Introduction
In recent decades, apoptosis has been targeted as a
therapeutic strategy to induce cancer cell death; therefore,
several currently available anticancer drugs are designed to
activate the apoptotic pathway directly or indirectly. However,
numerous cancer cell types have acquired resistance to apoptosis by
altering apoptosis-associated signaling pathways (e.g., loss of
caspase 3), which has been a major obstacle to traditional
chemotherapy. Therefore, other types of programmed cell death (PCD)
have been explored. Dysregulation of PCD may induce various
diseases, including numerous types of cancer. Initially, cell death
has traditionally been divided into two types: Regulated cell
death, i.e., apoptosis, and unregulated and accidental cell death,
i.e., necrosis. However, more recent work has led to further
information regarding the concept of cell death. One conceptual
alteration is that another type of necrosis has been identified,
which is considered to be a type of PCD; this type of necrosis has
been named necroptosis (1-4). Therefore, it has been accepted that
two types of necrosis exist, unregulated necrosis and regulated
necroptosis. The present study focused on targeting necroptosis as
an alternative strategy for treating cancer.
Necroptosis is a caspase-independent form of cell
death, which is morphologically characterized by a loss of physical
integrity, including swelling of organelles, increase in cellular
volume, disruption of plasma and mitochondrial membranes, and
release of intracellular contents. The most well studied signaling
pathway for the induction of necroptosis is tumor necrosis
factor-mediated necroptosis, which is mainly mediated by activation
of three core regulators: Receptor-interacting serine/threonine
protein kinase (RIP)1, RIP3 and mixed lineage kinase domain-like
protein (MLKL) (1,5-9).
These regulators form three complexes (membrane-bound complex I,
complex II and necrosome) sequentially, by changing binding
partners (1,5-7,10).
Necroptosis can be induced by various types of stress, including
extreme nutrient deficiency. Unlike normal cells, cancer cells
require unusually large amounts of nutrients to support their
uncontrolled growth and proliferation; therefore, they are more
easily exposed to severe nutrient deficiencies compared with normal
cells. However, many cancer cells develop tolerance and overcome
nutrient limitations by increasing nutrient uptake through
alterations in metabolic signaling pathways or by activating
angiogenesis (11-16). Therefore, it is important to
elucidate these mechanisms, in order to develop novel therapeutic
approaches. Therefore, this study initially focused on p53 and
AMP-activated protein kinase (AMPK), as they are well known to
serve various important roles under nutrient-deprived conditions
and are considered promising therapeutic targets for cancer
treatment.
One of the most powerful tumor suppressors, p53, is
considered an appealing target for effective cancer therapy.
Notably, approximately half of human cancers are known to have
defective p53 expression, and p53 has been reported to be involved
in several types of cell death [e.g., apoptosis,
caspase-independent apoptosis (CIA), autophagy-mediated cell death,
necroptosis, entosis, anoikis, paraptosis, pyroptosis, ferroptosis,
mitotic catastrophe and efferocytosis] (17). It is widely accepted that p53
sensitizes normal cells to stressful conditions, including nutrient
starvation. Mutations or deletions of p53 in cancer cells may
overcome cell death pathways; therefore, these cells may exhibit
resistance to chemotherapeutic drugs. Considering its vital roles
in the regulation of cell death in response to various stressors,
determining the role of p53 in the regulation of necroptosis may
present a novel therapeutic strategy for cancer treatment.
AMPK also serves several vital roles in the
regulation of metabolism and energy homeostasis. Cellular stress,
such as nutrient starvation, induces the activation of AMPK, which
is important for restoring intracellular energy balance. Activated
AMPK inhibits energy-consuming biosynthetic processes and activates
ATP biosynthesis (18). The most
well known function of AMPK is its involvement in autophagy, in
which it directly or indirectly promotes autophagy. In normal
cells, AMPK acts as a metabolic tumor suppressor protein by
activating autophagy to remove damaged or unnecessary proteins and
organelles (19-25). However, it also functions as an
oncogenic protein by protecting cancer cells from cell death by
supporting their uncontrolled growth and proliferation (22,26,27).
In contrast to its well-known function in autophagy, few studies
have reported on its role in necroptosis.
The present study aimed to determine how cancer
cells evade necroptosis under nutrient starvation, and revealed
that enhanced AMPK activation may suppress necroptosis in p53-null
cells (p53−/−) but not in wild type cells
(p53+/+) under the same conditions.
Materials and methods
Cell lines, cell culture and cell
treatment
The colorectal carcinoma cell lines, HCT116
p53+/+ and HCT116 p53−/−, were a gift from Dr
B. Vogelstein (Johns Hopkins University, Baltimore, MD, USA)
(28). The cells were cultured in
Roswell Park Memorial Institute (RPMI)-1640 medium (WelGENE, Inc.,
Gyeongsan, South Korea) supplemented with 10% fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and
1% Antibiotic-Antimycotic solution (cat. no. 15240-06; Gibco;
Thermo Fisher Scientific, Inc.). Both cell lines were grown at 37°C
in a humidified atmosphere containing 5% CO2 and 95%
air. Dorsomorphin dihydrochloride (DM; cat. no. sc-361173; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA) and
5-aminoimidazole-4-carboxamide ribonucleotide (AICAR; cat. no.
BML-EI330; Enzo Life Sciences, Inc., Farmingdale, NY, USA) were
used to inhibit and activate AMPK, respectively. Cells were treated
with 0, 5 and 10 µM DM for 48 h, or with 0, 75, 150 and 300 µM
AICAR for 48 h at 37°C.
Cell viability and proliferation
analysis
Cell viability was analyzed using the Cell
Viability, Proliferation & Cytotoxicity Assay kit (EZ-CYTOX,
cat. no. EZ-3000; DoGenBio, Seoul, South Korea), according to the
manufacturer's protocol, in three replicates. Briefly, HCT116
p53+/+ and HCT116 p53−/− cells
(~2×104 cells/well) were seeded in 96-well plates for 24
h, and the media were replaced with RPMI 1640 media with or without
10% FBS for 24, 48, or 72 h. Cell viability was evaluated by
measuring absorbance at 450 nm using a Gemini XPA Microplate
Reader. Cell proliferation was analyzed by Trypan blue (EBT-001;
NanoEnTek, Inc., Seoul, South Korea) staining under the same
conditions; 10 µl cells (~105 cells) and 10 µl Trypan
blue were mixed in 1.5 ml microcentrifuge tubes at room temperature
and were then placed into an EVE™ cell counting slide (NanoEnTek,
Inc.), in order to count the number of live or dead cells. Cell
proliferation was determined by counting the number of live cells
at various time points (0, 24, 48 and 72 h) after culturing in
complete media (CM) or serum-starved media (SS). Morphological
images of cells were captured using an optical microscope with IS
capture software (KI-400F; Korea Lab Tech, Seongnam, South Korea)
at ×100 magnification.
Analysis of necroptosis, apoptosis and
autophagy
Necroptosis, apoptosis and autophagy were detected
using western blotting for the detection of the following
corresponding biomarkers: Cyclophilin (Cyp)A and high mobility
group box 1 (HMGB1) for necroptosis (29); cleaved poly (ADP-ribose) polymerase
(PARP) for apoptosis; and p62/sequestome 1 (SQSTM1) and
microtubule-associated protein 1A/1B-light chain 3 (LC3) for
autophagy. Necroptosis was also analyzed using the Green
Fluorescent Protein-Certified Apoptosis/Necrosis Detection kit
(cat. no. ENZ-51002; Enzo Life Sciences, Inc.), according to the
manufacturer's protocol. This kit contained 7-amino-actinomycin D
(7-AAD) and Annexin V, which were used to detect necroptosis and
apoptosis, respectively.
Western blotting
HCT116 p53+/+ and HCT116
p53−/− cells (~5x106/ml) were centrifuged, (Smart R17;
Hanil Scientific, Inc., Gimpo, South Korea) at 3,000 × g for 3 min
at 4°C, washed in ice-cold PBS and lysed in
radioimmunoprecipitation assay (RIPA) lysis buffer (50 mM Tris, pH
7.5; 48 mM NaCl; 1% Triton X-100 and 1 mM EGTA) with 0.5 mM
Na3VO4, 1 mM DTT and 1 µl premade protease
inhibitor cocktail III (cat. no. P-1512; AG Scientific, Inc., San
Diego, CA, USA)/ml RIPA solution. Protein concentration was
quantified using a protein assay kit (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA), and 10 or 30 µg protein was loaded into each
lane. Subsequently, proteins were separated by 10 or 12% SDS-PAGE
and were then transferred onto an Immobilon-P®
polyvinylidene fluoride transfer membrane (cat. no. IPVH00010; EMD
Millipore, Billerica, MA, USA). Membranes were then blocked using
5% skimmed milk in PBS for 30-60 min at 20-25°C, and incubated
overnight at 4°C with the primary antibodies. After washing with
Tris-buffered saline containing 1% Tween-20 (cat. no. T1027;
Biosesang, Seongnam, Korea), membranes were incubated with the
corresponding secondary antibodies: Anti-rabbit (cat. no. 7074S,
1:5,000; Cell Signaling Technology, Inc., Danvers, MA, USA),
anti-mouse (cat. no. sc-516102, 1:5,000; Santa Cruz Biotechnology,
Inc.) and anti-goat (cat. no. sc-2020, 1:5,000; Santa Cruz
Biotechnology, Inc.). Immunodetection was performed using the
PowerOpti-ECL western blotting detection reagent (cat. no. LR01-02;
BioNote, Inc., Hwaseong, South Korea). The antibodies used in this
study were as follows: Mouse monoclonal anti-p53 (cat. no. sc-126,
1:10,000; Santa Cruz Biotechnology, Inc.), rabbit monoclonal
anti-AMPKα (cat. no. 2603P, 1:3,000) and anti-phosphorylated
(p)-AMPKα (Thr172) (cat. no. 2535S, 1:3,000) (both Cell Signaling
Technology, Inc.), rabbit polyclonal anti-CypA (cat. no. BML-SA296,
1:3,000), rabbit anti-HMGB1 (1:3,000, cat. no. ALX-210-964) (both
Enzo Life Sciences, Inc.), rabbit polyclonal anti-PARP (cat. no.
9542S, 1:3,000), rabbit poly-clonal anti-p62/SQSTM1 (cat. no. 5114,
1:3,000) (both Cell Signaling Technology, Inc.), mouse monoclonal
anti-LC3 (cat. no. ALX-803-082, 1:2,500; Enzo Life Sciences, Inc.)
and mouse monoclonal anti-β-actin (cat. no. sc-47778, 1:5,000;
Santa Cruz Biotechnology, Inc.). β-actin was used as a loading
control. To measure the extracellular levels of necroptosis
biomarkers (CypA and HMGB1), 80 µl media were collected from 10 ml
cell culture media and mixed with 20 µl 5X SDS-PAGE loading buffer
(cat. no. S2002; Biosesang), of which 20 µl was separated by 12%
SDS-PAGE and underwent western blotting. The results of western
blot analysis were semi-quantified using ImageJ software (version
1.51u; https://imagej.nih.gov/ij/) supplied by
the National Institutes of Health (Bethesda, MD, USA) and relative
intensity compared with β-actin is presented in the bar graphs.
Measurement of reactive oxygen species
(ROS)
HCT116 p53+/+ and HCT116
p53−/− cells were cultured in RPMI media with or without
10% FBS for 24, 48 or 72 h. The cells were then collected in 15 ml
conical tubes by centrifugation at ~200 × g for 2 min.
Subsequently, ~5x104 cells/50 µl PBS were added to each
well of a 96-well plate and 50 µl 200 µM
2',7'-dichlo-rodihydrofluorescein diacetate (DCFH-DA; cat. no.
D399; Invitrogen; Thermo Fisher Scientific, Inc.) was added into
each well using a multichannel-pipette (PIPETMAN; Gilson
Incorporated, Middleton, WI, USA), in order to achieve a final
concentration of 100 µM DCFH-DA ROS levels were detected by
measuring fluorescence at an excitation wavelength of 485 nm and an
emission wavelength of 535 nm every 5 min for 30 min with a Gemini
XPA microplate reader.
Measurement of mitochondrial membrane
potential (MMP)
MMP was assessed using a Mito-ID Membrane Potential
Cytotoxicity kit (cat. no. ENZ-51019; Enzo Life Sciences, Inc.),
according to the manufacturer's protocol. Briefly, HCT116
p53+/+ and HCT116 p53−/− cells
(~2×105 cells/well) were cultured in 96-well plates (BD
Falcon; BD Biosciences, Franklin Lakes, NJ, USA) in RPMI media with
or without 10% FBS for 48 or 72 h. Subsequently, Mito-ID membrane
potential dye loading solution was added to each well and was
incubated for 3 h at room temperature. MMP was assessed by
measuring the resulting fluorescence with a Gemini XPA microplate
reader, at an excitation wavelength of 480 nm and an emission
wavelength of 590 nm.
Measurement of cellular ATP levels
Cellular ATP levels were measured using the
Luminescent ATP Detection Assay kit (cat. no. ab113849; Abcam,
Cambridge, MA, USA), according to the manufacturer's protocol.
Briefly, HCT116 p53+/+ and HCT116 p53−/−
cells were seeded at ~2×104/well in 96-well plates for
24 h. Subsequently, cells were incubated in RPMI media with or
without 10% FBS and were cultured for 24 h. Detergent solution (50
µl) was then added to each well and the plate was agitated for 5
min at 600-700 rpm in an orbital shaker, in order to lyse the
cells. Subsequently, 50 µl reconstituted substrate solution was
added and the plate was agitated for a further 5 min. After 10 min
in the dark, cellular ATP levels were measured by detecting
luminescence using the GloMax® Discover multimode
microplate reader (Promega Corporation, Madison, WI, USA).
Statistical analysis
Data are presented as the means ± standard deviation
from three independent experiments. S tatistical analysis was
performed using Student's t-test to compare two different groups or
one-way analysis of variance, followed by Bonferroni's multiple
comparisons test, to compare multiple groups. SPSS statistics
version 23 was used to analyze data (IBM Corporation, Armonk, NY,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
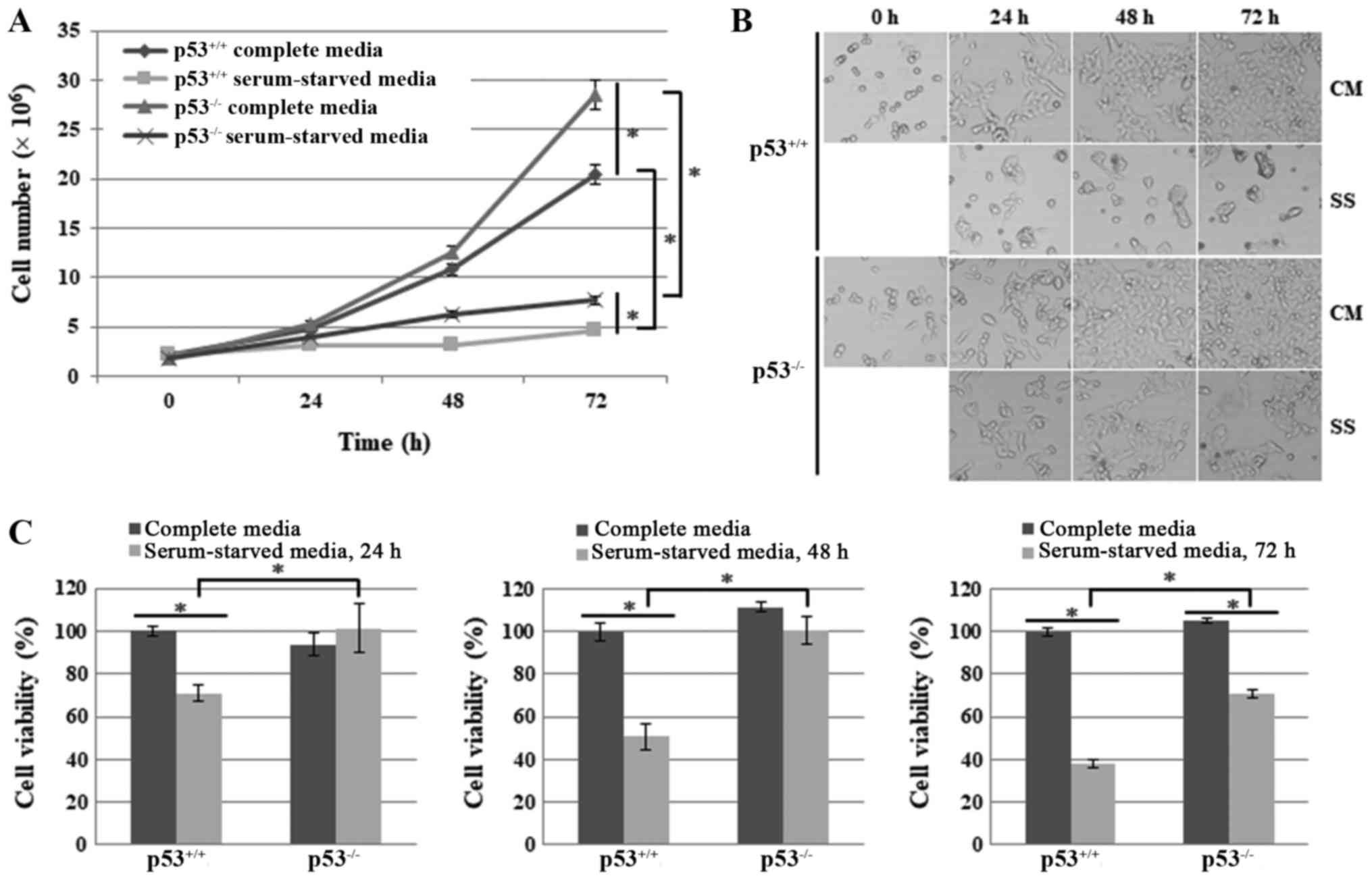
Cell proliferation is increased and cell
death is reduced in HCT116 p53−/− human colon cancer
cells under nutrient/serum starvation
To explore the effects of p53 on necroptosis under
nutrient/serum starvation, the present study compared cell
proliferation and cell death between HCT116 p53+/+ and
HCT116 p53−/− cells after culturing them in CM or SS for
24, 48 or 72 h. HCT116 p53−/− cells exhibited a higher
growth rate compared with HCT116 p53+/+ cells under both
conditions (Fig. 1A). Cell images
were also obtained under an optical microscope. Microscope images
revealed that, in response to serum starvation, HCT116
p53+/+ cells stopped proliferating and exhibited the
typical morphological alterations of cell death, including
detachment from the bottom of the plate and generation of
round-shaped, shrunk or swollen cells, whereas HCT116
p53−/− cells exhibited far fewer morphological
alterations, although proliferation was greatly decreased (Fig. 1B). The cell viability assay also
revealed a lower level of cell growth and viability in HCT116
p53+/+ cells compared with HCT116 p53−/−
cells following serum deprivation for 24, 48 and 72 h (Fig. 1C). Taken together, these results
suggested that p53 may suppress cell proliferation and enhance cell
death under the stress of nutrient/serum starvation.
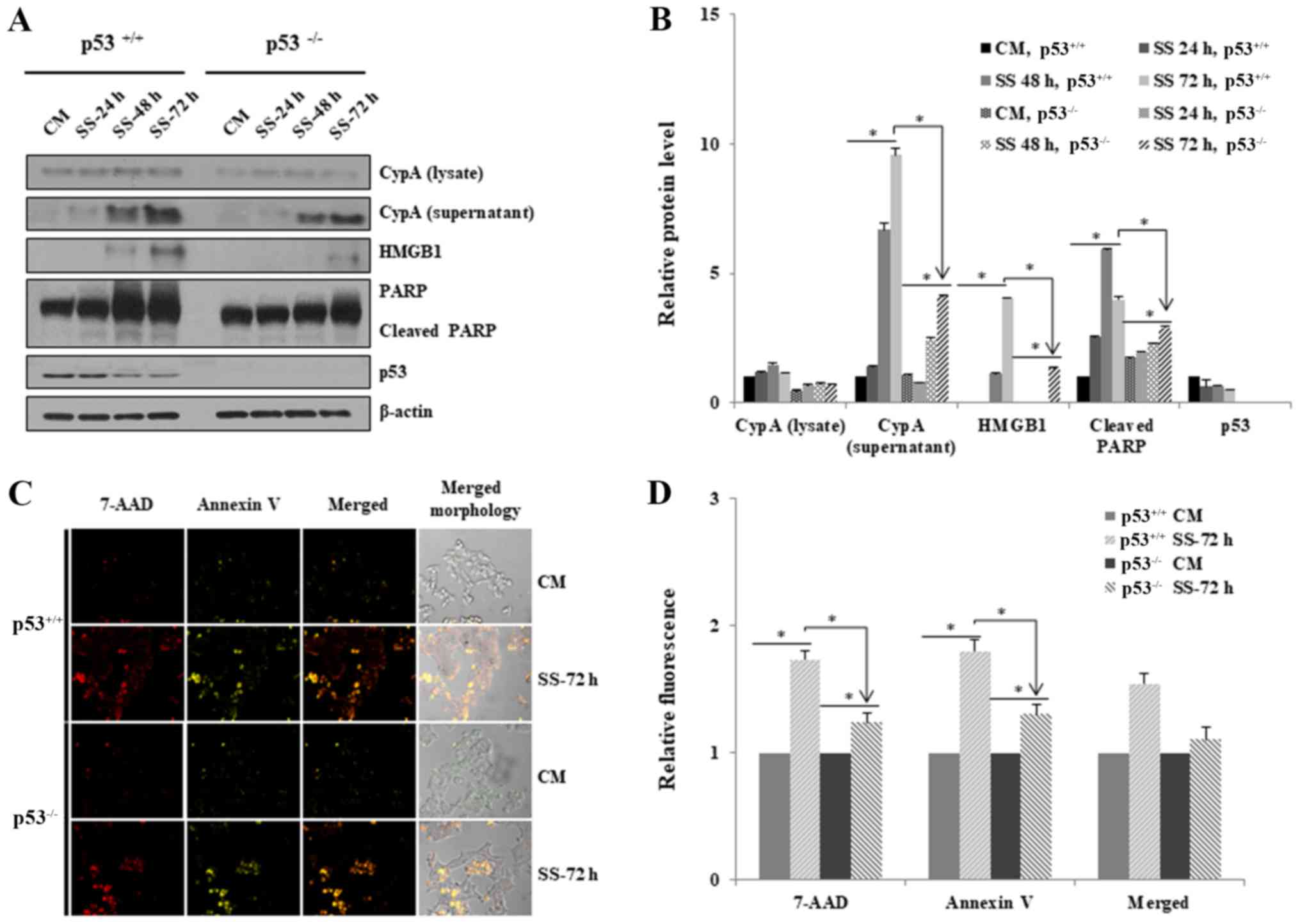
Necroptosis is reduced in HCT116
p53−/− cells in response to nutrient/serum
starvation
HCT116 p53−/− cells exhibited reduced
cell death under serum deprivation. To investigate whether loss of
p53 exerts inhibitory effects on necroptosis, the extracellular
levels of necroptosis biomarkers, CypA and HMGB1, were detected
using western blotting after HCT116 p53+/+ and HCT116
p53−/− cells were grown in RPMI media with or without
serum for 24, 48 and 72 h (Fig.
2). The export of both proteins into the extracellular media
was significantly increased after 48 h serum starvation in both
cell lines, indicating the induction of necroptosis (Fig. 2A and B). However, the extracellular
levels were much higher for HCT116 p53+/+ cells than for
HCT116 p53−/− cells after 72 h, thus indicating that
necroptosis was suppressed in HCT116 p53−/− cells
(Fig. 2A and B). In addition, PARP
cleavage was increased in HCT116 p53+/+ cells compared
with in HCT116 p53−/− cells, thus suggesting that the
loss of p53 suppressed apoptotic cell death, as well as necroptosis
(Fig. 2A and B). The expression
levels of p53 were decreased in a time-dependent manner in HCT116
p53+/+ cells under the conditions of serum starvation
(Fig. 2A and B).
The induction of necroptosis and apoptosis in HCT116
p53+/+ and HCT116 p53−/− cells was examined
by confocal microscopy; 7-AAD and Annexin V staining were used to
detect necroptosis and apoptosis, respectively (Fig. 2C and D). In response to serum
starvation for 72 h, marked 7-AAD and Annexin V fluorescence was
detected in both cell lines, thus indicating that necroptosis and
apoptotic cell death pathways were induced; much higher levels of
necroptosis and apoptosis were detected in HCT116 p53+/+
cells compared with HCT116 p53−/− cells (Fig. 2C and 2D).
Taken together, these results suggested that the
absence of p53 in HCT116 cells may suppress necroptosis to a higher
degree than in p53+/+ cells under serum starvation.
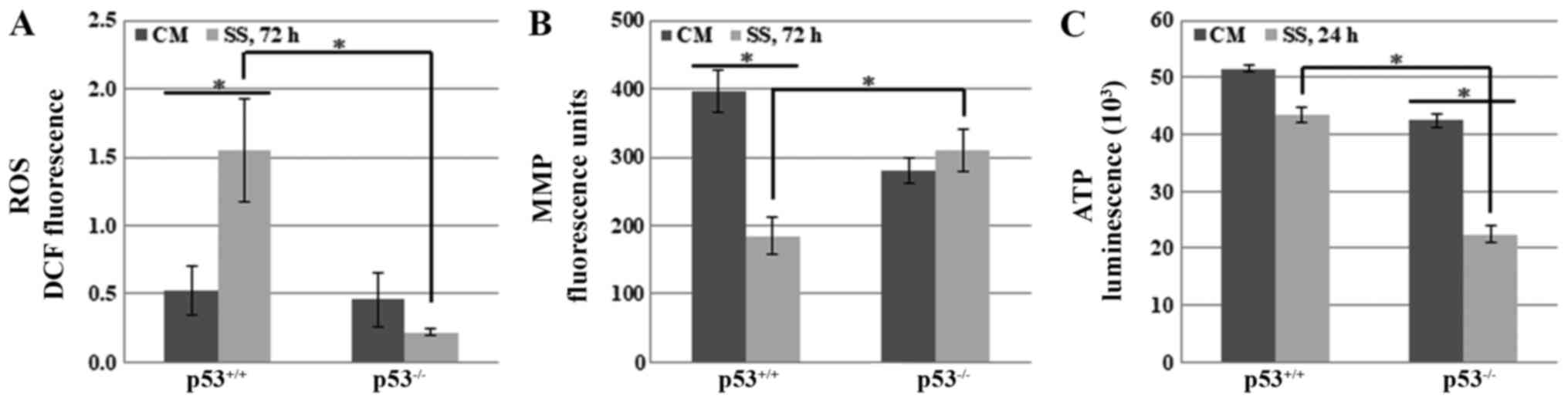
Effects of p53 on mitochondria-mediated
necroptosis under serum starvation
To determine the role of p53 in necroptosis, it was
hypothesized that p53 may induce it by regulating mitochondrial
functions, including the production of ROS, maintenance of the MMP,
or generation of ATP. These functions are understood to be strongly
associated with several types of cell death; very high levels of
ROS, and low levels of MMP and ATP, are known to accelerate cell
death pathways (30-39). Therefore, the present study
detected the cellular levels of ROS and ATP, as well as the MMP, in
HCT116 p53+/+ and HCT116 p53−/− cells under
serum starvation.
Cellular ROS levels were detected in HCT116
p53+/+ and HCT116 p53−/− cells cultured in
media with or without serum for various durations. The results
revealed that there was no significant difference between the cell
lines until 48 h (data not shown). However, ROS levels were
significantly increased after 72 h in HCT116 p53+/+
cells, whereas they were slightly decreased in HCT116
p53−/− cells (Fig. 3A).
These data suggested that p53-mediated necroptosis, at least in
part, may occur by increasing cellular ROS levels under serum
starvation.
Another mitochondrial mediator of cell death, MMP,
was also compared between HCT116 p53+/+ and HCT116
p53−/− cells. Cells were grown in SS for 24, 48 and 72
h. MMP was markedly decreased after 72 h in HCT116
p53+/+ cells, whereas no significant difference was
observed in HCT116 p53−/− cells (Fig. 3B). These data indicated that p53
may induce necroptosis by decreasing MMP in response to serum
starvation.
Notably, several types of cell death are accompanied
by ATP depletion (40-43). Therefore, the effects of p53 on
cellular ATP levels were assessed in response to serum starvation.
The results indicated that ATP levels were decreased in both cell
lines. However, it was unexpectedly observed that ATP levels were
more markedly decreased in HCT116 p53−/− cells, whereas
they were only slightly reduced in HCT116 p53+/+ cells
after 24 h (Fig. 3C).
Taken together, these findings supported the
hypothesis that p53 may accelerate necroptosis, at least partially,
by increasing ROS levels and decreasing MMP in HCT116 cells under
serum starvation.
AMPK activation is increased in
p53−/− cells under serum starvation
It is well known that nutrient starvation (e.g.,
depletion of glucose, amino acids or serum) leads to decreased
intracellular ATP levels and that further extreme depletion may
eventually induce cell death. Based on the importance of cellular
ATP levels with regards to cell survival, it was expected that ATP
levels would be lower in HCT116 p53+/+ cells, due to the
higher levels of necroptosis. However, the present data revealed
that ATP levels were lower in HCT116 p53−/− cells in
both CM and SS (Fig. 3C). In
addition, much higher levels of proliferation were detected in
HCT116 p53−/− cells compared with in HCT116
p53+/+ cells under serum starvation (Fig. 1). Therefore, it was hypothesized
that ATP levels may be lower in p53−/− cells due to
uncontrolled rapid cell proliferation and growth, which may cause
much higher consumption of ATP. ATP depletion is the main signal
that induces AMPK activation by phosphorylation of Thr172 (44-46).
The present study demonstrated that serum starvation markedly
increased phosphorylation of AMPK at Thr172 in HCT116
p53−/− cells compared with in HCT116 p53+/+
cells. Even though there was a slight variation in the expression
levels of total AMPK (Fig. 4A and
B), the ratio of p-AMPK (Thr172) to total AMPK (p-AMPK/AMPK)
indicated the significant increase in levels of AMPK
phosphorylation (Fig. 4B).
Therefore, it was hypothesized that AMPK activation may be involved
in necroptotic cell death in a p53-dependent manner in response to
serum depletion.
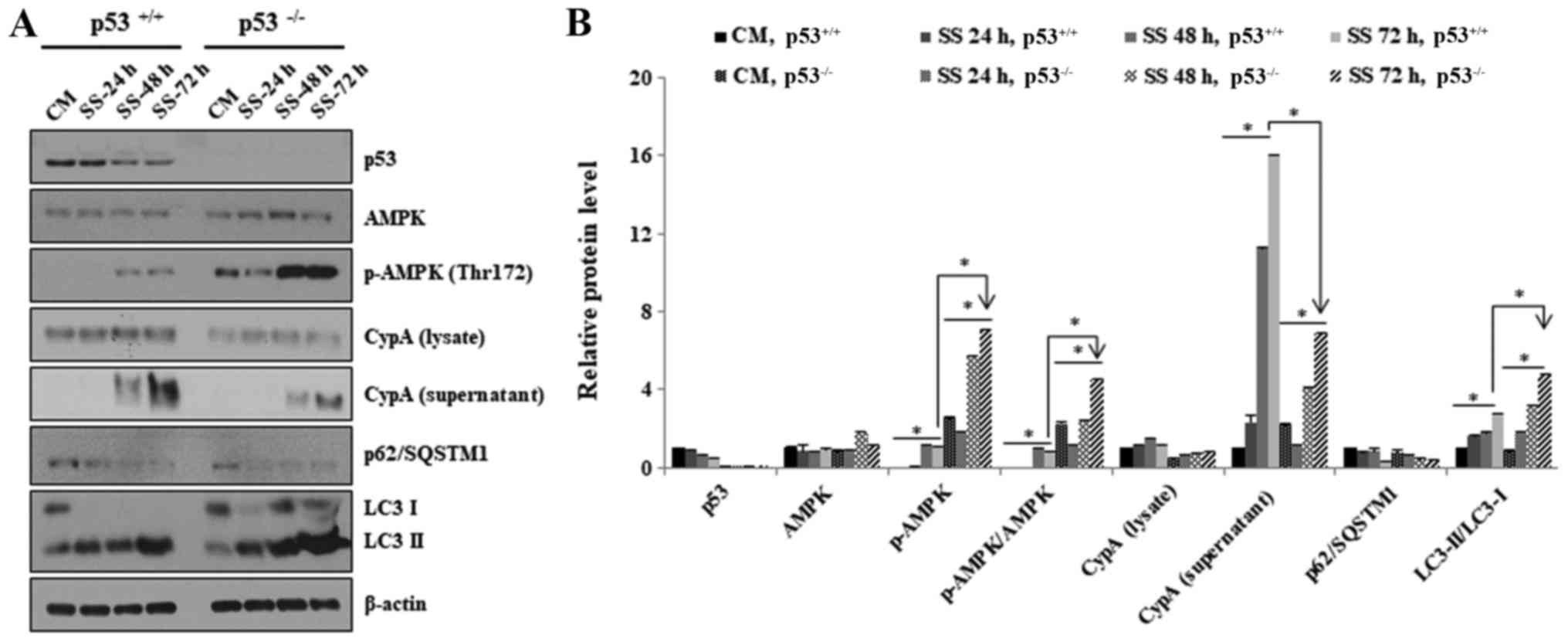 | Figure 4Negative effects of p53 on AMPK
activation in response to serum starvation. (A) HCT116
p53+/+ and HCT116 p53−/− cells were grown in
CM or SS for 24, 48 and 72 h. Western blotting was used to
determine the expression levels of AMPK, p-AMPK (Thr172), CypA,
p62/SQSTM1 and LC3. β-actin was used as a loading control. All
experiments were performed independently at least three times and
representative data are shown. (B) Western blots were
semi-quantified using ImageJ. *P<0.05. The ratio of
p-AMPK (Thr172) to total AMPK is shown as p-AMPK/AMPK. AMPK,
AMP-activated protein kinase; CM, complete media; CypA, cyclophilin
A; LC3, microtubule-associated protein 1A/1B-light chain 3; p,
phosphorylated; SQSTM1, sequestome 1; SS, serum-starved media. |
A well-known function of activated AMPK is the
induction of autophagy, as one of the master regulators.
Accordingly, autophagy was analyzed by detecting two well-known
biomarkers, p62/SQSTM1 and LC3. The results revealed that serum
deficiency decreased the expression levels of p62/SQSTM1 and
increased the conversion ratio of LC3-I to LC3-II in HCT116
p53−/− and HCT116 p53+/+ cells, thus
indicating autophagic induction (Fig.
4A and B). However, p62/SQSTM1 protein levels were decreased to
a slightly higher level in HCT116 p53−/− cells after 24
h (Fig. 4A and B). In addition,
the conversion ratio from LC3-I to LC3-II was also greater in
HCT116 p53−/− cells after 48 h (Fig. 4A and B). These data suggested that
AMPK may be activated in HCT116 p53−/− cells at a higher
level compared with in HCT116 p53+/+ cells.
Taken together, these results indicated that AMPK
activation was highly induced in p53−/− compared with in
p53+/+ cells, probably due to reduced ATP levels.
AMPK activation alleviates necroptosis in
HCT116 p53−/− cells but not in HCT116 p53+/+
cells under serum starvation
To investigate whether activated AMPK is responsible
for the inhibition of necroptotic cell death in p53−/−
cells, the effects of AMPK activation on necroptosis were examined
using an AMPK inhibitor and AMPK activator.
AMPK activity was suppressed using the potent and
selective AMPK inhibitor, DM. HCT116 p53+/+ and HCT116
p53−/− cells were exposed to CM or SS accompanied by DM
at various doses (0, 5 and 10 µM) for 48 h (Fig. 5A and B). Phosphorylation of AMPK at
residue Thr172 was significantly decreased following treatment with
5 and 10 μM DM, thus indicating that AMPK activation was
successfully inhibited (Fig. 5A and
B). Notably, the export of CypA was markedly increased in
HCT116 p53−/− cells in response to DM, even in CM
(Fig. 5A and B); in addition, no
significant differences were detected in CypA export between HCT116
p53+/+ and HCT116 p53−/− cells following DM
treatment (Fig. 5A and B). These
findings strongly suggested that serum starvation-induced AMPK
activation effectively suppressed necroptosis in the
p53−/− cells.
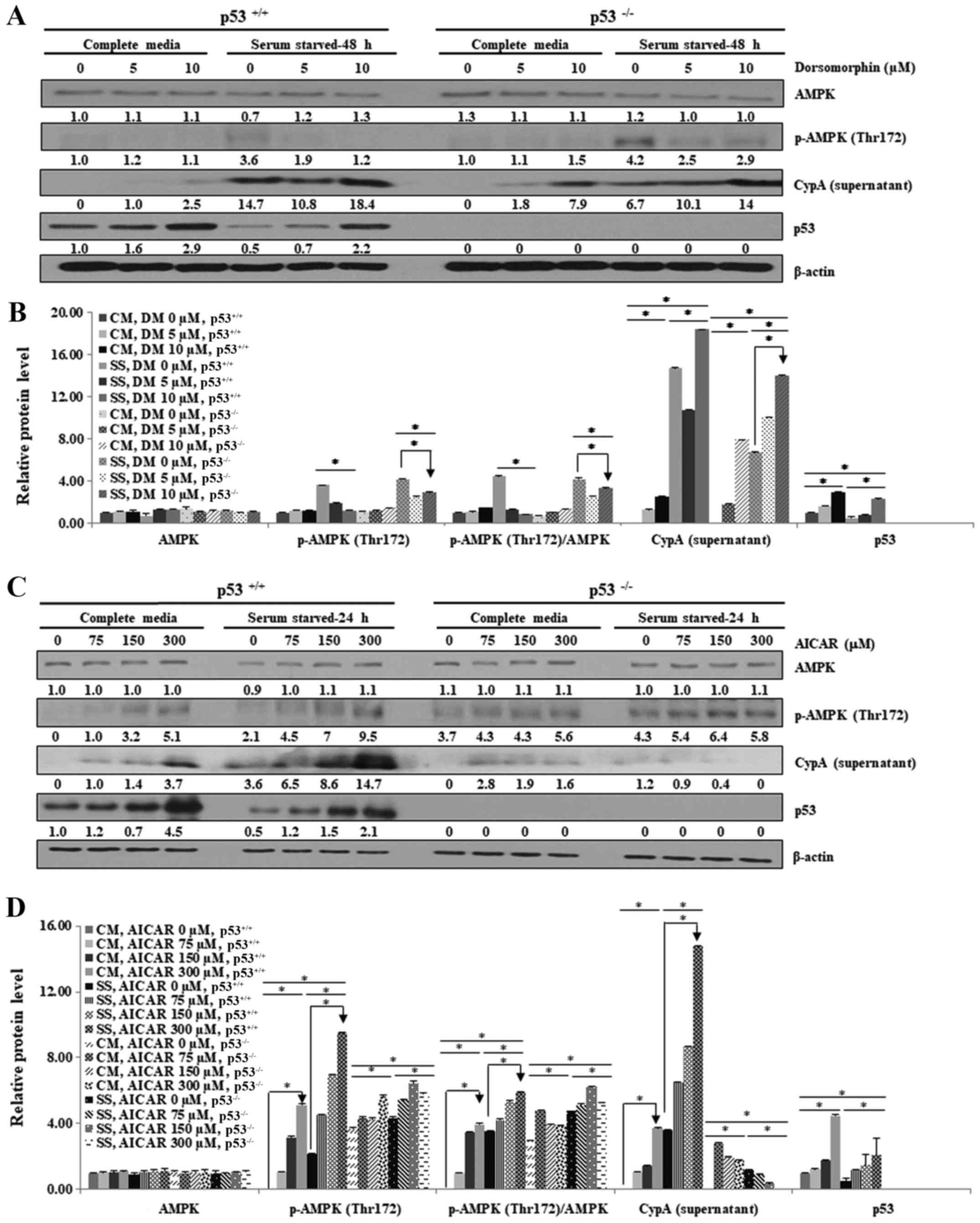 | Figure 5AMPK activation alleviates
necroptosis in HCT116 p53−/− cells but not in HCT116
p53+/+ cells under serum starvation. HCT116
p53+/+ and HCT116 p53−/− cells were cultured
in CM or SS for the indicated durations. Cells were treated with (A
and B) 0, 5 and 10 µM DM for 48 h or (C and D) 0, 75, 150 and 300
µM AICAR for 48 h. (A and C) Lysates were obtained and analyzed by
western blotting, in order to detect phosphorylation of AMPK on the
Thr172 residue, and the expression levels of CypA and p53. β-actin
was used as a loading control. (B and D) Western blotting results
were semi-quantified using ImageJ. The experiment was performed
independently at least three times and representative data are
shown. *P<0.05. Numbers represent relative intensity
versus β-actin. The ratio of p-AMPK (Thr172) to total AMPK is shown
as p-AMPK/AMPK. AICAR, 5-aminoimidazole-4-carboxamide
ribonucleotide; AMPK, AMP-activated protein kinase; CM, complete
media; CypA, cyclophilin A; DM, dorsomorphin dihydrochloride; p,
phosphorylated; SS, serum-starved media. |
Secondly, AMPK activation was enhanced by AICAR,
which mimics the effects of AMP (21,47-49).
HCT116 p53+/+ and HCT116 p53−/− cells were
cultured in CM and SS with various concentrations of AICAR (75, 150
and 300 µM) for 48 h (Fig. 5C and
D). The results revealed that phosphorylation of AMPK at
residue Thr172 was increased in HCT116 p53+/+ cells
following AICAR treatment, thus indicating that AMPK activity was
successfully enhanced (Fig. 5C and
D). AMPK activation was consistently high in HCT116
p53−/− cells even prior to AICAR treatment. AICAR
treatment slightly increased CypA export into the extracellular
media in HCT116 p53+/+ and HCT116 p53−/−
cells maintained in CM (Fig. 5C and
D). Notably, extracellular CypA levels were not decreased but
were markedly increased in HCT116 p53+/+ cells, whereas
no alteration was detected in HCT116 p53−/− cells under
SS, indicating that enhanced AMPK activation by AICAR can inhibit
necroptosis only in HCT116 p53−/− cells (Fig. 5C and D). These findings strongly
suggested that AMPK activation may suppress necroptosis only in
p53−/− cells in response to serum starvation.
The present study revealed that inhibition and
activation of AMPK promoted necroptosis in HCT116 p53+/+
cells under serum starvation, in which p53 protein expression
levels were increased (Fig. 5).
This may be because abnormal AMPK activity triggered upregulation
of p53 and induced necroptosis.
Taken together, these observations suggested that
AMPK activation through phosphorylation of Thr172 enhanced
inhibition of necroptosis in p53−/− cells, but not in
p53+/+ cells, under nutrient/serum starvation.
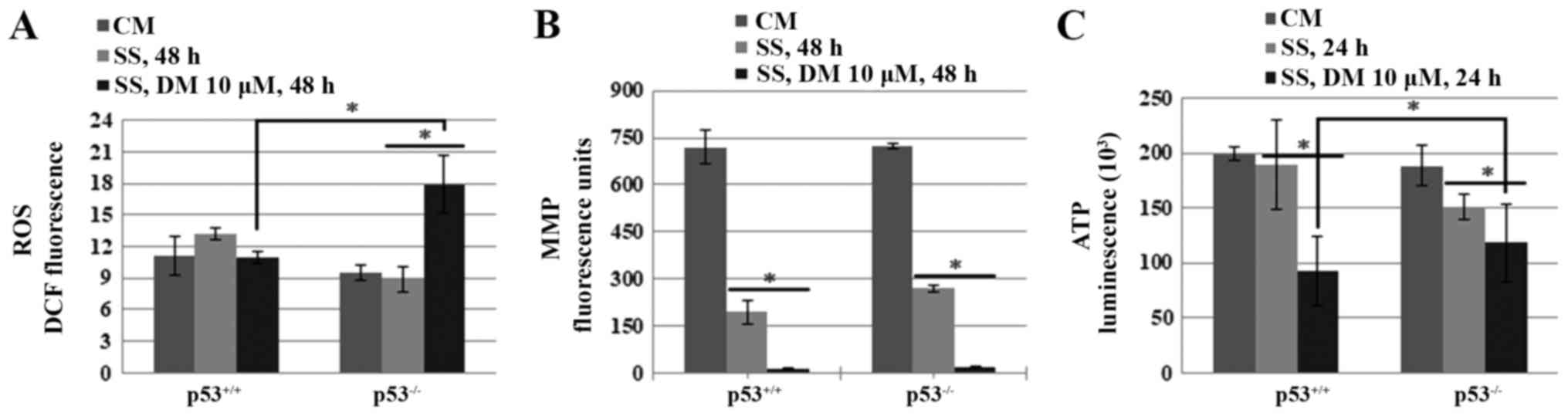
AMPK inhibits necroptosis through the
suppression of ROS generation in p53−/− cells
To address how AMPK activation suppresses
necroptosis in HCT116 p53−/− cells, the effects of AMPK
on cellular levels of ROS and ATP, and on the MMP, were assessed in
HCT116 p53+/+ and HCT116 p53−/− cells. The
results indicated that AMPK inhibition by DM significantly
increased ROS generation in HCT116 p53−/− cells
(Fig. 6A). However, no significant
difference was detected in MMP between the HCT116 p53+/+
and HCT116 p53−/− cells, thus indicating that reductions
in MMP by AMPK inhibition are independent of p53 (Fig. 6B). Furthermore, DM reduced ATP
levels more than SS alone in both cell lines; however, ATP levels
were slightly higher in HCT116 p53−/− cells compared
with in HCT116 p53+/+ cells following DM treatment
(Fig. 6C).
Taken together, these data indicated that highly
activated AMPK may be at least partially responsible for the
inhibition of necroptosis via suppression of ROS generation in
p53−/− cells.
Discussion
Escape from cell death, particularly apoptosis, is a
serious problem in current cancer research. Therefore, it might be
considered an effective therapeutic strategy to trigger several
types of cell death at the same time. The present study focused on
necroptosis, in addition to apoptosis, with the eventual aim of
identifying a novel class of combined targeted therapeutics. The
present study revealed that AMPK serves a different role in the
regulation of necroptosis in the presence and absence of p53. It
was demonstrated that a much lower level of necroptosis was
detected in p53−/− cells, and serum starvation-induced
necroptosis was suppressed by enhancing AMPK activation.
p53 is involved in the regulation of several types
of cell death, including apoptosis, CIA, autophagy-mediated cell
death, necroptosis, entosis, anoikis, paraptosis, pyroptosis,
ferroptosis, mitotic catastrophe and efferocytosis (17). Among these types of cell death,
several studies have reported vital roles for p53 in necroptosis.
Tu et al demonstrated that p53 can activate necroptosis in
response to ROS-induced DNA damage through the activation of the
lysosomal cysteine protease, cathepsin Q (50). Furthermore, it has been suggested
that p53 accumulates in the mitochondrial matrix, complexes with
CypD, and induces necroptosis in response to oxidative stress
(31,32). Vaseva et al suggested that
p53 causes necroptosis by controlling the opening of the
mitochondrial permeability transition pore (32). Montero et al reported that
p53 triggers necroptosis by increasing PARP-1 activity, and
inducing depletion of NAD and ATP (33). In spite of these studies, the role
of p53 in necroptosis remains unclear and more information is
required.
There are discordant results regarding the functions
of p53 in cell death regulation. Different combinations of master
regulators may explain it, each of which has various functions,
such as AMPK. The present results revealed that serum
starvation-induced AMPK activation suppressed necroptosis in
p53−/− cells but not in p53+/+ cells. In
contrast to these data, two research groups proposed that AMPK
induced necroptosis and apoptosis (51-53).
For example, Koo et al suggested that AMPK activation,
resulting from impaired mitochondrial oxidative phosphorylation,
induces necroptosis in human lung epithelial cells (51). Other research groups revealed that
AMPK activation induces apoptosis in liver cells by activating
signaling pathways, including c-Jun N-terminal kinase and caspase-3
(52,53). However, they did not consider the
effects of p53 in their research. It has been reported that the
metabolic environmental conditions and pathways that activate AMPK
differentially regulate cell proliferation or viability in cancer
and normal cells (19,54). Only a few studies have reported on
the highly complex interrelationship between p53 and AMPK in cell
death. For example, Okoshi et al proposed that AMPK
activation induces p53-dependent apoptosis in response to energetic
stress (53). Huang et al
revealed that AMPK inhibition by DM induces apoptosis in skin
cancer cells via a p53-dependent pathway (55). The same effect of DM was reported
in human colorectal cancer cells, with regards to the induction of
apoptosis and necroptosis (18).
Lee et al also demonstrated that AMPK induces p53
acetylation via phosphorylation and inactivation of sirtuin 1 in
liver cancer cells (56). In
addition, p53 can regulate autophagy, even though its regulation is
controversial, indicating an effect of p53 on AMPK (57-59).
However, the exact mechanism underlying the interaction between p53
and AMPK remains elusive.
The present study demonstrated that autophagy was
induced at a higher level in HCT116 p53−/− cells,
probably due to enhanced AMPK activation. Numerous studies have
reported that autophagy protects cells from cell death under
various types of cell death-inducing stress, such as anticancer
treatment and severe nutrient starvation (60-62).
These data suggested that increased activation of AMPK in HCT116
p53−/− cells may induce higher levels of autophagy and
eventually suppress other types of cell death in response to serum
starvation; however, the association between autophagy and
necroptosis requires further study.
RIP1-RIP3-MLKL is known to be a core signaling
pathway in necroptosis. The RIP1-RIP3-MLKL-dependent necroptosis
machinery may be considered a primary mechanism for suppressing
tumorigenesis and cancer progression. Cancer cells may suppress
necroptosis by downregulating or inducing functional mutations in
RIP1, RIP3 and MLKL. Previous studies indicated that RIP1
expression levels are decreased in only a few cancer cells, whereas
RIP3 expression is downregulated in numerous cancer cells (5,63-65).
For example, HeLa cervical cancer cells are well known for their
resistance to necroptosis, and they express normal levels of RIP1
but no RIP3 expression (66,67).
Notably, HCT116 cells have also been reported to lack RIP3 protein
(5,68). However, in the present study,
HCT116 p53+/+ cells underwent necroptosis under the
stress of serum starvation; therefore, it remains to be determined
as to how HCT116 cells undergo necroptosis. Elucidating the
mechanism underlying necroptosis will be of great interest.
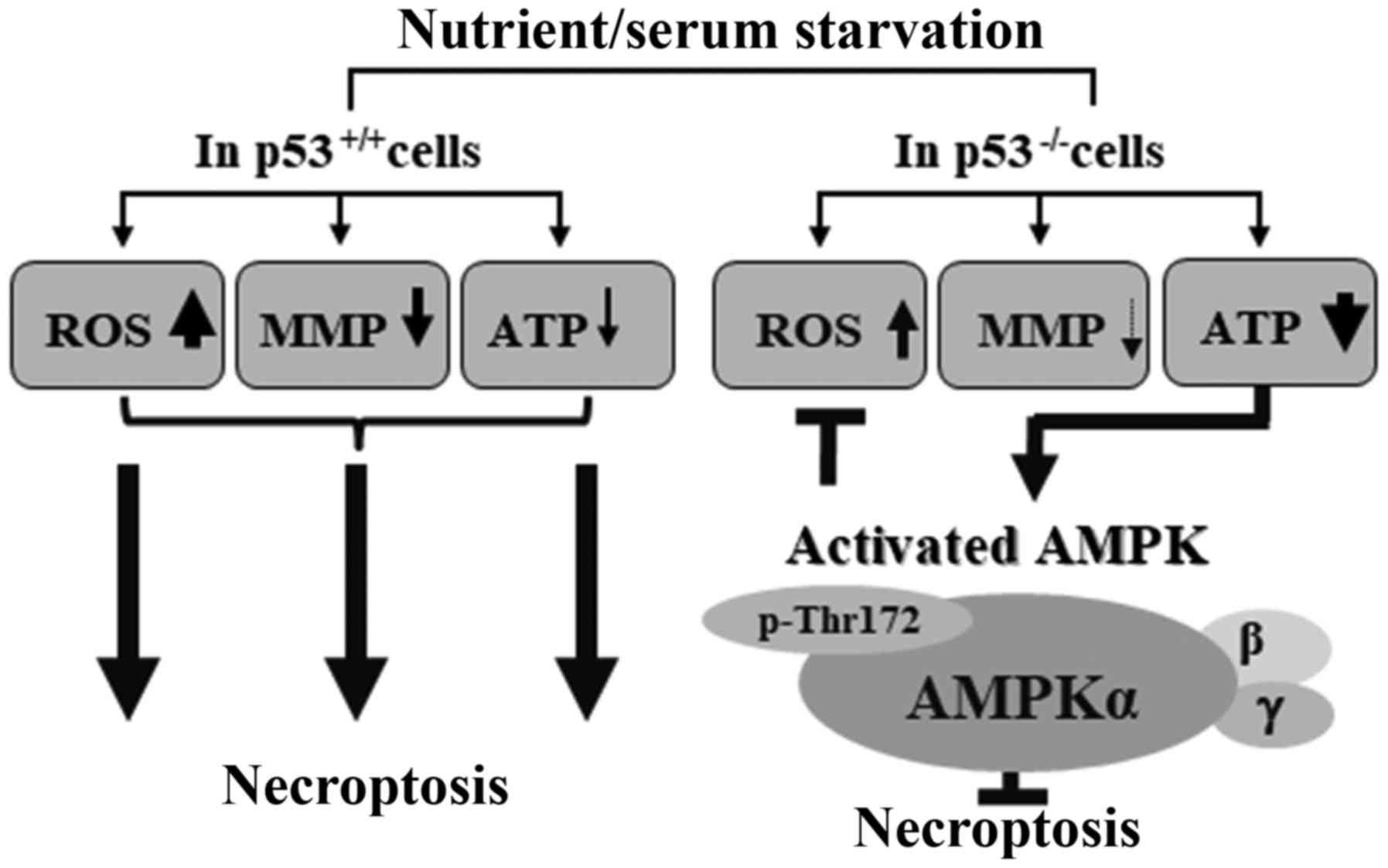
In conclusion, the present study proposed a
plausible model, in which activation of AMPK through
phosphorylation of Thr172 may inhibit necroptosis in the absence of
p53 in response to serum starvation (Fig. 7). This study proposed that AMPK may
be a combined target to efficiently kill cancer cells with
defective p53 under low-nutrient conditions. These data provided
information regarding a signaling network between p53 and AMPK in
necroptosis, which may be provoked in response to serum
starvation.
Funding
The present study was supported by Sookmyung Women's
University (2015).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
DDTL conducted the majority of the experiments,
analyzed data and drafted the manuscript. SJ conceptualized the
study, developed the general experimental design, analyzed data and
revised the manuscript. NTNQ was involved in the use of ImageJ,
statistical analysis and manuscript preparation. ZS assisted in
developing the study design and preparing the manuscript. BSL
contributed to confocal analysis and preparation of the manuscript.
SK, HGL and HJL assisted in experimental preparation, cell
culturing and maintenance, and preparation of manuscript. MSL made
substantial contributions to conception and experimental design,
revised the manuscript and gave final approval of the version to be
published. All authors read and approved the manuscript and agree
to be accountable for all aspects of this research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
PCD
|
programmed cell death
|
|
MMP
|
mitochondrial membrane potential
|
|
ROS
|
reactive oxygen species
|
|
AMPK
|
AMP-activated protein kinase
|
|
DCFH-DA
|
2',7'-dichlorodihydrofluorescein
diacetate
|
|
DM
|
dorsomorphin dihydrochloride
|
|
AICAR
|
5-aminoimidazole-4-carboxamide
ribonucleotide
|
Acknowledgments
The authors of this study are grateful to Dr. B.
Vogelstein (Johns Hopkins University, Baltimore, MD, USA) for
generously donating the colorectal carcinoma cell lines, HCT116
p53+/+ and HCT116 p53−/−, for use in the
experiments.
References
|
1
|
Pasparakis M and Vandenabeele P:
Necroptosis and its role in inflammation. Nature. 517:311–320.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vanden Berghe T, Linkermann A,
Jouan-Lanhouet S, Walczak H and Vandenabeele P: Regulated necrosis:
The expanding network of non-apoptotic cell death pathways. Nat Rev
Mol Cell Biol. 15:135–147. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vandenabeele P, Galluzzi L, Vanden Berghe
T and Kroemer G: Molecular mechanisms of necroptosis: An ordered
cellular explosion. Nat Rev Mol Cell Biol. 11:700–714. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Teng X, Degterev A, Jagtap P, Xing X, Choi
S, Denu R, Yuan J and Cuny GD: Structure-activity relationship
study of novel necroptosis inhibitors. Bioorg Med Chem Lett.
15:5039–5044. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Moriwaki K, Bertin J, Gough PJ, Orlowski
GM and Chan FK: Differential roles of RIPK1 and RIPK3 in
TNF-induced necroptosis and chemotherapeutic agent-induced cell
death. Cell Death Dis. 6:e16362015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
de Almagro MC and Vucic D: Necroptosis:
Pathway diversity and characteristics. Semin Cell Dev Biol.
39:56–62. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xu YZ, Kanagaratham C, Youssef M and
Radzioch D: New Frontiers in Cancer Chemotherapy-Targeting Cell
Death Pathways. In Cell Biology - New Insights. Najman S:
IntechOpen. pp. 93–140. 2016
|
|
8
|
Dondelinger Y, Hulpiau P, Saeys Y,
Bertrand MJM and Vandenabeele P: An evolutionary perspective on the
necroptotic pathway. Trends Cell Biol. 26:721–732. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Belizário J, Vieira-Cordeiro L and Enns S:
Necroptotic Cell Death Signaling and Execution Pathway: Lessons
from Knockout Mice. Mediators Inflamm. 2015:1280762015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chan FKM, Luz NF and Moriwaki K:
Programmed necrosis in the cross talk of cell death and
inflammation. Annu Rev Immunol. 33:79–106. 2015. View Article : Google Scholar :
|
|
11
|
Avril T, Vauléon E and Chevet E:
Endoplasmic reticulum stress signaling and chemotherapy resistance
in solid cancers. Oncogenesis. 6:e3732017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Palorini R, Votta G, Pirola Y, De Vitto H,
De Palma S, Airoldi C, Vasso M, Ricciardiello F, Lombardi PP,
Cirulli C, et al: Protein Kinase A Activation Promotes Cancer Cell
Resistance to Glucose Starvation and Anoikis. PLoS Genet.
12:e10059312016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Izuishi K, Kato K, Ogura T, Kinoshita T
and Esumi H: Remarkable tolerance of tumor cells to nutrient
deprivation: Possible new biochemical target for cancer therapy.
Cancer Res. 60:6201–6207. 2000.PubMed/NCBI
|
|
14
|
Sato K, Tsuchihara K, Fujii S, Sugiyama M,
Goya T, Atomi Y, Ueno T, Ochiai A and Esumi H: Autophagy is
activated in colorectal cancer cells and contributes to the
tolerance to nutrient deprivation. Cancer Res. 67:9677–9684. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Esumi H, Izuishi K, Kato K, Hashimoto K,
Kurashima Y, Kishimoto A, Ogura T and Ozawa T: Hypoxia and nitric
oxide treatment confer tolerance to glucose starvation in a
5'-AMP-activated protein kinase-dependent manner. J Biol Chem.
277:32791–32798. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim SM, Nguyen TT, Ravi A, Kubiniok P,
Finicle BT, Jayashankar V, Malacrida L, Hou J, Robertson J, Gao D,
et al: PTEN deficiency and AMPK activation promote nutrient
scavenging and anabolism in prostate cancer cells. Cancer Discov.
8:866–883. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ranjan A and Iwakuma T: Non-Canonical Cell
Death Induced by p53. Int J Mol Sci. 17:20682016. View Article : Google Scholar
|
|
18
|
Liu X, Chhipa RR, Nakano I and Dasgupta B:
The AMPK inhibitor compound C is a potent AMPK-independent
antiglioma agent. Mol Cancer Ther. 13:596–605. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hardie DG: AMPK and autophagy get
connected. EMBO J. 30:634–635. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Luo Z, Zang M and Guo W: AMPK as a
metabolic tumor suppressor: Control of metabolism and cell growth.
Future Oncol. 6:457–470. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mihaylova MM and Shaw RJ: The AMPK
signalling pathway coordinates cell growth, autophagy and
metabolism. Nat Cell Biol. 13:1016–1023. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liang J and Mills GB: AMPK: A contextual
oncogene or tumor suppressor? Cancer Res. 73:2929–2935. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Faubert B, Boily G, Izreig S, Griss T,
Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, et
al: AMPK is a negative regulator of the Warburg effect and
suppresses tumor growth in vivo. Cell Metab. 17:113–124. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hardie DG: AMP-activated protein kinase:
An energy sensor that regulates all aspects of cell function. Genes
Dev. 25:1895–1908. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li W, Saud SM, Young MR, Chen G and Hua B:
Targeting AMPK for cancer prevention and treatment. Oncotarget.
6:7365–7378. 2015.PubMed/NCBI
|
|
26
|
Jeon SM, Chandel NS and Hay N: AMPK
regulates NADPH homeostasis to promote tumour cell survival during
energy stress. Nature. 485:661–665. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kato K, Ogura T, Kishimoto A, Minegishi Y,
Nakajima N, Miyazaki M and Esumi H: Critical roles of AMP-activated
protein kinase in constitutive tolerance of cancer cells to
nutrient deprivation and tumor formation. Oncogene. 21:6082–6090.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bunz F, Dutriaux A, Lengauer C, Waldman T,
Zhou S, Brown JP, Sedivy JM, Kinzler KW and Vogelstein B:
Requirement for p53 and p21 to sustain G2 arrest after DNA damage.
Science. 282:1497–1501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Christofferson DE and Yuan J: Cyclophilin
A release as a biomarker of necrotic cell death. Cell Death Differ.
17:1942–1943. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lin Y, Choksi S, Shen H-M, Yang Q-F, Hur
GM, Kim YS, Tran JH, Nedospasov SA and Liu ZG: Tumor necrosis
factor-induced nonapoptotic cell death requires
receptor-interacting protein-mediated cellular reactive oxygen
species accumulation. J Biol Chem. 279:10822–10828. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dashzeveg N and Yoshida K: Cell death
decision by p53 via control of the mitochondrial membrane. Cancer
Lett. 367:108–112. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vaseva AV, Marchenko ND, Ji K, Tsirka SE,
Holzmann S and Moll UM: p53 opens the mitochondrial permeability
transition pore to trigger necrosis. Cell. 149:1536–1548. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Montero J, Dutta C, van Bodegom D,
Weinstock D and Letai A: p53 regulates a non-apoptotic death
induced by ROS. Cell Death Differ. 20:1465–1474. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tsujimoto Y and Shimizu S: Role of the
mitochondrial membrane permeability transition in cell death.
Apoptosis. 12:835–840. 2007. View Article : Google Scholar
|
|
35
|
Eguchi Y, Shimizu S and Tsujimoto Y:
Intracellular ATP levels determine cell death fate by apoptosis or
necrosis. Cancer Res. 57:1835–1840. 1997.PubMed/NCBI
|
|
36
|
Tatsumi T, Shiraishi J, Keira N, Akashi K,
Mano A, Yamanaka S, Matoba S, Fushiki S, Fliss H and Nakagawa M:
Intracellular ATP is required for mitochondrial apoptotic pathways
in isolated hypoxic rat cardiac myocytes. Cardiovasc Res.
59:428–440. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liou G-Y and Storz P: Reactive oxygen
species in cancer. Free Radic Res. 44:479–496. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Redza-Dutordoir M and Averill-Bates DA:
Activation of apoptosis signalling pathways by reactive oxygen
species. Biochim Biophys Acta. 1863:2977–2992. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Marchi S, Giorgi C, Suski JM, Agnoletto C,
Bononi A, Bonora M, De Marchi E, Missiroli S, Patergnani S, Poletti
F, et al: Mitochondriaros crosstalk in the control of cell death
and aging. J Signal Transduct. 2012:3296352012. View Article : Google Scholar
|
|
40
|
Fulda S: The mechanism of necroptosis in
normal and cancer cells. Cancer Biol Ther. 14:999–1004. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kaczmarek A, Vandenabeele P and Krysko DV:
Necroptosis: The release of damage-associated molecular patterns
and its physiological relevance. Immunity. 38:209–223. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dasgupta A, Nomura M, Shuck R and Yustein
J: Cancer's Achilles' Heel: Apoptosis and Necroptosis to the
Rescue. Int J Mol Sci. 18:232016. View Article : Google Scholar
|
|
43
|
González-Juarbe N, Gilley RP, Hinojosa CA,
Bradley KM, Kamei A, Gao G, Dube PH, Bergman MA and Orihuela CJ:
Pore-Forming Toxins Induce Macrophage Necroptosis during Acute
Bacterial Pneumonia. PLoS Pathog. 11:e10053372015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Steinberg GR and Kemp BE: AMPK in Health
and Disease. Physiol Rev. 89:1025–1078. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Oakhill JS, Chen Z-P, Scott JW, Steel R,
Castelli LA, Ling N, Macaulay SL and Kemp BE: β-Subunit
myristoylation is the gatekeeper for initiating metabolic stress
sensing by AMP-activated protein kinase (AMPK). Proc Natl Acad Sci
USA. 107:19237–19241. 2010. View Article : Google Scholar
|
|
46
|
Yung MMH, Ngan HYS and Chan DW: Targeting
AMPK signaling in combating ovarian cancers: Opportunities and
challenges. Acta Biochim Biophys Sin (Shanghai). 48:301–317. 2016.
View Article : Google Scholar
|
|
47
|
Wang W, Yang X, López de Silanes I,
Carling D and Gorospe M: Increased AMP:ATP ratio and AMP-activated
protein kinase activity during cellular senescence linked to
reduced HuR function. J Biol Chem. 278:27016–27023. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Grahame Hardie D: Regulation of
AMP-activated protein kinase by natural and synthetic activators.
Acta Pharm Sin B. 6:1–19. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Smith AC, Bruce CR and Dyck DJ: AMP kinase
activation with AICAR simultaneously increases fatty acid and
glucose oxidation in resting rat soleus muscle. J Physiol.
565:537–546. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tu HC, Ren D, Wang GX, Chen DY, Westergard
TD, Kim H, Sasagawa S, Hsieh JJD and Cheng EHY: The p53-cathepsin
axis cooperates with ROS to activate programmed necrotic death upon
DNA damage. Proc Natl Acad Sci USA. 106:1093–1098. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Koo MJ, Rooney KT, Choi ME, Ryter SW, Choi
AMK and Moon JS: Impaired oxidative phosphorylation regulates
necroptosis in human lung epithelial cells. Biochem Biophys Res
Commun. 464:875–880. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Meisse D, Van de Casteele M, Beauloye C,
Hainault I, Kefas BA, Rider MH, Foufelle F and Hue L: Sustained
activation of AMP-activated protein kinase induces c-Jun N-terminal
kinase activation and apoptosis in liver cells. FEBS Lett.
526:38–42. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Okoshi R, Ozaki T, Yamamoto H, Ando K,
Koida N, Ono S, Koda T, Kamijo T, Nakagawara A and Kizaki H:
Activation of AMP-activated protein kinase induces p53-dependent
apoptotic cell death in response to energetic stress. J Biol Chem.
283:3979–3987. 2008. View Article : Google Scholar
|
|
54
|
Ferretti AC, Tonucci FM, Hidalgo F, Almada
E, Larocca MC and Favre C: AMPK and PKA interaction in the
regulation of survival of liver cancer cells subjected to glucose
starvation. Oncotarget. 7:17815–17828. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Huang SW, Wu CY, Wang YT, Kao JK, Lin CC,
Chang CC, Mu SW, Chen YY, Chiu HW, Chang CH, et al: p53 modulates
the AMPK inhibitor compound C induced apoptosis in human skin
cancer cells. Toxicol Appl Pharmacol. 267:113–124. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lee CW, Wong LL, Tse EY, Liu HF, Leong VY,
Lee JM, Hardie DG, Ng IO and Ching YP: AMPK promotes p53
acetylation via phosphorylation and inactivation of SIRT1 in liver
cancer cells. Cancer Res. 72:4394–404. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Maiuri MC, Galluzzi L, Morselli E, Kepp O,
Malik SA and Kroemer G: Autophagy regulation by p53. Curr Opin Cell
Biol. 22:181–185. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Scherz-Shouval R, Weidberg H, Gonen C,
Wilder S, Elazar Z and Oren M: p53-dependent regulation of
autophagy protein LC3 supports cancer cell survival under prolonged
starvation. Proc Natl Acad Sci USA. 107:18511–18516. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Tasdemir E, Chiara Maiuri M, Morselli E,
Criollo A, D'Amelio M, Djavaheri-Mergny M, Cecconi F, Tavernarakis
N and Kroemer G: A dual role of p53 in the control of autophagy.
Autophagy. 4:810–814. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Fitzwalter BE and Thorburn A: Recent
insights into cell death and autophagy. FEBS J. 282:4279–4288.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Boya P, González-Polo R-A, Casares N,
Perfettini J-L, Dessen P, Larochette N, Métivier D, Meley D,
Souquere S, Yoshimori T, et al: Inhibition of macroautophagy
triggers apoptosis. Mol Cell Biol. 25:1025–1040. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Nikoletopoulou V, Markaki M, Palikaras K
and Tavernarakis N: Crosstalk between apoptosis, necrosis and
autophagy. Biochim Biophys Acta. 1833:3448–3459. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Nugues AL, El Bouazzati H, Hétuin D,
Berthon C, Loyens A, Bertrand E, Jouy N, Idziorek T and Quesnel B:
RIP3 is down-regulated in human myeloid leukemia cells and
modulates apoptosis and caspase-mediated p65/RelA cleavage. Cell
Death Dis. 5:e13842014. View Article : Google Scholar
|
|
64
|
Koo G-B, Morgan MJ, Lee D-G, Kim W-J, Yoon
J-H, Koo JS, Kim SI, Kim SJ, Son MK, Hong SS, et al:
Methylation-dependent loss of RIP3 expression in cancer represses
programmed necrosis in response to chemotherapeutics. Cell Res.
25:707–725. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yang C, Li J, Yu L, Zhang Z, Xu F, Jiang
L, Zhou X and He S: Regulation of RIP3 by the transcription factor
Sp1 and the epigenetic regulator UHRF1 modulates cancer cell
necroptosis. Cell Death Dis. 8:e30842017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Su Z, Yang Z, Xie L, DeWitt JP and Chen Y:
Cancer therapy in the necroptosis era. Cell Death Differ.
23:748–756. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wang Z, Jiang H, Chen S, Du F and Wang X:
The mitochondrial phosphatase PGAM5 functions at the convergence
point of multiple necrotic death pathways. Cell. 148:228–243. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Brown MF, Leibowitz BJ, Chen D, He K, Zou
F, Sobol RW, Beer-Stolz D, Zhang L and Yu J: Loss of caspase-3
sensitizes colon cancer cells to genotoxic stress via
RIP1-dependent necrosis. Cell Death Dis. 6:e17292015. View Article : Google Scholar : PubMed/NCBI
|