1. Introduction
The mammalian fibroblast growth factor (FGF) family
includes 22 members, which can be subdivided into six subgroups
based on sequence similarities, biochemical properties and
evolutionary relationships (1).
FGF8, FGF17 and FGF18 are classified as the FGF8 subfamily, since
these three proteins share 60-80% amino acid sequence identities
and similar receptor-binding properties (2,3).
FGF8 subfamily members are highly conserved between
humans and mice (Fig. 1). FGF8,
reported as an androgen-induced growth factor, was originally
isolated from the conditioned medium of the mouse mammary carcinoma
cell line SC-3 (4,5). Human FGF8 isoform a and b show 100%
similarity in protein sequence with murine FGF8 isoform a and b,
respectively (6). However, human
FGF8e and FGF8f isoforms share 98% identity with corresponding
murine isoforms (7). Both FGF17
and FGF18 were originally isolated from mouse embryos. Similar to
FGF8, 98.6% and 99% homology has been identified between human and
murine FGF17 and FGF18, respectively (6,8).
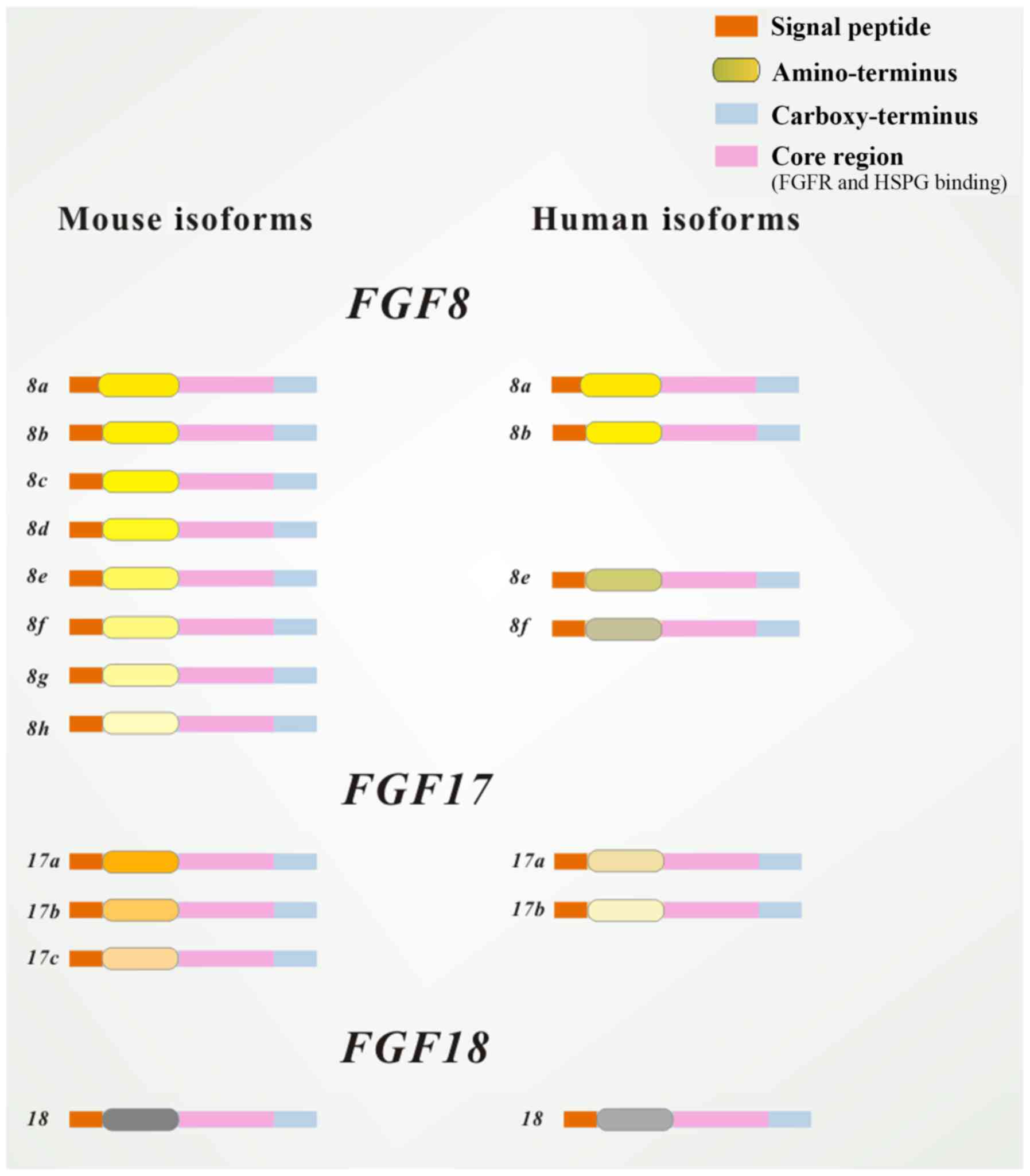 | Figure 1Structure of isoforms of FGF8
subfamily members. All peptides contain an FGFR and HSPG binding
site in the core region and a cleavable secreted signal sequence in
the amino-terminal (marked in different colors). Human isoforms are
very similar to their murine counterparts in both the N- and
C-terminus. Mouse FGF8 has eight isoforms: FGF8a, FGF8b, FGF8c,
FGF8d, FGF8e, FGF8f, FGF8g and FGF8h; human FGF8 has only four
isoforms: FGF8a, FGF8b, FGF8e and FGF8f. Mouse FGF17 has three
isoforms: FGF17a, FGF17b and FGF17c; human FGF17 has only two
isoforms: FGF17a and FGF17b. FGF, fibroblast growth factor; FGFR,
FGF receptor; HSPG, heparan sulfate proteoglycan. |
It is documented that FGF8 and FGF17 are expressed
in the heart, limb, kidney and central nervous system (CNS), and in
craniomaxillofacial development, whereas FGF18 is expressed in the
cartilage and palate. The pivotal roles of FGFs and their binding
receptors (FGF receptors, FGFRs) in development and pathogenic
processes are thought to be due to the increasing functional
diversities of the signaling systems enabled by them (9).
All FGFs share a conserved structure, including an
internal core region encompassing 120-125 amino acids, which is
essential for binding to FGFR, as well as a carboxy-terminal and an
amino-terminal (Fig. 1) (10). FGF8 subfamily members have a
hydrophobic amino-terminal (~22 amino acids), which is a typical
cleavable signal peptide (11).
Interestingly, the amino-terminal sequences of FGF17 and FGF18 are
more closely conserved to each other, than to FGF8 (6). Variations in the N-terminal tails of
FGF8 subfamily members may play a role in their distinct biological
functions (12). The core region
structure of all FGFs is a conserved β-trefoil fold consisting of
12 antiparallel β-strands (β1-β12). However, the alternatively
spliced βF and βG strands of FGF8 subfamily members harbor the
primary receptor binding sequences and give them binding affinities
to specific FGFRs (13,14).
The complexity of FGF8 mRNA splicing was reported
previously. The first exon of FGF8 gene in mice contains at least
four different coding sequences that can be alternatively spliced
to produce eight secreted isoforms (a-h). However, there are only
four isoforms (a, b, e and f) in humans because of a terminator
sequence in exon 1B of the FGF8 gene. The shortest FGF8 isoform,
FGF8a, contains the core region, which is conserved in all FGF8
isoforms. All other FGF8 isoforms have an extra sequence, which is
different for each isoform, flanking the common core region
(15). Nevertheless, FGF8 variants
share identical reading frames and the same peptide sequence in
their carboxy-terminal domains (5,16).
None of the alternative-splicing events alters the sequence of the
conserved FGF core domain (11).
Human FGF17 gene codes for two isoforms: FGF17a and
FGF17b. FGF17a is not thought to be involved in FGF signal
transduction, since FGFRs are not activated in NIH3T3 cells treated
with FGF17a (17).
2. Signal transduction
FGF8 receptors and signal
transduction
FGF8 subfamily members share a similar receptor
binding specificity. They exhibit high binding affinities with IIIc
variants of FGFR1-3 and the non-spliced FGFR4 (3), while only a weak affinity is observed
with IIIb variants (13,18). Furthermore, it has been
demonstrated that FGF8 and FGF17 have a higher binding affinity
with FGFR3c and FGFR4, and a weaker binding affinity with FGFR1c.
Likewise, FGF18 preferentially binds to FGFR3c rather than FGFR2c
(3).
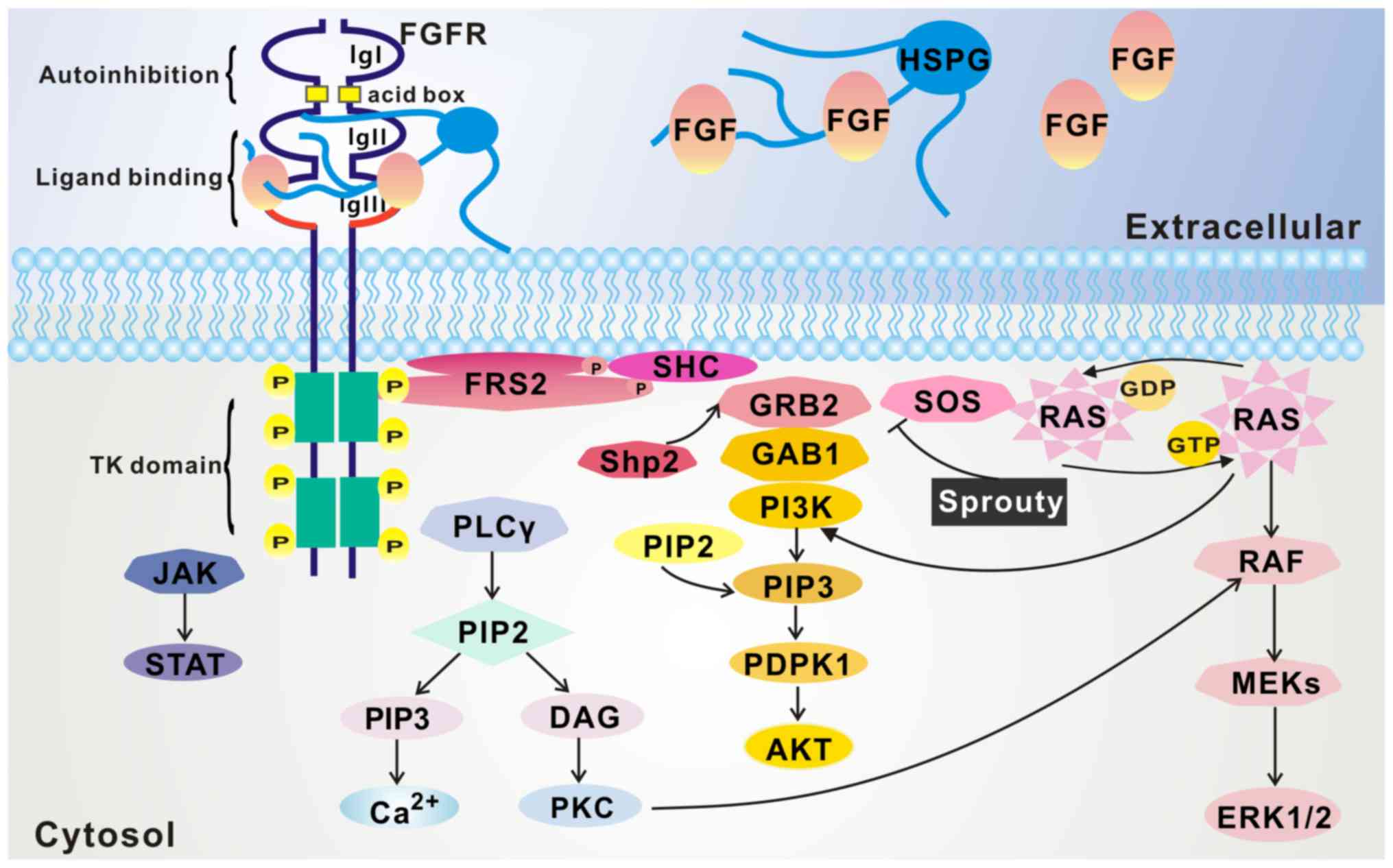
Binding with FGF ligands leads to a conformational
shift in the FGFR that activates the intracellular kinase domain,
resulting in intermolecular transphosphorylation of the tyrosine
kinase domains and the intracellular tail. FGFR substrate 2 (FRS2)
and phospholipase Cγ (PLCγ), which are key adaptor proteins
phosphorylated by FGFRs, are then recruited to the FGFR
intracellular tail, phosphorylated by FGFRs, and in turn initiate
intracellular signal cascades. The FRS2/Ras/mitogen-activated
protein kinase (MAPK) pathway, PLCγ/Ca2+ pathway, and
phosphoinositide-3-kinase (PI3K)/AKT pathway are considered to be
major downstream effector pathways of the FGF8 subfamily (Fig. 2) (19).
Ras/MAPK pathway
FGFRs can directly phosphorylate several tyrosine
sites on the FRS2 protein after binding with FGFs (20). These phosphorylation sites are
recognized by the adapter protein growth factor receptor-bound
protein 2 (Grb2), a small adaptor molecule, which forms a complex
with the guanine nucleotide exchange factor Son of Sevenless (SOS)
via its SH3 domain, allowing SOS to activate Ras. Activation of Ras
triggers a series of downstream effector proteins, including Raf,
MAPK kinase (MEK) and MAPK, and the latter finally enter the
nucleus and phosphorylates transcription factors, including c-Myc
and activation protein-1 (21,22).
PI3K/Akt pathway
Besides SOS, Grb2 is able to recruit the adaptor
protein Grb2-associated binding protein 1 (Grb1) to the complex,
once Grb2 is activated by FGFR-phosphorylated FRS2α. This leads to
activation of PI3K, which eventually activates AKT (2,23).
PLCγ/Ca2+ pathway
The PLCγ/Ca2+ pathway is activated when
PLCγ directly binds to an autophosphorylated tyrosine in the
C-terminal tail of FGFRs, resulting in PLCγ phosphorylation and
activation. Activated PLCγ hydrolyzes
phosphatidylino-sitol-4,5-biphosphate (PIP2) to produce two second
messengers: Phosphatidylinositol-3,4,5-triphosphate (PIP3) and
diacylglycerol (DAG). PIP3, in turn, stimulates the release of
intracellular calcium, while DAG activates calcium-dependent
downstream signaling of protein kinase C (23,24).
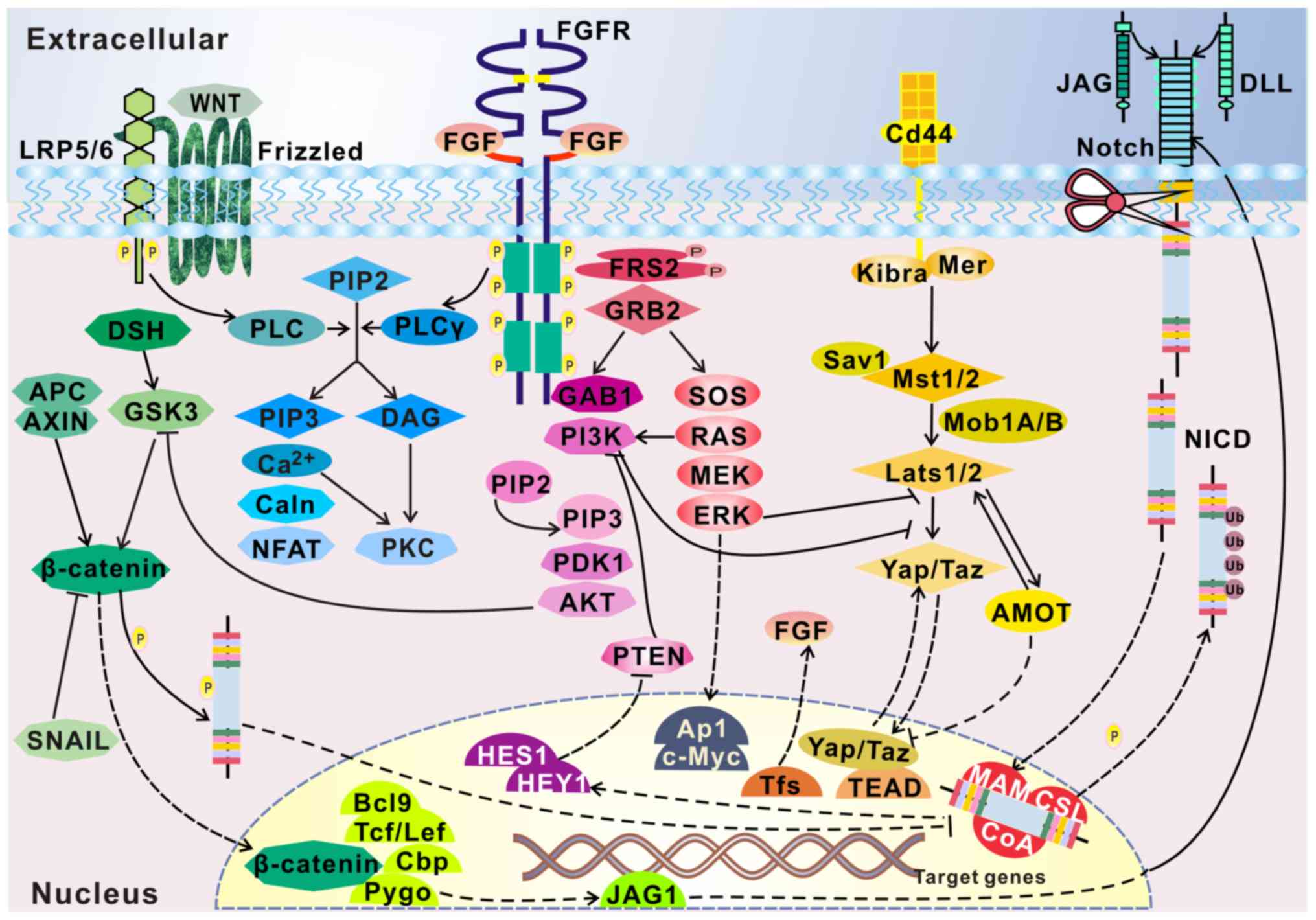
Several other pathways are also found to be
activated by FGFRs, such as the signal transducer and activator of
transcription signaling pathway, the Wnt signaling pathway
associated with tooth development (25), and the Sonic hedgehog (Shh)
signaling pathway involved in the palate (26). These pathways interact with other
growth factor pathways, forming a network that regulates cell
behavior (Fig. 3).
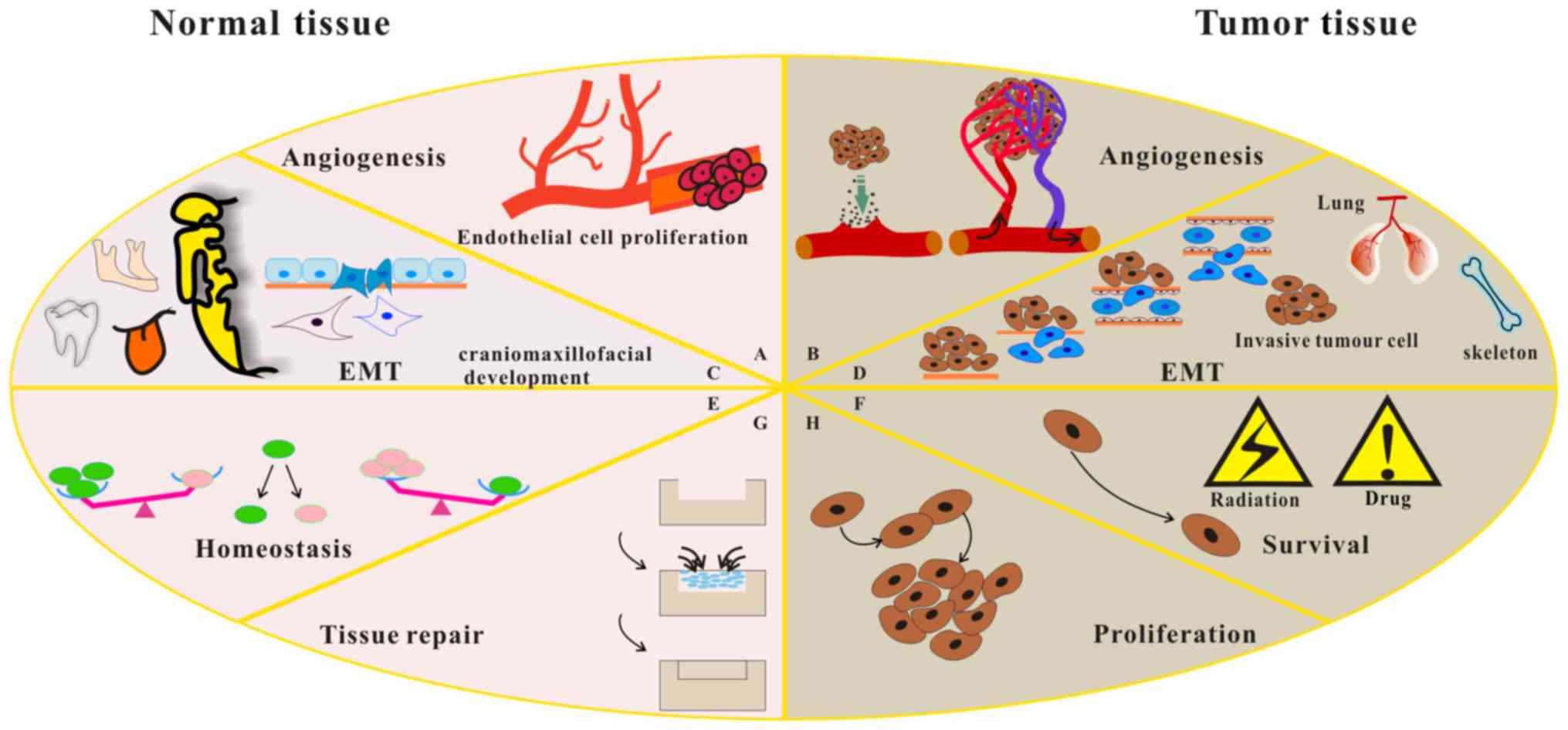
3. Regulation of cell phenotype by FGF8
subfamily members
Previous studies have suggested that FGFs play
important roles in morphogenesis, angiogenesis and development of a
variety of cells. FGFs bind to FGFRs with different affinities,
eliciting a wide range of biological responses, including cellular
proliferation, differentiation, migration, adhesion and survival
(Fig. 4) (18,23,27).
4. Epithelial-to-mesenchymal transition in
oral-maxillofacial development
Due to the critical role of FGF8 and FGF18 in embryo
development, germline inactivation of FGF8 and FGF18 genes causes
embryonic or perinatal death in mice and the loss of all embryonic
mesoderm and endoderm-derived structures (3,5,28).
FGF17 knockout mice are viable but exhibit impaired hindbrain
development, suggesting that FGF17 may play a less critical role
than FGF8 or FGF18 (29,30).
FGF8 subfamily members in craniofacial
development
The mechanisms responsible for craniofacial
development are partially understood (31). In mammals, the first branchial arch
(BA1) develops a number of craniofacial skeletal elements,
including teeth, the mandible and maxilla, the lateral skull wall,
and parts of the tongue and other soft-tissue derivatives (32).
FGF8 is considered to be an epithelial
cell-originating factor that regulates gene expression when
patterning of the mandibular mesenchyme occurs during BA1
development (Fig. 4) (32,33).
FGF8 is involved in the development of a large proximal region of
the BA1 primordium, either promoting mesenchymal cell survival or
inducing BA1 morphogenesis (32).
It has been identified that FGF8 positively regulates Sprouty gene
expression in the BA1. A reduced level of Sprouty homolog 2 (Spry2)
in mutant BA1 mesenchyme may partially compensate for reduced Ras
activity due to the inactivation of FGF8 by depressing other
receptor tyrosine kinase pathways normally inhibited by Sprouty,
and thereby promote cell survival (22,32).
FGF8 is expressed in the outmost lateral regions of each pharyngeal
pouch and exhibits a high level of expression in the overlying
surface ectoderm of the mandibular and maxillary prominences. It is
reported that FGF8 mutations affect signaling in the BA1 ectoderm
and result in failure in developing BA1 structures (34). Newborn mice harboring these mutants
lack most BA1-derived structures, except those that develop from
the distalmost region of BA1, including mandibular incisors
(32).
The signal transduction pathways activated by FGF8
are implicated in early steps of craniofacial development (28). FGF8, −17 and −18 have been
demonstrated to be key regulators in craniofacial structure
formations, including tooth, palate, mandible and salivary gland
(35,36). Thus, FGF8 subfamily members may
play different roles in different developmental contexts (22,32).
Regulating odontogenesis
In the initial stages of tooth development, FGF
signaling is involved in the dental epithelium and the invagination
of the dental epithelium into the underlying mesenchyme (37). FGF8 is expressed in both the
epithelium and mesenchyme, promoting initiation and patterning of
tooth morphogenesis (38). FGF17
is expressed in the epithelium, while FGF18 is expressed in the
mesenchyme (28,38,39).
The expression of pituitary homeobox 2 (Pitx2), a
marker for the dental lamina band, is restricted to the dental
epithelium of both molars and incisors throughout odontogenesis
(37,40,41).
Bone morphogenetic protein 4 (BMP4) and FGF8 may act as feedback
regulators to control Pitx2 expression during early odontogenesis.
FGF8 positively regulates Pitx2 expression and BMP4 exerts the
opposite effect (40,42). In the absence of FGF8, Pitx2
expression in the oral epithelium is diminished, thereby affecting
tooth development (43,44). In addition, by activating the
dental mesenchymal markers Barx1 and Pax9 in the mesenchyme, the
coordination of FGF8 and BMP4 signaling pathways determines not
only the proto-molar or proto-incisor fate of mandibular
mesenchymal cells, but also the tooth emergence sites by regulating
the migration of the tooth germinal cells (37,42,45).
Conditional FGF8 gene knockout in the ectoderm leads to
significantly decreased expression of Barx1 and Pax9 in the
presumptive molar region, and thus molar teeth are not formed and
rudimentary incisors develop instead (37,46-52).
FGF18 is mainly expressed in the mesenchyme. As the
epithelium within the dental lamina continues to form the
epithelial bud, FGF18 continues to be expressed in the mesenchyme.
In the incisor at the bell stage, FGF18 expression is detected in
the mesenchyme of the cervical loop. FGF18 expression is also
detected at the buccal side of the mesenchyme at the lamina stage.
In addition, FGF18 is involved in the regulation of odontoblasts in
the latter stages of molar tooth development and may regulate
dentin matrix formation and/or mineralization in both the crown and
root formation (37,39).
FGF8 subfamily members in tongue
development
FGF8 and FGF18 are associated with the generation
and early morphogenesis of the tongue (53). For rodents, the formation of the
tongue begins at E11, when two lateral tongue buds derive from the
branchial arch (35,54). These two buds merge to shape into
the tongue primordium at E11.5, and cell proliferation is followed
by cell differentiation in both the local epithelium and
mesenchyme, causing the rapid enlargement of the tongue (55,56).
Tongue epithelium differentiation begins at E13, initiated from a
single circumvallate papilla and numerous fungiform papillae, which
is followed by the differentiation of filiform papillae and foliate
papillae at approximately E15 (54). Both FGF8 and FGF18 are expressed in
the tongue epithelium at relatively constant levels during early
tongue development, whereas FGF17 is undetectable (18). FGF8 is strongly expressed in the
dorsal tongue epithelium and also more diffusely in the underlying
tongue mesenchyme. However, FGF8 expression is downregulated before
morphologically detectable gustatory papillae initiation (53), suggesting that FGF8 might be less
important during these stages. A similar expression pattern is
found in FGF18 during tongue development. FGF18 is highly expressed
in the lingual margin of the anterior half of the tongue, and
weakly expressed in the posterior half. Like in other organs, FGF18
is thought to stimulate proliferation of both mesenchymal and
epithelial cells during tongue development (35).
Regulation of skeletal development
Mandibular development from BA1 is regulated by the
interaction of the mesenchyme with the endoderm and ectoderm. FGF8
expression is spatially restricted to the ectoderm precursors of
the proximal mandible and its expression is regulated by adjacent
endoderm-derived signals (17).
FGF8 is pivotal for the survival and migration of mesenchymal cells
in BA1. FGF8 overexpression due to ectopic activation in
CNC-derived mesenchymal cells inhibits tissue differentiation and
organogenesis in the craniofacial region, instead of impairing
migration. Therefore, FGF8 expression does not affect the
odontogenic fate of the CNC-derived mesenchymal cells (46). If the expression of FGF8 is
conditionally lost in the ectoderm of BA1, the arch is markedly
reduced in size, resulting in almost complete loss of the
BA1-derived skeletal structures and tooth agenesis (28).
The expression of FGF18 persists in mesenchymal
cells and osteoblasts during mandibular bone development. FGF18 is
thought to positively regulate osteogenesis and negatively regulate
chondrogenesis (18,57-59).
However, FGF17 expression is only observed in the maxilla (17).
FGF8 subfamily members in salivary gland
branching morphogenesis
The development of the embryonic submandibular
salivary gland (SMG) begins with an invagination of the original
oral epithelium into undifferentiated mandibular mesenchyme, which
requires epithelial-mesenchymal transition. FGF8 and its cognate
receptor, FGFR-2c (IIIc), are essential for branching morphogenesis
(26,60,61).
FGF8 hypomorphic mice, which have defective FGF8
throughout embryogenesis, develop hypoplastic SMGs. FGF8 ectoderm
conditional mutants, with silenced FGF8 expression in the BA1
ectoderm, exhibit ontogenic arrest followed by involution and
eventually disappear at E18.5. SMG aplasia in FGF8 conditional
mutants indicates that FGF8 signaling is essential for salivary
gland branching morphogenesis. Notably, the presence of an initial
SMG bud is observed in FGF8 mutants, suggesting that the initial
bud formation is independent of FGF8 (61).
5. Aberrant regulation of FGF8 subfamily
members in oral-maxillofacial diseases
During early craniomaxillofacial morphogenesis, an
important role of the FGF8 subfamily is to control
epithelial-mesenchymal transition (EMT) (5). Errors during this complex process can
cause craniofacial anomalies (Fig.
5). Interestingly, EMT is also implicated in the progression of
invasive metastasis of malignant tumor cells (62). For instance, FGF8 is reactivated
and overexpressed in ovarian, breast and prostate cancers, and
further promotes tumor invasion and metastasis (5,63,64).
The subsequent sections will focus on the aberrant
signaling of FGF8 subfamily resulting in diseases of the oral and
maxillofacial regions.
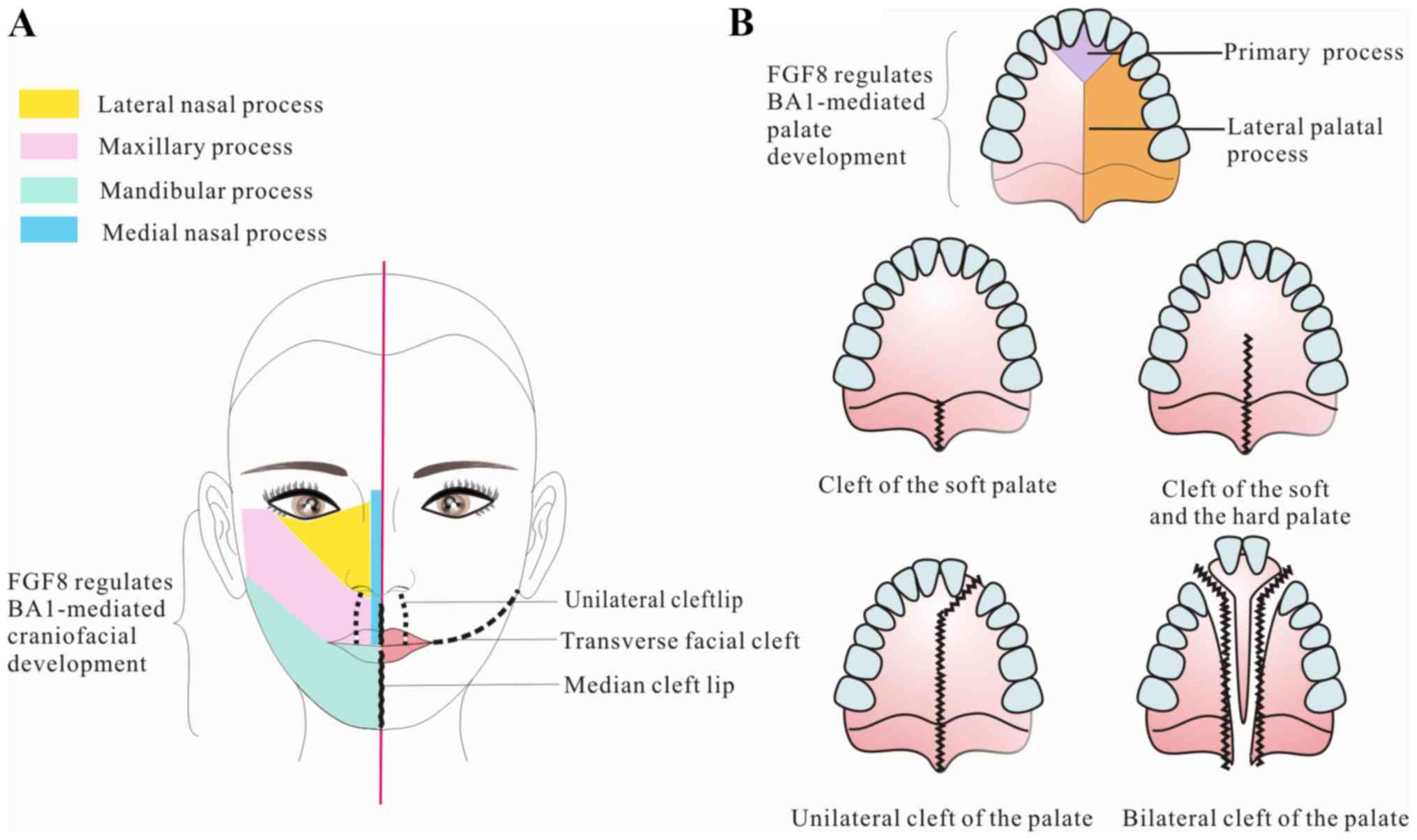
FGF8 subfamily members and cleft lip
and/or palate (CLP)
CLP is among the most commonly observed congenital
malformations. CLP can affect feeding, speech, hearing and dental
function. Although the condition can be surgically repaired to
different degrees, lots of patients maybe still suffer lifelong
psychosocial effects from the malformation (65).
During the final stages of palatogenesis, two palate
shelves undergo mesenchyme proliferation, expansion and elevation,
then dissolution of the epithelium and midline fusion. Failure to
undergo any of these processes results in a palatal cleft (Fig. 5) (66).
Genome-wide analysis has suggested that FGF8
mutations are involved in the development of CLP. For instance,
D73H missense mutation in the FGF8 gene was found in a CLP patient.
Neither parent of the patient had the variant allele, suggesting
that the mutation arose in the patient. Structural analysis
revealed that D73 is located in the docking domain for binding with
FGFR2 IIIc, and the side chain of D73 creates three hydrogen bonds
connecting with the FGFR. This mutation reduces the binding
affinity of FGF8 with its cognate receptors, probably by both
destabilizing the conformation of the N-terminus of FGF8 and
subsequently eliminating hydrogen bonding with receptors (30,67).
The FGF8 D73H mutation is the first disease-associated mutation in
the coding region of FGF8. The patient with this mutation appeared
to have non-syndromic CLP with no additional phenotypes (67).
T-box 1 (Tbx1) and transcription factor activator
protein 2 (Tfap2) are FGF8-associated transcriptional mediators of
developmental abnormalities that control palatal elongation and
elevation. In Tbx1-null mice, FGF8 expression is markedly
downregulated in the palate epithelium (68). Mutations in the Tfap2a gene induce
upregulated FGF8 expression, resulting in cleft palate by changing
growth and morphogenesis. Reducing FGF8 expression compensates
these morphogenic changes, and decreases the incidence of cleft
palate in mice. Thus, it is reasonable to infer that loss of Tbx1
and Tfap2 results in aberrant proliferation and apoptosis in
palatal cells, probably through altering FGF8 expression (65).
By contrast, CLP in mice caused by enhanced cell
apoptosis in the malformed mandible and tongue may be associated
with the prominent decrease of FGF8 expression in the ectoderm
(25,65). In addition, Sp8 is the zinc finger
transcription factor gene, which is considered as a strong
candidate gene for causing non-syndromic CLP (69). Reduced expression of murine FGF8
and FGF17 in the anterior neural ridge and olfactory pit signaling
centers caused by mutations of Sp8 could also lead to CLP (70).
Cleft palate is also observed in FGF18 null mice,
but the mechanisms remain unclear (65). Shh signaling is reported to play a
key role in palate development by regulating the expression of
Foxf1 and Foxf2 of the Fox family transcription factors (26,71,72).
Foxf1 and Foxf2 regulate Shh transduction and repress FGF18
expression in the palatal mesenchyme during the palatal shelf
growth. Notably, Foxf2 mutants exhibit decreased Shh expression but
enhanced FGF18 expression. These results suggest a
Shh-Foxf-FGF18-Shh negative feedback loop in the reciprocal
epithelial-mesenchymal signaling molecular network controlling
palatogenesis (26,58,72).
FGF8 subfamily members and
ciliopathies
Ciliopathies are a wide class of human disorders
commonly characterized by craniofacial dysmorphology. The most
frequent craniofacial phenotype in ciliopathies is a high arched
palate with a prominent median groove, but with the roof of the
mouth remaining intact across the midline. The disease is
associated with secondary dental anomalies, including postnatal
gingival swelling and molar crowding, which greatly influence
speech and quality of life. Tabler et al (73) reported that the primary cause of
ciliopathic high arched palate is excessive neural crest cells
producing an enlarged BA1 and maxillary hyperplasia in early
embryogenesis.
Fuzzy (Fuz) is a central regulator of vertebrate
ciliogenesis (74). Ciliopathic
Fuz mutant mice indicate that the mechanistic basis of this
phenotype originates from dysregulated Gli processing, which in
turn leads to craniofacial FGF8 overexpression. More specifically,
loss of Gli3 can result in increased cranial FGF8 gene
transcription, resulting in a dramatic increase in FGF signaling.
Notably, at E9.5 in Fuz mutants, FGF8 distribution is significantly
expanded, and exhibits a mediolateral expansion within the
mandibular and maxillary prominences. This expansion is maintained
in mutant maxillae at E10.5. It is believed that excessive FGF
signals drive the maxillary hyperplasia that is the basis of the
palate defects, and by contrast, downregulation of FGF8 ameliorates
the maxillary phenotypes (73,75).
It has been suggested that the most likely reason
for craniofacial defects in Fuz mutant mice is increased expression
of FGF8. High arched palate is also a common feature of FGF
hyperactivation syndromes, raising the new possibilities that
clinical diagnosis and treatment of high arched palate should also
be reconsidered in this developmental and molecular context
(73).
FGF8 subfamily members and agnathia
Agnathia is a malformation characterized by agenesis
of the mandible. As an isolated abnormality, agnathia is rare, with
an estimated incidence of 1 in 70,000 newborns. The abnormality
includes ventral microstomia, malpositioning of the external ears
and persistence of the buccopharyngeal membrane. These conditions
are usually lethal, as the airway cannot be established (34). Mechanistic studies suggest a role
of BMP and FGF8 in BA1 patterning, including mandibular development
(76,77).
It has been identified that ectodermal FGF8
expression could be either activated or repressed by BMP4 in a
dose-dependent fashion, and that high BMP4 levels repress FGF8,
while low signaling levels promote FGF8 transcription. Differential
effects of BMP4 on FGF8 expression are also observed in the
proximal and distal mandible (77,78).
FGF8 is known to be expressed in the epithelium of the proximal
region and BMP4 in the distal region (78). In the distal mandibular ectoderm,
the absence of BMP4 antagonists, chordin and noggin, results in
high levels of BMP4, strongly downregulating FGF8 expression and
increasing cell death during mandibular outgrowth, thus leading to
mandibular hypoplasia (76). On
the other hand, a low level of BMP4 is needed to maintain FGF8
expression, which is required for proximal mandible formation.
Thus, this concentration and position-dependent relationship
between BMP4 and FGF8 is necessary for appropriate proximodistal
patterning of the mandible (34).
FGF8 and odontogenic tumors (OTs)
OTs are a special category of neoplasms discovered
exclusively in the jawbones or related soft tissues, stemming from
tooth-forming apparatus or their remnants (79). OTs include solid multicystic
ameloblastoma (SMA) from epithelial origin, ameloblastic fibroma
(AF) from mixed origin, and odontogenic myxoma (OM) from
ectomesenchymal origin (80).
The pathogenesis of OTs is a complex process, where
certain steps are similar to the odontogenic processes. Expression
of either tumor initiating factors or tumor progression factors in
OTs exhibits a striking resemblance to those expressed during
odontogenesis (79). Since FGF8
plays a major role in odontogenesis, it is believed that FGF8 may
affect OT development.
FGF8 has been identified to be expressed in both
epithelial and mesenchymal tumor tissues to varying degrees,
suggesting that it is involved in epithelial and mesenchymal
interactions (81). FGF8 is
expressed in different odontogenic tumors with a distinct intensity
according to the tumor's properties. Higher expression of FGF in
invasive types may suggest a resemblance to proliferative stages of
odontogenesis. However, in milder cases, a lower level of
expression is observed, which may be parallel with different stages
of odontogenesis (36). Swarup
et al (36) analyzed the
expression of FGF8 in OTs and tooth development. Dental organs of
various odontogenic stages and 30 OTs, including 10 cases of SMA,
10 cases of AF and 10 cases of OM, were evaluated in both
mesenchymal and epithelial tissues by immunohistochemistry. Among
all OTs, the epithelial tissue of SMA exhibited the strongest
signal for FGF8, whereas the mesenchymal FGF8 signal was highest in
OM. However, the overall reactivity for FGF8 in AF was low.
Therefore, increased FGF8 expression may exert a marked effect on
tumor initiation and progression by inducing odontogenic epithelium
changes in SMA. Upregulation of FGF8 was associated with OM and was
likely to have induced an aggressive phenotype. Limited data on
upregulation of FGF8 expression have been reported in
myxoinflammatory fibroblastic sarcoma (82) and other tumors. Thus, FGF8 may be
associated with the initiation and progression of OM, but further
investigation is required.
FGF8 subfamily members and oral squamous
cell carcinoma (OSCC)
Oral cancer is one of the most common cancer types
in the world. More than 90% of oral cancer cases are OSCC, which is
associated with severe deformity and high mortality due to poor
prognosis and strong potential for metastasis. Despite improvements
in treatment, the clinical outcome remains unacceptable, with a
5-year survival rate of approximately 60% over the last decade
(83,84).
Low-density lipoprotein receptor-related protein 6
(LRP6) is reported to be an essential Wnt coreceptor for activating
the canonical Wnt/β-catenin signaling pathway (85), and FGF8 is a potential downstream
gene of the Wnt pathway (86). It
was previously demonstrated that FGF8 is present in an LRP6-related
protein-protein interaction network by bioinformatics analyses, and
that FGF8 expression exerts a positive synergistic effect with its
upstream oncogene, LRP6, in OSCC (84).
FGF8 is required for LRP6-induced proliferation in
OSCC cell lines (84,87-89).
Knockdown of FGF8 by short interfering RNAs significantly inhibits
endogenous FGF8 expression, and subsequently abolishes LRP6-induced
cell proliferation in OSCC cell lines. Additionally, using FGF8
immunostaining methods, the immunostaining signals of FGF8 are
paralleled by LRP6 in corresponding OSCC tissue slides.
Furthermore, co-immunofluorescent staining in OSCC tissues revealed
that strong FGF8 signals are frequently observed in cells with
strong LRP6 signals. Collectively, this suggests that
overexpression of LRP6 triggers FGF8 expression in OSCC cell lines,
and the two genes act synergistically (84).
Notably, compared with LRP6 expression alone, it has
been identified that concurrent expression of FGF8 and LRP6 could
be a better predictor of OSCC patient outcome. Patients with high
expression levels of both LRP6 and FGF8 exhibit even shorter
overall survival time compared with patients with high LRP6 or FGF8
expression alone. Similarly, concurrently low LRP6 and FGF8
expression is associated with a better survival rate compared with
low LRP6 or FGF8 expression alone (84). A similar association between
simultaneous expression of LRP6/FGF8 and patient outcome has also
been identified in tongue cancer (90).
Genome-editing technology in
FGF8-associated diseases
The CRISPR-Cas9 system, as the most effective
genome-editing tool, has been successfully applied in diverse
organisms (91). Disease-causing
mutations in FGF8 subfamily members introduced by a CRISPR-Cas9
system have been reported in a lamprey model. CRISPR/Cas9-mediated
disruption of FGF8/17/18 in the sea lamprey (Petromyzon
marinus) produced mutant F0 embryos, with most of the injected
embryos developing into complete or partial mutants (92). The ability of the CRISPR-Cas9
system to create large numbers of mutant embryos without inbred
lines not only provides new possibilities for studying development
in model organisms with life histories that prohibit the generation
of mutant lines, but also suggests a potential application in
treating human diseases. However, biosecurity risks of this
technology, including off-target effects and other unknown side
effects, must be carefully estimated before clinical usage. The
question thus remains whether CRISPR/Cas9 approaches can be used as
a 'silver bullet' solution for the management of FGF8-associated
oral diseases (93).
6. Conclusions
In recent decades, a large number of studies have
described the relevance of the FGF8 subfamily in human embryonic
development. Aberrant FGF signaling transduction, caused by
activating mutations, increased expression or abnormal isoform
splicing is observed in the development of oral-maxillofacial
diseases. Abnormal regulation of the FGF8 signaling pathway has
also been implicated in the development of oral cancer. Targeting
FGF8 subfamily members may provide a novel and promising strategy
for the treatment of oral diseases. Further work is still required
to illustrate the detailed mechanisms underlying the roles of the
FGF8 subfamily in the development of oral disease.
Funding
This work was supported by the Nonprofit Industry
Research Specific Fund of National Health and Family Planning
Commission of China (grant no. 201502018 to QC), the National
Natural Science Foundation of China (grant nos. 81520108009 and
81621062 to QC and grant no. 81672674 to RL), 111 Project of MOE
B14038 (grant awarded to QC), and the Young Elite Scientist
Sponsorship Program by CAST 2016QNRC001 (grant awarded to RL).
Availability of data and materials
Not applicable.
Authors' contributions
RL and QC contributed to the conception of the
study. YH wrote the main part of the manuscript. All authors read
and approved the final manuscript. YY and ST helped perform the
analysis with constructive discussion.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Belov AA and Mohammadi M: Molecular
mechanisms of fibroblast growth factor signaling in physiology and
pathology. Cold Spring Harb Perspect Biol. 5:52013. View Article : Google Scholar
|
|
2
|
Böttcher RT and Niehrs C: Fibroblast
growth factor signaling during early vertebrate development. Endocr
Rev. 26:63–77. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Estienne A and Price CA: The fibroblast
growth factor 8 family in the female reproductive tract.
Reproduction. 155:R53–R62. 2018. View Article : Google Scholar
|
|
4
|
Liu R, Huang S, Lei Y, Zhang T, Wang K,
Liu B, Nice EC, Xiang R, Xie K, Li J, et al: FGF8 promotes
colorectal cancer growth and metastasis by activating YAP1.
Oncotarget. 6:935–952. 2015.
|
|
5
|
Mattila MM and Härkönen PL: Role of
fibroblast growth factor 8 in growth and progression of hormonal
cancer. Cytokine Growth Factor Rev. 18:257–266. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Katoh M and Katoh M: Comparative genomics
on FGF8, FGF17, and FGF18 orthologs. Int J Mol Med. 16:493–496.
2005.PubMed/NCBI
|
|
7
|
Gemel J, Gorry M, Ehrlich GD and MacArthur
CA: Structure and sequence of human FGF8. Genomics. 35:253–257.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hoshikawa M, Ohbayashi N, Yonamine A,
Konishi M, Ozaki K, Fukui S and Itoh N: Structure and expression of
a novel fibroblast growth factor, FGF-17, preferentially expressed
in the embryonic brain. Biochem Biophys Res Commun. 244:187–191.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Eswarakumar VP, Lax I and Schlessinger J:
Cellular signaling by fibroblast growth factor receptors. Cytokine
Growth Factor Rev. 16:139–149. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Popovici C, Roubin R, Coulier F and
Birnbaum D: An evolutionary history of the FGF superfamily.
BioEssays. 27:849–857. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Itoh N and Ornitz DM: Functional
evolutionary history of the mouse Fgf gene family. Dev Dyn.
237:18–27. 2008. View Article : Google Scholar
|
|
12
|
Beenken A and Mohammadi M: The FGF family:
Biology, pathophysiology and therapy. Nat Rev Drug Discov.
8:235–253. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang X, Ibrahimi OA, Olsen SK, Umemori H,
Mohammadi M and Ornitz DM: Receptor specificity of the fibroblast
growth factor family. The complete mammalian FGF family. J Biol
Chem. 281:15694–15700. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Olsen SK, Li JY, Bromleigh C, Eliseenkova
AV, Ibrahimi OA, Lao Z, Zhang F, Linhardt RJ, Joyner AL and
Mohammadi M: Structural basis by which alternative splicing
modulates the organizer activity of FGF8 in the brain. Genes Dev.
20:185–198. 2006. View Article : Google Scholar :
|
|
15
|
Cretekos CJ, Deng JM, Green ED and
Rasweiler JJ: Isolation, genomic structure and developmental
expression of Fgf8 in the short-tailed fruit bat, Carollia
perspicillata. Int J Dev Biol. 51:333–338. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Crossley PH and Martin GR: The mouse Fgf8
gene encodes a family of polypeptides and is expressed in regions
that direct outgrowth and patterning in the developing embryo.
Development. 121:439–451. 1995.PubMed/NCBI
|
|
17
|
Xu J, Lawshe A, MacArthur CA and Ornitz
DM: Genomic structure, mapping, activity and expression of
fibroblast growth factor 17. Mech Dev. 83:165–178. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Haque T, Nakada S and Hamdy RC: A review
of FGF18: Its expression, signaling pathways and possible functions
during embryogenesis and post-natal development. Histol
Histopathol. 22:97–105. 2007.
|
|
19
|
Goetz R and Mohammadi M: Exploring
mechanisms of FGF signalling through the lens of structural
biology. Nat Rev Mol Cell Biol. 14:166–180. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kouhara H, Hadari YR, Spivak-Kroizman T,
Schilling J, Bar-Sagi D, Lax I and Schlessinger J: A lipid-anchored
Grb2-binding protein that links FGF-receptor activation to the
Ras/MAPK signaling pathway. Cell. 89:693–702. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sternberg PW and Alberola-Ila J:
Conspiracy theory: RAS and RAF do not act alone. Cell. 95:447–450.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Thisse B and Thisse C: Functions and
regulations of fibroblast growth factor signaling during embryonic
development. Dev Biol. 287:390–402. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Turner N and Grose R: Fibroblast growth
factor signalling: From development to cancer. Nat Rev Cancer.
10:116–129. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Haugsten EM, Wiedlocha A, Olsnes S and
Wesche J: Roles of fibroblast growth factor receptors in
carcinogenesis. Mol Cancer Res. 8:1439–1452. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jin YR, Turcotte TJ, Crocker AL, Han XH
and Yoon JK: The canonical Wnt signaling activator, R-spondin2,
regulates cranio-facial patterning and morphogenesis within the
branchial arch through ectodermal-mesenchymal interaction. Dev
Biol. 352:1–13. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu J, Liu H, Lan Y, Aronow BJ,
Kalinichenko VV and Jiang R: A Shh-Foxf-Fgf18-Shh molecular circuit
regulating palate development. PLoS Genet. 12:e10057692016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Laestander C and Engström W: Role of
fibroblast growth factors in elicitation of cell responses. Cell
Prolif. 47:3–11. 2014. View Article : Google Scholar
|
|
28
|
Jaskoll T, Witcher D, Toreno L, Bringas P,
Moon AM and Melnick M: FGF8 dose-dependent regulation of embryonic
submandibular salivary gland morphogenesis. Dev Biol. 268:457–469.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cormier S, Leroy C, Delezoide AL and Silve
C: Expression of fibroblast growth factors 18 and 23 during human
embryonic and fetal development. Gene Expr Patterns. 5:569–573.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Itoh N and Ornitz DM: Fibroblast growth
factors: From molecular evolution to roles in development,
metabolism and disease. J Biochem. 149:121–130. 2011. View Article : Google Scholar :
|
|
31
|
Nie X, Luukko K and Kettunen P: FGF
signalling in craniofacial development and developmental disorders.
Oral Dis. 12:102–111. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Trumpp A, Depew MJ, Rubenstein JL, Bishop
JM and Martin GR: Cre-mediated gene inactivation demonstrates that
FGF8 is required for cell survival and patterning of the first
branchial arch. Genes Dev. 13:3136–3148. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Haworth KE, Wilson JM, Grevellec A,
Cobourne MT, Healy C, Helms JA, Sharpe PT and Tucker AS: Sonic
hedgehog in the pharyngeal endoderm controls arch pattern via
regulation of Fgf8 in head ectoderm. Dev Biol. 303:244–258. 2007.
View Article : Google Scholar
|
|
34
|
Schmotzer CL and Shehata BM: Two cases of
agnathia (otocephaly): With review of the role of fibroblast growth
factor (FGF8) and bone morphogenetic protein (BMP4) in patterning
of the first branchial arch. Pediatr Dev Pathol. 11:321–324. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Du W, Prochazka J, Prochazkova M and Klein
OD: Expression of FGFs during early mouse tongue development. Gene
Expr Patterns. 20:81–87. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Swarup N, Nayak MT, Chowdhary Z,
Mehendiratta M, Khatana S, Choi SJ and Sagolsem C: Evaluation and
immunolocalization of BMP4 and FGF8 in odontogenic cyst and tumors.
Anal Cell Pathol. 2018:12045492018. View Article : Google Scholar
|
|
37
|
Li CY, Prochazka J, Goodwin AF and Klein
OD: Fibroblast growth factor signaling in mammalian tooth
development. Odontology. 102:1–13. 2014. View Article : Google Scholar
|
|
38
|
Jernvall J and Thesleff I: Reiterative
signaling and patterning during mammalian tooth morphogenesis. Mech
Dev. 92:19–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Baba O, Ota MS, Terashima T, Tabata MJ and
Takano Y: Expression of transcripts for fibroblast growth factor 18
and its possible receptors during postnatal dentin formation in rat
molars. Odontology. 103:136–142. 2015. View Article : Google Scholar
|
|
40
|
St Amand TR, Zhang Y, Semina EV, Zhao X,
Hu Y, Nguyen L, Murray JC and Chen Y: Antagonistic signals between
BMP4 and FGF8 define the expression of Pitx1 and Pitx2 in mouse
tooth-forming anlage. Dev Biol. 217:323–332. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mucchielli ML, Mitsiadis TA, Raffo S,
Brunet JF, Proust JP and Goridis C: Mouse Otlx2/RIEG expression in
the odontogenic epithelium precedes tooth initiation and requires
mesenchyme-derived signals for its maintenance. Dev Biol.
189:275–284. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tucker AS, Matthews KL and Sharpe PT:
Transformation of tooth type induced by inhibition of BMP
signaling. Science. 282:1136–1138. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lu MF, Pressman C, Dyer R, Johnson RL and
Martin JF: Function of Rieger syndrome gene in left-right asymmetry
and craniofacial development. Nature. 401:276–278. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lin CR, Kioussi C, O'Connell S, Briata P,
Szeto D, Liu F, Izpisúa-Belmonte JC and Rosenfeld MG: Pitx2
regulates lung asymmetry, cardiac positioning and pituitary and
tooth morphogenesis. Nature. 401:279–282. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tsikandelova R, Mladenov P, Planchon S,
Kalenderova S, Praskova M, Mihaylova Z, Stanimirov P, Mitev V,
Renaut J and Ishkitiev N: Proteome response of dental pulp cells to
exogenous FGF8. J Proteomics. 183:14–24. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shao M, Liu C, Song Y, Ye W, He W, Yuan G,
Gu S, Lin C, Ma L, Zhang Y, et al: FGF8 signaling sustains
progenitor status and multipotency of cranial neural crest-derived
mesenchymal cells in vivo and in vitro. J Mol Cell Biol. 7:441–454.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lin D, Huang Y, He F, Gu S, Zhang G, Chen
Y and Zhang Y: Expression survey of genes critical for tooth
development in the human embryonic tooth germ. Dev Dyn.
236:1307–1312. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Porntaveetus T, Otsuka-Tanaka Y, Basson
MA, Moon AM, Sharpe PT and Ohazama A: Expression of fibroblast
growth factors (Fgfs) in murine tooth development. J Anat.
218:534–543. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Agarwal A, Gundappa M, Miglani S and Nagar
R: Asyndromic hypodontia associated with tooth morphology
alteration: A rare case report. J Conserv Dent. 16:269–271. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Neubüser A, Peters H, Balling R and Martin
GR: Antagonistic interactions between FGF and BMP signaling
pathways: A mechanism for positioning the sites of tooth formation.
Cell. 90:247–255. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tucker AS, Yamada G, Grigoriou M, Pachnis
V and Sharpe PT: Fgf-8 determines rostral-caudal polarity in the
first branchial arch. Development. 126:51–61. 1999.
|
|
52
|
Bae JM, Clarke JC, Rashid H, Adhami MD,
McCullough K, Scott JS, Chen H, Sinha KM, de Crombrugghe B and
Javed A: Specificity protein 7 is required for proliferation and
differentiation of ameloblasts and odontoblasts. J Bone Miner Res.
33:1126–1140. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Jung HS, Oropeza V and Thesleff I: Shh,
Bmp-2, Bmp-4 and Fgf-8 are associated with initiation and
patterning of mouse tongue papillae. Mech Dev. 81:179–182. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Paulson RB, Hayes TG and Sucheston ME:
Scanning electron microscope study of tongue development in the
CD-1 mouse fetus. J Craniofac Genet Dev Biol. 5:59–73.
1985.PubMed/NCBI
|
|
55
|
Nagata J and Yamane A: Progress of cell
proliferation in striated muscle tissues during development of the
mouse tongue. J Dent Res. 83:926–929. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Nie X: Apoptosis, proliferation and gene
expression patterns in mouse developing tongue. Anat Embryol
(Berl). 210:125–132. 2005. View Article : Google Scholar
|
|
57
|
Liu Z, Xu J, Colvin JS and Ornitz DM:
Coordination of chondrogenesis and osteogenesis by fibroblast
growth factor 18. Genes Dev. 16:859–869. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ohbayashi N, Shibayama M, Kurotaki Y,
Imanishi M, Fujimori T, Itoh N and Takada S: FGF18 is required for
normal cell proliferation and differentiation during osteogenesis
and chondrogenesis. Genes Dev. 16:870–879. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ellsworth JL, Berry J, Bukowski T, Claus
J, Feldhaus A, Holderman S, Holdren MS, Lum KD, Moore EE, Raymond
F, et al: Fibroblast growth factor-18 is a trophic factor for
mature chondrocytes and their progenitors. Osteoarthritis
Cartilage. 10:308–320. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
MacArthur CA, Lawshé A, Xu J,
Santos-Ocampo S, Heikinheimo M, Chellaiah AT and Ornitz DM: FGF-8
isoforms activate receptor splice forms that are expressed in
mesenchymal regions of mouse development. Development.
121:3603–3613. 1995.PubMed/NCBI
|
|
61
|
Jaskoll T and Melnick M: Embryonic
salivary gland branching morphogenesis. Branching Morphogenesis.
Springer; Boston, MA: pp. 160–175. 2011
|
|
62
|
Kang Y and Massagué J:
Epithelial-mesenchymal transitions: Twist in development and
metastasis. Cell. 118:277–279. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Tanaka A, Furuya A, Yamasaki M, Hanai N,
Kuriki K, Kamiakito T, Kobayashi Y, Yoshida H, Koike M and Fukayama
M: High frequency of fibroblast growth factor (FGF) 8 expression in
clinical prostate cancers and breast tissues, immunohistochemically
demonstrated by a newly established neutralizing monoclonal
antibody against FGF 8. Cancer Res. 58:2053–2056. 1998.PubMed/NCBI
|
|
64
|
Ishibe T, Nakayama T, Okamoto T, Aoyama T,
Nishijo K, Shibata KR, Shima Y, Nagayama S, Katagiri T, Nakamura Y,
et al: Disruption of fibroblast growth factor signal pathway
inhibits the growth of synovial sarcomas: Potential application of
signal inhibitors to molecular target therapy. Clin Cancer Res.
11:2702–2712. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Weng M and Chen Z, Xiao Q, Li R and Chen
Z: A review of FGF signaling in palate development. Biomed
Pharmacother. 103:240–247. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Goudy S, Law A, Sanchez G, Baldwin HS and
Brown C: Tbx1 is necessary for palatal elongation and elevation.
Mech Dev. 127:292–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Riley BM, Mansilla MA, Ma J, Daack-Hirsch
S, Maher BS, Raffensperger LM, Russo ET, Vieira AR, Dodé C,
Mohammadi M, et al: Impaired FGF signaling contributes to cleft lip
and palate. Proc Natl Acad Sci USA. 104:4512–4517. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Arnold JS, Werling U, Braunstein EM, Liao
J, Nowotschin S, Edelmann W, Hebert JM and Morrow BE: Inactivation
of Tbx1 in the pharyngeal endoderm results in 22q11DS
malformations. Development. 133:977–987. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Juriloff DM and Harris MJ: Mouse genetic
models of cleft lip with or without cleft palate. Birth Defects Res
A Clin Mol Teratol. 82:63–77. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kasberg AD, Brunskill EW and Potter SS:
SP8 regulates signaling centers during craniofacial development.
Dev Biol. 381:312–323. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Rice R, Connor E and Rice DP: Expression
patterns of Hedgehog signalling pathway members during mouse palate
development. Gene Expr Patterns. 6:206–212. 2006. View Article : Google Scholar
|
|
72
|
Taneyhill LA, Hoover-Fong J, Lozanoff S,
Marcucio R, Richtsmeier JT and Trainor PA: The society for
craniofacial genetics and developmental biology 38th annual
meeting. Am J Med Genet A. 170:1732–1753. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Tabler JM, Barrell WB, Szabo-Rogers HL,
Healy C, Yeung Y, Perdiguero EG, Schulz C, Yannakoudakis BZ,
Mesbahi A, Wlodarczyk B, et al: Fuz mutant mice reveal shared
mechanisms between ciliopathies and FGF-related syndromes. Dev
Cell. 25:623–635. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Gray RS, Abitua PB, Wlodarczyk BJ,
Szabo-Rogers HL, Blanchard O, Lee I, Weiss GS, Liu KJ, Marcotte EM,
Wallingford JB, et al: The planar cell polarity effector Fuz is
essential for targeted membrane trafficking, ciliogenesis and mouse
embryonic development. Nat Cell Biol. 11:1225–1232. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ferrante MI, Zullo A, Barra A, Bimonte S,
Messaddeq N, Studer M, Dollé P and Franco B: Oral-facial-digital
type I protein is required for primary cilia formation and
left-right axis specification. Nat Genet. 38:112–117. 2006.
View Article : Google Scholar
|
|
76
|
Stottmann RW, Anderson RM and Klingensmith
J: The BMP antagonists Chordin and Noggin have essential but
redundant roles in mouse mandibular outgrowth. Dev Biol.
240:457–473. 2001. View Article : Google Scholar
|
|
77
|
Liu W, Selever J, Murali D, Sun X, Brugger
SM, Ma L, Schwartz RJ, Maxson R, Furuta Y and Martin JF:
Threshold-specific requirements for Bmp4 in mandibular development.
Dev Biol. 283:282–293. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Mackenzie BA, Wolff R, Lowe N, Billington
CJ Jr, Peterson A, Schmidt B, Graf D, Mina M, Gopalakrishnan R and
Petryk A: Twisted gastrulation limits apoptosis in the distal
region of the mandibular arch in mice. Dev Biol. 328:13–23. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Kumamoto H: Molecular pathology of
odontogenic tumors. Oral Pathol Med. 35:65–74. 2006. View Article : Google Scholar
|
|
80
|
El-Naggar AK, Chan J, Takata T, Grandis JR
and Slootweg PJ: The fourth edition of the head and neck World
Health Organization blue book: Editors' perspectives. Hum Pathol.
66:10–12. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Kallioniemi A: Bone morphogenetic protein
4-a fascinating regulator of cancer cell behavior. Cancer Genet.
205:267–277. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Hallor K, Sciot RJ, Staaf J, Heidenblad M,
Rydholm A, Bauer HC, Aström K, Domanski HA, Meis JM, Kindblom LG,
et al: Two genetic pathways, t (1;10) and amplification of 3p11-12,
in myxoinflammatory fibroblastic sarcoma, haemosiderotic
fibrolipomatous tumour, and morphologically similar lesions. J
Pathol. 217:716–727. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Xie X, Wang Z, Chen F, Yuan Y, Wang J, Liu
R and Chen Q: Roles of FGFR in oral carcinogenesis. Cell Prolif.
49:261–269. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Yuan Y, Xie X, Jiang Y, Wei Z, Wang P,
Chen F, Li X, Sun C, Zhao H, Zeng X, et al: LRP6 is identified as a
potential prognostic marker for oral squamous cell carcinoma via
MALDI-IMS. Cell Death Dis. 8:e3035. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Bryja V, Andersson ER, Schambony A, Esner
M, Bryjová L, Biris KK, Hall AC, Kraft B, Cajanek L, Yamaguchi TP,
et al: The extracellular domain of Lrp5/6 inhibits noncanonical Wnt
signaling in vivo. Mol Biol Cell. 20:924–936. 2009. View Article : Google Scholar :
|
|
86
|
Canning CA, Lee L, Irving C, Mason I and
Jones CM: Sustained interactive Wnt and FGF signaling is required
to maintain isthmic identity. Dev Biol. 305:276–286. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Patel SA, Barnes A, Loftus N, Martin R,
Sloan P, Thakker N and Goodacre R: Imaging mass spectrometry using
chemical inkjet printing reveals differential protein expression in
human oral squamous cell carcinoma. Analyst. 134:301–307. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Lemieux E, Cagnol S, Beaudry K, Carrier J
and Rivard N: Oncogenic KRAS signalling promotes the Wnt/β-catenin
pathway through LRP6 in colorectal cancer. Oncogene. 34:4914–4927.
2015. View Article : Google Scholar
|
|
89
|
Bryja V, Andersson ER, Schambony A, et al:
The extracellular domain of Lrp5/6 inhibits noncanonical Wnt
signaling in vivo. Mol Biol Cell. 20:924–936. 2009. View Article : Google Scholar :
|
|
90
|
Guo Y, Ren MS, Shang C, Zhu L and Zhong M:
MTSS1 gene regulated by miR-96 inhibits cell proliferation and
metastasis in tongue squamous cellular carcinoma Tca8113 cell line.
Int J Clin Exp Med. 8:15441–15449. 2015.PubMed/NCBI
|
|
91
|
Ceasar SA, Rajan V, Prykhozhij SV, Berman
JN and Ignacimuthu S: Insert, remove or replace: A highly advanced
genome editing system using CRISPR/Cas9. Biochim Biophys Acta.
1863:2333–2344. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Square T, Romášek M, Jandzik D, Cattell
MV, Klymkowsky M and Medeiros DM: CRISPR/Cas9-mediated mutagenesis
in the sea lamprey, Petromyzon marinus: a powerful tool for
understanding ancestral gene functions in vertebrates. Development.
142:4180–4187. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Webber BL, Raghu S and Edwards OR:
Opinion: Is CRISPR-based gene drive a biocontrol silver bullet or
global conservation threat. Proc Natl Acad Sci USA.
112:10565–10567. 2015. View Article : Google Scholar
|