Introduction
Oral cancer refers any cancerous cells that are
located in the oral cavity. It is a type of head and neck cancer,
accounting for most head and neck cancers and leading to
>145,400 cases/year of mortality globally (1). Oral squamous cell carcinoma (OSCC) is
the most common type of malignancy in the oral cavity (2). Conventional treatment of OSCC
includes surgery, radiotherapy and chemotherapy(3). Although the clinical outcome of
patients with OSCC has gradually improved in the last few years,
the prognosis of patients with advanced-stage disease remains poor,
reflecting limited advances in present understanding of the
pathogenesis of this disorder (4).
Therefore, it is necessary to develop novel therapeutic approaches
for patients with advanced and unresectable OSCC.
Vincristine (VCR), a naturally occurring
Vinca alkaloid, is a classical microtubule-destabilizing
agent (5). It is widely used for
hematologic malignancies and certain solid tumors (6-8)
However, despite the proven antitumor activity of VCR treatment,
the efficacy of chemotherapy is often limited by rapid emergence of
acquired resistance and patient relapse following initial
response.
Tumors consist of a complex microenvironment
composed of cancer cells, stroma and immune cells (9). Drug resistance is a complex process
involving reciprocal interplay between different types of cells.
Secreted proteins are responsible for the cross-talk among cells,
which may facilitate drug resistance in tumors (10-12).
Several studies have indicated that soluble
mediators from the microenvironment can promote cancer growth and
therapy resistance (13-15). Amphiregulin (AREG) (16), a ligand of the epidermal growth
factor (EGF) receptor (EGFR), is synthesized as a transmembrane
precursor that undergoes a series of proteolytic steps to produce
mature forms for secretion (16).
AREG has been reported to induce oncogenic effects in numerous
cancer cell types, including breast, liver, pancreatic and
colorectal cancer cells (17-20)
and to be implicated in drug resistance (10,18,21,22);
however, the effects of AREG and its mechanisms of action in OSCC
cells remain unknown. Understanding the complex mechanisms
underlying its effects may reveal potential therapeutic
opportunities.
Antibody arrays possess valuable applications in
cancer research to identify biomarkers or molecules that are
potentially relevant for diagnosis, prognosis, treatment and drug
development (23). To elucidate
the association between secreted proteins and VCR resistance in
OSCC cells, a VCR-resistant SAS subline (SAS-VCR) was established
by exposure to an increasing drug concentration gradient.
Conditioned medium (CM) was collected from parental and
VCR-resistant cells, and the secreted proteins were assessed using
an antibody array. In the present study, comprehensive secretion
profiling was performed to provide novel insight in the mechanisms
of VCR resistance. Understanding the relationship between secreted
proteins and drug resistance may contribute to the development of
novel therapeutic strategies and biomarkers in OSCC.
Materials and methods
Chemicals, reagents and antibodies
VCR, 5-fluorouracil (5-FU), cisplatin, MTT and
dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (Merck
KGaA). VCR was dissolved in sterile PBS and diluted in cell culture
medium to the required concentration prior to use. 5-FU was
dissolved in DMSO. Cisplatin was dissolved in dimethylformamide. A
potent glycogen synthase kinase-3 (GSK-3) inhibitor, LY2090314
(Selleck Chemicals), was dissolved in DMSO. The antibodies used in
this study were as follows: Cleaved poly (ADP-ribose) polymerase
(1:1,000; PARP; cat. no. 9541) was purchased from Cell Signaling
Technology, Inc.; AREG (1:200; cat. no. sc-74501), Bcl-2 (1:200;
cat. no. sc-7382), phosphorylated (p)-GSK-3β (1:200; cat. no.
sc-135653) and GSK-3β (1:200; cat. no. sc-9166) were purchased from
Santa Cruz Biotechnology, Inc.; α-tubulin (1:10,000; cat. no.
05-829) was purchased from EMD Millipore. Human recombinant AREG
(rAREG) was purchased from R&D Systems, Inc.
Cell culture
The human OSCC cell lines, SAS and SCC9, were kindly
provided by Dr Ming-Chang Hsieh (Chung Shan Medical University
Hospital, Taichung, Taiwan). All cells were cultured in DMEM/F12
medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with
10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific,
Inc.), 1% penicillin-streptomycin (10,000 U/ml penicillin and 10
mg/ml streptomycin) and 2 mM glutamine, and maintained at 37°C in a
humidified atmosphere of 5% CO2. To investigate the
mechanism of VCR resistance in OSCC, SAS-VCR and SCC9-VCR cells
were established over ~6 months by gradually increasing the
concentration of VCR in the culture medium by 0.5 to 16 nM and
0.125 to 4 nM, respectively.
Cell viability assay and treatments
An MTT assay was conducted to evaluate the effects
of VCR on the viability of OSCC cells. Briefly, cells were seeded
into the wells of 96-well plates at a density of 5,000 cells in 100
µl of culture medium. Following overnight incubation to
allow the attachment of cells, cells were incubated with 0-64 nM
VCR in serum-free medium at 37°C. At 0-48 h time intervals
following VCR treatment, 30 µl of MTT (5 mg/ml) was added to
each well and incubated for a further 4 h at 37°C. The supernatant
was then discarded, and 100 µl DMSO was added to each well
to dissolve the formazan crystals. The optical density was
evaluated by measuring the absorbance, with a test wavelength of
490 nm and a reference wavelength of 630 nm.
To investigate whether CM can enhance VCR
resistance, CM was collected as described in the 'Collection of CM'
section. SAS cells were activated by CM from SAS and SAS-VCR cells
for 8 h, and then treated with 8 µM VCR for 24 h.
To further determine the relationship between AREG
and VCR resistance, SAS cells were pretreated with SAS-VCR-CM in
combination with or without AREG-neutralizing antibody (1:500) for
8 h, and then treated with 8 µM VCR for 24 h.
For the effect of rAREG on VCR sensitivity, SAS
cells were pretreated with rAREG (50 ng/ml) for 4 h and then
stimulated with 16 µM VCR for 48 h. SCC9 cells were
pretreated with rAREG (100 ng/ml) for 4 h and then stimulated with
4 µM VCR for 24.
To analyze whether serum starvation can induce
expression of AREG, 5×105 cells/well were seeded in
6-well plates and cultured overnight. Following attachment, cells
were washed twice with PBS, and then serum-free DMEM/F12 was added.
Cells were analyzed by MTT and western blot assays after 12, 24 and
48 h.
To investigate whether inhibition of GSK3-β
activation can block AREG-induced VCR resistance, cells were
pretreated with LY2090314 (20 nM) for 30 min, followed by treatment
with rAREG (50 ng/ml) for 4 h and then treatment with 4 µM
VCR for 24 h. Cell viability was assessed by an MTT assay.
To investigate the sensitivity of SAS and SAS-VCR
cells to 5-FU and cisplatin, cells were treated with 0-160
µM 5-FU for 48 h or 0-20 µM cisplatin for 24 h, and
then analyzed using an MTT assay.
Colony formation assay
Cells were cultured a 6-well plate at a density of
5×104 cells/well with regular medium. Cells were treated
with the 0-64 nM VCR concentrations for 24 h. Then, the cells were
seeded at 1×105 cells/well into a separate 6-well plate.
Cells were allowed to grow until colonies were visible (5-6 days)
and then fixed with methanol for 30 min and stained with 0.5%
crystal violet for 30 min at room temperature (RT). Images of the
colonies were acquired using a digital camera. Colonies were
counted using ImageJ 1.52a software (National Institutes of
Health).
Western blot analysis
Cells were lysed in RIPA buffer containing protease
inhibitor cocktail (Thermo Fisher Scientific, Inc.). The protein
concentration was determined using a Pierce™ BCA Protein Assay kit
(Thermo Fisher Scientific, Inc.). Proteins from the total cell
lysates or CM (40 µg/lane) were separated by via 8-15%
SDS-PAGE and electrotransferred onto PVDF membranes. The membranes
were blocked with 5% skim milk for 1 h at RT and then probed with
the indicated primary antibodies for 1 h at RT. Following three
washes with TBS-0.1% Tween 20, the membranes were incubated with
the HRP-conjugated anti-mouse (cat. no. 20102) or anti-rabbit IgG
antibody (cat. no. 20202; both 1:5,000; Leadgene Biomedical, Inc.)
for 1 h at RT. The blots were visualized using ECL reagent
(PerkinElmer, Inc.) and autoradiography.
Collection of CM
Cells seeded in 10-cm dishes were grown to 80%
confluence and then washed with PBS twice. Cells were subsequently
incubated in serum-free media for 48 h. CM was collected via gentle
aspiration and then centrifuged at 875 × g at RT for 10 min to
remove cell debris. The CM was further concentrated using
Amicon® Ultra 15 ml centrifugal filters with a 3-kDa
cut-off (EMD Millipore) at 4,000 × g and 4°C to a total volume of
~150-200 ul. The CM was aliquoted and stored at −20°C prior to
use.
Growth factor human antibody array
A human growth factor antibody array (cat. no.
ab134002; Abcam) was used according to the manufacturer's
protocols; all reagents listed below were included in this array
unless otherwise specified. Briefly, the concentrated CM (200
µg total protein) was mixed with blocking buffer and
incubated with membranes at 4°C overnight. Membranes were then
washed and incubated with biotin-conjugated anti-cytokines for 2 h
at RT, followed by washing and incubation with HRP-conjugated
strep-tavidin for 2 h at RT. Membranes were then washed again, and
bound antibodies were visualized using ECL reagents and
autoradiography. The relative expression was determined using
UN-SCAN-IT gel 6.1 software (Silk Scientific, Inc.). The average
signal of a pair of duplicate spots was normal-ized using negative
control spots as a background value. The relative intensities in
SAS-VCR cells were determined by comparing the corresponding
signals to SAS cells.
Oncomine database
The Oncomine Cancer Microarray database (http://www.oncomine.org/) was used to study the
expression levels of AREG in human oral tumor and normal tissues
obtained from separate individuals. The gene expression data were
log transformed, median centered per array, and the standard
deviation was normalized to 1 per array. A gene was considered as
overexpressed when its mean value in tumor samples was
significantly increased compared with its mean value in normal
tissue as determined using a t-test (P≤0.05) and the fold change
was ≥1.5.
Lentivirus infection and short hairpin
(sh)RNA knockdown
The pLKO.1-puro-based lentiviral plasmids containing
TRCN0000117995-shAREG (sequence:
5′-CCGGGAACGAAAGAAACTTCGACAACTCGAGTTGTCGAAGTTTCTTTCGTTCTTTTTG-3′)
and pLKO.1-shScramble (sequence:
5′-CCGGCCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGGTTTTT-3′)
were obtained from National RNAi Core Facility (Academia Sinica,
Taipei, Taiwan). All plasmids (4 µg lentiviral plasmid; 4
µg pCMVΔR8.91; 0.4 µg pMD; all Academia Sinica) were
cotransfected into 293T cells (5×105; cat. no. 632180;
Clontech Laboratories, Inc.) in 6-cm dishes using TurboFect (Thermo
Fisher Scientific, Inc.), according to the manufacturer's
protocols. The lentivirus-containing supernatants were harvested at
48 h post-transfection. SAS-VCR cells (1×105) were
infected using the lentivirus-containing supernatant (12,762
RIU/µl). For stable cell lines, the infected cells were
selected by puromycin (5 µg/ml) within 1 week.
AREG overexpression
An AREG overexpression plasmid (pCMV3-AREG) and
negative control (pCMV3) were purchased from Sino Biological Inc.
Plasmids were transfected into the two OSCC cell lines using
TurboFect according to the manufacturer's protocol. Cells
(1×105) were seeded in each well of a 24-well plate.
After culturing for 24 h, cells were transiently transfected under
optimized transfection conditions. Briefly, 1 µg of DNA
plasmid DNA was diluted in 100 µl of serum-free DMEM/F12,
and mixed with 2 µl of transfection reagent followed by
incubation at RT for 20 min. The mixture was then added dropwise to
the cells and incubated for an additional 72 h at 37°C in a
humidified atmosphere and 5% CO2.
Statistical analysis
All values represent the mean ± SEM from at least 3
independent experiments. Student's t-test was used when comparing
two independent groups. Statistical comparisons of >2 groups
were performed using one-way analysis of variance (ANOVA) with
Bonferroni's post hoc test. In all cases, P<0.05 was
considered to indicate a statistically significant difference.
Results
Establishment and characterization of
VCR-resistant SAS cells
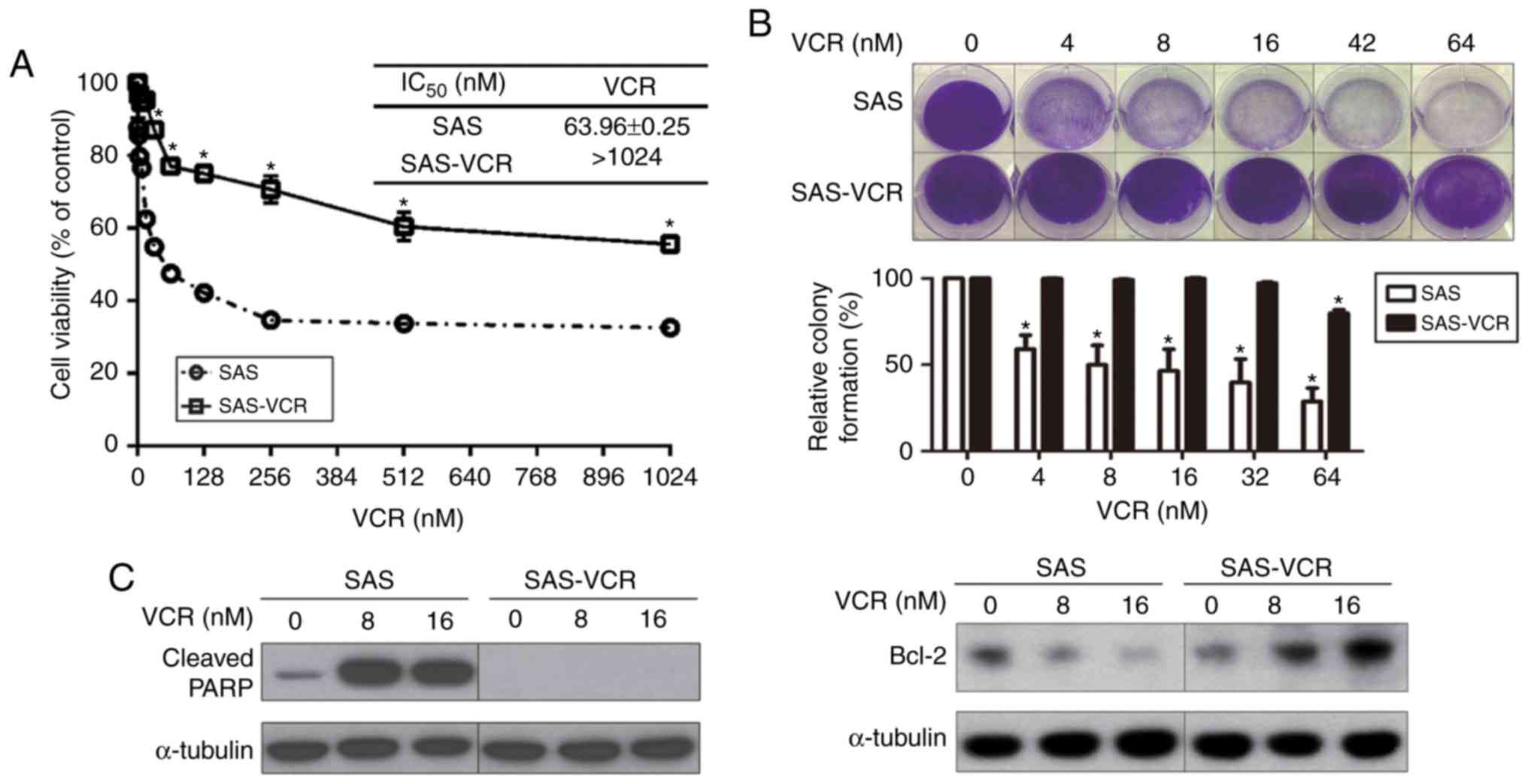
As presented in Fig.
1A, SAS-VCR cells (IC50 >1,024 nM) were more
resistant to VCR than their respective parental SAS cells
(IC50=63.96±0.25 nM). In addition, a colony formation
assay was also conducted. As presented in Fig. 1B, the colony numbers of SAS cells
were significantly decreased compared with control treatment in
dose-dependent manner; however, the SAS-VCR colony number was
significantly decreased compared with the control only in the high
dose group (64 nM). Next, the effects of VCR on the expression of
cleaved PARP, a marker of apoptosis, and Bcl-2 (an antiapoptotic
protein) were evaluated via western blotting. As shown in Fig. 1C, the expression of cleaved PARP
was notably induced in VCR-treated SAS cells. In contrast, the
expression of cleaved PARP was not observed in SAS-VCR cells
following exposure to 8 or 16 nM VCR for 24 h, indicating that
SAS-VCR cells were highly resistant to VCR compared with SAS cells.
The expression of Bcl-2 was increased in a dose-dependent manner in
VCR-treated SAS-VCR cells, whereas the expression of Bcl-2 was
decreased in a dose-dependent manner in VCR-treated SAS cells.
These results indicated that SAS-VCR cells were resistant to VCR
compared with SAS cells. Furthermore, the sensitivity of the
SAS-VCR cell line to 5-FU and cisplatin was also explored, and it
was revealed that only resistance to 5-FU was observed in these
cells, suggesting a potential link between resistance to VCR and
5-FU resistance (Fig. S1).
Comparison of growth factor profiles in
the secretomes of SAS and SAS-VCR cells
To evaluate whether the secreted proteins from
resistant cells were associated with the induction of drug
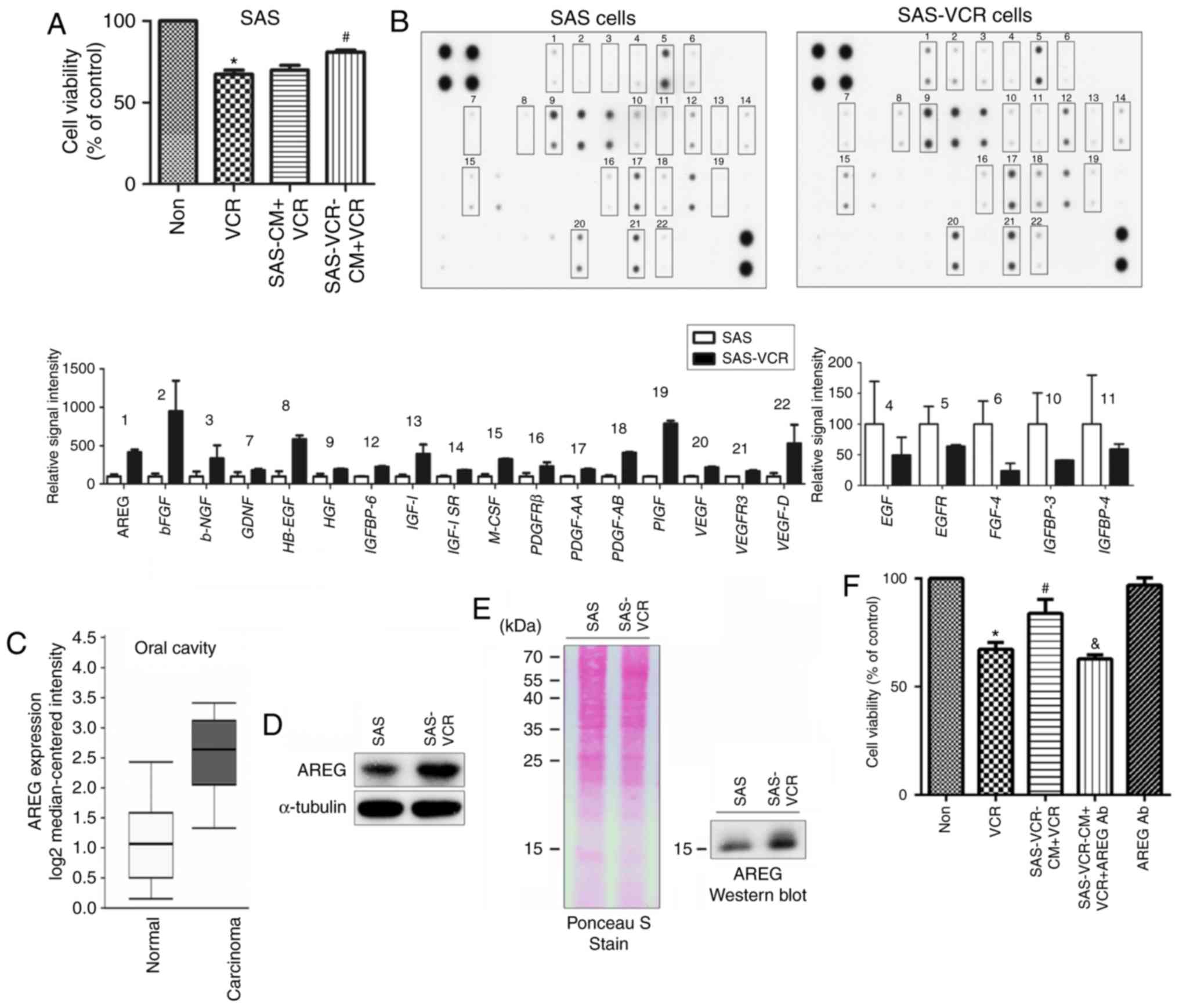
resistance, CM was obtained from SAS and SAS-VCR cells. Notably,
the CM from SAS-VCR cells significantly increased VCR resistance
when applied to parental SAS cells (Fig. 2A). Subsequently, antibody arrays
were used to analyze the differences in the secretomes of CM from
SAS and SAS-VCR cell lines. As presented in Fig. 2B, a total of 22 secreted proteins
were identified whose expression was changed >1.5-fold between
the two media. The levels of 17 secreted proteins were increased,
and those of 5 secreted proteins was decreased in SAS-VCR. Of
these, the levels of AREG, basic fibroblast growth factor,
heparin-binding EGF-like growth factor, platelet-derived growth
factor-AB, placental growth factor and vascular endothelial growth
factor D secretion were most notably upregulated (>4-fold) in
SAS-VCR CM, indicating that the secreted proteins may be important
mediators of VCR resistance in OSCC cells.
AREG is highly expressed and secreted to
promote VCR resistance in OSCC cells
To determine the clinical relevance of these
secreted proteins, the Oncomine database was employed to select the
appropriate target for further study. The results revealed that
there were only clinical data concerning the expression of AREG;
its expression was significantly upregulated in carcinoma tissue
compared normal oral cavity tissue (Fig. 2C). In addition, among the six
proteins, numerous studies indicated that AREG serves a critical
role in OSCC (24-26); however, its role in VCR sensitivity
is yet to be described in the literature. To further confirm the
antibody array results, western blotting was performed to determine
if AREG was highly expressed and secreted in SAS-VCR cells. As
shown in Fig. 2D, the expression
of AREG in the cell lysates was analyzed, and the results indicated
that expression of AREG was increased in SAS-VCR cells compared
with in SAS cells. In addition, the levels of AREG in CM were also
significantly elevated for SAS-VCR cells compared with SAS cells
(Fig. 2E). To provide further
evidence that AREG mediates VCR sensitivity, AREG activity was
blocked using a neutralizing antibody. The results showed that
pretreatment of CM with neutralizing antibodies against AREG
restored VCR sensitivity in SAS cells (Fig. 2F).
AREG also modulates VCR sensitivity in
SCC9 cells
To further validate the role of AREG in VCR
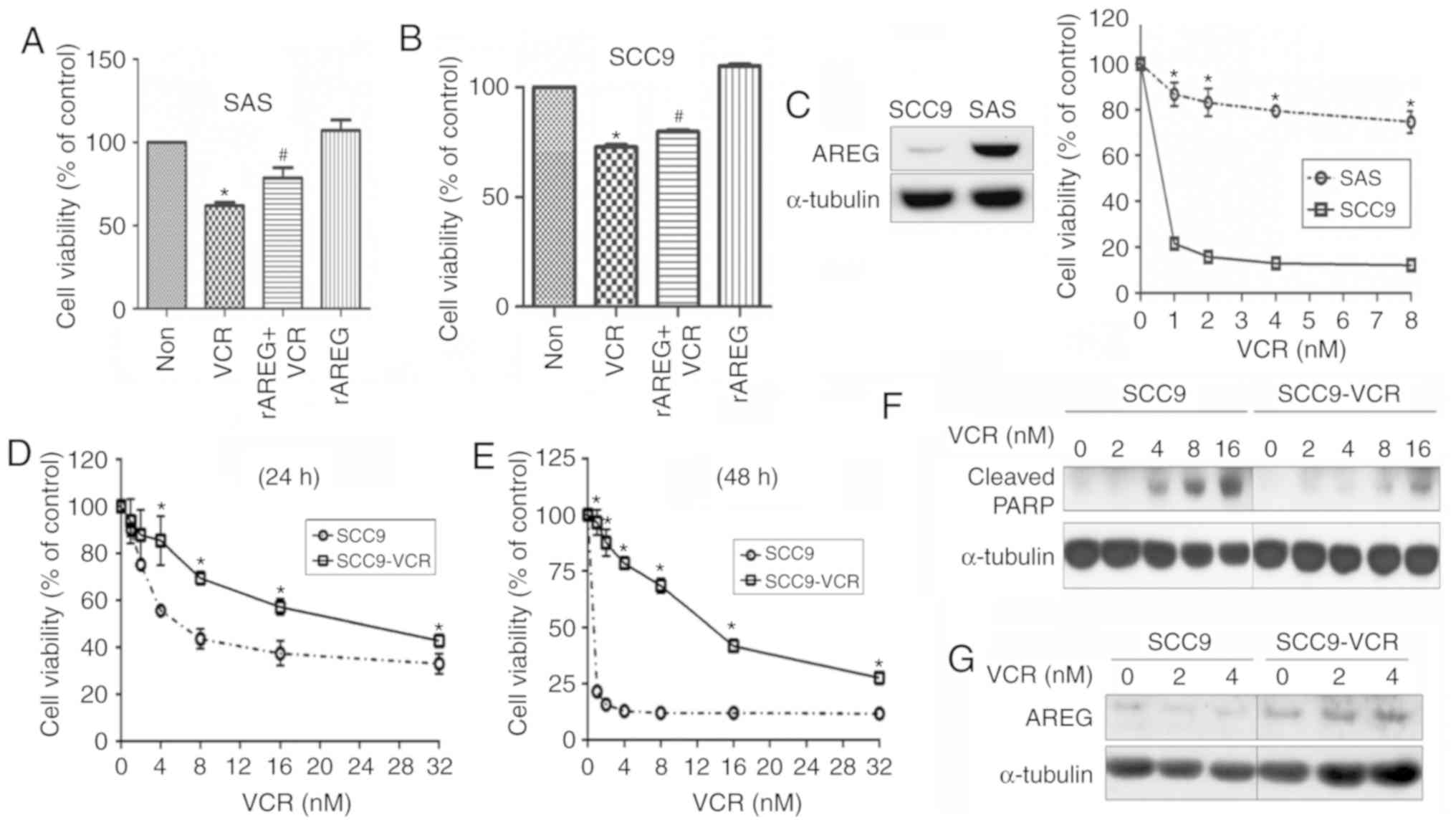
sensitivity, SAS cells were pretreated with rAREG and then
stimulated with VCR. The results showed that AREG markedly
increased resistance to VCR in SAS cells (Fig. 3A). Similarly, rAREG also enhanced
VCR resistance in SCC9 cells (Fig.
3B). Furthermore, the expression levels of AREG and VCR
resistance in the two cell lines were measured. The results showed
that the expression of AREG was higher in the more VCR-resistant
cell line, SAS (Fig. 3C). To
clarify whether AREG expression is associated with VCR sensitivity
in SCC9 cells, a VCR-resistant SCC9 subline termed SCC9-VCR was
established. According to the results of the MTT assay, SCC9-VCR
cells were significantly more viable following VCR treatment
compared with SCC9 cells, as assessed at 24 and 48 h (Fig. 3D and E). Furthermore, SCC9 cells
exhibited increased expression of cleaved PARP compared with
SCC9-VCR cells following VCR treatment (Fig. 3F). To further evaluate the
association between AREG levels and VCR resistance, SCC9 and
SCC9-VCR cells were treated with increasing doses of VCR, and then
the expression of AREG was analyzed via western blotting. The
results revealed that SCC9-VCR cells exhibited upregulated AREG
expression compared with SCC9 cells. Furthermore, an increase in
the expression levels of AREG was observed in SCC9-VCR cells, but
not SCC9 cells, after treatment with increasing doses of VCR
(Fig. 3G).
Knockdown of AREG restores VCR
sensitivity, and overexpression of AREG confers resistance to
VCR
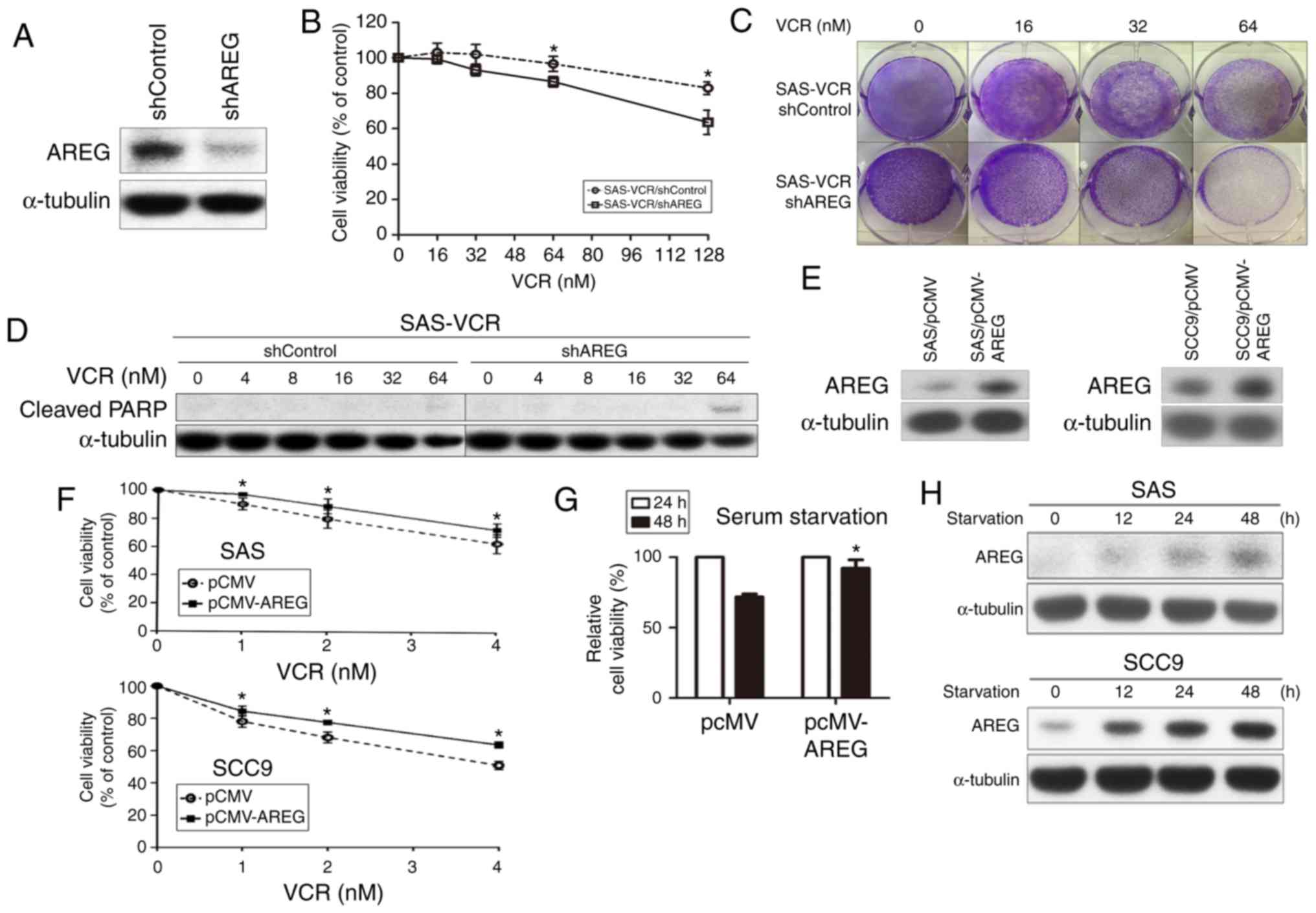
To determine the impact of AREG expression on VCR
sensitivity, AREG expression was suppressed in SAS-VCR cells using
a lentivirus-mediated RNA interference system. The knockdown
efficiency of shAREG was evaluated via western blotting (Fig. 4A). Silencing AREG expression
resulted in elevated sensitivity to VCR in a dose-dependent manner
compared with shControl cells, as determined by MTT (Fig. 4B) and colony formation assays
(Fig. 4C). Furthermore, AREG
knockdown notably promoted cell apoptosis in SAS-VCR cells, as
evidenced by an increase in the cleavage of PARP at a concentration
of 64 nM VCR for 24 h (Fig. 4D).
The results indicated that the knockdown of AREG may increase
VCR-induced apoptosis in OSCC cells. To verify the relevance of
AREG in mediating resistance to VCR, cells were then transiently
trans-fected with AREG expression vector or control vector. AREG
was overexpressed in SAS and SCC9 cells following transfection, as
determined via western blotting (Fig.
4E). As presented in Fig. 4F,
overexpression of AREG in SAS and SCC9 cells significantly
increased resistance to VCR, as assessed using an MTT assay.
Furthermore, whether AREG overexpression protected against
starvation-induced death in OSCC cells was analyzed. The result
revealed that overexpression of AREG significantly increased the
viability of serum-starved SAS cells at 48 h compared with cells
transfected with a control vector (Fig. 4G). Furthermore, whether serum
starvation can affect the expression of AREG, which may prevent
cell death and promote resistance to harsh environments such as
drug treatment, was evaluated. As presented in Fig. 4H, serum deprivation notably induced
AREG expression in SAS and SCC9 cells in a time-dependent manner.
These results indicated that AREG was involved in protecting OSCC
cells against various stresses, including VCR treatment.
AREG regulates VCR sensitivity in OSCC
cells via activation of GSK-3β
A previously study reported that AREG can modulate
the GSK-3β pathway to regulate cell functions; GSK-3β is known to
be a potential therapeutic target for cancer treatment (27). In addition, it has been
demonstrated that targeting the GSK-3β pathway may be beneficial
for the treatment of oral cancer (28). Therefore, the activation of GSK-3β
was analyzed after treatment of SAS and SAS-VCR cells with
increasing concentrations of VCR. The results showed that VCR
induced a dose-dependent decrease in GSK-3β phosphorylation in SAS
cells, whereas GSK-3β maintained sustained activation in SAS-VCR
cells (Fig. 5A). In addition, a
marked downregulation of p-GSK-3β was also observed in VCR-treated
SAS-VCR/shAREG cells compared with SAS-VCR/shControl cells
(Fig. 5B). To further confirm that
AREG can indeed activate the GSK-3β pathway, GSK-3β phosphorylation
was directly analyzed in response to rAREG. The results revealed
that treatment of SAS cells with rAREG can induce an increase in
the phosphorylation of GSK-3β in a time-dependent manner (Fig. 5C). Then, a GSK-3β inhibitor,
LY2090314, was used to interfere with AREG-induced GSK-3β
activation and observe whether blocking GSK-3β activation would
affect the role of AREG in VCR resistance. It was demonstrated that
rAREG induced a significant increase in VCR resistance; however,
this effect was significantly attenuated by LY2090314 (Fig. 5D). These findings suggested that
AREG can regulate the activation of GSK-3 to promote VCR resistance
in OSCC cells.
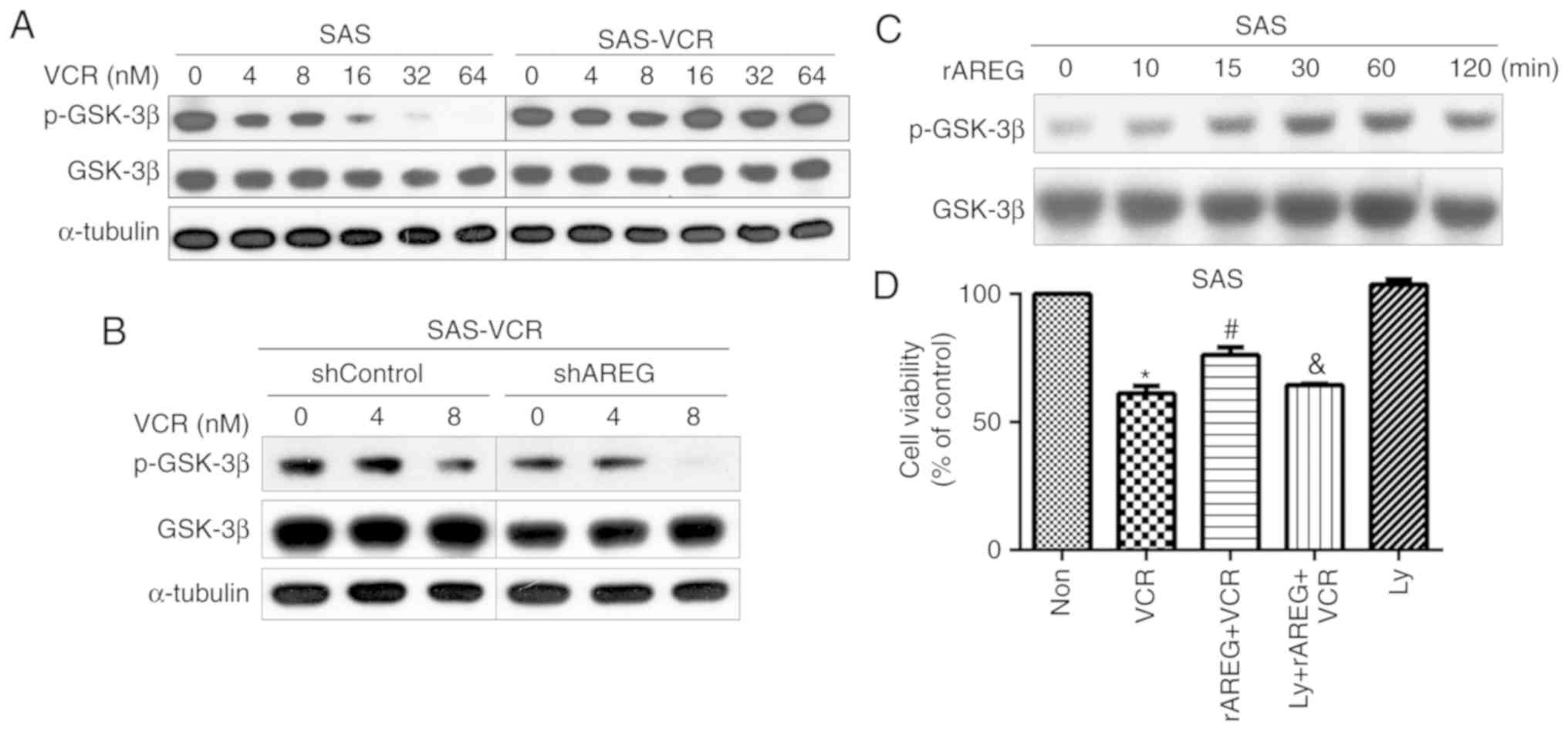 | Figure 5GSK3-β signaling pathways may be
involved in the effects of AREG on VCR sensitivity. (A) SAS and
SAS-VCR cells were treated with different concentrations of VCR
(0-64 nM) for 24 h. (B) SAS-VCR/shControl and SAS-VCR/shAREG cells
were exposed to various concentrations of VCR for 24 h (C) SAS
cells were incubated with rAREG (50 ng/ml) for the indicated time
intervals. p-GSK3-β and GSK3-β were analyzed via western blotting.
(D) Cells were pretreated with Ly (20 nM) for 30 min, followed by
treatment with rAREG (50 ng/ml) for 4 h, and then treatment with 4
µM VCR for 24 h. Cell viability was evaluated by an MTT
assay. Data are presented as the mean ± SEM (n=5).
*P<0.05 vs. Non; #P<0.05 vs. VCR;
&P<0.05 vs. rAREG + VCR. AREG, amphiregulin;
GSK-3β, glycogen synthase kinase-3β; Ly, LY2090314; Non, untreated
cells; p, phosphorylated; r, recombinant; SAS-VCR, VCR-resistant
SAS cells; VCR, vincristine. |
Discussion
A substantial body of evidence has revealed that
upregulated expression of AREG is associated with cancer
progression in a wide variety of cancers, including lung (29,30),
breast (17,31), ovarian (32,33),
liver (18,34), pancreatic (19,35)
and colorectal cancers (20,36).
At present, however, there has been no study into the function of
ARGE in relation to drug resistance in OSCC. In the present study,
an antibody array was performed to explore the secretion profile of
OSCC cells, and it was revealed that AREG was highly expressed and
secreted in VCR-resistant cells. In addition, it was also suggested
that the GSK-3β pathway may be involved in AREG-induced VCR
resistance. These findings may aid the development of novel
therapeutic targets for OSCC treatment and improved prognosis.
Chemotherapy is widely used in the treatment of
OSCC, and VCR is a classic microtubule-destabilizing agent that is
effective and widely used in hematological malignancies and certain
solid tumors (37). As VCR
exhibits substantial anticancer activity, it may be a potential
treatment for OSCC. However, several studies have indicated that
high-dose VCR is associated with a significant risk of severe
gastrointestinal toxicity; mortality from treatment-related
toxicity has been previously reported in patients (38,39).
Therefore, combining VCR with other agents or molecules is may
improve the clinical management of oral cancers. Certain drugs with
antitumor activity have been reported to increase the VCR
sensitization of VCR-resistant oral epidermoid carcinoma cells
(40-42). Though great efforts have been made
in developing novel anticancer drugs with increased curative
potential or the ability to reverse drug resistance, the results
remain satisfactory, due to either a lack of potency or
unacceptable side effects.
Proteins secreted, shed or leaking from cancer
cells, collectively termed the cancer secretome, are considered
promising biomarkers, as they may be detectable in blood or other
body fluids (43-45). Previous studies indicated that
cancer cells can secrete soluble mediators in response to drug
therapy, which contribute to the promotion of drug resistance and
tumor progression (46,47). In addition, secreted proteins are
also considered good candidate serological tumor markers, as they
are released by the cells and thus exhibit the greatest possibility
of entering the circulation (48).
Therefore, secreted proteins are regarded as a rich source of
potential markers and drug targets for cancer treatment (49).
AREG is an EGF-like ligand that has been identified
as a ligand responsible for EGFR-ERK signaling activation, which
can lead to cancer progression (19,50-52).
Regarding drug sensitivity, AREG is upregulated in non-responding
patients compared with patients who do respond to gefitinib
(21). In animal models, a
previous study has shown that AREG silencing can reduce the size of
tumor growth and increase drug sensitivity in glioma cells
(53). As AREG is a secreted
protein, it can enter the circulatory system. A separate study
showed that circulating AREG could be clinically relevant as an
indicator of unfavorable response to gefitinib in NSCLC (54). In Taiwan, oral cancer accounts for
the fourth highest incidence of malignancy in males (55). Numerous studies indicate that EGFR
is frequently overexpressed in human OSCC (56-59).
High EGFR expression has been associated with
resistance to chemotherapeutic agents used in the treatment of
OSCC, including cisplatin, 5-FU, cyclophosphamide and doxoru-bicin,
suggesting that EGFR signaling may be a promising target for OSCC
therapy (58-60). Cetuximab is a chimeric IgG1-human
antibody targeted against the extracellular domain of EGFR
(61). It was approved by the FDA
in 2006 as a component of combination therapy along with radiation
and/or chemotherapy to treat OSCC; however, clinical use of
cetuximab is limited, as EGFR expression levels have not been
associated with response levels to cetuximab (62,63).
At present, in OSCC, clinically relevant mechanisms of cetuximab
resistance have not been clearly elucidated.
GSK-3β, a multifunctional serine/threonine kinase,
was originally discovered as a key regulator of glycogen metabolism
(64). Accumulating evidence
suggests that it involved in tumorigenesis, migration, invasion,
chemotherapy and drug resistance (65,66).
Therefore, GSK-3β has emerged as a potential therapeutic target for
different types of cancers (67,68).
Recent research in OSCC has shown that GSK-3β can regulate matrix
metalloproteinase-9 activity to promote cancer progression and
invasion (69). In clinical
specimens, another study also showed that the levels of p-GSK-3β
and GSK-3β in OSCC tissues are upregulated compared with in
controls, and are positively associated with tumor metastasis and
poor survival in patients (70).
Although a number of studies have indicated that multidrug
resistance can be reversed by inhibiting GSK-3β (71-73),
whether this occurs in OSCC remains unclear. The relationship of
GSK-3β with the drug treatment of OSCC remains to be further
explored in future studies.
In conclusion, to improve understanding of the
mechanisms underlying drug resistance in OSCC, a VCR-resistant OSCC
cell line was established, and the secreted proteins were analyzed
using an antibody microarray. This study is the first, to our
knowledge, to characterize changes in the secretome of
VCR-resistant OSCC cells. The results indicated that AREG was
highly expressed and secreted in VCR-resistant cells compared with
VCR-sensitive cells. Pretreatment with exogenous rAREG markedly
increased drug resistance against VCR in OSCC cells. Furthermore,
knockdown of AREG increased VCR sensitivity, whereas overexpression
of AREG further promoted VCR resistance. The results indicated that
AREG contributes to VCR resistance in OSCC cells. Additionally, it
was also demonstrated that the GSK-3β pathway may be involved in
AREG-induced VCR resistance. These findings may provide valuable
insight for the development of effective treatments against
OSCC.
Supplementary Data
Acknowledgments
The author would like to thank Ms Yu-Ping Wu and Ms
Wen-Ling Li at the National Chiayi University for their
assistance.
Funding
This work was supported by the Ministry of Science
and Technology (grant no. 107-2314-B-415-002-) and grant nos.
107-CCH-HCR-046 and 107-CCH-HCR-047 from Changhua Christian
Hospital, Taiwan, R.O.C.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JCC was involved in the study design, data analysis
and drafting the manuscript. MJH and YHC performed the experiments
and data acquisition. INL, CH and YJK contributed to data analysis
and interpretation.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Curado MP and Hashibe M: Recent changes in
the epidemiology of head and neck cancer. Curr Opin Oncol.
21:194–200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Scully C and Bagan J: Oral squamous cell
carcinoma overview. Oral Oncol. 45:301–308. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schwam ZG and Judson BL: Improved
prognosis for patients with oral cavity squamous cell carcinoma:
Analysis of the National Cancer Database 1998-2006. Oral Oncol.
52:45–51. 2016. View Article : Google Scholar
|
|
5
|
Dumontet C and Jordan MA:
Microtubule-binding agents: A dynamic field of cancer therapeutics.
Nat Rev Drug Discov. 9:790–803. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Silverman JA, Reynolds L and Deitcher SR:
Pharmacokinetics and pharmacodynamics of vincristine sulfate
liposome injection (VSLI) in adults with acute lymphoblastic
leukemia. J Clin Pharmacol. 53:1139–1145. 2013.PubMed/NCBI
|
|
7
|
Yan Z, Zhu ZL, Qian ZZ, Hu G, Wang HQ, Liu
WH and Cheng G: Pharmacokinetic characteristics of vincristine
sulfate liposomes in patients with advanced solid tumors. Acta
Pharmacol Sin. 33:852–858. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Olasz L, Orsi E, Marko T and Szalma J:
Induction chemotherapy response and recurrence rates in correlation
with N0 or N+ stage in oral squamous cell cancer (OSCC). Cancer
Metastasis Rev. 29:607–611. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Villanueva J and Herlyn M: Melanoma and
the tumor microenvi-ronment. Curr Oncol Rep. 10:439–446. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen JC, Lee IN, Huang C, Wu YP, Chung CY,
Lee MH, Lin MH and Yang JT: Valproic acid-induced amphiregulin
secretion confers resistance to temozolomide treatment in human
glioma cells. BMC Cancer. 19:7562019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Milman N, Ginini L and Gil Z: Exosomes and
their role in tumorigenesis and anticancer drug resistance. Drug
Resist Updat. 45:1–12. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Siveen KS, Raza A, Ahmed EI, Khan AQ,
Prabhu KS, Kuttikrishnan S, Mateo JM, Zayed H, Rasul K, Azizi F, et
al: The role of extracellular vesicles as modulators of the tumor
microenvironment, metastasis and drug resistance in colorectal
cancer. Cancers (Basel). 11. pii: E746. 2019, View Article : Google Scholar
|
|
13
|
Wilson TR, Fridlyand J, Yan Y, Penuel E,
Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, et al:
Widespread potential for growth-factor-driven resistance to
anticancer kinase inhibitors. Nature. 487:505–509. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Straussman R, Morikawa T, Shee K,
Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J,
Frederick DT, et al: Tumour micro-environment elicits innate
resistance to RAF inhibitors through HGF secretion. Nature.
487:500–504. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Acharyya S, Oskarsson T, Vanharanta S,
Malladi S, Kim J, Morris PG, Manova-Todorova K, Leversha M, Hogg N,
Seshan VE, et al: A CXCL1 paracrine network links cancer
chemoresistance and metastasis. Cell. 150:165–178. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Berasain C and Avila MA: Amphiregulin.
Semin Cell Dev Biol. 28:31–41. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Willmarth NE and Ethier SP: Autocrine and
juxtacrine effects of amphiregulin on the proliferative, invasive,
and migratory properties of normal and neoplastic human mammary
epithelial cells. J Biol Chem. 281:37728–37737. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Berasain C, Castillo J, Perugorria MJ,
Prieto J and Avila MA: Amphiregulin: A new growth factor in
hepatocarcinogenesis. Cancer Lett. 254:30–41. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yotsumoto F, Fukami T, Yagi H, Funakoshi
A, Yoshizato T, Kuroki M and Miyamoto S: Amphiregulin regulates the
activation of ERK and Akt through epidermal growth factor receptor
and HER3 signals involved in the progression of pancreatic cancer.
Cancer Sci. 101:2351–2360. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kuramochi H, Nakajima G, Kaneko Y,
Nakamura A, Inoue Y, Yamamoto M and Hayashi K: Amphiregulin and
Epiregulin mRNA expression in primary colorectal cancer and
corresponding liver metastases. BMC Cancer. 12:882012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen JC, Huang C, Lee IN, Wu YP and Tang
CH: Amphiregulin enhances cell migration and resistance to
doxorubicin in chon-drosarcoma cells through the MAPK pathway. Mol
Carcinog. 57:1816–1824. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tokunaga S, Nagano T, Kobayashi K,
Katsurada M, Nakata K, Yamamoto M, Tachihara M, Kamiryo H, Yokozaki
H and Nishimura Y: Amphiregulin as a novel resistance factor for
amru-bicin in lung cancer cells. Anticancer Res. 37:2225–2231.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Haab BB: Antibody arrays in cancer
research. Mol Cell Proteomics. 4:377–383. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tsai ST, Yang KY, Jin YT, Lin YC, Chang MT
and Wu LW: Amphiregulin as a tumor promoter for oral squamous cell
carcinoma: Involvement of cyclooxygenase 2. Oral Oncol. 42:381–390.
2006. View Article : Google Scholar
|
|
25
|
Zhang J, Wang Y, Chen X, Zhou Y, Jiang F,
Chen J, Wang L and Zhang WF: MiR-34a suppresses amphiregulin and
tumor metastatic potential of head and neck squamous cell carcinoma
(HNSCC). Oncotarget. 6:7454–7469. 2015.PubMed/NCBI
|
|
26
|
Gao J, Ulekleiv CH and Halstensen TS:
Epidermal growth factor (EGF) receptor-ligand based molecular
staging predicts prognosis in head and neck squamous cell carcinoma
partly due to deregulated EGF-induced amphiregulin expression. J
Exp Clin Cancer Res. 35:1512016. View Article : Google Scholar
|
|
27
|
Wang S, Zhang Y, Wang Y, Ye P, Li J, Li H,
Ding Q and Xia J: Amphiregulin confers regulatory T cell
suppressive function and tumor invasion via the EGFR/GSK-3β/Foxp3
Axis. J Biol Chem. 291:21085–21095. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mishra R: Glycogen synthase kinase 3 beta:
Can it be a target for oral cancer. Mol Cancer. 9:1442010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Addison CL, Ding K, Zhao H, Le Maître A,
Goss GD, Seymour L, Tsao MS, Shepherd FA and Bradbury PA: Plasma
transforming growth factor alpha and amphiregulin protein levels in
NCIC Clinical Trials Group BR.21. J Clin Oncol. 28:5247–5256. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hsu YL, Huang MS, Cheng DE, Hung JY, Yang
CJ, Chou SH and Kuo PL: Lung tumor-associated dendritic
cell-derived amphireg-ulin increased cancer progression. J Immunol.
187:1733–1744. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
McBryan J, Howlin J, Napoletano S and
Martin F: Amphiregulin: Role in mammary gland development and
breast cancer. J Mammary Gland Biol Neoplasia. 13:159–169. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
D'Antonio A, Losito S, Pignata S, Grassi
M, Perrone F, De Luca A, Tambaro R, Bianco C, Gullick WJ, Johnson
GR, et al: Transforming growth factor alpha, amphiregulin and
cripto-1 are frequently expressed in advanced human ovarian
carcinomas. Int J Oncol. 21:941–948. 2002.PubMed/NCBI
|
|
33
|
Freimann S, Ben-Ami I, Hirsh L, Dantes A,
Halperin R and Amsterdam A: Drug development for ovarian
hyper-stimulation and anti-cancer treatment: Blocking of
gonadotropin signaling for epiregulin and amphiregulin
biosynthesis. Biochem Pharmacol. 68:989–996. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Castillo J, Erroba E, Perugorria MJ,
Santamaría M, Lee DC, Prieto J, Avila MA and Berasain C:
Amphiregulin contributes to the transformed phenotype of human
hepatocellular carcinoma cells. Cancer Res. 66:6129–6138. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ebert M, Yokoyama M, Kobrin MS, Friess H,
Lopez ME, Büchler MW, Johnson GR and Korc M: Induction and
expression of amphiregulin in human pancreatic cancer. Cancer Res.
54:3959–3962. 1994.PubMed/NCBI
|
|
36
|
Yamada M, Ichikawa Y, Yamagishi S,
Momiyama N, Ota M, Fujii S, Tanaka K, Togo S, Ohki S and Shimada H:
Amphiregulin is a promising prognostic marker for liver metastases
of colorectal cancer. Clin Cancer Res. 14:2351–2356. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gidding CE, Kellie SJ, Kamps WA and de
Graaf SS: Vincristine revisited. Crit Rev Oncol Hematol.
29:267–287. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Corbett R, Pinkerton R, Pritchard J,
Meller S, Lewis I, Kingston J and McElwain T: Pilot study of
high-dose vincristine, etoposide, carboplatin and melphalan with
autologous bone marrow rescue in advanced neuroblastoma. Eur J
Cancer. 28A:1324–1328. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gordon SJ, Pearson AD, Reid MM and Craft
AW: Toxicity of single-day high-dose vincristine, melphalan,
etoposide and carboplatin consolidation with autologous bone marrow
rescue in advanced neuroblastoma. Eur J Cancer. 28A:1319–1323.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xi GM, Sun B, Jiang HH, Kong F, Yuan HQ
and Lou HX: Bisbibenzyl derivatives sensitize vincristine-resistant
KB/VCR cells to chemotherapeutic agents by retarding P-gp activity.
Bioorg Med Chem. 18:6725–6733. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yan YX, Li WZ, Huang YQ and Liao WX: The
COX-2 inhibitor Celecoxib enhances the sensitivity of KB/VCR oral
cancer cell lines to Vincristine by down-regulating P-glycoprotein
expression and function. Prostaglandins Other Lipid Mediat.
97:29–35. 2012. View Article : Google Scholar
|
|
42
|
Yuan Z, Wang H, Hu Z, Huang Y, Yao F, Sun
S and Wu B: Quercetin inhibits proliferation and drug resistance in
KB/VCR oral cancer cells and enhances its sensitivity to
vincristine. Nutr Cancer. 67:126–136. 2015. View Article : Google Scholar
|
|
43
|
Canto LMD, Cury SS, Barros-Filho MC,
Kupper BEC, Begnami MDFS, Scapulatempo-Neto C, Carvalho RF, Marchi
FA, Olsen DA, Madsen JS, et al: Locally advanced rectal cancer
transcriptomic-based secretome analysis reveals novel biomarkers
useful to identify patients according to neoadjuvant
chemoradiotherapy response. Sci Rep. 9:87022019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lin M, Zhou C, He S, Yu H, Guo T, Ye J,
Feng X and Bian X: The research advances of exosomes in esophageal
cancer. Biomark Med. 13:685–695. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Osborne DG, Domenico J, Luo Y, Reid AL,
Amato C, Zhai Z, Gao D, Ziman M, Dinarello CA, Robinson WA and
Fujita M: Interleukin-37 is highly expressed in regulatory T cells
of melanoma patients and enhanced by melanoma cell secretome. Mol
Carcinog. 58:1670–1679. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Obenauf AC, Zou Y, Ji AL, Vanharanta S,
Shu W, Shi H, Kong X, Bosenberg MC, Wiesner T, Rosen N, et al:
Therapy-induced tumour secretomes promote resistance and tumour
progression. Nature. 520:368–372. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Amrutkar M, Aasrum M, Verbeke CS and
Gladhaug IP: Secretion of fibronectin by human pancreatic stellate
cells promotes chemoresistance to gemcitabine in pancreatic cancer
cells. BMC Cancer. 19:5962019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kulasingam V and Diamandis EP: Strategies
for discovering novel cancer biomarkers through utilization of
emerging technologies. Nat Clin Pract Oncol. 5:588–599. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
May M: From cells, secrets of the
secretome leak out. Nat Med. 15:8282009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wang X, Masri S, Phung S and Chen S: The
role of amphiregulin in exemestane-resistant breast cancer cells:
Evidence of an auto-crine loop. Cancer Res. 68:2259–2265. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chen JC, Chen YJ, Lin CY, Fong YC, Hsu CJ,
Tsai CH, Su JL and Tang CH: Amphiregulin enhances alpha6beta1
integrin expression and cell motility in human chondrosarcoma cells
through Ras/Raf/MEK/ERK/AP-1 pathway. Oncotarget. 6:11434–11446.
2015.PubMed/NCBI
|
|
52
|
Kakiuchi S, Daigo Y, Ishikawa N, Furukawa
C, Tsunoda T, Yano S, Nakagawa K, Tsuruo T, Kohno N, Fukuoka M, et
al: Prediction of sensitivity of advanced non-small cell lung
cancers to gefitinib (Iressa, ZD1839). Hum Mol Genet. 13:3029–3043.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lorente M, Carracedo A, Torres S, Natali
F, Egia A, Hernández-Tiedra S, Salazar M, Blázquez C, Guzmán M and
Velasco G: Amphiregulin is a factor for resistance of glioma cells
to cannabinoid-induced apoptosis. Glia. 57:1374–1385. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ishikawa N, Daigo Y, Takano A, Taniwaki M,
Kato T, Hayama S, Murakami H, Takeshima Y, Inai K, Nishimura H, et
al: Increases of amphiregulin and transforming growth factor-alpha
in serum as predictors of poor response to gefitinib among patients
with advanced non-small cell lung cancers. Cancer Res.
65:9176–9184. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kao SY and Lim E: An overview of detection
and screening of oral cancer in Taiwan. Chin J Dent Res. 18:7–12.
2015.PubMed/NCBI
|
|
56
|
Chiang WF, Liu SY, Yen CY, Lin CN, Chen
YC, Lin SC and Chang KW: Association of epidermal growth factor
receptor (EGFR) gene copy number amplification with neck lymph node
metastasis in areca-associated oral carcinomas. Oral Oncol.
44:270–276. 2008. View Article : Google Scholar
|
|
57
|
Huang SF, Chuang WY, Chen IH, Liao CT,
Wang HM and Hsieh LL: EGFR protein overexpression and mutation in
areca quid-associated oral cavity squamous cell carcinoma in
Taiwan. Head Neck. 31:1068–1077. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Huang SF, Chien HT, Cheng SD, Chuang WY,
Liao CT and Wang HM: EGFR copy number alterations in primary
tumors, metastatic lymph nodes, and recurrent and multiple primary
tumors in oral cavity squamous cell carcinoma. BMC Cancer.
17:5922017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Huang SF, Chien HT, Chuang WY, Lai CH,
Cheng SD, Liao CT and Wang HM: Epidermal growth factor receptor
intron-1 CA repeat polymorphism on protein expression and clinical
outcome in Taiwanese oral squamous cell carcinoma. Sci Rep.
7:49632017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Rubin Grandis J, Melhem MF, Gooding WE,
Day R, Holst VA, Wagener MM, Drenning SD and Tweardy DJ: Levels of
TGF-alpha and EGFR protein in head and neck squamous cell carcinoma
and patient survival. J Natl Cancer Inst. 90:824–832. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Harding J and Burtness B: Cetuximab: An
epidermal growth factor receptor chemeric human-murine monoclonal
antibody. Drugs Today (Barc). 41:107–127. 2005. View Article : Google Scholar
|
|
62
|
Bonner JA, Harari PM, Giralt J, Azarnia N,
Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J, et al:
Radiotherapy plus cetuximab for squamous-cell carcinoma of the head
and neck. N Engl J Med. 354:567–578. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Vermorken JB, Mesia R, Rivera F, Remenar
E, Kawecki A, Rottey S, Erfan J, Zabolotnyy D, Kienzer HR, Cupissol
D, et al: Platinum-based chemotherapy plus cetuximab in head and
neck cancer. N Engl J Med. 359:1116–1127. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Doble BW and Woodgett JR: GSK-3: Tricks of
the trade for a multi-tasking kinase. J Cell Sci. 116:1175–1186.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Luo J: Glycogen synthase kinase 3beta
(GSK3beta) in tumorigen-esis and cancer chemotherapy. Cancer Lett.
273:194–200. 2009. View Article : Google Scholar
|
|
66
|
Domoto T, Pyko IV, Furuta T, Miyashita K,
Uehara M, Shimasaki T, Nakada M and Minamoto T: Glycogen synthase
kinase-3β is a pivotal mediator of cancer invasion and resistance
to therapy. Cancer Sci. 107:1363–1372. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Maqbool M and Hoda N: GSK3 inhibitors in
the therapeutic development of diabetes, cancer and
neurodegeneration: Past, present and future. Curr Pharm Des.
23:4332–4350. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Walz A, Ugolkov A, Chandra S, Kozikowski
A, Carneiro BA, O'Halloran TV, Giles FJ, Billadeau DD and Mazar AP:
Molecular pathways: Revisiting glycogen synthase kinase-3β as a
target for the treatment of cancer. Clin Cancer Res. 23:1891–1897.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Pramanik KK, Nagini S, Singh AK, Mishra P,
Kashyap T, Nath N, Alam M, Rana A and Mishra R: Glycogen synthase
kinase-3β mediated regulation of matrix metalloproteinase-9 and its
involvement in oral squamous cell carcinoma progression and
invasion. Cell Oncol (Dordr). 41:47–60. 2018. View Article : Google Scholar
|
|
70
|
Matsuo FS, Andrade MF, Loyola AM, da Silva
SJ, Silva MJB, Cardoso SV and de Faria PR: Pathologic significance
of AKT, mTOR, and GSK3β proteins in oral squamous cell
carcinoma-affected patients. Virchows Arch. 472:983–997. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Huang GL, Shen DY, Cai CF, Zhang QY, Ren
HY and Chen QX: β-escin reverses multidrug resistance through
inhibition of the GSK3β/β-catenin pathway in cholangiocarcinoma.
World J Gastroenterol. 21:1148–1157. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Ugolkov A, Gaisina I, Zhang JS, Billadeau
DD, White Kozikowski A, Jain S, Cristofanilli M, Giles F,
O'Halloran T, et al: GSK-3 inhibition overcomes chemoresistance in
human breast cancer. Cancer Lett. 380:384–392. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Ugolkov A, Qiang W, Bondarenko G, Procissi
D, Gaisina I, James CD, Chandler J, Kozikowski A, Gunosewoyo H,
O'Halloran T, et al: Combination treatment with the GSK-3 inhibitor
9-ING-41 and CCNU cures orthotopic chemoresistant glioblastoma in
patient-derived xenograft models. Transl Oncol. 10:669–678. 2017.
View Article : Google Scholar : PubMed/NCBI
|