Introduction
Cancer upregulated gene 2 (CUG2), a candidate
oncogene, is commonly upregulated in various types of cancer,
including ovarian, liver, colon and lung cancers, and plays a
crucial role in tumorigenesis (1).
Some studies have identified CUG2 as a novel centromeric component
required for appropriate kinetochore functioning during cell
division (2,3). In a previous study, in an NIH3T3 cell
transplantation model, CUG2 was found to exert an oncogenic effect
similar to that of mutant Ras (1).
CUG2 overexpression activates Ras and mitogen-activated protein
kinases (MAPKs), including p38 MAPK, which facilitates oncolytic
retroviral replication (4). CUG2
has been suggested to induce epithelial-mesenchymal transition
(EMT) through transforming growth factor (TGF)-β signaling
(5). A recent study reported that
CUG2 enhanced epidermal growth factor receptor (EGFR) expression to
induce doxorubicin resistance by activating the signal transducer
and activator of transcription 1 (Stat1)/histone deacetylase 4
(HDAC4) signaling axis, which involves the TGF-β signaling pathway
(6).
The Wnt/β-catenin signaling pathway plays an
important role in cell proliferation, differentiation and
oncogenesis (7,8). Particularly, the abnormal
upregulation of Wnt/β-catenin activity is frequently detected as an
early event in a number of types of cancer (9). The Wnt signal is initiated by the
interaction of Wnt proteins (Wnt1, Wnt3a and Wnt8) with the
Frizzled receptor and low-density lipoprotein receptor-related
protein 5/6 coreceptors (9). This
signal is then transduced through disheveled protein to negatively
regulate glycogen synthase kinase 3β (GSK3β), which results in the
cytoplasmic accumulation of β-catenin. The accumulated β-catenin is
then translocated into the nucleus where it complexes with T cell
factor/lymphocyte enhancer factor (TCF/LEF) family of transcription
factors to activate the expression of β-catenin-responsive genes
such as cyclin D1, c-Jun, c-Myc and
peroxisome proliferator-activated receptor-δ (10-13).
A number of types of cancer exhibit the accumulation of β-catenin
and the consequent activation of TCF/LEF-dependent gene
transcription (14-16).
In quiescent cells, β-catenin is maintained in the
cytoplasm at low levels. This is facilitated by its interaction
with scaffolding proteins, such as adenomatous polyposis coli and
axin, and with protein kinases, such as casein kinase 1a and GSK3β,
which phosphorylate β-catenin at Ser45 and Ser33/Ser37/Thr41,
respectively, leading to its ubiquitination and proteasomal
degradation (17-19). Wnt and other growth stimuli induce
GSK3β phosphorylation, resulting in the inactivation of β-catenin
phosphorylation at Ser33/Ser37/Thr41, its stabilization, and its
subsequent translocation to the nucleus (20). Previous studies have demonstrated
that protein kinase A (PKA) also stabilizes β-catenin by
phosphorylating it at Ser675 (21,22).
The present study examined whether the
overexpression of CUG2, a novel oncogene, affects the Wnt/β-catenin
signaling pathway, which is essential for tumorigenesis. We found
that CUG2 overexpression increased β-catenin activity and
stability, which was regulated by never in mitosis gene A-related
kinase 2 (NEK2). Treatment with CGK062 targeting β-catenin through
PKCα inhibited CUG2-induced cancer stem cell (CSC)-like phenotypes,
thus impairing tumor formation in vivo. Taken together, the
findings of this study provide new evidence to suggest that CUG2
overexpression contributes to tumor formation through
NEK2/β-catenin signaling.
Materials and methods
Cell culture and CUG2 plasmid
construction
Human lung cancer-derived A549 cells were obtained
from the American Type Culture Collection (ATCC, Manassas, VA,
USA). As previously described (1),
cDNA of CUG2 was inserted into the pCDNA3.1/Myc-His vector
(Invitrogen/Thermo Fisher Scientific, Waltham, MA, USA) using the
BamHI and XhoI sites. A549 parental cells was
transfected with pCDNA3.1/Myc-His vector or pCDNA3.1/Myc-His-CUG2
using Lipofectamine 2000 (Invtrogen/Thermo Fisher Scentific). Cells
stably expressing the vector alone (A549-VEC) or wild-type CUG2
(A549-CUG2) were selected under 1,000 µg/ml G418
(Sigma-Aldrich, St. Louis, MO, USA) for 30 days and maintained in
RPMI-1640 medium supplemented with 10% FBS, 1% penicillin, 1%
streptomycin and 500 µg/ml G418 at 37°C in a humidified
atmosphere of 5% CO2.
Reagents and antibodies
Anti-NEK2 antibodies (610593) for used in western
blot analysis and immunofluorescence microscopywere purchased from
BD Biosciences (San Jose, CA, USA) and those (sc-55601) for
performing NEK2 kinase assay were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). Anti-ALDH1 antibodies (611194)
were acquired from BD Biosciences. Antibodies against Smad2/3
(#5678), phosphorylated Smad2 (#3108), β-catenin (#9562), and
phosphorylated β-catenin (Ser33/Ser37/Thr41, Ser45, or Ser675;
#9561, #9564, or #4176, respectively) were purchased from Cell
Signaling Biotechnology (Danvers, MA, USA). Antibodies against
E-cadherin (ab15148), N-cadherin (ab18203), vimentin, Snail
(ab180714), Twist (ab175430), Bmi1 (ab126783), Sox2 (ab97959),
octamer-binding transcription factor 4 (Oct4; ab109183),
Kruppel-like factor 4 (Klf4; ab129473) and Nanog (ab109250) were
obtained from Abcam (Cambridge, MA, USA). Anti-β-actin antibody
(sc-4778) and anti-Sp1 antibody (sc-17824) were obtained from Santa
Cruz Biotechnology, and wortmannin, MG132, H89, PP2 and
bisindolylmaleimide I (BMI) were purchased from Calbiochem (San
Diego, CA, USA). Wnt3a was purchased from R&D Systems Inc.
(Minneapolis, MN, USA). CGK062 was prepared as previously described
(23).
Cellular fractionation
As previously described (24), cells cultured in 100-mm plates were
washed and harvested with ice-cold PBS and cell pellets were lysed
in 800 µl TTN buffer [20 mM Tris-HCl (pH 7.4), 0.05% Triton
X-100, 150 mM NaCl, 1 mM EDTA, 1 mM DTT, 10% glycerol, 0.5 mM PMSF
and 1X protease inhibitor cocktail] on ice for 20 min, followed by
centrifugation at 10,000 × g for 15 min. The supernatant obtained
was used as a soluble fraction. Pellets, which were used as an
insoluble fraction, were solubilized in 800 µl RIPA buffer
[50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1 mM DTT, 1%
NP-40, 0.5% deoxycholic acid, 0.1% SDS, 10% glycerol, 0.5 mM PMSF
and 1X protease inhibitor cocktail] on ice for 30 min and were
centrifuged at 12,000 × g for 15 min. Thereafter, the supernatants
were used for the nuclear extracts.
Western blot analysis and
immunoprecipitation
Cells were harvested and lysed in a lysis buffer
containing 1% NP-40 and protease inhibitors (Sigma-Aldrich). For
performing western blot analysis, proteins from whole cell lysates
were resolved by performing SDS-PAGE (10% or 12% gel) and were
transferred onto nitrocellulose membranes. Primary antibodies were
used at a dilution of 1:1,000 or 1:2,000, and horseradish
peroxidase-conjugated goat anti-mouse (12-349; Merck-Millipore,
Billerica, MA, USA) or anti-rabbit (12-348; Merck-Millipore)
antibodies were used at a dilution of 1:2,000 in 5% non-fat dry
milk. After washing, the membranes were examined by performing an
enhanced chemiluminescence assay by using Image Quant LAS 4000 Mini
(GE-Healthcare, Tokyo, Japan). For immunoprecipitation, cells were
harvested after 48 h of transfection, and the cell debris was
removed by centrifugation at 10,000 × g for 10 min at 4°C. Cell
lysates were pre-cleared with 25 µl of protein A/G agarose
and incubated with the appropriate primary antibody and protein A/G
agarose for 1 h at 4°C. Following 3 washes with lysis buffer, the
precipitates were resolved on SDS-PAGE gels and analyzed by
immunoblotting with the appropriate antibodies.
siRNA transfection
Before performing siRNA transfection, the cells were
trypsinized and cultured overnight to achieve 60-70% confluence.
Subsequently, the cells were incubated with a transfection mixture
containing premade β-catenin, NEK2 and GSK3β and PKCα siRNAs
(Bioneer, Daejeon, Korea) or a negative control siRNA (Bioneer) and
Lipofectamine 2000 (Invitrogen/Thermo Fisher Scientific) for 6 h,
rinsed with a medium containing 10% FBS, and incubated at 37°C for
48 h before harvesting. To confirm the effect of the silencing
protein levels, two types of premade siRNAs were used.
Reverse transcription-polymerase chain
reaction (RT-PCR)
For performing RT-PCR, total RNA was extracted from
the cells using an RNeasy protect cell mini kit (Qiagen, Valencia,
CA, USA), in accordance with the manufacturer’s instructions.
Subsequently, 3 µg total RNA were reversed transcribed into
cDNA using Superscript II reverse transcriptase (Invitrogen/Thermo
Fisher Scientific). PCR was performed using specific primers
described elsewhere (25,26). cDNAs obtained for each sample were
diluted, and PCR was performed using an optimized cycle number.
β-actin mRNA was used as an internal standard. The relative ratio
of β-catenin and β-actin mRNA levels was expressed after measuring
band intensity with Multi Gauge Ver. 2.1 (Fuji Photo Film,
Japan).
Immunofluorescence microscopy
For performing immunofluorescence microscopy, cells
grown on coverslips were fixed with 4% paraformaldehyde for 15 min,
permeabilized with cold acetone for 15 min, blocked with 10% goat
serum for 30 min, and treated with anti-NEK2 and
anti-phosphorylated β-catenin antibodies (Ser33/Ser37/Thr41;
dilution, 1:100) for 30 min at room temperature. Following
incubation, the cells were washed extensively with PBS, incubated
with Alexa Fluor 488-conjugated goat anti-rabbit antibody (A11008)
and Alexa Fluor 594-conjugated goat anti-mouse antibody (A11005)
(dilution, 1:500; Thermo Fisher Scientific), respectively, in PBS
for 30 min at room temperature; and washed 3 times with PBS. For
performing nuclear staining, the cells were incubated with
4′,6-diamidino-2-phenylindole (DAPI) for 5 min in the dark and were
washed 3 times with PBS. Subsequently, cover-slips with stained
cells were mounted on slides by using PBS containing 10% glycerol
and were imaged using a fluorescence microscope (Zeiss Axio
Observer D1, Oberkochen, German).
Luciferase reporter assays
A549-CUG2 cells were transfected with TGF-β promoter
(phTG5 and 7) (27), Top-Flash
(containing 2 sets of 3 copies of the TCF binding sites), or
Fop-Flash (carrying the mutated TCF binding sites) vectors using
Lipofectamine 2000. To normalize the transfection efficiency, a
pGK-βgal vector expressing β-galactosidase under the control of the
phosphoglucokinase promoter was included in the transfection
mixture. At 48 h after the transfection, the cells were washed with
cold PBS and lysed in a lysis solution [25 mM Tris (pH 7.8), 2 mM
EDTA, 2 mM DTT, 10% glycerol and 1% Triton X-100] and the
luciferase activity was measured using a luminometer and luciferase
kit (Promega, Madison, WI, USA).
Invasion assay
Invasion assays were performed using 48-well Boyden
chambers (Neuroprobe, Gaithersburg, MD, USA), as previously
described (28). Lower wells of
the chamber were filled with a standard culture medium. The chamber
was assembled using polycarbonate filters (Neuroprobe) coated with
Matrigel. Cells cultured in a serum-free medium (5×104
cells/well) were seeded in the upper compartment of the chamber and
were incubated at 37°C for 24 h. The cells that invaded through the
membrane were fixed with methanol (cat. no. 34860; Sigma-Aldrich)
for 10 min at room temperature, followed by staining with
hematoxylin (cat. no. HHS16, Sigma-Aldrich) for 10 min.
Subsequently, the cells were counterstained with eosin (cat. no.
HT110132; Sigma-Aldrich) for 15 sec. Cell migration was quantified
by counting the number of migrated cells under a phase-contrast
microscope (CKX31-11PHP; Olympus, Tokyo, Japan).
Wound healing assay
Cell migration was assessed by performing a wound
healing assay, as previously described (29). Briefly, the cells were cultured in
6-well plates (5×105 cells/well). When the cells reached
90% confluence, a single wound was created in the center of the
cell monolayer using a P-200 pipette tip. At 0 and 24 h following
incubation at 37°C, wound closure areas were visualized using a
phase-contrast microscope (CKX31-11 PHP; Olympus) at ×100
magnification.
Sphere formation assay
Cells were cultured in 24-well ultra-low attachment
plates in a serum-free medium supplemented with 5 µg/ml
insulin, 0.4% bovine serum albumin, 10 ng/ml basic fibroblast
growth factor and 20 ng/ml human recombinant EGF for 6 days. The
size and number of spheroids formed were analyzed using a light
microscope (CKX31-11 PHP; Olympus). Sphere formation was determined
by counting the number of spheroids with a size of >50 µm
using NIS-Element F3.0 program (Nikon, Tokyo, Japan).
NEK2 kinase assay
Cell lysates were pre-cleared with 25 µl
protein A/G agarose and were incubated with anti-NEK2 antibody and
protein A/G agarose for 1 h at 4°C. After washing 3 times with a
lysis buffer, precipitates obtained were used for detecting NEK2. A
reaction mixture containing the NEK2-containing immunocomplex (15
µl), recombinant GST-β-catenin (1 µl, 1 mg/1 ml; Sino
Biological Inc., Beijing, China), 10 mM ATP (2 µl;
Sigma-Aldrich), and reaction buffer [2 µl, 10X; 100 mM
Tris-acetate (pH 7.5), 100 mM MgCl2, 500 mM NaCl and 10
mM DTT] was incubated at 30°C for 1 h. The reaction was terminated
using SDS loading buffer, and the reaction mixture was loaded onto
a 10% SDS-polyacrylamide gel. The phosphorylation of GST-β-catenin
at Ser33/Ser37 was determined by performing western blot analysis
with a corresponding antibody.
Animal experiments
All animal experiments were conducted in accordance
with the Laboratory Animal Resources Guide for the Care and Use of
Laboratory Animals. All animal study protocols were approved by the
Pusan National University Animal Care and Use Committee
(PNU-2017-1541). Balb/C nude mice (12 mice in total; 6 male and 6
female, 4 weeks old, weighing 18-20 g) were purchased from Orient
Bio Inc. (Seongnam, Korea) and were maintained under specific
pathogen-free conditions (a 12-h light/12-h dark cycle, at 22°C,
and 50-55% humidity with free access to diet and tap water).
Subsequently, A549-CUG2 cells (1×106 cells/mouse, 0.1 ml
suspension in 50% PBS and 50% Matrigel; Corning, Bedford, MA, USA)
were subcutaneously injected into the right flanks of the mice, and
the mice were divided into 2 groups (6 mice/group). At 3 days
following transplantation, the mice were intraperitoneally injected
with CGK062 (100 mg/kg body weight), and tumor formation and body
weight were monitored for 30 days. As mock treatment, DMSO (25
µl) dissolved in PBS (125 µl) was used for every
injection. The tumor volume was calculated weekly using the
following formula: V (mm3) = 0.5 × length (mm) ×
width2 (mm2).
TUNEL assay
The mice were euthanized, and their tumor tissues
were fixed in 4% paraformaldehyde in PBS, dehydrated using ethanol
and xylene, and embedded in paraffin. Subsequently, the
paraffin-embedded tissues were cut into 5-µm-thick sections,
mounted on poly-L-lysine-coated glass slides and analyzed using the
ApopAlert DNA fragment kit (Takara, Mountain View, CA, USA),
according to the manufacturer’s instructions. TUNEL assay involves
the catalytic addition of green fluorescein-labeled dUTPs to the
3′-OH end of a DNA fragment by using a terminal deoxynucleotidyl
transferase. The resultant green fluorescein-labeled DNA was
assessed in 10 random fields using a fluorescence microscope (Zeiss
Axio Observer D1, Oberkochen, German).
Statistical analysis
All data are presented as the means ± standard
deviation (SD). Multiple groups were compared by one-way analysis
of variance followed by Dunnett’s post-hoc test and differences
between two groups were analyzed using an unpaired t-test with
GraphPad Prism software. A P-value <0.05 was considered to
indicate a statistically significant difference.
Results
CUG2 upregulates β-catenin, which induces
CSC-like phenotypes in A549 lung cancer cells
As it was well known that Wnt/β-catenin signaling
plays an important role in oncogenesis (7,8), we
examined whether CUG2 overexpression affects Wnt/β-catenin
signaling. We found that human A549 lung cancer cells
constitutively expressing CUG2 (A549-CUG2) (5) exhibited a higher β-catenin expression
in the cytosolic and nuclear fraction than the A549 cells
expressing an empty vector (A549-VEC), although the protein levels
of β-catenin between the A549-CUG2 and A549-VEC cells were
indistinguishable in the whole cell lysates due to the abundance of
β-catenin protein (Fig. 1A). The
A549-CUG2 cells also had higher levels of β-catenin gene
transcripts than the A549-VEC cells (Fig. 1B). We then introduced a Top-Flash
luciferase reporter vector controlled by TCF-4 protein to examine
whether the CUG2-induced increase in β-catenin levels promotes more
interaction between β-catenin and TCF-binding element in the
nucleus. The overexpression of CUG2 enhanced β-catenin-mediated
transcriptional activity, whereas the overexpression of CUG2 and
the Fop-Flash vector carrying a mutant TCF-binding site did not
enhance β-catenin-mediated transcriptional activity (Fig. 1C). Collectively, these results
suggest that CUG2 overexpression induces the transcriptional
activity of β-catenin, increases the level of β-catenin and
activates its downstream targets.
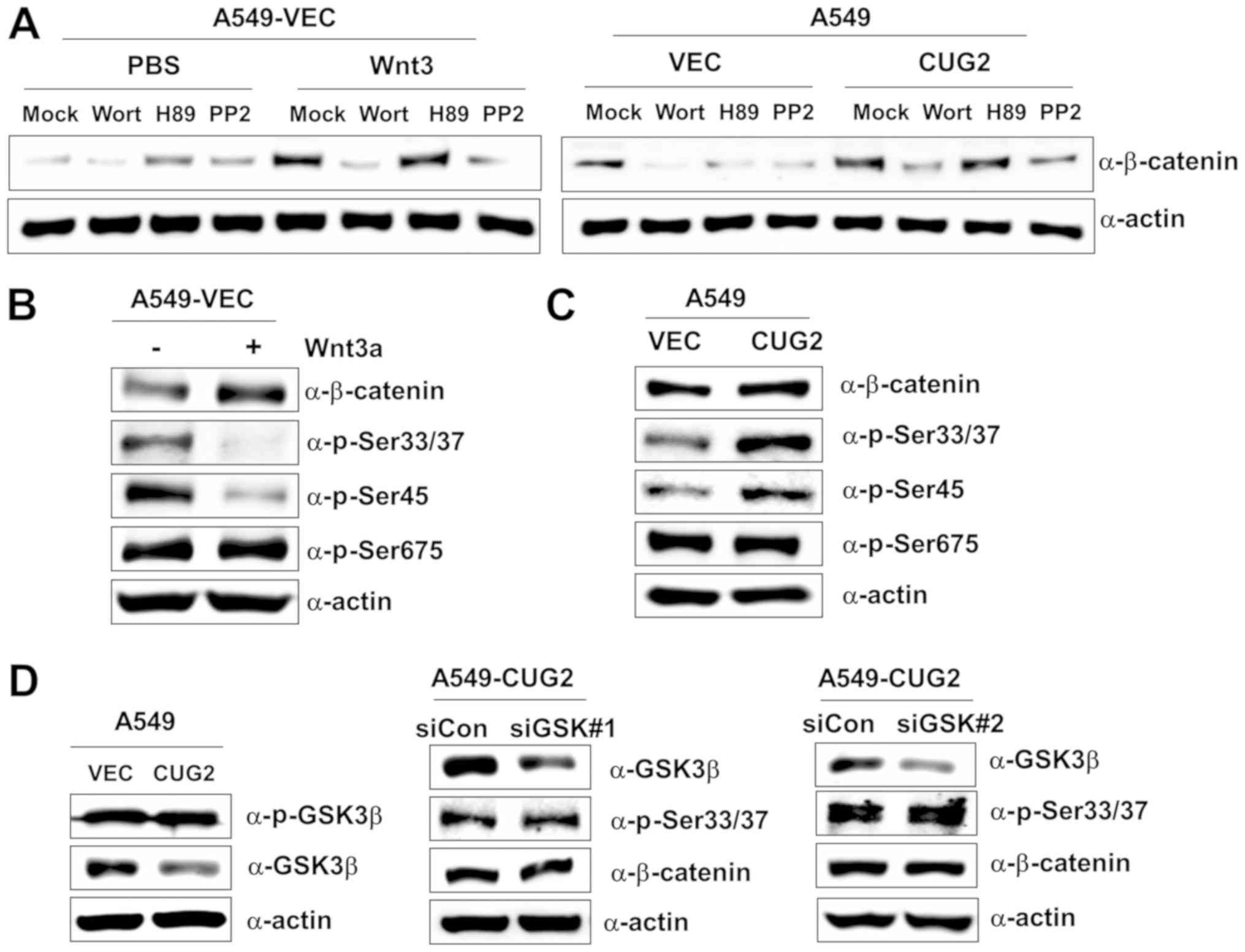
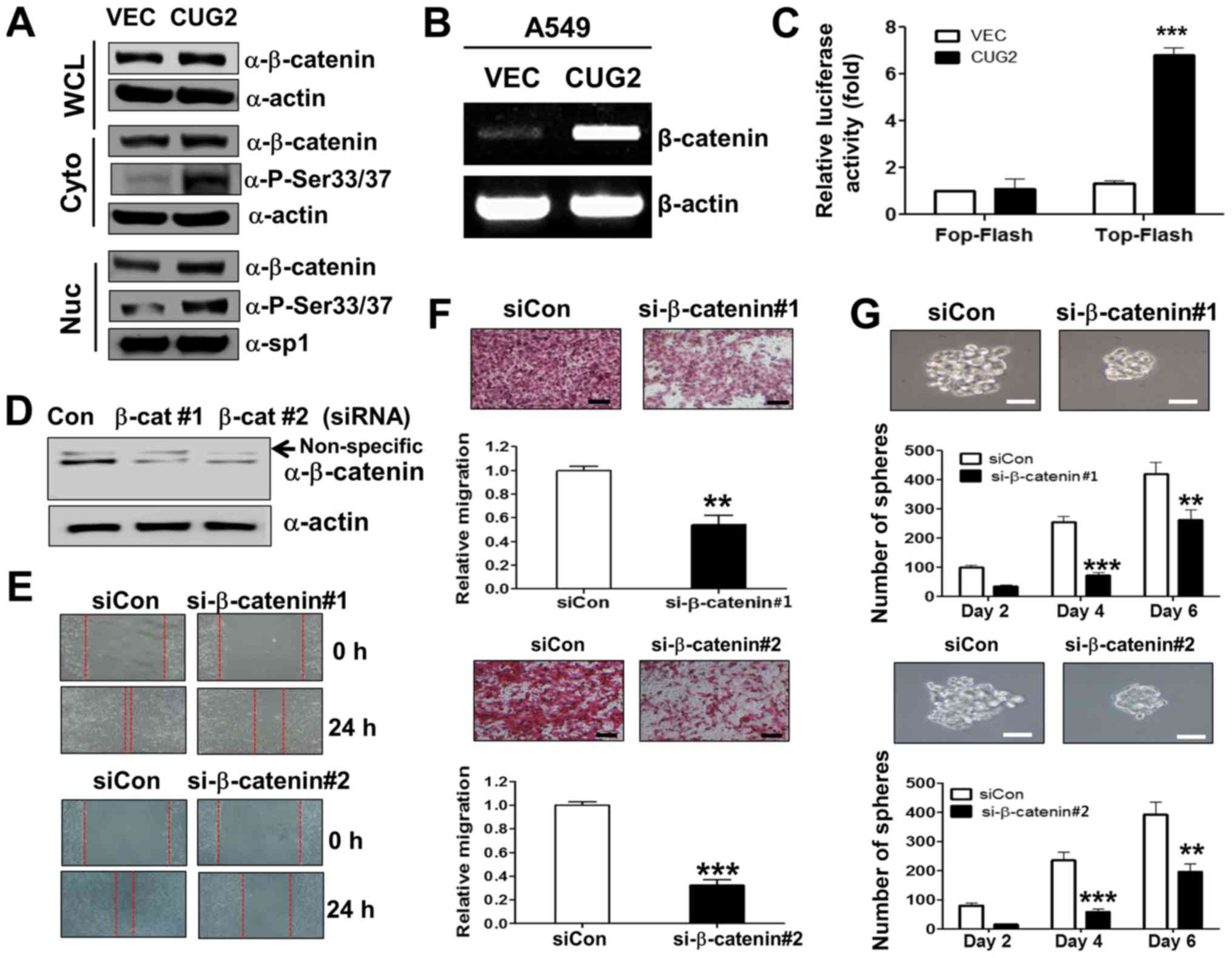 | Figure 1β-catenin is essential for
CUG2-induced CSC-like phenotypes. (A) A549-VEC and A549-CUG2 cells
were separated into cytosolic and nuclear fractions. β-catenin
expression was determined by performing western blot analysis with
an anti-β-catenin antibody. Phosphorylation states of β-catenin
following treatment were detected using antibodies against
Ser33/Ser37/Thr41, Ser45 and Ser675 of β-catenin. The cytosolic and
nuclear fractions were confirmed by detecting actin and Sp1,
respectively. (B) Total RNAs (3 µg) were isolated from the
A549-VECand A549-CUG2 cells, and cDNAs were synthesized using
reverse transcriptase II. β-catenin gene sequences were amplified
using specific primers by using an optimized PCR cycle and were
visualized on 1.5% agarose gels following ethidium bromide
staining. β-actin was used as an internal control. (C) A549-VEC and
A549-CUG2 cells were transfected with the Top-Flash (1 µg)
or Fop-Flash (1 µg) luciferase reporter vector and were
harvested at 48 h after the transfection. Transfection efficiency
was normalized with that of the β-galactosidase reporter vector
pGK-βgal (1 µg) during the measurement of luciferase
activity. Results are an average of three experiments; bars
indicate the means ± SD (***P<0.001, A549-VEC vs
A549-CUG2 cells). (D) Transfection of efficiency of β-catenin
siRNAs (#1 or #2) was confirmed by western blot analysis. (E)
Migration of A549-CUG2 cells was measured with the wound healing
assay at 24 h after control or β-catenin siRNAs (#1 and #2)
transfection. Wound closure areas were monitored using a
phase-contrast microscope at ×100 magnification, and the assay was
repeated twice. (F) Invasion of A549-CUG2 cells transfected with
control or β-catenin siRNAs (#1 and #2) was determined using the
48-well Boyden chamber. The chamber was assembled using
Matrigel-coated polycarbonate filters. Scale bar indicates 100
µm, and the assay was repeated twice. Each assay was
performed in triplicate, and error bars indicate the means ± SD
(***P<0.001, control vs. β-catenin siRNA). (G)
A549-CUG2 cells (1,000 cells per well) transfected with control or
β-catenin siRNAs (#1 and #2) were seeded in ultra-low attachment
plates for 2, 4, or 6 days. The assay was performed in triplicate,
and error bars indicate the means ± SD (**P<0.01 and
***P<0.001, control vs. β-catenin siRNA). Scale bars
indicate 50 µm. CUG2, cancer-upregulated gene 2. |
We then examined whether upregulated β-catenin is
closely associated with CUG2-induced CSC-like phenotypes. For this
purpose, we examined whether the suppression of β-catenin
expression impedes CUG2-induced EMT and sphere formation, which are
the features of CSC phenotypes. We found that β-catenin gene
knockdown using a specific siRNA (#1 or #2; Fig. 1D) reduced the wound healing and
invasive ability of the A549-CUG2 cells compared with that of the
cells transfected with a control siRNA (Fig. 1E and F). This result indicates that
upregulated β-catenin expression is involved in CUG2-induced EMT.
Furthermore, β-catenin silencing using specific siRNAs (#1 or #2)
decreased the number and size of A549-CUG2 cell spheroids compared
with that of the cells transfected with the control siRNA (Fig. 1G). Thus, these results suggest that
β-catenin plays a crucial role in promoting CUG2-induced CSC-like
phenotypes.
The CUG2-induced phosphorylation of
β-catenin differs from that induced by Wnt3a
We then wished to determine whether the
CUG2-mediated upregulation of β-catenin expression emulates
activation patterns induced via the Wnt3a signaling pathway. The
Akt, PKA and Src signaling proteins directly or indirectly regulate
Wnt/β-catenin signaling; thus, we examined their effects on
β-catenin expression in the presence of CUG2 overexpression. The
A549-VEC cells were treated with wortmannin (an Akt inhibitor), H89
(a PKA inhibitor), or PP2 (a Src inhibitor) in the presence of
Wnt3a. Wortmannin and PP2 treatments inhibited β-catenin expression
in the A549-VEC cells, although H89 treatment did not (Fig. 2A). Similarly, wortmannin and PP2
treatments decreased β-catenin expression in the presence of CUG2
overexpression, although H89 treatment did not (Fig. 2A). These results suggest the
presence of shared pathways between Wnt3a- and CUG2-mediated
signaling in regulating β-catenin. To delineate the two signaling
pathways, we examined the phosphorylation pattern of β-catenin.
Wnt3a treatment decreased the phosphorylation of β-catenin at
Ser33/Ser37 and Ser45 compared with mock treatment, although it did
not affect its phosphorylation at Ser675 (Fig. 2B). By contrast, CUG2 overexpression
increased the phosphorylation of β-catenin at Ser33/Ser37 and
Ser45, although it did not affect its phosphorylation at Ser675,
similar to Wnt3a treatment (Fig.
2C). This result indicates the distinct regulation of β-catenin
by the Wnt3a- and CUG2-mediated signaling pathways. As GSK3β
phosphorylates β-catenin at Ser33/Ser37 to promote its degradation
(17-19), we examined the role of GSK3β in the
phosphorylation of β-catenin at Ser33/Ser37 in the A549-CUG2 cells.
We found that the A549-CUG2 cells exhibited a decreased GSK3β
expression compared with the A549-VEC cells, but similar
phosphorylated GSK3β levels (Fig.
2D). Moreover, the suppression of GSK3β expression using a
specific siRNA (#1 or #2) did not affect the levels of β-catenin
and phosphorylation at Ser33/Ser37 in the A549-CUG2 cells (Fig. 2D), indicating that CUG2-induced
β-catenin phosphorylation is independent of GSK3β.
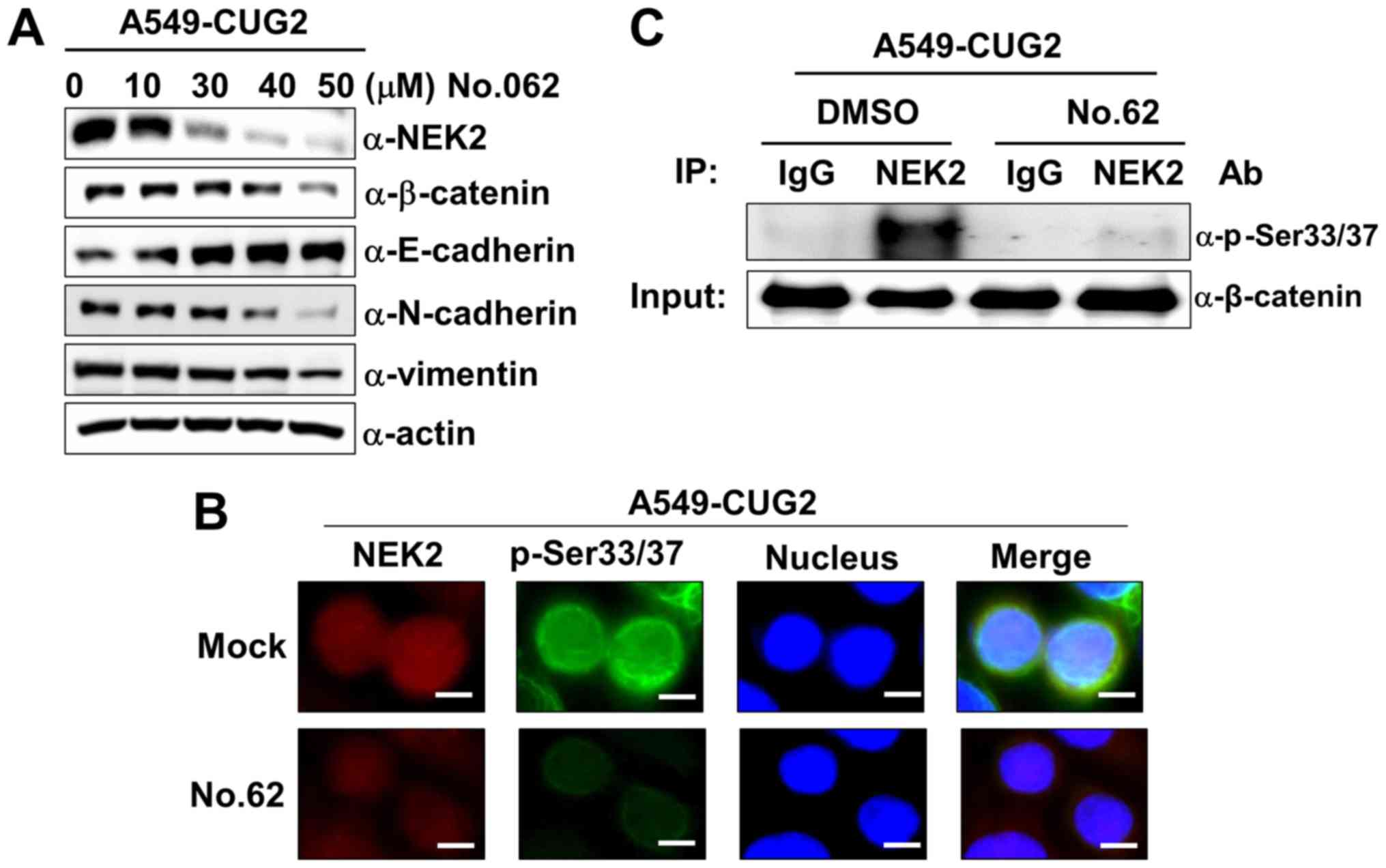
CUG2-mediated NEK2 activation is involved
in the phosphorylation of β-catenin at Ser33/Ser37
Subsequently, when we explored the mechanism
underlying the phosphorylation of β-catenin at Ser33/Ser37 in the
presence of CUG2 overexpression. A previous study reported that
NEK2 phosphorylates β-catenin at the same regulatory sites
(Ser33/Ser37) and binds to β-catenin (30). By preventing E3 ligase β-TrCP from
accessing β-catenin, NEK2 contributes to the stabilization of
β-catenin (30). Thus, in this
study, we measured the NEK2 expression levels in the A549-VEC and
A549-CUG2 cells. The A549-CUG2 cells exhibited higher NEK2 levels
than the A549-VEC cells (Fig. 3A).
To explore the association between NEK2 and β-catenin, we examined
the localization of NEK2 and β-catenin phosphorylated at
Ser33/Ser37 (p-β-catenin), as well as the interaction between NEK2
and β-catenin. Although p-β-catenin was detected in both the
cytoplasm and nucleus, p-β-catenin was preferentially detected in
the nucleus (Fig. 1A). This result
was confirmed by performing immunofluorescence microscopy with
Alexa 488-conjugated anti-p-β-catenin antibody (Fig. 3B). We also found that NEK2
exhibited greater localization to the nucleus than to the
cytoplasm, essentially overlapping with the localization of
p-β-catenin (Fig. 3B). Abundant
NEK2 levels were detected in the immunocomplexes of A549-CUG2 cells
pulled down by using anti-β-catenin antibody, supporting the
co-localization of NEK2 and β-catenin (Fig. 3C). The immunocomplexes that were
pulled down by anti-NEK2 antibody from the lysates of the A549-VEC
or A549-CUG2 cells were incubated with purified GST-β-catenin and
ATP. The lysates of the A549-CUG2 cells exhibited a greater NEK2
kinase activity than those of the A549-VEC cells (Fig. 3D). To confirm the role of NEK2 in
the phosphorylation of β-catenin at Ser33/Ser37, we examined
whether NEK2 silencing indeed decreases the phosphorylation of
β-catenin at Ser33/Ser37. We found that NEK2 silencing using a
specific siRNA (#1 or #2) evidently reduced the phosphorylation of
β-catenin at Ser33/Ser37 and Ser45, but very slightly at Ser675
(Fig. 3E and 3F). NEK2 slightly decreased the level of
total β-catenin (Fig. 3E). Taken
together, these results indicate that CUG2 overexpression increases
the expression and kinase activity of NEK2 responsible for the
phosphorylation of β-catenin at Ser33/Ser37.
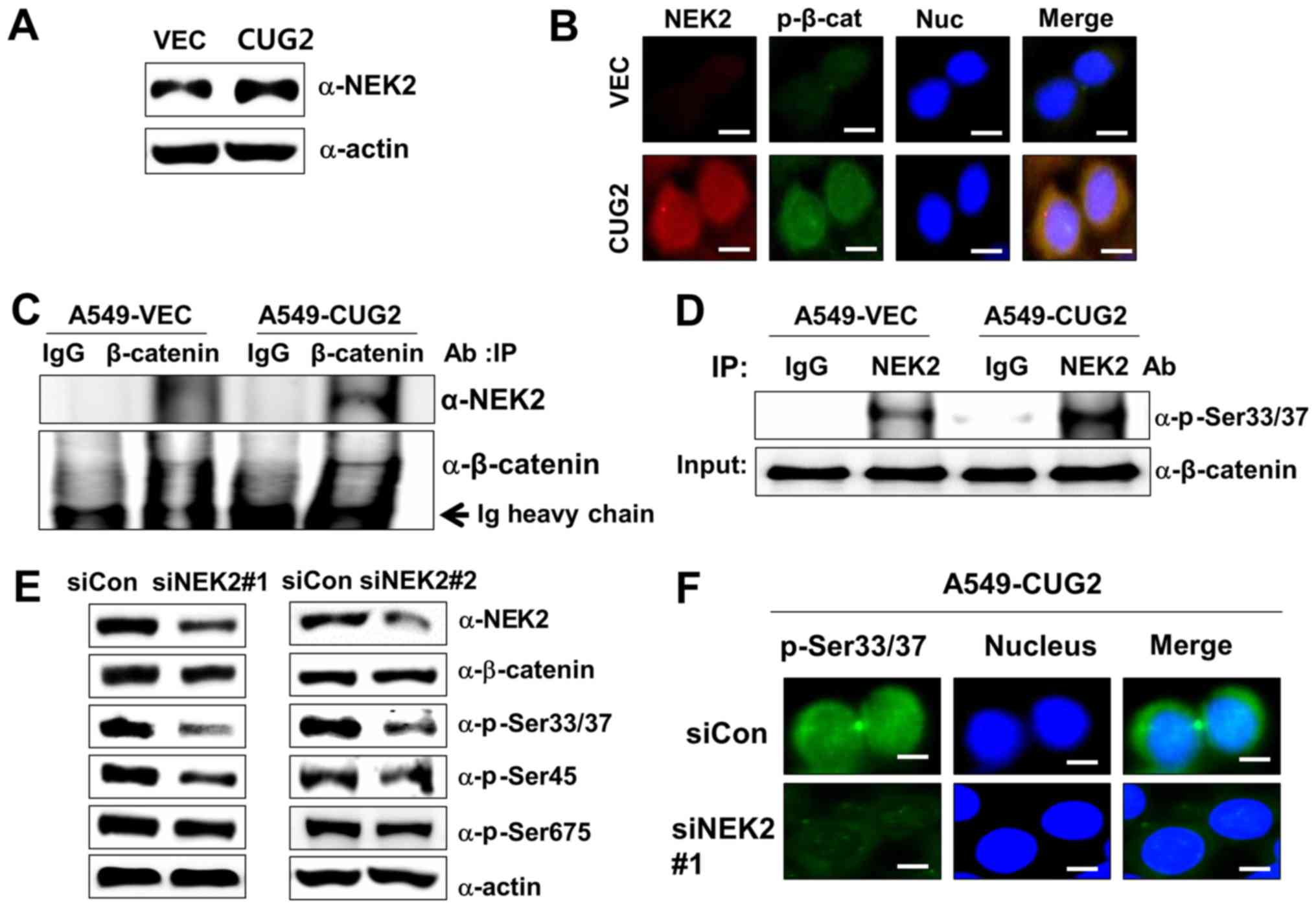 | Figure 3CUG2-activated NEK2 is responsible
for the phosphorylation of β-catenin at Ser33/Ser37. (A) Lysates of
A549-VEC and A549-CUG2 cells were separated by performing SDS-PAGE
on 10% gels, and NEK2 was detected by performing western blot
analysis with anti-NEK2 antibody. (B) Levels of NEK2 and β-catenin
phosphorylated at Ser33/Ser37 (p-β-catenin; p-Ser33/Ser37) in
A549-VEC and A549-CUG2 cells were determined by performing
immunofluorescence microscopy with Alexa Fluor 594-conjugated goat
anti-mouse IgG (red) and Alexa Fluor 488-conjugated goat
anti-rabbit IgG (green), respectively. For performing nuclear
staining, DAPI was added before mounting in glycerol. Scale bar
indicates 10 µm. (C) β-catenin was pulled down from the
lysates of A549-VEC and A549-CUG2 cells by using an anti-β-catenin
antibody. NEK2 present in the immunoprecipitates was detected using
an anti-NEK2 antibody, and β-catenin present in the
immunoprecipitates was detected as a loading control by using an
anti-β-catenin antibody. (D) NEK2 was pulled down from A549-VEC and
A549-CUG2 cells by using an anti-NEK2 antibody or isotype IgG as a
control. The reaction mixture for the NEK2 kinase assay, including
immunoprecipitates as NEK2 kinase, recombinant GST-β-catenin as a
substrate, ATP, and a reaction buffer, was incubated at 30°C for 1
h. NEK2 kinase activity was analyzed by determining the
phosphorylation of GST-β-catenin at Ser33/Ser37 by performing
immunoblotting, and β-catenin levels were examined as a loading
control. (E) Lysates of A549-CUG2 cells transfected with the
control or NEK2 siRNAs (#1 and #2) were separated by performing
SDS-PAGE on 10% gels. Phosphorylation states of β-catenin following
treatment were detected using antibodies against Ser33/Ser37/Thr41,
Ser45 and Ser675 of β-catenin. (F) Following transfection with
control or NEK2 siRNA#1, the levels of p-β-catenin (p-Ser33/Ser37)
in A549-CUG2 cells were detected by performing immunofluorescence
microscopy with Alexa Fluor 488-conjugated goat anti-rabbit IgG
(green). For performing nuclear staining, DAPI was added before
mounting in glycerol. Scale bar indicates 10 µm. CUG2,
cancer-upregulated gene 2; NEK2, never in mitosis gene A-related
kinase 2. |
CGK062 treatment promotes the degradation
of β-catenin through PKCα in the presence of CUG2
overexpression
Our previous study reported that the small chemical
molecule CGK062 activates PKCα, leading to the phosphorylation of
β-catenin at Ser33/Ser37 for its rapid degradation (23). Therefore, in this study, we
examined whether CGK062 induces β-catenin degradation in the
presence of CUG2 overexpression. We found that CGK062 treatment
reduced the protein levels of β-catenin and β-catenin-mediated
promoter activity in a dose-dependent manner (Fig. 4A and B). To determine whether
CGK062 treatment affects the mRNA levels of β-catenin, we performed
RT-PCR. We found that CGK062 did not decrease the mRNA levels of
β-catenin (Fig. 4C). To examine
whether the CGK062-induced β-catenin degradation is dependent on a
proteasome-mediated pathway, we treated the cells with MG132, a
proteasome inhibitor, in the presence of CGK062. CGK062 treatment
alone reduced the β-catenin levels; however, MG132 treatment
prevented the CGK062-mediated β-catenin degradation, suggesting
that the CGK062-induced β-catenin degradation is dependent on a
proteasome-mediated pathway (Fig.
4D). To confirm that PKCα is involved in CGK062-induced
β-catenin degradation, the A549-CUG2 cells were treated with BMI, a
PKC inhibitor, or PKCα siRNA in the presence of CGK062. Treatment
with both BMI and PKCα siRNA (#1 or #2) inhibited the
CGK062-induced β-catenin degradation (Fig. 4E), indicating that CGK062 treatment
suppresses the CUG2-induced enhancement of β-catenin
expression.
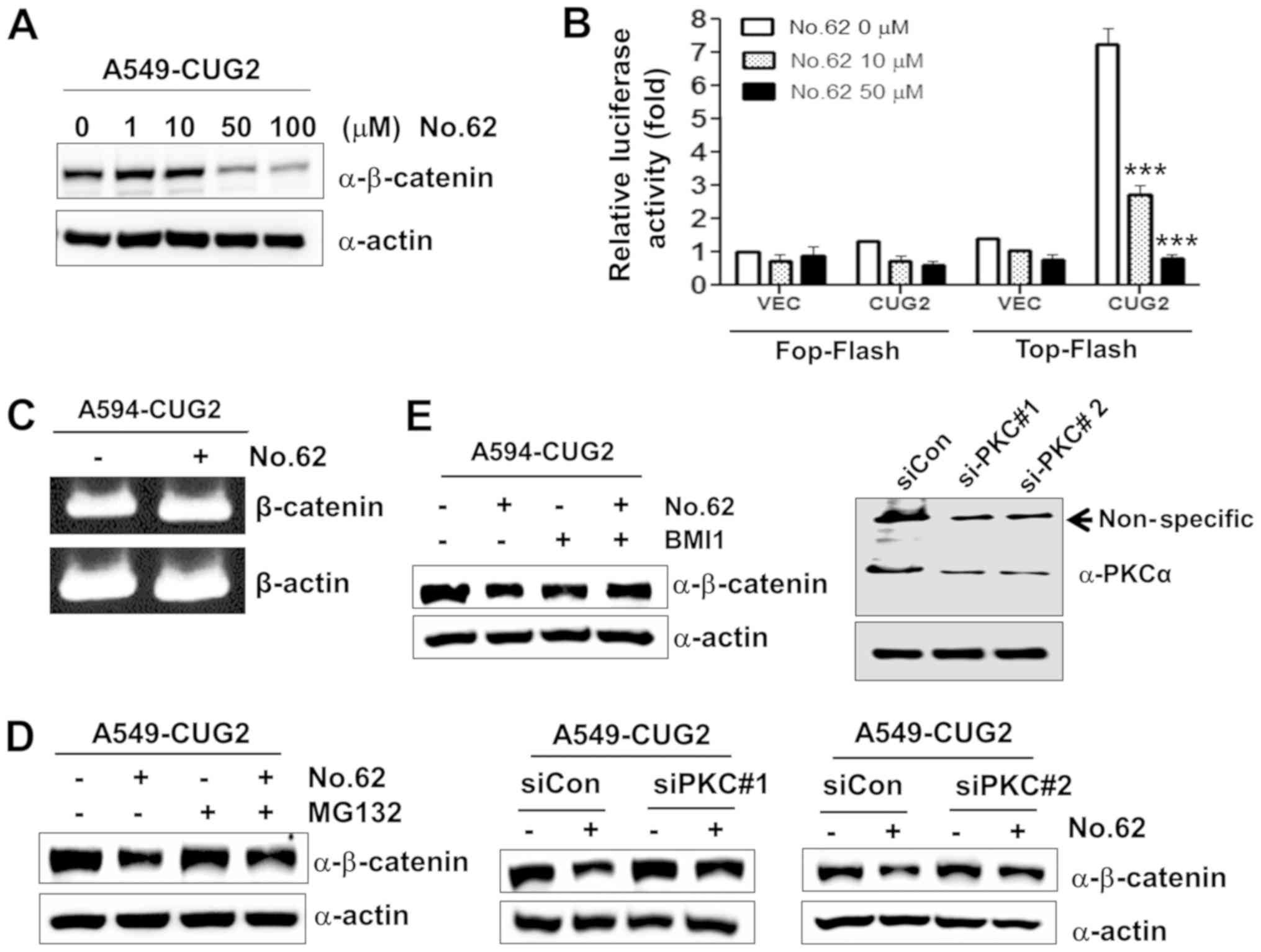 | Figure 4CGK062 treatment destabilizes
β-catenin in a proteasome- and PKCα- dependent manner. (A)
A549-CUG2 cells were treated with CGK062 (0, 1, 10, 50 and 100
µM) for 24 h, and β-catenin levels were measured by
performing western blot analysis with an anti-β-catenin antibody.
(B) A549-CUG2 cells were transfected with the Top-Flash (1
µg) or Fop-Flash (1 µg) luciferase reporter vector in
the presence of CGK062 (50 µM) and were harvested at 48 h
following transfection. Transfection efficiency was normalized with
that of the β-galactosidase reporter vector pGK-βgal (1 µg)
during the measurement of luciferase activity. Results shown are an
average of 3 experiments; bars indicate the means ± SD
(***P<0.001, 0 vs. 10 µM, 0 vs. 50 µM).
(C) Total RNAs (3 µg) were isolated from A549-CUG2 cells
treated with or without CGK062 (50 µM), and cDNAs were
synthesized using reverse transcriptase II. β-catenin gene
sequences were amplified using specific primers by using an
optimized PCR cycle and were visualized on 1.5% agarose gels
following ethidium bromide staining. β-actin was used as an
internal control. (D) A549-CUG2 cells were treated with MG132 (1
µM) for 8 h in the absence or presence of CGK062 (50
µM) and were harvested. β-catenin levels were measured by
performing western blot analysis with an anti-β-catenin antibody.
(E) A549-CUG2 cells were treated with BMI (7.5 µM), a
specific PKCα inhibitor, or were transfected with a control and
PKCα siRNAs (#1 or #2) in the absence or presence of CGK062 (50
µM) for 24 h. Transfection efficiency of PKCα siRNAs (#1 or
#2) was confirmed by western blot analysis. β-catenin levels were
measured by performing western blot analysis with an anti-β-catenin
antibody. CUG2, cancer-upregulated gene 2; PKC, protein kinase
C. |
CGK062 treatment inhibits NEK2 expression
and kinase activity in A549-CUG2 cells
As CGK062 treatment decreased the β-catenin levels,
we wished to examine whether it also affects NEK2 expression in the
presence of CUG2 overexpression. Surprisingly, we found that CGK062
treatment, but not DMSO treatment, reduced NEK2 expression in a
dose-dependent manner in the A549-CUG2 cells (Fig. 5A). The results of
immunofluorescence microscopy also revealed that CGK062 treatment
reduced the p-β-catenin and NEK2 staining levels, supporting the
results of western blot analysis (Fig.
5B). Moreover, CGK062 treatment decreased NEK2 kinase activity
in vitro (Fig. 5C).
Although the mechanisms underlying the effects of CGK062 on NEK2
are unknown, our results indicate that CGK062 affects both
β-catenin and NEK2.
 | Figure 5CGK062 treatment decreases NEK2
expression and kinase activity in A549-CUG2 cells. (A) Lysates of
A549-CUG2 cells treated with CGK062 (0, 10, 30, 40 and 50
µM) for 24 h were separated by performing SDS-PAGE on 10%
gels. The expression of NEK2, β-catenin, E-cadherin, N-cadherin and
vimentin was detected by performing western blot analysis with
corresponding antibodies. (B) Levels of NEK2 and p-β-catenin
(p-Ser33/Ser37) in A549-CUG2 cells treated with CGK062 (50
µM) or DMSO as Mock were detected by performing
immunofluorescence microscopy with Alexa Fluor 594-conjugated goat
anti-mouse IgG (red) and Alexa Fluor 488-conjugated goat
anti-rabbit IgG (green), respectively. For performing nuclear
staining, DAPI was added before mounting in glycerol. Scale bar
indicates 10 µm. (C) NEK2 was pulled down from A549-VEC and
A549-CUG2 cells treated with CGK062 (50 µM) or DMSO by using
the anti-NEK2 antibody or isotype IgG as a control. The reaction
mixture for the NEK2 kinase assay was incubated at 30°C for 1 h.
NEK2 kinase activity was analyzed by detecting the phosphorylation
of GST-β-catenin at Ser33/Ser37 by performing western blot
analysis, and β-catenin levels were examined as a loading control.
CUG2, cancer-upregulated gene 2; NEK2, never in mitosis gene
A-related kinase 2. |
CGK062 treatment inhibits CUG2-induced
CSC-like phenotypes
In light of our above-mentioned finding that
β-catenin is involved in CUG2-induced CSC-like phenotypes (Fig. 1), we examined the hypothesis that
CGK062 treatment impedes the formation of CUG2-induced CSC-like
phenotypes. For this purpose, we evaluated the recovery and
invasion of A549-CUG2 cells after wound induction and found that
CGK062 treatment reduced cell recovery after wound induction and
invasion into the lower wells (Fig. 6A
and B). To determine the CGK062-induced inhibition of these EMT
phenomena, we examined the biochemical features of EMT, such as the
E-cadherin, N-cadherin and vimentin levels, in the A549-CUG2 cells
treated with CGK062. As expected, CGK062 treatment decreased the
N-cadherin and vimentin levels, but increased the E-cadherin
levels, which is opposite of that observed during EMT induction
(Fig. 5A). Moreover, as TGF-β
signaling is closely involved in EMT and Wnt/β-catenin signaling
(31), we examined whether CGK062
treatment impairs TGF-β signaling. We found that CGK062 treatment
decreased phosphorylated the Smad2, Snail and Twist levels
(Fig. 6C), indicating the
existence of a crosstalk between TGF-β and β-catenin signaling.
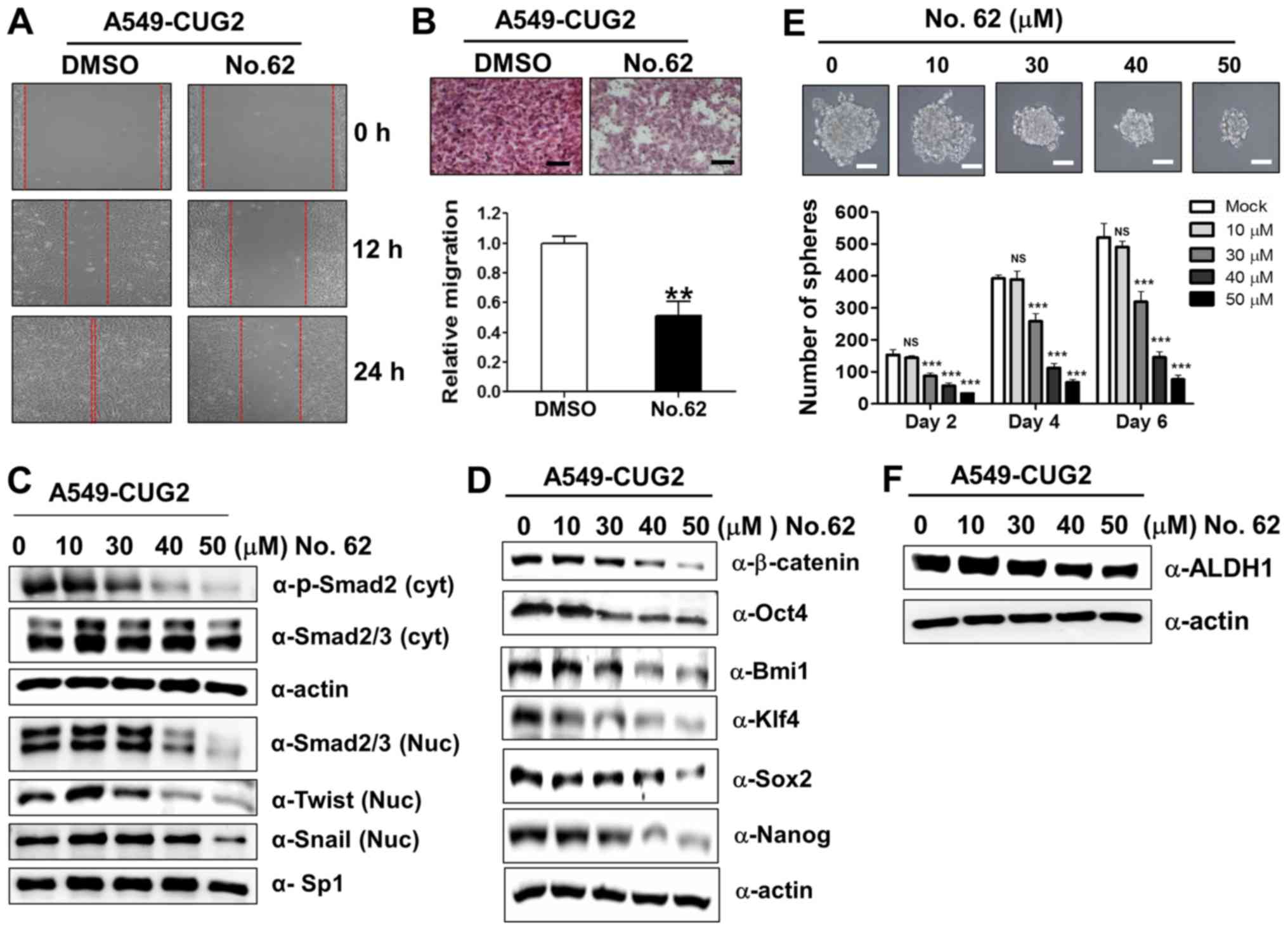 | Figure 6CGK062 treatment inhibits
CUG2-induced CSC-like phenotypes. (A) Migration of A549-CUG2 cells
treated with CGK062 (50 µM) or DMSO (control) was measured
by performing wound healing assay. Wound closure areas were
monitored using a phase-contrast microscope at ×100 magnification,
and the assay was repeated twice. (B) Invasion of A549-CUG2 cells
treated with CGK062 (50 µM) or DMSO was determined using the
48-well Boyden chamber. The chamber was assembled using
Matrigel-coated polycarbonate filters. Scale bar indicates 100
µm, and the assay was repeated twice. Each assay was
performed in triplicate, and error bars indicate the means ± SD
(**P<0.01, DMSO vs. CGK062). (C, D and F) Lysates of
A549-CUG2 cells treated with CGK062 (0, 10, 30, 40 and 50
µM) or DMSO were separated by performing SDS-PAGE on 10%
gels, and expression of phosphorylated Smad2, Smad2/3, Twist,
Snail, Oct4, Bmi1, Klf4, Sox2, Nanog and ALDH1 was determined by
performing western blot analysis with corresponding antibodies. (E)
A549-CUG2 cells treated with CGK062 (0, 10, 30, 40 and 50
µM) or DMSO (1,000 cells per well) were seeded in ultra-low
attachment plates for 2, 4 or 6 days. The assay was performed in
triplicate, and error bars indicate the means ± SD (ns; not
significant, DMSO vs. CGK062 at 10 µM,
***P<0.001, DMSO vs. CGK062 at 30, 40 and 50
µM). Scale bars indicate 50 µm. CUG2,
cancer-upregulated gene 2. |
Furthermore, as we hypothesized that CGK062
treatment enabled to interrupt CUG2-induced stemness on the basis
of results from Figs. 1 and
4, we measured the expression
levels of transcription factors or co-factors associated with
stemness and the number and size of A549-CUG2 cell spheroids
following CGK062 treatment. We found that CGK062 treatment
decreased the Bmi1, Klf4, Oct4, Sox2 and Nanog levels (Fig. 6D). The effect of CGK062 was clearly
detected from 30 µM. Moreover, CGK062 treatment
significantly decreased the number and size of A549-CUG2 cell
spheroids compared with mock treatment (Fig. 6E). We then examined the expression
of aldehyde dehydrogenase 1 (ALDH1), a biochemical feature of
stemness, and found that CGK062 treatment decreased the level of
this protein (Fig. 6F). Taken
together, these results suggest that CGK062 treatment inhibits
CSC-like phenotypes in A549-CUG2 cells.
CGK062 treatment impairs CUG2-mediated
tumor formation in transplanted nude mice
Finally, we examined whether CGK062 treatment
inhibits CUG2-induced tumor formation in vivo. For this,
Balb/C nude mice were subcutaneously transplanted with A549-CUG2
cells into their right flanks and were intraperitoneally injected
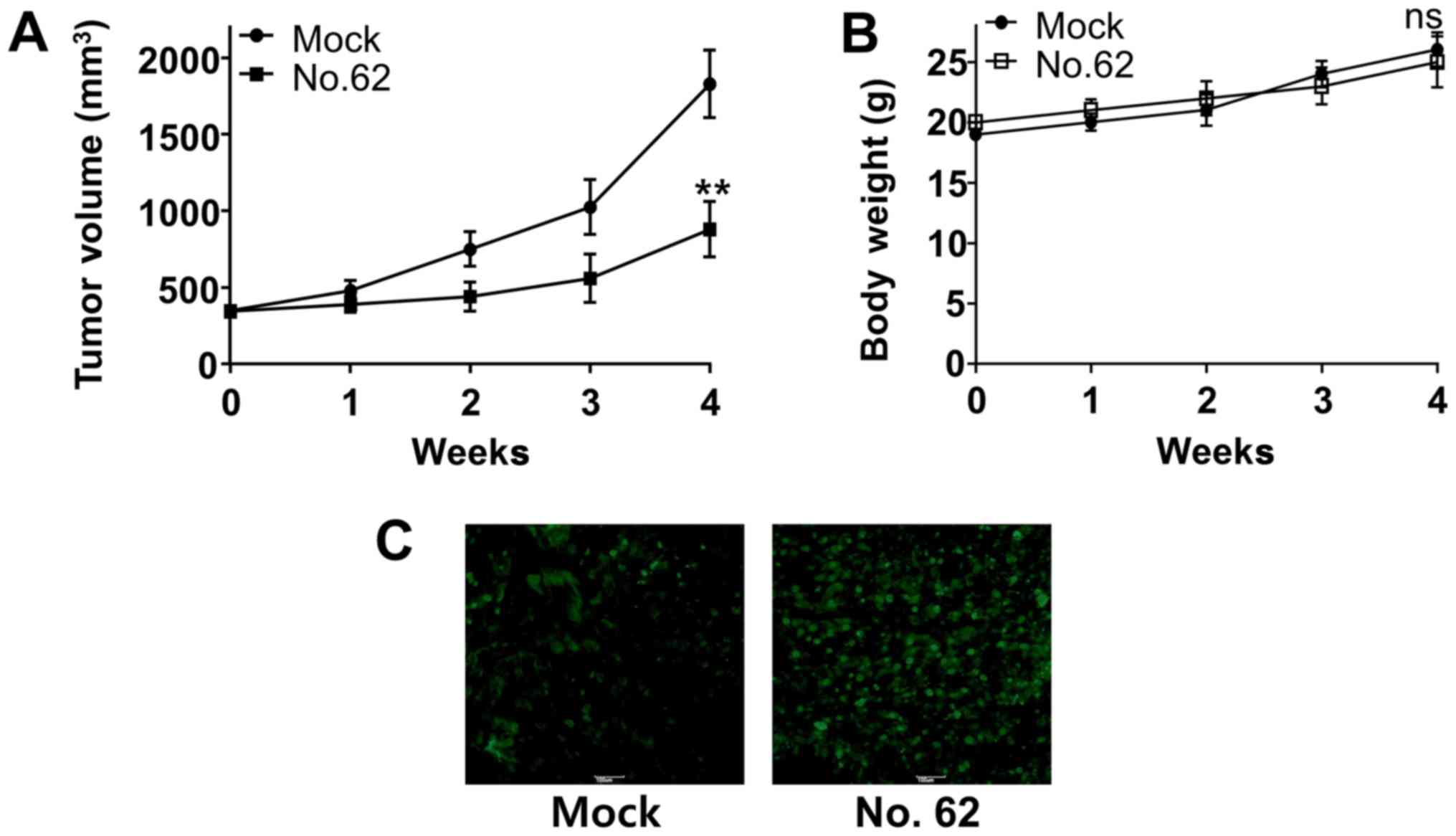
with CGK062 for 30 days. We found that CGK062 treatment
significantly decreased CUG2-induced tumor formation in Balb/C nude
mice, although mock treatment did not (Fig. 7A). Moreover, no significant change
in body weights was observed in the mock- or CGK062-treated mice
during the treatment period (Fig.
7B). We then performed TUNEL assay to determine the presence of
apoptotic cells in tumor tissues isolated from mock- or
CGK062-treated mice and found higher numbers of stained cells in
the tumor tissues isolated from CGK062-treated mice than in those
isolated from mock-treated mice (Fig.
7C). These results indicate that CGK062 exerts a cytotoxic
effect on tumorigenic A549-CUG2 lung cancer cells in
vivo.
Discussion
In the present study, we found that CUG2
specifically activated and stabilized β-catenin to induce the
features of malignant cancer, including increased cell migration,
aggressive invasion, enforced sphere formation and tumor formation
in vivo. Moreover, we found that the CUG2-induced
phosphorylation of β-catenin followed a different pattern compared
with that induced by Wnt3a. Surprisingly, despite the involvement
of the Akt and Src signaling molecules in both CUG2- and
Wnt3a-mediated signaling pathways, CUG2 stabilized β-catenin by
inducing its hyperphosphorylation at Ser33/Ser37, which is
traditionally suggested to induce the ubiquitination and
proteasomal degradation of β-catenin. As expected, Wnt3a did not
phosphorylate β-catenin at Ser33/Ser37 in the presence of CUG2
overexpression. These results suggest that CUG2 recruits or
activates a signaling molecule to prevent the predicted ubiquitin
E3 ligase-mediated degradation of β-catenin. Growing evidence
indicates that a long form of FLICE/caspase 8 inhibitory protein
(cFLIP) stabilizes β-catenin (32). Furthermore, another study reported
that acetyltransferase p300/CBP-associated factor (PCAF) directly
acetylated and stabilized β-catenin and that protease-activated
receptor-1 (PAR-1) stabilized β-catenin through Gα13 independently
of Wnt signaling (33). Herein, we
focused on NEK2 as it physically interacts with and phosphorylates
β-catenin at Ser33/Ser27 (30). We
found that CUG2 enhanced NEK2 expression. NEK2 silencing reduced
the phosphorylation of β-catenin at Ser33/Ser37, but did not
decrease the level of total β-catenin. As the interaction of NEK2
with β-catenin interrupts the binding of GSK3β to β-catenin
(30), we expected that
NEK2 knockdown facilitated the binding of GSK3β to
β-catenin, leading to its phosphorylation at Ser33/Ser37 and
subsequent degradation through the E3 ligase β-TrCP. However, we
did not observe any change in the β-catenin levels. Moreover, GSK3β
inhibition or silencing did not increase the β-catenin levels. In
our next study, we aim to examine whether the long form of cFLIP,
PCAF, or PAR-1 participates in β-catenin stabilization in the
presence of CUG2 overexpression. Furthermore, we aim to determine
the mechanisms underlying the CUG2-induced increase in NEK2
expression in our future studies.
During interphase, centrosomes are held together by
a proteinaceous linker. At the onset of mitosis, this linker is
dissembled to facilitate centrosome separation and bipolar spindle
formation (34). NEK2 is
implicated to be involved in this process, which is known as
centrosome disjunction (34).
Besides its cellular effects, NEK2 overexpression activates
Ras-Src, PI3 kinase, and Wnt signaling pathways to promote
metastasis (35). Consistently,
aberrant NEK2 expression has been reported in various cancers,
including hepatocellular carcinoma (36), non-small cell lung (37), colon (38), brain (39), and ovarian cancers (40). Based on these lines of clinical
evidence, small-molecule drugs have been designed or screened for
targeting the potentially oncogenic NEK2 (41-43).
Notably, treatment with CGK062, which destabilizes
β-catenin through PKCα, reduced the NEK2 levels. Although the
molecular mechanisms underlying this finding are unclear, this
result suggests that NEK2 expression is affected by the β-catenin
levels. Thus, our next assignment will aim to illustrate how CGK062
decreases the protein levels of NEK2. A recent study synthesized
(+)-decursin derivatives substituted with cinnamoyl- and phenyl
propiony groups using (+)-CGK062 as a leading compound (44). The decursin derivatives inhibited
Wnt3a-induced β-catenin response transcription and enhanced
degradation of β-catenin, leading to the suppression of cyclin D1
and c-Myc expression (44). Other
synthetic decursin derivatives also exhibited suppression of
androgen receptor signaling (45).
In conclusion, the findings of this study
demonstrated that CUG2 overexpression increased the phosphorylation
of β-catenin at Ser33/Ser37 through the elevated NEK2 expression
and activity. This event provided the resistance of β-catenin to E3
ligase for degradation. Consequently, the upregulated β-catenin was
involved in CUG2-induced CSC-like phenotypes. However, treatment
with CGK062 reduced the protein levels of β-catenin through NEK2.
We thus suggest that CGK062 may be used as a potential drug against
CUG2-overexpressing lung cancer cells.
Funding
This study was supported by the Basic Research
Program of the National Research Foundation funded by the Korean
government (NRF-2016R 1D1A1B03930168). This study was also
supported by Korea Institute for Advancement of Technology (KIAT)
in Republic of Korea (N0002310, Construction Project of Supporting
Center for Commercializing Customized Nano-mold-based
Technologies).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors’ contributions
SK, SO, SSK and YHC conceived and designed the
experiments. SK, JM, AYY, MRL, JEK, DYH and SEY performed the
experiments. HYK and SSK were involved in data analysis and
compilation. SK and YHC wrote the manuscript. All authors have read
and approved the manuscript and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
All animal experiments were conducted in accordance
with the Laboratory Animal Resources Guide for the Care and Use of
Laboratory Animals. All animal study protocols were approved by the
Pusan National University Animal Care and Use Committee
(PNU-2017-1541).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they no competing
interests.
Acknowledgments
The authors would like to thank Dr Charles C. Chung
(Rochester General Hospital, Rochester, NY, USA) for proofreading
this manuscript.
References
|
1
|
Lee S, Gang J, Jeon SB, Choo SH, Lee B,
Kim YG, Lee YS, Jung J, Song SY and Koh SS: Molecular cloning and
functional analysis of a novel oncogene, cancer-upregulated gene 2
(CUG2). Biochem Biophys Res Commun. 360:633–639. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hori T, Amano M, Suzuki A, Backer CB,
Welburn JP, Dong Y, McEwen BF, Shang WH, Suzuki E, Okawa K, et al:
CCAN makes multiple contacts with centromeric DNA to provide
distinct pathways to the outer kinetochore. Cell. 135:1039–1052.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim H, Lee M and Lee S, Park B, Koh W, Lee
DJ, Lim DS and Lee S: Cancer-upregulated gene 2 (CUG2), a new
component of centromere complex, is required for kinetochore
function. Mol Cells. 27:697–701. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Park EH, Park EH, Cho IR, Srisuttee R, Min
HJ, Oh MJ, Jeong YJ, Jhun BH, Johnston RN, Lee S, et al: CUG2, a
novel oncogene confers reoviral replication through Ras and p38
signaling pathway. Cancer Gene Ther. 17:307–314. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kaowinn S, Kim J, Lee J, Shin DH, Kang CD,
Kim DK, Lee S, Kang MK, Koh SS, Kim SJ, et al: Cancer upregulated
gene 2 induces epithelial-mesenchymal transition of human lung
cancer cells via TGF-β signaling. Oncotarget. 8:5092–5110. 2017.
View Article : Google Scholar
|
|
6
|
Kaowinn S, Jun SW, Kim CS, Shin DM, Hwang
YH, Kim K, Shin B, Kaewpiboon C, Jeong HH, Koh SS, et al: Increased
EGFR expression induced by a novel oncogene, CUG2, confers
resistance to doxorubicin through Stat1-HDAC4 signaling. Cell Oncol
(Dordr). 40:549–561. 2017. View Article : Google Scholar
|
|
7
|
Peifer M and Polakis P: Wnt signaling in
oncogenesis and embryogenesis - a look outside the nucleus.
Science. 287:1606–1609. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huelsken J and Birchmeier W: New aspects
of Wnt signaling pathways in higher vertebrates. Curr Opin Genet
Dev. 11:547–553. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Giles RH, van Es JH and Clevers H: Caught
up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta.
1653:1–24. 2003.PubMed/NCBI
|
|
10
|
Tetsu O and McCormick F: Beta-catenin
regulates expression of cyclin D1 in colon carcinoma cells. Nature.
398:422–426. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
He TC, Sparks AB, Rago C, Hermeking H,
Zawel L, da Costa LT, Morin PJ, Vogelstein B and Kinzler KW:
Identification of c-MYC as a target of the APC pathway. Science.
281:1509–1512. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
He TC, Chan TA, Vogelstein B and Kinzler
KW: PPARdelta is an APC-regulated target of nonsteroidal
anti-inflammatory drugs. Cell. 99:335–345. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Saadeddin A, Babaei-Jadidi R, Spencer-Dene
B and Nateri AS: The links between transcription, beta-catenin/JNK
signaling, and carcinogenesis. Mol Cancer Res. 7:1189–1196. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Damsky WE, Curley DP, Santhanakrishnan M,
Rosenbaum LE, Platt JT, Gould Rothberg BE, Taketo MM, Dankort D,
Rimm DL, McMahon M, et al: β-catenin signaling controls metastasis
in Braf-activated Pten-deficient melanomas. Cancer Cell.
20:741–754. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu L, Zhu XD, Wang WQ, Shen Y, Qin Y, Ren
ZG, Sun HC and Tang ZY: Activation of beta-catenin by hypoxia in
hepatocellular carcinoma contributes to enhanced metastatic
potential and poor prognosis. Clin Cancer Res. 16:2740–2750. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim
NH, Cha SY, Ryu JK, Choi YJ, Kim J, et al: A Wnt-Axin2-GSK3beta
cascade regulates Snail1 activity in breast cancer cells. Nat Cell
Biol. 8:1398–1406. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rubinfeld B, Albert I, Porfiri E, Fiol C,
Munemitsu S and Polakis P: Binding of GSK3beta to the
APC-beta-catenin complex and regulation of complex assembly.
Science. 272:1023–1026. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Aberle H, Bauer A, Stappert J, Kispert A
and Kemler R: beta-catenin is a target for the ubiquitin-proteasome
pathway. EMBO J. 16:3797–3804. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu C, Li Y, Semenov M, Han C, Baeg GH,
Tan Y, Zhang Z, Lin X and He X: Control of beta-catenin
phosphorylation/degradation by a dual-kinase mechanism. Cell.
108:837–847. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
van Noort M, Meeldijk J, van der Zee R,
Destree O and Clevers H: Wnt signaling controls the phosphorylation
status of beta-catenin. J Biol Chem. 277:17901–17905. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hino S, Tanji C, Nakayama KI and Kikuchi
A: Phosphorylation of beta-catenin by cyclic AMP-dependent protein
kinase stabilizes beta-catenin through inhibition of its
ubiquitination. Mol Cell Biol. 25:9063–9072. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Taurin S, Sandbo N, Qin Y, Browning D and
Dulin NO: Phosphorylation of beta-catenin by cyclic AMP-dependent
protein kinase. J Biol Chem. 281:9971–9976. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gwak J, Lee JH, Chung YH, Song GY and Oh
S: Small molecule-based promotion of PKCα-mediated β-catenin
degradation suppresses the proliferation of CRT-positive cancer
cells. PLoS One. 7:e466972012. View Article : Google Scholar
|
|
24
|
Habelhah H, Takahashi S, Cho SG, Kadoya T,
Watanabe T and Ronai Z: Ubiquitination and translocation of TRAF2
is required for activation of JNK but not of p38 or NF-kappaB. EMBO
J. 23:322–332. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Akimov IA, Chernolovskaya EL, Spitsyna YE,
Ryabchikova EI and Zenkova MA: Silencing of Her2, CCNB1 and PKC
Genes by siRNA Results in Prolonged Retardation of Neuroblastoma
Cell Division. Acta Naturae. 3:29–39. 2011.
|
|
26
|
Jung JK, Kwun HJ, Lee JO, Arora P and Jang
KL: Hepatitis B virus X protein differentially affects the
ubiquitin-mediated proteasomal degradation of beta-catenin
depending on the status of cellular p53. J Gen Virol. 88:2144–2154.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim SJ, Glick A, Sporn MB and Roberts AB:
Characterization of the promoter region of the human transforming
growth factor-beta 1 gene. J Biol Chem. 264:402–408.
1989.PubMed/NCBI
|
|
28
|
Lee Y, Kim SJ, Park HD, Park EH, Huang SM,
Jeon SB, Kim JM, Lim DS and Koh SS: PAUF functions in the
metastasis of human pancreatic cancer cells and upregulates CXCR4
expression. Oncogene. 29:56–67. 2010. View Article : Google Scholar
|
|
29
|
Liang CC, Park AY and Guan JL: In vitro
scratch assay: A convenient and inexpensive method for analysis of
cell migration in vitro. Nat Protoc. 2:329–333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mbom BC, Siemers KA, Ostrowski MA, Nelson
WJ and Barth AI: Nek2 phosphorylates and stabilizes β-catenin at
mitotic centrosomes downstream of Plk1. Mol Biol Cell. 25:977–991.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Steinway SN, Zañudo JG, Ding W, Rountree
CB, Feith DJ, Loughran TP Jr and Albert R: Network modeling of TGFβ
signaling in hepatocellular carcinoma epithelial-to-mesenchymal
transition reveals joint sonic hedgehog and Wnt pathway activation.
Cancer Res. 74:5963–5977. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nakagiri S, Murakami A, Takada S, Akiyama
T and Yonehara S: Viral FLIP enhances Wnt signaling downstream of
stabilized beta-catenin, leading to control of cell growth. Mol
Cell Biol. 25:9249–9258. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ge X, Jin Q, Zhang F, Yan T and Zhai Q:
PCAF acetylates {beta}-catenin and improves its stability. Mol Biol
Cell. 20:419–427. 2009. View Article : Google Scholar :
|
|
34
|
Mardin BR, Lange C, Baxter JE, Hardy T,
Scholz SR, Fry AM and Schiebel E: Components of the Hippo pathway
cooperate with Nek2 kinase to regulate centrosome disjunction. Nat
Cell Biol. 12:1166–1176. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Das TK, Dana D, Paroly SS, Perumal SK,
Singh S, Jhun H, Pendse J, Cagan RL, Talele TT and Kumar S:
Centrosomal kinase Nek2 cooperates with oncogenic pathways to
promote metastasis. Oncogenesis. 2:e692013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang X, Fan Q, Li Y, Yang Z, Yang L, Zong
Z, Wang B, Meng X, Li Q, Liu J, et al: Transforming growth
factor-beta1 suppresses hepatocellular carcinoma proliferation via
activation of Hippo signaling. Oncotarget. 8:29785–29794.
2017.PubMed/NCBI
|
|
37
|
Zhong X, Guan X, Liu W and Zhang L:
Aberrant expression of NEK2 and its clinical significance in
non-small cell lung cancer. Oncol Lett. 8:1470–1476. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lu L, Zhai X and Yuan R: Clinical
significance and prognostic value of Nek2 protein expression in
colon cancer. Int J Clin Exp Pathol. 8:15467–15473. 2015.
|
|
39
|
Liu H, Liu B, Hou X, Pang B, Guo P, Jiang
W, Ding Q, Zhang R, Xin T, Guo H, et al: Overexpression of
NIMA-related kinase 2 is associated with poor prognoses in
malignant glioma. J Neurooncol. 132:409–417. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu X, Gao Y, Lu Y, Zhang J, Li L and Yin
F: Upregulation of NEK2 is associated with drug resistance in
ovarian cancer. Oncol Rep. 31:745–754. 2014. View Article : Google Scholar
|
|
41
|
Coxon CR, Wong C, Bayliss R, Boxall K,
Carr KH, Fry AM, Hardcastle IR, Matheson CJ, Newell DR,
Sivaprakasam M, et al: Structure-guided design of purine-based
probes for selective Nek2 inhibition. Oncotarget. 8:19089–19124.
2017. View Article : Google Scholar :
|
|
42
|
Xi JB, Fang YF, Frett B, Zhu ML, Zhu T,
Kong YN, Guan FJ, Zhao Y, Zhang XW, Li HY, et al: Structure-based
design and synthesis of imidazo[1,2-a]pyridine derivatives as novel
and potent Nek2 inhibitors with in vitro and in vivo antitumor
activities. Eur J Med Chem. 126:1083–1106. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Frett B, Brown RV, Ma M, Hu W, Han H and
Li HY: Therapeutic melting pot of never in mitosis gene a related
kinase 2 (Nek2): A perspective on Nek2 as an oncology target and
recent advancements in Nek2 small molecule inhibition. J Med Chem.
57:5835–5844. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lee JH, Kim MA, Park S, Cho SH, Yun E, O
YS, Kim J, Goo JI, Yun MY, Choi Y, et al: Synthesis and evaluation
of (+)-decursin derivatives as inhibitors of the Wnt/β-catenin
pathway. Bioorg Med Chem Lett. 26:3529–3532. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang Y, Shaik AA, Xing C, Chai Y, Li L,
Zhang J, Zhang W, Kim SH, Lü J and Jiang C: A synthetic decursin
analog with increased in vivo stability suppresses androgen
receptor signaling in vitro and in vivo. Invest New Drugs.
30:1820–1829. 2012. View Article : Google Scholar
|