Introduction
Bladder cancer is the ninth most commonly diagnosed
cancer, with an estimated 382,700 new cases and 150,300 mortalities
recorded worldwide in 2008 (1).
Approximately 90% of bladder cancer cases are urothelial carcinoma
(UC), and 70-85% of TCC are at stages Ta, T1 and carcinoma in
situ. In total, 50-70% of UC cases at these stages will
relapse, although there are conservative measures such as
transurethral and intravesical treatment (2). The molecular mechanisms that control
the development and progression of bladder cancer remain to be
elucidated.
Previous studies have demonstrated that a number of
intracellular signaling molecules have key roles in the progression
of bladder cancer (3,4). It is well-known that the
phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) signaling
pathway is activated in bladder cancer (3). This pathway can be misaligned by
losing phosphatase and tensin homolog, and mutations in
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit α
of PI3K are correlated with fibroblast growth factor receptor 3
mutations in low-grade and early-stage bladder cancer (4-7).
Nuclear factor (NF)-κB acts as a transcription regulator mediating
interleukin (IL)-5, IL-8, and IL-28A expression in muscle-invasive
bladder cancer, and the hypoxic microenvironment activates the
NF-κB pathway, promoting the invasion and migration of T24 cells
(8,9). These signaling pathways serve
critical roles in bladder cancer progression; however, the
mechanism of AKT and NF-κB activation in cancer cells by the tumor
microenvironment remains to be elucidated.
Tumorigenesis depends on many signaling pathways,
and many types of molecules, such as extracellular matrix
components, cytokines and growth factors. Epidermal growth factor
receptor (EGFR) and its ligands are overexpressed in bladder
cancer, and are correlated with tumor grade and stage (10). As a feature of the tumor
microenvironment, tumor angiogenesis has an important role in
cancer progression, and these are complex processes in which novel
angiogenesis occurs in response to interactions between cancer
cells and endothelial cells (ECs), and is mediated by cytokines and
growth factors (11).
Angiogenesis-related cytokines and growth factors secreted by
cancer cells or stromal cells can directly bind to their receptors
on vascular ECs and stimulate angiogenesis by promoting endothelial
sprouting, differentiation and survival (11,12).
It has recently been demonstrated that cancer cells and vascular
ECs secrete growth factors that enhance the proliferation and
migration of both cells by mediating interactions between them
(13-15). As such, vascular endothelial growth
factor (VEGF) secreted by tumor or stromal cells specifically binds
to its receptors, VEGF receptor(R)-1 and VEGFR-2, on EC; the
PI3K/AKT and extracellular signal-regulated kinase (ERK) signaling
pathways were reported to be induced by the binding of VEGF and its
receptor (16,17). However, the mechanisms of these
interactions in bladder cancer tissue remain unclear.
Previous studies have demonstrated that cytokines
secreted by vascular endothelial or cancer cells can induce
proliferation, migration and invasion (13,14).
In the present study, it was hypothesized that cytokines or growth
factors secreted by vascular ECs or cancer cells into the tumor
microenvironment can promote bladder cancer cell proliferation,
migration and invasion. The present study demonstrated that the
VEGFR-2 signaling pathway was activated in EC-secreted EGFR ligands
via the interaction between bladder cancer cells and ECs;
furthermore, vascular EC-secreted EGFR ligands binding to their
receptors on bladder cancer cells induced proliferation, migration
and invasion through EGFR signaling and induced the secretion of
CXC chemokines from bladder cancer cells, to enhance EC
recruitment.
Materials and methods
Cell culture and co-culture
T24 and 253J human bladder cancer cells and human
umbilical vein ECs (HUVECs) were obtained from the American Type
Culture Collection (Manassas, VA, USA) and cultured in Dulbecco's
modified Eagle's medium (DMEM) supplemented with 10% fetal bovine
serum (FBS) (both from Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) in an atmosphere with 5% CO2 at
37°C.
In order to mimic the interaction between a cancer
cell and EC in the tumor microenvironment, 0.4-µm pore
diameter chambers of a 6-well plate (EMD Millipore, Billerica, MA,
USA) were used; 50,000 T24/253J cells were added to the lower
chambers of the 6-well plate, and 50,000 HUVECs were added to the
upper chambers, and cultured in DMEM (Invitrogen; Thermo Fisher
Scientific, Inc.) supplemented with 10% FBS (Invitrogen; Thermo
Fisher Scientific, Inc.) in an atmosphere with 5% CO2 at
37°C for 72 h. The EGFR inhibitor lapatinib, the VEGFR inhibitor ZM
323881 HCl and the CXCR2 inhibitor SB225002 were obtained from
Selleck Chemicals (Houston, TX, USA); the NF-κB inhibitor PDTC was
obtained from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany),
dissolved in dimethyl sulfoxide (DMSO) and stored at −20°C.
Western blotting
Following 72 h of co-culture, bladder cancer cell
and vascular EC protein lysates were isolated with
radioimmunoprecipitation assay buffer [50 mM Tris (pH 8.0), 150 mM
NaCl, 0.1% SDS, 1% NP40 and 0.5% sodium deoxycholate containing
proteinase inhibitor 1% cocktail and 1 mM phenylmethane sulfonyl
fluoride, both from Sigma-Aldrich; Merck KGaA]. The protein
concentration was calculated using a NanoDrop 1000
Spectrophotometer (Thermo Fisher Scientific, Inc.). Protein lysates
were separated via 10% SDS-PAGE. Membranes were blocked with 5%
non-fat milk in 1X TBS containing 0.3% Tween-20 (TBST), incubated
with primary antibodies (Table I)
overnight at 4°C and washed with 1X TBST (pH 7.6). Membranes
incubated with IRDye®-conjugated goat anti-rabbit (cat.
no. 926-32211) or goat anti-mouse secondary antibodies (cat. no.
926-68070; both LI-COR Biosciences, Lincoln, NE, USA) diluted at
1:1,000 in 5% skimmed milk were then applied for 1 h at room
temperature, followed by washing as described previously, in a dark
room, drying with neutral absorbent paper and scanning with an
Odyssey detection system (LI-COR Biosciences). GAPDH was used as
the loading control.
 | Table IPrimary antibodies used for western
blotting. |
Table I
Primary antibodies used for western
blotting.
| Primary antibodies
(dilution) | Catalogue
number | Supplier |
|---|
| Anti-EGFR | D38B1 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-ErbB2 | D8F12 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-ErbB3 | D22C5 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-ErbB4 | 111B2 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-p-EGFR
(Tyr1068) | D7A5 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-p-ErbB2
(Tyr1221/1222) | 6B12 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-p-ErbB3
(Tyr1289) | D1B5 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-p-ErbB4
(Tyr1284) | 21A9 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-AKT (pan) | 11E7 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-p-AKT
(Ser473) | D9E | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-ERK1/2 | 137F5 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-p-ERK1/2
(Thr202/Tyr204) | D13.14.4E | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-STAT3 | D3Z2G | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-p-STAT3
(Tyr705) | D3A7 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-NF-κB p65 | D14E12 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-p-NF-κB
p65 | 93H1 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-MMP-2 | D2O4T | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-MMP-9 | D6O3H | Cell Signaling
Technology, Inc., Danvers, MA, USA |
|
Anti-N-cadherin | D4R1H | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-ZEB1 | D80D3 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-Survivin | 71G4B7 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-GAPDH | KC-5G4 | Kangchen BioTech
Co., Ltd., Shanghai, China |
| Anti-CXCR2 | ab14935 | Abcam, Cambridge,
UK |
| Anti-p-VEGFR2
(Tyr1059) | D5A6 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-p-VEGFR2
(Tyr1175) | D5B11 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-p-VEGFR2
(Tyr996) | 2474 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
| Anti-VEGFR2 | D5B1 | Cell Signaling
Technology, Inc., Danvers, MA, USA |
Following 48 h of co-culture, the medium was
extracted and supplemented with FBS-free DMEM, and the upper
chambers were removed and placed into a 6-well plate. The EGFR
inhibitor lapatinib (10 µM in medium) or NF-κB inhibitor
PDTC (5 µM in medium) were added to the lower chambers, and
the VEGFR inhibitor ZM 323881 HCl (10 nM in medium) or the
inhibitor CXCR2 SB225002 (2 µM in medium) were added to the
upper chambers for a further 24 h at 37°C prior to protein or total
mRNA extraction.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA of the cells was isolated using RNAfast
200 reagent (Shanghai Fastagen Biotechnology Co., Ltd., Shanghai,
China) and quantitated by measuring the absorbance at a wavelength
of 260 nm. The RNA (2 µg) sample was reverse transcribed
with 5X PrimeScript RT Master Mix (2 µl; Takara
Biotechnology Co., Ltd., Dalian, China), RNase-free dH2O
(7 µl) and total RNA (1 µl) were mixed and reacted at
37°C for 16 min and 85°C for 5 sec. qPCR was then performed using
the SYBR Premix Ex Taq™ II system (Takara Biotechnology Co., Ltd.)
and the Bio-Rad CFX96™ Real-time system (Bio-Rad Laboratories,
Inc.). SYBR Premix Ex Taq II (12.5 µl), 1 µl sense
primer (10 µM), 1 µl anti-sense primer (10 µM)
2 µl cDNA solution and 8.5 µl RNase-free water were
mixed together. The following thermocycling protocol was used with
three stages, including pre-degeneration for 95°C for 30 sec, one
repeat; PCR amplification at 95°C for 5 sec followed by 60°C for 30
sec, 40 repeats; and dissociation at 95°C for 15 sec followed by
60°C for 30 sec and 95°C for 15 sec. GAPDH was used as the loading
control to balance the quantity of the samples, and the gene
expression was normalized to the GAPDH to calculate relative
expression level using the 2−ΔΔCq method (18). The gene-specific primers used are
listed in Table II.
| Table IIPrimers used for reverse
transcription-quantitative polymerase chain reaction. |
Table II
Primers used for reverse
transcription-quantitative polymerase chain reaction.
| Gene ID | Gene | Primers | Sequence
(5′-3′) |
|---|
| 2597 | GAPDH | F |
GGAGCGAGATCCCTCCAAAAT |
| R |
GGCTGTTGTCATACTTCTCATGG |
| 1950 | EGF | F |
TCCTCACCCGATAATGGTGGA |
| R |
CCAGGAAAGCAATCACATTCCC |
| 7039 | TNFA | F |
AGATAGACAGCAGCCAACCCTGA |
| R |
CTAGGGCCATTCTGCCCATC |
| 1839 | HBEGF | F |
ATCGTGGGGCTTCTCATGTTT |
| R |
TTAGTCATGCCCAACTTCACTTT |
| 685 | BTC | F |
CTAGGTGCCCCAAGCAATACA |
| R |
GCAGACACCGATGACCAAAATA |
| 374 | AREG | F |
GTGGTGCTGTCGCTCTTGATA |
| R |
CCCCAGAAAATGGTTCACGCT |
| 2069 | EREG | F |
GGACAGTGCATCTATCTGGTGG |
| R |
TTGGTGGACGGTTAAAAAGAAGT |
| 255324 | EPGN | F |
ATGGCTTTGGGAGTTCCAATATC |
| R |
TCCTTCTATGTTGTCAGCTTGC |
| 7422 | VEGF-A | F |
AGGGCAGAATCATCACGAAGT |
| R |
AGGGTCTCGATTGGATGGCA |
| 7424 | VEGF-C | F |
GAGGAGCAGTTACGGTCTGTG |
| R |
TCCTTTCCTTAGCTGACACTTGT |
| 2919 | CXCL1 | F |
AACCGAAGTATAGCCACAC |
| R |
GTTGGATTTGTCACTGTTCAGC |
| 6374 | CXCL5 | F |
AGCTGCGTTGCGTTTGTTTAC |
| R |
TGGCGAACACTTGCAGATTAC |
| 3576 | CXCL8 | F |
ACTGAGAGTGATTGAGAGTGGAC |
| R |
AACCCTCTGCACCCAGTTTTC |
Colony formation assay
Chambers (0.4-µm pore diameter) were obtained
from EMD Millipore. A total of 1,000 T24/253J cells were added to
the lower chambers of the 6-well plate, and 1,000 HUVECs were added
to the upper chambers, then cultured in DMEM supplemented with 10%
FBS in an atmosphere with 5% CO2 at 37°C. In the control
group, 1,000 T24/253J cells were added to the lower chambers of the
6-well plate, and the upper chambers were not seeded with HUVECs.
Following 14 days, the plates were washed with PBS, fixed in 4%
formalin, stained with crystal violet solution for 15 min at room
temperature, and washed with PBS to remove the excess dye. Cells
counted from five randomly selected fields were counted via light
microscopy at ×100 magnification.
MTT assay
Briefly, a 24-well Transwell plate was used
(0.4-µm pore diameter; EMD Millipore, Schaffhausen,
Switzerland) for MTT assay. Following 48 h of co-culture, the upper
chambers were removed, 100 µl 5 mg/ml MTT solution was added
to each well, and the plate was then incubated at 37°C for a
further 4 h. Thereafter, the medium was aspirated and 1,000
µl DMSO was added to each well. The microtitre plate was
placed on a shaker in order to dissolve the dye. After the formazan
crystals had dissolved, the absorbance was determined
spectrophotometrically at 490 nm on an ELX800 UV universal
microplate reader (BioTek Instruments, Inc., Winooski, VT,
USA).
Migration and invasion assay
Cell migration and invasion was assessed using a
Transwell Boyden chamber assay. The chambers (8-µm pore
diameter) were obtained from EMD Millipore. For migration assay,
following co-culture 48 h later, 400 µl FBS-free DMEM
suspension with 10,000 T24 cells and 50,000 253J cells were added
to the upper chamber in the 24-well plate, and 800 µl DMEM
without FBS was added to the lower chambers. Following 24 h
incubation at 5% CO2 and 37°C, cells were fixed with 4%
formalin for 30 min at 37°C, stained with crystal violet (0.01% in
ethanol) for 10 min at room temperature, washed three times and
counted under an inverted light microscope. Five random sections
imaged for each well at ×200 magnification and the average number
of cells was calculated. For analysis of invasion, suspension in
the upper chambers contained 60 µl mixture [FBS-free
DMEM:Matrigel (Sigma-Aldrich; Merck KGaA) = 12:1], and either
10,000 T24 or 5,000 253J cells, the incubation time was 36 h, and
all other steps were the same as for the migration analysis.
HUVEC recruitment
In order to monitor the HUVEC recruitment of cancer
cell lines, Boyden chambers were used (8-µm pore diameter;
EMD Millipore). A total of 10,000 T24 cells and 50,000 253J cells,
which educated by co-culture or control for 36 h, was seeded into
the 24-well plate until its adhesion to the bottom, followed by
planting 10,000 HUVECs into the upper chambers. Following
incubation for 24 h at 37°C, the number of HUVECs counted as the
Boyden chamber assay.
Statistical analysis
All statistical analyses were performed using
GraphPad Prism version 5.0 software (GraphPad Software, Inc., La
Jolla, CA, USA). All data were reported as the mean ± standard
error, and significant differences were measured using an unpaired
two-sided Student's t-test or differences in each group were
analyzed by one-way analysis of variance, followed by Dunnett's
t-test for separate comparisons. Kaplan-Meier analysis was used to
estimate cluster of differentiation (CD)31 and EGFR protein
expression in The Cancer Genome Atlas (TCGA) bladder cancer
PRAD_exp_HiSeqV2 (n=345) dataset (http://firebrowse.org/). Based on the mean number of
CD31 protein expression, 0.023, samples were divided into two
groups and the group with >0.023 had the lower survival ratio.
Based on the mean number of EGFR protein expression, -0.003248,
samples were divided into two groups, and the group with
>-0.003248 had the lower survival ratio. Spearman's correlation
analysis was used to identify and correlation between EGFR pY1068
expression and AKT pS473 and NF-κB p65 pS536 expression in TCGA
dataset. P<0.05 was considered to indicate a statistically
significant difference.
Results
Interactions between ECs and bladder
cancer cells increase cell viability and malignancy
Research of bladder cancer has demonstrated that
angiogenesis has an important role in bladder cancer progression
(19). It was demonstrated that
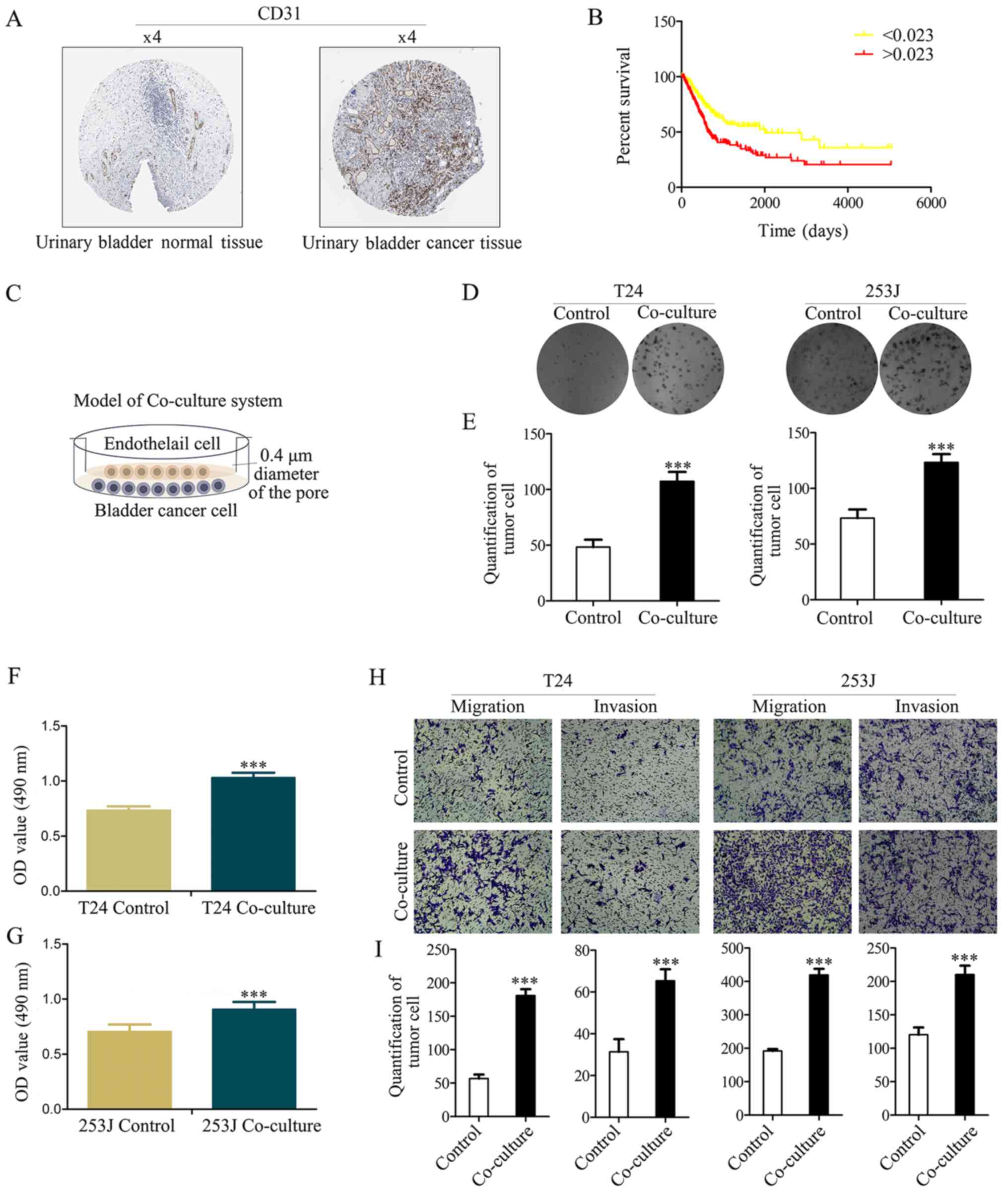
CD31 had a strong positive expression in bladder cancer, and a weak
negative expression in normal bladder tissue (Fig. 1A). Furthermore, Kaplan-Meier
analysis was used to estimate CD31 protein expression in the TCGA
bladder cancer PRAD_exp_HiSeqV2 (n=345) dataset, which indicated
that CD31 expression is an independent risk factor for patients
with bladder cancer (based on the mean number of CD31 protein
expression, 0.023, samples are divided into two groups, and the
group >0.023 has a lower survival ratio; P<0.001; Fig. 1B).
It has previously been demonstrated that the number
of ECs are increased according to tumor progression and are
negatively associated with the prognosis of bladder cancer
(19). In the present study it was
hypothesized that ECs serve other direct or indirect roles in
bladder cancer apart from the known metabolism-associated roles
(such as oxygen supply). The Boyden chamber system (0.4-µm
pore diameter) was used to co-culture bladder cancer cells
(T24/253J) with ECs (HUVECs) to mimic the tumor microenvironment
(Fig. 1C). Colony-formation
(Fig. 1D and E) and MTT assays
(Fig. 1F and G) indicated that
co-culture contributes to cancer cell viability. The results of the
Boyden chamber assay indicated that the co-culture resulted in the
enhanced malignancy of T24/253J cells (Fig. 1H and I).
Co-culture treatment activates EGFR
signaling in bladder cancer cells
Overexpression of ErbB family proteins has been
reported in a number of studies on bladder cancer, which indicated
that there was a significant association between clinical outcome
and tumor grade (20). EGFR
protein expression was analyzed in clinical specimens from the
human protein atlas (www.proteinatlas.org). It was demonstrated that EGFR
had strong positive expression in bladder cancer and weak negative
expression in normal bladder tissues (Fig. 2A). Furthermore, Kaplan-Meier
analysis was used to estimate EGFR protein expression in the TCGA
bladder cancer PRAD_exp_HiSeqV2 (n=345) dataset (http://firebrowse.org/), which indicated that EGFR
expression is a risk factor for patients with bladder cancer
(Fig. 2B). To obtain insight into
the role of the EGFR family in bladder cancer cells, EGFR family
expression in response to the co-culture of HUVEC and T24/253J
cells was determined. Western blotting revealed that EGFR signaling
was induced by co-culture treatment (Fig. 2C). In addition, it was demonstrated
that downstream EGFR signaling was induced, including AKT, signal
transducer and activator of transcription factor 3 (STAT3), ERK and
NF-κB. It was also attempted to analyze the gene expression of
matrix metalloproteinase (MMP)2, MMP9, zinc finger E-box-binding
homeobox (ZEB)-1, survivin and N-cadherin following co-culture,
using western blotting (Fig. 2D).
As presented in Fig. 2E,
co-culture upregulated MMP2, MMP9, ZEB-1, survivin and N-cadherin
gene expression. Spearman's rank correlation analysis was performed
in the TCGA dataset, which revealed a significant positive
correlation between the EGFR pY1068 and AKT pS473, and NF-κB p65
pS536 (Fig. 2F and G).
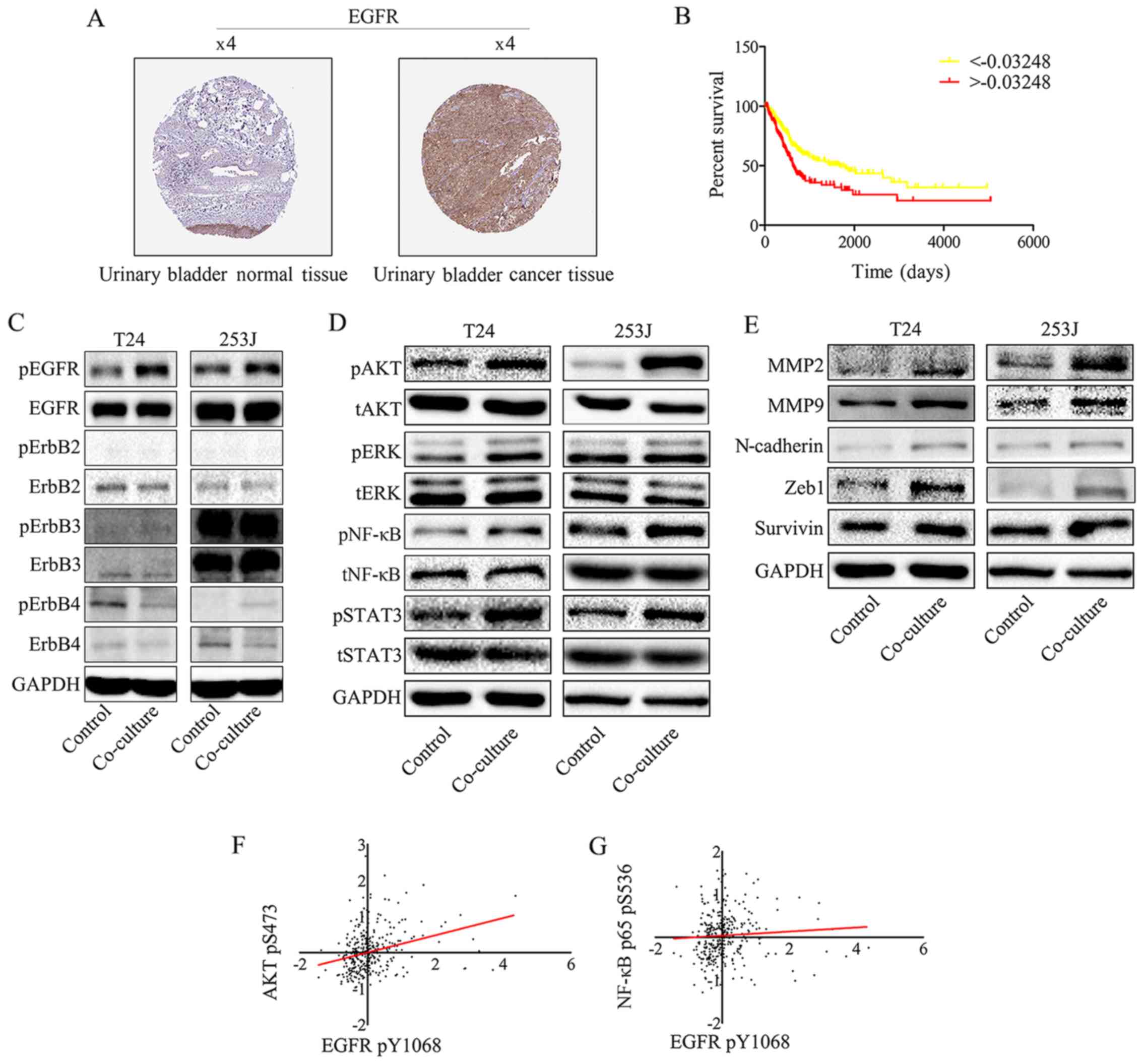 | Figure 2EGFR signaling in bladder cancer
cells is triggered by EGFR ligands secreted by endothelial cells
and promotes bladder cancer progression. (A) EGFR expression in
normal bladder tissue and bladder carcinoma specimens. Images were
taken from the Human Protein Atlas (http://www.protein-atlas.org) online database. (B)
Kaplan-Meier analysis estimated EGFR protein expression in TCGA
bladder cancer PRAD_exp_HiSeqV2 (n=345) dataset (http://firebrowse.org/), and indicated that EGFR
expression is an independent risk factor for patients with bladder
cancer (based on the mean number of EGFR protein expression
-0.003248, samples were divided into two groups, the group
>-0.003248 has the lower survival ratio;
***P<0.001). (C) EGFR signaling was induced by
co-culture treatment. Western blot analysis of EGFR family protein
expression following co-culture treatment revealed that EGFR pY1068
was upregulated compared with the control group. (D) Downstream
EGFR signaling was activated by co-culture treatment. Western blot
analysis indicates that co-culture induces downstream EGFR
signaling: AKT pS473, NF-κB p65 pS536, STAT3 pY705 and pERK1/2
(Thr202/Tyr204) were all upregulated. (E) Western blotting
indicated that the co-culture treatment of T24/253J leads to
upregulation of MMP2, MMP9, ZEB-1, survivin and N-cadherin.
Correlation between EGFR pY1068 expression and (F) AKT pS473 and
(G) NF-κB p65 pS536 expression in TCGA dataset. EGFR, epidermal
growth factor receptor; AKT, protein kinase B; NF, nuclear factor;
STAT3, signal transducer and activator of transcription factor 3;
ERK, extracellular signal-regulated kinase; p, phosphorylated; MMP,
matrix metalloprotein; ZEB, zinc finger E-box-binding homeobox;
TCGA, The Cancer Genome Atlas; t, total. |
Inhibition of EGFR signaling in T24/253J
cells attenuates co-culture induced malignancy and
proliferation
In order to demonstrate the mechanism of EGFR
signaling in the co-culture system, lapatinib, an inhibitor of EGFR
and Her2, was used to inhibit this signaling pathway. Lapatinib
resulted in the attenuation of EGFR signaling activated by
co-culture compared with co-culture + 0.5% DMSO (Fig. 3A), which was accompanied by
attenuated malignancy (Fig. 3B and
C). In addition, this signaling inhibition ameliorated the
co-culture induced proliferative ability of T24/253J cells
(Fig. 3D-G). This indicated that
the EGFR pathway was a key regulator of interactions between
bladder cancer cells and ECs. Furthermore, the inhibition of EGFR
signaling also led to the reversal of epithelial-mesenchymal
transition (EMT) marker expression including the downregulation of
MMP2, MMP9, N-cadherin, ZEB-1 and survivin (Fig. 3H). These results are in accordance
with those from a previous study (10), which indicated that the EGFR
pathway was a key EMT regulator.
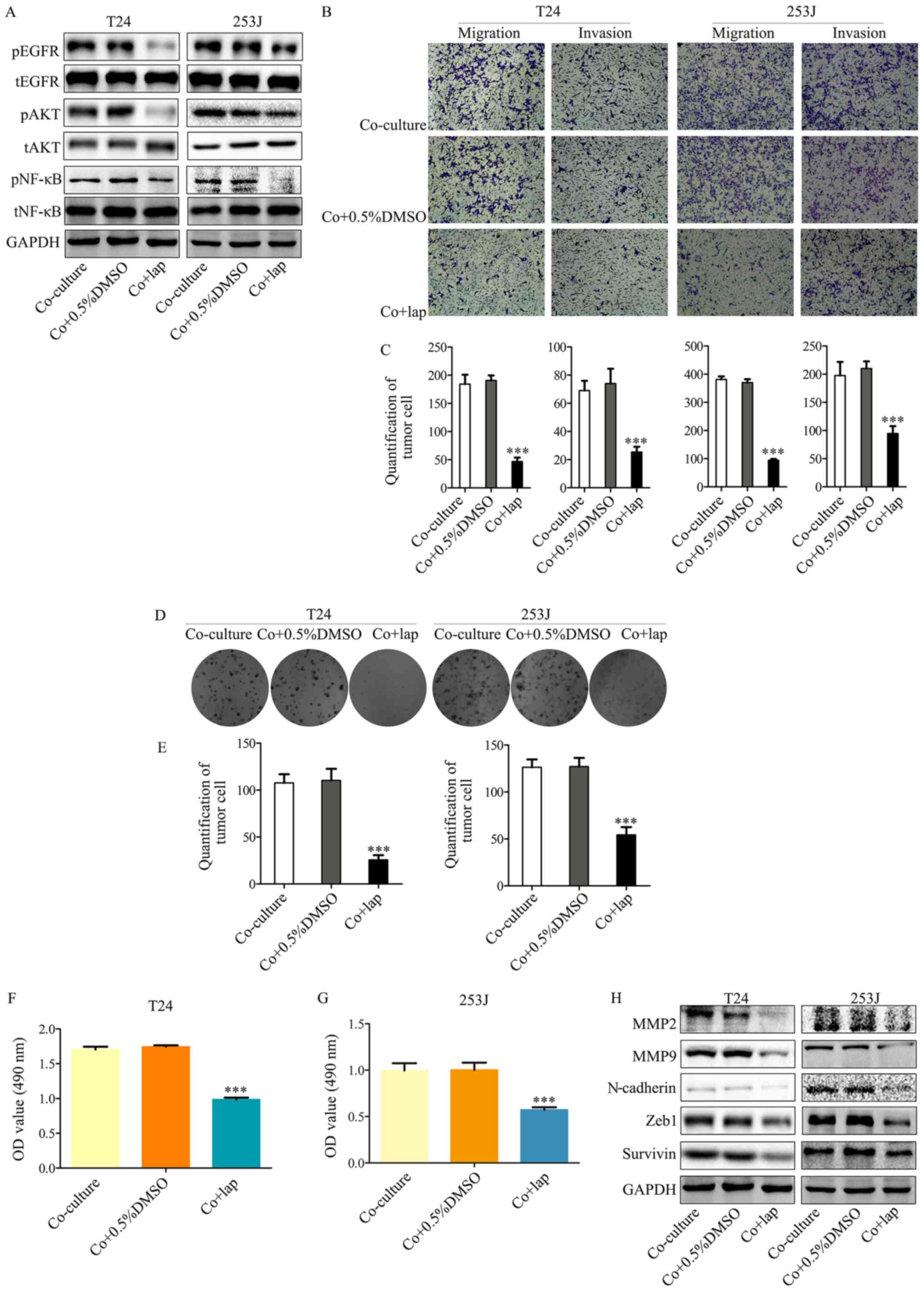 | Figure 3Inhibition of EGFR signaling in
bladder cancer cells by lapatinib in a co-culture system abrogates
co-culture induced cancer cell malignancy and proliferation. (A)
Western blot analysis indicates that downstream EGFR signaling was
inhibited by co-culture treatment with lapatinib. (B and C)
Transwell migration assays indicated that co-culture induced
malignancy of T24/253J is attenuated in the presence of lapatinib
(magnification, ×200). (D and E) Colony formation assay indicated
that the co-culture induced enhanced proliferation of T24/253J is
ameliorated in the absence of EGFR signaling (magnification, ×200).
(F and G) MTT assay determination of bladder cancer T24/253J cell
proliferation following co-culture treatment with lapatinib. (H)
Western blot analysis indicates that in co-culture system the
proteins MMP2, MMP9, ZEB-1, survivin and N-cadherin were
downregulated by inhibited EGFR signaling. ***P<0.001
vs. co-culture and co+0.5%DMSO. EGFR, epidermal growth factor
receptor; co+0.5%DMSO, co-culture + 0.5% dimethyl sulfoxide; MMP,
matrix metalloprotein; ZEB, zinc finger E-box-binding homeobox;
co+lap, co-culture + lapatinib; OD, optical density; p,
phosphorylated; t, total; AKT, protein kinase B; NF, nuclear
factor. |
Co-culture system activates VEGFR2
signaling in EC and upregulates EGFR ligand expression
Both EGFR and its ligands are transmembrane
proteins. Previous studies have provided evidence that
ligand-receptor binding activates the cytoplasmic tyrosine kinase
domains of EGFR, resulting in signal transduction to promote cell
proliferation, migration, differentiation and survival (21). The present results indicated that
the co-culture system activates EGFR signaling. In order to
elucidate the mechanism underlying this phenomenon, EGFR ligands
were monitored in control and co-culture systems. As indicated, the
EGFR ligands except for heparin-binding EGF-like growth factor,
have different expression profiles in HUVECs, when compared with in
T24 or 253J cells (Fig. 4A and B).
This indicated that there is a feature of the co-culture system
that may contribute to the upregulation of EGFR ligands.
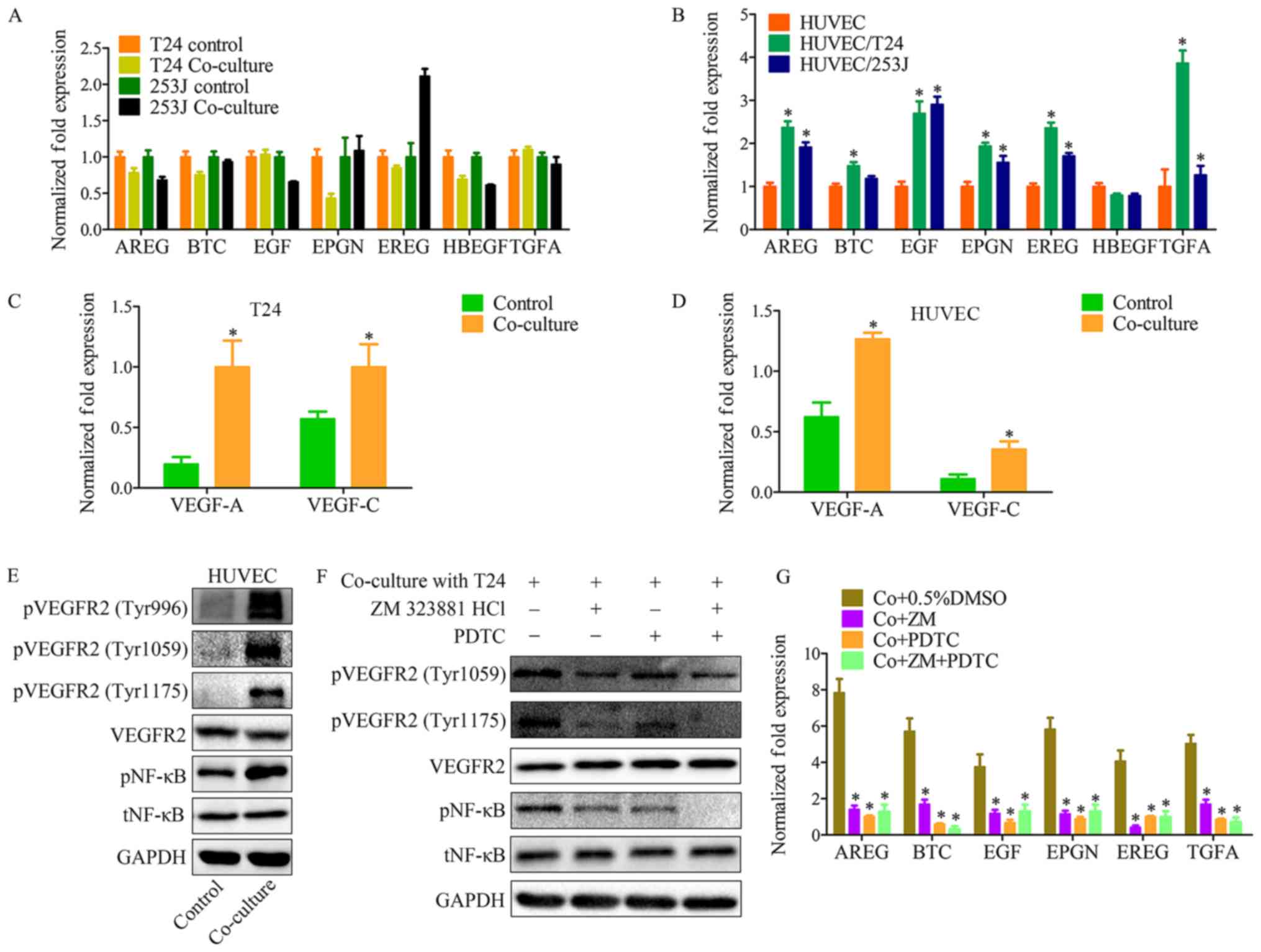 | Figure 4The VEGFR2 pathway of endothelial
cells is triggered by VEGF-A and VEGF-C in co-culture system, and
induces EGFR ligand upregulation. (A) RT-qPCR for screening EGFR
ligands in T24/253J following co-culture with HUVECs. EREG are
significantly elevated in 253J by co-culture. (B) RT-qPCR for
screening EGFR ligands in HUVECs following co-culture with
T24/253J. Except for HBEGF, EGFR ligands are significantly elevated
in HUVEC after co-culture; co-culture vs. control,
*P<0.05; RT-qPCR for screening VEGF-A and VEGFR-C in
(C) T24 and (D) HUVECs following co-culture; co-culture vs.
control, *P<0.05. (E) Western blot analysis
demonstrated that VEGFR2 signaling was induced in HUVECs by
co-culture with T24. pVEGFR2-Tyr996, pVEGFR2-Tyr1059, and
pVEGFR2-Tyr1175 were upregulated compared with the control group,
and the downstream NF-κB pathway was upregulated compared with the
control group. (F) Western blot analysis indicates that in the
co-culture system, VEGFR2 signaling inhibition by ZM 323881 HCL and
NF-κB pathway was inhibited by PDTC. (G) RT-qPCR for screening the
expression of EGFR ligands in HUVECs following co-culture with
T24/253J, followed by inhibition of the EGFR-NF-κB pathway.
*P<0.05 vs. co+0.5%DMSO. VEGF, vascular endothelial
growth factor; R, receptor; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; EGFR,
epidermal growth factor receptor; HUVEC, human umbilical vein
endothelial cell; HBEGF, heparin-binding EGF-like growth factor;
NF, nuclear factor; Co+0.5%DMSO, co-culture + 0.5% dimethyl
sulfoxide; Co+ZM, co-culture+ZM 323881 HCl; Co+PDTC,
co-culture+PDTC; Co+ZM+PDTC, co-culture+ZM 323881 HCl+PDTC; p,
phosphorylated; t, total; EGF, epidermal growth factor; AREG,
amphiregulin; EREG, epiregulin; BTC, betacellulin; TNFA, tumor
necrosis factor α; EPGN, epithelial mitogen. |
It has previously been demonstrated that VEGFs and
VEGFRs have important roles in both physiological vascular
development and pathological diseases, for example, tumor
angiogenesis. VEGFR-2 (Flk-1/KDR) is expressed in ECs and lymphatic
ECs (22). VEGFR-2 has three
ligands: VEGF-A, VEGF-C and VEGF-D. These ligands bind to VEGFR to
induce activation of intracellular signaling cascades results in
proliferation, migration, survival and increased permeability
(23). With the co-culture system
it was demonstrated that both T24 cancer cells and HUVECs have a
high level of VEGF-A and VEGF-C expression compared with the
control group (Fig. 4C and D). In
addition, VEFGR2 protein expression was analyzed in the co-culture
system, which revealed that VEGFR2 signaling in HUVECs was induced
by co-culture (Fig. 4E and F). A
number of techniques, such as cDNA microarray analysis, have
previously been used to identify genes that are upregulated in ECs
following stimulation with VEGF (23). The present findings suggest that
the VEGFR2 signaling pathway has an effect on the expression of
those EGFR ligands. Education of HUVECs by co-culture was repeated
in the presence of ZM 323881 HCl (an inhibitor of VEGFR2) and PDTC,
and RT-qPCR was used to assess the expression of EGFR ligands in
tumor cells (Fig. 4G).
Co-culture enhances EC recruitment
through the EGFR-NF-κB-CXCL1/5/8-CXCR2 signaling pathways
In the present study it was demonstrated that
T24/253J cells educated by co-culture exhibited enhanced HUVEC
recruitment ability compared with the control group (Fig. 5A and B). It is known that the CXC
chemokine has an important role in the processes of tumor
angiogenesis and cell recruitment. Subsequently, it was observed
that CXCL1, CXCL5 and CXCL8 were upregulated in cancer cells when
co-cultured with HUVECs (Fig. 5C),
and that CXCR2 (the receptor for CXCL1/5/8) was upregulated in
HUVECs when co-cultured with cancer cells (Fig. 5D). In addition, CXCR2 was inhibited
in HUVECs by co-culturing for 36 h with SB225002 (an inhibitor of
CXCR2); the results indicated that the co-culture system enhanced
the EC recruitment ability through the CXC chemokine and its
receptor (Fig. 5E and F). However,
the mechanisms involved in the upregulation of CXCL1, CXCL5 and
CXCL8 in cancer cells are not fully understood. Our previous study
has demonstrated that the EGFR signaling pathway and its downstream
signaling was activated by co-culture in cancer cells. This
previous study also revealed that B cell lymphoma-2 upregulated CXC
chemokine expression in ECs through the NF-κB pathway (24); it is known that the NF-κB pathway
has a key role in the transcription of several cytokines and growth
factors (25). These observations
led to the hypothesis that in co-culture system, EGFR signaling
induces the expression of CXCL1/5/8 in cancer cells and enhances EC
recruitment ability. To test this hypothesis, the EGFR signaling
and downstream NF-κB pathway were inhibited; the results
demonstrated that the co-culture induced enhanced HUVEC recruitment
of T24/253J cells was attenuated in the absence of the EGFR/NF-κB
signaling pathway (Fig. 5G-I). In
addition, inhibition of the EGFR/NF-κB signaling pathway in bladder
cancer cells though the co-culture system downregulated the gene
expression of CXCL1/5/8 (Fig. 5J and
K).
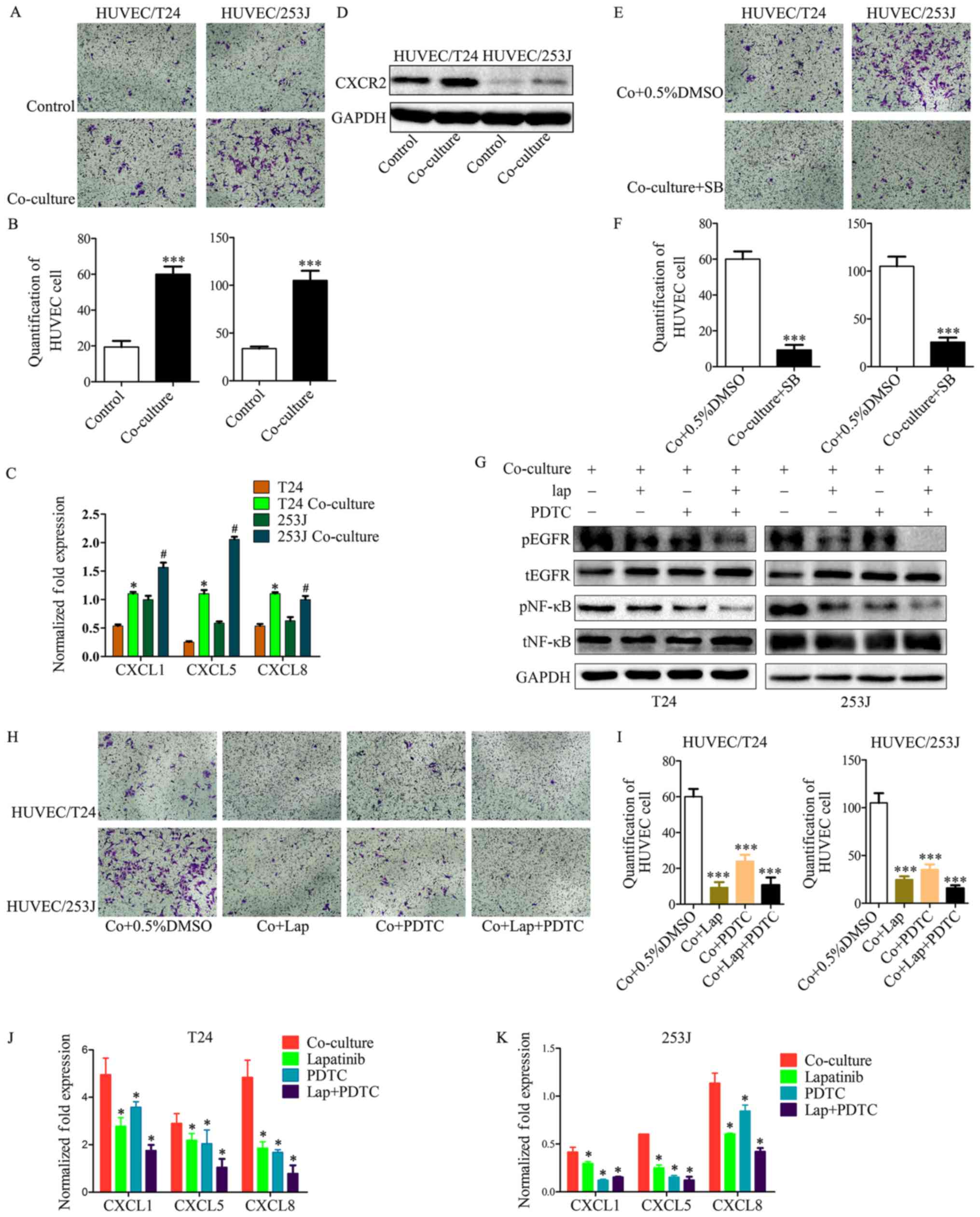 | Figure 5Cancer cell treatment by the
co-culture system led to enhanced HUVEC recruitment. (A and B)
T24/253J treatment by co-culture with HUVECs and enhanced HUVEC
recruitment (magnification, ×200). ***P<0.001 vs.
control. (C) RT-qPCR for screening the expression of CXCL1, CXCL5
and CXCL8 in T24/253J following co-culture with HUVECs.
*P<0.05 vs. T24; #P<0.05 vs. 253J. (D)
Western blot analysis indicates that the co-culture of HUVECs leads
to the upregulation of CXCR2. (E and F) Inhibition of the CXCR2
pathway following co-culture treatment in HUVECs, and reduced HUVEC
recruitment (magnification, ×200). ***P<0.001 vs.
Co+0.5%DMSO. (G) Western blot analysis indicates inhibition of the
EGFR-NF-κB pathway in T24/253J following co-culture with HUVECs. (H
and I) Inhibition of the EGFR-NF-κB pathway in T24/253J following
co-culture with HUVECs, and reduced endothelial cell recruitment
(magnification, ×200). ***P<0.001 vs. Co+0.5%DMSO. (J
and K) RT-qPCR for screening the expression of CXCL1, CXCL5 and
CXCL8 in T24/253J following co-culture with HUVECs, followed by
inhibition of the EGFR-NF-κB pathway. *P<0.05 vs.
co-culture. HUVEC, human umbilical vein endothelial cell;
co+0.5%DMSO, co-culture + 0.5% dimethyl sulfoxide; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; EGFR,
epidermal growth factor receptor; NF, nuclear factor; SB, SB225002;
lap, lapatinib; p, phosphorylated; t, total. |
Discussion
The present data suggest a novel site for vascular
EC and cancer cell interaction in the tumor microenvironment. It
was demonstrated that EC has an active role in promoting cancer
cell proliferation, survival, migration and invasion. A number of
previous reports suggested a role for macrophages (26,27)
and neutrophils (28) in the
progression of bladder cancer. However, the role of ECs in the
tumor microenvironment in bladder cancer is only beginning to be
unveiled. The present study provides evidence for ECs as key
players in bladder cancer progression.
The present results suggest that ECs have an active
role in bladder cancer. It was demonstrated that EGFR was
phosphorylated in bladder cancer cells by the EC-secreted EGFR
ligands in the co-culture system. This finding is in accordance
with observations from other reports that have characterized growth
factors as critical in cancer progression (29,30).
These findings demonstrated that EGFR ligands, including epidermal
growth factor (EGF), amphiregulin, epiregulin, betacellulin, tumor
necrosis factor-α and epithelial mitogen are upregulated in EC by
co-culturing (28,29). EGFR overexpression has been
identified in a variety of tumors, including non-small cell lung
cancer (31), breast cancer
(32), prostate cancer (33) and bladder cancer (34). The expression of EGFR ligands and
EGFR is correlated with cancer proliferation, survival, and
metastasis. For example, cancer cell proliferation, migration, and
invasion are increased through the EGFR-Ras/Raf/MEK/ERK and
EGFR-PI3K/AKT pathways (35). A
previous study has demonstrated that vascular ECs secrete CXCL8,
IL-6 and EGF following co-culture with head and neck squamous cell
carcinoma cells. It was observed that the primary effect of
EC-derived IL-6 was the activation of STAT3, the primary effect of
CXCL8 was on the activity of AKT, and the primary effect of EGF was
on ERK activity in head and neck squamous cell carcinoma cells
(15). However, the present
results indicated that AKT and NF-κB pathways were downstream of
EGFR signaling, and can be activated by EGFR signaling in the
co-culture system.
In the tumor vasculature, VEFGR2 expression is
upregulated compared with normal vasculature (22). In addition, VEGFR2 expression is a
prognostic marker for the clinical outcome of patients (36). It has been demonstrated previously
that stimulation with VEGF upregulated a number of genes in
vascular EC. For example, Ets-1, MMP1 and Fil-1 were stimulated by
VEGF via VEGFR2 signaling (37).
In the present study, it was demonstrated that VEGFR2 signaling was
activated by co-culture, as was downstream NF-κB. VEGFR2 was
inhibited in the vascular ECs following co-culture education, and
the results indicated that the expression of EGFR ligands is
regulated through the VEGFR2/NF-κB pathway in the co-culture
system. The findings also demonstrated that the expression of both
VEGF-A and VEGF-C in cancer cells and vascular ECs is upregulated
following co-culture treatment. The mechanism behind this remains
to be elucidated. The ELR+ chemokine has been reported
to induce vascular EC migration in vitro and promote
angiogenesis in vivo (38).
A previous study revealing that the ELR+ chemokines
CXCL5 and CXCL8 bind to CXCR2 and induce neovascularization, and
that the neutralization of CXCL5 or CXCL8 using specific
neutralizing antibodies against these small molecules may inhibit
the chemokine mediated angiogenesis (39).
In the present study, the expression levels of
CXCL1, CXCL5, and CXCL8 were upregulated in bladder cancer cells by
co-culture treatment, and the expression of CXCR2 was upregulated
in ECs. Following co-culture treatment CXCR2 was inhibited using
SB225002. These results indicated that vascular EC interactions
with bladder cancer cells induce vascular EC recruitment though the
CXCL1/CXCL5/CXCL8-CXCR2 pathway. Multiple signaling pathways
contribute to CXCL1, CXCL5, and CXCL8 regulation. EGFR ligands, for
example transforming growth factor-α and amphiregulin, induce CXCL8
expression in bronchial epithelial cells and mediate cigarette
smoke-induced CXCL8 expression through an autocrine loop (40,41).
CXCL1 is critical in cancer progression and a previous study
revealed that EGF activation of the PI3K pathway induced the
expression of CXCL1 (42).
Furthermore, the expression of CXCL5 has been reported to be
upregulated by NF-κB family members, or by the p53 pathway
(25, 43). In the present study, the activation
of EGFR signaling in the co-culture system was inhibited using
lapatinib, and the results indicated that EGFR ligands were
involved in the regulation of CXCL1, CXCL5 and CXCL8. In addition,
the EGFR pathway and/or the NF-κB pathway were inhibited in the
co-culture system, and the results indicated that CXCL1, CXCL5 and
CXCL8 were upregulated in bladder cancer cells through the EGFR
pathway. A summary of the present study is presented in Fig. 6.
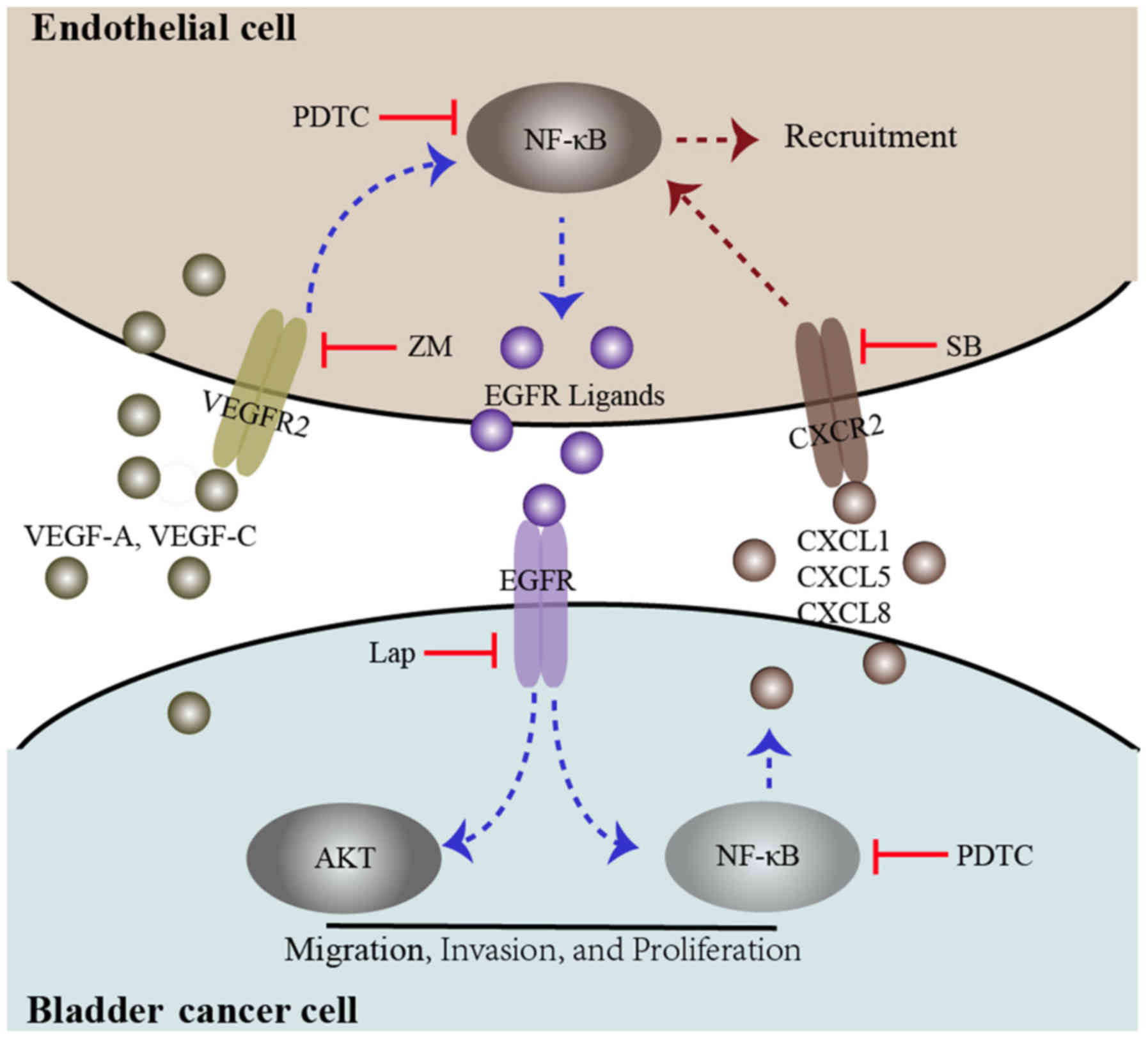 | Figure 6Diagram proposing a model for the
interaction between bladder cancer cells and endothelial cells. In
the co-culture system, both bladder cancer cells and endothelial
cells secrete the VEGFR2 ligands VEGF-A and VEGF-C, which induce
VEGFR2 signaling and downstream NF-κB signaling, promoting EGFR
ligand expression. These events may be inhibited by a VEGFR2
inhibitor, ZM and an NF-κB inhibitor (PDTC). EGFR signaling in
bladder cancer cells was triggered by EGFR ligands secreted by
endothelial cells, which induces phosphorylation of AKT and NF-κB.
These events enhance bladder cancer migration, invasion, and
proliferation. Furthermore, activated EGFR signaling in bladder
cancer cells could enhance endothelial cell recruitment through the
upregulation of CXCL1, CXCL5 and CXCL8. These events could be
inhibited by an EGFR inhibitor, lap, PDTC and a CXCR2 inhibitor,
SB. VEGF, vascular endothelial growth factor; R, receptor; NF,
nuclear factor; EGFR, epidermal growth factor receptor; ZM, ZM
323881 HCL; AKT, protein kinase B; lap, lapatinib; SB,
SB225002. |
In conclusion, the present study demonstrated that
interactions of bladder cancer cells with vascular ECs enhance
vascular EC recruitment by cancer cells through the CXC chemokine
and CXCR2 signaling pathways. The recruited vascular ECs interact
with bladder cancer cells and tissues to promote cancer progression
through the EGFR signaling pathway. The present study may also
illuminate mechanisms by which the tumor microenvironment promotes
malignant progression in other cancer types, including colon,
prostate and breast cancer.
Funding
This study was supported by grants from the National
Natural Science Foundation of China (grant no. 81572520) and the
Key Science and Technology Program of Shaanxi Province, China
(grant no. 2015SF176).
Availability of data and materials
The analyzed datasets generated during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZH wrote the main manuscript. ZH, MZ, GC and YY
performed the experiments. JF, ZG and ZH designed the study. WW, XW
and PZ performed data analysis. ZH and JF contributed to manuscript
revisions. All authors reviewed the manuscript. All authors read
and approved the final manuscript
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicts of
interest.
Acknowledgments
Not applicable.
References
|
1
|
Chavan S, Bray F, Lortet-Tieulent J,
Goodman M and Jemal A: International variations in bladder cancer
incidence and mortality. Eur Urol. 66:59–73. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Smith ZL and Guzzo TJ: Urinary markers for
bladder cancer. F1000Prime Rep. 5:212013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Calderaro J, Rebouissou S, de Koning L,
Masmoudi A, Hérault A, Dubois T, Maille P, Soyeux P, Sibony M, de
la Taille A, et al: PI3K/AKT pathway activation in bladder
carcinogenesis. Int J Cancer. 134:1776–1784. 2014. View Article : Google Scholar
|
|
4
|
Ching CB and Hansel DE: Expanding
therapeutic targets in bladder cancer: The PI3K/Akt/mTOR pathway.
Lab Invest. 90:1406–1414. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
López-Knowles E, Hernández S, Malats N,
Kogevinas M, Lloreta J, Carrato A, Tardón A, Serra C and Real FX:
PIK3CA mutations are an early genetic alteration associated with
FGFR3 mutations in superficial papillary bladder tumors. Cancer
Res. 66:7401–7404. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu X, Obata T, Khan Q, Highshaw RA, De
Vere White R and Sweeney C: The phosphatidylinositol-3 kinase
pathway regulates bladder cancer cell invasion. BJU Int.
93:143–150. 2004. View Article : Google Scholar
|
|
7
|
Oka N, Tanimoto S, Taue R, Nakatsuji H,
Kishimoto T, Izaki H, Fukumori T, Takahashi M, Nishitani M and
Kanayama HO: Role of phosphatidylinositol-3 kinase/Akt pathway in
bladder cancer cell apoptosis induced by tumor necrosis
factor-related apoptosis-inducing ligand. Cancer Sci. 97:1093–1098.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mukherjee N, Houston TJ, Cardenas E and
Ghosh R: To be an ally or an adversary in bladder cancer: The NF-κB
story has not unfolded. Carcinogenesis. 36:299–306. 2015.
View Article : Google Scholar
|
|
9
|
Guan Z, Ding C, Du Y, Zhang K, Zhu JN,
Zhang T, He D, Xu S, Wang X and Fan J: HAF drives the switch of
HIF-1α to HIF-2α by activating the NF-κB pathway, leading to
malignant behavior of T24 bladder cancer cells. Int J Oncol.
44:393–402. 2014. View Article : Google Scholar
|
|
10
|
Bennasroune A, Gardin A, Aunis D, Crémel G
and Hubert P: Tyrosine kinase receptors as attractive targets of
cancer therapy. Crit Rev Oncol Hematol. 50:23–38. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Folkman J: Role of angiogenesis in tumor
growth and metastasis. Semin Oncol. 29(Suppl 16): 15–18. 2002.
View Article : Google Scholar
|
|
12
|
Sparmann A and Bar-Sagi D: Ras-induced
interleukin-8 expression plays a critical role in tumor growth and
angiogenesis. Cancer Cell. 6:447–458. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Warner KA, Miyazawa M, Cordeiro MMR, Love
WJ, Pinsky MS, Neiva KG, Spalding AC and Nör JE: Endothelial cells
enhance tumor cell invasion through a crosstalk mediated by CXC
chemokine signaling. Neoplasia. 10:131–139. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zeng Q, Li S, Chepeha DB, Giordano TJ, Li
J, Zhang H, Polverini PJ, Nor J, Kitajewski J and Wang CY:
Crosstalk between tumor and endothelial cells promotes tumor
angiogenesis by MAPK activation of Notch signaling. Cancer Cell.
8:13–23. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Z, Dong Z, Lauxen IS, Filho MS and
Nör JE: Endothelial cell-secreted EGF induces epithelial to
mesenchymal transition and endows head and neck cancer cells with
stem-like phenotype. Cancer Res. 74:2869–2881. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ferrara N, Hillan KJ, Gerber HP and
Novotny W: Discovery and development of bevacizumab, an anti-VEGF
antibody for treating cancer. Nat Rev Drug Discov. 3:391–400. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shaulian E and Karin M: AP-1 as a
regulator of cell life and death. Nat Cell Biol. 4:E131–E136. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
19
|
Fus LP and Górnicka B: Role of
angiogenesis in urothelial bladder carcinoma. Cent European J Urol.
69:258–263. 2016.PubMed/NCBI
|
|
20
|
Rajjayabun PH, Keegan PE, Lunec J and
Mellon JK: erbB receptor expression patterns in human bladder
cancer. Urology. 66:196–200. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nicholson RI, Gee JM and Harper ME: EGFR
and cancer prognosis. Eur J Cancer. 37(Suppl 4): pp. S9–S15. 2001,
View Article : Google Scholar
|
|
22
|
Plate KH, Breier G, Millauer B, Ullrich A
and Risau W: Up-regulation of vascular endothelial growth factor
and its cognate receptors in a rat glioma model of tumor
angiogenesis. Cancer Res. 53:5822–5827. 1993.PubMed/NCBI
|
|
23
|
Holmes K, Roberts OL, Thomas AM and Cross
MJ: Vascular endothelial growth factor receptor-2: Structure,
function, intracellular signalling and therapeutic inhibition. Cell
Signal. 19:2003–2012. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kaneko T, Zhang Z, Mantellini MG, Karl E,
Zeitlin B, Verhaegen M, Soengas MS, Lingen M, Strieter RM, Nunez G,
et al: Bcl-2 orchestrates a cross-talk between endothelial and
tumor cells that promotes tumor growth. Cancer Res. 67:9685–9693.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Richmond A: Nf-kappa B, chemokine gene
transcription and tumour growth. Nat Rev Immunol. 2:664–674. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Miyake M, Hori S, Morizawa Y, Tatsumi Y,
Nakai Y, Anai S, Torimoto K, Aoki K, Tanaka N, Shimada K, et al:
CXCL1-mediated interaction of cancer cells with tumor-associated
macrophages and cancer-associated fibroblasts promotes tumor
progression in human bladder cancer. Neoplasia. 18:636–646. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tian YF, Tang K, Guan W, Yang T, Xu H,
Zhuang QY and Ye ZQ: OK-432 suppresses proliferation and metastasis
by tumor associated macrophages in bladder cancer. Asian Pac J
Cancer Prev. 16:4537–4542. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lin C, Lin W, Yeh S, Li L and Chang C:
Infiltrating neutrophils increase bladder cancer cell invasion via
modulation of androgen receptor (AR)/MMP13 signals. Oncotarget.
6:43081–43089. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gunes S, Sullu Y, Yegin Z, Buyukalpelli R,
Tomak L and Bagci H: ErbB receptor tyrosine kinase family
expression levels in urothelial bladder carcinoma. Pathol Res
Pract. 209:99–104. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Appert-Collin A, Hubert P, Crémel G and
Bennasroune A: Role of ErbB receptors in cancer cell migration and
invasion. Front Pharmacol. 6:2832015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gately K, Forde L, Cuffe S, Cummins R, Kay
EW, Feuerhake F and O'Byrne KJ: High coexpression of both EGFR and
IGF1R correlates with poor patient prognosis in resected
non-small-cell lung cancer. Clin Lung Cancer. 15:58–66. 2014.
View Article : Google Scholar
|
|
32
|
Lee HJ, Seo AN, Kim EJ, Jang MH, Kim YJ,
Kim JH, Kim SW, Ryu HS, Park IA, Im SA, et al: Prognostic and
predictive values of EGFR overexpression and EGFR copy number
alteration in HER2-positive breast cancer. Br J Cancer.
112:103–111. 2015. View Article : Google Scholar :
|
|
33
|
Day KC, Lorenzatti Hiles G, Kozminsky M,
Dawsey SJ, Paul A, Broses LJ, Shah R, Kunja LP, Hall C, Palanisamy
N, et al: HER2 and EGFR overexpression support metastatic
progression of prostate cancer to bone. Cancer Res. 77:74–85. 2017.
View Article : Google Scholar :
|
|
34
|
Lipponen P and Eskelinen M: Expression of
epidermal growth factor receptor in bladder cancer as related to
established prognostic factors, oncoprotein (c-erbB-2, p53)
expression and long-term prognosis. Br J Cancer. 69:1120–1125.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lo HW and Hung MC: Nuclear EGFR signalling
network in cancers: Linking EGFR pathway to cell cycle progression,
nitric oxide pathway and patient survival. Br J Cancer. 94:184–188.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fine BA, Valente PT, Feinstein GI and Dey
T: VEGF, flt-1, and KDR/flk-1 as prognostic indicators in
endometrial carcinoma. Gynecol Oncol. 76:33–39. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sato Y, Kanno S, Oda N, Abe M, Ito M,
Shitara K and Shibuya M: Properties of two VEGF receptors, Flt-1
and KDR, in signal transduction. Ann NY Acad Sci. 902:201–205;
discussion 205-207. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Strieter RM, Polverini PJ, Kunkel SL,
Arenberg DA, Burdick MD, Kasper J, Dzuiba J, Van Damme J, Walz A,
Marriott D, et al: The functional role of the ELR motif in CXC
chemokine-mediated angiogenesis. J Biol Chem. 270:27348–27357.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Addison CL, Daniel TO, Burdick MD, Liu H,
Ehlert JE, Xue YY, Buechi L, Walz A, Richmond A and Strieter RM:
The CXC chemokine receptor 2, CXCR2, is the putative receptor for
ELR+ CXC chemokine-induced angiogenic activity. J
Immunol. 165:5269–5277. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Subauste MC and Proud D: Effects of tumor
necrosis factor-alpha, epidermal growth factor and transforming
growth factor-alpha on interleukin-8 production by, and human
rhinovirus replication in, bronchial epithelial cells. Int
Immunopharmacol. 1:1229–1234. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Richter A, O'Donnell RA, Powell RM,
Sanders MW, Holgate ST, Djukanović R and Davies DE: Autocrine
ligands for the epidermal growth factor receptor mediate
interleukin-8 release from bronchial epithelial cells in response
to cigarette smoke. Am J Respir Cell Mol Biol. 27:85–90. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Moscova M, Marsh DJ and Baxter RC: Protein
chip discovery of secreted proteins regulated by the
phosphatidylinositol 3-kinase pathway in ovarian cancer cell lines.
Cancer Res. 66:1376–1383. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yeudall WA, Vaughan CA, Miyazaki H,
Ramamoorthy M, Choi MY, Chapman CG, Wang H, Black E, Bulysheva AA,
Deb SP, et al: Gain-of-function mutant p53 upregulates CXC
chemokines and enhances cell migration. Carcinogenesis. 33:442–451.
2012. View Article : Google Scholar
|