Introduction
With ~1.4 million new cases every year worldwide,
colorectal cancer is the third most common type of cancer with high
incidence and mortality rates (1,2).
Currently, surgery with chemotherapy is the main treatment for
colorectal cancer, but the 5-year survival rate of patients with
colorectal cancer remains low (3).
Therefore, identifying novel anticancer drugs is necessary.
An effective treatment strategy for colorectal
cancer is to induce cell death (4). A number of studies have demonstrated
that cell death mainly includes four major morphologically
processes: Necrosis, apoptosis, autophagy and pyroptosis (5,6).
Autophagy is an important process in evolutionarily conserving
cellular material and energy flow. It is a normal physiological
process of metabolizing cellular material in evolved eukaryotic
organisms and is one form of type II programmed cell death
(7,8). Autophagy enables cells to resist
metabolic stress caused by the lack of nutrition and excessive
proliferation of cells, and may promote the survival of tumor
cells. For example, autophagy is an important cause of drug
resistance in cancer cells during chemotherapy. However, excessive
autophagy may lead to autophagic cell death (9). Therefore, the degree of autophagy
determines whether the cell survives or dies.
Autophagy and apoptosis are both important
mechanisms determining the survival or death of cancer cells.
Cross-talk of autophagy and apoptosis has been documented in the
tumorigenesis and progression of cancer (10), while the interplay between the two
pathways in colorectal cancer has not yet been comprehensively
summarized. Although autophagy and apoptosis are significantly
different in metabolic pathways and morphology, their signaling
pathways are inextricably linked (11,12).
According to different regulation modes, their interaction can be
roughly classified into three types: Cooperative, antagonistic and
promotion relationships (13).
Reactive oxygen species (ROS) are produced by
aerobic cells in the process of metabolism. Previous studies have
demonstrated that ROS can affect a series of intracellular signal
pathways (14). ROS regulate the
balance cell death dependent on their concentration (15). Low level ROS can activate
transcription factors and promote cell proliferation,
differentiation and autophagy (16). However, moderate and high
concentrations of ROS can induce apoptosis and autophagic cell
death (17,18). It has previously been demonstrated
that ROS activate the mitochondrial apoptotic pathway (19). In addition, ROS have been
demonstrated to activate autophagic death signaling pathway through
AMP-activated protein kinase (AMPK)/mechanistic target of rapamycin
(mTOR) signaling pathway (20).
Previous studies have demonstrated that many anticancer drugs can
stimulate the production of ROS in cancer cells, and then lead to
cell apoptosis and autophagic cell death (21,22).
α-hederin, a monodesmosidic triterpenoid saponin, is
a major active ingredient isolated from the leaves of ivy
(Hedera helix L.) or Nigella sativa. Previous studies
have demonstrated that α-hederin has anticancer properties, which
may be attributed to the inhibition of proliferation and motility
(23,24), induction of apoptosis (25,26),
membrane permeabilization (27),
inhibition of epithelial to mesenchymal transition (28) and morphologic changes (27). α-Hederin can activate the
mitochondrial apoptotic pathway by increasing ROS (24,26).
Our previous study has reported that α-hederin can induce apoptosis
in colorectal cancer cells (29).
Based on the effects of ROS on autophagy, it was suggested that
α-hederin may induce autophagy through ROS.
In the present study, to determine whether α-hederin
can induce autophagy in colorectal cancer cells, the effects of
α-hederin on colorectal cancer cells were investigated. As such,
its effects on autophagy and the underlying mechanisms were
explored. In addition, the interplay between the apoptosis and
autophagy in colorectal cancer was also explored.
Materials and methods
Cells and reagents
HCT116 and HCT8 human colorectal cancer cells were
purchased from the Type Culture Collection of the Chinese Academy
of Sciences (Shanghai, China). Both cell lines were cultured in
RPMI-1640 medium supplemented with 10% (v/v) fetal bovine serum
(both from Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA), 100 µg/ml streptomycin and 100 U/ml penicillin at 37°C
and 5% CO2. α-hederin was purchased from Sigma-Aldrich;
Merck KGaA (Darmstadt, Germany). ROS inhibitor N-acetyl-L-cysteine
(NAC), oxidation-activated fluorescent dye DCHF-DA and Hoechst
33258 were purchased from Beyotime Institute of Biotechnology
(Haimen, China). Autophagy inhibitor 3-MA was purchased from
Sigma-Aldrich; Merck KGaA.
Antibodies
Anti-light chain (LC)3 (12135-1-AP), anti-caspase-9
(10380-1-AP), anti-caspase-3 (19677-1-AP), anti-P62 (18420-1-AP)
and anti-Beclin 1 (11306-1-AP) antibodies were purchased from
ProteinTech Group, Inc. (Chicago, IL, USA). Anti-phosphorylated
(p)-mTOR (#2971), anti-mTOR (#2972), anti-β-actin (8456),
anti-p-Unc-51 like autophagy activating kinase 1 (ULK1; #4634),
anti-ULK1 (#4773), anti-p-P70S6K (#9205), anti-P70S6K (#9202),
anti-poly(ADP-ribose) polymerase (PARP; #9532), anti-p-AMPK
(#2531), anti-AMPK (#2532), anti-B cell lymphoma (Bcl)-2 (#2872),
anti-Bcl-2 associated X protein (Bax; #2772), anti-Bcl-xl (#2762),
horseradish peroxidase (HRP)-linked anti-rabbit immunoglobulin
(Ig)G (#7074) and HRP-linked anti-mouse IgG (#7076) were obtained
from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Flow cytometry assays
Flow cytometry assays were performed on a
FACSCalibur system (Becton-Dickinson and Company, Franklin Lakes,
NJ, USA). Apoptosis rate assay was performed using an Annexin
V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis
detection kit (BD Biosciences, San Jose, CA, USA). Cells were
digested using trypsin, washed twice with cold PBS and then
incubated with Annexin V-FITC and PI at room temperature for 30
min. Subsequently, the apoptosis rate was determined using flow
cytometry. Intracellular ROS levels were determined through flow
cytometry using DCHF-DA, an oxidation-activated fluorescent dye.
Cells in dishes were loaded with PBS containing 10 µM
DCHF-DA for 20 min at 37°C. Then, cells were washed thrice with
cold PBS, digested with trypsin and washed twice with cold PBS in
turn. ROS assay was performed using a FACSCalibur flow cytometer
(Becton-Dickinson and Company) and the data were analyzed using
FlowJo software, version 7.6 (Tree Star, Inc., Ashland, OR,
USA).
Hoechst 33258 staining
Cells were digested, dispersed and plated in a
96-well plate at a density of 1×105 cells/well. Cells
were incubated with Hoechst 33258 (0.5 µg/ml; Beyotime
Institute of Biotechnology) for 3 min at room temperature. Washed 3
times with PBS, cells were observed and photographed directly under
a microscope.
Cell viability assay
Cells were digested, dispersed and plated in a
96-well plate at a density of 1×105 cells/well. Cell
viability was analyzed using Cell Counting Kit-8 (Dojindo Molecular
Technologies, Inc., Kumamoto, Japan) according to the
manufacturer's protocol. Absorbances of the wells were read at 450
nm using a plate reader (Bio-Rad Laboratories, Inc., Hercules, CA,
USA).
Colony formation assay
Single cells were seeded into a 35-mm cell culture
dish at a density of 1,000 cells/well and cultured with α-hederin
at concentrations of 0, 10 and 60 µM for 14 days. The medium
was replaced every 2 days, a total of six times. Ultimately, cells
were treated in turn with 4% paraformaldehyde for 20 min and
crystal for 20 min, and were washed with PBS at least twice in room
temperature. The colonies were counted, and images were captured.
Colonies were determined as those able to form a colony consisting
of ≥50 cells.
Western blot analysis
Cells were lysed using cell lysis buffer (Beyotime
Institute of Biotechnology) with Protease Inhibitor Cocktail (100X;
Sangon Biotech Co., Ltd., Shanghai, China) and 1 mM
phenylmethylsulfonyl fluoride (Sigma-Aldrich; Merck KGaA).
Following centrifugation at 4°C and 12,000 × g for 15 min, protein
concentration in the supernatant of the lysates was determined
using the Bradford Coomassie Blue G-250 method. Proteins (40
µg per lane) were separated using 10% SDS-PAGE, and
transferred onto polyvinylidene difluoride (PVDF) membranes (EMD
Millipore, Billerica, MA, USA) in sequence. Thereafter, PVDF
membranes were blocked with 5% bovine serum albumin (Sigma-Aldrich;
Merck KGaA) in TBS containing 0.05% Tween-20 at room temperature
for 2 h. Subsequently, PVDF membranes were incubated with primary
antibodies at 1:1,000 in TBS containing 0.05% Tween-20 and 5%
bovine serum albumin overnight at 4°C for 16 h. Next, PVDF
membranes were incubated with corresponding HRP-conjugated
secondary antibodies (1:1,000) at room temperature for 2 h.
Finally, membranes were visualized using an enhanced
chemiluminescence Western blot detection system (EMD Millipore). In
the present study, all relative protein concentrations were
normalized to that of β-actin.
Lentivirus and cell fluorescence
analysis
LC3 lentivirus and mutation lentivirus were
purchased from Shanghai GeneChem Co., Ltd. (Shanghai, China). The
LC3 lentivirus consisted of the red fluorescent protein Stub-RFP,
green fluorescent protein Sens-GFP, and autophagy marker protein
LC3. HCT116 and HCT8 cells were infected with these lentiviruses to
establish four cells line stably expressing LC3 (HCT116 LC3, HCT8
LC3) or LC3-mutation (HCT116 LC3-mu, HCT8 LC3-mu).
At first, cells were digested, dispersed and plated
in a 24-well plate. When cells reached 60% confluence, the medium
was replaced with infection mixture. The mixture contained
RPMI-1640 medium, lentiviruses (MOI=100) and polybrene (5
µg/ml; Shanghai GeneChem Co., Ltd.). Following incubation
for 24 h, the mixture was replaced with normal medium containing
FBS. These established cell lines were used to detect the
occurrence of autophagy. In the absence of autophagy, the
RFP-GFP-LC3 fusion protein diffuses in the cytoplasm. When
autophagy is formed, it translocates into the autophagosome
membrane. The fusion protein will form bright fluorescent spots,
which can be observed under fluorescence microscope. Fluorescent
dot aggregation was observed via confocal laser microscopy. One
spot is equivalent to an autophagosome, and the activity of
autophagy can be evaluated by counting. As the control,
RFP-GFP-LC3-mu fusion protein could not participate in the
formation of autophagosome membrane.
RNA interference
Small interfering (si)RNA for the human AMPK gene
(Gene ID: 94557300) was synthesized by Biomics Biotechnologies Co.,
Ltd. (Nantong, China). The AMPK siRNA sequence was
5′-GAUAUCAGGGAACAUGAAUdTdT-3′. The control sequence was
5′-UAAGGCUAUGAAGAGAUAC-3′. Cells were transfected with the
aforementioned siRNAs at a concentration of 50 nM using
Lipofectamine 3000 (Life Technologies; Thermo Fisher Scientific,
Inc.) for 48 h.
Electron microscopy
After being treated with 10 µM α-hederin for
24 h, HCT116 and HCT8 cells were digested with trypsin and washed
twice with cold PBS. Subsequently, these cells were fixed overnight
using 3% glutaraldehyde in 0.1 M phosphate buffer (pH 7.2) at 4°C
and post-fixed in 1% OsO4 buffer for 1 h at 4°C. After
being dehydrated in a graded series of ethanol, the cells were
embedded in spur resin at 56°C overnight. The spur resins were cut
into ultrathin sections (60 nm) and stained with saturated
solutions of uranyl acetate and lead citrate at room temperature
for 10 min. Finally, the autophagosomes in these cells were
observed using an electron microscope.
In vivo tumor xenograft model
A total of 10 male nude mice (BALB/c nu/nu; age, 5
weeks) were purchased from Shanghai SLAC Laboratory Animal Co.,
Ltd. (Shanghai, China). The initial body weight of all nude mice
was ~18 g. Nude mice were bred in specific pathogen-free (SPF)
conditions at the Laboratory Animals Breeding and Research Centre,
Shanghai University of Traditional Chinese Medicine (Shanghai,
China). The housing conditions for mice were as follows: Room
temperature, 20-26°C and 12-h light/dark cycle. The mice received 5
g food and 100 ml water per 100 g body weight per day. Two million
HCT116 cells in 0.1 ml PBS were injected into subcutaneous tissues
of each mouse. After 2 weeks, mice were weighed and randomly
divided into control group and α-hederin group (n=5/group).
α-Hederin group mice were injected intraperitoneally with α-hederin
(2 mg/kg body weight) and the control group were injected
intraperitoneally with the same volume of PBS (control) every 3
days for 3 weeks. Finally, the tumor-bearing mice were sacrificed
and the tumors were excised and weighed. The protocol was approved
by the Ethics Committee of Shuguang Hospital affiliated with
Shanghai University of Traditional Chinese Medicine.
Immunohistochemistry assay
All tumors from tumor xenograft model were
formalin-fixed for 24 h at room temperature, embedded in paraffin,
sectioned serially to 5-µm thickness and mounted on glass
slides. Subsequently, immunohistochemical staining of LC3 was
carried out using a standard protocol (30). Anti-LC3 antibody (18725-1-AP; 1:50)
for immunohistochemistry was purchased from ProteinTech Group, Inc.
HRP-labeled goat anti-human IgG (A0201; 1:50) was purchased from
Beyotime Institute of Biotechnology. Anti-LC3 antibody and
HRP-labeled goat anti-human IgG were all diluted in PBS containing
5% bovine serum albumin (Sigma-Aldrich; Merck KGaA). Meanwhile,
serial sections were stained with hematoxylin and eosin
(H&E).
Statistical analysis
All data were subjected to a single-factor analysis
of variance with post hoc Fisher's least significant different
test, or Student's t-test using the SPSS 13.0 software package
(SPSS, Inc., Chicago, IL, USA). Data are presented as the mean ±
standard deviation. P<0.05 was considered to indicate a
statistically significant difference.
Results
α-hederin inhibits cell viability and
proliferation of colorectal cancer cells
Previous studies have suggested that α-hederin has
anticancer properties against different cancers (24,27,29,31-36),
including colorectal cancer (29).
To evaluate the effect of α-hederin on the proliferation of
colorectal cancer cells, its effect on the viability of two
colorectal cancer cell lines (HCT116 and HCT8) was tested.
Furthermore, colony formation assay was performed to determine cell
proliferation.
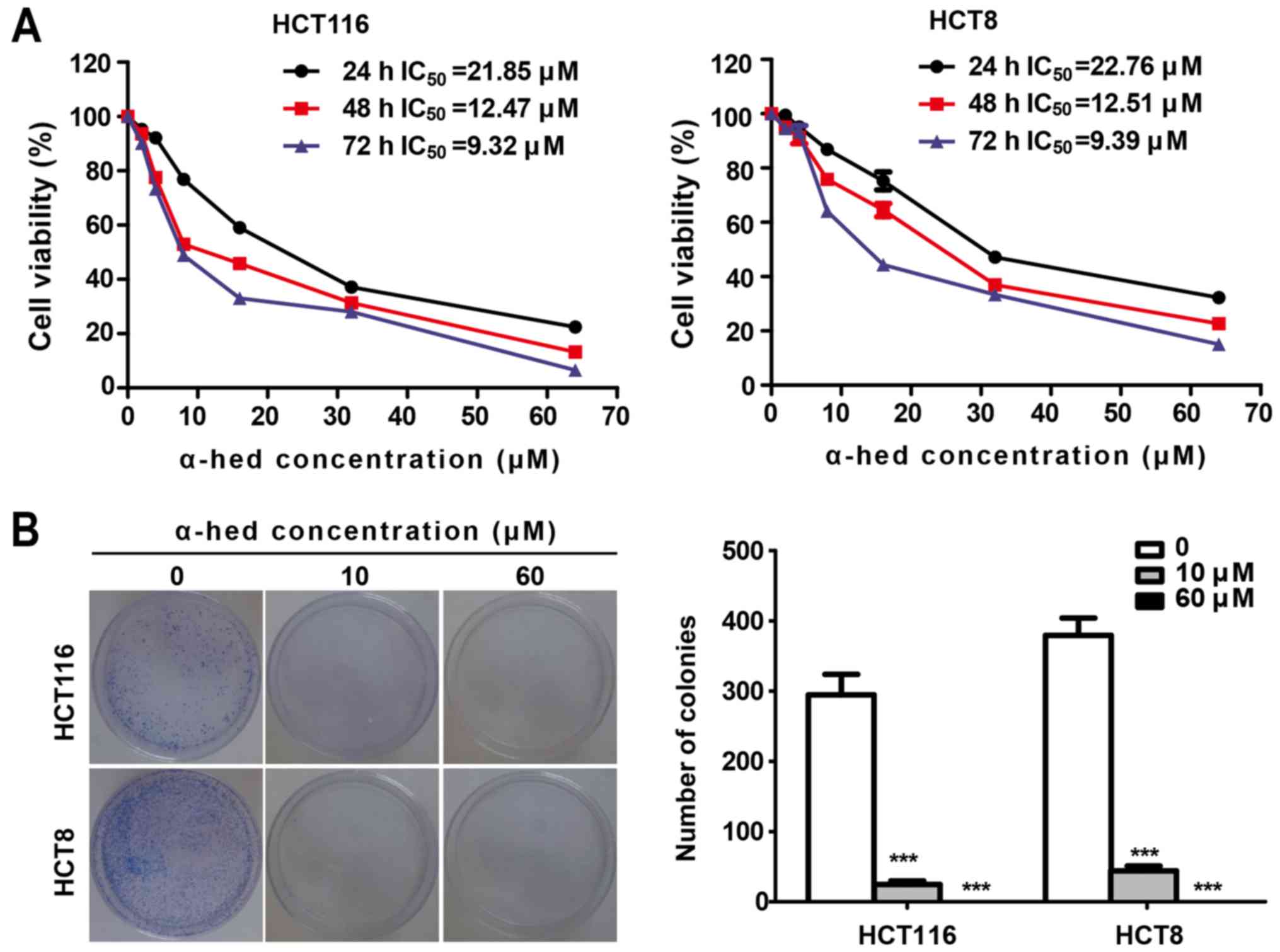
In cell viability assay, HCT116 and HCT8 wells were
divided into seven groups according to α-hederin concentration (0,
2, 4, 8, 16, 32 and 64 µM). Each group contained six wells.
As presented in Fig. 1A, the cell
viability of the two cell lines decreased significantly with the
increase of α-hederin concentration. The half maximal inhibitory
concentration (IC50) of α-hederin in HCT116 was 21.85,
12.47 and 9.32 µM for 24, 48 and 72 h, respectively.
Similarly, IC50 values of α-hederin in HCT8 cells
treated for 24, 48 and 72 h were 22.76, 12.51 and 9.39 µM,
respectively. Therefore, α-hederin decreased the viability of both
colorectal cancer cell lines in a dose- and time-dependent manner.
Colony formation assay using the adhesive culture system was used
to analyze the antiproliferation effects of α-hederin. It was
demonstrated that α-hederin treatment resulted in a dose-dependent
decrease in the efficiency of colony formation (Fig. 1B). α-Hederin concentrations at 10
µM caused a significant decreased in the number of colonies
in both colorectal cancer cell lines (P<0.001). Furthermore,
after increasing α-hederin concentrations to 60 µM, no
colonies were observed.
α-hederin induces apoptosis in colorectal
cancer cells
Previous studies have demonstrated that α-hederin
may induce cell apoptosis (27,37).
In the present study, α-hederin-induced apoptosis rates of
colorectal cancer cells were determined using flow cytometry with
Annexin V/PI (Fig. 2A) and
microscopy using Hoechst 33258 staining (Fig. 2B). Flow cytometry results indicated
that the α-hederin-induced (0, 10 and 60 µM) apoptosis rates
were 5.51±1.18, 8.97±1.69 and 21.75±2.85% in HCT116 cells.
Apoptosis rates in HCT8 cells were 3.54±1.3, 7.66±1.89 and
15.66±2.36% (Fig. 2A). Compared
with control, 10 µM α-hederin induced higher levels of
apoptosis in HCT116 and HCT8 cells (P<0.01), with 60 µM
α-hederin inducing even greater levels of apoptosis (P<0.001).
In addition, Hoechst 33258 staining exhibited results similar to
those for flow cytometry (Fig.
2B).
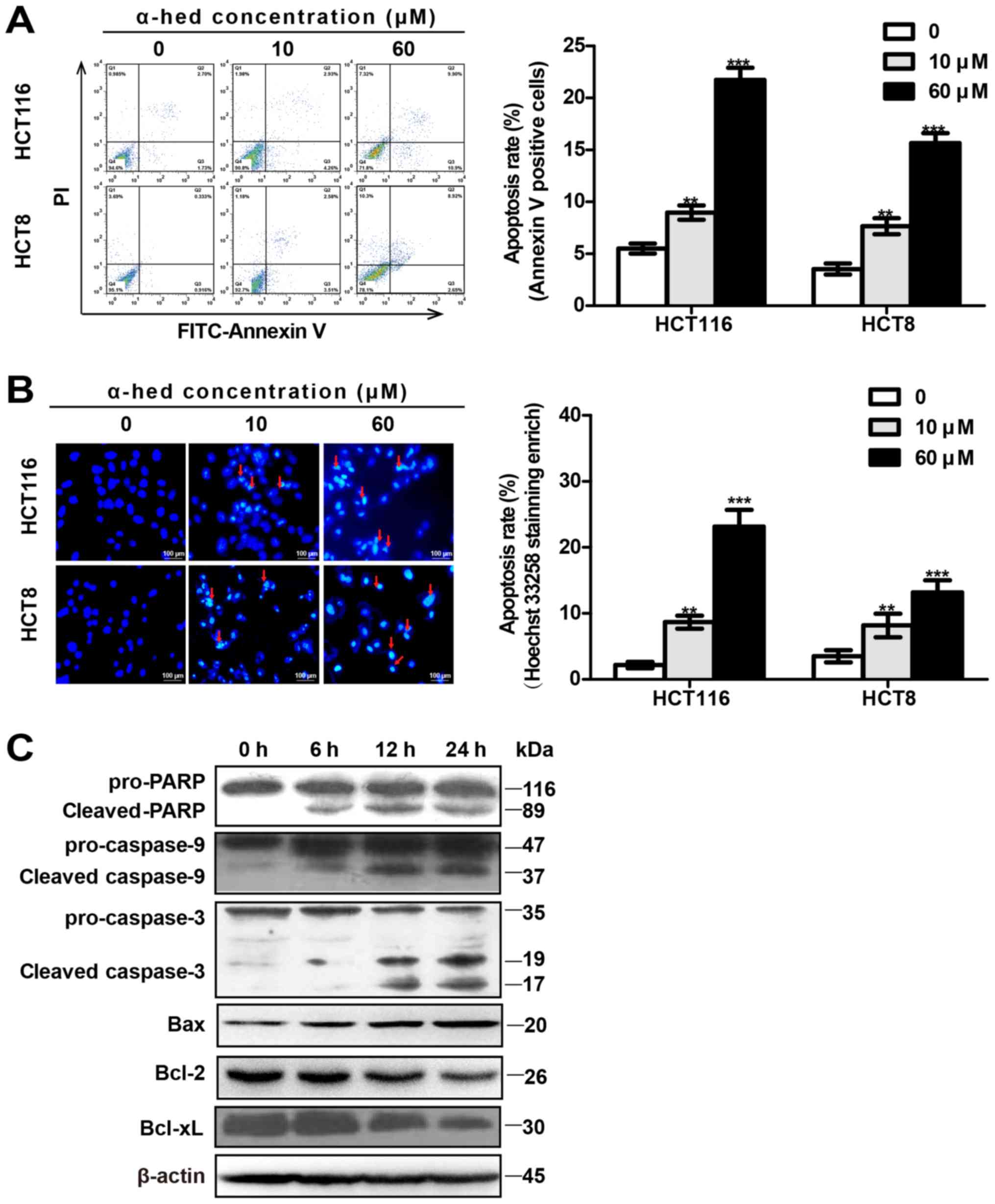 | Figure 2α-hederin induces colorectal cancer
cell apoptosis. (A) Apoptosis analysis of α-hederin-treated cells
through flow cytometry using Annexin V/PI. HCT8 and HCT116 cells
were treated with 0, 10 and 60 µM α-hederin for 24 h. (B)
Apoptosis analysis of α-hederin-treated cells through microscopy
using Hoechst 33258 staining (magnification, ×200). Following
treatment with α-hederin for 24 h, Hoechst 33258 staining was used
to identify apoptotic cells (red arrows). Histograms on the right
represent apoptosis rates following Hoechst 33258 staining
enrichment. (C) HCT116 cells were treated with 10 µM
α-hederin for 6, 12 and 24 h, after which their lysates were
analyzed by western blotting for mitochondrial apoptosis pathway
protein levels using anti-PARP, anti-cas-pase-9, anti-caspase-3,
anti-Bax, anti-Bcl-2, and anti-Bcl-xL antibody.
**P<0.01, ***P<0.001 vs. 0 µM.
PI, propidium iodide; α-hed, α-hederin; FITC, fluorescein
isothiocyanate; PARP, poly (ADP-ribose) polymerase; Bcl, B cell
lymphoma; Bax, Bcl associated X protein. |
The mitochondrial signaling pathway is one of the
major pathways involved in apoptosis (38). To observe the effect of α-hederin
on the mitochondrial signal pathway, the expression of
mitochondrial apoptosis-related proteins was measured in HCT116
cells. As presented in Fig. 2C,
compared with the control, the expression of activated caspase-9
(cleaved caspase-9), activated caspase-3 (cleaved caspase-3), PARP
degradation (cleaved PARP), and Bax were all increased in HCT116
cells treated with α-hederin. Furthermore, α-hederin increased
these proteins expression of HCT116 cells in a time-dependent
manner. Conversely, α-hederin decreased the expression of Bcl-2 and
Bcl-xl in a time-dependent manner.
α-hederin induces autophagy in colorectal
cancer cells
Analysis of Figs.
1A and 2A indicated that the
apoptosis rate did not match the inhibition rate of α-hederin on
colorectal cancer cell viability. This suggested that
α-hederin-induced cell death may have involved other mechanisms.
Autophagy rates in HCT116 and HCT8 cells were determined using LC3
lentivirus (39). As a negative
control, mutant LC3 (LC3-mu) was used as it could not bind to
autophagosomes and form bright fluorescent spots. As presented in
Fig. 3A, α-hederin induced LC3 but
not LC3-mu aggregation in HCT116 and HCT8 cells. Compared with
controls, 10 µM α-hederin exhibited significantly higher
autophagic rate in HCT116 and HCT8 cells (P<0.001). The presence
of autophagosomes in HCT116 cells was also evaluated using an
electron microscope. As presented in Fig. 3B, autophagosomes were present in
α-hederin-treated HCT116 and HCT8 cells. This result further
supported that α-hederin induced autophagy in colorectal cancer
cells.
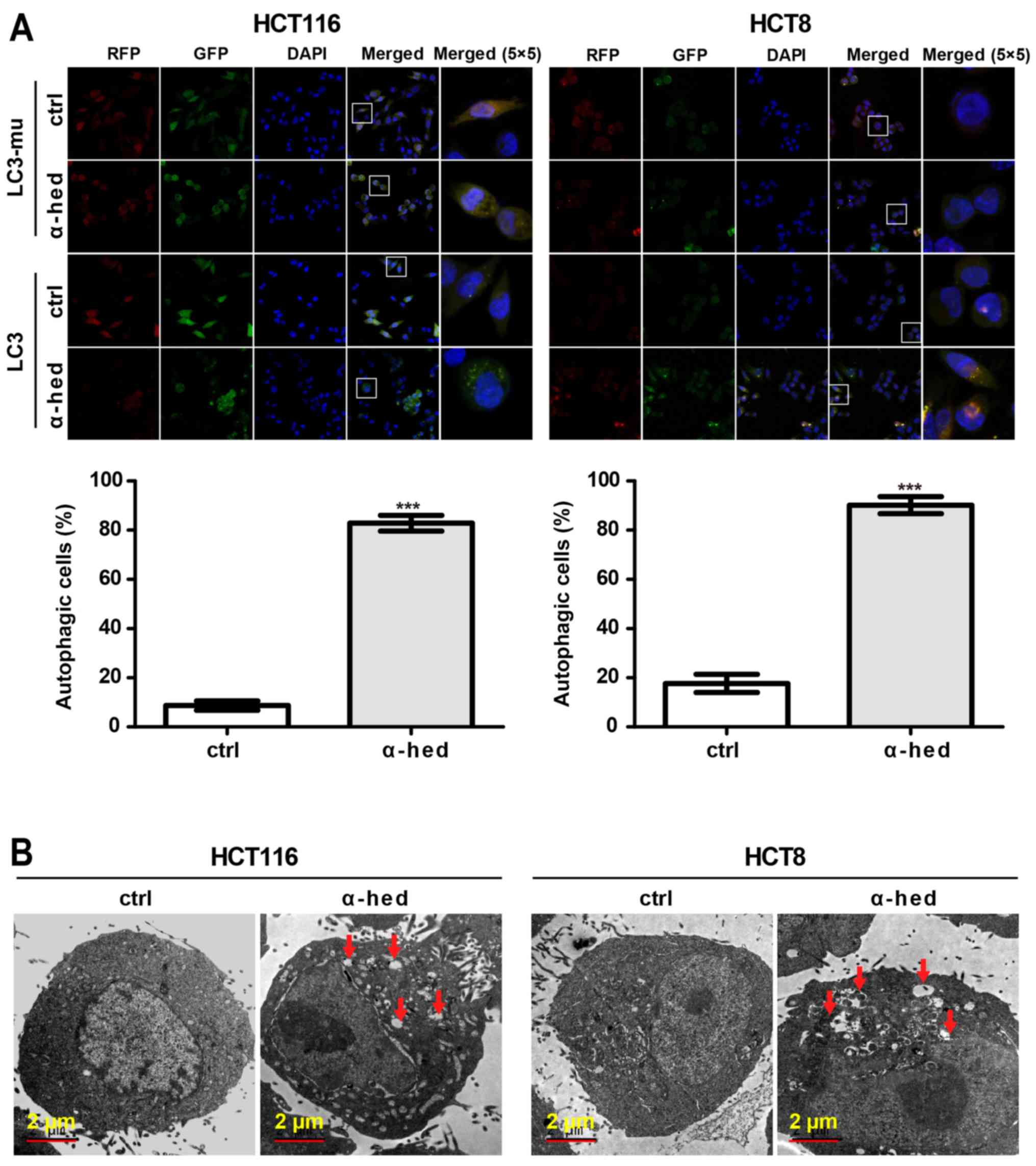 | Figure 3α-hederin induces autophagy in
colorectal cancer cells. (A) HCT116 and HCT8 cells were infected
with LC3 and LC3-mu. After these cells were treated with 10
µM α-hederin for 24 h, autophagy rate assay was then
performed. Images were captured using laser confocal microscopy.
Magnification of (RFP, GFP, DAPI and Merge magnification, ×400;
Merge 5×5 magnification, ×10,000. Histograms present autophagy
rates according to the proportion of highlighted cells. (B) Normal
and autophagic HCT116 and HCT8 cells were photographed using an
electron microscope. Red arrows in the graphs represent
autophagosomes. ***P<0.001 vs. ctrl. LC3, light chain
3 lentivirus; LC3-mu, mutation light chain 3 lentivirus; α-hed,
α-hederin; ctrl, control. |
Effect of α-hederin on LC3 expression in
vivo
In vitro results had demonstrated that
α-hederin could induce autophagy in colorectal cancer cells. To
investigate the inducing autophagy effect of α-hederin in
vivo, a subcutaneous xenograft model of HCT116 cells in nude
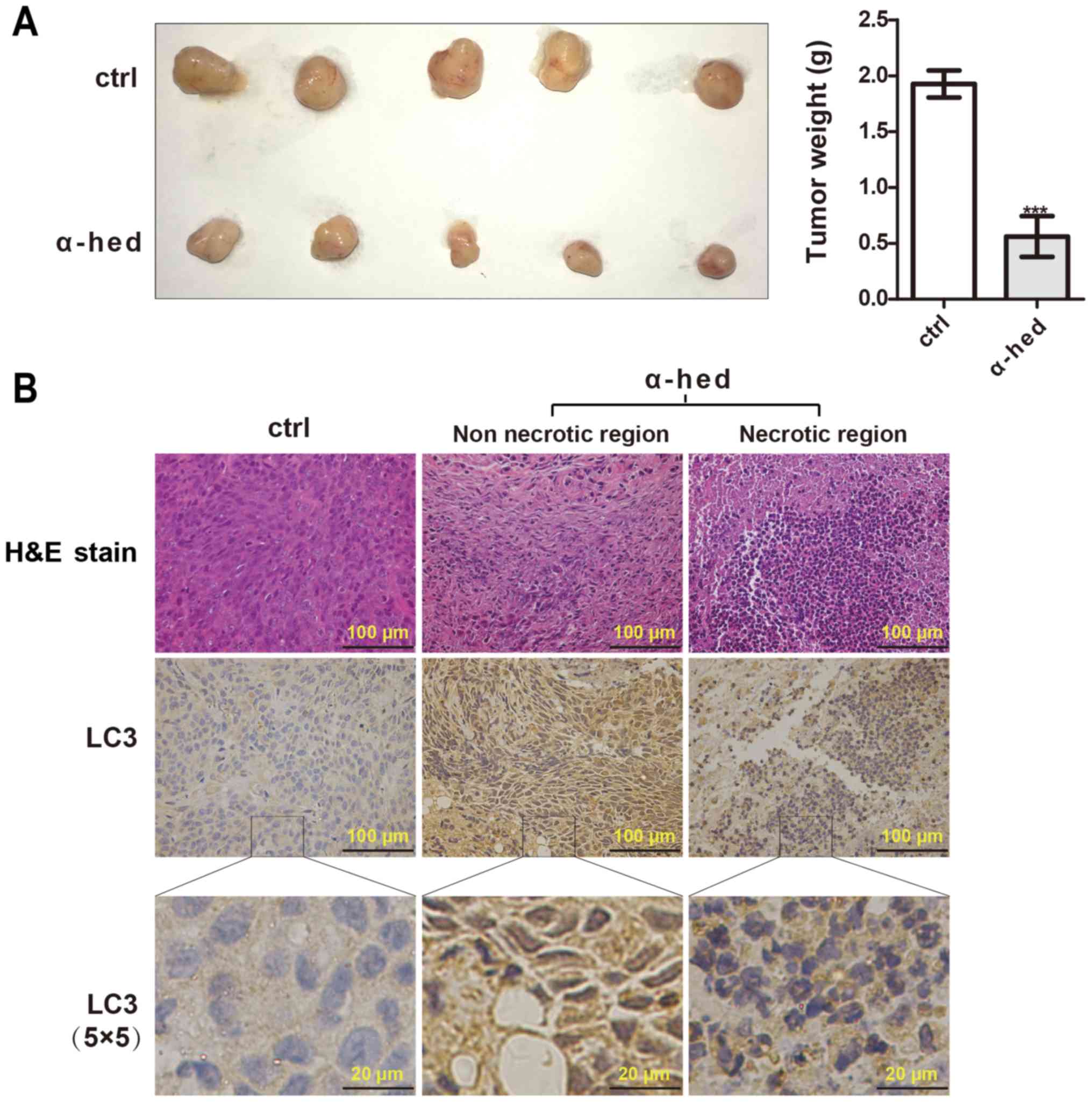
mice was used. As presented in Fig.
4A, α-hederin significantly inhibited tumor growth compared
with the control. According to the results of H&E staining
(Fig. 4B), tumors treated with
α-hederin exhibited marked necrosis. LC3 puncta was assessed using
immunohistochemistry to evaluate the effect of α-hederin on
autophagy in vivo. As presented in Fig. 4B, the presence of LC3 puncta was
observed in samples treated with α-hederin. In addition, the
necrotic area also exhibited highly aggregated LC3 puncta. While,
the control exhibited significant diffuse cytoplasmic staining
without puncta. These results suggested that α-hederin could
inhibit tumorigenicity through promoting autophagy of colorectal
cancer cells in vivo.
α-hederin induces autophagy of colorectal
cancer cells through the AMPK/mTOR pathway
Given that dephosphorylation of p-mTOR and
degradation of LC3 I to LC3 II are the major mechanisms involved in
autophagy (40), LC3 II protein
levels were used to determine the extent of cell autophagy
(41). After treating HCT116 cells
with α-hederin for 24 h, cell lysates were used to detect p-mTOR
and LC3 II protein levels. As presented in Fig. 5A, an increase in α-hederin
concentration resulted in a gradual increase in LC3 II levels but a
gradual decrease in p-mTOR protein levels. HCT116 cells were also
treated with 10 µM α-hederin for 6, 12 and 24 h. The results
demonstrated that, over time, α-hederin caused a gradual decrease
in p-mTOR, p-ULK1, p-P70S6K and P62 protein levels but a gradual
increase in p-AMPK and beclin-1 protein levels (Fig. 5B).
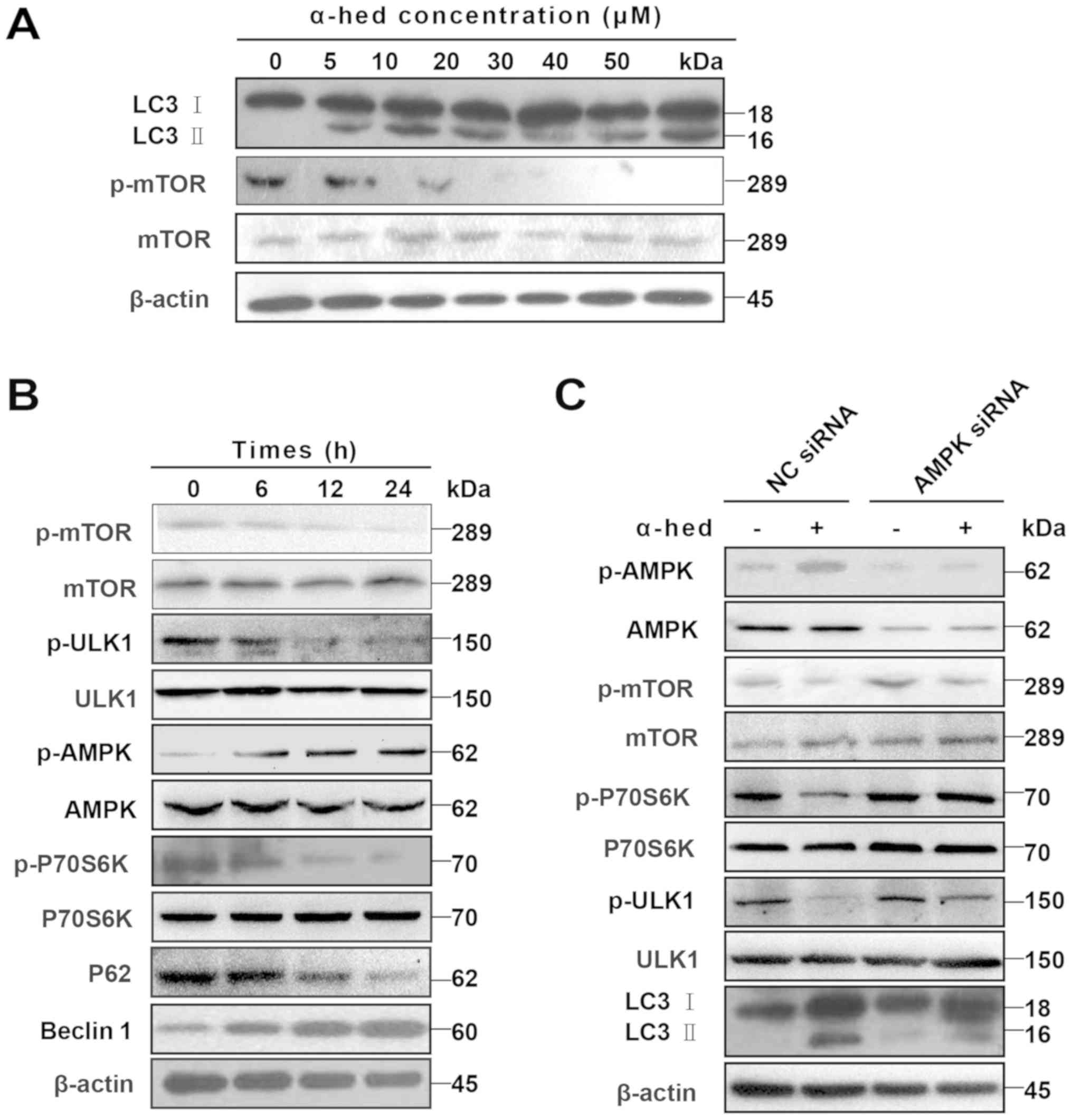 | Figure 5AMPK/mTOR pathway participated in
α-hederin-induced autophagy. (A) α-hederin upregulated LC3 II
levels and inhibited p-mTOR in a dose-dependent manner. (B) After
HCT116 cells were treated with 10 µM α-hederin for 6, 12 and
24 h, expression levels of p-mTOR, mTOR, p-ULK1, ULK1, p-AMPK,
AMPK, p-P70S6K, P70S6K, P62 and beclin1 were determined using
specific antibodies. (C) HCT116 cells were treated with AMPK siRNA
and NC siRNA for 3 days, with α-hederin being added during the last
2 days. The expression levels of p-AMPK, AMPK, p-mTOR, mTOR,
p-ULK1, ULK1, p-P70S6K, P70S6K and LC3 were then evaluated using
western blotting. AMPK, AMP-activated protein kinase; mTOR,
mechanistic target of rapamycin; LC3, light chain 3; p,
phosphorylated; ULK1, Unc-51 like autophagy activating kinase 1;
siRNA, small interfering RNA; NC, normal control; α-hed,
α-hederin. |
AMPK/mTOR is a major signaling pathway involved in
autophagy (42). In this signaling
pathway, AMPK serves as the activator of autophagy. AMPK activation
induces dephosphorylation of mTOR, which separates it from the ULK1
complex. The subsequent dephosphorylation of ULK1 then initiates
autophagy (43). To verify the
role of the AMPK/mTOR pathway in α-hederin-induced autophagy, the
expression of autophagy-related signals was detected in HCT116
cells treated with AMPK siRNA. It was demonstrated that AMPK siRNA
restored the expression of p-mTOR, p-P70S6K and p-ULK1, which had
been decreased by α-hederin (Fig.
5C). Results for p-AMPK indicated that although α-hederin
increased LC3 II, AMPK knockdown did not restore LC3 II.
ROS-dependent AMPK activation by
α-hederin
Previous studies have demonstrated that ROS is a
major factor in α-hederin-induced apoptosis (37,44).
ROS production is also important for AMPK/mTOR pathway activation.
To verify the role of ROS in α-hederin-induced AMPK/mTOR pathway
activation, oxidation-activated fluorescent dye DCHF-DA was used to
detect intracellular ROS using flow cytometry. HCT116 cells were
treated with 10 µM each of α-hederin and ROS inhibitor NAC
for 24 h at 37°C then incubated in RPMI-1640 medium supplemented
with 5 µmol/ml DCHF-DA for 30 min at room temperature.
Fig. 6A demonstrated that although
α-hederin increased ROS levels, ROS inhibitor NAC reversed this
effect. Furthermore, NAC inhibited α-hederin-induced AMPK/mTOR
pathway activation (Fig. 6B).
Subsequently, the effects of NAC and α-hederin on apoptosis were
detected by flow cytometry. As presented in Fig. 6C, NAC alone did not affect the
apoptosis rate but NAC reduced the apoptosis rate treated by
α-hederin in HCT116 cells (P<0.001). Additional, results from
confocal microscopy demonstrated that NAC could inhibit
α-hederin-induced autophagy (Fig.
6D).
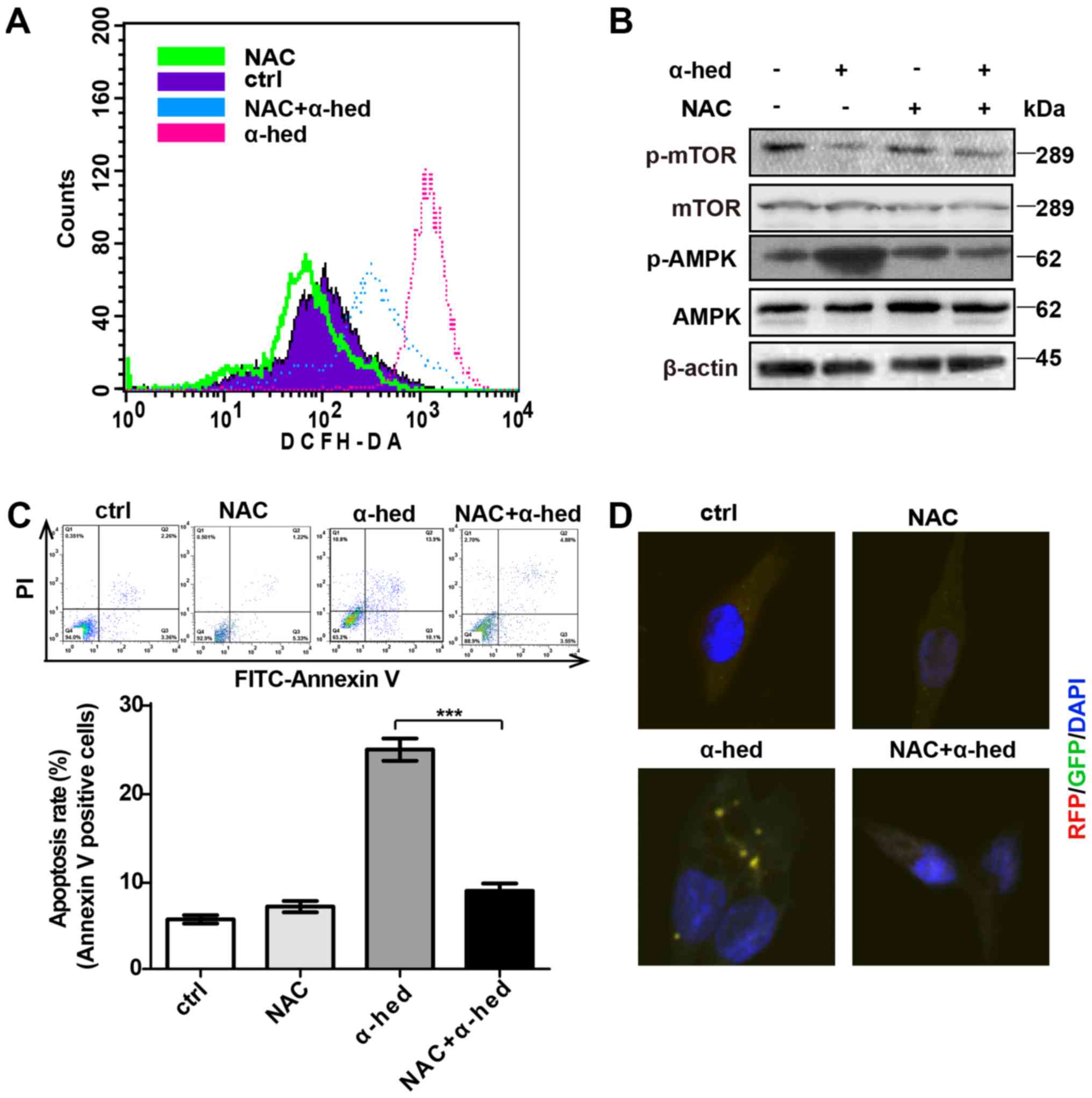 | Figure 6α-hederin-induced AMPK pathway
activation depended on ROS. (A) After being treated with 10
µM α-hederin or ROS inhibitor NAC (10 µM) for 24 h,
HCT116 cells were dyed with oxidation-activated fluorescent dye
DCHF-DA and analyzed using flow cytometry to detect intracellular
ROS. (B) After being treated with 10 µM α-hederin or ROS
inhibitor NAC (10 µM) for 24 h, western blotting was used to
test HCT116 cell lysates for p-mTOR and p-AMPK expression levels.
Following treatment with α-hederin, NAC and NAC plus α-hederin for
24 h, (C) The apoptosis rate of HCT116 cells was tested by flow
cytom-etry. (D) Light chain 3 fluorescence of HCT116 cells was
photographed via confocal microscopy (magnification, ×400).
***P<0.001. AMPK, AMP-activated protein kinase; ROS,
reactive oxygen species; mTOR, mechanistic target of rapamycin; p,
phosphorylated; α-hed, α-hederin; ctrl, control; PI, propidium
iodide; FITC, fluorescein isothiocyanate. |
Autophagy and apoptosis comprise the
major mechanisms by which α-hederin induces colorectal cancer cell
death
To clarify the interplay between apoptosis and
autophagy induced by α-hederin in colorectal cancer, the effects of
autophagy inhibitor 3-MA on apoptosis and autophagy were
investigated. HCT116 cells were treated with 2 mM 3-MA for 48 h to
evaluate the effect of 3-MA alone on cell viability. The results
indicated that 2 mM 3-MA alone had no significant effect on cell
viability of HCT116 cells (Fig.
7A). Then, HCT116 cells were treated with α-hederin (2, 4, 8,
16, 32 and 64 µM) or α-hederin plus 2 mM 3-MA for 48 h,
after which cell viability was tested. As presented in Fig. 7B, 2 mM 3-MA significantly reduced the effect
of α-hederin on cell viability (P<0.05). Furthermore, HCT116
cells were treated with 2 mM 3-MA, 10 mM α-hederin and 2 mM 3-MA
with 10 mM α-hederin for 48 h. As presented in Fig. 7C, 3-MA had no significant effect on
α-hederin-induced apoptosis. Finally, apoptosis rate was detected
by flow cytometry. It was also demonstrated that 3-MA blocked the
α-hederin-induced autophagosome formation (Fig. 7D).
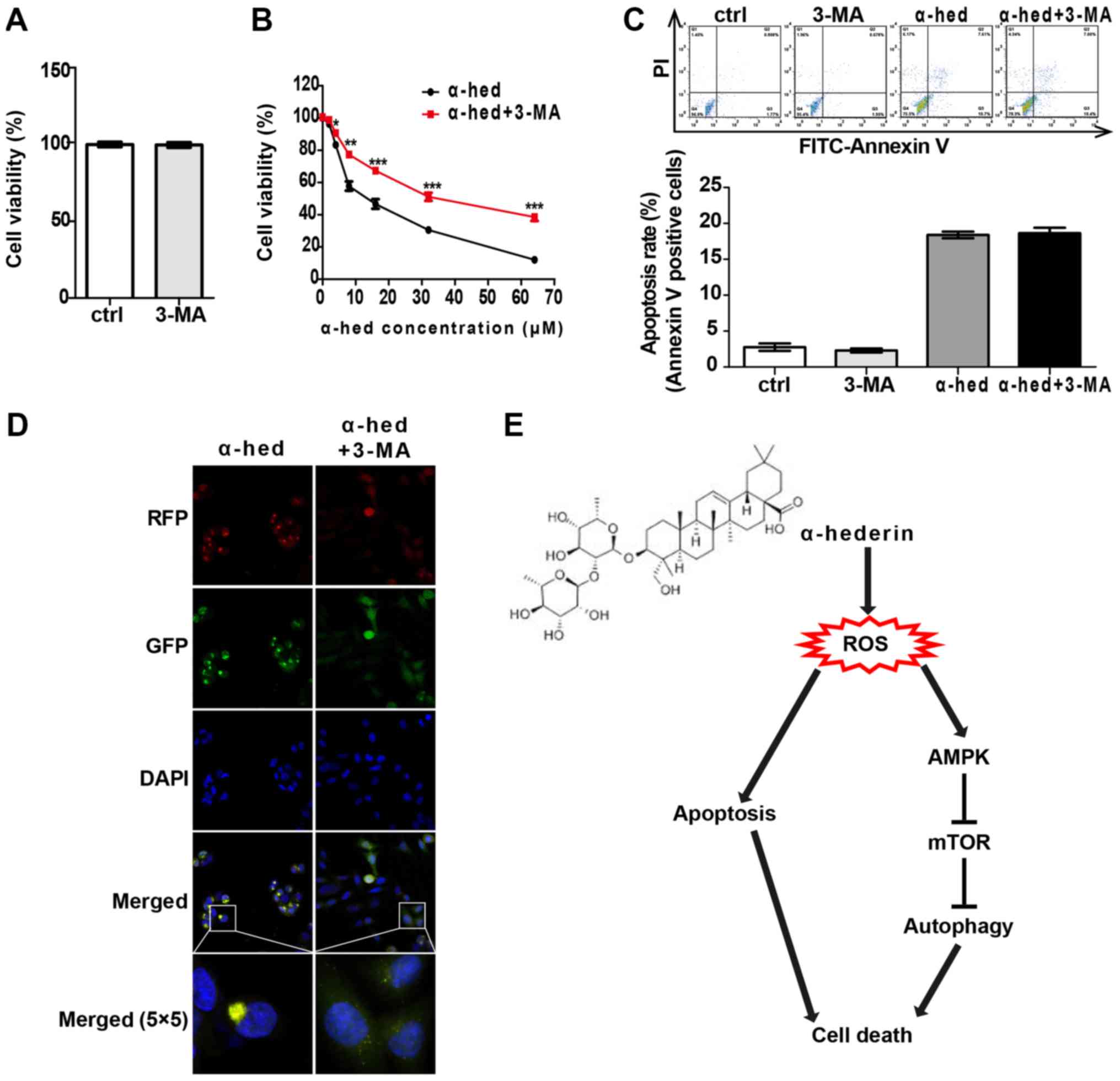 | Figure 7Cell death induced by α-hederin
depended on apoptosis and autophagy. (A) HCT116 was treated with 2
mM 3-MA for 24 h. CCK8 was used to evaluate the effects of 2 mM
3-MA alone on the cell viability. (B) HCT116 was incubated with 10
µM α-hederin or 10 µM α-hederin plus 2 mM 3-MA for 24
h. Thereafter, CCK8 assay was used to detect cell viability. (C)
HCT116 was incubated with 10 µM α-hederin, 2 mM 3-MA or 10
µM α-hederin plus 2 mM 3-MA for 24 h. Apoptosis assay by
flow cytometry was used to determine the effect of 3-MA on
α-hederin induced apoptosis. (D) Light chain 3 lentivirus-infected
HCT116 cells were treated with 10 µM α-hederin or 10
µM α-hederin plus 2 mM 3-MA for 24 h. Autophagosomes were
then photographed using laser confocal microscopy (RFP, GFP, DAPI
and Merge magnification, ×400; Merge 5×5 magnification, ×10,000).
(E) Model of the mechanism for α-hederin induced colorectal cancer
cell death. *P<0.05, **P<0.01,
***P<0.001 vs. α-hed. CCK8, cell counting kit 8;
ctrl, control; α-hed, α-hederin; PI, propidium iodide; FITC,
fluorescein isothiocyanate; ROS, reactive oxygen species. |
Discussion
α-Hederin, a novel type of drug derived from the
leaves of ivy or Nigella sativa, had exhibited strong
anticancer activities in recent studies. Previous studies had
demonstrated that α-hederin could induce cell apoptosis (23,26,27,37).
In the present study, it was demonstrated that α-hederin activated
the mitochondrial apoptosis signal pathway in colorectal cancer
cells. However, the results also demonstrated that the rate of
apoptosis induced by α-hederin was not consistent with its
inhibitory effects on colorectal cancer cells, suggesting that
other mechanisms may be involved in the anticancer activity of
α-hederin.
Previous studies had demonstrated that autophagy
regulated by α-hederin was differently in different cells:
α-hederin induced autophagy in neuronal PC12 cell (45) but inhibited autophagy in non-small
cell lung cancer cells (44). In
the present study, autophagosomes were observed in two colorectal
cancer cell lines (HCT116 and HCT8) treated with α-hederin by
fluorescence microscopy using LC3 lentivirus and electron
microscopy. In view of the high toxicity of α-hederin, low-dose (2
mg/kg body weight) α-hederin was used for intraperitoneal
injection. The results also demonstrated that α-hederin could
induce autophagy in colorectal cancer cells in vivo.
The mTOR pathway is a typical signaling pathway that
regulates cell autophagy (40).
LC3, a subunit of the microtubule-associated proteins involved in
the formation of autophagic vacuoles, is regarded as an autophagy
marker (41). It was demonstrated
that α-hederin was able to decrease the expression of p-mTOR and
increase the ratio of LC3II/I in a concentration-dependent manner.
Which suggested that α-hederin could activate autophagy mediated by
the mTOR signaling pathway.
AMPK signaling is one major regulatory signal of
mTOR signaling pathway and autophagy (46). In addition, α-hederin is able to
induce AMPK/mTOR dependent autophagy and promote the degradation of
neurodegenerative mutant disease protein (45). The expression of AMPK/mTOR
signaling pathway related proteins and autophagy-related proteins
were also detected. The results demonstrated that α-hederin could
induce AMPK-mTOR dependent autophagy in HCT116 cells. In addition,
this autophagy could be blocked by the AMPK siRNA. It suggested
that the AMPK/mTOR signaling pathway served a major role in
α-hederin-induced autophagy.
ROS production has been regarded as the ideal
anticancer mechanism for many drugs (37,47)
given that it activates both apoptosis and autophagy to kill cancer
cells (48). In the present study,
ROS levels of HCT116 cells treated with α-hederin were determined
using DCHF-DA. The results demonstrated that although α-hederin
increased ROS levels, NAC reversed this effect. The results of
western blotting demonstrated that NAC blocked the
α-hederin-induced activation of the AMPK/mTOR signaling pathway.
This suggested that the α-hederin-induced AMPK/mTOR signaling
pathway activation depended on ROS production. In addition, NAC
could reverse the apoptosis and autophagy induced by α-hederin.
These results suggested that increasing ROS may serve a key role
for the anticancer effects of α-hederin.
Autophagy is one of the major mechanisms involved in
cancer cell apoptosis and drug resistance during the treatment of
various cancers (49). This is
primarily because drugs stimulate the cell's protective autophagy
and inhibit cell apoptosis (50).
However, a number of studies have demonstrated that certain drugs
cause not only excessive autophagy in specific tumor cells, but
also autophagic cell death (51,52),
making autophagy their main anticancer mechanism. Therefore,
considering that different drugs act on different cells, their
autophagic and apoptotic outcomes may be different. This may have
been caused by differing activation of the signaling pathway
(53). In the present study, it
was demonstrated that autophagy inhibitor 3-MA significantly
reduced the anticancer effects of α-hederin. Furthermore, 3-MA had
no significant effects on α-hederin-induced apoptosis in HCT116
cells. These results suggest that autophagy and apoptosis are
equally important mechanisms through which α-hederin exerts its
anticancer effects.
In conclusion, the present findings suggest that
α-hederin may stimulate ROS production in colorectal cancer cells
and activate both autophagy and apoptosis, both of which increase
the anticancer effects of α-hederin. Autophagy is produced by the
ROS-activated AMPK/mTOR signaling pathway (Fig. 7E). With its strong anticancer
effect and multiple anticancer mechanisms, α-hederin may
potentially become a more reliable anticancer drug. However, due to
its strong toxicity (54-57), protein absorption (58) and hemolytic effect (59), further molecular modifications and
nanotarget drug carriers should be applied during the anticancer
study of α-hederin.
Funding
The present study was supported by the International
Cooperation Key Project of the National Natural Science Foundation
of China (grant no. 81520108031), the National Natural Science
Foundation of China (grant no. 81473478), Shanghai Academic
Research Leader (grant no. 16XD1403600), Shanghai Rising-Star
Program (grant no. 16QA1403700), Municipal Human Resources
Development Program for Outstanding Leaders in Medical Disciplines
in Shanghai (grant no. 2017BR031) and Shanghai University of
Traditional Chinese Medicine (grant no. 18LK042).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article or are available from the
corresponding author on reasonable request.
Authors' contributions
JS, YF, YW, QJ, GC, LS, YW, YH, JZ and QL performed
the experiments and data analysis, and wrote the manuscript. JS
made substantial contributions to the design of the present study
and acquired experimental materials. JZ and QL served a key role in
guiding the subject, providing funding and writing the manuscript.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The animal protocol was approved by the Ethics
Committee of Shuguang Hospital affiliated with Shanghai University
of Traditional Chinese Medicine.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Center MM, Jemal A, Smith RA and Ward E:
Worldwide variations in colorectal cancer. CA Cancer J Clin.
59:366–378. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel R, Desantis C and Jemal A:
Colorectal cancer statistics, 2014. CA Cancer J Clin. 64:104–117.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Koehler BC, Jäger D and Schulze-Bergkamen
H: Targeting cell death signaling in colorectal cancer: Current
strategies and future perspectives. World J Gastroenterol.
20:1923–1934. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Green DR and Llambi F: Cell death
signaling. Cold Spring Harb Perspect Biol. 7:a0060802015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kroemer G, Galluzzi L, Vandenabeele P,
Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS,
Golstein P, Green DR, et al: Nomenclature Committee on Cell Death
2009: Classification of cell death: Recommendations of the
Nomenclature Committee on Cell Death 2009. Cell Death Differ.
16:3–11. 2009. View Article : Google Scholar
|
|
7
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: A double-edged sword. Science. 306:990–995.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ng G and Huang J: The significance of
autophagy in cancer. Mol Carcinog. 43:183–187. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Codogno P and Meijer AJ: Autophagy and
signaling: Their role in cell survival and cell death. Cell Death
Differ. 12(Suppl 2): 1509–1518. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Su M, Mei Y and Sinha S: Role of the
crosstalk between autophagy and apoptosis in cancer. J Oncol.
2013:1027352013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT,
Liu B and Bao JK: Programmed cell death pathways in cancer: A
review of apoptosis, autophagy and programmed necrosis. Cell
Prolif. 45:487–498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ou L, Lin S, Song B, Liu J, Lai R and Shao
L: The mechanisms of graphene-based materials-induced programmed
cell death: A review of apoptosis, autophagy, and programmed
necrosis. Int J Nanomedicine. 12:6633–6646. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Qian HR, Shi ZQ, Zhu HP, Gu LH, Wang XF
and Yang Y: Interplay between apoptosis and autophagy in colorectal
cancer. Oncotarget. 8:62759–62768. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Reczek CR and Chandel NS: ROS-dependent
signal transduction. Curr Opin Cell Biol. 33:8–13. 2015. View Article : Google Scholar :
|
|
15
|
Marchi S, Giorgi C, Suski JM, Agnoletto C,
Bononi A, Bonora M, De Marchi E, Missiroli S, Patergnani S, Poletti
F, et al: Mitochondria-ros crosstalk in the control of cell death
and aging. J Signal Transduct. 2012:3296352012. View Article : Google Scholar
|
|
16
|
Na AR, Chung YM, Lee SB, Park SH, Lee MS
and Yoo YD: A critical role for Romo1-derived ROS in cell
proliferation. Biochem Biophys Res Commun. 369:672–678. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li L, Tan J, Miao Y, Lei P and Zhang Q:
ROS and autophagy: Interactions and molecular regulatory
mechanisms. Cell Mol Neurobiol. 35:615–621. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Scherz-Shouval R and Elazar Z: Regulation
of autophagy by ROS: Physiology and pathology. Trends Biochem Sci.
36:30–38. 2011. View Article : Google Scholar
|
|
19
|
Xie J, Xu Y, Huang X, Chen Y, Fu J, Xi M
and Wang L: Berberine-induced apoptosis in human breast cancer
cells is mediated by reactive oxygen species generation and
mitochondrial-related apoptotic pathway. Tumour Biol. 36:1279–1288.
2015. View Article : Google Scholar
|
|
20
|
Li GH, Lin XL, Zhang H, Li S, He XL, Zhang
K, Peng J, Tang YL, Zeng JF, Zhao Y, et al: Ox-Lp(a) transiently
induces HUVEC autophagy via an ROS-dependent PAPR-1-LKB1-AMPK-mTOR
pathway. Atherosclerosis. 243:223–235. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee MS, Tsai CW, Wang CP, Chen JH and Lin
HH: Anti-prostate cancer potential of gossypetin via inducing
apoptotic and autophagic cell death. Mol Carcinog. 56:2578–2592.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sun ZL, Dong JL and Wu J: Juglanin induces
apoptosis and autophagy in human breast cancer progression via
ROS/JNK promotion. Biomed Pharmacother. 85:303–312. 2017.
View Article : Google Scholar
|
|
23
|
Sun D, Shen W, Zhang F, Fan H, Tan J1, Li
L, Xu C, Zhang H, Yang Y and Cheng H: α-Hederin arrests cell cycle
at G2/M checkpoint and promotes mitochondrial apoptosis by blocking
nuclear factor-B signaling in colon cancer cells. BioMed Res Int.
2548378:20182018.
|
|
24
|
Gumushan-Aktas H and Altun S: Effects of
Hedera helix L. extracts on rat prostate cancer cell proliferation
and motility. Oncol Lett. 12:2985–2991. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Claereboudt EJS, Eeckhaut I, Lins L and
Deleu M: How different sterols contribute to saponin tolerant
plasma membranes in sea cucumbers. Sci Rep. 8:108452018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li J, Wu DD, Zhang JX, Wang J, Ma JJ, Hu X
and Dong WG: Mitochondrial pathway mediated by reactive oxygen
species involvement in α-hederin-induced apoptosis in
hepatocellular carcinoma cells. World J Gastroenterol.
24:1901–1910. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lorent JH, Léonard C, Abouzi M, Akabi F,
Quetin-Leclercq J and Mingeot-Leclercq MP: α-Hederin induces
apoptosis, membrane permeabilization and morphologic changes in two
cancer cell lines through a cholesterol-dependent mechanism. Planta
Med. 82:1532–1539. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun D, Shen W, Zhang F, Fan H, Xu C, Li L,
Tan J, Miao Y, Zhang Yang Y, et al: α-Hederin inhibits interleukin
6-induced epithelial-to-mesenchymal transition associated with
disruption of JAK2/STAT3 signaling in colon cancer cells. Biomed
Pharmacother. 101:107–114. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sun J, Liu T and Xu J: Improving the
anticancer activity of α-hederin by physically encapsulating it
with targeted micelles assembled from amphiphilic block copolymers.
J Drug Deliv Sci Technol. 35:252–259. 2016. View Article : Google Scholar
|
|
30
|
Sun J, Xu K, Qiu Y, Gao H, Xu J, Tang Q
and Yin P: Bufalin reverses acquired drug resistance by inhibiting
stemness in colorectal cancer cells. Oncol Rep. 38:1420–1430. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhu R, Zhang CG, Liu Y, Yuan ZQ, Chen WL,
Yang SD, Li JZ, Zhu WJ, Zhou XF, You BG, et al: CD147 monoclonal
antibody mediated by chitosan nanoparticles loaded with α-hederin
enhances antineoplastic activity and cellular uptake in liver
cancer cells. Sci Rep. 5:179042015. View Article : Google Scholar
|
|
32
|
Cheng L, Xia TS, Wang YF, Zhou W, Liang
XQ, Xue JQ, Shi L, Wang Y, Ding Q and Wang M: The anticancer effect
and mechanism of α-hederin on breast cancer cells. Int J Oncol.
45:757–763. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cheng L, Xia TS, Wang YF, Zhou W, Liang
XQ, Xue JQ, Shi L, Wang Y and Ding Q: The apoptotic effect of D
Rhamnose β-hederin, a novel oleanane-type triterpenoid saponin on
breast cancer cells. PLoS One. 9:e908482014. View Article : Google Scholar
|
|
34
|
Randhawa MA and Alghamdi MS: Anticancer
activity of Nigella sativa (black seed) - a review. Am J Chin Med.
39:1075–1091. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rooney S and Ryan MF: Effects of
alpha-hederin and thymoquinone, constituents of Nigella sativa, on
human cancer cell lines. Anticancer Res. 25:2199–2204.
2005.PubMed/NCBI
|
|
36
|
Rooney S and Ryan MF: Modes of action of
alpha-hederin and thymoquinone, active constituents of Nigella
sativa, against HEp-2 cancer cells. Anticancer Res. 25:4255–4259.
2005.PubMed/NCBI
|
|
37
|
Swamy SM and Huat BT: Intracellular
glutathione depletion and reactive oxygen species generation are
important in alpha-hederin-induced apoptosis of P388 cells. Mol
Cell Biochem. 245:127–139. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Suen DF, Norris KL and Youle RJ:
Mitochondrial dynamics and apoptosis. Genes Dev. 22:1577–1590.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gump JM, Staskiewicz L, Morgan MJ, Bamberg
A, Riches DW and Thorburn A: Autophagy variation within a cell
population determines cell fate through selective degradation of
Fap-1. Nat Cell Biol. 16:47–54. 2014. View Article : Google Scholar
|
|
40
|
Dunlop EA and Tee AR: mTOR and autophagy:
A dynamic relationship governed by nutrients and energy. Semin Cell
Dev Biol. 36:121–129. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tanida I, Ueno T and Kominami E: LC3 and
Autophagy. Methods Mol Biol. 445:77–88. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kou B, Liu W, Xu X, Yang Y, Yi Q, Guo F,
Li J, Zhou J and Kou Q: Autophagy induction enhances
tetrandrine-induced apoptosis via the AMPK/mTOR pathway in human
bladder cancer cells. Oncol Rep. 38:3137–3143. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zha QB, Zhang XY, Lin QR, Xu LH, Zhao GX,
Pan H, Zhou D, Ouyang DY, Liu ZH and He XH: Cucurbitacin E induces
autophagy via downregulating mTORC1 signaling and upregulating AMPK
activity. PLoS One. 10:e01243552015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhan Y, Wang K, Li Q, Zou Y, Chen B, Gong
Q, Ho HI, Yin T, Zhang F, Lu Y, et al: The novel autophagy
inhibitor alpha-hederin promoted paclitaxel cytotoxicity by
increasing reactive oxygen species accumulation in non-small cell
lung cancer cells. Int J Mol Sci. 19:32212018. View Article : Google Scholar :
|
|
45
|
Wu AG, Zeng W, Wong VK, Zhu YZ, Lo AC, Liu
L and Law BY: Hederagenin and α-hederin promote degradation of
proteins in neurodegenerative diseases and improve motor deficits
in MPTP-mice. Pharmacol Res. 115:25–44. 2017. View Article : Google Scholar
|
|
46
|
Roach PJ: AMPK -> ULK1 -> autophagy.
Mol Cell Biol. 31:3082–3084. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mao X, Yu CR, Li WH and Li WX: Induction
of apoptosis by shikonin through a ROS/JNK-mediated process in
Bcr/Abl-positive chronic myelogenous leukemia (CML) cells. Cell
Res. 18:879–888. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ghavami S, Eshragi M, Ande SR, Chazin WJ,
Klonisch T, Halayko AJ, McNeill KD, Hashemi M, Kerkhoff C and Los
M: S100A8/A9 induces autophagy and apoptosis via ROS-mediated
cross-talk between mitochondria and lysosomes that involves BNIP3.
Cell Res. 20:314–331. 2010. View Article : Google Scholar
|
|
49
|
Chen S, Rehman SK, Zhang W, Wen A, Yao L
and Zhang J: Autophagy is a therapeutic target in anticancer drug
resistance. Biochim Biophys Acta. 1806:220–229. 2010.PubMed/NCBI
|
|
50
|
Xu L, Qu XJ, Liu YP, Xu YY, Liu J, Hou KZ
and Zhang Y: Protective autophagy antagonizes oxaliplatin-induced
apoptosis in gastric cancer cells. Chin J Cancer. 30:490–496. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang N, Feng Y, Zhu M, Tsang CM, Man K,
Tong Y and Tsao SW: Berberine induces autophagic cell death and
mitochondrial apoptosis in liver cancer cells: The cellular
mechanism. J Cell Biochem. 111:1426–1436. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kanzawa T, Zhang L, Xiao L, Germano IM,
Kondo Y and Kondo S: Arsenic trioxide induces autophagic cell death
in malignant glioma cells by upregulation of mitochondrial cell
death protein BNIP3. Oncogene. 24:980–991. 2005. View Article : Google Scholar
|
|
53
|
Zhai B, Hu F, Jiang X, Xu J, Zhao D, Liu
B, Pan S, Dong X, Tan G, Wei Z, et al: Inhibition of Akt reverses
the acquired resistance to sorafenib by switching protective
autophagy to autophagic cell death in hepatocellular carcinoma. Mol
Cancer Ther. 13:1589–1598. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Bang SC, Seo HH, Shin HR, Lee KC, Hoang TA
and Jung SH: A convenient preparation of a disaccharide motif and
its role in the cytotoxicity of the triterpenoid saponin,
alpha-hederin. Arch Pharm Res. 31:555–561. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Jeong HG and Park HY: The prevention of
carbon tetrachloride-induced hepatotoxicity in mice by
alpha-hederin: Inhibiton of cytochrome P450 2E1 expression. Biochem
Mol Biol Int. 45:163–170. 1998.PubMed/NCBI
|
|
56
|
Gaillard Y, Blaise P, Darré A, Barbier T
and Pépin G: An unusual case of death: Suffocation caused by leaves
of common ivy (Hedera helix). Detection of hederacoside C,
α-hederin, and hederagenin by LC-EI/MS-MS. J Anal Toxicol.
27:257–262. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Delmas F, Di Giorgio C, Elias R, Gasquet
M, Azas N, Mshvildadze V, Dekanosidze G, Kemertelidze E and
Timon-David P: Antileishmanial activity of three saponins isolated
from ivy, alpha-hederin, beta-hederin and hederacolchiside A1, as
compared to their action on mammalian cells cultured in vitro.
Planta Med. 66:343–347. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Danloy S, Quetin-Leclercq J, Coucke P, De
Pauw-Gillet MC, Elias R, Balansard G, Angenot L and Bassleer R:
Effects of alpha-hederin, a saponin extracted from Hedera helix, on
cells cultured in vitro. Planta Med. 60:45–49. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Takechi M and Yasuo T: Structure-activity
relationships of the saponin α-hederin. Phytochemistry. 29:451–452.
1990. View Article : Google Scholar
|