Introduction
Pancreatic ductal adenocarcinoma (PDAC) is
associated with a high mortality rate, as it is often diagnosed at
an advanced stage and is resistant to current therapies (1,2).
Current treatment strategies largely comprise surgical and
chemotherapy regimens, which have yielded only modest improvements
in survival. Notably, survival of patients with PDAC has shown
little improvement in the last four decades (3). Therefore, novel targeted therapies
are urgently required for the treatment of patients with these
conditions. Metastatic disease is often treated with the
chemotherapeutic DNA synthesis inhibitor gemcitabine, in
combination with the small molecule inhibitor tyrosine kinase
inhibitor (TKI) erlotinib (4,5).
Erlotinib acts as an inhibitor of the human epidermal growth factor
(EGF) receptor type 1 receptor (EGFR), which is overexpressed in
several types of cancer, including PDAC (6). EGFR activation stimulates downstream
signaling pathways that promote proliferation and metastasis
(3). Clinically, erlotinib plus
gemcitabine treatment provides a modest increase in patient outcome
over gemcitabine alone (5).
However, further preclinical and clinical studies are required to
address the significant problem of resistance that develops in
response to several targeted therapies, also known as acquired
resistance (7). One such
drug-resistance mechanism activated during erlotinib treatment is
the signal transducer and activator of transcription 3 (STAT3)
pathway, which promotes proliferation, as well as differentiation,
survival, inflammation and angiogenesis (8). Previous studies on lung and
pancreatic cancer cells combining STAT3 inhibition with
EGFR-targeted therapy exhibit increased efficacy (9,10).
Activating mutations of KRAS proto-oncogene, GTPase
(KRAS), and inactivating mutations of the tumor suppressor genes
cyclin-dependent kinase (CDK) inhibitor 2A (CDKN2A; also known as
p16INK4a or p16), tumor protein p53 and SMAD family member 4 have
been reported to promote carcinogenesis in PDAC (2). In particular, CDKN2A is most commonly
inactivated by a homozygous deletion that leads to p16INK4a loss of
function in >90% of PDAC cases (11,12).
Inactivation of CDKN2A/p16 is believed to be an early event in
pancreatic cancer progression, since its inactivation is detected
in 40% of precursor pancreatic intraepithelial neoplastic lesions
(13,14). In addition, CDKN2A has been
identified as a gatekeeper gene in PDAC, which indicates its
importance in this cancer type (15). Furthermore, recent evidence has
suggested that the progression of PDAC may be due to high genomic
instability in the form of chromothripsis, and CDKN2A has been
identified as one of the genes lost by this mechanism (16). Finally, while KRAS mutation is
thought to be the first and most frequent genetic disruption in
PDAC, it has been reported that oncogenic KRAS function is
controlled by the tumor suppressor function of p16INK4a (17). Therefore, downregulation of
p16INK4a together with oncogenic activation of KRAS may cooperate
to promote pancreatic tumorigenesis (18). p16INK4a blocks cell cycle
progression by interacting with and inhibiting CDK4/6, thus
resulting in reduced phosphorylation of the retinoblastoma (Rb)
protein. Unphosphorylated Rb associates with the E2F transcription
factor to inhibit the G1 to S transition (19). Treatments that target Rb
phosphorylation in cancer cells have been developed and exhibit
efficacy in Rb-positive cells. For example, palbociclib is an
orally active CDK4/6-specific inhibitor that causes cell cycle
arrest in PDAC and other cancer cell types (20-23).
Notably, palbociclib was the first CDK4/6 inhibitor approved by the
United States Food and Drug Administration for the treatment of
advanced breast cancer in women with estrogen receptor-positive
human epidermal growth factor receptor 2-negative disease (24). Notwithstanding the development of
resistance that occurs in response to palbociclib, clinical trials
testing CDK4/6 inhibitors for efficacy in PDAC are underway.
A novel approach has been developed that targets the
Rb hyperphosphorylation present in cancer cells. Protein
phosphatase 1 (PP1) is the major Rb phosphatase (25), and specificity toward substrates is
imparted onto PP1 by various interacting proteins (26,27).
In proliferating cells, PP1 is associated with a regulatory protein
known as phosphatase nuclear targeting subunit (PNUTS) (28,29).
Our previous study demonstrated that PNUTS inhibits PP1 activity
toward Rb (30). It was further
revealed that PNUTS blocks Rb binding to PP1, thus identifying the
PNUTS:PP1 complex as a putative cancer drug target (31,32).
PNUTS dissociation from PP1, which permits Rb dephosphorylation,
occurs due to alterations in protein kinase A-mediated
phosphorylation of PNUTS (33).
Our previous study reported that when cells are exposed to stress,
including hypoxia or treatment with chemotherapeutic drugs, PNUTS
dissociates from PP1 and Rb is dephosphorylated (34). Furthermore, small interfering RNA
(siRNA)-mediated PNUTS depletion in breast and colon cancer cells
leads to an increase in PP1 phosphatase activity towards Rb, Rb
dephosphorylation on several sites, and a 3-4-fold increase in
apoptosis (35). Another study
revealed that apoptosis induced by PNUTS depletion involved the
phosphatase and tensin homolog tumor suppressor (36). However, in our previous studies the
ability of PNUTS depletion to induce apoptosis was demonstrated to
be dependent on Rb expression. For example, PNUTS knockdown has no
effect on Rb-null Saos2 cells; however, sensitivity is restored
upon stable expression of Rb (35). That the effect of PNUTS knockdown
requires phosphorylated (p)-Rb has been revealed in studies of
non-transformed cells. In cells that do not express
hyperphosphorylated Rb, for example in MCF10A breast and CCD-18Co
colon cells, PNUTS depletion has no effect on cell number (35). Furthermore, in a study using
non-transformed epithelial breast or breast cancer cells grown in
3D culture, it was demonstrated that the effect of PNUTS knockdown
is dependent on the expression of hyperphosphorylated Rb. PNUTS
depletion reduces the number of MCF7 and MDA-MB-231 breast cancer
cells (containing hyperphosphorylated Rb) but does not affect
non-transformed MCF10A cells that lack hyperphosphorylated Rb or
MDA-MB-468 breast cancer cells that are Rb-null (37). Therefore, our laboratory has
developed a method to activate the Rb phosphatase targeting Rb
phosphorylation in cells via knockdown of the PP1-binding protein,
PNUTS.
The present study aimed to determine the effects of
activating phosphatase activity toward Rb in pancreatic cancer
cells. The results demonstrated that PNUTS depletion caused
apoptosis in p16-deficient pancreatic cancer cells, but had no
effect on p16-positive pancreatic cancer cells or human pancreatic
ductal epithelial cells. The effects of palbociclib treatment were
compared with those of PNUTS depletion with regards to cell number,
and an additive effect was detected for the two treatments, with
palbociclib inhibiting proliferation and PNUTS depletion inducing
apoptosis. Using the currently utilized clinical treatments for
PDAC, TKIs erlotinib and gefitinib, cell proliferation was measured
in response to these treatments in combination with PNUTS
depletion, in order to evaluate the usefulness of targeting Rb
phosphorylation in reducing pancreatic cancer cell proliferation.
Finally, it was revealed that activation of phosphatase in
combination with erlotinib does not stimulate drug resistance in
pancreatic cancer cells.
Materials and methods
Cell culture
Cell culture materials were obtained from Gibco;
Thermo Fisher Scientific, Inc. (Waltham, MA, USA), unless otherwise
indicated. The Panc1, MIAPaCa-2 and Capan-2 cell lines were
obtained from the American Type Culture Collection (ATCC, Manassas,
VA, USA) and were used within 4 months of receipt. Panc1 and
MIAPaCa-2 cells were grown in Dulbecco's modified Eagle's medium
supplemented with 10% fetal bovine serum (FBS), 100 U/ml
penicillin, 100 µg/ml streptomycin and 2 mM glutamine (PSG).
Capan-2 cells were grown in McCoys 5A media (ATCC) supplemented
with 10% FBS and PSG. The H6c7 human pancreatic duct epithelial
cell line was obtained from Kerafast, Inc. (Boston, MA, USA) and
was used within 4 months of purchase. H6c7 cells were grown in
keratinocyte serum-free media containing EGF and bovine pituitary
extract (cat. no. 17005042; Gibco; Thermo Fisher Scientific, Inc.).
Cells were routinely maintained at 37°C in a humidified atmosphere
containing 5% CO, and were split 2-3 times weekly to maintain
sub-confluent cultures. Cultures were routinely tested for
mycoplasma contamination using the MycoFluor™ Mycoplasma Detection
kit (Invitrogen; Thermo Fisher Scientific, Inc.).
siRNA transfection and
immunoblotting
PNUTS depletion was performed using Dharmafect II
(GE Healthcare Dharmacon, Inc., Lafayette, CO, USA). The RNA
oligonucleotides used were generated based on the human mRNA for
PNUTS, and the sequences were as follows: PNUTS RNA interference
(RNAi) sequence 5, 5'-CAGCUAAACUGGUGAAGCA-3'; PNUTS RNAi sequence
7, 5'-CCUAAUGCCACCAAAGAGA-3'; or nontargeting RNAi (siCONTROL
non-targeting siRNA ≥1; GE Healthcare Dharmacon, Inc.). The
nontargeting RNAi has at least four mismatches with all known
human, mouse and rat genes, which is sufficient to eliminate
nonspecific silencing of genes with similar sequences. Sequence 5
was used in all experiments shown. Rb depletion was performed using
SignalSilence® Rb siRNA1 (cat. no. 6451) from Cell
Signaling Technology, Inc. (Danvers, MA, USA). Transfection
mixtures (final concentration, 100 nM RNA) were added to 40-60%
confluent Panc1, MIA PaCa-2, Capan-2 or H6c7 cells. After 48 h, the
transfection mixtures were removed from the cells and the cells
underwent cell counting and immunoblotting. Cell extracts were
prepared after washing in ice-cold Tris-buffered saline [TBS; 25 mM
Tris-HCl (pH 8.0) and 150 mM NaCl] and were lysed for 15 min at 4°C
in EBC buffer [50 mM Tris-HCl (pH 8.0), 120 mM NaCl and 0.5%
Nonidet P-40] containing 10 µg/ml protease inhibitors
aprotinin and phenylmethylsulfonylfluoride. The lysates were
cleared by centrifugation at 13,000 × g for 10 min at 4°C. Protein
concentration was determined using the Bradford protein assay.
Electrophoresis was performed using 4-20% gradient
SDS-polyacrylamide gels containing 30 µg total cell protein
in each sample lane. Following electrophoresis, the proteins were
transferred to nitrocellulose membranes. Residual protein binding
sites on the nitrocellulose membranes were blocked by incubation
with TBS-0.5% Tween-20 (TBST) containing 4% non-fat dry milk for
30-60 min at room temperature (RT). Subsequently, the
nitrocellulose membranes were incubated in TBST containing 2%
non-fat dry milk and 1 µg/ml primary antibody overnight at
4°C. Antibodies were all used at a concentration of 1 µg/ml.
After three washes with TBST (10 min/wash), the nitrocellulose
membranes were probed with horseradish peroxidase-conjugated
anti-immunoglobulin G antibodies [1:2,000; cat. nos. 1031-05
(anti-mouse) and 4050-05 (anti-rabbit); SouthernBiotech,
Birmingham, AL, USA] for 1.5 h at RT and developed using enhanced
chemiluminescence detection (Pierce; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. The following
primary antibodies were used in this study: p16 (cat. no. 554079),
cyclin D3 (cat. no. 610279) and PNUTS (cat. no. 611060) (all from
BD Biosciences, Franklin Lakes, NJ, USA); p-Rb 807/811 (cat. no.
8516), cleaved poly(ADP-ribose) polymerase (Parp; cat. no. 9541),
c-jun (cat. no. 9165), p-c-jun (cat. no. 2361), EGFR (cat. no.
4267), p-EGFR (cat. no. 3777), STAT3 (cat. no. 4904), p-STAT3 (cat.
no. 9145), proliferating cell nuclear antigen (PCNA; cat. no.
13110) (all from Cell Signaling Technology, Inc.); β-actin (cat.
no. A1978; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany); Rb (cat.
no. sc-102) and minichromosome maintenance complex component 7
(mcm7; cat. no. sc-9966) (both from Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA).
Cell number and apoptosis assays
Erlotinib, gefitinib and palbociclib were obtained
from Selleck Chemicals (Houston, TX, USA), and were used within 3
months of receipt. Erlotinib and gefitinib were dissolved in
dimethyl sulfoxide (DMSO), palbociclib was dissolved in water.
Dose-response curves were generated that identified the
concentration of each drug required to reduce cell numbers between
20 and 60% after a 24-h treatment: 40 µM erlotinib, 4
µM gefitinib and 20 µM palbociclib. These
concentrations are within the range of half maximal inhibitory
concentrations reported to inhibit proliferation of pancreatic
cancer cells in previous studies (21,38-40).
Panc1 or MIAPaCa-2 cells were plated (3,000/well) in 96-well plates
and allowed to proliferate for 24 h. Treated cells were subjected
to PNUTS depletion for 48 h and control cells were treated with
nontargeting control (NTC) RNA. After 2 days, cells were treated
with erlotinib, gefitinib or palbociclib for 24 h, or with a
vehicle control (DMSO for erlotinib/gefitinib, water for
palbociclib). Cell number was measured using the CellTiter Glo-2
assay (Promega Corporation, Madison, WI, USA). Apoptosis was
measured using the Cell Death Detection ELISA (cat. no.
11544675001; Roche Diagnostics, Indianapolis, IN, USA), which
detects degraded DNA released from the nucleus into the cytoplasm.
Both assays were performed according to the manufacturers'
protocols. Briefly, in the apoptosis assay, 104 cells
from each condition were lysed and subjected to a slow-spin
centrifugation (300 × g for 5 min at 4°C) to pellet nuclei.
Extracts from the cytoplasmic fraction were used to detect
fragmented DNA and quantified on a microplate reader at 405 nm
(Promega Corporation).
Statistical analysis
All experiments performed in this study were
conducted at least three times. Numerical data are presented as the
means ± standard deviation. All data from each experiment were
analyzed by Student's t-test or one-way analysis of variance with
Tukey's post hoc test for multiple comparisons using SPSS 19.0 (IBM
Corp., Armonk, NY USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
PNUTS depletion in pancreatic cancer
cells reduces cell number by activation of apoptosis
Hyperphosphorylation of Rb is a hallmark of most
types of human cancer; therefore, we devised a method to target Rb
phosphorylation by phosphatase activation. Our previous studies
revealed that activation of phosphatase by PNUTS depletion is
dependent on Rb (35,37). The function of p16 is disrupted in
the majority of pancreatic carcinoma cases (11); in the present study it was
demonstrated that the effects of PNUTS depletion on cell number
were dependent on p16 expression in pancreatic cancer cells. Three
pancreatic cancer cell types were used in this study: MIAPaCa-2 and
Panc1 cells, which carry a homozygous deletion of the p16 gene; and
Capan-2 cells, which are p16-positive (41-43).
These cells have been extensively used to model pancreatic cancer.
For comparison, experiments were also performed using the human
pancreatic duct epithelial cell line H6c7 (44). As shown in Fig. 1A, Capan-2 and non-transformed H6c7
cells expressed p16, whereas MIAPaCa-2 and Panc1 cells were
p16-deficient. p-Rb was also detected in the three cancer cell
types, but was absent in the non-transformed cell line. As shown in
Fig. 1B, Rb dephosphorylation
initiated by PNUTS knockdown reduced cell number by 55% in
MIAPaCa-2 and by 30% in Panc-1 cancer cells that contain
hyperphosphorylated Rb; however, it had no effect on cell number in
p16-positive Capan-2 cancer cells and in non-transformed H6c7 cells
that lack hyperphosphorylated Rb. As shown in Fig. 1C, Rb was dephosphorylated in
response to PNUTS depletion, and as shown in Fig. 1D, Rb depletion partially reversed
the reduction in cell number induced by PNUTS depletion, indicating
that the reduction in cell number due to PNUTS depletion may be
Rb-dependent. Transfection with Rb siRNA alone was also confirmed
to be successful (data not shown). The reduction in cell number
observed may be due to cell proliferation arrest or an increase in
apoptotic cell death. Assays using Panc1 and MIAPaCa-2 pancreatic
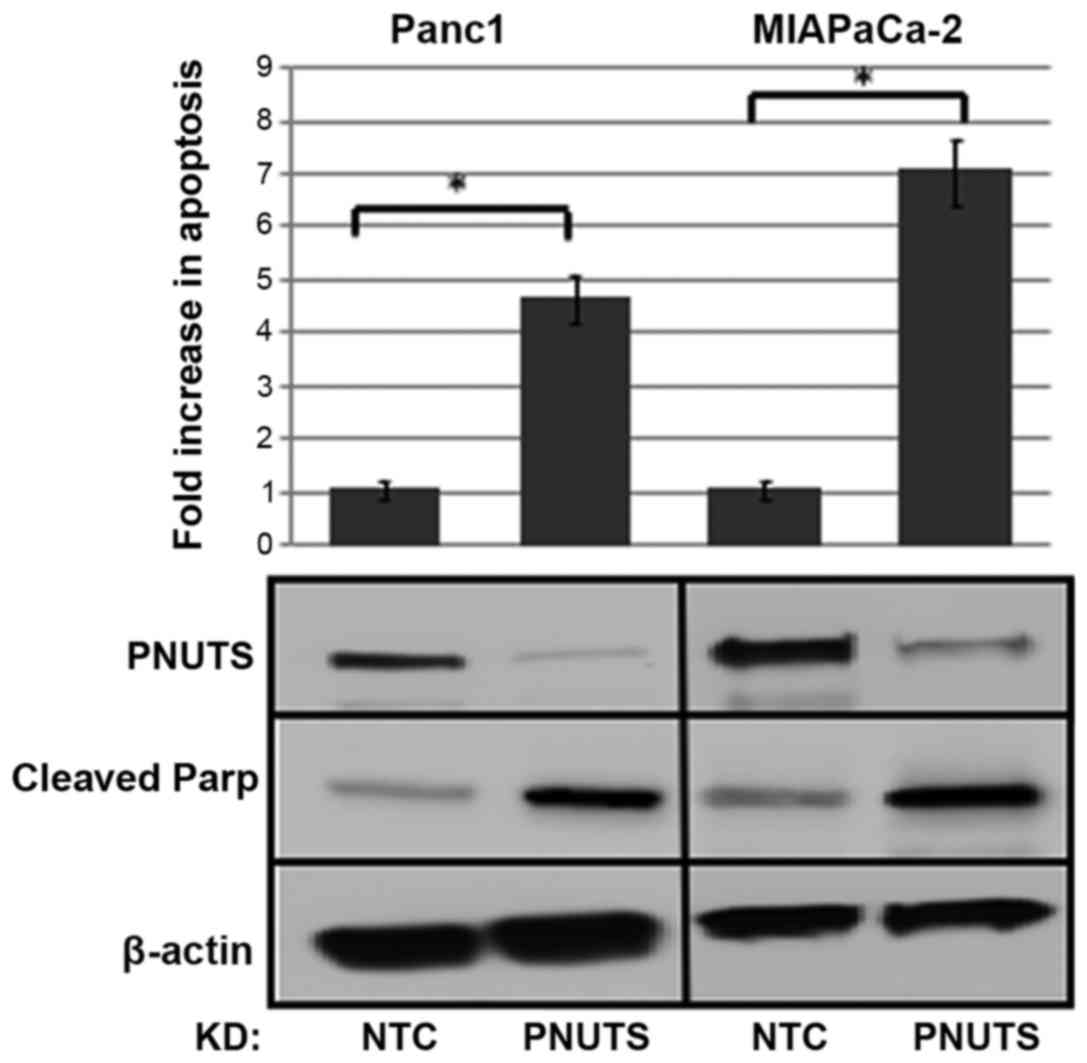
cancer cells revealed that apoptosis was increased by ~4-fold due
to PNUTS depletion in Panc1 cells and by ~7-fold in MIAPaCa-2
cells. Furthermore, the appearance of cleaved Parp in response to
PNUTS depletion confirmed that apoptosis was induced (Fig. 2).
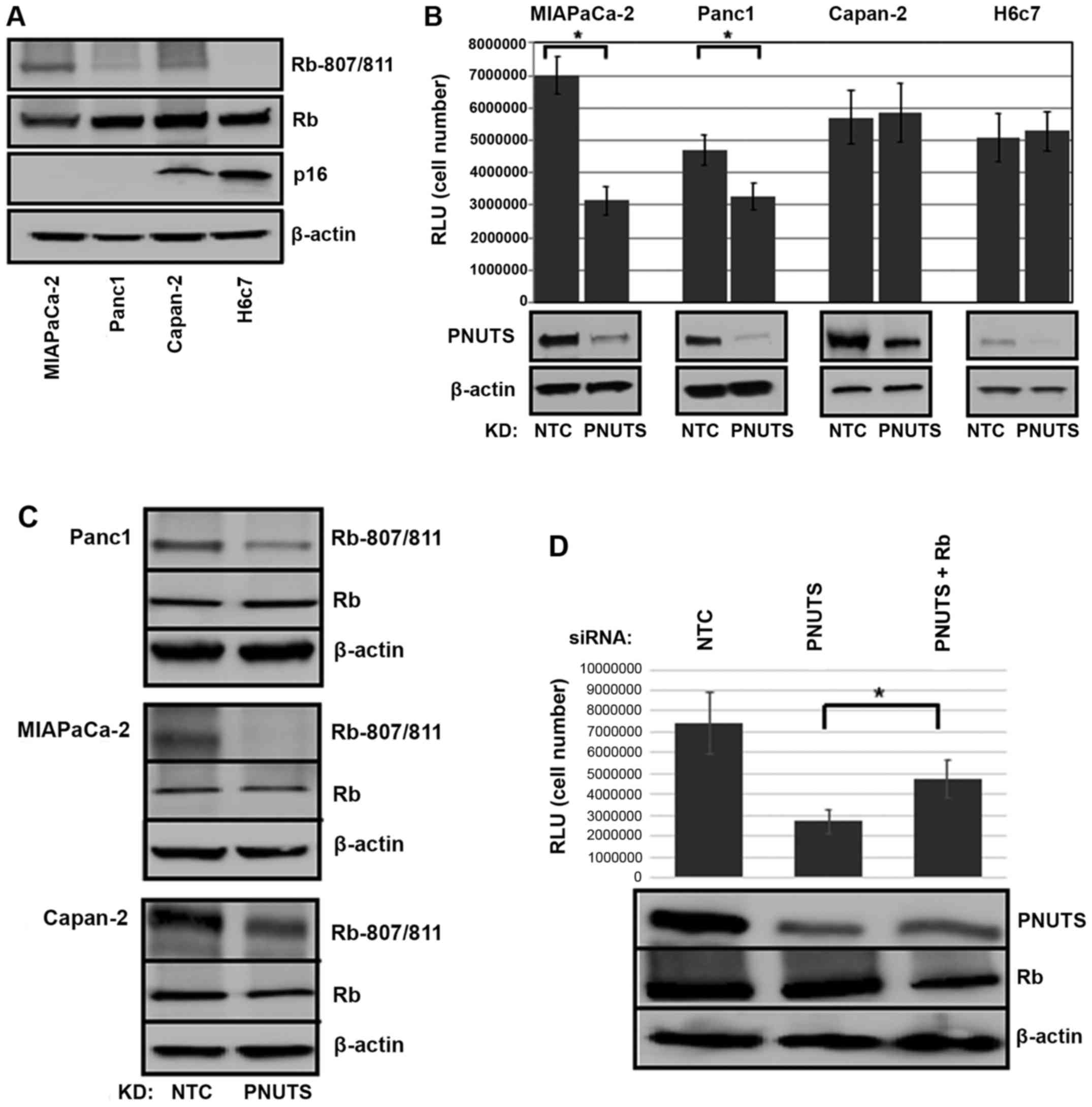 | Figure 1(A) Immunoblotting was performed on
whole cell lysates from MIAPaCa-2, Panc1, Capan-2 and H6c7 cell
lines. Antibodies against p-Rb (S807/811), total Rb and p16 were
used; β-actin was used as a loading control. Results shown are
representative of three separate experiments. (B) MIAPaCa-2, Panc-1
and Capan-2 pancreatic cancer cells, and H6c7 normal pancreatic
duct epithelial cells were subjected to PNUTS depletion for 48 h.
Cell number was measured using the CellTiter Glo assay, with RLU
proportional to cell number. Data shown are representative of three
separate experiments. Results are expressed as the means ± standard
deviation, n=8 replicates/experiment. *P<0.05,
Student’s t-test. (C) Panc1, MIAPaCa-2 and Capan-2 cells were
analyzed by immunoblotting with antibodies against p-Rb
(Rb-807/811) or total Rb (Rb). (D) MIAPaCa-2 cells were transfected
with PNUTS siRNA, or PNUTS and Rb siRNA. Cell number was measured
using the CellTiter Glo assay. Data shown are representative of
three separate experiments. *P<0.05, one-way analysis
of variance and Tukey honestly significant difference test. KD,
knockdown; NTC, nontargeting control; p, phosphorylated; PNUTs,
phosphatase nuclear targeting subunit; Rb, retinoblastoma; RLU,
relative light units; siRNA, small interfering RNA. |
Comparison of CDK inhibition with PNUTS
depletion in pancreatic cancer cells
Since Rb is phosphorylated on 16 amino acid sites
in vivo, it is difficult to elucidate the effects of
phosphatases and kinases on the phosphorylation of Rb. To the best
of our knowledge, the specific CDK responsible for each
modification has only been partially elucidated to date (45). Furthermore, there appear to be
overlapping functions of CDKs with regards to specific Rb sites.
Our previous study demonstrated that activation of phosphatase
toward Rb causes dephosphorylation of Rb at certain sites, and that
these are distinct from those affected by the CDK2/5 inhibitor
roscovitine (46). To compare the
effects of CDK4/6 inhibition with that of PNUTS depletion on cell
number in pancreatic cancer cells, it was first determined that 20
µM (24-h treatment) of palbociclib was an appropriate
concentration to cause a 30-50% drop in cell number, and
immunoblotting confirmed that Rb was dephosphorylated using this
concentration (data not shown). The efficacy of palbociclib to
reduce Panc1 cell number was comparable to the efficacy of PNUTS
depletion (Fig. 3). To examine the
combination of palbociclib and PNUTS knockdown, 48 h following
PNUTS depletion (or NTC transfection), the medium was replaced with
either 20 µM palbociclib or fresh medium (control). Finally,
after 24 h, cells were counted using the CellTiter Glo assay. PNUTS
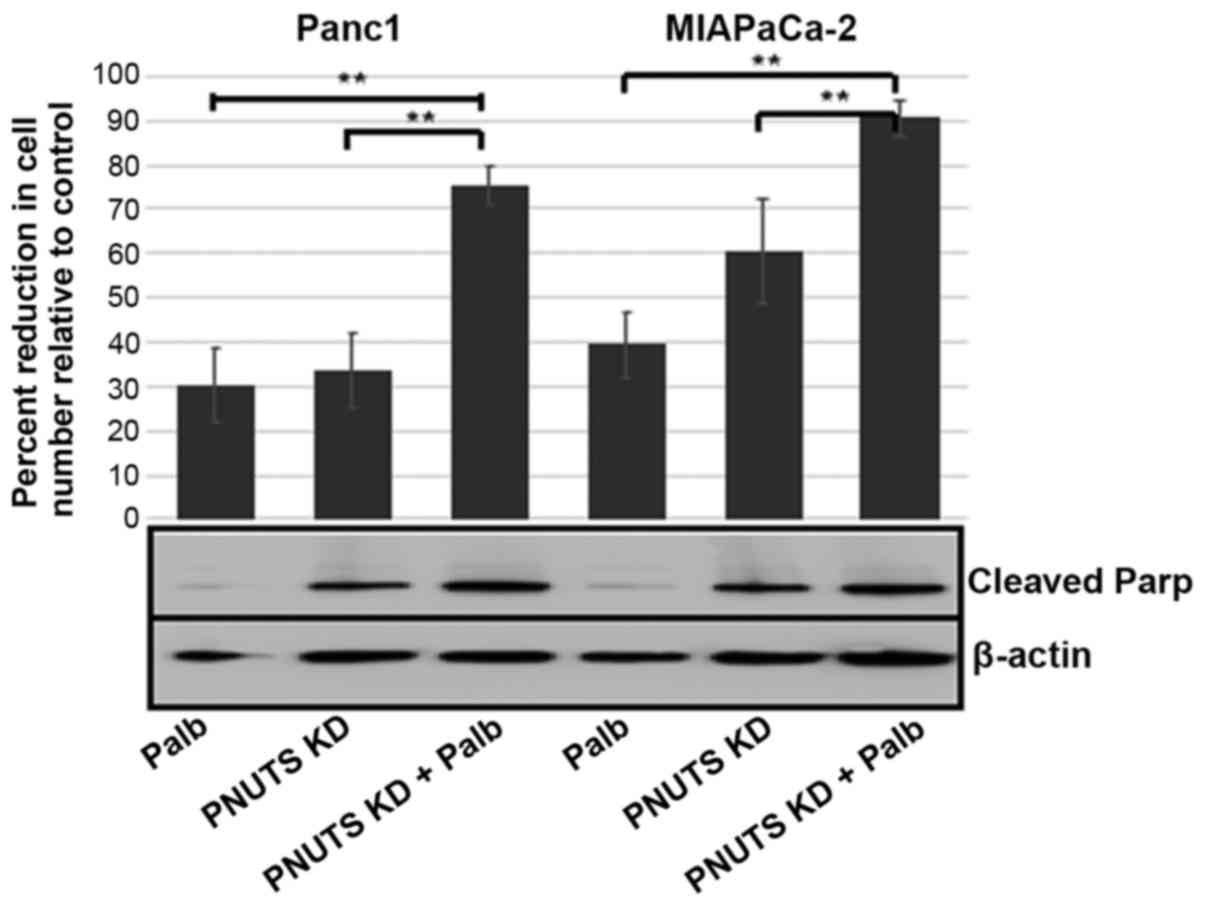
depletion or palbociclib treatment each reduced cell number
relative to controls by 30% in Panc1 cells; a higher efficiency was
observed in response to PNUTS depletion in MIAPaCa-2 cells.
However, when combining the two treatments, cell number was reduced
by 75-90% in the two cell lines. In addition, while PNUTS depletion
induced Parp cleavage, palbociclib treatment did not. These results
suggested that the inhibition of CDK activity towards Rb and
activation of phosphatase activity towards Rb are not functionally
equivalent, and may influence Rb phosphorylation at distinct
sites.
Differential phosphorylation of Rb may impart
distinctive consequences in cells. Using Panc1 cells, the effects
of 20 µM palbociclib and PNUTS depletion on proteins that
control the processes of proliferation and apoptosis were
determined. As shown in Fig. 4,
palbociclib treatment induced a decrease in proteins involved in
proliferation (cyclin D3, mcm7 and PCNA), but did not increase the
expression of proteins involved in apoptosis (cleaved Parp, c-jun
and p-c-jun), thus suggesting that palbociclib inhibited
proliferation but had no effect on cell death. Conversely, PNUTS
depletion led to an increase in apoptosis-associated proteins but
did not affect proliferation-associated proteins.
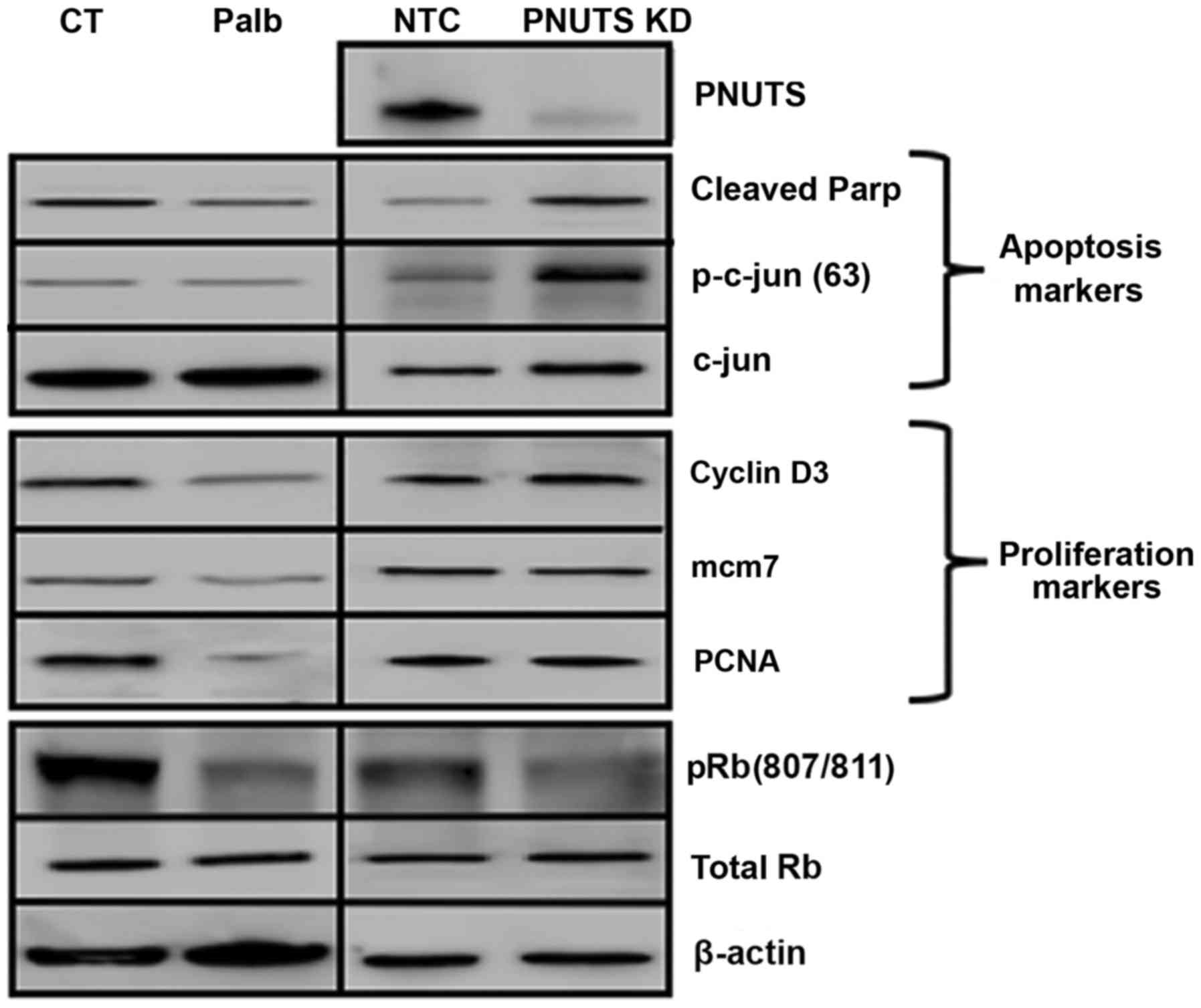 | Figure 4Panc1 cells were treated with Palb
(20 µM) for 24 h, or were transfected with NTC or PNUTS
siRNA for 48 h. Subsequently, immunoblotting was conducted to
analyze the expression of markers of proliferation and apoptosis.
Rb phosphorylation (S807/S811), total Rb, β-actin and PNUTS
expression was also detected. Data shown are representative of
three independent experiments. CT, control; KD, knockdown; mcm7,
minichromosome maintenance complex component 7; NTC, nontargeting
control; Palb, palbociclib; p, phosphorylated; Parp,
poly(ADP-ribose) polymerase; PCNA, proliferating cell nuclear
antigen; PNUTs, phosphatase nuclear targeting subunit; Rb,
retinoblastoma. |
Effects of PNUTS depletion combined with
erlotinib or gefitinib
It was speculated that erlotinib, acting as an
inhibitor of EGFR, blocked proliferation of cancer cells;
therefore, the combination of erlotinib with PNUTS depletion, which
induced apoptosis, may be an efficacious method to reduce
pancreatic cancer cell numbers. For these experiments, gefitinib
was also employed, which is an additional clinical treatment that
blocks proliferation by inhibiting EGFR (47). Although MIAPaCa-2 and Panc1 cells
are considered erlotinib-insensitive compared with other cancer
cell lines, with the concentrations of erlotinib and gefitinib
used, activation of EGFR was inhibited, as demonstrated by reduced
phosphorylation (Fig. 5A).
Subsequently, MIAPaCa-2 and Panc1 cells were subjected to 48 h of
PNUTS depletion, followed by 24-h treatment with either DMSO,
erlotinib or gefitinib. In both cell lines, when compared to each
treatment alone, the combination of PNUTS depletion plus EGFR
inhibition resulted in a significant decrease in cell number
relative to the control (Fig. 5B and
C). Concomitantly, increased apoptosis was indicated by Parp
cleavage, thus suggesting that targeting the Rb tumor suppressor
protein in combination with EGFR inhibition may be a rational
strategy to reduce pancreatic cancer cell growth.
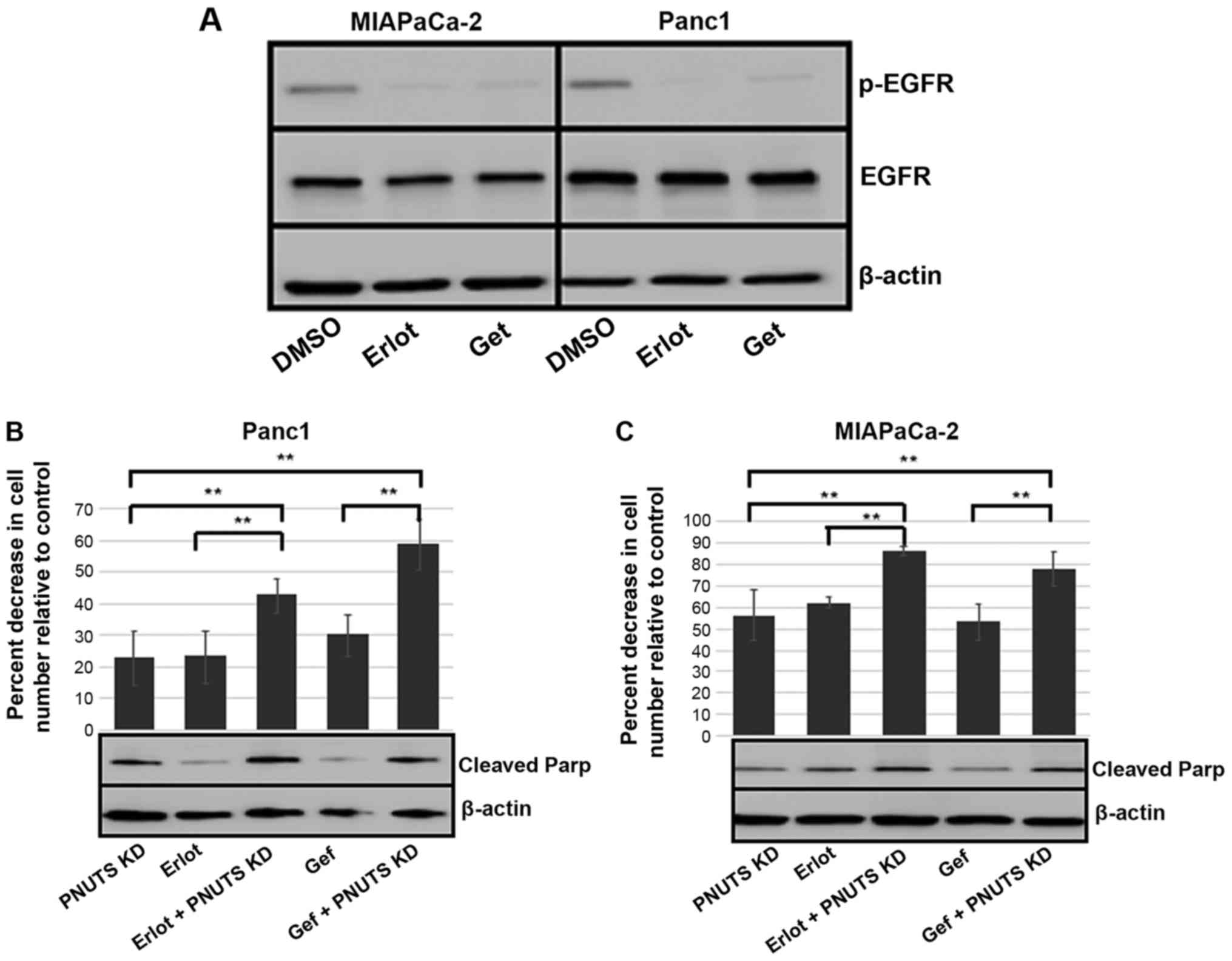 | Figure 5(A) MIAPACa-2 and Panc1 cells were
treated with Erlot (40 µM), Gef (4 µM) or DMSO
(control). After 24 h, immunoblotting was performed on cell lysates
obtained from cells. EGFR, p-EGFR and β-actin expression is shown.
Results are representative of two separate experiments. (B) Panc1
and (C) MIAPaCa-2 cells were subjected to PNUTS depletion for 48 h
and/or Erlot or Gef treatment, and cell counting assays were
performed using CellTiter Glo. The percentage reduction in cell
number is shown relative to nontargeting control/DMSO controls.
Results are expressed as the means ± standard deviation, n=8
replicates/experiment. **P<0.01, one-way analysis of
variance and Tukey honestly significant difference test. Data shown
are representative of three independent experiments. Cleaved Parp
and β-actin expression was determined by immunoblotting. DMSO,
dimethyl sulfoxide; EGFR, human epidermal growth factor receptor
type 1 receptor; Erlot, erlotinib; Gef, gefitinib; p,
phosphorylated; Parp, poly(ADP-ribose) polymerase; PNUTs,
phosphatase nuclear targeting subunit. |
Effects of PNUTS depletion and erlotinib
or gefitinib on the STAT3 pathway
Because STAT3 pathway activation is often present in
cancer cells, and since erlotinib has been reported to induce
resistance in lung and pancreatic cancer via STAT3 activation,
which leads to increased cell survival (9,10),
the present study determined the effects of a combination of
erlotinib and PNUTS depletion on STAT3 activation by
immunoblotting. As shown in Fig.
6, erlotinib in Panc1 and MIAPaCa-2 cells reduced
phosphorylation of EGFR, as expected. STAT3 appeared to be
constitutively activated in Panc1 and MIAPaCa-2 cells (DMSO and NTC
groups), and stimulated to various extents by erlotinib,
palbociclib and PNUTS depletion, as evidenced by phosphorylation of
STAT3. Although the various treatments affected STAT3
phosphorylation to different degrees, the combination of erlotinib
with palbociclib resulted in STAT3 pathway activation, whereas the
combination of erlotinib with PNUTS depletion did not activate
STAT3, thus suggesting that this strategy may be an effective
method to target EGFR and Rb phosphorylation without the
development of resistance in pancreatic cancer cells.
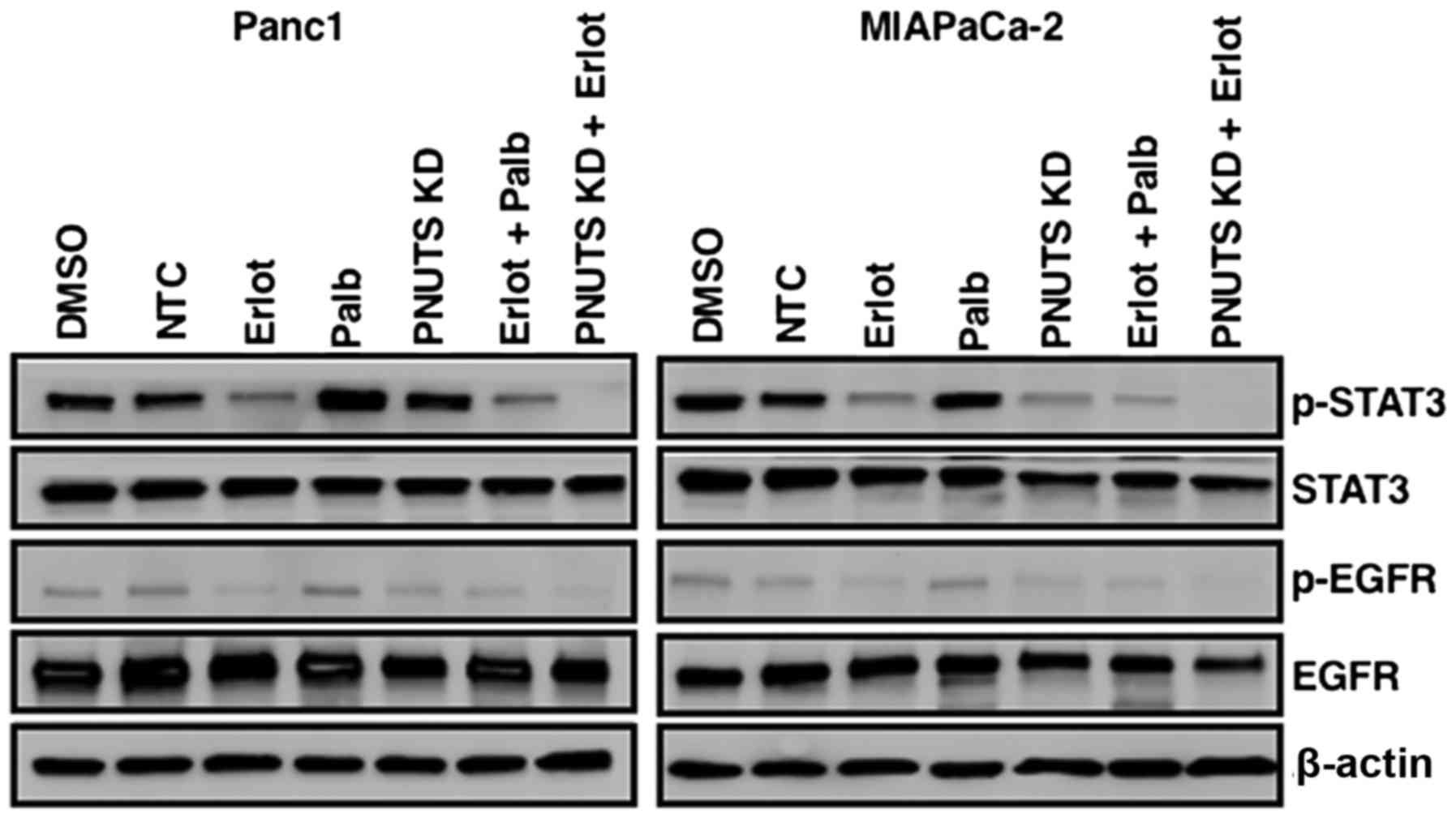 | Figure 6Panc1 or MIAPaCa-2 cells were treated
with DMSO, erlotinib, NTC, PNUTS KD, palbociclib, or combinations
of erlotinib + palbociclib or erlotinib + PNUTS knockdown. STAT3,
p-STAT3, EGFR, p-EGFR and β-actin expression was determined by
immunoblotting. Data shown are representative of three separate
experiments. DMSO, dimethyl sulfoxide; EGFR, human epidermal growth
factor receptor type 1 receptor; Erlot, erlotinib; KD, knockdown;
NTC, nontargeting control; Palb, palbociclib; p, phosphorylated;
PNUTs, phosphatase nuclear targeting subunit; STAT3, signal
transducer and activator of transcription 3. |
Discussion
Resistance to targeted therapy is a major issue in
cancer, and resistance to the small molecule EGFR TKIs erlotinib
and gefitinib has been investigated thoroughly in non-small cell
lung cancer (NSCLC) (48). In
NSCLC, marked initial responses to TKIs are attenuated by 6-12
months when patients experience tumor progression due to acquired
resistance (49,50). Approximately 50% of the acquired
resistance to erlotinib and gefitinib is due to mutations of the
TKI-binding site of EGFR; however, resistance may also be due to
compensatory growth promoting pathways that are activated by
treatment. For example, activation of STAT3 in NSCLC occurs in
response to erlotinib treatment and promotes resistance (9). In human pancreatic cancer tumors,
STAT3 activation is exhibited in the majority of cases (51,52).
STAT3 is activated by phosphorylation by Janus-activated kinases,
interleukin-6, EGFRs and Src kinases. Upon phosphorylation, STAT3
dimerization stimulates gene transcription. STAT3 activation
consequently promotes pancreatic tumorigenesis, cell invasion and
metastatic potential (53,54). Notably, high expression of active
STAT3 is correlated with poor prognosis (55,56).
The importance of the STAT3 pathway in treatment resistance has
been confirmed by the fact that acquired resistance of pancreatic
cancer cells to EGFR TKIs, including erlotinib, can be abrogated by
STAT3 inhibition (10).
In p16-negative cancer, hyperphosphorylated Rb is
the target of CDK4/6 inhibitors; however, acquired resistance often
develops in response to CDK4/6 inhibition. Several mechanisms have
been proposed to be involved in CDK inhibition resistance; for
example, biomarkers of treatment resistance include loss of Rb
function, hyperactivity of the cyclin E-CDK2 axis and increased
CDK6 activity (57-59). In addition, two recent studies
demonstrated a role for the mechanistic target of rapamycin (mTOR)
pathway in acquired resistance to CDK4/6 inhibition. Dysregulation
of mTOR is a hallmark of adaptive resistance to several targeted
therapies (60). In experiments
using pancreatic cancer cells, CDK4/6 inhibition leads to mTOR
complex 1 activation and metabolic alterations, including increased
mitochondrial mass, and upregulation of glycolysis and oxidative
metabolism (61). In other
studies, it has been reported that CDK4/6 inhibition leads to
activation of the pro-survival kinase protein kinase B (62). Finally, promotion of epithelial to
mesenchymal transition by CDK4/6 inhibition has been demonstrated
to contribute to drug resistance in pancreatic cancer (22,63).
Dysregulation of the tumor suppressor protein Rb is
commonly found in human cancer. Hyperphosphorylation of Rb, rather
than mutation of Rb, is thought to promote tumorigenesis. The CDKs
that accomplish Rb phosphorylation are regulated by cyclin binding
and by the association of endogenous CDK inhibitors, such as
p16INK4a. Overexpression of cyclin D1/CDK4 or the loss of p16INK4a
proteins are common genetic alterations in cancer that result in
hyperphosphorylation of Rb (64,65).
It has been reported that phosphorylation of Rb stimulates
proliferation, blocks apoptosis and promotes invasion (37,66-69).
By targeting PNUTS, an inhibitor of phosphatase activity toward Rb,
dephosphorylation of Rb in cancer cells can be achieved. The
present study demonstrated that PNUTS depletion in p16-negative
pancreatic cancer cells reduced cell numbers, due to stimulation of
apoptosis. In addition, non-transformed pancreatic duct epithelial
cells that express p16 remained impervious to PNUTS depletion. When
PNUTS depletion was compared with palbociclib-induced CDK4/6
inhibition, it was revealed that the combination of these
treatments reduced cell numbers more effectively than either
treatment alone. These results suggested that activation of
phosphatase compared with inhibition of kinase are not functionally
equivalent, perhaps by affecting different subsets of Rb
phosphorylation sites, and thus Rb activity. Specific patterns of
Rb phosphorylation may lead to distinctive Rb protein-binding
abilities that lead to functional consequences in the cell. The
present study demonstrated that, in Panc1 pancreatic cancer cells,
CDK4/6 inhibition with palbociclib reduced proliferation; however,
PNUTS depletion-mediated phosphatase activation resulted in
apoptosis. It may be the case that in response to these two
treatments the Rb phosphorylation pattern is altered such that in
one case proliferation is inhibited, whereas in the other apoptosis
is stimulated. This may be due to Rb interaction with
transcriptional regulators that promote either proliferation arrest
or apoptosis.
Because the TKIs (erlotinib and gefitinib) that
block EGFR in pancreatic cancer cells inhibit proliferation and are
used in the clinic, the present study tested how a combination of
these TKIs with PNUTS depletion-mediated Rb phosphatase activation
would affect p16-positive pancreatic cancer cells. It was revealed
that in MIAPaCa-2 and Panc1 cells, which are considered
erlotinib-resistant, targeting Rb phosphorylation by this method
alongside EGFR inhibition resulted in a greater reduction in cell
number than either treatment alone. Furthermore, whereas activation
of the STAT3 pathway was constitutive in these pancreatic cancer
cells, the combination of erlotinib with PNUTS depletion abrogated
activation of this pathway, thus suggesting that targeting Rb
phosphatase activity may be a strategy that circumvents
erlotinib-induced acquired resistance. Additional studies may
further elucidate the usefulness of this strategy in the clinical
setting.
Funding
This work was supported by the National Cancer
Institute of the National Institutes of Health (grant nos.
R15CA182723 and R15CA231372) awarded to NAK.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
NAK initiated the work, performed the apoptosis
experiments, analyzed data and co-wrote the manuscript with NAT.
NAT compared the effects of PNUTS knockdown with those of
palbociclib on cell proliferation, and performed the PNUTS
depletion + gefitinib study. RGA performed and analyzed the
erlotinib + PNUTS knockdown experiments. BD performed and analyzed
the PNUTS knockdown study using cell proliferation and
immunoblotting assays. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar
|
|
2
|
Ryan DP, Hong TS and Bardeesy N:
Pancreatic adenocarcinoma. N Engl J Med. 371:1039–1049. 2014.
View Article : Google Scholar
|
|
3
|
Philip PA and Lutz MP: Targeting Epidermal
Growth factor receptor-related signaling pathways in pancreatic
cancer. Pancreas. 44:1046–1052. 2015. View Article : Google Scholar
|
|
4
|
Yang ZY, Yuan JQ, Di MY, Zheng DY, Chen
JZ, Ding H, Wu XY, Huang YF, Mao C and Tang JL: Gemcitabine plus
erlotinib for advanced pancreatic cancer: A systematic review with
meta-analysis. PLoS One. 8:e575282013. View Article : Google Scholar :
|
|
5
|
Moore MJ, Goldstein D, Hamm J, Figer A,
Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, et al:
National Cancer Institute of Canada Clinical Trials Group:
Erlotinib plus gemcitabine compared with gemcitabine alone in
patients with advanced pancreatic cancer: A phase III trial of the
National Cancer Institute of Canada Clinical Trials Group. J Clin
Oncol. 25:1960–1966. 2007. View Article : Google Scholar
|
|
6
|
Troiani T1, Martinelli E, Capasso A,
Morgillo F, Orditura M, De Vita F and Ciardiello F: Targeting EGFR
in pancreatic cancer treatment. Curr Drug Targets. 13:802–810.
2012. View Article : Google Scholar
|
|
7
|
Nedaeinia R, Avan A, Manian M, Salehi R
and Ghayour- Mobarhan M: EGFR as a potential target for the
treatment of pancreatic cancer: Dilemma and controversies. Curr
Drug Targets. 15:1293–1301. 2014. View Article : Google Scholar
|
|
8
|
Zhao C, Li H, Lin HJ, Yang S, Lin J and
Liang G: Feedback activation of STAT3 as a cancer drug-resistance
mechanism. Trends Pharmacol Sci. 37:47–61. 2016. View Article : Google Scholar
|
|
9
|
Lee H-J, Zhuang G, Cao Y, Du P, Kim HJ and
Settleman J: Drug resistance via feedback activation of Stat3 in
oncogene-addicted cancer cells. Cancer Cell. 26:207–221. 2014.
View Article : Google Scholar
|
|
10
|
Ioannou N, Seddon AM, Dalgleish A,
Mackintosh D, Solca F and Modjtahedi H: Acquired resistance of
pancreatic cancer cells to treatment with gemcitabine and
HER-inhibitors is accompanied by increased sensitivity to STAT3
inhibition. Int J Oncol. 48:908–918. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schutte M, Hruban RH, Geradts J, Maynard
R, Hilgers W, Rabindran SK, Moskaluk CA, Hahn SA, Schwarte-Waldhoff
I, Schmiegel W, et al: Abrogation of the Rb/p16 tumor-suppressive
pathway in virtually all pancreatic carcinomas. Cancer Res.
57:3126–3130. 1997.
|
|
12
|
Hustinx SR, Leoni LM, Yeo CJ, Brown PN,
Goggins M, Kern SE, Hruban RH and Maitra A: Concordant loss of MTAP
and p16/CDKN2A expression in pancreatic intraepithelial neoplasia:
Evidence of homozygous deletion in a noninvasive precursor lesion.
Mod Pathol. 18:959–963. 2005. View Article : Google Scholar
|
|
13
|
Fukushima N, Sato N, Ueki T, Rosty C,
Walter KM, Wilentz RE, Yeo CJ, Hruban RH and Goggins M: Aberrant
methylation of preproenkephalin and p16 genes in pancreatic
intraepithelial neoplasia and pancreatic ductal adenocarcinoma. Am
J Pathol. 160:1573–1581. 2002. View Article : Google Scholar
|
|
14
|
Gerdes B, Ramaswamy A, Kersting M, Ernst
M, Lang S, Schuermann M, Wild A and Bartsch DK: p16(INK4a)
alterations in chronic pancreatitis-indicator for high-risk lesions
for pancreatic cancer. Surgery. 129:490–497. 2001. View Article : Google Scholar
|
|
15
|
Makohon-Moore A and Iacobuzio-Donahue CA:
Pancreatic cancer biology and genetics from an evolutionary
perspective. Nat Rev Cancer. 16:553–565. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Notta F, Chan-Seng-Yue M, Lemire M, Li Y,
Wilson GW, Connor AA, Denroche RE, Liang SB, Brown AM, Kim JC, et
al: A renewed model of pancreatic cancer evolution based on genomic
rearrangement patterns. Nature. 538:378–382. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rabien A, Sanchez-Ruderisch H, Schulz P,
Otto N, Wimmel A, Wiedenmann B and Detjen KM: Tumor suppressor
p16INK4a controls oncogenic K-Ras function in human pancreatic
cancer cells. Cancer Sci. 103:169–175. 2012. View Article : Google Scholar
|
|
18
|
Chang Z, Ju H, Ling J, Zhuang Z, Li Z,
Wang H, Fleming JB, Freeman JW, Yu D, Huang P, et al: Cooperativity
of oncogenic K-ras and downregulated p16/INK4A in human pancreatic
tumorigenesis. PLoS One. 9:e1014522014. View Article : Google Scholar :
|
|
19
|
Li J, Poi MJ and Tsai MD: Regulatory
mechanisms of tumor suppressor P16(INK4A) and their relevance to
cancer. Biochemistry. 50:5566–5582. 2011. View Article : Google Scholar
|
|
20
|
Franco J, Witkiewicz AK and Knudsen ES:
CDK4/6 inhibitors have potent activity in combination with pathway
selective therapeutic agents in models of pancreatic cancer.
Oncotarget. 5:6512–6525. 2014. View Article : Google Scholar
|
|
21
|
Heilmann AM, Perera RM, Ecker V, Nicolay
BN, Bardeesy N, Benes CH and Dyson NJ: CDK4/6 and IGF1 receptor
inhibitors synergize to suppress the growth of p16INK4A-deficient
pancreatic cancers. Cancer Res. 74:3947–3958. 2014. View Article : Google Scholar
|
|
22
|
Liu F and Korc M: Cdk4/6 inhibition
induces epithelial-mesenchymal transition and enhances invasiveness
in pancreatic cancer cells. Mol Cancer Ther. 11:2138–2148. 2012.
View Article : Google Scholar
|
|
23
|
Witkiewicz AK, Borja NA, Franco J, Brody
JR, Yeo CJ, Mansour J, Choti MA, McCue P and Knudsen ES: Selective
impact of CDK4/6 suppression on patient-derived models of
pancreatic cancer. Oncotarget. 6:15788–15801. 2015. View Article : Google Scholar
|
|
24
|
Finn RS, Martin M, Rugo HS, Jones S, Im
SA, Gelmon K, Harbeck N, Lipatov ON, Walshe JM, Moulder S, et al:
Palbociclib and Letrozole in advanced breast cancer. N Engl J Med.
375:1925–1936. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nelson DA, Krucher NA and Ludlow JW: High
molecular weight protein phosphatase type 1 dephosphorylates the
retinoblastoma protein. J Biol Chem. 272:4528–4535. 1997.
View Article : Google Scholar
|
|
26
|
Bollen M, Peti W, Ragusa MJ and Beullens
M: The extended PP1 toolkit: Designed to create specificity. Trends
Biochem Sci. 33:113–121. 2003.
|
|
27
|
Kolupaeva V and Janssens V: PP1 and PP2A
phosphatases - cooperating partners in modulating retinoblastoma
protein activation. FEBS J. 280:627–643. 2013. View Article : Google Scholar
|
|
28
|
Allen PB, Kwon YG, Nairn AC and Greengard
P: Isolation and characterization of PNUTS, a putative protein
phosphatase 1 nuclear targeting subunit. J Biol Chem.
273:4089–4095. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kreivi JP, Trinkle-Mulcahy L, Lyon CE,
Morrice NA, Cohen P and Lamond AI: Purification and
characterisation of p99, a nuclear modulator of protein phosphatase
1 activity. FEBS Lett. 420:57–62. 1997. View Article : Google Scholar
|
|
30
|
Udho E, Tedesco VC, Zygmunt A and Krucher
NA: PNUTS (phosphatase nuclear targeting subunit) inhibits
retinoblastoma-directed PP1 activity. Biochem Biophys Res Commun.
297:463–467. 2002. View Article : Google Scholar
|
|
31
|
Choy MS, Hieke M, Kumar GS, Lewis GR,
Gonzalez-DeWhitt KR, Kessler RP, Stein BJ, Hessenberger M, Nairn
AC, Peti W, et al: Understanding the antagonism of retinoblastoma
protein dephosphorylation by PNUTS provides insights into the PP1
regulatory code. Proc Natl Acad Sci USA. 111:4097–4102. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hoekstra E, Peppelenbosch MP and Fuhler
GM: Meeting report Europhosphatase 2015: Phosphatases as drug
targets in cancer. Cancer Res. 76:193–196. 2016. View Article : Google Scholar
|
|
33
|
Kim YM, Watanabe T, Allen PB, Kim YM, Lee
SJ, Greengard P, Nairn AC and Kwon YG: PNUTS, a protein phosphatase
1 (PP1) nuclear targeting subunit. Characterization of its PP1- and
RNA-binding domains and regulation by phosphorylation. J Biol Chem.
278:13819–13828. 2003. View Article : Google Scholar
|
|
34
|
Krucher NA, Rubin E, Tedesco VC, Roberts
MH, Sherry TC and De Leon G: Dephosphorylation of Rb (Thr-821) in
response to cell stress. Exp Cell Res. 312:2757–2763. 2006.
View Article : Google Scholar
|
|
35
|
De Leon G, Sherry TC and Krucher NA:
Reduced expression of PNUTS leads to activation of Rb-phosphatase
and caspase- mediated apoptosis. Cancer Biol Ther. 7:833–841. 2008.
View Article : Google Scholar
|
|
36
|
Kavela S, Shinde SR, Ratheesh R,
Viswakalyan K, Bashyam MD, Gowrishankar S, Vamsy M, Pattnaik S, Rao
S, Sastry RA, et al: PNUTS functions as a proto-oncogene by
sequestering PTEN. Cancer Res. 73:205–214. 2013. View Article : Google Scholar
|
|
37
|
Egger JV, Lane MV, Antonucci LA, Dedi B
and Krucher NA: Dephosphorylation of the Retinoblastoma protein
(Rb) inhibits cancer cell EMT via Zeb. Cancer Biol Ther.
17:1197–1205. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pino MS, Shrader M, Baker CH, Cognetti F,
Xiong HQ, Abbruzzese JL and McConkey DJ: Transforming growth factor
alpha expression drives constitutive epidermal growth factor
receptor pathway activation and sensitivity to gefitinib (Iressa)
in human pancreatic cancer cell lines. Cancer Res. 66:3802–3812.
2006. View Article : Google Scholar
|
|
39
|
Buck E, Eyzaguirre A, Haley JD, Gibson NW,
Cagnoni P and Iwata KK: Inactivation of Akt by the epidermal growth
factor receptor inhibitor erlotinib is mediated by HER-3 in
pancreatic and colorectal tumor cell lines and contributes to
erlotinib sensitivity. Mol Cancer Ther. 5:2051–2059. 2006.
View Article : Google Scholar
|
|
40
|
Ali S, Banerjee S, Ahmad A, El-Rayes BF,
Philip PA and Sarkar FH: Apoptosis-inducing effect of erlotinib is
potentiated by 3,3'-diindolylmethane in vitro and in vivo using an
orthotopic model of pancreatic cancer. Mol Cancer Ther.
7:1708–1719. 2008. View Article : Google Scholar
|
|
41
|
Deer EL, Gonzalez-Hernandez J, Coursen JD,
Shea JE, Ngatia J, Scaife CL, Firpo MA and Mulvihill SJ: Phenotype
and genotype of pancreatic cancer cell lines. Pancreas. 39:425–435.
2010. View Article : Google Scholar :
|
|
42
|
Gradiz R, Silva HC, Carvalho L, Botelho MF
and Mota-Pinto A: MIA PaCa-2 and PANC-1 - pancreas ductal
adenocarcinoma cell lines with neuroendocrine differentiation and
somatostatin receptors. Sci Rep. 6:216482016. View Article : Google Scholar
|
|
43
|
Loukopoulos P, Kanetaka K, Takamura M,
Shibata T, Sakamoto M and Hirohashi S: Orthotopic transplantation
models of pancreatic adenocarcinoma derived from cell lines and
primary tumors and displaying varying metastatic activity.
Pancreas. 29:193–203. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Furukawa T, Duguid WP, Rosenberg L,
Viallet J, Galloway DA and Tsao MS: Long-term culture and
immortalization of epithelial cells from normal adult human
pancreatic ducts transfected by the E6E7 gene of human papilloma
virus 16. Am J Pathol. 148:1763–1770. 1996.PubMed/NCBI
|
|
45
|
Macdonald JI and Dick FA:
Posttranslational modifications of the retinoblastoma tumor
suppressor protein as determinants of function. Genes Cancer.
3:619–633. 2012. View Article : Google Scholar
|
|
46
|
De Leon G, Cavino M, D'Angelo M and
Krucher NA: PNUTS knockdown potentiates the apoptotic effect of
Roscovitine in breast and colon cancer cells. Int J Oncol.
36:1269–1275. 2010.PubMed/NCBI
|
|
47
|
Mohammed A, Janakiram NB, Li Q, Madka V,
Ely M, Lightfoot S, Crawford H, Steele VE and Rao CV: EGFR
inhibitor gefitinib prevents progression of pancreatic lesions to
carcinoma in a conditional LSL-KrasG12D/+ transgenic mice model.
Cancer Prev Res (Phila). 3:1417–1426. 2010. View Article : Google Scholar
|
|
48
|
Lin Y, Wang X and Jin H: EGFR-TKI
resistance in NSCLC patients: Mechanisms and strategies. Am J
Cancer Res. 4:411–435. 2014.
|
|
49
|
Paez JG, Jänne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et
al: EGFR mutations in lung cancer: Correlation with clinical
response to gefitinib therapy. Science. 304:1497–1500. 2004.
View Article : Google Scholar
|
|
50
|
Jackman D, Pao W, Riely GJ, Engelman JA,
Kris MG, Jänne PA, Lynch T, Johnson BE and Miller VA: Clinical
definition of acquired resistance to epidermal growth factor
receptor tyrosine kinase inhibitors in non-small-cell lung cancer.
J Clin Oncol. 28:357–360. 2010. View Article : Google Scholar
|
|
51
|
Scholz A, Heinze S, Detjen KM, Peters M,
Welzel M, Hauff P, Schirner M, Wiedenmann B and Rosewicz S:
Activated signal transducer and activator of transcription 3
(STAT3) supports the malignant phenotype of human pancreatic
cancer. Gastroenterology. 125:891–905. 2003. View Article : Google Scholar
|
|
52
|
Toyonaga T, Nakano K, Nagano M, Zhao G,
Yamaguchi K, Kuroki S, Eguchi T, Chijiiwa K, Tsuneyoshi M and
Tanaka M: Blockade of constitutively activated Janus kinase/signal
transducer and activator of transcription-3 pathway inhibits growth
of human pancreatic cancer. Cancer Lett. 201:107–116. 2003.
View Article : Google Scholar
|
|
53
|
Corcoran RB, Contino G, Deshpande V,
Tzatsos A, Conrad C, Benes CH, Levy DE, Settleman J, Engelman JA
and Bardeesy N: STAT3 plays a critical role in KRAS-induced
pancreatic tumorigenesis. Cancer Res. 71:5020–5029. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Fofaria NM and Srivastava SK: STAT3
induces anoikis resistance, promotes cell invasion and metastatic
potential in pancreatic cancer cells. Carcinogenesis. 36:142–150.
2015. View Article : Google Scholar
|
|
55
|
Wu X, Tang W, Marquez RT, Li K, Highfill
CA, He F, Lian J, Lin J, Fuchs JR, Ji M, et al: Overcoming
chemo/radio-resistance of pancreatic cancer by inhibiting STAT3
signaling. Oncotarget. 7:11708–11723. 2016.
|
|
56
|
Zhang X, Ren D, Wu X, Lin X, Ye L, Lin C,
Wu S, Zhu J, Peng X and Song L: miR-1266 contributes to pancreatic
cancer progression and chemoresistance by the STAT3 and NF-kB
signaling pathways. Mol Ther Nucleic Acids. 11:142–158. 2018.
View Article : Google Scholar
|
|
57
|
Finn RS, Dering J, Conklin D, Kalous O,
Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, et al: PD
0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially
inhibits proliferation of luminal estrogen receptor-positive human
breast cancer cell lines in vitro. Breast Cancer Res. 11:R772009.
View Article : Google Scholar
|
|
58
|
Dean JL, Thangavel C, McClendon AK, Reed
CA and Knudsen ES: Therapeutic CDK4/6 inhibition in breast cancer:
Key mechanisms of response and failure. Oncogene. 29:4018–4032.
2010. View Article : Google Scholar
|
|
59
|
Yang C, Li Z, Bhatt T, Dickler M, Giri D,
Scaltriti M, Baselga J, Rosen N and Chandarlapaty S: Acquired CDK6
amplification promotes breast cancer resistance to CDK4/6
inhibitors and loss of ER signaling and dependence. Oncogene.
36:2255–2264. 2017. View Article : Google Scholar
|
|
60
|
Guri Y and Hall MN: mTOR signaling confers
resistance to targeted cancer drugs. Trends Cancer. 2:688–697.
2016. View Article : Google Scholar
|
|
61
|
Franco J, Balaji U, Freinkman E,
Witkiewicz AK and Knudsen ES: Metabolic reprogramming of pancreatic
cancer mediated by CDK4/6 inhibition elicits unique
vulnerabilities. Cell Rep. 14:979–990. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhang J, Xu K, Liu P, Geng Y, Wang B, Gan
W, Guo J, Wu F, Chin YR, Berrios C, et al: Inhibition of Rb
phosphorylation leads to mTORC2 mediated activation of AKT. Mol
Cell. 62:929–942. 2016. View Article : Google Scholar
|
|
63
|
Arumugam T, Ramachandran V, Fournier KF,
Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey
DJ and Choi W: Epithelial to mesenchymal transition contributes to
drug resistance in pancreatic cancer. Cancer Res. 69:5820–5828.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sherr CJ: Cancer cell cycles. Science.
274:1672–1677. 1996. View Article : Google Scholar
|
|
65
|
Mittnacht S: The retinoblastoma
protein--from bench to bedside. Eur J Cell Biol. 84:97–107. 2005.
View Article : Google Scholar
|
|
66
|
Sherr CJ and McCormick F: The RB and p53
pathways in cancer. Cancer Cell. 2:103–112. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Harbour JW and Dean DC: The Rb/E2F
pathway: Expanding roles and emerging paradigms. Genes Dev.
14:2393–2409. 2000. View Article : Google Scholar
|
|
68
|
Thangavel C, Boopathi E, Liu Y, Haber A,
Ertel A, Bhardwaj A, Addya S, Williams N, Ciment SJ, Cotzia P, et
al: RB loss promotes prostate cancer metastasis. Cancer Res.
77:982–995. 2017. View Article : Google Scholar :
|
|
69
|
Arima Y, Hayashi H, Sasaki M, Hosonaga M,
Goto TM, Chiyoda T, Kuninaka S, Shibata T, Ohata H, Nakagama H, et
al: Induction of ZEB proteins by inactivation of RB protein is key
determinant of mesenchymal phenotype of breast cancer. J Biol Chem.
287:7896–7906. 2012. View Article : Google Scholar
|