Introduction
Breast cancer is the most common type of cancer and
the leading cause of cancer-associated mortality among women
worldwide (1). Breast cancer is a
heterogenous disease which can be divided into immunohistochemical
and molecular subtypes (2,3). The most frequent subtype expresses
estrogen receptor (ERα), which can be targeted by anti-estrogens or
aromatase inhibitors (2). Less
frequent subtypes are human epidermal growth factor receptor 2
(Her-2)-positive breast cancers, which are vulnerable to Her2
inhibitors (4) and triple-negative
breast cancers (TNBCs), which are devoid of ERα, Her2 and
progesterone receptor. For TNBCs, selective drugs are still not
available (5). Breast cancers also
exhibit intratumoral heterogeneity (6), part of which is caused by cancer stem
cells (CSCs) (7). CSCs have been
linked to tumor initiation, metastasis and drug resistance
(8).
Drug resistance is a major challenge in the
treatment of patients with breast cancer (9,10).
Different mechanisms leading to drug resistance are known.
Endocrine resistance often involves the activation of the
phosphoinositol-3-kinase (PI3K)/AKT and/or
Ras/Raf/mitogen-activated protein kinase
(MEK-1)/extracellular-signal-regulated kinase (ERK)
kinase-1)/ERK1/2 pathways, which are both able to activate ERα
independently of estrogen (11).
The activation of these pathways can occur intrinsically or
extrinsically. By interacting with breast cancer cells, stromal
cells, such as mesenchymal stem cells (MSCs) and
carcinoma-associated fibroblasts (CAFs), can extrinsically activate
these pathways and thereby promote the development of endocrine
resistance (12).
Recently, the authors demonstrated that MSCs and
CAFs are able to induce resistance to the selective ERα
down-regulator, fulvestrant, in breast cancer cells by increasing
the expression of the atypical inhibitor of nuclear factor-κB (IκB)
family member B-cell leukemia/lymphoma 3 (Bcl-3) (13). Bcl-3, whose expression can also be
induced by the withdrawal of estrogen (14), has been associated with higher
metastatic activities in breast cancer (15). Bcl-3 is a potent activator of the
nuclear factor (NF)-κB pathway (16), a pathway that has also been linked
to endocrine resistance (17). In
addition, the activity of Bcl-3 is positively regulated by the
PI3K/AKT and Ras/Raf/MEK-1/ERK1/2 signaling pathways (18).
The stromal cell-induced increase in Bcl-3
expression has been shown to be causally linked to a prior
downregulation of the insulin-like growth factor binding protein 5
(IGFBP5) level (13). A main
function of IGFBP5 is to regulate the availability of insulin-like
growth factor (IGF) for the interaction of this growth factor with
its receptor, IGF1 receptor (IGF1R) (19). In addition, IGFBP5 can also act
without targeting IGF (19). Such
an IGF-independent effect may be responsible for the regulation of
Bcl-3 by IGFBP5. There is evidence to indicate that IGFBP5 plays an
important role in breast cancer progression and that it is of
prognostic value (20,21). Notably, IGFBP5 expression is higher
in ERα-positive tumors (20).
The stromal cell-induced desensitization of MCF-7
cells to fulvestrant does not require direct contact between
stromal and breast cancer cells (13); however, it can be fully
recapitulated by conditioned medium (CM) derived from stromal
cells. CM can also mimic all effects of stromal cells on signaling
pathways, namely the Janus kinase 2 (JAK2)/signal transducer and
activator of transcription 3 (STAT3), PI3K/AKT and the
Ras/Raf/MEK-1/ERK1/2 pathways, as well as on the expression of
proteins, namely Bcl-3, integrin β1, IGF1R and carbonic anhydrase
IX (CAIX). This suggests that factors secreted by stromal cells are
responsible for the desensitization to fulvestrant and for
alterations in protein expression.
Interleukin (IL)-6 belongs to the IL-6 family of
cytokines (22). By forming a
complex with the IL-6 receptor (IL-6R) and glycoprotein 130 (gp130)
(23), IL-6 activates JAKs
(24). Downstream targets of JAKs
are STATs, MAPK and the PI3K/AKT pathways, whereby the activation
of STAT3 is a major cellular response to IL-6 (25). Soluble IL-6R allows trans
signaling. In trans signaling, an extracellular complex of
IL-6 and IL-6R activates gp130-expressing targets cells (24). Since, in this case, the target
cells do not need to express IL-6R by themselves, the number of
cells that can respond to IL-6 increases. IL-6 is primarily
secreted by leukocytes to regulate hematopoietic cells involved in
inflammation and adaptive immunity (22). In addition, IL-6 acts on
non-hematopoietic cells, such as fibroblasts, adipocytes,
endothelial and epithelial cells and may, when deregulated, lead to
the development of certain diseases, such as fibrosis. Epithelial
cells benefit from the survival-promoting activity of IL-6, helping
damaged epithelia to be repaired (26). Intriguingly, IL-6 also supports the
survival of premalignant epithelial cells, which links IL-6 to
cancer progression. Strikingly, IL-6 has often been found to be
upregulated in the bodily fluids of cancer patients (27) and activated STAT3 is a common
feature of numerous cancer types (28). IL-6 has been associated with
inflammation and multidrug resistance in cancer (29,30).
In breast cancer, IL-6 has been found to induce resistance to the
anti-estrogen tamoxifen and the Her2 antibody trastuzumab and has
been shown to contribute to chemoresistance (12). Evidence for a role of IL-6 in
maintaining cancer stem cell activity in breast cancer has also
been provided (8). IL-6 is able to
increase the cancer stem cell population and, along with it, the
expression of crucial stemness factors, such as octamer-binding
transcription factor 4 (Oct4) (31). IL-6 also induces
epithelial-to-mesenchymal transition (32,33),
which promotes cancer stem cell activity (34). The ability of IL-6 to induce drug
resistance has been found to be linked to its stemness-supporting
activity (35,36). IL-6 has further been shown to be
involved in a cytokine network between MSCs, CSCs and non-CSC
breast cancer cells (37). Based
on the assumption that CSCs are the likely drivers of metastasis
(38), it is noteworthy that IL-6
serum levels are higher in breast cancer patients with metastatic
disease (39).
Given its multiple effects on cancer progression,
IL-6 has been discussed as a promising target for drug intervention
in breast cancer (40,41). IL-6- or IL-6R-directed drugs are
already routinely used for treatment of diseases with excessive
IL-6 expression, such as inflammatory arthritis (22) and could therefore be made available
for cancer treatment. Since the major source of IL-6 are MSCs and
CAFs in cancer (12), in this
study, the potential of recombinant IL-6 to mimic the effects of
stromal cells on fulvestrant resistance and on the expression and
activities of those proteins which may be involved therein was
examined.
This study demonstrates that IL-6 is the mediator of
the majority of the CAF-CM-induced effects on protein expression
and on STAT3 phosphorylation, although not on PI3K/AKT pathway
activity. It is further demonstrated that IL-6 participates in
CAF-CM-induced fulvestrant resistance in 3D spheroid cultures, but
not in 2D adherent cultures. In addition, it was found that IL-6
likely contributes to the growth-inhibitory effects of CAF-CM on
ERα-negative breast cancer cells.
Materials and methods
Cell lines and agents
MCF-7, BT474, T47D, SKBR3 and MDA-MB-231 cells,
which were authenticated by SNP analysis (Genolytic), were
propagated in RMPI-1640 supplemented with 10% fetal calf serum (PAN
Biotech). The MCF-7 subline, AnD5 cells, and the generation of
CAF-CM have been described previously (42). One part CAF-CM was mixed with four
parts fresh medium/serum (20% CAF-CM). Recombinant human IL-6
(rhIL-6) was purchased from PeproTech and reconstituted in water as
recommended by the provider. Fulvestrant (LKT Laboratories) was
added to the cells at a final concentration of 1 µM.
RNA interference
The p110α (pik3ca)-specific siRNA siPIK
(5′-GUACAGGACUUCCGAAGAA-3′), siBcl-3 (5′-UGGUC UUCUCUCCGCAUCA-3′)
and the control siRNA and Firefly luciferase-siRNA siLuc
(5′-CUUACGCUG AGUACUUCGA-3′), were purchased from Eurofins MWG.
Transfections were performed by electroporation by using a Bio-Rad
GenePulserX-Cell as previously described (42). Briefly, following electroporation
using a Bio-Rad GenePulserX-cell (250 V, 800 µF), cells were
seeded on a 10 cm culture dish and incubated for 3 days to allow
the siRNA to downregulate its specific target.
Western blot analysis
Protein extractions and western blot analysis were
carried out as previously described (42). Blots were incubated with primary
and secondary antibodies at room temperature for 1 h. The primary
antibodies used are listed below. Rabbit polyclonal antibodies:
Anti-p(S473)-AKT (1:2,000, D9E, #4060, Cell Signaling Technology),
anti-Bcl-3 (1:1,000, C-14, sc-185, Santa Cruz Biotechnology),
anti-ERα (1:2,000, HC-20, sc-543, Santa Cruz Biotechnology),
anti-p(Thr202, Tyr204)-ERK1/2 and anti-ERK1/2 (both 1:2,000, #9101
and #9102, Cell Signaling Technology), anti-IGF1Rβ (1:2,000, #3027,
Cell Signaling Technology), anti-protein kinase Cα (PKCα; 1:2,000,
C-20, sc-208, Santa Cruz Biotechnology), anti-p(Tyr705)-STAT3
(1:1,000, D3A7, #9145, Cell Signaling Technology) and anti-STAT3
(1:1,000, 79D7, #4904, Cell Signaling Technology); rabbit
monoclonal antibodies: Anti-PI3 linase p110α (1:1,000, C73F8,
#4249, Cell Signaling Technology), anti-Her2 (1:1,000, 29D8, #2165,
Cell Signaling Technology), anti-NF-κB1 p105/p50 (1:1,000, D4P4D,
#13586, Cell Signaling Technology), anti-p21 Waf1/Cip1 (1:1,000,
12D1, #2947, Cell Signaling Technology), anti-integrin β1 (1:2,000,
EPR1040Y, ab134179, Abcam), anti-ABCG2 (1:1,000, EPR20080,
ab207732, Abcam), anti-Ki67 (1:2,000, EPR3610, ab92742, Abcam),
anti-c-Myc (1:500, EP121, AC-0116, Epitomics) and
anti-poly(ADP-ribose) polymerase 1 (PARP-1; cleaved 25 kDa;
1:10,000, #1051-1, Epitomics); mouse monoclonal antibodies:
Anti-(pan) AKT (1:2,000, 40D4, #2920, Cell Signaling Technology).
Anti-CAIX antibody was kindly provided by Professor S. Pastorekova
(Slovak Academy of Sciences). Secondary antibody conjugates
(anti-rabbit/anti-mouse horse radish peroxidase, 1:2,000, #7074 and
#7076) were purchased from Cell Signaling Technology. Protein
loading was examined by either staining proteins with Coomassie
Blue (Blue G, SERVA Electrophoresis) or with Fast Green (MERCK).
Incubations with either staining agent were carried out at room
temperature for 1 h. Antibodies against housekeeping proteins were
not used for this purpose, since they are not reliable markers for
protein loading (43,44).
Immunocytochemistry
Immunocytochemical analysis of formaldehyde-fixed
and paraffin-embedded 3D cell aggregates was carried out as
previously described (45).
Growth/survival assays
In 3D and high-density 2D cultures, the mass of
living cells was determined by an ATP-based assay (Vialight Plus
kit, Lonza). For spheroid formation in 3D suspension cultures, the
cells were incubated in ultra-low attachment 96-well microplates
(Corning) at a density of 5×103 cells/well. For
incubation in high-density 2D cultures, the cells were seeded at a
density of 1×104 per well (24-well plate). The cells
were then exposed to fulvestrant and/or CAF-CM or left untreated
for 3-7 days as indicated. Following the removal of the growth
medium, the 2D-cultured cells were lysed by the addition of a
mixture of 100 µl PBS and 50 µl lysis buffer to each
well. After mixing 50 µl of the lysate with 50 µl
luciferase stock solution, the luciferase activity was measured in
a Sirius luminometer (Berthold). For measuring ATP in 3D-cultured
cells, 50 µl of the lysis buffer was directly added to the
100-µl culture medium the cells were grown in. For examining
cell growth in low-cell density 2D cultures, the cells were seeded
at a density of 3×104 cells per ø 10 cm dish. Following
treatment either the average size (MCF-7, MDA-MB-231 cells) or the
average cell number (SKBR3 cells) of individual colonies was
determined. Colony size was measured using an AxioCAM MRc5 camera
and the AxioVision R 4.5 imaging software (Zeiss) as described
previously (45). For each
condition, 50 randomly selected colonies were examined. For
measuring spheroid/aggregate size in 3D suspension cultures, the
area occupied by the spheroid or aggregate was measured. Of note,
some cell lines did not form regularly shaped spheroids. Instead
they formed randomly clusters (T47D) or discs (MDA-MB-231, SKBR3)
(data not shown).
Reverse transcription-quantitative PCR
(RT-qPCR)
RNA isolation, cDNA synthesis and quantitative
(q)PCR were carried out as described previously (46). Briefly, NucleoSpin RNA II (Macherey
& Nagel) and Superscript II (Invitrogen; Thermo Fisher
Scientific) were used for RNA isolation and cDNA synthesis,
respectively. Following the addition of ABsolute qPCR SYBR-Green
Fluorescein Mix (Thermo Fisher Scientific), qPCRs were run on a
Bio-Rad iCycler and analyzed using iQ5 Optical System software
version 2.1. Relative RNA levels of genes were calculated by the
comparative Cq (2−∆∆Cq) method using glyceraldehyde
3-phosphate dehydrogenase (GAPDH) and hypoxanthine-guanine
phos-phoribosyltransferase (HPRT) as reference genes for
normalization (47). Primers for
Bcl-3, fibroblast growth factor (FGF)18, GAPDH, HPRT, IGFBP5,
kinesin family member 12 (KIF12), kelch like family member 4
(KLHL4), Kallikrein-11 (KLK11), RAB30, receptor activity modifying
protein 3 (RAMP3), selenoprotein P (SEPP1), transforming growth
factor β receptor III (TGFBR3), transmembrane protein 26 (TMEM26),
UDP-glucuronosyltransferase 2B15 (UGT2B15) and yippee like 1
(YPEL1) were as previously described (13). The primers used for the detection
of aldehyde dehydroge-nase 1 family, member A1 (ALDH1A1), aldehyde
dehydrogenase 3 family, member A1 (ALDH3A1) and STAT3 were as
follows: ALDH1A1 (forward 5′→3′, CAAAGAAGC TGCCGGGAAA and reverse
5′→3′, TCCAAGCTCCAGGGT CACC), ALDH3A1 (forward 5′→3′,
GTCCCTGAGACCACG GAGC and reverse 5′→3′, CCCGTGTACAGGATATGGTCG) and
STAT3 (forward 5′→3′, GGACAATATCATTGACCTTGT GAAAA and reverse
5′→3′, CTTCGTTCCAAAGGGCCAG).
Antibody array
To identify proteins that are abundant in CAF-CM,
the Human Obesity Antibody Array I (RayBiotech) was used,
containing 62 different antibodies. Among the proteins, which can
be detected by this array are a number of ILs, stromal cell-derived
factor 1 (SDF-1) and TGFβ. The incubation of this array with CAF-CM
and control medium/serum was carried out according to the
manufacturer's instructions.
Statistical analyses
Data obtained from colony growth assays were
analyzed by Kruskal-Wallis test followed by pairwise comparison
with Bonferroni-corrected test. All other statistical analyses were
carried out by using one-way ANOVA. The Bonferroni correction was
applied for post-hoc analysis. A P-value <0.05 was considered to
indicate a statistically significant difference.
Results
IL-6 is a major mediator of the effects
of CAF-CM on protein expression and phosphorylation in MCF-7
cells
By selecting an antibody array that was able to
detect the majority of factors reported to be secreted by CAFs
(48), it was found that, in the
CAF-CM preparations in this study, IL-6 was the most prominent
component among other proteins [IL-8, CC-chemokine ligand (CCL)2
and CCL7, and tissue inhibitor of metallopro-teinases (TIMP)1 and
TIMP3] (Fig. 1A).
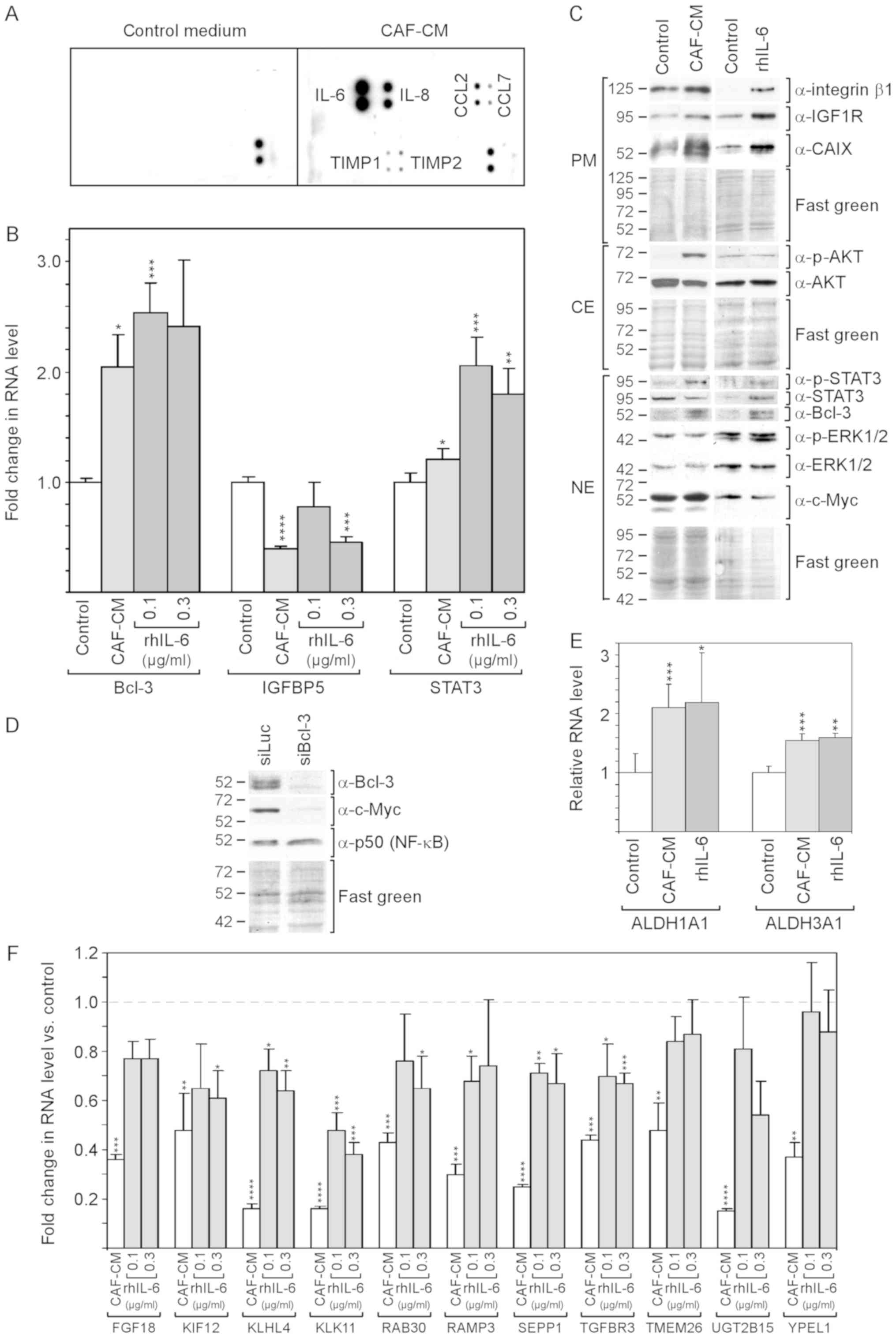 | Figure 1rhIL-6 recapitulates many of the
effects of CAF-CM on protein expression in MCF-7 cells. (A)
Antibody arrays of CAF-CM and control medium. (B) Effects of CAF-CM
and rhIL-6 on relative RNA expression on Bcl-3, IGFBP5 and STAT3 as
measured by RT-qPCR. (C) Western blot analyses of protein extracts
derived from cells treated with either CAF-CM or rhIL-6 (0.3
µg/ml) or of untreated cells (control). Cells were incubated
for 3 days before plasma membrane (PM), cytosolic (CE) and nuclear
(NE) proteins were extracted. (D) Effect of a Bcl-3-specific siRNA
(siBcl3) on the protein expression of Bcl-3, c-Myc and p50 (NF-κB)
as determined by western blot analyses of nuclear protein extracts.
(E) Effects of CAF-CM and rhIL-6 (0.3 µg/ml) on the relative
RNA expression of ALDH1A1 and ALDH1A3 as measured by RT-qPCR. (F)
Effect of CAF-CM and rhIL-6 (0.1 and 0.3 µg/ml) on the
relative RNA expression of 11 CAF-CM-responsive genes. (B, E and F)
Each bar represents the average values of 3 independent
experiments. Statistical analyses were carried out by ANOVA and the
Bonferroni post-hoc test. Asterisk(s) indicate statistical
significance in comparison to the control. *P<0.05,
**P<0.01, ***P<0.005 and
****P<0.001. rhIL-6, recombinant human interleukin 6;
CAF-CM, carcinoma-associated fibroblast-conditioned medium;
ALDH1A1, aldehyde dehydrogenase 1 family, member A1; ALDH1A3,
aldehyde dehydrogenase 3 family, member A1; STAT3, signal
transducer and activator of transcription 3. |
Of note, SDF-1, vascular endothelial growth factor
(VEGF), platelet-derived growth factor (PDGF), tumor necrosis
factor (TNF) and interferon (IFN)γ could not be detected in
considerable amounts. rhIL-6 was used to determine to which extent
IL-6 is able to mimic the effects of CAF-CM on MCF-7 cells.
rhIL-6 was as potent as CAF-CM in increasing Bcl-3
RNA and protein expression (Fig. 1B
and C). Of note, several bands of Bcl-3 were visible in the
western blot analysis, which likely represent Bcl-3 isoforms with a
different phosphorylation status (49). In the CAF-CM-treated MCF-7 cells,
Bcl-3 expression is linked to IGFBP5 downregulation (13), and in rhIL-6-treated cells, STAT3
has been shown to drive Bcl-3 expression (49). In this study, rhIL-6 was found to
be as capable as CAF-CM in activating STAT3 (Fig. 1C) and, above that, even increased
STAT3 mRNA expression (Fig. 1B).
However, at 0.1 µg/ml, it failed to mimic the downregulating
effect of CAF-CM on IGFBP5 expression (Fig. 1B). This suggests that, in contrast
to CAF-CM, rhIL-6 upregulates Bcl-3 in MCF-7 cells through STAT3
and not through IGFBP5.
The authors have previously reported that Bcl-3
modulates the expression of IGF1R and CAIX (13). The levels of both could be
increased in a similar manner by CAF-CM and rhIL-6 (Fig. 1C). Another potential target of
Bcl-3 is c-Myc (50), a mitogenic
oncoprotein (51). After it was
confirmed that c-Myc expression also depends on Bcl-3 in MCF-7
cells (Fig. 1D), the effect of
CAF-CM and rhIL-6 on the c-Myc level was examined. However, neither
agent interfered with c-Myc expression (Fig. 1C) suggesting that the basal level
of Bcl-3 is sufficient to maintain high c-Myc levels. It was also
determined whether Bcl-3 modulates the expression of p50 (NF-κB), a
Bcl-3-binding transcription factor through which Bcl-3 activates
transcription (52). However, p50
expression was not affected by Bcl-3 knockdown (Fig. 1D).
Integrin β1, another protein whose expression can be
upregulated by CAF-CM, also exhibited a higher level in the
presence of rhIL-6 (Fig. 1C). In
addition, the RNA expression levels of the cancer stem cell
markers, ALDH1A1 and ALDH1A3 (8)
were increased in a similar manner by CAF-CM and rhIL-6 (Fig. 1E). However, rhIL-6 failed to
increase AKT phosphorylation (Fig.
1C). In addition, it was not able to down-regulate a number of
genes to the same extent as CAF-CM (Fig. 1F), whereby two of these genes
(TMEM26 and YPEL1) exhibited no response to rhIL-6 at all. Of note,
the 11 genes depicted in Fig. 1F
shared the ability to react to inhibitors of the PI3K/AKT pathway
by increasing their expression (data not shown). This links the
weaker effect of rhIL-6 on these genes to its inability to induce
AKT phosphorylation.
On the whole, these data demonstrate that rhIL-6
recapitulates a number of the effects of CAF-CM on RNA and protein
expression in MCF-7 cells. However, it fails to activate the
PI3K/AKT pathway and has a weaker or no effect on a number of
PI3K/AKT-responsive genes.
rhIL-6 partly mimics CAF-CM-induced
fulvestrant resistance in 3D, but not in 2D cultures
The PI3K/AKT pathway and Bcl-3 play important roles
in CAF-induced anti-estrogen resistance (13,53).
The PI3K/AKT pathway is able to phosphorylate ERα and restore ERα
activity in the presence of anti-estrogens. In addition, it
increases Bcl-3 protein stability and fosters its nuclear
localization (18). As
demonstrated above, CAF-CM and rhIL-6 increased the expression of
Bcl-3, but only CAF-CM was able to activate the PI3K/AKT pathway.
We therefore examined whether rhIL-6 is capable of inducing
fulvestrant resistance to the same extent as CAF-CM. To this end,
MCF-7 cells were grown in the absence and presence of fulvestrant
for 5 or 7 days at high or low density, respectively, and the mass
of viable cells was determined by an ATP/luciferase-based assay
(high density) or average colony size (low density). In the absence
of fulvestrant, CAF-CM negatively affected growth in high-density
cultures and had a slight positive effect in low-density cultures,
whereas rhIL-6 had no effect (Fig. 2A
and B). In the presence of fulvestrant, CAF-CM potently
increased cell mass both in low- and high-density cultures, whereas
rhIL-6 had no effect at the concentration of 0.01 and 0.1
µg/ml, and slightly increased cell mass at 0.3 µg/ml.
This indicates that, in contrast to CAF-CM, rhIL-6 is unable to
induce fulvestrant resistance.
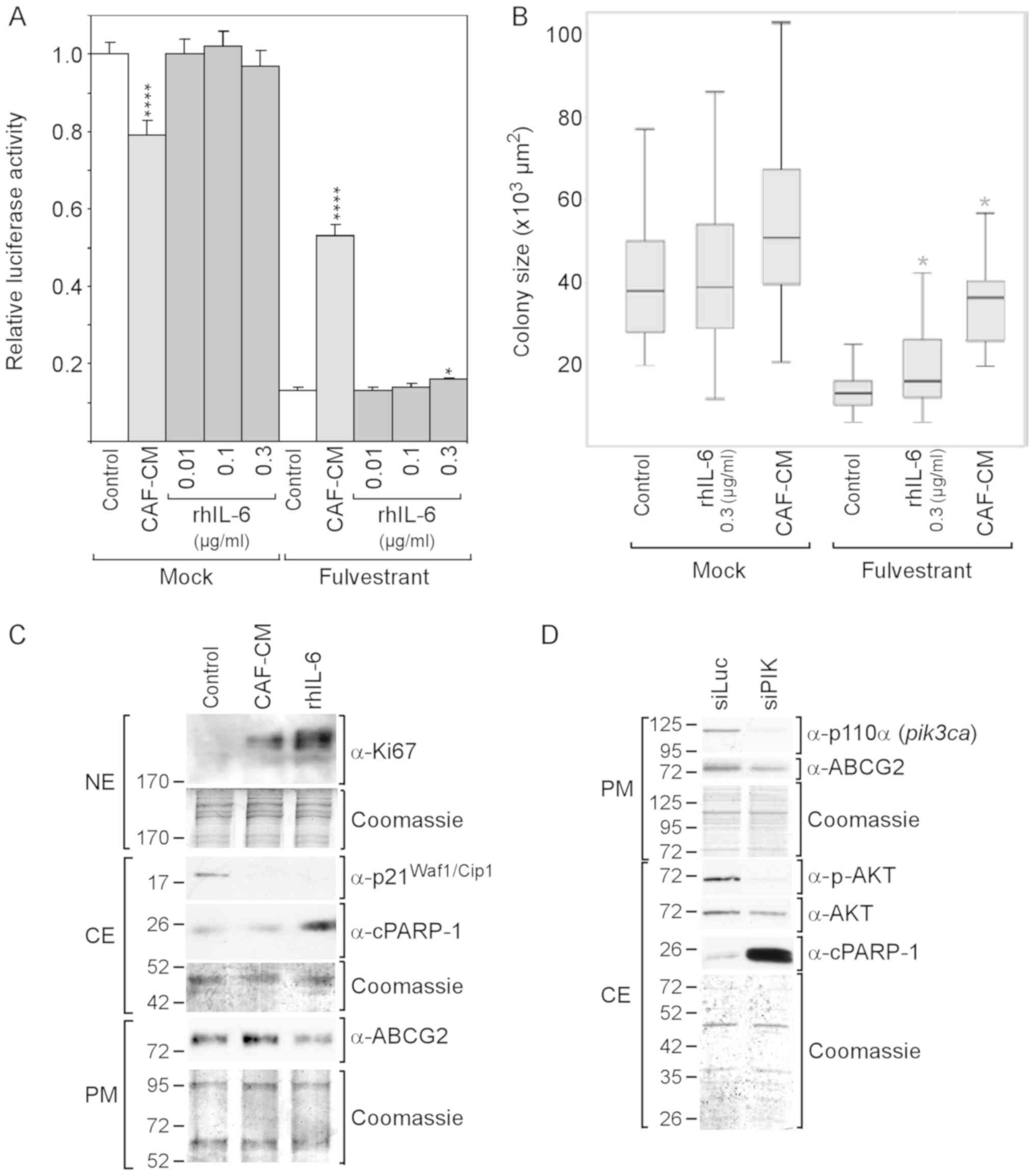 | Figure 2rhIL-6 fails to mimic the
growth-stimulatory effect of CAF-CM on MCF-7 cells in the presence
of fulvestrant. (A and B) Cells were incubated for 6 days in the
presence or absence of fulvestrant and exposed to either CAF-CM or
rhIL-6 (at concentrations as indicated) or to none of these two
agents after cells have been seeded at (A) high or (B) low density.
Viability/growth was either measured by (A) an ATP/luciferase-based
assay or (B) examined by determining the average size of 50
randomly selected individual colonies. Statistical analyses were
either performed by (A) ANOVA or (B) by the Kruskal-Wallis test and
the Bonferroni post-hoc test. Asterisk(s) indicate statistical
significance in comparison to the control. *P<0.05
and ****P<0.001. (C) Cells were incubated for 7 days
in the presence of fulvestrant and exposed to either CAF-CM, rhIL-6
(0.3 µg/ml) or none of these agents, before plasma membrane
(PM), cytosolic (CE) and nuclear proteins (NE) were extracted and
examined by western blot analysis for the proteins as indicated.
(D) Effects of a PI3KCA (p110α)-specific siRNA (siPIK) on the
levels of p110α, ABCG2, p-AKT, AKT and cPARP-1 as determined by
western blot analysis. rhIL-6, recombinant human interleukin 6;
CAF-CM, carcinoma-associated fibroblast-conditioned medium;
cPARP-1, cleaved poly(ADP-ribose) polymerase-1; ABCG2, ATP-binding
cassette transporter G2. |
To examine whether CAF-CM and rhIL-6 differently
affect proliferation and/or apoptosis in the presence of
fulvestrant, the proliferation marker Ki67 and the apoptosis marker
cPARP, which is the 25-kDa fragment of cleaved PARP1, were
analyzed. Both CAF-CM and rhIL-6 increased the Ki67 levels in the
fulvestrant-treated MCF-7 cells and, along with this, abrogated the
expression of cytoplasmic p21Waf1/Cip1, a marker of both
senescence and apoptosis resistance (54,55).
Unlike CAF-CM, rhIL-6 increased cPARP-1 expression (Fig. 2C). This suggests that, in contrast
to CAF-CM, rhIL-6 exhibited pro-apoptotic activity in MCF-7 cells,
which is consistent with the findings of a previous study
demonstrating that, at 5 ng/ml, rhIL-6 induced apoptosis-linked DNA
laddering in these cells (56).
Along with the increased expression of cPARP-1, the level of ABCG2,
a multidrug resistance protein acting as drug efflux pump (57), was found to be reduced (Fig. 2C). This suggest that the
pro-apoptotic activity of rhIL-6 is linked to a decline in the
activity of ABCG2.
Subsequently, it was determined whether the PI3K/AKT
pathway is involved in this process by using siRNA directed to the
PI3K subunit p110α. This siRNA completely abolished p110α
expression and markedly reduced AKT phosphorylation (Fig. 2D). It also potently increased
cPARP-1 expression and downregulated ABCG2 expression. These data
suggest that the PI3K/AKT pathway is important for protecting MCF-7
cells against apoptosis and that ABCG2 plays a role in this process
(Fig. 2D).
These data suggest that, in the presence of
fulvestrant, CAF-CM increased cell growth by stimulating
proliferation. While rhIL-6 shares with CAF-CM the ability to
stimulate proliferation, it also acts in a pro-apoptotic manner,
which seems to lead to a steady state between proliferation and
apoptosis and therefore prevents growth. The data further suggest
that the pro-apoptotic activity of rhIL-6 is linked to ABCG2
downregulation and its inability to raise AKT activity.
The effects of CAF-CM and rhIL-6 on the sensitivity
of MCF-7 cells to fulvestrant in 3D spheroid cultures were then
analyzied for two reasons. These culture conditions more likely
resemble in vivo conditions (57) and cells in 3D cultures react
differently to external stimuli as compared to cells in 2D cultures
(59,60). In 3D cultures, MCF-7 cells form
regularly-shaped spheroids (Fig.
S1A). Spheroid formation is complete after three days of
incubation. Thereafter, the spheroids become larger (Fig. S1A), which coincides with cell
growth (Fig. S1B) and a high
expression of Ki67 (Fig. S1C).
Later, growth ceases, possibly since cells die in the center of the
spheroid, as indicated by cPARP-1 expression (61). In the presence of fulvestrant,
cells form smaller spheroids, which later on appear disheveled
(Fig. S1A). These morphological
changes were accompanied by progressive cell death (Fig. S1B).
To assess the effects of CAF-CM and rhIL-6 on
spheroid-assembled MCF-7 cells, cell growth and spheroid size were
measured. In the absence of fulvestrant, neither CAF-CM nor rhIL-6
affected cell growth (Fig. 3A). In
the presence of fulvestrant, both CAF-CM and rhIL-6 increased cell
mass; however, the effect of CAF-CM was more potent (3.5- vs.
1.9-fold). The difference became even more pronounced, when the
incubation time was extended to 11 days (5.5- vs. 2.4-fold) and 14
days (10- vs. 3.4-fold) (Fig. 3B).
Spheroid size was increased by CAF-CM both in absence and presence
of fulvestrant; however, rhIL-6 only weakly enlarged the spheroids
in the absence of fulvestrant and had no effect on spheroid size in
the presence of fulvestrant (Fig.
3C).
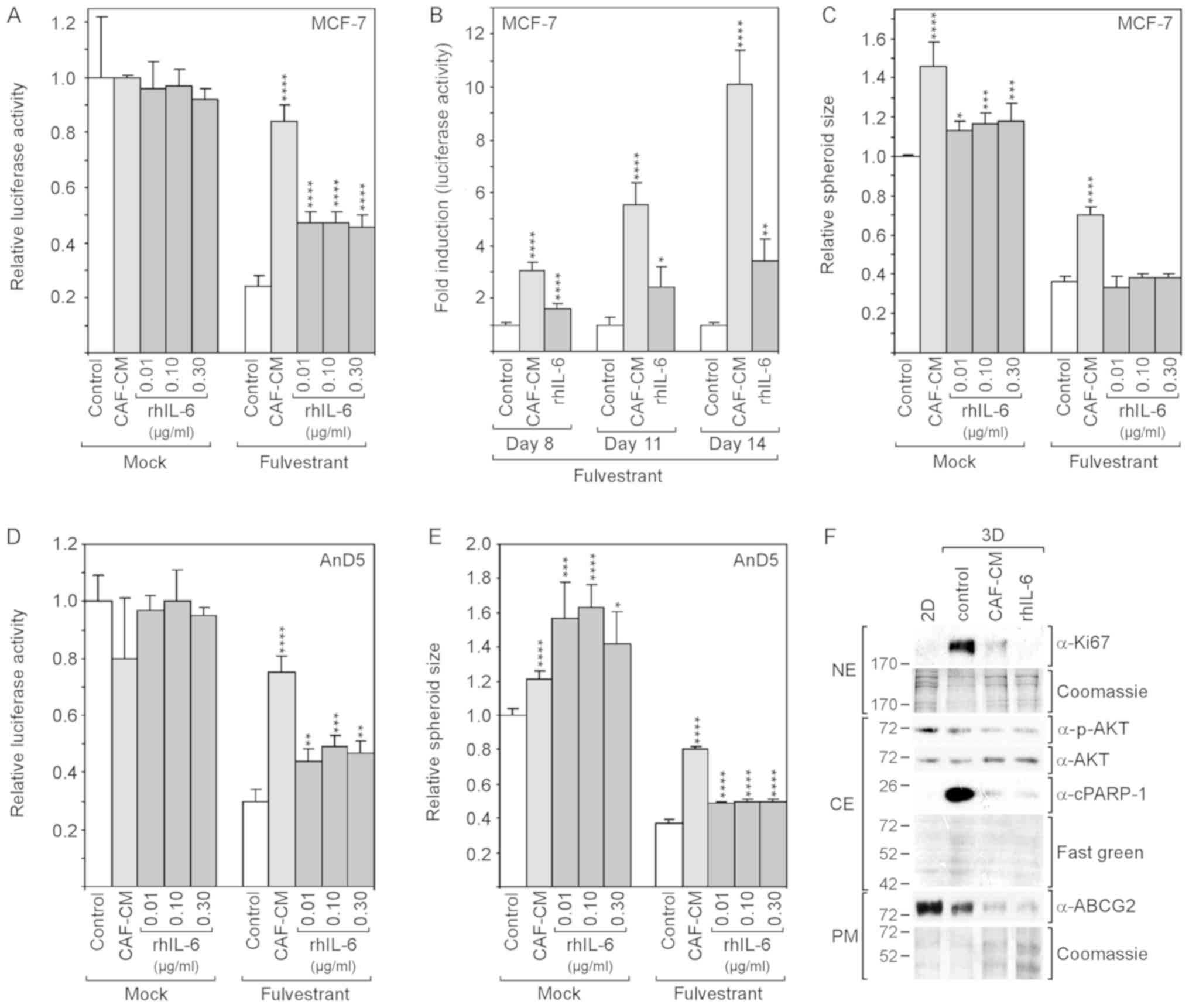 | Figure 3rhIL-6 partly mimics the
growth-stimulatory effects of CAF-CM in 3D cultures of MCF-7 and
AnD5 cells. (A-C) MCF-7 cells were incubated for (A and C) 6 days
or (B) for 8, 11 and 14 days in the presence or absence of
fulvestrant and exposed to either CAF-CM or rhIL-6 (A and C, at
concentrations as indicated; B, at 0.3 μg/ml) or to none of
these two agents. (A and B) Viability was measured by an
ATP/luciferase-based assay, whereby, in (B) fold induction is
shown, calculated relative to the control value for each time
point. (C) Spheroid size was measured as described in the Materials
and methods. (D and E) AnD5 cells were incubated for 3 days in the
presence or absence of fulvestrant and exposed to either CAF-CM or
rhIL-6 (at concentrations as indicated) or to none of these two
agents. (D) Viability was measured by an ATP/luciferase-based
assay. (E) Spheroid size was measured as described in the Materials
and methods. (A-E) Statistical analyses were carried out by ANOVA
and the Bonferroni post-hoc test. Asterisk(s) indicate statistical
significance in comparison to the control. *P<0.05,
**P<0.01, ***P<0.005 and
****P<0.001. (F) Western blot analysis of plasma
membrane (PM), cytosolic (CE) and nuclear protein extracts (NE)
derived from 2D- or 3D-cultured MCF-7 cells after 7-day-incubation
in the presence of fulvestrant and exposure to CAF-CM, rhIL-6 (0.3
μg/ml) or to none of these agents (control). rhIL-6,
recombinant human interleukin 6; CAF-CM, carcinoma-associated
fibroblast-conditioned medium; cPARP-1, cleaved poly(ADP-ribose)
polymerase-1; ABCG2, ATP-binding cassette transporter G2. |
These experiments were repeated with the AnD5 cell
line (42). This MCF-7 subline
forms smaller spheroids and develops them more rapidly than the
parental MCF-7 cell line (data not shown). The results obtained
with the AnD5 cells were similar to those obtained with the
parental cell line, with the exception that, in the absence of
fulvestrant, rhIL-6 affected spheroid size more potently than
CAF-CM and that, in the presence of fulvestrant, rhIL-6 increased
spheroid size, although still not as efficiently as CAF-CM
(Fig. 3D and E).
The examination of the Ki67 and cPARP-1 levels after
7 days of fulvestrant treatment revealed that the levels of both
proteins were higher in the 3D cultures compared to their levels in
2D cultures, and that CAF-CM and rhIL-6 reduced the levels of both
proteins (Fig. 3F). While the
effects of CAF-CM and rhIL-6 were similar on cPARP-1 expression,
rhIL-6 had a more potent suppressive effect on Ki67 expression.
These data suggest that, in 3D cultures, CAF-CM- and rhIL-6-induced
fulvestrant resistance is based on their anti-apoptotic activities.
The residual proliferative activity in the presence of CAF-CM and
rhIL-6 may as well play a role. If so, the more potent suppressive
effect of rhIL-6 on Ki67 expression may explain why rhIL-6 is not
as effective as CAF-CM in inducing fulvestrant resistance. The
examination of p-AKT and ABCG2 expression revealed that the
anti-apoptotic effect of CAF-CM and rhIL-6 was not linked to these
proteins. In the presence of CAF-CM and rhIL-6, the levels of both
proteins were rather reduced (Fig.
3F). This suggests that, in 3D cultures, the PI3K/AKT pathway
and ABCG2 are not involved in the regulation of apoptosis.
Collectively, these data suggest that CAF-CM induces
fulvestrant resistance in 2D and 3D cultures through different
mechanisms, one by inducing proliferation, the other by inhibiting
apoptosis, respectively. To a large extent, rhIL-6 was able to
mimic the protective effect of CAF-CM against fulvestrant in 3D
cultures, likely due to the fact that it was as able as CAF-CM to
block apoptosis. By contrast, in 2D cultures, rhIL-6 shares with
CAF-CM a mitogenic effect; however, by also acting in a
pro-apoptotic manner under these culture conditions, it prevents
cell growth and instead induces a steady-state between cell
proliferation and cell death.
rhIL-6 fails to induce fulvestrant
resistance also in other ERα-positive cell lines in 2D
cultures
The question of whether the findings that were
obtained with the MCF-7 cells are also applicable for other
ERα-positive cell lines was then examined. The BT474 and T47D cell
lines were selected, which have been previously found by the
authors to respond to CAF-CM (13), both in terms of protein expression
patterns and survival in the presence of fulvestrant. Both cell
lines exhibited an increased STAT3 phosphorylation in response to
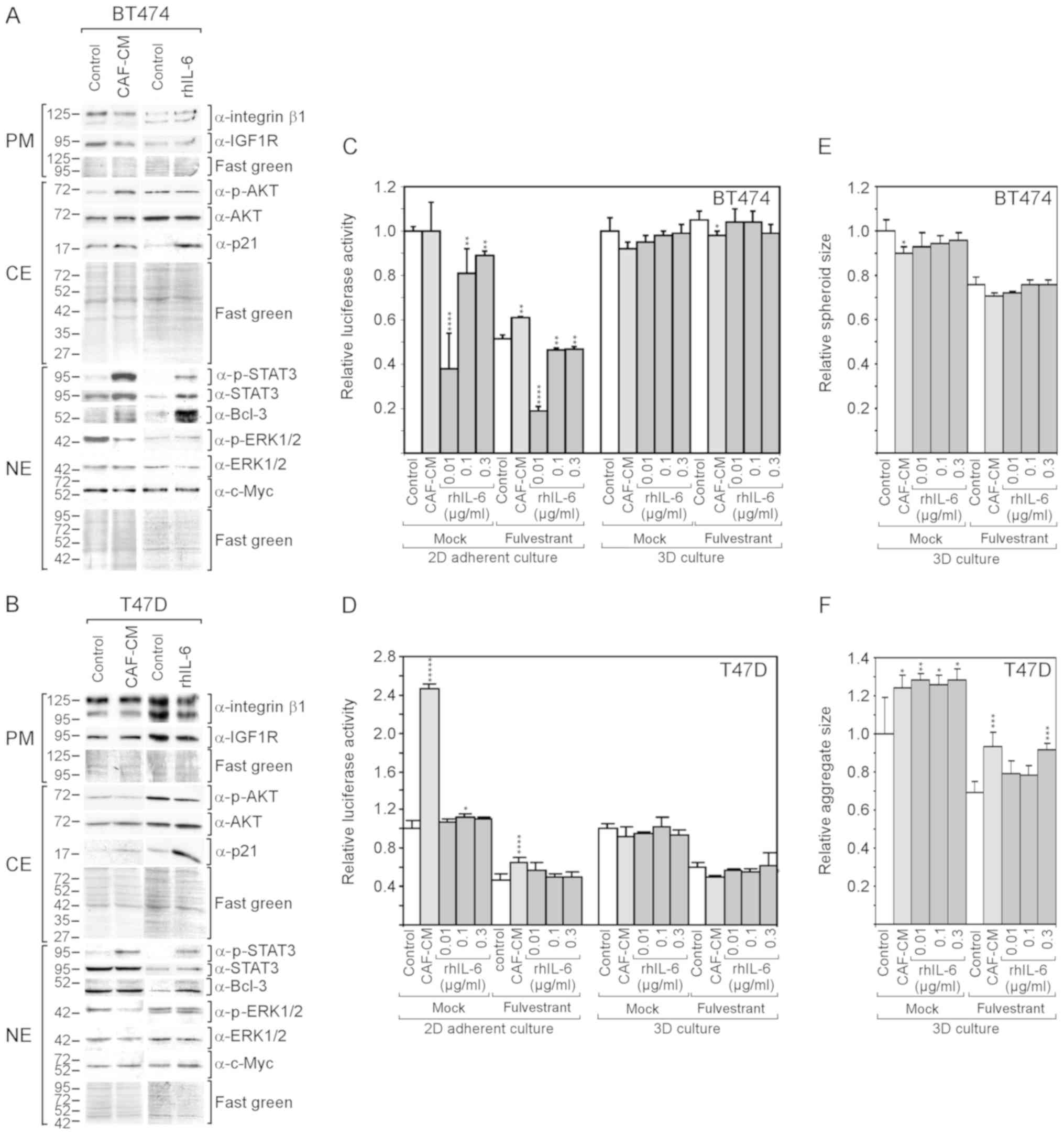
CAF-CM and rhIL-6 (Fig. 4A and B).
In the BT474 cells, both CAF-CM and rhIL-6 upregulated Bcl-3
expression, although the effect of rhIL-6 was more potent. In the
T47D cells, only rhIL-6 increased the Bcl-3 level. In neither case
was a gain in the Bcl-3 level accompanied by a substantial change
in c-Myc or IGF1R expression; nor did CAIX expression increase to
detectable levels (data not shown). In addition, neither CAF-CM nor
rhIL-6 upregulated the level of integrin β1. Different effects of
CAF-CM and rhIL-6 were observed on cytoplasmic p21, ERK1/2 and AKT
phosphorylation. In both cell lines, rhIL-6, but not CAF-CM,
substantially increased the level of cytoplasmic p21, while CAF-CM,
but not rhIL-6, decreased ERK1/2 phosphorylation. Importantly, as
was observed with the MCF-7 cells, rhIL-6 also failed to mimic the
positive effect of CAF-CM on AKT phosphorylation in BT474
cells.
As was previously demonstrated (13), CAF-CM increased the resistance of
BT474 and T47D cells to fulvestrant in 2D cultures (Fig. 4C and D), although these effects
were not as potent as with the MCF-7 cells (Fig. 2A). Again, with rhIL-6, these
results could not be reproduced.
In the absence of fulvestrant, CAF-CM had no effect
on BT474 cell growth, while it potently increased the growth of
T47D cells. By contrast, rhIL-6 failed to mimic the
growth-stimulatory effect on the T47D cells and even had a
growth-inhibitory effect on BT474 cells (Fig. 4C and D).
In 3D cultures, neither CAF-CM nor rhIL-6 had a
substantial effect on the growth of these cells (Fig. 4C and D). The spheroid size of the
BT474 cells and aggregate size of the T47D cells (which do not form
uniformly shaped spheroids) were affected by CAF-CM and rhIL-6 in a
similar manner. Both agents slightly reduced the average spheroid
size of the BT474 cells and increased the average aggregate size of
the T47D cells (Fig. 3E and
F).
Collectively, these data demonstrate that rhIL-6
also fails to mimic the growth-stimulatory effects of CAF-CM on
BT474 and T47D cells in 2D cultures. In the case of the BT474
cells, this failure may again be linked to the inability of rhIL-6
to increase AKT phosphorylation. Additionally, cytoplasmic p21 may
play a role, which was highly upregulated in both BT474 and T47D
cells in response to rhIL-6, but not in response to CAF-CM.
rhIL-6 mimics the majority of the
growth-inhibitory effects of CAF-CM on ERα-negative cell lines
Furthermore, ERα-negative cell lines
(triple-negative MDA-MB-231 and Her2-positive SKBR3 cells) were
analyzed. The lack of ERα expression in MDA-MB-231 and SKBR3 cells
and Her2 expression in SKBR3 cells were confirmed by western blot
analysis (Fig. S2). Of note, the
MDA-MB-231 cells exhibited a high basal activity of STAT3 (Fig. S2), which is the consequence of
their high production of IL-6 (61) and an IL-6/IL-6 receptor autocrine
loop (63). This cell line also
exhibited the highest basal level of integrin β1 of all the cell
lines tested (Fig. S2). However,
due to their ERα deficiency, no effect of fulvestrant on MDA-MB-231
and SKBR3 cells was expected; for reasons of completeness, the
analyses were also carried out in the presence of fulvestrant. In
addition, since these cells grow rapidly, growth in 2D cultures was
examined at a low density and either the average size of individual
colonies (MDA-MB-231) or the average number per colony (SKBR3) was
determined.
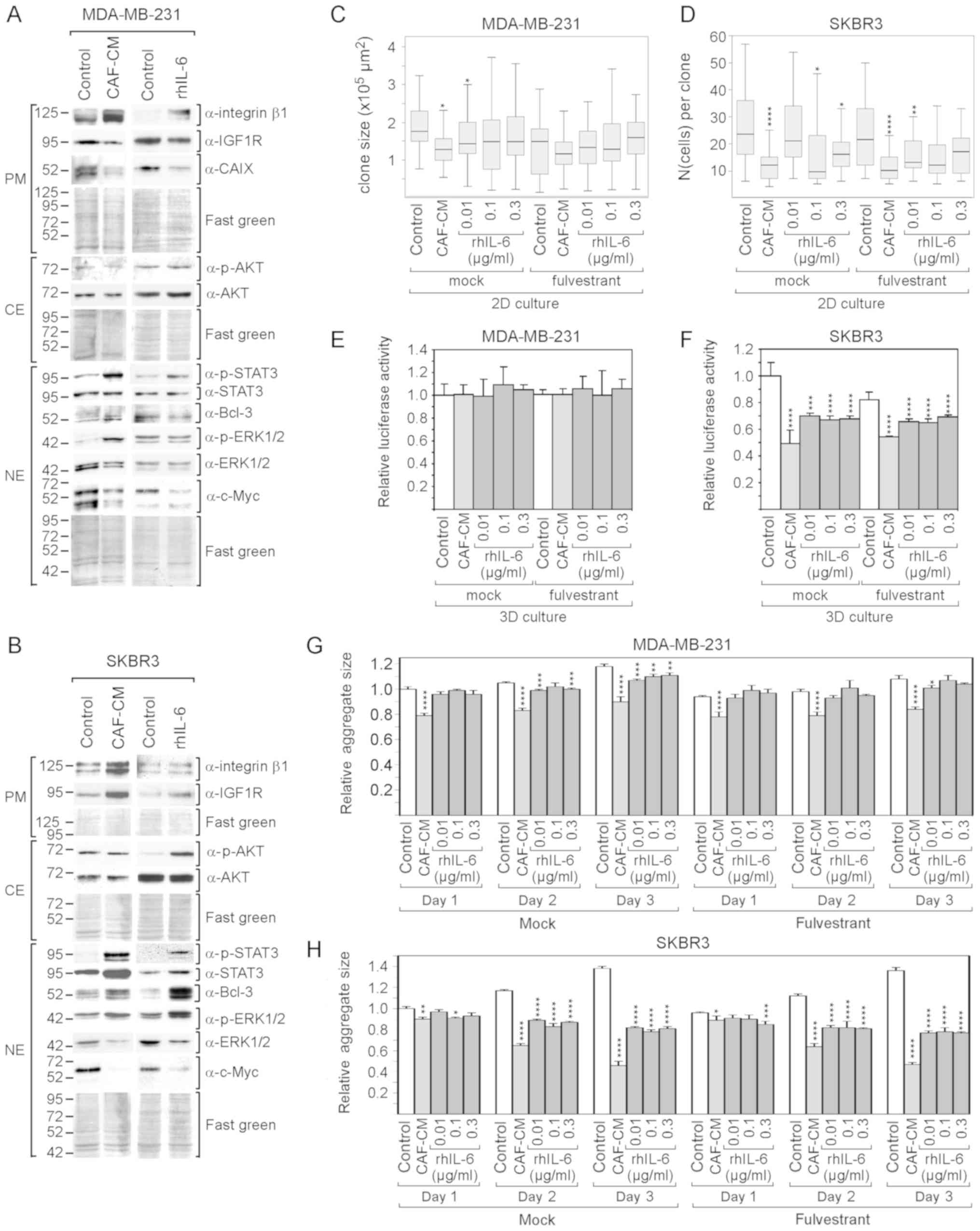
Despite the high basal levels of p-STAT3 and
integrin β1 in the MDA-MB-231 cells, CAF-CM and rhIL-6 were able to
further upregulate the levels of both proteins in these cells
(Fig. 5A). By contrast, none of
the two agents induced Bcl-3 expression and both even negatively
affected the expression of IGF1R and CAIX. In SKBR3 cells, CAF-CM
and rhIL-6 similarly upregulated STAT3 phosphorylation, Bcl-3 and
IGF1R expression, while only CAF-CM enhanced integrin β1 expression
(Fig. 5B). In both cell lines,
CAF-CM increased ERK1/2 phosphorylation and decreased total ERK1/2
levels, while having no effect on AKT phosphorylation (Fig. 5A and B). The effects on ERK1/2 were
mimicked by rhIL-6 in the SKBR3, but not in the MDA-MB-231 cells.
In addition, in the SKBR3 cells, rhIL-6 increased AKT
phosphorylation. Of note, in both cell lines, CAF-CM, as well as
rhIL-6, potently decreased the expression of c-Myc.
Cell growth analysis revealed that both CAF-CM and
rhIL-6 reduced the growth of MDA-MB-231and SKBR3 cells in 2D
cultures (Fig. 5C and D). With the
SKBR3 cells, but not with the MDA-MB-231 cells, this
growth-inhibitory effect was also observed in 3D cultures (Fig. 5E and F). In addition, the average
sizes of the 3D SKBR3 cell aggregates decreased significantly,
while the aggregates of the MDA-MB-231 cells, which form discs
rather than spheroids (data not shown), remained unaltered
(Fig. 5G and H). In general, the
negative effects of CAF-CM on the growth of MDA-MB-231 and SKBR3
cells were more potent than the corresponding effects of
rhIL-6.
Collectively, these data demonstrate that rhIL-6
also mimics the majority of the effects of CAF-CM on protein
expression in MDA-MB-231 and SKBR3 cells. rhIL-6 also shares the
growth-inhibitory effects of CAF-CM on both cell lines, without
being as potent as CAF-CM. The decline in growth activity may be
linked to the potent reduction in c-Myc expression as inflicted by
both agents in these cell lines. In the case of the MDA-MB-231
cells, additionally, the decline in IGF1R and CAIX may play a role
here. c-Myc, IGF1R and CAIX have been reported to be key players
driving MDA-MB-231 cell proliferation (64-66).
Discussion
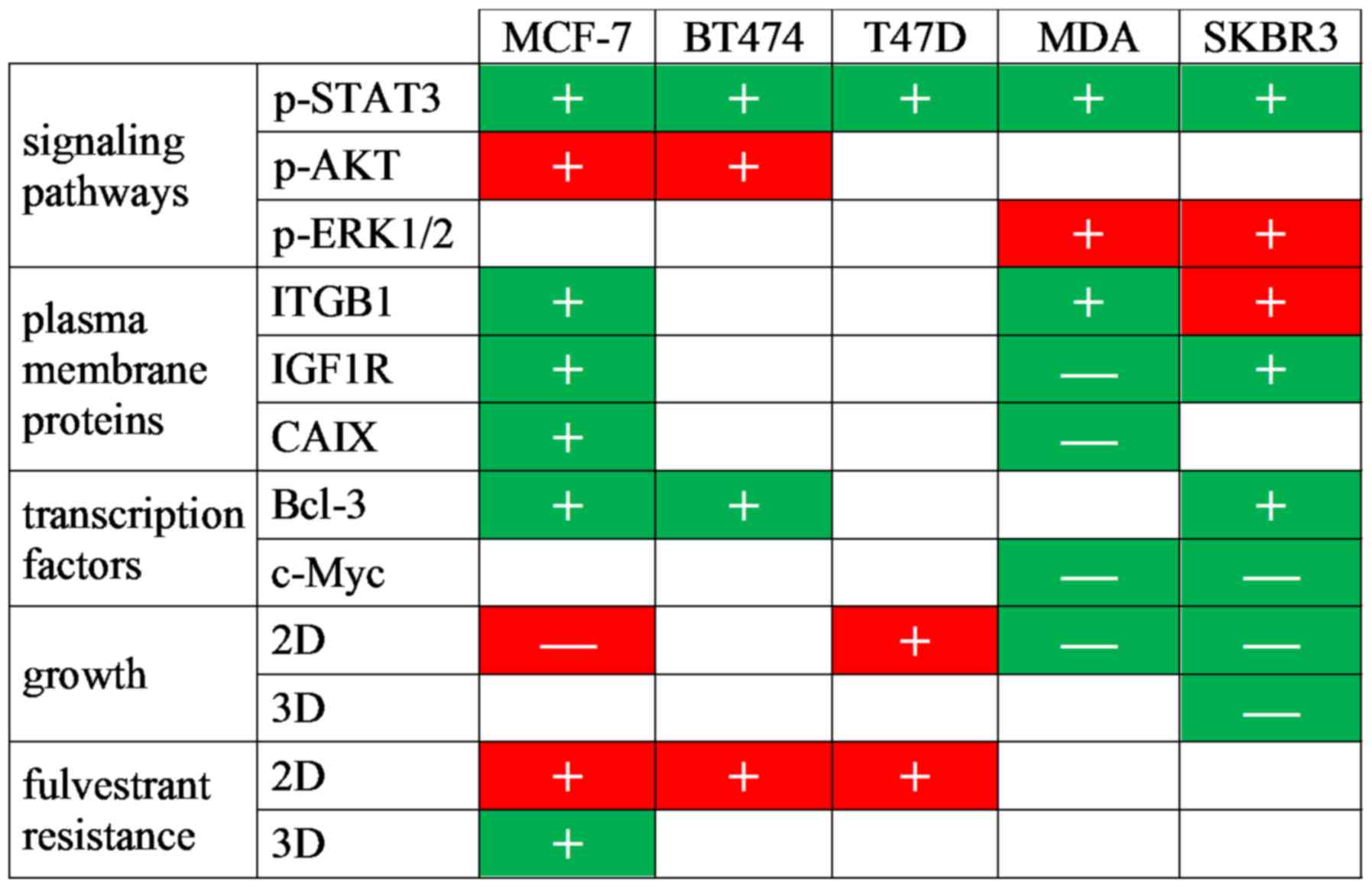
The findings of this study suggest that IL-6 is not
only a major component of CAF-CM, but also mimics the majority of
the effects of CAF-CM on protein expression in both ERα-positive
and -negative breast cancer cells (Fig. 6). In agreement with previous
reports that IL-6 is a classical activator of STAT3 (24,25,67),
this study found that an increase in the p-STAT3 level was a common
response of all tested breast cancer cell lines to rhIL-6. This
effect was also observed with CAF-CM, suggesting that IL-6 is the
general mediator of the STAT3-activating effect of CAF-CM. In most
cases, rhIL-6 also shared with CAF-CM the ability to increase the
expression of Bcl-3. Bcl-3 gene expression can be upregulated by
rhIL-6 through STAT3 (49), which
may suggest that the increase in STAT3 activity was responsible for
the increase in Bcl-3 expression in response to rhIL-6 and CAF-CM.
However, in MCF-7 cells, the CAF-CM-induced decrease in IGFBP5
expression was shown to be the major mediator of Bcl-3 upregulation
(13). Since rhIL-6 had no effect
on IGFBP5 expression at a lower concentration, it is likely that
CAF-CM and rhIL-6 induced Bcl-3 expression through two different
mechanisms: CAF-CM by decreasing IGFBP5 expression, rhIL-6 by
upregulating STAT3 activity. A downregulatory effect of CAF-CM on
the IGFBP5 level was as also observed in BT474 and SKBR3 cells
(data not shown). RNA interference confirmed that, also in these
cells, the downregulation of IGFBP5 caused an increase in Bcl-3
protein expression (data not shown). Of note, in MDA-MB-231 cells,
IGFBP5 RNA was barely detectable (data not shown) and basal STAT3
activity was the highest of all cell lines tested (Fig. S2). In these cells, neither CAF-CM
nor rhIL-6 increased the Bcl-3 level.
There were a number of other effects on protein
expression shared by CAF-CM and rhIL-6, which may be related to
their abilities to increase the p-STAT3 and Bcl-3 levels. Among
these is IGF1R, a major driver of the PI3K pathway in MCF-7 cells
and important for ERα-driven proliferation (68,69).
In MCF-7 cells, Bcl-3 mediates the CAF-CM-induced expression of
IGF1R (13). In SKBR3 cells, the
IGF1R levels were also found to increase along with Bcl-3. However,
in BT474 and T47D cells, a higher Bcl-3 expression did not coincide
with higher IGF1R levels and, in the MDA-MB-231 cells, the IGF1R
level even decreased in response to CAF-CM and rhIL-6, while no
change in Bcl-3 expression was observed. This suggests that IGF1R
expression is regulated by CAF-CM and rhIL-6 in an either
Bcl-3-dependent or -independent manner. In tongue squamous cell
carcinoma cells, the IGF1R promoter has been reported to be
regulated by c-Myc (70). Hence,
in MDA-MB-231 cells, the CAF-CM- and rhIL-6-induced loss of c-Myc
may be responsible for the decrease in the IGF1R level. However, in
the other cell lines tested, c-Myc expression was not associated
with IGF1R expression. In MCF-7 cells, c-Myc expression was closely
linked to Bcl-3 expression (Fig.
1D), which is consistent with the observation that, in colon
cancer cells, Bcl-3 increases the expression and stability of c-Myc
(50). However, the CAF-CM- and
rhIL-6-induced increase in Bcl-3 expression did not lead to a
higher expression of c-Myc (Fig.
1C), arguing against the possibility that in MCF-7 cells, Bcl-3
increased IGF1R expression indirectly through c-Myc.
Another protein, whose expression was modulated by
both CAF-CM and rhIL-6 was CAIX, which is important for the growth
of MCF-7 and MDA-MB-231 cells (65,71).
In MCF-7 cells, Bcl-3 and, to a minor extent, also STAT3 contribute
to the CAF-CM-induced increase in CAIX expression (13). The classical activator of CAIX
expression is hypoxia-inducible factor 1α (HIF1α) (72). CAF-CM has been shown to induce the
expression of HIF1α in MCF-7 cells under normoxic conditions,
although not as potently as the hypoxia mimetic, CoCl2
(13). NF-κB, through which Bcl-3
activates genes (16), and STAT3
have been shown to be able to upregulate HIF1α expression under
normoxic conditions (73,74). In MDA-MB-231 cells, in which CAF-CM
and rhIL-6 increased the p-STAT3 level, while leaving the Bcl-3
level unaffected, CAIX expression decreased (Fig. 5A). This suggests that additional
factors are involved in CAIX regulation. In the other cell lines
tested, the CAIX levels were not detectable neither in the absence
of CAF-CM or rhIL-6 (Fig. S2),
nor in their presence (data not shown).
Integrin β1, a protein linked to anti-estrogen
resistance (42,53), also exhibits a higher expression in
response to CAF-CM and rhIL-6 in some of the breast cancer cell
lines tested (MCF-7, SKBR3 and MDA-MB-231). The expression of both
the slower migrating, N-glycosylated (75) and the rapidly migrating,
unglycosylated forms of integrin β1 were similarly affected,
suggesting that CAF-CM and rhIL-6 did not modulate the
glycosylation of this protein. The regulation of integrin β1
expression is not yet well understood. In MCF-7 cells, neither
STAT3 nor Bcl-3 affect integrin β1 expression positively and
exhibit a rather downregulatory effect (13). The transcription factor, forkhead
box protein M1 (FoxM1), has recently been identified as a potential
regulator of integrin β1 expression in TNBC cells (76). However, in SKBR3 cells, CAF-CM and
rhIL-6 decreased the level of FoxM1 (data not shown), while CAF-CM
increased the level of integrin β1, and rhIL-6 did not affect
integrin β1 expression. This argues against an involvement of FoxM1
in CAF-CM- and rhIL-6-induced integrin β1 expression. Another
candidate that potentially may have contributed to the increase in
integrin β1 expression is TMEM26. Not only does its knockdown
increase integrin β1 expression (75), but CAF-CM also downregulated its
expression in MCF-7 cells (Fig.
1F). Furthermore, an inverse association between TMEM26 and
integrin β1 levels was also found with BT474 and SKBR3 cells (data
not shown). Hence, a downregulated TMEM26 level may have
contributed to the stimulatory effect of CAF-CM on integrin β1
expression. However, it does not explain the effect of rhIL-6 on
integrin β1, as rhIL-6 fails to downregulate TMEM26 (Fig. 1F).
Although rhIL-6 mimicked almost all the effects of
CAF-CM on protein expression, in most cases, it failed to
recapitulate the effects of CAF-CM on ERK1/2 and AKT
phosphorylation. In the BT474 and T47D cells, CAF-CM decreased and,
in the MDA-MB-231 and SKBR3 cells, it increased ERK1/2
phosphorylation, while, only in the SKBR3 cells, rhIL-6 was able to
mimic the effect of CAF-CM. Likewise, CAF-CM, but not rhIL-6
increased the phosphorylation of AKT in the MCF-7 and BT474 cells.
The failure of rhIL-6 (10 ng/ml) to modulate AKT and ERK1/2
activities in breast cancer cell lines has also been demonstrated
by others (40).
The failure to activate the PI3K/AKT pathway in
MCF-7 and BT474 cells coincided with the inability of rhIL-6 to
mimic CAF-CM-induced fulvestrant resistance in 2D cultures. The
authors demonstrate that, in MCF-7 cells, rhIL-6 induced apoptosis
in 2D cultures, probably by downregulating ABCG2 expression, and
that the apoptotic activity and ABCG2 expression are under the
control of the PI3K/AKT pathway. This suggests a connection between
the failure of rhIL-6 to activate the PI3K/AKT pathway and its
pro-apoptotic activity. Apart from stimulating apoptosis, rhIL-6 is
as capable as CAF-CM in stimulating proliferation in the presence
of fulvestrant. Hence, a likely scenario is that CAF-CM induces
fulvestrant resistance by promoting proliferation, while rhIL-6
fails to foster growth in the presence of fulvestrant by causing a
steady-state between cell proliferation and apoptotic cell death.
The PI3K/AKT pathway is not only important in regulating apoptosis,
but is also able to phosphorylate ERα, thereby rendering the
activity of ERα independent of estrogen (11). Furthermore, the PI3K/AKT pathway
can activate Bcl-3 (18). These
PI3K/AKT pathway-related activities may as well play a role in
CAF-CM-induced fulvestrant resistance.
CAF-CM also promoted the growth of T47D cells in the
presence of fulvestrant. T47D cells differ from the MCF-7 and BT474
cells, as they exhibit a higher growth activity in response to
CAF-CM also in the absence of fulvestrant. The effect on cell
growth in the absence of fulvestrant was more potent than that in
its presence. Neither of these effects were mimicked by rhIL-6.
These findings are supported by those of other studies. It was
previously demonstrated that co-cultures with CAFs upregulated the
proliferation of T47D cells (77),
whereas, at concentrations between 20 pg/ml and 100 ng/ml, rhIL-6
downregulated growth of these cells (56,78,79).
The p-AKT levels did not differ in the presence of CAF-CM and
rhIL-6 (Fig. 4B), ruling out the
possibility that the PI3K/AKT pathway was involved. In addition,
both agents upregulated the phosphorylation of STAT3, a factor that
T47D cells rely on for growth (80,81),
in a similar manner. However, unlike CAF-CM, rhIL-6 potently
increased the expression of cytoplasmic p21 and Bcl-3 (Fig. 4B). The activation of NF-κB, as can
be induced by Bcl-3 (16), can
lead to the reduced growth of T47D cells (82). In addition, IKKβ (IκB kinase β),
which activates NF-κB by triggering its dissociation from IκB, was
found to cause the cytoplasmic p21 level in breast cancer cells to
rise (83). Since cytoplasmic p21
is associated with cellular senescence (54), rhIL-6 may have induced senescence
through a Bcl-3/cytoplasmic p21 route. It should be noted that
cytoplasmic p21 has also been linked to apoptosis protection
(55). However, the data of this
study suggest that, at least in MCF-7 cells, the cytoplasmic p21
level is an indicator of proliferative and not apoptotic activity
(Fig. 2C).
By summarizing the data on the growth activity of
ERα-positive breast cancer cells in 2D cultures, it becomes clear
that all the growth-promoting effects of CAF-CM could not be
recapitulated by rhIL-6 and must therefore be mediated by a CAF-CM
component other than IL-6. By contrast, in 3D cultures, the
growth-stimulatory effects of CAF-CM on MCF-7 and AnD5 cells in the
presence of fulvestrant were mimicked by rhIL-6 to a large extent
(Fig. 3A and B). Of note, the
mechanism through which CAF-CM induces fulvestrant resistance
differs between 2D and 3D cultures. In 2D cultures, CAF-CM promotes
growth in the presence of fulvestrant by stimulating proliferation,
whereas in 3D cultures this is achieved by the inhibition of
apoptosis. rhIL-6 shares with CAF-CM the anti-apoptotic effect and
is therefore also able to induce fulvestrant resistance in 3D
cultures. Hence, there are opposite effects of rhIL-6 on apoptosis
in 2D vs. 3D cultures, whereby the regulation of apoptosis in 2D
cultures is dependent on the PI3K/AKT pathway, whereas, in 3D
cultures, it is not. In contrast to 3D-cultured MCF-7 cells,
neither the 3D-cultured BT474 nor 3D-cultured T47D cells responded
to CAF-CM or rhIL-6 by a considerable change in growth activity.
These data again emphasize that responses to external factors can
be quite different between 2D and 3D cultures.
In contrast to the ERα-positive breast cancer cell
lines tested, the ERα-negative cell lines MDA-MB-231 and SKBR3
responded to CAF-CM in 2D cultures by a decrease in growth
activity. The CAF-CM-treated SKBR3 cells also exhibited a lower
growth activity in 3D cultures. Depending on the concentration
used, all these growth-inhibitory actions were more or less
mimicked by rhIL-6. Notably, CAF-CM and rhIL-6 potently decreased
c-Myc expression in both cell lines. Since c-Myc is essential for
cell cycling in MDA-MB-231, as well as in SKBR3 cells (66,84),
the loss of c-Myc is the likely reason for the reduced growth
activity as inflicted by CAF-CM and rhIL-6. The loss of c-Myc does
not seem to be connected to Bcl-3. In the SKBR3 cells, the c-Myc
levels were decreased, while the Bcl-3 levels were increased; in
the MDA-MB-231 cells, the c-Myc levels decreased, while the Bcl-3
levels remained unaltered. In the MDA-MB-231 cells, the loss of
c-Myc may be related to the decrease in the levels of IGF1R and
CAIX, which are both important for MDA-MB-231 cell proliferation
(64,65).
Differences in the reactivity to CAF-CM and rhIL-6
in terms of growth activity in 2D as opposed to 3D culture have
also been found with MDA-MB-231 cells, demonstrating that
ERα-negative breast cancer cells are react differently between 2D
and 3D cultures. This was also shown in a previous study, in which
the B-Raf inhibitor, RAF-265, was found to be more effective in
inhibiting the growth of 3D-cultured than that of 2D-cultured
MDA-MB-231 cells (60). Likewise,
the growth inhibition of SKBR3 cells by the Her2 inhibitor,
lapatinib, has been shown to be more pronounced in 3D as compared
to 2D cultures (85).
Although the stroma is the major source of IL-6
expression in primary breast cancers (86), breast cancer cells also secrete
IL-6, whereby TNBC cells produce the highest amounts (56,62,87).
For instance, MDA-MB-231 cells secrete a ~1,000-fold greater amount
of IL-6 than MCF-7 cells (88).
MDA-MB-231 cells use the endogenous production of IL-6 to stimulate
a IL-6/IL-6 receptor loop for keeping STAT3 activity high (63). In fact, their p-STAT3 level was the
highest of all p-STAT3 levels in the breast cancer cell lines we
have tested (Fig. S2).
Nevertheless, CAF-CM and rhIL-6 were able to further increase STAT3
activity (Fig. 5A) suggesting that
the amount of IL-6 they secrete does not reach the maximum
concentration for optimal stimulation of STAT3 activity. The
inhibition of the IL-6/IL-6R loop or STAT3 activity has been shown
to lead to the decreased proliferation and the increased apoptosis
of MDA-MB-231 cells (63,89). On the other hand, the addition of
rhIL-6 to MDA-MB-231 cells has been demonstrated to decrease
proliferation, while not having any effect on apoptosis (56). In this study, rhIL-6 also acted
growth-inhibitory on MDA-MB-231 cells in 2D cultures. As discussed
above, the downregulation of the c-Myc, IGF1R and CAIX levels may
be the reasons for the rhIL-6-induced growth inhibition.
IL-6 plays a role in tissue remodeling and can
stimulate both reprogramming and senescence (90). In this study, at the concentrations
of 0.1 and 0.3 µg/ml, rhIL-6 was found to recapitulate the
effects of CAF-CM on the expression of CSC-related ALDH1A1 and
ALDH3A1 in MCF-7 cells (Fig. 1E).
This is in agreement with earlier findings indicating that 0.1
µg/ml rhIL-6 increased ALDEFLOUR activity in SUM159 cells
(37).
In conclusion, the findings of this study suggest
that, in all cell lines tested, CAF-CM component IL-6 mediates
CAF-CM-induced STAT3 phosphorylation and the majority of the
changes in protein expression pattern. However, almost all
CAF-CM-induced alterations in ERK1/2 and AKT activities are not
mediated by IL-6. The failure to activate AKT seems to be linked to
its inability to mimic CAF-CM-induced fulvestrant resistance in
common adherent 2D cultures. Yet, it likely mediates CAF-CM-induced
fulvestrant resistance in 3D cultures. This suggests that the
ability of IL-6 to participate in the acquisition of fulvestrant
resistance depends on culture conditions. Notably, IL-6 acts in a
pro-apoptotic manner in 2D and in an anti-apoptotic manner in 3D
cultures. Differences in the apoptotic activities of cells in 2D
and 3D cultures have also been found by others (91). Tissue architecture has a potent
effect on the behavior of cells. One reason may be that cells
become polarized in 3D cultures, but not in 2D cultures. Given that
cells in tissues are organized in three and not in two dimensions,
the data obtained with 3D cultures are more likely to resemble the
in vivo situation. The Kaplan-Meier-Plotter (http://kmplot.com/analysis/index.php?p=service&default=true)
allows the assessment in-silico of the importance of IL-6 on the
outcome of patients based on its mRNA level. Such a survival
analysis of 494 ERα+/Her2− breast cancer
samples for IL-6 mRNA revealed no significant difference in
relapse-free survival at high and low levels of IL-6 mRNA (data not
shown). However, IL-6 mRNA levels may not necessarily reflect the
levels of secreted IL-6. Therefore, future studies on primary ERα+
breast cancer samples, comparing low and high IL-6 protein
expresser, are warranted.
Supplementary Data
Acknowledgments
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
AD performed most of the growth assays and RNA
interference experiments and carried out Western blot analyses. TL
performed most of the Western blot analyses and contributed to the
growth assay studies. BL performed antibody arrays and RT-qPCR
analyses. AD, TL and BL contributed to the interpretation of the
results. JD designed the study, interpreted the data and wrote the
manuscript. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Davies C, Godwin J, Gray R, Clarke M,
Cutter D, Darby S, McGale P, Pan HC, Taylor C, Wang YC, et al Early
Breast Cancer Trialists' Collaborative Group (EBCTCG): Relevance of
breast cancer hormone receptors and other factors to the efficacy
of adjuvant tamoxifen: Patient-level meta-analysis of randomised
trials. Lancet. 378:771–784. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Prat A and Perou CM: Deconstructing the
molecular portraits of breast cancer. Mol Oncol. 5:5–23. 2011.
View Article : Google Scholar
|
|
4
|
Hudis CA: Trastuzumab - mechanism of
action and use in clinical practice. N Engl J Med. 357:39–51. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Omarini C, Guaitoli G, Pipitone S,
Moscetti L, Cortesi L, Cascinu S and Piacentini F: Neoadjuvant
treatments in triple-negative breast cancer patients: Where we are
now and where we are going. Cancer Manag Res. 10:91–103. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wagner J, Rapsomaniki MA, Chevrier S,
Anzeneder T, Langwieder C, Dykgers A, Rees M, Ramaswamy A, Muenst
S, Soysal SD, et al: A Single-Cell Atlas of the Tumor and Immune
Ecosystem of Human Breast Cancer. Cell. 177:1330–1345.e18. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hong D, Fritz AJ, Zaidi SK, van Wijnen AJ,
Nickerson JA, Imbalzano AN, Lian JB, Stein JL and Stein GS:
Epithelial-to-mesenchymal transition and cancer stem cells
contribute to breast cancer heterogeneity. J Cell Physiol.
233:9136–9144. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dittmer J: Breast cancer stem cells:
Features, key drivers and treatment options. Semin. Cancer Biol.
53:59–74. 2018. View Article : Google Scholar
|
|
9
|
Bailey TA, Luan H, Clubb RJ, Naramura M,
Band V, Raja SM and Band H: Mechanisms of Trastuzumab resistance in
ErbB2-driven breast cancer and newer opportunities to overcome
therapy resistance. J Carcinog. 10:282011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Musgrove EA and Sutherland RL: Biological
determinants of endocrine resistance in breast cancer. Nat Rev
Cancer. 9:631–643. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Osborne CK and Schiff R: Mechanisms of
endocrine resistance in breast cancer. Annu Rev Med. 62:233–247.
2011. View Article : Google Scholar
|
|
12
|
Dittmer J and Leyh B: The impact of tumor
stroma on drug response in breast cancer. Semin Cancer Biol.
31:3–15. 2015. View Article : Google Scholar
|
|
13
|
Leyh B, Dittmer A, Lange T, Martens JW and
Dittmer J: Stromal cells promote anti-estrogen resistance of breast
cancer cells through an insulin-like growth factor binding protein
5 (IGFBP5)/B-cell leukemia/lymphoma 3 (Bcl-3) axis. Oncotarget.
6:39307–39328. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pratt MA, Bishop TE, White D, Yasvinski G,
Ménard M, Niu MY and Clarke R: Estrogen withdrawal-induced
NF-kappaB activity and bcl-3 expression in breast cancer cells:
Roles in growth and hormone independence. Mol Cell Biol.
23:6887–6900. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen X, Cao X, Sun X, Lei R, Chen P, Zhao
Y, Jiang Y, Yin J, Chen R, Ye D, et al: Bcl-3 regulates TGFβ
signaling by stabi-lizing Smad3 during breast cancer pulmonary
metastasis. Cell Death Dis. 7:e25082016. View Article : Google Scholar
|
|
16
|
Schuster M, Annemann M, Plaza-Sirvent C
and Schmitz I: Atypical IκB proteins - nuclear modulators of NF-κB
signaling. Cell Commun Signal. 11:232013. View Article : Google Scholar
|
|
17
|
Sas L, Lardon F, Vermeulen PB, Hauspy J,
Van Dam P, Pauwels P, Dirix LY and Van Laere SJ: The interaction
between ER and NFκB in resistance to endocrine therapy. Breast
Cancer Res. 14:2122012. View Article : Google Scholar
|
|
18
|
Wang VY, Li Y, Kim D, Zhong X, Du Q,
Ghassemian M and Ghosh G: Bcl3 Phosphorylation by Akt, Erk2, and
IKK is required for its transcriptional activity. Mol Cell.
67:484–497.e5. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Baxter RC: IGF binding proteins in cancer:
Mechanistic and clinical insights. Nat Rev Cancer. 14:329–341.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mita K, Zhang Z, Ando Y, Toyama T,
Hamaguchi M, Kobayashi S, Hayashi S, Fujii Y, Iwase H and Yamashita
H: Prognostic significance of insulin-like growth factor binding
protein (IGFBP)-4 and IGFBP-5 expression in breast cancer. Jpn J
Clin Oncol. 37:575–582. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Becker MA, Hou X, Harrington SC, Weroha
SJ, Gonzalez SE, Jacob KA, Carboni JM, Gottardis MM and Haluska P:
IGFBP ratio confers resistance to IGF targeting and correlates with
increased invasion and poor outcome in breast tumors. Clin Cancer
Res. 18:1808–1817. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
West NR: Coordination of Immune-Stroma
Crosstalk by IL-6 Family Cytokines. Front Immunol. 10:10932019.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Boulanger MJ, Chow DC, Brevnova EE and
Garcia KC: Hexameric structure and assembly of the
interleukin-6/IL-6 alpha-receptor/gp130 complex. Science.
300:2101–2104. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schaper F and Rose-John S: Interleukin-6:
Biology, signaling and strategies of blockade. Cytokine Growth
Factor Rev. 26:475–487. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Avalle L, Pensa S, Regis G, Novelli F and
Poli V: STAT1 and STAT3 in tumorigenesis: A matter of balance.
JAK-STAT. 1:65–72. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Grivennikov S, Karin E, Terzic J, Mucida
D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H,
Eckmann L, et al: IL-6 and Stat3 are required for survival of
intestinal epithelial cells and development of colitis-associated
cancer. Cancer Cell. 15:103–113. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lacina L, Brábek J, Král V, Kodet O and
Smetana K Jr: Interleukin-6: A molecule with complex biological
impact in cancer. Histol Histopathol. 34:125–136. 2019.
|
|
28
|
Hodge DR, Hurt EM and Farrar WL: The role
of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer.
41:2502–2512. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Morrow RJ, Etemadi N, Yeo B and Ernst M:
Challenging a Misnomer? The Role of Inflammatory Pathways in
Inflammatory Breast Cancer. Mediators Inflamm. 2017:47548272017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ghandadi M and Sahebkar A: Interleukin-6:
A Critical Cytokine in Cancer Multidrug Resistance. Curr Pharm Des.
22:518–526. 2016. View Article : Google Scholar
|
|
31
|
Kim SY, Kang JW, Song X, Kim BK, Yoo YD,
Kwon YT and Lee YJ: Role of the IL-6-JAK1-STAT3-Oct-4 pathway in
the conversion of non-stem cancer cells into cancer stem-like
cells. Cell Signal. 25:961–969. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sullivan NJ, Sasser AK, Axel AE, Vesuna F,
Raman V, Ramirez N, Oberyszyn TM and Hall BM: Interleukin-6 induces
an epithelial-mesenchymal transition phenotype in human breast
cancer cells. Oncogene. 28:2940–2947. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yadav A, Kumar B, Datta J, Teknos TN and
Kumar P: IL-6 promotes head and neck tumor metastasis by inducing
epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling
pathway. Mol Cancer Res. 9:1658–1667. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Brooks MD, Burness ML and Wicha MS:
Therapeutic Implications of Cellular Heterogeneity and Plasticity
in Breast Cancer. Cell Stem Cell. 17:260–271. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Korkaya H, Kim GI, Davis A, Malik F, Henry
NL, Ithimakin S, Quraishi AA, Tawakkol N, D'Angelo R, Paulson AK,
et al: Activation of an IL6 inflammatory loop mediates trastuzumab
resistance in HER2+ breast cancer by expanding the cancer stem cell
population. Mol Cell. 47:570–584. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Saha S, Mukherjee S, Khan P, Kajal K,
Mazumdar M, Manna A, Mukherjee S, De S, Jana D, Sarkar DK, et al:
Aspirin Suppresses the Acquisition of Chemoresistance in Breast
Cancer by Disrupting an NFκB-IL6 Signaling Axis Responsible for the
Generation of Cancer Stem Cells. Cancer Res. 76:2000–2012. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu S, Ginestier C, Ou SJ, Clouthier SG,
Patel SH, Monville F, Korkaya H, Heath A, Dutcher J, Kleer CG, et
al: Breast cancer stem cells are regulated by mesenchymal stem
cells through cytokine networks. Cancer Res. 71:614–624. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dittmer J: Mechanisms governing metastatic
dormancy in breast cancer. Semin Cancer Biol. 44:72–82. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang GJ and Adachi I: Serum interleukin-6
levels correlate to tumor progression and prognosis in metastatic
breast carcinoma. Anticancer Res. 19(2B): 1427–1432.
1999.PubMed/NCBI
|
|
40
|
Casneuf T, Axel AE, King P, Alvarez JD,
Werbeck JL, Verhulst T, Verstraeten K, Hall BM and Sasser AK:
Interleukin-6 is a potential therapeutic target in interleukin-6
dependent, estrogen receptor-a-positive breast cancer. Breast
Cancer (Dove Med Press). 8:13–27. 2016.
|
|
41
|
Zhang W, Guo J, Li S, Ma T, Xu D, Han C,
Liu F, Yu W and Kong L: Discovery of monocarbonyl curcumin-BTP
hybrids as STAT3 inhibitors for drug-sensitive and drug-resistant
breast cancer therapy. Sci Rep. 7:463522017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dittmer A and Dittmer J: Long-term
exposure to carcinoma-associated fibroblasts makes breast cancer
cells addictive to integrin β1. Oncotarget. 9:22079–22094. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dittmer A and Dittmer J: Beta-actin is not
a reliable loading control in Western blot analysis.
Electrophoresis. 27:2844–2845. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Moritz CP: Tubulin or Not Tubulin: Heading
Toward Total Protein Staining as Loading Control in Western Blots.
Proteomics. 17:172017. View Article : Google Scholar
|
|
45
|
Dittmer A, Schunke D and Dittmer J: PTHrP
promotes homotypic aggregation of breast cancer cells in
three-dimensional cultures. Cancer Lett. 260:56–61. 2008.
View Article : Google Scholar
|
|
46
|
Oerlecke I, Bauer E, Dittmer A, Leyh B and
Dittmer J: Cyclic AMP enhances TGFβ responses of breast cancer
cells by upregulating TGFβ receptor I expression. PLoS One.
8:e542612013. View Article : Google Scholar
|
|
47
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
48
|
Kalluri R: The biology and function of
fibroblasts in cancer. Nat Rev Cancer. 16:582–598. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Brocke-Heidrich K, Ge B, Cvijic H, Pfeifer
G, Löffler D, Henze C, McKeithan TW and Horn F: BCL3 is induced by
IL-6 via Stat3 binding to intronic enhancer HS4 and represses its
own transcription. Oncogene. 25:7297–7304. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Liu Z, Jiang Y, Hou Y, Hu Y, Cao X, Tao Y,
Xu C, Liu S, Wang S, Wang L, et al: The IκB family member Bcl-3
stabilizes c-Myc in colorectal cancer. J Mol Cell Biol. 5:280–282.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bretones G, Delgado MD and León J: Myc and
cell cycle control. Biochim Biophys Acta. 1849:506–516. 2015.
View Article : Google Scholar
|
|
52
|
Massoumi R, Chmielarska K, Hennecke K,
Pfeifer A and Fässler R: Cyld inhibits tumor cell proliferation by
blocking Bcl-3-dependent NF-kappaB signaling. Cell. 125:665–677.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Pontiggia O, Sampayo R, Raffo D, Motter A,
Xu R, Bissell MJ, Joffé EB and Simian M: The tumor microenvironment
modulates tamoxifen resistance in breast cancer: A role for soluble
stromal factors and fibronectin through β1 integrin. Breast Cancer
Res Treat. 133:459–471. 2012. View Article : Google Scholar
|
|
54
|
Sánchez-Pérez Y, Chirino YI,
Osornio-Vargas AR, Herrera LA, Morales-Bárcenas R, López-Saavedra
A, González-Ramírez I, Miranda J and García-Cuellar CM: Cytoplasmic
p21(CIP1/WAF1), ERK1/2 activation, and cytoskeletal remodeling are
associated with the senescence-like phenotype after airborne
particulate matter (PM(10)) exposure in lung cells. Toxicol Lett.
225:12–19. 2014. View Article : Google Scholar
|
|
55
|
Yousefi B, Rahmati M and Ahmadi Y: The
roles of p53R2 in cancer progression based on the new function of
mutant p53 and cytoplasmic p21. Life Sci. 99:14–17. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chiu JJ, Sgagias MK and Cowan KH:
Interleukin 6 acts as a paracrine growth factor in human mammary
carcinoma cell lines. Clin Cancer Res. 2:215–221. 1996.PubMed/NCBI
|
|
57
|
Kathawala RJ, Gupta P, Ashby CRJ Jr and
Chen ZS: The modulation of ABC transporter-mediated multidrug
resistance in cancer: A review of the past decade. Drug Resist
Updat. 18:1–17. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Dontu G, Abdallah WM, Foley JM, Jackson
KW, Clarke MF, Kawamura MJ and Wicha MS: In vitro propagation and
transcriptional profiling of human mammary stem/progenitor cells.
Genes Dev. 17:1253–1270. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
DuFort CC, Paszek MJ and Weaver VM:
Balancing forces: Architectural control of mechanotransduction. Nat
Rev Mol Cell Biol. 12:308–319. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Dittmer A, Fuchs A, Oerlecke I, Leyh B,
Kaiser S, Martens JW, Lützkendorf J, Müller L and Dittmer J:
Mesenchymal stem cells and carcinoma-associated fibroblasts
sensitize breast cancer cells in 3D cultures to kinase inhibitors.
Int J Oncol. 39:689–696. 2011.PubMed/NCBI
|
|
61
|
Dittmer A, Hohlfeld K, Lützkendorf J,
Müller LP and Dittmer J: Human mesenchymal stem cells induce
E-cadherin degradation in breast carcinoma spheroids by activating
ADAM10. Cell Mol Life Sci. 66:3053–3065. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sasser AK, Sullivan NJ, Studebaker AW,
Hendey LF, Axel AE and Hall BM: Interleukin-6 is a potent growth
factor for ER-alpha-positive human breast cancer. FASEB J.
21:3763–3770. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Bharti R, Dey G, Ojha PK, Rajput S,
Jaganathan SK, Sen R and Mandal M: Diacerein-mediated inhibition of
IL-6/IL-6R signaling induces apoptotic effects on breast cancer.
Oncogene. 35:3965–3975. 2016. View Article : Google Scholar
|
|
64
|
Klinakis A, Szabolcs M, Chen G, Xuan S,
Hibshoosh H and Efstratiadis A: Igf1r as a therapeutic target in a
mouse model of basal-like breast cancer. Proc Natl Acad Sci USA.
106:2359–2364. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ward C, Meehan J, Mullen P, Supuran C,
Dixon JM, Thomas JS, Winum JY, Lambin P, Dubois L, Pavathaneni NK,
et al: Evaluation of carbonic anhydrase IX as a therapeutic target
for inhibition of breast cancer invasion and metastasis using a
series of in vitro breast cancer models. Oncotarget. 6:24856–24870.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Chen CR, Kang Y and Massagué J: Defective
repression of c-myc in breast cancer cells: A loss at the core of
the transforming growth factor beta growth arrest program. Proc
Natl Acad Sci USA. 98:992–999. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Friedrich K, Dolznig H, Han X and Moriggl
R: Steering of carcinoma progression by the YIN/YANG interaction of
STAT1/STAT3. Biosci Trends. 11:1–8. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Marconett CN, Singhal AK, Sundar SN and
Firestone GL: Indole-3-carbinol disrupts estrogen receptor-alpha
dependent expression of insulin-like growth factor-1 receptor and
insulin receptor substrate-1 and proliferation of human breast
cancer cells. Mol Cell Endocrinol. 363:74–84. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Gaben AM, Sabbah M, Redeuilh G, Bedin M
and Mester J: Ligand-free estrogen receptor activity complements
IGF1R to induce the proliferation of the MCF-7 breast cancer cells.
BMC Cancer. 12:2912012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Sun J, Lu Z, Deng Y, Wang W, He Q, Yan W
and Wang A: Up-regulation of INSR/IGF1R by C-myc promotes TSCC
tumorigenesis and metastasis through the NF-κB pathway. Biochim
Biophys Acta Mol Basis Dis. 1864(5 Pt A): 1873–1882. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Ansari MF, Idrees D, Hassan MI, Ahmad K,
Avecilla F and Azam A: Design, synthesis and biological evaluation
of novel pyridine-thiazolidinone derivatives as anticancer agents:
Targeting human carbonic anhydrase IX. Eur J Med Chem. 144:544–556.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Sedlakova O, Svastova E, Takacova M,
Kopacek J, Pastorek J and Pastorekova S: Carbonic anhydrase IX, a
hypoxia-induced catalytic component of the pH regulating machinery
in tumors. Front Physiol. 4:4002014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Jung YJ, Isaacs JS, Lee S, Trepel J and
Neckers L: IL-1beta-mediated up-regulation of HIF-1alpha via an
NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between
inflammation and oncogenesis. FASEB J. 17:2115–2117. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Wang K, Zhu X, Zhang K, Yin Y, Chen Y and
Zhang T: Interleukin-6 contributes to chemoresistance in MDA-MB-231
cells via targeting HIF-1a. J Biochem Mol Toxicol. 32:e220392018.
View Article : Google Scholar
|
|
75
|
Nass N, Dittmer A, Hellwig V, Lange T,
Beyer JM, Leyh B, Ignatov A, Weiβenborn C, Kirkegaard T,
Lykkesfeldt AE, et al: Expression of transmembrane protein 26
(TMEM26) in breast cancer and its association with drug response.
Oncotarget. 7:38408–38426. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Hamurcu Z, Kahraman N, Ashour A and
Ozpolat B: FOXM1 transcriptionally regulates expression of integrin
β1 in triple-negative breast cancer. Breast Cancer Res Treat.
163:485–493. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Gache C, Berthois Y, Martin PM and Saez S:
Positive regulation of normal and tumoral mammary epithelial cell
proliferation by fibroblasts in coculture. In. Vitro Cell Dev Biol
Anim. 34:347–351. 1998. View Article : Google Scholar
|
|
78
|
Chen L, Shulman LM and Revel M: IL-6
receptors and sensitivity to growth inhibition by IL-6 in clones of
human breast carcinoma cells. J Biol Regul Homeost Agents.
5:125–136. 1991.PubMed/NCBI
|
|
79
|
Badache A and Hynes NE: Interleukin 6
inhibits proliferation and, in cooperation with an epidermal growth
factor receptor autocrine loop, increases migration of T47D breast
cancer cells. Cancer Res. 61:383–391. 2001.PubMed/NCBI
|
|
80
|
Ikeda O, Sekine Y, Mizushima A, Nakasuji
M, Miyasaka Y, Yamamoto C, Muromoto R, Nanbo A, Oritani K,
Yoshimura A, Matsuda T, et al: Interactions of STAP-2 with Brk and
STAT3 participate in cell growth of human breast cancer cells. J
Biol Chem. 285:38093–38103. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Proietti C, Salatino M, Rosemblit C,
Carnevale R, Pecci A, Kornblihtt AR, Molinolo AA, Frahm I, Charreau
EH, Schillaci R, et al: Progestins induce transcriptional
activation of signal transducer and activator of transcription 3
(Stat3) via a Jak- and Src-dependent mechanism in breast cancer
cells. Mol Cell Biol. 25:4826–4840. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Segev DL, Ha TU, Tran TT, Kenneally M,
Harkin P, Jung M, MacLaughlin DT, Donahoe PK and Maheswaran S:
Mullerian inhibiting substance inhibits breast cancer cell growth
through an NFkappa B-mediated pathway. J Biol Chem.
275:28371–28379. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Ping B, He X, Xia W, Lee DF, Wei Y, Yu D,
Mills G, Shi D and Hung MC: Cytoplasmic expression of p21CIP1/WAF1
is correlated with IKKβ overexpression in human breast cancers. Int
J Oncol. 29:1103–1110. 2006.PubMed/NCBI
|
|
84
|
Neve RM, Sutterlüty H, Pullen N, Lane HA,
Daly JM, Krek W and Hynes NE: Effects of oncogenic ErbB2 on G1 cell
cycle regulators in breast tumour cells. Oncogene. 19:1647–1656.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Weigelt B, Lo AT, Park CC, Gray JW and
Bissell MJ: HER2 signaling pathway activation and response of
breast cancer cells to HER2-targeting agents is dependent strongly
on the 3D micro-environment. Breast Cancer Res Treat. 122:35–43.
2010. View Article : Google Scholar
|
|
86
|
Hugo HJ, Lebret S, Tomaskovic-Crook E,
Ahmed N, Blick T, Newgreen DF, Thompson EW and Ackland ML:
Contribution of Fibroblast and Mast Cell (Afferent) and Tumor
(Efferent) IL-6 Effects within the Tumor Microenvironment. Cancer
Microenviron. 5:83–93. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Mao Y, Zhang Y, Qu Q, Zhao M, Lou Y, Liu
J, Huang O, Chen X, Wu J and Shen K: Cancer-associated fibroblasts
induce trastuzumab resistance in HER2 positive breast cancer cells.
Mol Biosyst. 11:1029–1040. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Gallo M, Frezzetti D, Roma C, Chicchinelli
N, Barbieri A, Arra C, Scognamiglio G, Botti G, De Luca A and
Normanno N: RANTES and IL-6 cooperate in inducing a more aggressive
phenotype in breast cancer cells. Oncotarget. 9:17543–17553. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Singh S, Murillo G, Chen D, Parihar AS and
Mehta RG: Suppression of Breast Cancer Cell Proliferation by
Selective Single-Domain Antibody for Intracellular STAT3. Breast
Cancer (Auckl). 12:11782234177508582018.
|
|
90
|
Mosteiro L, Pantoja C, de Martino A and
Serrano M: Senescence promotes in vivo reprogramming through
p16INK4a and IL-6. Aging Cell. 17:172018. View Article : Google Scholar
|
|
91
|
Correia AL and Bissell MJ: The tumor
microenvironment is a dominant force in multidrug resistance. Drug
Resist Updat. 15:39–49. 2012. View Article : Google Scholar : PubMed/NCBI
|