Introduction
Lung cancer, the most common type of cancer
worldwide, and non-small cell lung cancer (NSCLC), is the primary
subtype, accounting for 80% of all new lung cancer cases (1,2).
Despite current advances in chemotherapy and molecular targeted
therapy, the 5-year survival rate of patients with NSCLC remains
<15%. Resistance to therapies in lung cancer is closely
associated with the dysregulation of oncogenes or tumor suppressor
genes, all of which involve changes in the biological
characteristics of cancer cells, cell growth, apoptosis, migration,
invasion, etc (3). Although
cisplatin (Cis) has long been a cornerstone agent in anticancer
chemotherapy (4), many cancer
cells, including lung cancer, may develop and acquire the
resistance to Cis, and other cancers are naturally resistance to
this drug, which is an obstacle to successful chemotherapy
(5). Furthermore, Cis
cytotoxicity, such as nephrotoxicity and neurotoxicity, is a common
side-effect of Cis (6). Therefore,
novel therapeutic strategies for lung malignancies using combined
agents with distinct molecular mechanisms are considered more
promising for the enhanced therapeutic efficacy to improve the
survival rate.
Human pentraxin-3 (PTX3) is a multimeric
glycoprotein with eight subunits that comprise 381 amino acids
(7). Among the different species,
the primary sequence of PTX3 is highly conserved, sharing 92% of
consensus amino acid residues between human and murine sequences,
indicating strong evolutionary pressure to maintain its
function/structure relationships (8). Human pentraxin-3 family is divided
into long and short subfamilies. Similar to other members of the
long pentraxin subfamily, PTX3 is constructed of a distinctive
C-terminal domain, and N-terminal region with homology to short
pentraxin, C-reactive protein (CRP) and serum amyloid P component
(SAP) (9,10). PTX3 is involved in the inflammatory
process. The increased PTX3 was reported in chronic kidney
diseases, endotoxin shock, sepsis and vasculitis (11). Furthermore, the level of PTX3
reflects the severity of inflammation. It is considered that
tumorigenesis is an inflammation-related disease. An elevated PTX3
expression is noted in glioma, liposarcoma, prostate cancer, lung
cancer and breast cancer (12-16).
It is closely associated with cell proliferation, metastasis,
pathological grading and drug sensitivity (13,17,18).
The glycosylation of proteins is an important
post-translational modification, and the resulting glycoproteins
participate in a number of key biological processes, including cell
growth, differentiation and immune modulation, etc (19). Altered glycosylation has been found
in inflammatory diseases and many types of cancer (20-22).
Recent advances in the evaluation of protein glycosylation and cell
malignancy have gained the attention of many researchers, with the
aim to treat cancer or other diseases with alterations in cellular
glycosylation (23), and
modifications in the glycan structures of proteins have been known
to be an indicator of inflammation and carcinogenesis (24,25).
Alterations in N-linked oligosaccharides on tumor cell surfaces are
associated with proliferative, migratory and invasive properties
(26). Particularly, the
C-terminal domain of PTX3 at Asn-220 has been identified as a
single N-linked glycosylation site that is completely occupied by
complex oligosaccharides (7),
generally fucosylated and sialylated bi-antennary sugars with a
minor fraction of tri- and tetra-antennary glycans. Interestingly,
the relative contents of bi-, tri- and tetra-antennary glycans, and
the level of sialylation have been shown to be highly variable
among PTX3 isolated from different cellular sources, suggesting
that the glyco-moiety of PTX3 is involved in a number of biological
activities (10). Based on
three-dimensional models, PTX3 oligosaccharides interact with the
polar and basic amino acids from the other proteins by surface
(i.e., Arg332 and Glu252), typically through terminal residues of
sialic acid (8). These types of
interactions are forfeited when sialic acid is removed, and protein
sites that are potentially relevant to ligand recognition and/or
modification of the PTX3 tertiary/quaternary structure become
attainable. Therefore, modifications in glycosylation have been
investigated as potential biomarkers for both chronic inflammatory
processes and cancer, associated with disease severity in certain
conditions (23). These
alterations in glycosylation are also known to regulate cell
signaling and may contribute to disease pathogenesis.
Tunicamycin (TM) is used to inhibit N-linked
oligosaccharide biosynthesis in cells (27). Existing reports indicate that
disruption of N-linked glycosylation inhibits downstream signaling
pathways, suggesting that TM is an alternative therapeutic approach
to reduce oncogenic signaling and drug resistance (25). TM has been demonstrated to enhance
the susceptibility of lung cancer cells to therapy, sensitize
resistant cancer cells to chemotherapy and to potentiate
Cis-induced cytotoxicity in human head and neck carcinoma (28). TM has also been reported to
decrease vascular endothelial growth factor (VEGF) expression in
nude mice and to inhibit angiogenesis (29). These findings indicate that the
TM-induced disruption of N-linked glycosylation has a considerable
advantage in targeting multiple glycoproteins and sensitizing
cancer cells to anticancer therapeutics.
The current study was designed to assess the
significance of PTX3 deglycosylation in the suppression of human
lung cancer. We examined PTX3 expression levels, which were
increased in human lung cancer tissues and cells, and dePTX3
suppressed lung cancer cell proliferation and migration. We also
found that the dePTX3 by TM enhanced the sensitivity of lung cancer
cells to Cis through AKT/NF-κB signaling pathway.
Materials and methods
Cell culture and human samples
Human lung cancer cell lines (A549 and H1299) were
purchased from the American Type Culture Collection (ATCC) and
SPCA1 cell line was purchased from the Cell Bank of Chinese Academy
of Sciences (Shanghai, China). The cells were cultured in RPMI-1640
medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml
penicillin and 100 µg/ml streptomycin (Gibco Life
Technologies/Thermo Fisher Scientific, Inc., Waltham, MA, USA) at
37°C in humidified air containing 5% CO2. A total of 34
patients (28 males and 6 females) with third-grade lung cancer and
42 normal healthy patients (26 males and 16 females) with a mean
age of 56 years were recruited (by lung biopsy or lobectomy) after
obtaining informed consent. Lung tissues and serum samples were
collected from January, 2015 to December, 2017. All lung cancer
paraffin-embedded sections and lung cancer tissues were obtained at
the First Affiliated Hospital of Dalian Medical University.
Reagents and antibodies
TM was purchased from Sigma-Aldrich (St. Louis, MO,
USA) which was dissolved in DMSO. Cis and peptide N-glycosidase F
(PNGase F) were purchased from Sigma-Aldrich. Cell Counting kit-8
(CCK-8) was obtained from Beyotime Biotechnology (Shanghai, China);
recombinant human PTX3 (rhPTX3) (#PROTP26022; Boster Biological
Technology Co., Ltd., Fremont, CA); NF-κB inhibitor (IKK-16), PI3K
inhibitor (GDC0941), MEK1/2 inhibitor, (MEK162) were purchased from
Selleck Chemicals (Shanghai, China). Antibodies against p-p65
(#3033), p65 (#8242), p-IKK (#2078) and IKK (#2682) were from Cell
Signaling Technology (Danvers, MA, USA); MMP-9 (#10375) was from
Proteintech (Wuhan, China); antibodies against AKT (#AA326), p-AKT
(T308) (#AA331) were from Beyotime Biotechnology; PTX3
(#13797-1-AP), poly (ADP-ribose) polymerase (PARP) (#AF-1657),
cleaved PARP (#AF-1567), Bcl2 (#12789-1-AP), Bax (#50599-2-Ig),
proliferating cell nuclear antigen (PCNA) (#24036-1-AP),
HRP-conjugated anti-rabbit and anti-mouse antibodies and GAPDH
(#10494-1-AP, #60004-1-Ig) antibodies were from Proteintech;
HRP-conjugated goat anti-rabbit secondary antibody (ZSGB-BIO,
Beijing, China).
Coomassie brilliant blue (CBB)
staining
The concentrated CBB R-250 (#20278; Thermo Fisher
Scientific, Inc.) solution [5% (w/v)] was prepared in distilled
water and stirred for more than 1 h. The supernatant of the CBB
solution, centrifuged at 7,000 × g for 5 min, was used to prepare
the staining solution. The gels were soaked in the staining
solution containing 0.1% (w/v) CBB R-250, 50% (v/v) methanol, and
10% (v/v) acetic acid at room temperature for 4 h. Subsequently,
the CBB-stained gels were agitated in the de-staining solution
containing 40% (v/v) methanol and 10% (v/v) acetic acid.
Immunohistochemistry
The excised tumor tissues were fixed in 5% formalin
for 24 h, embedded in paraffin and then sectioned (4 µm
thickness) in a standard manner. The sections were deparaffinized
in xylene followed by a graded series of ethanol (100, 95, 85 and
70%), and rehydrated in phosphate-buffered solution, pH 7.5. The
antibodies used for paraffin-embedded tissues were monoclonal
rabbit anti-PTX3 antibody, (#13797-1-AP; Proteintech). The primary
antibodies were diluted (1:50) in PBS containing 3% bovine serum
albumin (BSA). The sample slides were treated at 37°C for 30 min
with blocking solution (8% goat serum in PBS with 3% BSA) before
the primary antibody was applied. Endogenous peroxidase was blocked
by incubation with 3% hydrogen peroxide. Endogenous avidin and
biotin were blocked using the Avidin/Biotin Blocking kit (SP-9000;
ZSGB-BIO). Overnight incubation with the primary antibody was
followed by incubation with the respective biotinylated secondary
antibody, followed by the ABC reagent for signal amplification.
Between the incubation steps, the slides were washed in TBS.
3,3′-diami-nobenzidine (DAB kit; ZLI-9017; ZSGB-BIO) was used to
develop the color. The slides were counter-stained with
hematoxylin, mounted in Kaisers Glycerinegelatine (Merck, Germany),
and covered with a coverslip. Dark yellowish brown stain indicated
positive cells. Images of representative staining were captured
using a microscope (BX51; Olympus, Tokyo, Japan).
Immunofluorescent staining and lectin
fluorescent staining
The cells were cultured on glass coverslips.
Following 48 h of treatment with TM (0.5 µg/ml) and Cis (20
µM), the cells were fixed in 4% paraformaldehyde or cold
acetone for 30 min, followed by blocking with 1% goat serum for 2
h. Coverslips were incubated with PTX3 antibody at 4°C overnight
followed by incubation with TRITC-conjugated goat anti-rabbit IgG
(1:50 dilution, #6926-100; BioVision, Milpitas, CA, USA). For PHA
lectin fluorescent staining, the samples were incubated with PHA
(#B-1115; Vector Laboratories) for 1 h. After washing, the samples
were incubated with FITC-conjugated streptavidin for 45 min.
Following incubation with DAPI for 10 min, coverslips were dropped
with an anti-fade solution (Beyotime Biotechnology), and
photographed under a fluorescent microscope (BX51; Olympus, Tokyo,
Japan).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from the lung cancer cells
using TRIzol reagent (Takara, Dalian, China), and prepared for
complementary DNA (cDNA) synthesis. RNA was reverse transcribed
into cDNA using the Prime Script™ RT reagent kit (Takara, Tokyo,
Japan). The SYBR-Green PCR Master Mix kit (Transgene, Shenzhen,
China) was used according to the manufacturer’s instructions. The
conditions for qPCR were as follows: cycle 1: 95°C for 10 min;
cycle 2 (×40): 95°C for 10 sec and 58°C for 45 sec. Specific
primers were designed and prepared (Invitrogen/Thermo Fisher
Scientific, Inc.): which included PTX3, 5′-CATCCAGTGAGACCAATGAG-3′
(forward) and 5′-GTAGCCGCCAGTTCACCATT-3′ (reverse); GAPDH,
5′-GCTGTGTGGCAAAGTCCAA-3′ (forward) and 5′-GGTCAGGCTCCTGGAAGATA-3′
(reverse). Quantitative PCR reactions were performed with the
Applied Biosystems StepOne Real-time PCR system (Life Technologies,
Carlsbad, CA, USA). The gene expression was normalized to the GAPDH
level as an internal control, and calculated using the
2−ΔΔCq method (29).
Data were presented from 3 independent experiments.
ELISA
Human PTX3 ELISA kit (Boster Biological Technology
Co., Ltd., Fremont, CA, USA) was used to measure the PTX3 level in
human serum samples and cell culture supernatant according to the
manufacturer’s instructions. Sample concentrations were calculated
from a standard curve generated by dilutions of a known amount of
recombinant human PTX3 protein. Each standard or sample was assayed
in duplicate. Three internal quality control serum samples or
culture were tested in each assay to assess inter-assay
precision.
Cell viability assay
Cell viability was assessed by CCK-8 assay. Briefly,
the cells were seeded in 96-well plate at a density of 1,000
cells/well and incubated at 37°C for 24 to 96 h; the CCK-8
solution, 10 µl was added to each well, followed by
incubation for 2 h. The absorbance value of each well was measured
at 450 nm. Three wells were used in all experiments and repeated at
least 3 times.
Wound healing assay
Wound healing assay was performed following the drug
treatment. In brief, the cells were cultured in a 6-well plate
reaching at least 80% confluence. The cells were then incubated at
37°C for 0-48 h in the culture medium containing TM (0.5
µg/ml), Cis (20 µM), dePTX3. Cell migration was
observed using a light microscope and quantified by the relative
wound healing width as the distance of the cells migrated across
the injury line at 0 and 48 h.
Transwell migration and invasion
assay
For cell migration assay, transwell inserts (Corning
Costar, Cambridge, MA, USA) containing polycarbonate filters with 8
µm pores were not precoated with Matrigel matrix. For cell
invasion assay, the inserts were precoated with Matrigel matrix.
1×105 A549 and SPCA1 cells in 200 µl of
serum-free RPMI-1640 were added to the upper chamber, and the
medium with 10% FBS was added to the lower chamber. Following 16 h
of incubation, the cells on the lower surface of the filter were
fixed, stained and examined under an inverted microscope (Leica
Microsystems, Wetzlar, Germany). The number of migrated and invaded
cells in 5 random fields (magnification, ×100) from triplicate
filters was counted, and representative images and statistical
analysis were shown.
Colony formation assay
Cells were plated in 6-well plate at 250 cells per
well. Following incubation at 37°C for 15 days, the cells were
washed twice with PBS and stained with Giemsa (BDH Chemicals Ltd.,
Poole, UK) solution for 5 min. After 3 times of rinsing in sterile
water, the number of colonies containing ≥50 cells was counted
under an inverted microsrope.
Deglycosylation treatment
The A549 and SPCA1 cells were used to determine the
role of PTX3 glycosylation in lung cancer growth. PTX3 was
deglycosylated by mixing rhPTX3 (100 ng/ml) with PNGase F (500
U/ml) for 2 h at 37°C. Subsequently, the cells were treated with
dePTX3 and various concentrations of TM as indicated in Fig. 2.
Plasmid construction and site-directed
mutagenesis
A plasmid pcDNA3.1-PTX3 containing the human
full-length PTX3 cDNA (GenBank accession no. NM_002852.3) was used
to produce the PTX3 N-glycan site mutant. The mutated plasmid
(mPTX3) was constructed by replacement of asparagine 220 (Asn-220)
to glutamine according to the manufacturer’s instructions
(GenePharma, Shanghai, China). The mutation was further confirmed
by DNA sequencing. The cancer cells were transfected with mPTX3 by
using Lipofectamine 2000 (Invitrogen/Thermo Fisher Scientific,
Inc.).
Western blot analysis
The cells were washed with PBS (pH 7.4), and
incubated with 2× concentrated electrophoresis sample buffer (125
mM Tris-HCl, pH 6.8, 5% glycerol, 2% SDS, 1% β-mercaptoethanol) for
30 min on ice. Protein concentration was determined with Coomassie
protein assay reagent using bovine serum albumin as a standard.
Total protein from the whole cell lysates was separated by 12%
SDS-PAGE gel, and transferred electrophoretically onto PVDF
membranes, incubated with TBST (50 mM Tris HCl, pH 7.5, 0.15 M
NaCl, 0.1% Tween-20) containing 5% fat-free dry milk for 2 h,
followed by overnight incubation with the appropriate primary
antibodies at the dilutions recommended by the suppliers at 4°C.
Following incubation with an HRP-conjugated appropriate secondary
antibody, the enhanced electrochemical luminescence (ECL) detection
system (Bio-Rad Laboratories, Hercules, CA, USA) was used to
visualize immunoreactive bands. The primary antibodies to PTX3
(rabbit antibody, 1:200), MMP-9 (rabbit antibody, 1:200), AKT,
p-AKT (T308), p65, p-p65, Bcl2, Bax, cleaved PARP, IKK, p-IKK and
GAPDH (rabbit antibodies, 1:1,000) were used. The amount of each
protein was determined by the intensity of the band. To verify
equal protein loading, the GAPDH level in each lane was examined
with the anti-GAPDH antibody. Bands were semi-quantified with Image
software.
Analysis of cell apoptosis by flow
cytometry
The A549 and SPCA1 cells were cultivated in a 6-well
plate at a concentration of 2×105 cells/well and then
incubated at 37°C with TM (0.5 µg/ml) or Cis (20 µM)
alone or in combination for 24 h. Untreated cells were used as a
negative control. The cells were washed with PBS twice and followed
by trypsinization. The cells were stained with Annexin
V-FITC/propidium iodide (PI) apoptosis detection kit (Nanjing
KeyGen Biotech Co., Ltd., Nanjing, China) as per the manufacturer’s
recommendations. The fluorescence intensity of the samples was
determined by flow cytometry. The number of apoptotic cells in each
treatment was detected by flow cytometric analysis (BD Acuri™ C6;
BD Biosciences, San Jose, CA, USA). The data were presented as the
total number of apoptotic cells in each treatment. The whole
experiment was carried out in triplicate and the error bars were
calculated from the independently repeated experiments.
Statistical analysis
The experiments were repeated at least 3 times, and
the results were expressed as the means ± SEM. Data were analyzed
using the Student’s t-test and an analysis of variance (one-way
ANOVA) test followed by a Tukey’s post hoc test were applied to
determine the significant differences between groups. A P-value
<0.05 was considered as statistically significant. The
statistical analysis was performed using GraphPad-Prism 6 software
(GraphPad-Prism Software Inc., San Diego, CA, USA).
Results
PTX3 expression in human lung cancer
samples and cell lines
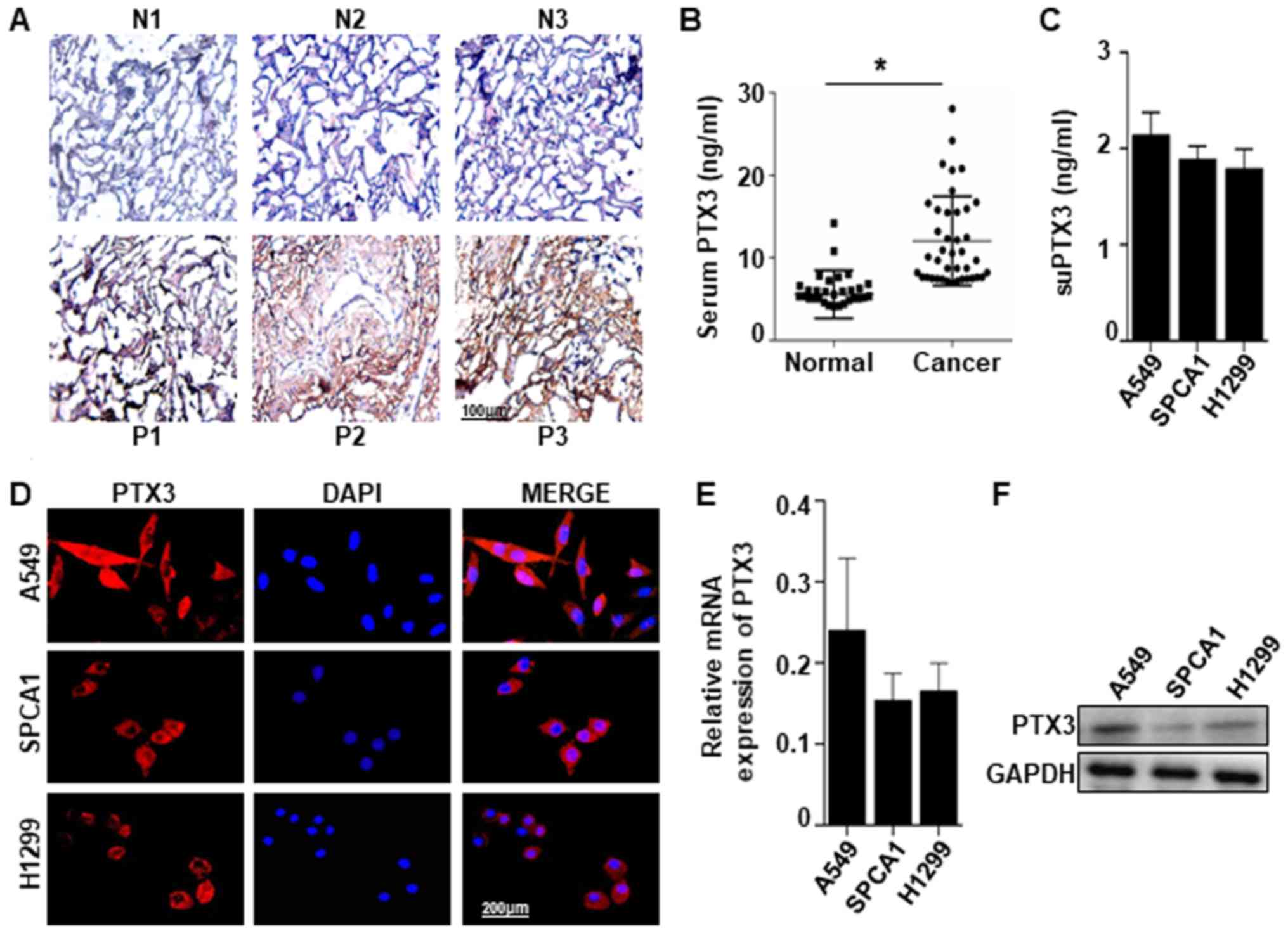
PTX3 expression was detected in human lung cancer
tissues and serum. Immunohistochemical staining of PTX3 protein
revealed a significant difference between the lung cancer tissues
and normal tissues. Positive PTX3 staining was observed in 24 of 34
(70.59%) lung cancer tissues compared with the normal tissues in 8
out of 42 (19.04%). Represented images were shown in Fig. 1A. PTX3 level detected by ELISA in
the serum of patients with lung cancer (12.01±0.85) was higher than
that in the normal controls (5.57±0.51) (Fig. 1B). PTX3 in the supernatant (suPTX3)
was also detected by ELISA in 3 types of lung cancer cells (A549,
SPCA1 and H1299) (Fig. 1C).
Comparatively, the expression of PTX3 was found in A549, SPCA1 and
H1299 cells examined by immunofluorescent staining, RT-qPCR and
western blot analysis (Fig. 1D-F).
These results suggest that the PTX3 expression level is high in
human lung cancer tissues, and PTX3 can be generated by lung cancer
cells.
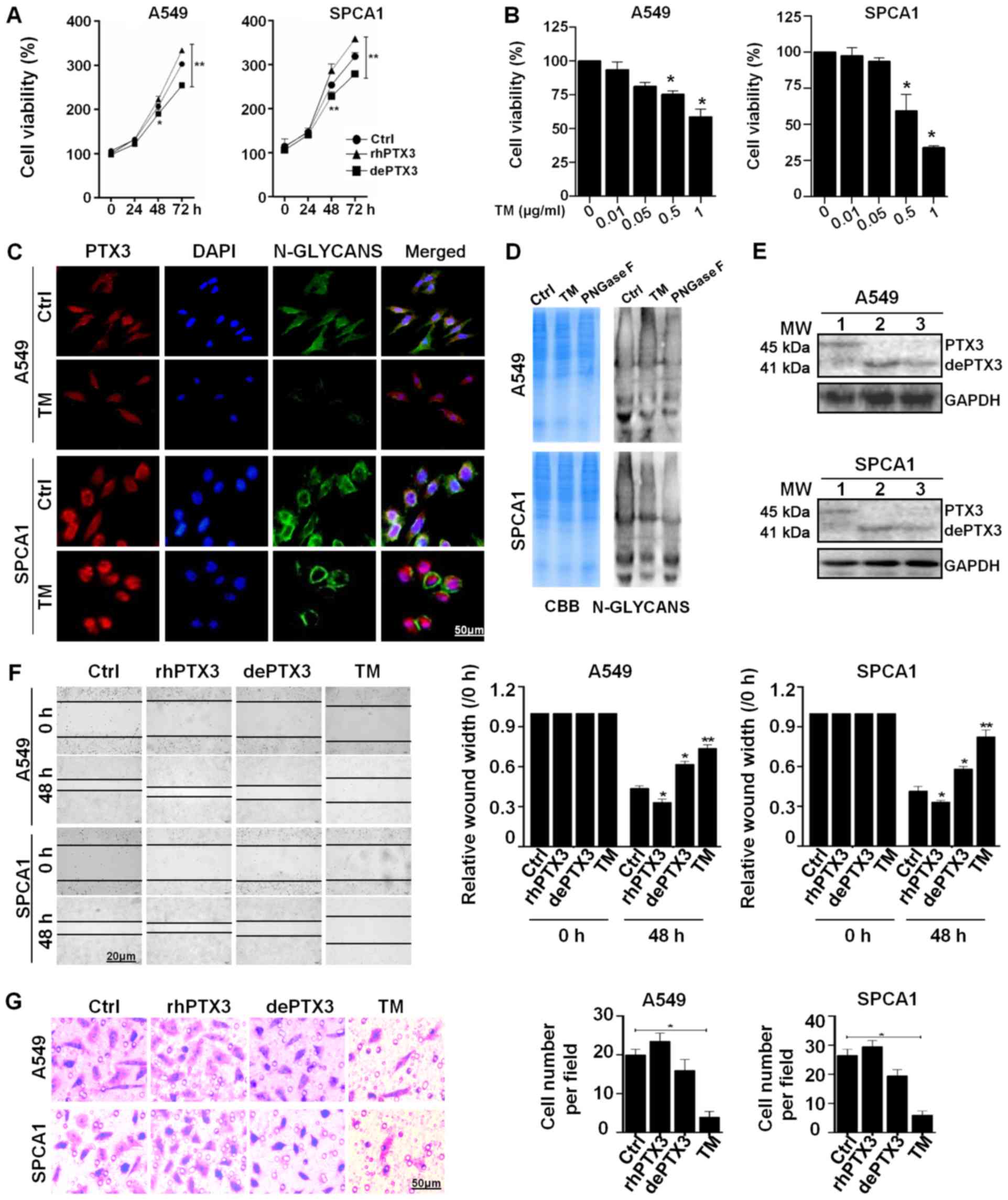
dePTX3 suppresses the growth and
migration of human lung cancer cells
The A549 and SPCA1 cells were used to determine the
role of PTX3 glycosylation in lung cancer growth. The glycosylated
rhPTX3 was deglycosylated by incubation with PNGase F (500 U/ml),
an enzyme that cleaves Asn-linked high mannose, as well as hybrid
and complex oligosaccharides for 2 h at 37°C. Lung cancer cells
were treated with rhPTX3 (100 ng/ml) or dePTX3, respectively. The
results of CCK-8 assay revealed that cell viability was
significantly inhibited following treatment with dePTX3, while
increased in response to rhPTX3 treatment in both A549 and SPCA1
cells. Differences in viability between the rhPTX3- and
dePTX3-treated cells were more significant after 48 and 72 h
incubation (Fig. 2A). We found
that treatment with 0.5 or 1 µg/ml TM significantly
inhibited the viability of A549 and SPCA1 cells in comparison with
the untreated control (Fig. 2B).
PHA is a lectin which can recognize and bind with the N-glycans of
glycoproteins. We further investigated the binding of
FITC-conjugated PHA to evaluate changes in N-deglycosylation in
response to TM. The A549 and SPCA1 cells treated with TM exhibited
decreased PHA-L binding compared to the untreated cells, as well as
a lower level of N-glycan PTX3 (Fig.
2C). A 12% SDS-PAGE gel stained with CBB revealed that both the
untreated cells and cells treated with TM or PNGase F enzyme had
comparable loading of proteins in each lane, and the lectin blot
showed that the untreated cells had stronger N-glycan bands than
the cells treated with TM (0.5 µg/ml) for 48 h and PNGase F
enzyme (500 U/ml) for 2 h (Fig.
2D). Treatment of the lung cancer cells with TM or rhPTX3 +
PNGase F incubation resulted in a decrease in the apparent
molecular mass of PTX3 approximately 4 kDa, from 45 to 41 kDa
(Fig. 2E) (lane 1, glycosylated
PTX3; lane 2, dePTX3 by TM; lane 3, dePTX3 by PNGase F). Moreover,
the results of wound healing and migration assays indicated that
rhPTX3 treatment significantly increased the migratory ability of
A549 and SPCA1 cells compared with the cells treated with dePTX3 or
TM (0.5 µg/ml) after 48 h (Fig.
2F and G). These results suggest that PTX3 deglycosylation
inhibits lung cancer cell growth and migration.
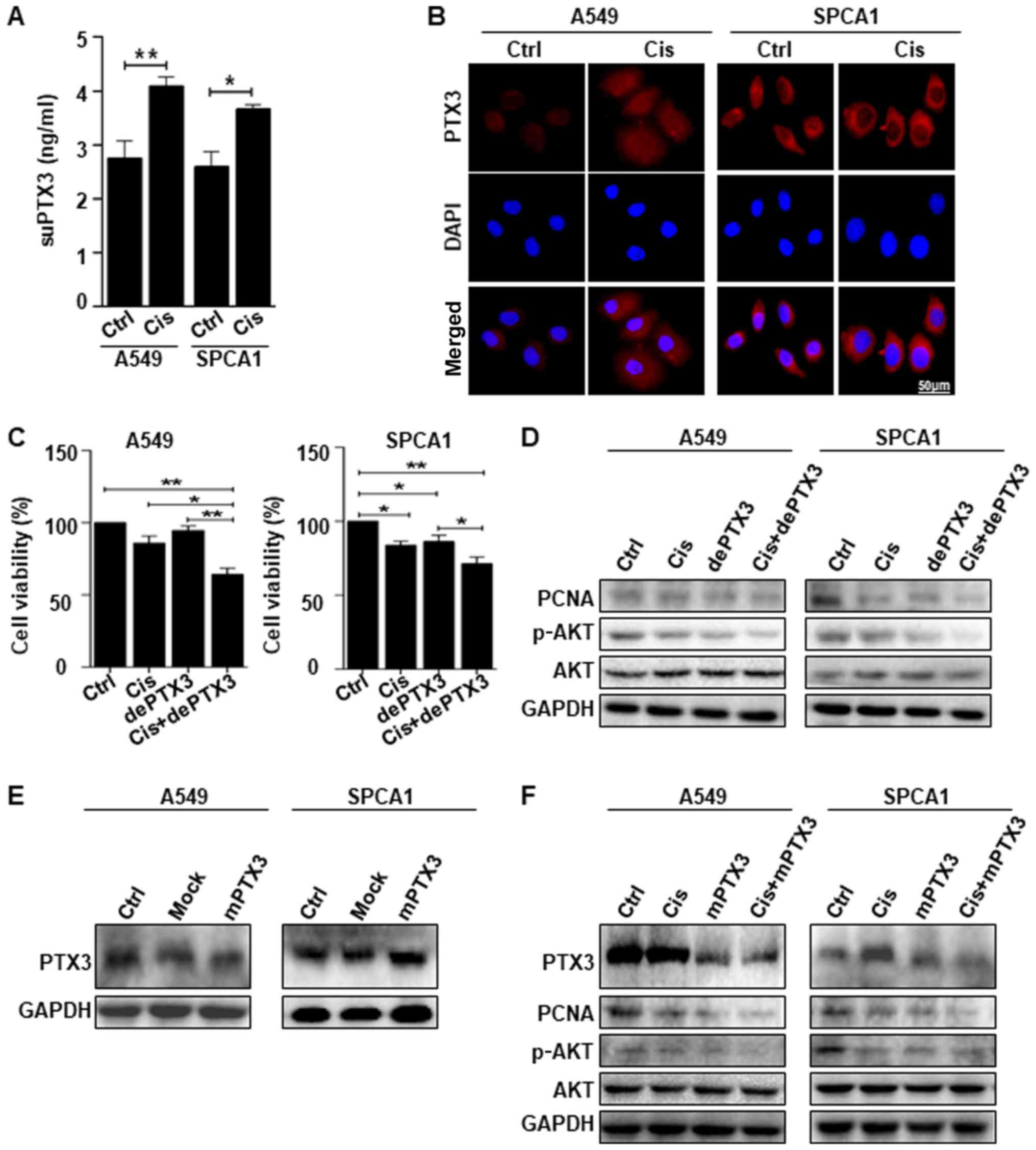
PTX3 deglycosylation enhances the
sensitivity of lung cancer cells to cisplatin treatment
The results of ELISA revealed that suPTX3 level was
significantly higher in the supernatant medium of Cis-treated A549
and SPCA1 cells than that in the control (Fig. 3A). The immunofluorescent staining
of PTX3 expression confirmed that the Cis-treated A549 and SPCA1
cells exhibited an increased expression of PTX3 compared to the
untreated cells. DAPI was used to stain the nuclei, as shown in
Fig. 3B. We further examined the
effects of PTX3 deglycosylation on the sensitivity of lung cancer
cells to Cis. Lung cancer cells were treated with Cis alone or in
combination with dePTX3. Cell viability was assessed by CCK-8
assay, which revealed that Cis suppressed the viability of A549 and
SPCA1 cells. However, following combined treatment with dePTX3 and
Cis, dePTX3 was found to significantly enhance the suppressive
effects of Cis on the viability of both cell lines, as shown in
Fig. 3C. Western blot analysis of
PCNA expression confirmed the increased anti-proliferative effects
of Cis following combined treatment with dePTX3 in both A549 and
SPCA1 (Fig. 3D). We also examined
cell signaling pathway-related proteins, specifically AKT
phosphorylation following treatment with Cis or combined treatment
with dePTX3. Combined treatment with Cis and dePTX3 for 48 h
suppressed AKT phosphorylation compared to Cis treatment alone, as
shown in Fig. 3D. These results
suggest that dePTX3 enhances the sensitivity of lung cancer cells
to Cis. We also sought to determine whether mPTX3 enhanced the
antitumor effects of Cis through an AKT-dependent pathway. First,
we examined the efficiency of the mutated PTX3 by western blot
analysis, which indicated a successful PTX3 mutation (Fig. 3E). As expected, combined treatment
with dePTX3 and Cis decreased PCNA expression and consistent
results were observed following the use of Cis and mPTX3 (Fig. 3F), and mPTX3 enhanced the antitumor
effects of Cis on the A549 and SPCA1 cells. This finding led us to
examine the role of PTX3 glycosylation in lung cancer
progression.
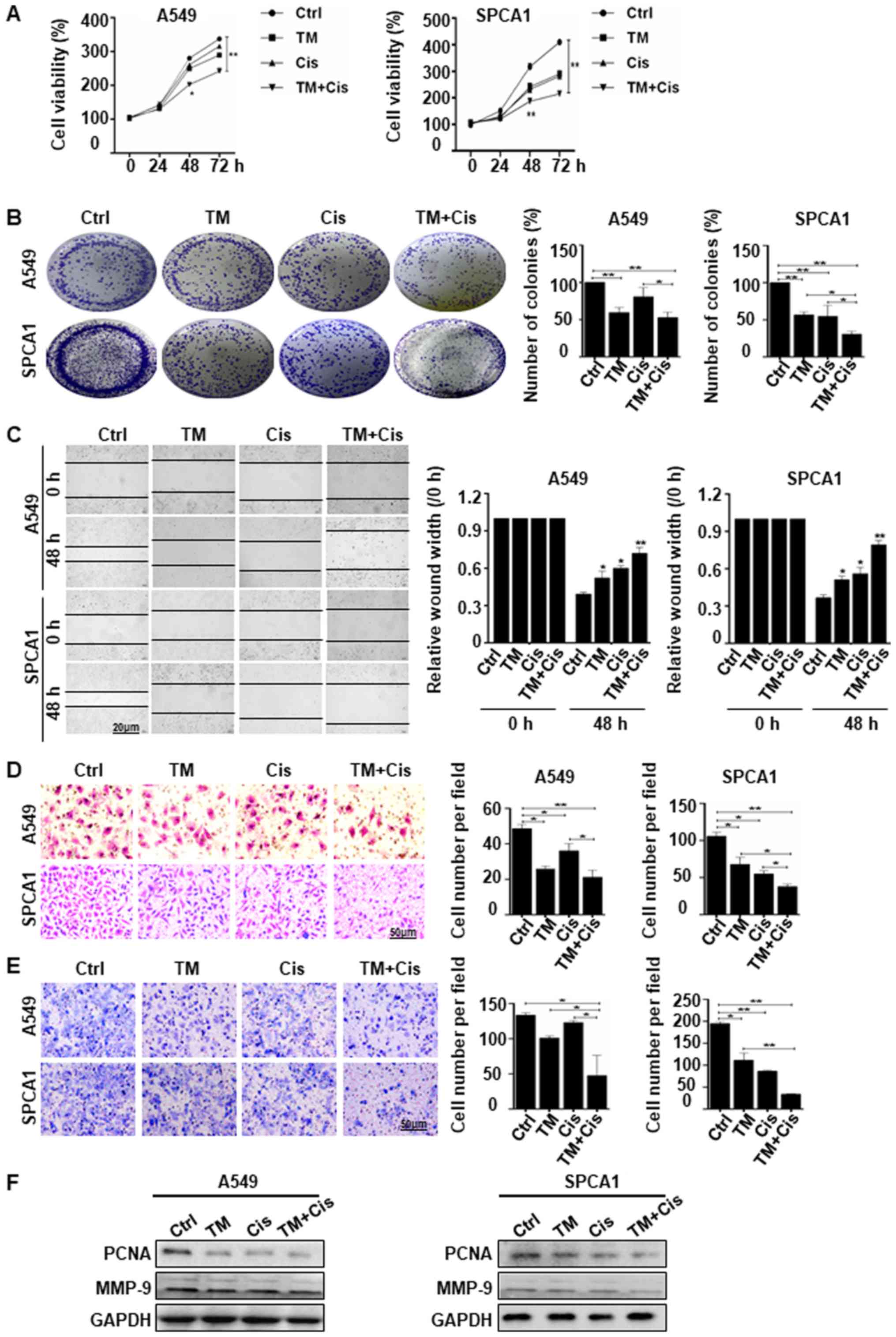
TM enhances the suppressive effects of
cisplatin in lung cancer cells
We used TM to enhance Cis drug sensitivity,
demonstrating the suppression of lung cancer growth and metastasis.
The results of CCK-8 assay revealed that combined treatment with TM
and Cis significantly suppressed the viability of both A549 and
SPCA1 cells in a time-dependent manner. Combined treatment
inhibited cell growth 2.0 fold in A549 cells and 2.5-fold in SPCA1
cells compared with no treatment cells (Fig. 4A). The results of colony formation
assay were consistent with CCK-8 assay, demonstrated a significant
decrease in cell viability in both cell lines following combined
treatment. Colony formation was reduced by 25 and 50% in A549 cells
and by 45 and 70% in SPCA1 cells treated with Cis alone or TM + Cis
(Fig. 4B). These results suggest
that TM potently enhanced the anti-proliferative effects of Cis on
lung cancer cells. Wound healing assay revealed that combined
treatment significantly decreased cell migration compared to no
treatment or individual TM or Cis treatment after 48 h (Fig. 4C). These results were consistent
with that of transwell migration and Matrigel invasion assay of
A549 and SPCA1 cells, which indicated that combined treatment
inhibited the invasion of the A549 and SPCA1 cells more
significantly than no treatment or treatment with TM or Cis alone
(Fig. 4D and E). Western blot
analysis of the proliferative marker, PCNA, also confirmed the
anti-proliferative activity in the A549 and SPCA1 cells following
combined treatment, with a decrease in PCNA expression compared to
the expression in the individually treated or untreated cells.
Furthermore, cells treated with the combined treatment expressed
lower level of MMP-9 than the untreated cells (Fig. 4F). The data indicate that TM
enhances the chemosensitivity of lung cancer cells by inhibiting
proliferation and metastasis.
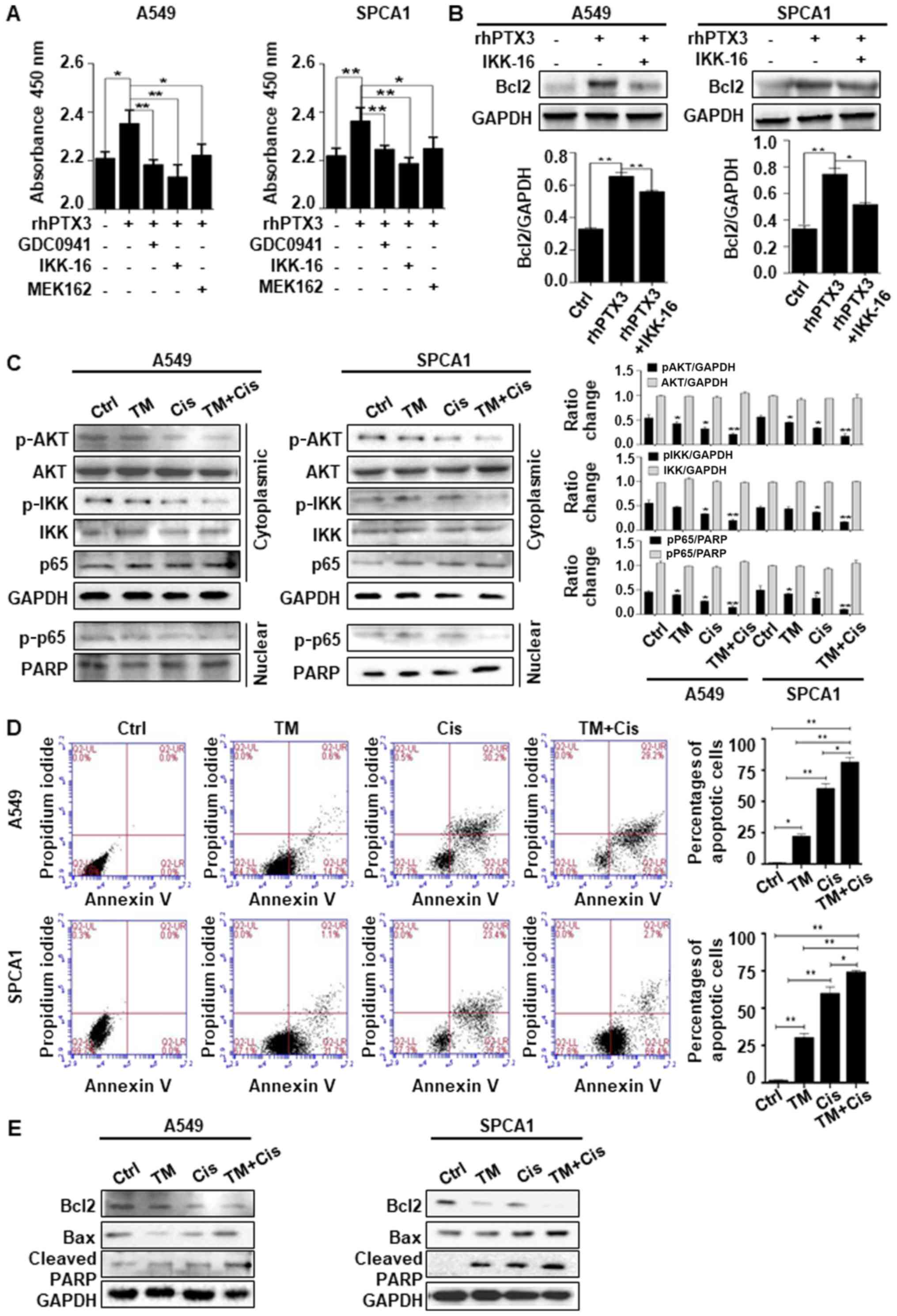
TM the sensitivity of lung cancer cells
to cisplatin-induced apoptosis through deactivating AKT/NF-κB
signaling pathway
We probed downstream signaling pathways of PTX3
using the inhibitors, including GDC0941 (an inhibitor of AKT
phosphorylation), MEK162 (an inhibitor of MEK1/2) and IKK-16 (an
inhibitor of NF-κB phosphorylation). The results of CCK-8 assay
revealed that treatment with rhPTX3 and the inhibitors targeting
AKT/NF-κB pathway led to an enhanced inhibition of the viability
and proliferation of A549 and SPCA1 cells compared to the cells
treated with rhPTX3 alone (Fig.
5A). The cells were treated with IKK-16 to determine whether
NF-κB was associated with the PTX3-mediated regulation of Bcl2. The
results indicated that treatment with IKK-16 inhibited the
PTX3-mediated upregulation of Bcl2 expression in both A549 and
SPCA1 cells (Fig. 5B). Taken
together, the evidence suggest that lung cancer cell survival is
positively regulated by PTX3, likely through AKT/NF-κB pathway and
Bcl2 upregulation. We then sought to determine whether TM enhanced
the anticancer effects of Cis through AKT/NF-κB-dependent pathway.
Western blot analysis of cytoplasmic protein extracts revealed that
combined treatment with TM and Cis decreased AKT and IKK
phosphorylation more significantly than TM or Cis drug treatment
alone. Likewise, in nuclear protein extracts, the phosphorylation
of p-p65 in the A549 and SPCA1 cells was decreased following
treatment (Fig. 5C). An Annexin V
FITC/PI apoptosis detection kit was used. The A549 and SPCA1 cells
were treated with TM, Cis or in combination for 24 h, and the
results of flow cytometry revealed that the percentage of total
apoptotic cells was increased significantly than treatment with TM
or Cis alone (Fig. 5D). Treatment
with TM alone led to an average of 20.3 and 30.5% total apoptotic
A549 and SPCA1 cells, respectively; Cis treatment alone led to an
average of 58.4 and 59.2% total apoptotic A549 and SPCA1 cells,
respectively; combined treatment with TM and Cis significantly
increased the percentage of apoptotic A549 and SPCA1 cells. The
total apoptotic A549 and SPCA1 cells were 80.3 and 73.2%,
respectively. These findings confirm that apoptosis is the
predominant pattern of cell death induced by combined treatment. We
also found that combined treatment with TM and Cis significantly
increased the expression level of apoptotic markers, including Bax
and cleaved PARP, and simultaneously decreased the expression of
Bcl2, compared with treatment with TM or Cis alone in both A549 and
SPCA1 cells (Fig. 5E). These
results suggest that the deglycosylation of PTX3 by TM in lung
cancer cells treated with Cis inhibits the activation of AKT/NF-κB
signaling pathway, promoting the apoptosis of lung cancer
cells.
Discussion
PTX3 is a potential marker of inflammation to detect
lung cancer. Recent studies have indicated a significant
association between serum PTX3 and cancer malignancy. A
proteomics-based study provided evidence that elevated level of
PTX3 in lung cancer patients as compared to healthy control by
ELISA (30). The increased PTX3
was also found in the serum of liposarcoma, pancreatic and breast
cancer (13,16,31).
Locatelli et al reported that PTX3 in glioma was
significantly correlated with tumor grade and severity assessed by
immunohistochemical staining (12). In the current study, the elevated
PTX3 level was detected in both human lung cancer serum and tissue
by ELISA and immunohistochemical staining. The consistent
alterations of PTX3 in serum and tissue of the cancer patients
indicated that serum PTX3 could represent the tissue pathogenesis.
Moreover, tumorigenesis has been considered as a chronic
inflammatory process, and the early release of inflammatory protein
PTX3 may be predisposed to the development of cancer. Therefore,
the detection of serum PTX3 can be applied as an early marker for
cancer diagnosis. Based on the facts in other labs and our results
that PTX level is linked to the growth, migration and invasion
capability, the inhibition of PTX3 may be a treatment target for
lung cancer.
Glycans alter protein structure and conformation and
as a result, modulate the functional activities of the glycoprotein
(32). Changes in cellular
glycosylation have recently been acknowledged as a key component of
cancer progression. Alterations in the glycosylation of
extracellular proteins do not only have a direct impact on cell
growth and survival, but also facilitate tumor-induced
immunomodulation, and hence metastasis (33). It has been demonstrated that
N-linked deglycosylation inhibits the growth of several types of
cancer cells (25). Oncogenic
roles for N-glycans on the cancer cell surface have been described
in breast cancer, colon cancer, prostate cancer, lung cancer,
hepatocellular carcinoma and gastric cancer (15,34-39).
Human PTX3 contains a single N-glycosylation site that is fully
occupied by complex oligosaccharides (7). The glycosylation of PTX3 has been
suggested to modulate PTX3 function during inflammation and tumor
development. Chi et al reported that the glycosylation of
PTX3 at Asn-220 was critical for its pro-tumor involvement
(18). Our results demonstrated
that tunicamycin (TM), which blocked N-glycan precursor
biosynthesis, enhanced the suppressive effects of Cis on lung
cancer cell proliferation and migration. TM and dePTX3 also
increased the suppressive effects of Cis on lung cancer cell
growth, migration and invasion compared to treatment with the
individual drugs. The inhibition of N-linked glycosylation
biosynthetic pathways may provide a novel diagnostic and
therapeutic target for cancer growth.
Cis is widely used as a chemotherapeutic drug in a
number of cancer treatments, and limited by acquired or intrinsic
resistance of cells to the drug (40,41).
Poor sensitivity to Cis is based on several mechanisms, including
diminished intracellular drug accumulation due to drug efflux or
metabolic inactivation, the inhibition of apoptosis, and improved
DNA damage repair in cancer cells (42). The elevated expression of cell
surface N-linked glycosylation has been reported to be associated
with drug resistance, and the inhibition of N-linked glycosylation
in breast cancer results in an elevated sensitivity to doxorubicin
(43-45). It has also been found that the
TM-induced inhibition of N-linked glycosylation enhances the
susceptibility of the multidrug-resistant ovarian cancer cells, to
vincristine, doxorubicin, and Cis (46). The increased apoptosis of breast
cancer cells has been reported following combined treatment with
Herceptin and TM (45). Similarly,
an enhanced sensitivity to Cis has been reported in head and neck
cancer following TM treatment (47). In this study, we found that Cis
treatment increased the expression of PTX3 in lung cancer cells,
which was associated with a reduced potency of its anticancer
effects on lung cancer. However, the deglycosylation of PTX3 by TM
in lung cancer cells functions synergistically with Cis. This study
demonstrates that alterations in PTX3 glycosylation lead to a
recovery of Cis chemosensitivity with respect to the suppression of
lung cancer growth and migration. To date there are limited studies
available on the role of dePTX3 in cancer therapy; this warrants
further investigation to elucidate the mechanisms through PTX3
glycosylation in cancer cells that is associated with a
chemosensitivity to treatment.
In this study, we found that combined treatment with
TM and Cis inhibited the survival and growth of lung cancer cells,
indicating that PTX3 deglycosylation decreased the resistance of
cancer cells to chemotherapeutic drugs. We further confirmed that
the deglycosylation of PTX3 inactivated AKT/NF-κB signaling
pathway. These results reveal that the induction of PTX3
deglycosylation by TM inhibited AKT/NF-κB pathway and contributed
to prevent NF-κB-associated tumor metastasis. In lung cancer, the
inhibition of NF-κB nuclear translocation also enhances apoptosis
(48). The findings of this study
were consistent with those of the previous studies. TM and Cis
treatment reduced AKT activation and inhibited NF-κB
phosphorylation, leading to the induction of apoptosis and
decreasing lung cancer cell growth, which resulted in the reduction
of the distinct survival pathways, AKT/NF-κB. These results suggest
that PTX3 secretion may be detrimental to the chemotherapeutic
efficacy of Cis in lung cancer treatment; however, dePTX3 augments
the efficacy of Cis by inhibiting the activation of AKT/NF-κB in
lung cancer.
In conclusion, the findings of this study suggest
that the deglycosylation of PTX3 by TM significantly sensitizes
lung cancer cells to Cis, and impairs cell growth and metastasis.
Our preliminary results also suggest that the manipulation of
N-linked glycosylation of PTX3 by TM in tumor cells may prove to be
a useful therapeutic strategy for successful chemotherapy in
combination with Cis against lung cancer or other solid
cancers.
Funding
This study was supported by the National Natural
Science Foundation of China Research Grants (nos. 31670810,
31870794, 81572881).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors’ contributions
BA and QY conceived and designed the study with
inputs from MNK. QY and BA were responsible for the supervision and
coordination of the project. BA, MNK, SK and MAN performed most of
the in vitro experiments. BA led the data analysis with
inputs from QY, MNK, QZ, YL and SK. BA and YL performed statistical
analysis of the data. The first draft of the manuscript was written
by BA and MNK then QY, MNK, SK, YL and MAN contributed to revise
and review the manuscript. All authors read and approved the final
manuscript before submission.
Ethics approval and consent to
participate
The use of human lung cancer serum and tissue was
reviewed and approved by the Ethics Committee of Dalian Medical
University, and was performed in accordance with the approved
guidelines. Informed consent was obtained from lung cancer patients
and samples were obtained from the First Affiliated Hospital of
Dalian Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Abbreviations:
|
PTX3
|
pentraxin-3
|
|
dePTX3
|
deglycosylated PTX3
|
|
PNGase F
|
peptide N-glycosidase F
|
|
rhPTX3
|
recombinant human pentraxin-3
|
|
TM
|
tunicamycin
|
|
Cis
|
cisplatin
|
References
|
1
|
O’Byrne KJ, Barr MP and Gray SG: The role
of epigenetics in resistance to Cisplatin chemotherapy in lung
cancer. Cancers (Basel). 3:1426–1453. 2011. View Article : Google Scholar
|
|
2
|
Zhuang H, Jiang W, Zhang X, Qiu F, Gan Z,
Cheng W, Zhang J, Guan S, Tang B, Huang Q, et al: Suppression of
HSP70 expression sensitizes NSCLC cell lines to TRAIL-induced
apoptosis by upregulating DR4 and DR5 and downregulating c-FLIP-L
expressions. J Mol Med (Berl). 91:219–235. 2013. View Article : Google Scholar
|
|
3
|
Lv XB, Liu L, Cheng C, Yu B, Xiong L, Hu
K, Tang J, Zeng L and Sang Y: SUN2 exerts tumor suppressor
functions by suppressing the Warburg effect in lung cancer. Sci
Rep. 5:179402015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Boulikas T: Molecular mechanisms of
cisplatin and its liposomally encapsulated form, Lipoplatin.
Lipoplatin as a chemotherapy and antiangiogenesis drug. Cancer Biol
Ther. 5:351–376. 2007.
|
|
5
|
Lin Y, Wang Z, Liu L and Chen L: Akt is
the downstream target of GRP78 in mediating cisplatin resistance in
ER stress-tolerant human lung cancer cells. Lung Cancer.
71:291–297. 2011. View Article : Google Scholar
|
|
6
|
Fuertes MA, Alonso C and Pérez JM:
Biochemical modulation of cisplatin mechanisms of action:
Enhancement of antitumor activity and circumvention of drug
resistance. Chem Rev. 103:645–662. 2003. View Article : Google Scholar
|
|
7
|
Bottazzi B, Inforzato A, Messa M,
Barbagallo M, Magrini E, Garlanda C and Mantovani A: The pentraxins
PTX3 and SAP in innate immunity, regulation of inflammation and
tissue remodelling. J Hepatol. 64:1416–1427. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Balhara J, Koussih L, Zhang J and Gounni
AS: Pentraxin 3: An immuno-regulator in the lungs. Front Immunol.
4:1272013. View Article : Google Scholar
|
|
9
|
Inforzato A, Rivieccio V, Morreale AP,
Bastone A, Salustri A, Scarchilli L, Verdoliva A, Vincenti S, Gallo
G, Chiapparino C, et al: Structural characterization of PTX3
disulfide bond network and its multimeric status in cumulus matrix
organization. J Biol Chem. 283:10147–10161. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Inforzato A, Reading PC, Barbati E,
Bottazzi B, Garlanda C and Mantovani A: The ‘sweet’ side of a long
pentraxin: How glycosylation affects PTX3 functions in innate
immunity and inflammation. Front Immunol. 3:4072013. View Article : Google Scholar
|
|
11
|
EL-Attar HA, Abaza MM, Gaber EW and
EL-sharkawy RM: Serum profiles of pentraxin-3 and high sensitivity
C - reactive protein in patients with chronic kidney disease
treated with or without hemodialysis. J Nephrol Ther. 7:12017.
|
|
12
|
Locatelli M, Ferrero S, Martinelli
Boneschi F, Boiocchi L, Zavanone M, Maria Gaini S, Bello L,
Valentino S, Barbati E, Nebuloni M, et al: The long pentraxin PTX3
as a correlate of cancer-related inflammation and prognosis of
malignancy in gliomas. J Neuroimmunol. 260:99–106. 2013. View Article : Google Scholar
|
|
13
|
Willeke F, Assad A, Findeisen P, Schromm
E, Grobholz R, von Gerstenbergk B, Mantovani A, Peri S, Friess HH,
Post S, et al: Overexpression of a member of the pentraxin family
(PTX3) in human soft tissue liposarcoma. Eur J Cancer.
42:2639–2646. 2006. View Article : Google Scholar
|
|
14
|
Stallone G, Cormio L, Netti GS, Infante B,
Selvaggio O, Fino GD, Ranieri E, Bruno F, Prattichizzo C,
Sanguedolce F, et al: Pentraxin 3: A novel biomarker for predicting
progression from prostatic inflammation to prostate cancer. Cancer
Res. 74:4230–4238. 2014. View Article : Google Scholar
|
|
15
|
Diamandis EP, Goodglick L, Planque C and
Thornquist MD: Pentraxin-3 is a novel biomarker of lung carcinoma.
Clin Cancer Res. 17:2395–2399. 2011. View Article : Google Scholar
|
|
16
|
Choi B, Lee EJ, Song DH, Yoon SC, Chung
YH, Jang Y, Kim SM, Song Y, Kang SW, Yoon SY, et al: Elevated
Pentraxin 3 in bone metastatic breast cancer is correlated with
osteolytic function. Oncotarget. 5:481–492. 2014. View Article : Google Scholar :
|
|
17
|
Argani H, Ghorbanihaghjo A, Panahi G,
Rashtchizadeh N, Safa J and Meimand SM: Serum Fetuin-A and
Pentraxin3 in hemodialysis and renal transplant patients. Clin
Biochem. 45:775–779. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chi JY, Hsiao YW, Li CF, Lo YC, Lin ZY,
Hong JY, Liu YM, Han X, Wang SM, Chen BK, et al: Targeting
chemotherapy-induced PTX3 in tumor stroma to prevent the
progression of drug-resistant cancers. Oncotarget. 6:23987–24001.
2015. View Article : Google Scholar
|
|
19
|
Vasconcelos-Dos-Santos A, Oliveira IA,
Lucena MC, Mantuano NR, Whelan SA, Dias WB and Todeschini AR:
Biosynthetic machinery involved in aberrant glycosylation:
Promising targets for developing of drugs against cancer. Front
Oncol. 5:1382015. View Article : Google Scholar
|
|
20
|
Song EY, Kang SK, Lee YC, Park YG, Chung
TH, Kwon DH, Byun SM and Kim CH: Expression of bisecting
N-acetylglucosaminyltransferase-III in human hepatocarcinoma
tissues, fetal liver tissues, and hepatoma cell lines of Hep3B and
HepG2. Cancer Invest. 19:799–807. 2001. View Article : Google Scholar
|
|
21
|
Mori S, Aoyagi Y, Yanagi M, Suzuki Y and
Asakura H: Serum N-acetylglucosaminyltransferase III activities in
hepatocellular carcinoma. J Gastroenterol Hepatol. 13:610–619.
1998. View Article : Google Scholar
|
|
22
|
Dube DH and Bertozzi CR: Glycans in cancer
and inflammation - potential for therapeutics and diagnostics. Nat
Rev Drug Discov. 4:477–488. 2005. View
Article : Google Scholar
|
|
23
|
Chatterjee BP, Mondal G and Chatterjee U:
Glycosylation of acute phase proteins: A promising disease
biomarker. Proc Natl Acad Sci, India - Sect B:Biol Sci. 84:865–874.
2014. View Article : Google Scholar
|
|
24
|
Reticker-Flynn NE and Bhatia SN: Aberrant
glycosylation promotes lung cancer metastasis through adhesion to
galectins in the metastatic niche. Cancer Discov. 5:168–181. 2015.
View Article : Google Scholar
|
|
25
|
Contessa JN, Bhojani MS, Freeze HH,
Rehemtulla A and Lawrence TS: Inhibition of N-linked glycosylation
disrupts receptor tyrosine kinase signaling in tumor cells. Cancer
Res. 68:3803–3809. 2008. View Article : Google Scholar
|
|
26
|
de Freitas Junior JCM and Morgado-Díaz JA:
The role of N-glycans in colorectal cancer progression: Potential
biomarkers and therapeutic applications. Oncotarget. 7:19395–19413.
2016.
|
|
27
|
Han X, Zhang X, Li H, Huang S, Zhang S,
Wang F and Shi Y: Tunicamycin enhances the antitumor activity of
trastuzumab on breast cancer in vitro and in vivo. Oncotarget.
6:38912–38925. 2015. View Article : Google Scholar
|
|
28
|
Ahsan A, Hiniker SM, Ramanand SG, Nyati S,
Hegde A, Helman A, Menawat R, Bhojani MS, Lawrence TS and Nyati MK:
Role of epidermal growth factor receptor degradation in
cisplatin-induced cytotoxicity in head and neck cancer. Cancer Res.
70:2862–2869. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Banerjee A, Lang JY, Hung MC, Sengupta K,
Banerjee SK, Baksi K and Banerjee DK: Unfolded protein response is
required in nu/nu mice microvasculature for treating breast tumor
with tunicamycin. J Biol Chem. 286:29127–29138. 2011. View Article : Google Scholar
|
|
30
|
Planque C, Kulasingam V, Smith CR, Reckamp
K, Goodglick L and Diamandis EP: Identification of five candidate
lung cancer biomarkers by proteomics analysis of conditioned media
of four lung cancer cell lines. Mol Cell Proteomics. 8:2746–2758.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kondo S, Ueno H, Hosoi H, Hashimoto J,
Morizane C, Koizumi F, Tamura K and Okusaka T: Clinical impact of
pentraxin family expression on prognosis of pancreatic carcinoma.
Br J Cancer. 109:739–746. 2013. View Article : Google Scholar
|
|
32
|
O’Connor SE and Imperiali B: Modulation of
protein structure and function by asparagine-linked glycosylation.
Chem Biol. 3:803–812. 1996. View Article : Google Scholar
|
|
33
|
Häuselmann I and Borsig L: Altered
tumor-cell glycosylation promotes metastasis. Front Oncol.
4:282014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kudo T, Nakagawa H, Takahashi M, Hamaguchi
J, Kamiyama N, Yokoo H, Nakanishi K, Nakagawa T, Kamiyama T,
Deguchi K, et al: N-glycan alterations are associated with drug
resistance in human hepatocellular carcinoma. Mol Cancer. 6:322007.
View Article : Google Scholar :
|
|
35
|
Tafani M, Russo A, Di Vito M, Sale P,
Pellegrini L, Schito L, Gentileschi S, Bracaglia R, Marandino F,
Garaci E, et al: Up-regulation of pro-inflammatory genes as
adaptation to hypoxia in MCF-7 cells and in human mammary invasive
carcinoma microenvironment. Cancer Sci. 101:1014–1023. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li WP, Zuber C, Heitz PU and Roth J:
Cytochemical staining for beta 1,6 branching of asparagine-linked
oligosaccharides in variants of metastatic human colon carcinoma
cells. Am J Pathol. 145:470–480. 1994.
|
|
37
|
Saldova R, Fan Y, Fitzpatrick JM, Watson
RW and Rudd PM: Core fucosylation and alpha2-3 sialylation in serum
N-glycome is significantly increased in prostate cancer comparing
to benign prostate hyperplasia. Glycobiology. 21:195–205. 2011.
View Article : Google Scholar
|
|
38
|
Liu L, Yan B, Huang J, Gu Q, Wang L, Fang
M, Jiao J and Yue X: The identification and characterization of
novel N-glycan-based biomarkers in gastric cancer. PLoS One.
8:e778212013. View Article : Google Scholar :
|
|
39
|
Saldova R, Reuben JM, Abd Hamid UM, Rudd
PM and Cristofanilli M: Levels of specific serum N-glycans identify
breast cancer patients with higher circulating tumor cell counts.
Ann Oncol. 22:1113–1119. 2011. View Article : Google Scholar
|
|
40
|
Li L, Khan MN, Li Q, Chen X, Wei J, Wang
B, Cheng JW, Gordon JR and Li F: G31P, CXCR1/2 inhibitor, with
cisplatin inhibits the growth of mice hepatocellular carcinoma and
mitigates high dose cisplatin-induced nephrotoxicity. Oncol Rep.
33:751–757. 2015. View Article : Google Scholar
|
|
41
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar
|
|
42
|
Tanida S, Mizoshita T, Ozeki K, Tsukamoto
H, Kamiya T, Kataoka H, Sakamuro D and Joh T: Mechanisms of
cisplatin-induced apoptosis and of cisplatin sensitivity: Potential
of BIN1 to act as a potent predictor of cisplatin sensitivity in
gastric cancer treatment. Int J Surg Oncol. 2012.862879:2012.
|
|
43
|
Liu X, Nie H, Zhang Y, Yao Y, Maitikabili
A, Qu Y, Shi S, Chen C and Li Y: Cell surface-specific N-glycan
profiling in breast cancer. PLoS One. 8:e727042013. View Article : Google Scholar
|
|
44
|
Ferreira JA, Peixoto A, Neves M, Gaiteiro
C, Reis CA, Assaraf YG and Santos LL: Mechanisms of cisplatin
resistance and targeting of cancer stem cells: Adding glycosylation
to the equation. Drug Resist Updat. 24:34–54. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Peiris D, Spector AF, Lomax-Browne H,
Azimi T, Ramesh B, Loizidou M, Welch H and Dwek MV: Cellular
glycosylation affects Herceptin binding and sensitivity of breast
cancer cells to doxorubicin and growth factors. Sci Rep.
7:430062017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hiss DC, Gabriels GA and Folb PI:
Combination of tunicamycin with anticancer drugs synergistically
enhances their toxicity in multidrug-resistant human ovarian
cystadenocarcinoma cells. Cancer Cell Int. 7:52007. View Article : Google Scholar
|
|
47
|
Noda I, Fujieda S, Seki M, Tanaka N,
Sunaga H, Ohtsubo T, Tsuzuki H, Fan GK and Saito H: Inhibition of
N-linked glycosylation by tunicamycin enhances sensitivity to
cisplatin in human head-and-neck carcinoma cells. Int J Cancer.
80:279–284. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen W, Li Z, Bai L and Lin Y: NF-kappaB,
a mediator for lung carcinogenesis and a target for lung cancer
prevention and therapy. Front Biosci. 16:1172–1185. 2011.
View Article : Google Scholar
|