P:
Nicotinic acetylcholine receptors in cancer: Multiple roles in
proliferation and inhibition of apoptosis. Trends Pharmacol Sci.
29:151–158. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nguyen VT, Hall LL, Gallacher G, Ndoye A,
Jolkovsky DL, Webber RJ, Buchli R and Grando SA: Choline
acetyltransferase, acetylcholinesterase, and nicotinic
acetylcholine receptors of human gingival and esophageal epithelia.
J Dent Res. 79:939–949. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Arredondo J, Chernyavsky AI, Jolkovsky DL,
Pinkerton KE and Grando SA: Receptor-mediated tobacco toxicity:
Cooperation of the Ras/Raf-1/MEK1/ERK and JAK-2/STAT-3 pathways
downstream of alpha7 nicotinic receptor in oral keratinocytes.
FASEB J. 20:2093–2101. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schuller HM: Is cancer triggered by
altered signalling of nicotinic acetylcholine receptors? Nat Rev
Cancer. 9:195–205. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nishioka T, Guo J, Yamamoto D, Chen L,
Huppi P and Chen CY: Nicotine, through upregulating pro-survival
signaling, cooperates with NNK to promote transformation. J Cell
Biochem. 109:152–161. 2010.
|
|
20
|
Guo J, Ibaragi S, Zhu T, Luo LY, Hu GF,
Huppi PS and Chen CY: Nicotine promotes mammary tumor migration via
a signaling cascade involving protein kinase C and CDC42. Cancer
Res. 68:8473–8481. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ye YN, Liu ESL, Shin VY, Wu WKK, Luo JC
and Cho CH: Nicotine promoted colon cancer growth via epidermal
growth factor receptor, c-Src, and 5-lipoxygenase-mediated signal
pathway. J Pharmacol Exp Ther. 308:66–72. 2004. View Article : Google Scholar
|
|
22
|
Nishioka T, Kim HS, Luo LY, Huang Y, Guo J
and Chen CY: Sensitization of epithelial growth factor receptors by
nicotine exposure to promote breast cancer cell growth. Breast
Cancer Res. 13:R1132011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Li H, Takayama S, Harada K, Okamoto
T, Iwama I, Fujii E, Ota A, Hidaka K, Kawano NY, et al: Nicotine
induces resistance to erlotinib via cross-talk between α 1 nAChR
and EGFR in the non-small cell lung cancer xenograft model. Lung
Cancer. 88:1–8. 2015. View Article : Google Scholar
|
|
24
|
Wang S, Takayama K, Tanaka K, Takeshita M,
Nakagaki N, Ijichi K, Li H and Nakanishi Y: Nicotine induces
resistance to epidermal growth factor receptor tyrosine kinase
inhibitor by α1 nicotinic acetylcholine receptor-mediated
activation in PC9 cells. J Thorac Oncol. 8:719–725. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhao W, Yan J, Gao L, Zhao J, Zhao C, Gao
C, Luo X and Zhu X: Cdk5 is required for the neuroprotective effect
of transforming growth factor-β1 against cerebral
ischemia-reperfusion. Biochem Biophys Res Commun. 485:775–781.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang S, Deng Z, Yao C, Huang P, Zhang Y,
Cao S and Li X: AT7867 Inhibits human colorectal cancer cells via
AKT-dependent and AKT-independent mechanisms. PLoS One.
12:e01695852017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
von Roemeling CA, Marlow LA, Kennedy WP,
Kennedy GT, Copland JA and Menefee ME: Preclinical evaluation of
the mTOR inhibitor, temsirolimus, in combination with the
epothilone B analog, ixabepilone in renal cell carcinoma. Am J
Cancer Res. 3:390–401. 2013.PubMed/NCBI
|
|
28
|
Minna JD: Nicotine exposure and bronchial
epithelial cell nicotinic acetylcholine receptor expression in the
pathogenesis of lung cancer. J Clin Invest. 111:31–33. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Heusch WL and Maneckjee R: Signalling
pathways involved in nicotine regulation of apoptosis of human lung
cancer cells. Carcinogenesis. 19:551–556. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kurai J, Chikumi H, Hashimoto K, Takata M,
Sako T, Yamaguchi K, Kinoshita N, Watanabe M, Touge H, Makino H, et
al: Therapeutic antitumor efficacy of anti-epidermal growth factor
receptor antibody, cetuximab, against malignant pleural
mesothelioma. Int J Oncol. 41:1610–1618. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Takada H, Ibaragi S, Eguchi T, Okui T,
Obata K, Masui M, Morisawa A, Takabatake K, Kawai H, Yoshioka N, et
al: Semaphorin 4D promotes bone invasion in head and neck squamous
cell carcinoma. Int J Oncol. 51:625–632. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Harrell MI, Iritani BM and Ruddell A:
Tumor-induced sentinel lymph node lymphangiogenesis and increased
lymph flow precede melanoma metastasis. Am J Pathol. 170:774–786.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bagdas D, Meade JA, Alkhlaif Y, Muldoon
PP, Carroll FI and Damaj MI: Effect of nicotine and alpha-7
nicotinic modulators on visceral pain-induced conditioned place
aversion in mice. Eur J Pain. Apr 10–2018.Epub ahead of print.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wild R, Fager K, Flefleh C, Kan D, Inigo
I, Castaneda S, Luo FR, Camuso A, McGlinchey K and Rose WC:
Cetuximab preclinical antitumor activity (monotherapy and
combination based) is not predicted by relative total or activated
epidermal growth factor receptor tumor expression levels. Mol
Cancer Ther. 5:104–113. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shibata T, Kan H, Murakami Y, Ureshino H,
Watari K, Kawahara A, Kage M, Hattori S, Ono M and Kuwano M: Y-box
binding protein-1 contributes to both HER2/ErbB2 expression and
lapatinib sensitivity in human gastric cancer cells. Mol Cancer
Ther. 12:737–746. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Matsui T, Ota T, Ueda Y, Tanino M and
Odashima S: Isolation of a highly metastatic cell line to lymph
node in human oral squamous cell carcinoma by orthotopic
implantation in nude mice. Oral Oncol. 34:253–256. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chikamatsu K, Reichert TE, Kashii Y, Saito
T, Kawashiri S, Yamamoto E and Whiteside TL: Immunotherapy with
effector cells and IL-2 of lymph node metastases of human
squamous-cell carcinoma of the head and neck established in nude
mice. Int J Cancer. 82:532–537. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nickell JR, Grinevich VP, Siripurapu KB,
Smith AM and Dwoskin LP: Potential therapeutic uses of mecamylamine
and its stereoisomers. Pharmacol Biochem Behav. 108:28–43. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shytle RD, Penny E, Silver AA, Goldman J
and Sanberg PR: Mecamylamine (Inversine): An old antihypertensive
with new research directions. J Hum Hypertens. 16:453–457. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Young HS, Herbette LG and Skita V:
α-bungarotoxin binding to acetylcholine receptor membranes studied
by low angle X-ray diffraction. Biophys J. 85:943–953. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dasgupta P, Rizwani W, Pillai S, Kinkade
R, Kovacs M, Rastogi S, Banerjee S, Carless M, Kim E, Coppola D, et
al: Nicotine induces cell proliferation, invasion and
epithelial-mesenchymal transition in a variety of human cancer cell
lines. Int J Cancer. 124:36–45. 2009. View Article : Google Scholar
|
|
42
|
Wang C, Niu W, Chen H, Shi N, He D, Zhang
M, Ge L, Tian Z, Qi M, Chen T, et al: Nicotine suppresses apoptosis
by regulating α7nAChR/Prx1 axis in oral precancerous lesions.
Oncotarget. 8:75065–75075. 2017.PubMed/NCBI
|
|
43
|
Arredondo J, Chernyavsky AI and Grando SA:
Nicotinic receptors mediate tumorigenic action of tobacco-derived
nitrosamines on immortalized oral epithelial cells. Cancer Biol
Ther. 5:511–517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fujiwara T, Eguchi T, Sogawa C, Ono K,
Murakami J, Ibaragi S, Asaumi JI, Okamoto K, Calderwood SK and
Kozaki KI: Anti-EGFR antibody cetuximab is secreted by oral
squamous cell carcinoma and alters EGF-driven mesenchymal
transition. Biochem Biophys Res Commun. 503:1267–1272. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ono K, Eguchi T, Sogawa C, Calderwood SK,
Futagawa J, Kasai T, Seno M, Okamoto K, Sasaki A and Kozaki KI:
HSP-enriched properties of extracellular vesicles involve survival
of metastatic oral cancer cells. J Cell Biochem. 119:7350–7362.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Togashi Y, Hayashi H, Okamoto K, Fumita S,
Terashima M, de Velasco MA, Sakai K, Fujita Y, Tomida S, Nakagawa
K, et al: Chronic nicotine exposure mediates resistance to EGFR-TKI
in EGFR-mutated lung cancer via an EGFR signal. Lung Cancer.
88:16–23. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kajizono M, Saito M, Maeda M, Yamaji K,
Fujiwara S, Kawasaki Y, Matsunaga H and Sendo T: Cetuximab-induced
skin reactions are suppressed by cigarette smoking in patients with
advanced colorectal cancer. Int J Clin Oncol. 18:684–688. 2013.
View Article : Google Scholar
|
|
48
|
Xu J, Huang H, Pan C, Zhang B, Liu X and
Zhang L: Nicotine inhibits apoptosis induced by cisplatin in human
oral cancer cells. Int J Oral Maxillofac Surg. 36:739–744. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Weller MA, Ward MC, Berriochoa C, Reddy
CA, Trosman S, Greskovich JF, Nwizu TI, Burkey BB, Adelstein DJ and
Koyfman SA: Predictors of distant metastasis in human
papillomavirus-associated oropharyngeal cancer. Head Neck.
39:940–946. 2017. View Article : Google Scholar : PubMed/NCBI
|