Introduction
Pancreatic cancer is the fourth leading cause of
cancer-related death, with a 5-year survival rate of <7%
(1). The only curative treatment
for this malignancy is complete resection, which is possible in
only 10-20% of patients (1-3). One
of the striking features of pancreatic cancer is its extremely
aggressive nature, with both local invasion and distant metastasis
usually already evident at presentation. Metastasis to regional
lymph nodes is one of the critical indicators of aggressive tumors.
Lymph node status is a powerful predictor of patient survival and
one of the crucial parameters used for staging tumors (4). These findings suggest that lymph node
metastasis has significant prognostic implications in pancreatic
cancer. However, the detailed mechanisms of lymph node metastasis
in pancreatic cancer remain not fully elucidated.
Single-cell migration via epithelial-mesenchymal
transition (EMT) is well known as a form of cell migration,
including cancer invasion (5).
Acquisition of the mesenchymal state is accompanied by E-cadherin
downregulation and vimentin upregulation, enabling cells to
dissociate from the epithelial tissue and migrate. EMT contributes
pathologically to cancer progression by enabling primary tumor
cells to break through the basal lamina and invade adjacent tissue,
leading to tumor dissemination (6). Collective cell migration is the
second principal mode of cell movement and is prevalent in multiple
types of cancer (7). Collective
cell migration differs from single cell migration in that cells
remain connected as they move. In histopathological sections, most
epithelial cancers display the hallmarks of collective invasion
into surrounding tissues, including intact cell-cell junctions, and
the expression of E-cadherin and other cadherins (8,9). In
fact, invasive cells are often observed as clusters in lymphatic
vessels pathologically (10).
Although the mechanism of single-tumor-cell invasion between
endothelial cells via EMT has been previously reported (11), the mechanism of collective invasion
in pancreatic cancer remains not well understood.
Recently, it was reported that the route of invasion
of metastatic tumor cells is associated with discontinuities in the
lymph vessel wall (10). Spheroids
of breast cancer cells formed large cell-free areas in the
lymphatic endothelial monolayer, termed 'circular
chemorepellent-induced defects' (CCIDs) (10). CCID formation is caused by the
migration of lymphatic endothelial cells (LECs), and not by their
apoptosis. Defects in the lymphatic endothelial layer may enable
cancer cells to enter the lymphatic duct (10). However, to the best of our
knowledge, no reports have described CCID formation ability and its
mechanism in pancreatic cancer.
S100 family proteins are small
Ca2+-binding proteins of the EF-hand type that have been
implicated in the regulation of a variety of intracellular and
extracellular processes, including cell proliferation,
differentiation, and intracellular signaling (12). S100P protein is a small isoform of
the S100 protein family that was isolated from the human placenta
(13). Its overexpression has been
detected in several tumors, including pancreatic cancer (14-17).
S100P has previously been demonstrated to regulate the
proliferation and survival of pancreatic cancer cells, as well as
to increase their migratory and invasive capabilities. In addition,
decreased metastatic potential was observed following S100P
silencing in an orthotropic mouse model (15). S100P facilitates the
transendothelial migration of pancreatic cancer cells (18), and was found to be significantly
upregulated in metastatic lymph nodes compared with the levels in
primary tumor by proteomic analysis and immunohistochemistry
(19). However, the molecular
mechanism underlying the role of S100P in lymph node metastasis is
not fully understood.
The present study demonstrated that spheroids of
human pancreatic cancer cells, and spheroids of cancer cells
established from a genetically engineered mouse model of pancreatic
cancer, caused CCID formation. Furthermore, S100P was identified as
a critical factor controlling this phenomenon.
Materials and methods
Patients and pancreatic tissues
Pancreatic cancer tissues were obtained from
patients who underwent pancreatic resection at the Kyushu
University Hospital between January 2010 and February 2012. The
mean age of the patients was 66 years (range 36-85 years), and 43%
were females. The study was approved by the Ethics Committee of
Kyushu University (reference no. 28-189) and conducted in
accordance with the Ethical Guidelines for Human Genome/Gene
Research enacted by the Japanese Government and the Helsinki
Declaration. Written informed consent was obtained from all
patients who agreed to the use of their samples in the present
research.
Immunohistochemistry
Primary pancreatic cancer tissues from patients were
evaluated by hematoxylin and eosin (H&E) staining. Expression
levels of cytokeratin-19 (CK-19), E-cadherin, vimentin, and the
lymphatic endothelial cell marker D2-40 were examined by
immunohistochemistry. The paraffin-embedded patient tissues were
sliced into 4-μm-thick section, deparaffinized in xylene,
and rehydrated through a graded series of ethanol concentrations.
Endogenous peroxidase activity was blocked by incubation with 3%
hydrogen peroxide in methanol for 30 min. Antigen retrieval was
performed by boiling in a microwave oven (citrate buffer, pH 6.0).
Sections were incubated with anti-CK19 antibody (1:500; cat. no.
sc376126), anti-E-cadherin (1:500; cat. no. sc8426) (both from
Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), anti-vimentin
(1:500; cat. no. ab92547; Abcam, Cambridge, MA, USA), anti-S100P
(1:500; cat. no. sc374547; Santa Cruz Biotechnology, Inc.), and
anti-D2-40 (cat. no. 413451; Nichirei Biosciences Inc., Tokyo,
Japan) overnight at 4°C. Sections were stained with EnVision
System-HRP Labeled Polymer Anti-Mouse (cat. no. K4001; Dako,
Carpinteria, CA, USA) at room temperature for 40 min. The labeled
antigens were visualized using 3,3-diaminobezidine
tetrahydrochloride as the chromogen at room temperature for 60 sec.
Staining was performed on serial sections. Images were acquired
using a confocal laser-scanning microscope (BZ-X700; Keyence
Corporation, Osaka, Japan).
Cells and culture conditions
Pancreatic cancer cell lines AsPC-1, BxPC-3,
Capan-1, CFPAC-1, Hs766T and SW1990 (American Type Culture
Collection, Manassas, VA, USA), Panc1 (RIKEN BioResource Center,
Tsukuba, Japan), MIAPaCa-2 and SUIT-2 (Japanese Collection of
Research Bioresources Cell Bank) were maintained in DMEM (cat. no.
D5523-10L; Sigma-Aldrich; Merck KGaG, Darmstadt, Germany)
supplemented with 10% FBS (cat. no. 10270; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), streptomycin (100
μg/ml), and penicillin (100 U/ml) at 37°C with humidified
90% air and 10% CO2. LECs (Japanese Collection of
Research Bioresources Cell Bank) were cultured in EGM2MV medium
(Lonza Group, Ltd., Basel, Switzerland) and were immortalized by
previously reported methods (20).
In all experiments, immortalized LECs were used. Human umbilical
vein endothelial cells (HUVECs) were obtained from Lonza Group,
Ltd. (cat. no. C2517A) and maintained in EBM-2 medium (Lonza Group,
Ltd.).
CCID formation assay
LECs were seeded in EGM2MV medium on 24-well plates
and allowed to grow to confluence. Spheroids were prepared using
cancer cells stained with CellTracker Green CMFDA (Thermo Fisher
Scientific, Inc.). Twelve spheroids were transferred into each
well. During the incubation period, frames were captured at 15-min
intervals with a confocal laser-scanning microscope (BZ-X700;
Keyence Corporation) and used to create a time-lapse video. The
CCID area was measured at 4 h following spheroid transfer using
ImageJ (v.1.48u; National Institutes of Health, Bethesda, MD,
USA).
Establishment of pancreatic cancer cells
from KPCL mice
KPCL (LSL-KrasG12D/+;
LSL-Trp53R172H/+; Pdx-1-Cre; ROSA26LSL-Luc/+)
transgenic mice were constructed as previously described (21). At the onset of distress and/or
abdominal distension, animals were sacrificed, and primary tumors
and metastatic tumors were excised. Pancreatic cancer cells from
KPCL mice were established in our laboratory from excised primary
tumors and metastatic tumors using the outgrowth method (22). Cancer cell lines derived from KPCL
mice were named as follows: i) number of the serial mouse that the
cell line was derived from; and ii) origin of the cancer cells (AC,
ascites-derived; PC, primary tumor-derived; LyC, lymph node
metastasis-derived; LC, liver metastasis-derived). Experimental
protocols involving animals were approved by the Animal Experiment
Committee, Graduate School of Medical Sciences, Kyushu University
(permit nos. A26-131-0 and A30-309-0).
Spheroid cohesion assay
Cancer cells were cultured in DMEM (Sigma-Aldrich;
Merck KGaA) with 10% FBS (Thermo Fisher Scientific, Inc.), rinsed
in PBS, trypsinized, and then prepared for spheroid formation and
image acquisition. The cells were distributed into
ultra-low-attachment-round bottomed 96-well plates (Corning, Inc.,
Corning, NY, USA) at a density of 1,000 cells/well. The plates were
centrifuged at 190 × g for 6 min. Following centrifugation, cells
were cultured as usual. Images were captured at 1 and 24 h later,
and the area of maximum horizontal section was measured using
ImageJ. Spheroid cohesion was compared at 24/1 h.
Adhesion assay
LECs (4×104/well) were cultured in
monolayers in 96-well collagen I-coated plates overnight. Collagen
I was used as the principal extracellular matrix molecule.
Pancreatic cancer cells were labeled with CellTracker Green CMFDA
(Thermo Fisher Scientific, Inc.). Pancreatic cancer cells
(4×104/well) were added to 96-well collagen I-coated
plates containing confluent LECs, and cells were incubated for 3 h
at 37°C. The plates were then washed three times with 200 μl
of PBS to remove the non-adherent tumor cells. The number of
adhered pancreatic cancer cells was determined in five random
fields at ×200 magnification using a confocal laser-scanning
microscope (BZ-X700).
Microarray analysis
Total RNA was isolated from cultured cells using a
High Pure RNA Isolation kit with DNase digestion (Roche Diagnostics
GmbH, Mannheim, Germany). RNA quality was evaluated using the
Agilent 2200 TapeStation system (Agilent Technologies, Inc., Santa
Clara, CA, USA) for microarray analysis. RNA was labeled and
hybridized to the Agilent SurePrint G3 Human Gene Expression
Microarray 8 × 60K Ver.3.0 (Agilent Technologies, Inc.). Data
analysis was performed using Feature Extraction software (Agilent
Technologies, Inc.).
Immunofluorescence staining
The paraffin-embedded patient tissues were sliced to
a thickness of 4 μm. These were the same tissues that were
used for immunohistochemistry. Endogenous peroxidase activity was
blocked with methanol containing 0.3% hydrogen peroxidase.
Subsequently, 3% hydrogen peroxide was applied as blocking reagent
at room temperature for 30 min. Antigen retrieval was performed by
boiling in a microwave oven (citrate buffer, pH 6.0). Sections were
incubated with rabbit anti-LYVE-1 (1:1,000; cat. no. ab14917;
Sigma-Aldrich; Merck KGaA) and mouse anti-S100P (1:500; cat. no.
sc374547; Santa Cruz Biotechnology, Inc.) antibodies overnight at
4°C. Sections were then incubated for 1 h with Alexa 546-conjugated
anti-mouse immunoglobulin G (IgG) and Alexa 488-conjugated
anti-rabbit IgG (both from Thermo Fisher Scientific, Inc.). Nuclear
DNA was counterstained with DAPI (0.05 mg/ml). Staining was
performed on serial sections. Images were acquired using a
fluorescence microscope (BZ-X700; Keyence Corporation).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cultured cells using a
High Pure RNA Isolation kit (cat. no. 11828665001; Roche
Diagnostics GmbH). The extracted RNA was quantified by the
absorbance at 260 nm, and its purity was evaluated by the 260/280
ratio of absorbance with an ND-1000 spectrophotometer (NanoDrop
Technologies; Thermo Fisher Scientific, Inc.). RT-qPCR was
performed using the iTaq Universal SYBR-Green One-Step kit and
CFX96 Touch Real-Time PCR Detection systems (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). Reactions were incubated at 50°C for 10
min; 95°C for 1 min; 95°C for 10 sec; 60°C for 30 sec for 39
cycles; 60°C for 5 sec; 95°C for 5 sec. The results were analyzed
using the 2-ΔΔCq method (23). Primers were purchased from Takara
Bio, Inc. The human ACTB gene and GAPDH gene were used as
endogenous controls. The following primers were used: S100P,
forward, 5′-GCACCATGACGGAACTAGAGACA-3′ and reverse,
5′-CAGGTCCTTGAGCAATTTATCCAC-3′; ACTB, forward,
5′-TGGCACCCAGCACAATGAA-3′ and reverse,
5′-CTAAGTCATAGTCCGCCTAGAAGCA-3′; GAPDH, forward,
5′-GCACCGTCAAGGCTGAGAAC-3′ and reverse,
5′-TGGTGAAGACGCCAGTGGA-3′.
Western blotting
Whole-cell lysates were prepared with PRO-PREP
solution (Intron Biotechnology, Inc.) from subconfluent cells. For
SDS-PAGE, 20 μg protein was separated by gel electrophoresis
on 4-15% Mini-PROTEAN TGX Precast gels (cat. no. 456-1084; Bio-Rad
Laboratories, Inc.) and transferred to Trans-Blot Turbo Mini PVDF
Transfer Packs (cat. no. 170-4156; Bio-Rad Laboratories, Inc.)
using a Trans-Blot Turbo Transfer Starter System (Bio-Rad
Laboratories, Inc.). The membranes were incubated with 5% milk as
blocking reagent at room temperature for 1 h. The membranes were
incubated overnight at 4°C with anti-S100P (1:200; cat. no.
sc374547; Santa Cruz Biotechnology, Inc.) and anti-β-actin (1:5000;
cat. no. ab8227; Abcam) antibodies, and then probed with
horseradish-peroxidase-conjugated-secondary antibodies (1:2,000;
cat. no. 7076; Cell Signaling Technology, Inc., Danvers, MA, USA)
for 1 h. Immunoblot detection was performed using chemiluminescence
with a ChemiDoc XRS System (Bio-Rad Laboratories, Inc.). Quantity
One software (version 4.6.6; Bio-Rad Laboratories, Inc.) was used
for densitometry.
Migration assay
Cell migration was assessed after incubation for 24
h using uncoated Transwell chambers. To assess the effect of S100P
on the migration of LECs, recombinant human S100P protein (Abcam),
the receptor for advanced glycation end-products (RAGE) antagonist
peptide (Tocris Bioscience, Bristol, UK), or recombinant human IL-6
protein (PeproTech, Inc., Rocky Hill, NJ, USA) was added to the
lower chambers in 750 μl of EGM2MV medium. LECs
(2×104/well) in 250 μl of EGM2MV medium were
seeded in each upper well and incubated for 24 h. Migrated cells at
the bottom of the chamber were fixed with 70% ethanol and stained
with H&E, and five random fields at ×200 magnification were
counted under a confocal microscope (BZ-X700).
Small interfering RNA (siRNA) silencing
of S100P
LECs at 90% confluence were transfected with siRNA
(Sigma-Aldrich; Merck KGaA) by electroporation using a Nucleofector
System (Lonza Group, Ltd.) according to the manufacturer's
recommendations. The following siRNAs directed against human S100P
were used in the study: siRNA-1, sense, 5′-AGGCUUCCUGCAGAGUGGA-3′
and antisense, 5′-UCCACUCUGCAGGAAGCCU-3′; siRNA-2, sense,
5′-GGAUGCCGUGGAUAAAUUG-3′ and antisense, 5′-CAAUUUAUCCACGGCAUCC-3′;
and siRNA-3, sense, 5′-CAAGGAUGCCGUGGAUAAA-3′ and antisense,
5′-UUUAUCCACGGCAUCCUUG-3′. To confirm knockdown specificity, a
negative control siRNA (cat. no. SIC001; Sigma-Aldrich; Merck KGaA)
was used. Transfected cells were used in subsequent experiments at
24-72 h post-transfection. S100P expression was assessed 72 h
post-transfection by RT-qPCR.
Statistical analysis
Results are presented as mean ± SD. Comparisons
between groups were performed using one-way ANOVA followed by the
Tukey-Kramer multiple comparisons test. Survival analyses were
conducted using the Kaplan-Meier method, and the curves were
compared using the log-rank test. All statistical analyses were
performed using JMP 12 software (SAS Institute, Inc., Cary, NC,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
Pancreatic cancer cells exist as clusters
in lymphatic ducts and lymph nodes
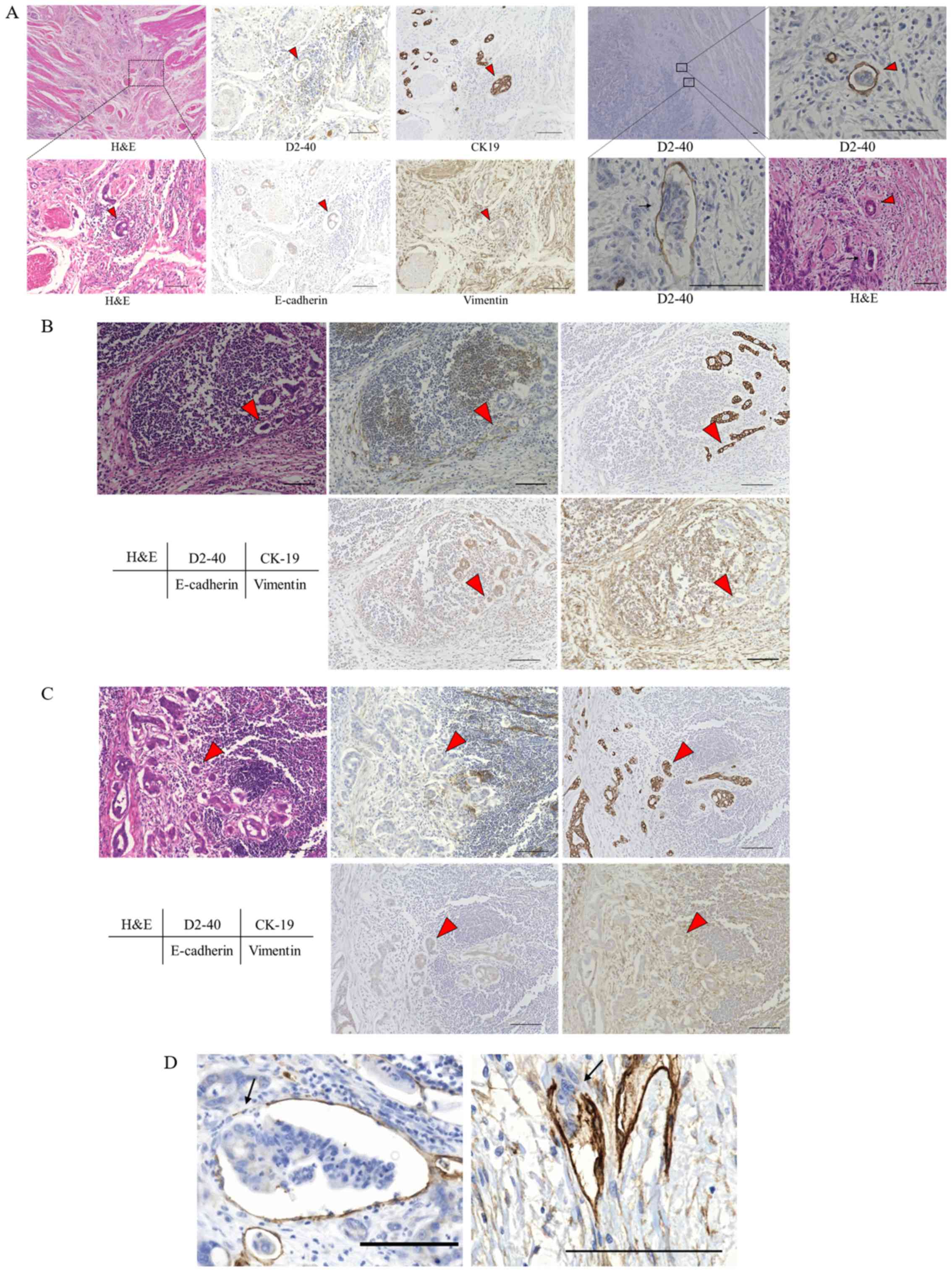
To investigate whether collective invasion occurs in
human pancreatic cancer tissue, human pancreatic cancer tissue
samples were stained with H&E, anti-D2-40 (a marker for
lymphatic endothelial cells) and anti-CK19 (a marker for cancer
cells). In addition, the tissues were stained for the epithelial
marker E-cadherin and the mesenchymal marker Vimentin.
Immunohistochemical staining of human pancreatic cancer tissues
revealed that clusters of tumor cells were present in the lymphatic
ducts around the primary tumor (Fig.
1A), and in the subcapsular sinuses (Fig. 1B) and the lymphatic cortex of the
lymph nodes (Fig. 1C). Thus,
embolic tumor cell clusters were observed in sites that are known
to be routes of lymphatic metastasis. These clusters of tumor cells
expressed epithelial markers but not mesenchymal markers (Fig. 1A-C). Clusters of cancer cells that
penetrated the wall of lymphatic ducts around the primary tumor
were frequently observed (Fig.
1D). These findings suggested that cancer cells invaded in the
form of clusters, and without having undergone EMT, to form lymph
node metastases.
Spheroids of pancreatic cancer cells
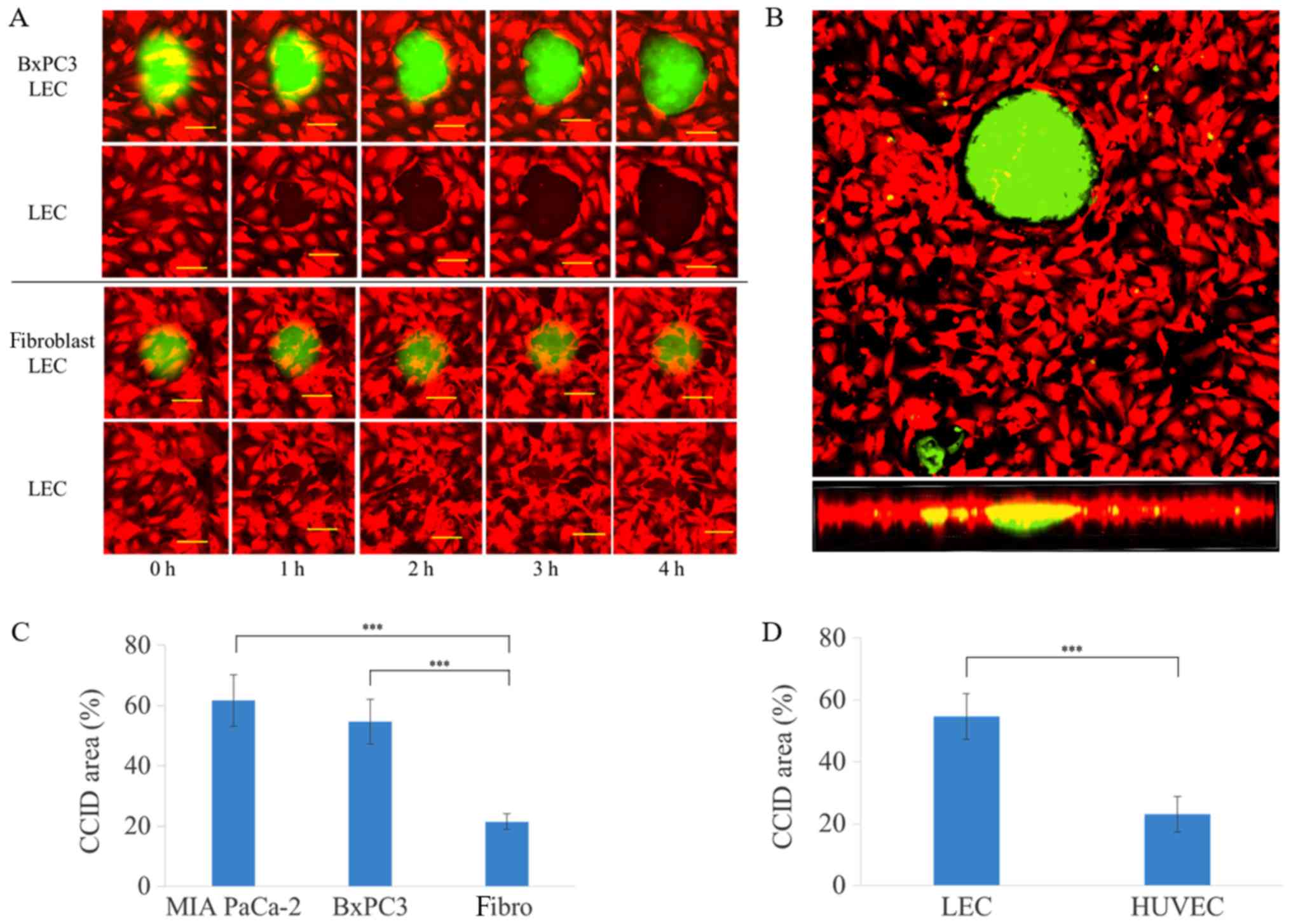
induce CCIDs in LEC monolayers
Spheroids of pancreatic cancer cells were placed on
top of lymphatic endothelial monolayers. LECs beneath these
spheroids migrated and formed large cell-free areas in the
lymphatic endothelial monolayer, called CCIDs (Fig. 2A). These in vitro CCIDs were
similar to the defects observed in the lymphovascular walls at the
sites of tumor cell invasion in human tissue. Observation of CCID
formation using a confocal microscope revealed that defects were
formed in the LEC layer immediately underneath spheroids (Fig. 2B). When spheroids of human
fibroblasts were used, CCIDs were smaller compared with those
formed with cancer cells (Fig. 2A and
C). CCID formation in LECs was more pronounced compared with
HUVECs (Fig. 2D).
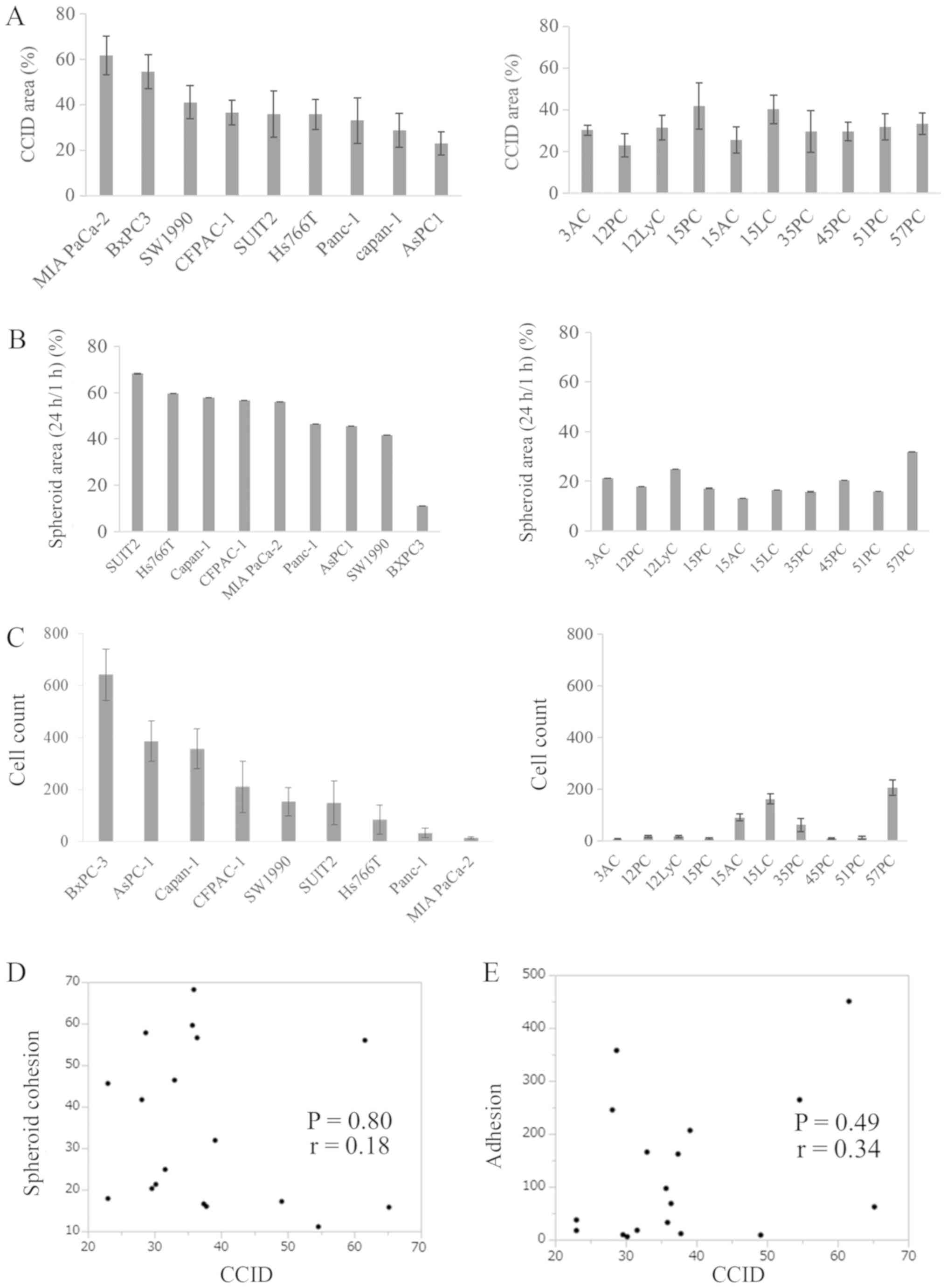
Neither spheroid cohesion nor adhesion to
LECs has a significant correlation with CCID formation
First, the spheroid cohesion ability of pancreatic
cancer cells and their ability to adhere to LECs were considered as
candidate factors related to CCID formation. CCID area (Fig. 3A), spheroid cohesion (Fig. 3B), and adhesion to LECs (Fig. 3C) were investigated using each cell
line (human pancreatic cancer cells and KPCL mouse-derived
pancreatic cancer cells). The CCID area differed for each cell line
(Fig. 3A). Similarly, spheroid
cohesion (Fig. 3B) and adhesion to
LECs (Fig. 3C) differed for each
cell line. The CCID area was not significantly different between
human and KPCL mouse-derived cell lines. Spheroid cohesion was
significantly higher in KPCL mouse-derived cell lines compared with
human cell lines (P<0.01). Adhesion to LECs was significantly
higher in human cell lines compared with KPCL mouse-derived cell
lines (P=0.02). However, they did not exhibit common features. The
correlation between each result and CCID was analyzed by Pearson
product-moment correlation coefficient. Neither spheroid cohesion
(P=0.80) nor adhesion to LECs (P=0.49) displayed a significant
correlation with CCID formation (Fig.
3D and E).
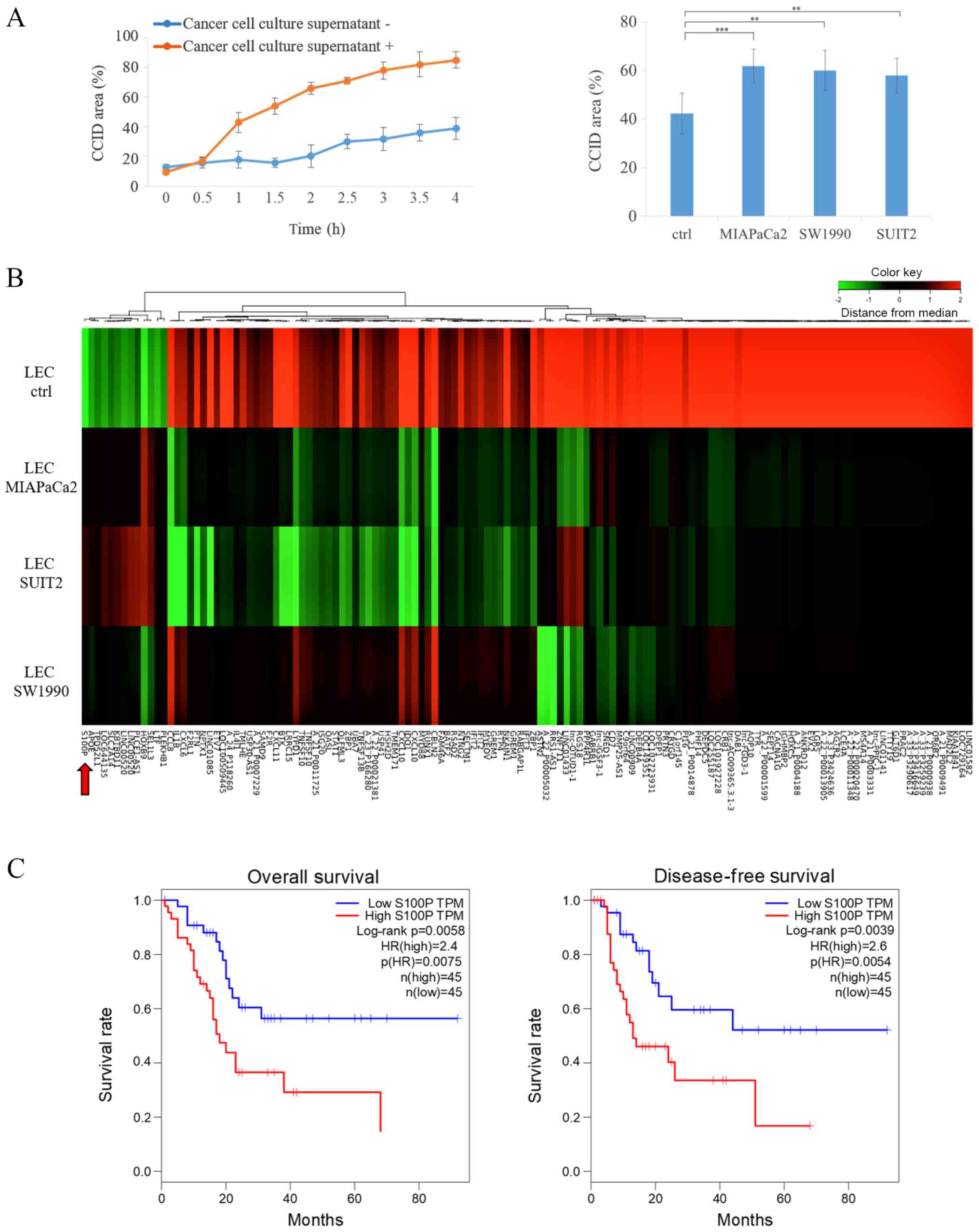
Treatment with cancer cell supernatant
increases CCID formation and S100P expression in LECs
To evaluate whether secreted cancer cell-derived
factors were involved in CCID formation of pancreatic cancer
spheroids, the effects of cancer cell supernatant on CCID formation
were investigated. When the cancer cells were subconfluent, the
medium was changed to serum-free DMEM and the supernatant was
collected after 48 h. Cancer cell culture supernatant (Fig. 4A left panel, from SUIT2 cells;
right panel, from MIAPaCa2, SW1990 and SUIT2 cells) was added in
LECs for 30 min and then removed. Serum-free DMEM was used in the
control group. Next, SUIT2 spheroids were placed on top of the
lymphatic endothelial monolayers. The results reveled that CCID
area was increased following treatment with cancer cell supernatant
(Fig. 4A). Similar results were
obtained using the culture supernatants of several lines of
pancreatic cancer cells (Fig. 4A).
To investigate how gene expression was altered in LECs following
treatment with cancer cell supernatant, microarray analysis was
performed using LECs with treatment of culture supernatants from
three pancreatic cancer cell lines. A heatmap analysis of gene
expression profiles revealed common variation in LEC samples
treated with culture supernatants from pancreatic cancer cells
(Fig. 4B). A total of 133 genes
were identified as differentially expressed in LECs following
treatment with culture supernatants from pancreatic cancer cells.
Out of these 133 genes, S100P was selected for further study. S100P
has been reported to be highly expressed in pancreatic cancer
(15) and is associated with the
cell cytoskeleton (24), migration
(24,25), and adhesion (25). In addition, The Cancer Genome Atlas
database was explored in regards to S100P expression, and the
results revealed that S100P expression was a significant prognostic
factor for pancreatic cancer (Fig.
4C), similar to previous reports (16,19).
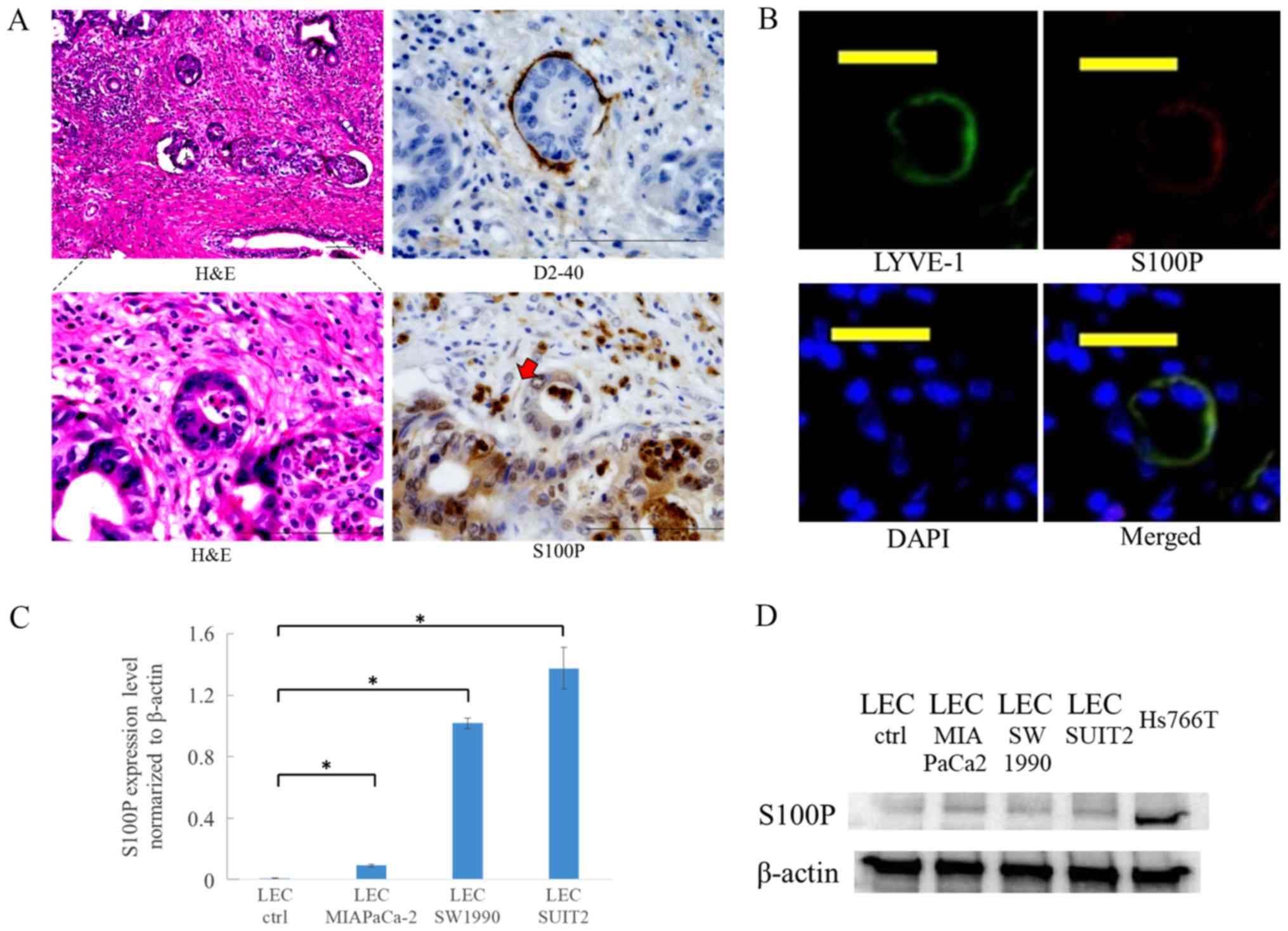
LECs express S100P in lymphatic vessels
around the primary tumor
To confirm whether lymphatic endothelial cells
express S100P in human tissues, serial sections from patient
pancreatic tumors were used for immunohistochemistry and
immunofluorescence staining. Although S100P was highly expressed in
tumor cells, it was also expressed in LECs at a site where a tumor
cluster was present inside the lymphatic vessel surrounding the
primary tumor (Fig. 5A and B).
Consistent with the results from the microarray analysis, S100P
mRNA expression increased in LECs following treatment with culture
supernatant from cancer cells for 48 h (Fig. 5C). S100P expression was also
increased at the protein level following treatment with the culture
supernatant (Fig. 5D).
S100P is involved in migration of LECs
and CCID formation
A previous report revealed that S100P mRNA
expression is increased in prostate cancer cells following
treatment with IL-6 (26). It has
also been reported that pancreatic cancer cells express higher
levels of IL-6 compared with normal human pancreatic ductal
epithelium cells (27). Therefore,
the changes in S100P expression were investigated in LECs following
treatment with IL-6. S100P expression in LECs increased following
IL-6 treatment (Fig. 6A). It has
been reported that S100P is associated with cell migration
(24,25). IL-6 treatment increased migration
in LECs (Fig. 6B) and CCID
formation (Fig. 6D). To confirm
that IL-6-induced LEC migration occurs via S100P, gene knockdown
studies were performed. S100P expression was silenced using siRNA
(Fig. 6C, left panel). The effect
of S100P knockdown on IL-6-induced LEC migration was then
investigated. IL-6-induced LEC migration was significantly reduced
following S100P silencing in LECs (Fig. 6C, right panel). Extracellular S100P
has been demonstrated to activate RAGE (15,28),
leading to the increased proliferation, invasion and migration of
cancer cells (28,29). Therefore, the migration ability of
LECs was evaluated following treatment with culture supernatant
from cancer cells, or recombinant human S100P protein with or
without RAGE antagonist peptide. Migration was increased by
treatment with culture supernatant from cancer cells or recombinant
human S100P protein, but this increase was suppressed following
treatment with RAGE antagonist peptide (Fig. 6E). CCID area was also increased
following treatment with recombinant human S100P protein, which was
suppressed by the addition of RAGE antagonist peptide (Fig. 6F).
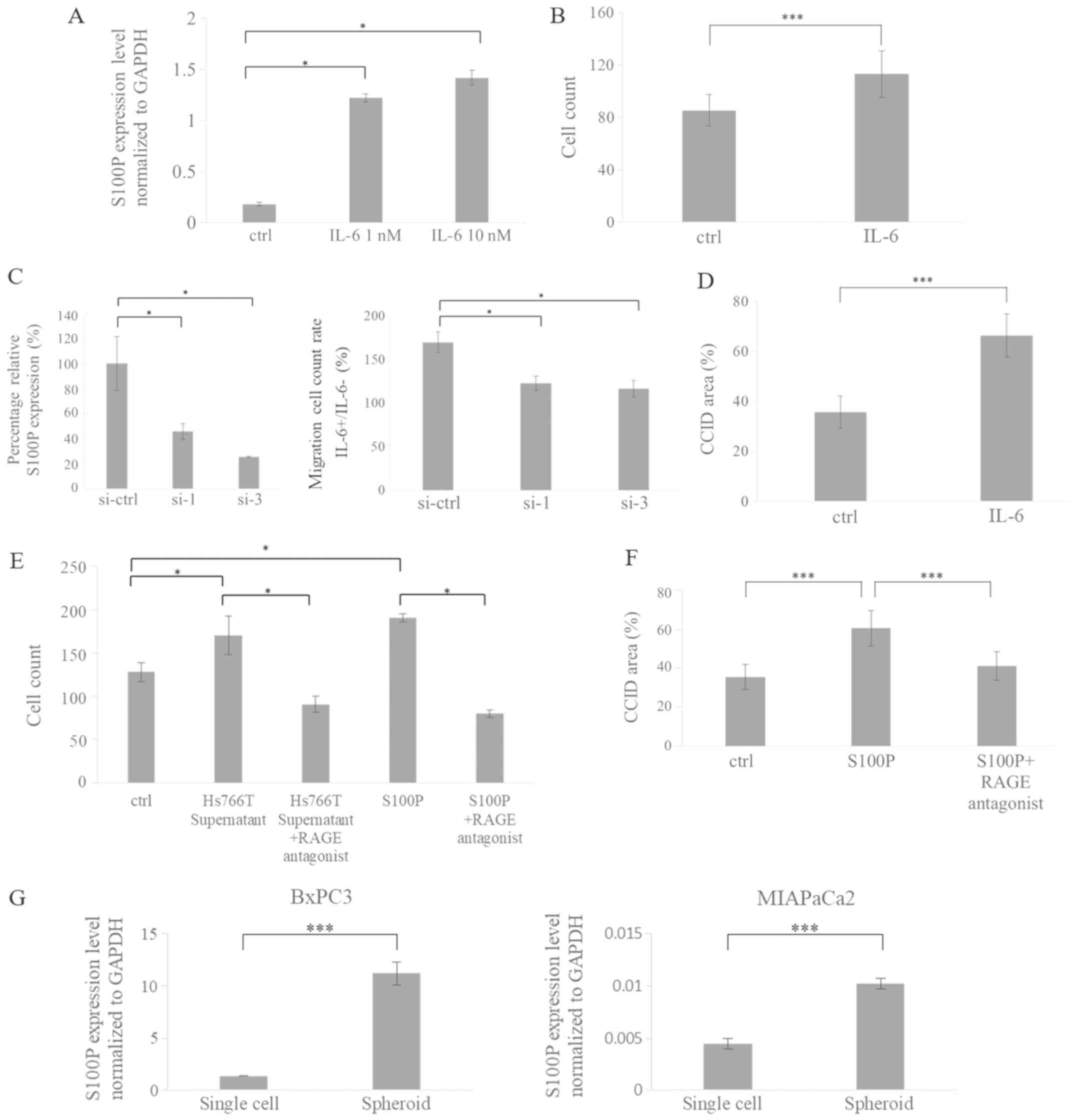 | Figure 6S100P is involved in migration of
LECs and CCID formation. (A) S100P mRNA expression in LECs
following treatment with IL-6 for 24 h. (B) LEC migration following
treatment with IL-6 (1 nM) was examined by Transwell migration
assay. (C) S100P mRNA levels were significantly reduced following
siRNA transfection (left panel). The IL-6-enhanced LEC migration
was reduced following S100P knockdown (right panel). (D) CCID area
following treatment with IL-6 (1 nM). (E) LEC migration following
treatment with culture supernatant from cancer cells, recombinant
human S100P protein (1 nM) and RAGE antagonist peptide (1
μg/ml). (F) CCID area following treatment with recombinant
human S100P protein (1nM) and RAGE antagonist peptide (1
μg/ml). (G) Comparison of S100P mRNA expression levels in
cancer cells from single-cell culture (normal plates) and from
spheroid culture (low-attachment plates) in two cell lines, BxPC3
and MIAPaCa2. *P<0.05 and ***P<0.001,
with comparisons indicated by brackets. S100P, S100 calcium binding
protein P; LECs, lymphatic endothelial cells; CCID, circular
chemorepellent-induced defect; IL, interleukin; si, small
interfering; RAGE, receptor for advanced glycation end-products;
ctrl, control. |
The expression profile of cancer cells
changes under spheroid culture conditions (10)
Therefore, pancreatic cancer cells were cultured in
spheroid culture conditions on low-attachment plates or in normal
culture conditions on normal plates, and then their S100P mRNA
expression levels were compared. Expression of S100P was higher in
the cancer cells cultured in low-attachment plates (spheroid
culture) compared with cells cultured in normal plates (single cell
culture; Fig. 6G). This finding
suggested that the expression and secretion of S100P were induced
by the spheroid structure, leading to enhanced CCID formation by
pancreatic cancer cell spheroids in LECs.
Discussion
The main findings of the present study were as
follows: i) In human pancreatic cancer tissue, there were clusters
of cancer cells that penetrated the wall of lymphatic ducts around
the primary tumor; ii) treatment with cancer cell culture
supernatant and IL-6 enhanced migration of LECs and CCID formation
in LECs; and iii) S100P promoted LEC migration and CCID formation
by pancreatic cancer cell spheroids in LECs.
In human pancreatic cancer tissue, clusters of
cancer cells that penetrated the wall of lymphatic ducts around the
primary tumor were observed by histology analysis. Clusters of
tumor cells were present at sites known to be routes of lymph node
metastasis (lymphatic vessels around the primary tumor, and
subcutaneous and lymphatic cortex of lymph nodes). These findings
suggested that there is a cluster-specific mechanism for lymphatic
metastasis in pancreatic cancer.
Previous reports have revealed that tumor cell
invasion to lymphatic ducts is crucial for the dissemination of
lymphatic metastatic tumors (10,30).
CCIDs enable the entire tumor bulk to penetrate vessels. The
present CCID assay resembles the pathological situation of
collective invasion in human pancreatic cancer. Therefore, this
assay is a valuable tool to quantitatively investigate the
cluster-specific mechanisms of lymph vessel invasion and to
elucidate the underlying molecular mechanisms. In the present
study, it was demonstrated that spheroids of pancreatic cancer
cells cause CCIDs in LEC monolayers. Time-lapse videos revealed
centrifugal migration of LECs beneath the BxPC3 spheroids. The
level of CCID formation induced by cancer cell spheroids was higher
than that induced by fibroblast spheroids, and cancer cell
spheroids exhibited higher CCID formation in LECs than in HUVECs.
These findings suggested that CCID formation is specific to
cluster-based collective invasion to lymphatic vessels, which is a
mechanism of lymphatic metastasis in pancreatic cancer.
In the present study, treatment with culture
supernatant from cancer cells enhanced CCID formation in LECs.
Therefore, to clarify the mechanisms involved, microarray analysis
was performed to investigate the changes in gene expression in LECs
following treatment with supernatant. A total of 133 genes were
differentially expressed following treatment with culture
supernatants from three pancreatic cancer cell lines. Among these
genes, S100P was selected for further study. To the best of our
knowledge, no reports have described S100P expression in LECs to
date. In the present study, S100P was demonstrated to be expressed
in LECs at a site at which there was a tumor cluster in lymphatic
vessels around the primary tumor, using immunohistochemistry and
immunofluorescence staining. S100P was not expressed in lymphatic
vessels distant from the tumor. These findings suggest that the
expression of S100P in LECs was increased by factors secreted by
cancer cells.
Previous reports have demonstrated that expression
of S100P is affected by several hormones [progesterone (31), androgens (32) and glucocorticoids (33)], multiple transcription factors
[bone morphogenetic protein 4, SMAD, STAT/cAMP responsive element
binding protein, and specificity protein/Kruppel-like factor
(34,35)] and cytokines (IL-6) (26). S100P mRNA expression was increased
in prostate cancer cells following treatment with IL-6 (26), and IL-6 has been demonstrated to be
associated with advanced tumor stage and poor survival in
pancreatic cancer (36,37). Higher serum levels of IL-6 are also
found in patients with pancreatic cancer than in healthy controls
(38). In addition, pancreatic
cancer cell lines express higher levels of IL-6 compared with
normal human pancreatic ductal epithelial cells (27). The present study demonstrated that
S100P expression in LECs increased following treatment with IL-6
and that IL-6 treatment increased LEC migration and CCID formation.
The IL-6-enhanced LEC migration was significantly reduced by S100P
knockdown in LECs. Therefore, these results indicated that S100P
partially mediated the IL-6-induced LEC migration. These findings
suggested that IL-6, which is known to be secreted from pancreatic
cancer cells and stromal cells, increased S100P expression in LECs
and regulated CCID formation.
In the present study, intracellular S100P expression
increased in LECs following treatment with culture supernatant from
pancreatic cancer cells. Intracellular S100P is released from cells
and extracellular S100P can activate RAGE, which are multiligand
transmembrane receptors of the immunoglobulin superfamily (15,28).
Therefore, the effect of extracellular S100P was investigated on
LEC migration and pancreatic cancer cell-induced CCID formation and
the results revealed that both were increased following treatment
with recombinant human S100P protein, as well as following
treatment with culture supernatant from pancreatic cancer cells.
Furthermore, this increase was suppressed by the addition of RAGE
antagonist peptide. These results suggest that the S100P/RAGE
signaling pathway controls the migration of LECs and the CCID
formation. Because the inhibitory effect of RAGE antagonist peptide
in the migration of LECs was significant, RAGE antagonist peptide
may inhibit the effects of S100P released not only from LECs but
also from the cancer cells themselves. In addition, S100P
expression was demonstrated to be increased in low attachment
plates (spheroid culture) compared with normal plates (single cell
culture), suggesting that high S100P expression was induced by
spheroid formation of pancreatic cancer cells, leading to enhanced
CCID formation by pancreatic cancer cell spheroids in LECs. Further
investigation of the mechanism of the S100P/RAGE axis is
necessary.
In the present study, the role of S100P in LECs was
not evaluated in vivo. Since S100P is not expressed in
rodents (39), it is difficult to
evaluate its role in mouse models. In summary, results from human
pancreatic cancer tissues revealed that cluster-based collective
invasion occured in lymphatic metastasis. The present in
vitro data demonstrated that spheroids of pancreatic cancer
cells caused CCID formation and that CCIDs in pancreatic cancer
were partly regulated by S100P, suggesting that S100P may be a
promising target to inhibit lymph node metastasis.
Funding
This work was supported in part by the Japan Society
for the Promotion of Science Grants-in-Aid for Scientific Research
and Scientific Research on Innovative Areas (grant nos. 17H04284,
17K19602, 16K10601, 16H05418 and 17K19605) and the Shinnihon
Foundation of Advanced Medical Treatment Research (grant no.
SN201705).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HN performed the majority of the experiments and
wrote the manuscript. KO and MN made notable contributions to the
design, data interpretation and the manuscript revision. AY, AS,
YA, SK, TA, SE, KK and TO performed experiments and established
experimental techniques. KS, TM, TO and KMiz helped to design and
plan experiments. KMiy, KN, YM and SI were involved in the
validation of data. All authors have read and approved the
manuscript.
Ethics approval and consent to
participate
Protocols involving the use of human tissues were
approved by the Ethics Committee of Kyushu University (reference
no. 28-189) and conducted in accordance with the Ethical Guidelines
for Human Genome/Gene Research enacted by the Japanese Government
and the Helsinki Declaration. Written informed consent was obtained
from all patients who agreed to the use of their samples in the
present research. Protocols involving the use of animals were
approved by the Animal Experiment Committee, Graduate School of
Medical Sciences, Kyushu University (permit nos. A26-131-0 and
A30-309-0).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
CCID
|
circular chemorepellent-induced
defect
|
|
LEC
|
lymphatic endothelial cell
|
|
EMT
|
epithelial-mesenchymal transition
|
|
H&E
|
hematoxylin and eosin
|
|
CK-19
|
cytokeratin-19
|
Acknowledgments
The authors thank E. Manabe and S. Sadatomi
(Department of Surgery and Oncology, Kyushu University
Hospital).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vincent A, Herman J, Schulick R, Hruban RH
and Goggins M: Pancreatic cancer. Lancet. 378:607–620. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Malvezzi M, Carioli G, Bertuccio P, Rosso
T, Boffetta P, Levi F, La Vecchia C and Negri E: European cancer
mortality predictions for the year 2016 with focus on leukaemias.
Ann Oncol. 27:725–731. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Basturk O, Saka B, Balci S, Postlewait LM,
Knight J, Goodman M, Kooby D, Sarmiento JM, El-Rayes B, Choi H, et
al: Substaging of lymph node status in resected pancreatic ductal
adenocarcinoma has strong prognostic correlations: Proposal for a
revised N classification for TNM staging. Ann Surg Oncol. 22(Suppl
3): S1187–S1195. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tam WL and Weinberg RA: The epigenetics of
epithelial-mesen-chymal plasticity in cancer. Nat Med.
19:1438–1449. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Friedl P, Hegerfeldt Y and Tusch M:
Collective cell migration in morphogenesis and cancer. Int J Dev
Biol. 48:441–449. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Alexander S, Koehl GE, Hirschberg M,
Geissler EK and Friedl P: Dynamic imaging of cancer growth and
invasion: A modified skin-fold chamber model. Histochem Cell Biol.
130:1147–1154. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Christiansen JJ and Rajasekaran AK:
Reassessing epithelial to mesenchymal transition as a prerequisite
for carcinoma invasion and metastasis. Cancer Res. 66:8319–8326.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kerjaschki D, Bago-Horvath Z, Rudas M,
Sexl V, Schneckenleithner C, Wolbank S, Bartel G, Krieger S, Kalt
R, Hantusch B, et al: Lipoxygenase mediates invasion of
intrameta-static lymphatic vessels and propagates lymph node
metastasis of human mammary carcinoma xenografts in mouse. J Clin
Invest. 121:2000–2012. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang J, Mani SA, Donaher JL, Ramaswamy S,
Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A and
Weinberg RA: Twist, a master regulator of morphogenesis, plays an
essential role in tumor metastasis. Cell. 117:927–939. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Donato R: S100: A multigenic family of
calcium-modulated proteins of the EF-hand type with intracellular
and extracellular functional roles. Int J Biochem Cell Biol.
33:637–668. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Becker T, Gerke V, Kube E and Weber K:
S100P, a novel Ca(2+)-binding protein from human placenta. cDNA
cloning, recombinant protein expression and Ca2+ binding
properties. Eur J Biochem. 207:541–547. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Crnogorac-Jurcevic T, Missiaglia E,
Blaveri E, Gangeswaran R, Jones M, Terris B, Costello E,
Neoptolemos JP and Lemoine NR: Molecular alterations in pancreatic
carcinoma: Expression profiling shows that dysregulated expression
of S100 genes is highly prevalent. J Pathol. 201:63–74. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Arumugam T, Simeone DM, Van Golen K and
Logsdon CD: S100P promotes pancreatic cancer growth, survival, and
invasion. Clin Cancer Res. 11:5356–5364. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ohuchida K, Mizumoto K, Egami T, Yamaguchi
H, Fujii K, Konomi H, Nagai E, Yamaguchi K, Tsuneyoshi M and Tanaka
M: S100P is an early developmental marker of pancreatic
carcinogenesis. Clin Cancer Res. 12:5411–5416. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Parkkila S, Pan PW, Ward A, Gibadulinova
A, Oveckova I, Pastorekova S, Pastorek J, Martinez AR, Helin HO and
Isola J: The calcium-binding protein S100P in normal and malignant
human tissues. BMC Clin Pathol. 8:22008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Barry S, Chelala C, Lines K, Sunamura M,
Wang A, Marelli-Berg FM, Brennan C, Lemoine NR and
Crnogorac-Jurcevic T: S100P is a metastasis-associated gene that
facilitates transendothelial migration of pancreatic cancer cells.
Clin Exp Metastasis. 30:251–264. 2013. View Article : Google Scholar
|
|
19
|
Naidoo K, Jones R, Dmitrovic B, Wijesuriya
N, Kocher H, Hart IR and Crnogorac-Jurcevic T: Proteome of
formalin-fixed paraffin-embedded pancreatic ductal adenocarcinoma
and lymph node metastases. J Pathol. 226:756–763. 2012. View Article : Google Scholar
|
|
20
|
Endo S, Nakata K, Ohuchida K, Takesue S,
Nakayama H, Abe T, Koikawa K, Okumura T, Sada M, Horioka K, et al:
Autophagy is required for activation of pancreatic stellate cells,
associated with pancreatic cancer progression and promotes growth
of pancreatic tumors in mice. Gastroenterology. 152:1492–1506.e24.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dosch JS, Ziemke EK, Shettigar A,
Rehemtulla A and Sebolt-Leopold JS: Cancer stem cell marker
phenotypes are reversible and functionally homogeneous in a
preclinical model of pancreatic cancer. Cancer Res. 75:4582–4592.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bachem MG, Schneider E, Gross H,
Weidenbach H, Schmid RM, Menke A, Siech M, Beger H, Grünert A and
Adler G: Identification, culture, and characterization of
pancreatic stellate cells in rats and humans. Gastroenterology.
115:421–432. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−ΔΔC(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
24
|
Austermann J, Nazmi AR, Müller-Tidow C and
Gerke V: Characterization of the Ca2+-regulated
ezrin-S100P interaction and its role in tumor cell migration. J
Biol Chem. 283:29331–29340. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Du M, Wang G, Ismail TM, Gross S, Fernig
DG, Barraclough R and Rudland PS: S100P dissociates myosin IIA
filaments and focal adhesion sites to reduce cell adhesion and
enhance cell migration. J Biol Chem. 287:15330–15344. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hammacher A, Thompson EW and Williams ED:
Interleukin-6 is a potent inducer of S100P, which is up-regulated
in androgen-refractory and metastatic prostate cancer. Int J
Biochem Cell Biol. 37:442–450. 2005. View Article : Google Scholar
|
|
27
|
Feurino LW, Zhang Y, Bharadwaj U, Zhang R,
Li F, Fisher WE, Brunicardi FC, Chen C, Yao Q and Min L: IL-6
stimulates Th2 type cytokine secretion and upregulates VEGF and
NRP-1 expression in pancreatic cancer cells. Cancer Biol Ther.
6:1096–1100. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Arumugam T, Simeone DM, Schmidt AM and
Logsdon CD: S100P stimulates cell proliferation and survival via
receptor for activated glycation end products (RAGE). J Biol Chem.
279:5059–5065. 2004. View Article : Google Scholar
|
|
29
|
Arumugam T, Ramachandran V, Gomez SB,
Schmidt AM and Logsdon CD: S100P-derived RAGE antagonistic peptide
reduces tumor growth and metastasis. Clin Cancer Res. 18:4356–4364.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nguyen CH, Senfter D, Basilio J, Holzner
S, Stadler S, Krieger S, Huttary N, Milovanovic D, Viola K,
Simonitsch-Klupp I, et al: NF-κB contributes to MMP1 expression in
breast cancer spheroids causing paracrine PAR1 activation and
disinte-grations in the lymph endothelial barrier in vitro.
Oncotarget. 6:39262–39275. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chandramouli A, Mercado-Pimentel ME,
Hutchinson A, Gibadulinová A, Olson ER, Dickinson S, Shañas R,
Davenport J, Owens J, Bhattacharyya AK, et al: The induction of
S100p expression by the Prostaglandin E (PGE signaling pathway in
colon cancer cells. Cancer Biol Ther2 2)/EP4 receptor.
10:1056–1066. 2010. View Article : Google Scholar
|
|
32
|
Averboukh L, Liang P, Kantoff PW and
Pardee AB: Regulation of S100P expression by androgen. Prostate.
29:350–355. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kino T, Manoli I, Kelkar S, Wang Y, Su YA
and Chrousos GP: Glucocorticoid receptor (GR) beta has intrinsic,
GRalpha-independent transcriptional activity. Biochem Biophys Res
Commun. 381:671–675. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hamada S, Satoh K, Hirota M, Fujibuchi W,
Kanno A, Umino J, Ito H, Satoh A, Kikuta K, Kume K, et al:
Expression of the calcium-binding protein S100P is regulated by
bone morpho-genetic protein in pancreatic duct epithelial cell
lines. Cancer Sci. 100:103–110. 2009. View Article : Google Scholar
|
|
35
|
Gibadulinova A, Tothova V, Pastorek J and
Pastorekova S: Transcriptional regulation and functional
implication of S100P in cancer. Amino Acids. 41:885–892. 2011.
View Article : Google Scholar
|
|
36
|
Ebrahimi B, Tucker SL, Li D, Abbruzzese JL
and Kurzrock R: Cytokines in pancreatic carcinoma: Correlation with
phenotypic characteristics and prognosis. Cancer. 101:2727–2736.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nixon AB, Pang H, Starr MD, Friedman PN,
Bertagnolli MM, Kindler HL, Goldberg RM, Venook AP and Hurwitz HI;
Alliance for Clinical Trials In Oncology: Prognostic and predictive
blood-based biomarkers in patients with advanced pancreatic cancer:
Results from CALGB80303 (Alliance). Clin Cancer Res. 19:6957–6966.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pop VV, Seicean A, Lupan I, Samasca G and
Burz CC: IL-6 roles - Molecular pathway and clinical implication in
pancreatic cancer - A systemic review. Immunol Lett. 181:45–50.
2017. View Article : Google Scholar
|
|
39
|
Shang X, Cheng H and Zhou R: Chromosomal
mapping, differential origin and evolution of the S100 gene family.
Genet Sel Evol. 40:241–264. 2008. View Article : Google Scholar
|