Introduction
The discovery of epidermal growth factor receptor
(EGFR)-activating mutations in lung adenocarcinoma and the
efficacy of EGFR-tyrosine kinases inhibitors (EGFR-TKIs) has
shifted the paradigm for lung cancer treatment from cytotoxic
chemotherapies to molecularly targeted therapies (1-3). The
majority of patients with lung adenocarcinoma harboring
EGFR-activating mutations, such as exon 19 deletion and
L858R, have significant positive responses to EGFR-TKIs, including
gefitinib, erlotinib, afatinib and osimertinib (4-7).
Therefore, EGFR-TKIs are currently the first choice of treatment
for lung adenocarcinoma with EGFR-activating mutations
(6,8-11).
However, despite favorable initial responses to EGFR-TKI treatment,
the majority of patients eventually acquire EGFR-TKI resistance and
develop recurrence within 1 year. The T790M mutation (12), MET amplification (13,14),
hepatocyte growth factor overexpression (15), transformation to small-cell lung
cancer (16),
epithelial-mesenchymal transition (EMT) (17-20)
and the excess secretion of cytokines such as interleukin-6 (IL)-6
(21) may contribute to the
acquisition of resistance to EGFR-TKIs. As primary tolerance or
acquired resistance is almost inevitable, the effectiveness of
EGFR-TKIs appears to be limited (22). Therefore, in order to obtain a
better understanding of the molecular events that result in
malignancy and drug resistance in lung adenocarcinoma, the
identification of novel diagnostic biomarkers is required, which
may also lead to the development of more effective evidence-based
therapeutics, and improved outcomes.
Recent large-scale transcriptome analyses with
next-generation sequencers have reported that the majority of the
genome is transcribed to non-coding RNAs that do not code for
protein, including small and long non-coding RNAs (lncRNAs)
(23). Although small non-coding
RNAs, such as microRNAs (miRNAs or miRs) negatively regulate gene
expression, few lncRNAs are well characterized (24,25).
lncRNAs are arbitrarily defined as non-coding RNAs >200
nucleotides in length (26).
Although lncRNAs were initially considered transcriptional noise,
recent studies have reported that they play critical roles in cell
growth, stem cell pluripotency and metabolism, as well as
functioning as regulatory molecules in multiple diseases (27-29).
Furthermore, lncRNAs have been shown to be associated with drug
resistance (29-32).
Recent studies have reported that the cellular roles
of lncRNAs are involved in regulating gene expression at the
transcriptional and post-transcriptional levels in various cellular
milieu and biological processes (33). As is the case for proteins, the
functions of lncRNAs depend on correct subcellular localization.
Typically, lncRNAs are responsible for the structural integrity of
the nucleus and can modulate expression of nearby genes, when
acting in cis in the nucleus, or distant genes, by interacting with
various intracellular biomolecules, such as proteins, RNAs and
DNAs, when acting in trans in the nucleus or cytoplasm (34,35). The
functions of lncRNAs are mediated through distinct modes, such as
signaling, scaffolds for protein-protein interactions, guides to
target elements and molecular decoys (36,37).
lncRNAs function as a molecular signal when they exhibit cell
type-specific expression and respond to various stimuli under tight
transcriptional controls. Their expression and presence can be used
as potential markers of important cellular functions (38). When lncRNAs function as scaffolds,
they provide a central platform for the assembly of multicomponent
molecular complexes, including complexes of ribonucleoproteins
(39). This function is important
for the precise control of the specificity and dynamics of
molecular interactions (40).
Guide lncRNAs interact with molecules, including ribonucleoprotein
complexes, to direct them to specific target genes (41). Finally, when lncRNAs function as
molecular decoys they limit the activity of regulatory factors by
binding to target molecules without exerting other functions.
Molecular decoy lncRNAs have been reported to regulate targets by
sequestering regulatory molecules, including transcription factors,
catalytic proteins and chromatin-modifying complexes, as well as
miRNAs (42,43). These types of decoy RNAs, which
bind competitively to miRNAs as substitutes of targeted mRNAs, thus
inhibiting their activity in the cytoplasm, are referred to as
competing endogenous RNAs (ceRNAs) (44).
Although an increasing number of lncRNAs have been
identified, the role of lncRNAs in EGFR-mutant lung adenocarcinoma
is poorly understood. It was thus hypothesized that lncRNAs
aberrantly expressed in EGFR-mutant lung adeno-carcinoma are
involved in cancer malignancy and EGFR-TKI resistance. Indeed,
recent experimental evidence suggests that lncRNA UCA1 is
upregulated in EGFR-mutant non-small cell lung cancer (NSCLC) and
induces acquired resistance to EGFR-TKIs by activating the AKT/mTOR
pathway and promoting EMT (32).
In this study, we investigated the clinical
importance of lncRNA expression in patients with EGFR-mutant
lung adenocarcinoma by using the expression profile of lncRNAs in
RNA-seq lung cancer datasets to identify a lncRNA associated with
malignancy and resistance to EGFR-TKIs. To determine the clinical
utility of detecting lncRNA expression in patients with
EGFR-mutant lung adenocarcinoma, the lncRNA expression level
in EGFR-mutant lung adenocarcinoma and the effects of its
expression on lung cancer cells were examined.
Materials and methods
Bioinformatics analysis of RNA-seq and
clinical data
RNA-seq and clinical datasets of The Cancer Genome
Atlas (TCGA) were downloaded from the Genomic Data Commons (GDC)
website (https://portal.gdc.cancer.gov) (45). RNA-seq datasets of the Cancer Cell
Line Encyclopedia (CCLE) were also obtained from the GDC website
(46). The R software package was
used to detect the differential expression of lncRNAs and to
construct the figures. The interactions of lncRNA-miRNA and
mRNA-miRNA were predicted using the miRDB website (http://www.mirdb.org) and TargetScan website
(http://www.targetscan.org/vert_72/).
Cell lines and reagents
A549 (CCL-185), H1299 (CRL-5803) and H1975
(CRL-5908) cells were obtained from the American Type Culture
Collection (ATCC®). 293 and PC9 cells were a kind gift
from Dr Yuichiro Kanno, Faculty of Pharmaceutical Sciences at Toho
University (Chiba, Japan). All non-small cell lung cancer (NSCLC)
cell lines were cultured in DMEM (Wako) supplemented with 10% fetal
bovine serum and antibiotics. To generate gefitinib-resistant PC9
cells (PC9-GR cells), PC9 cells were continuously exposed to
increasing concentrations of gefitinib (0.01-1 µM) for 6
months. The resistant cells were cultured in a gefitinib-free
medium for at least 5 days before all experiments. The cell lines
used in this study are listed in Table
I. Gefitinib, erlotinib, recombinant human EGF and IL-6 were
obtained from Wako. Osimertinib was purchased from MedChemExpress.
All drugs were dissolved in DMSO and stored at −80°C. Proteins were
stored at −20°C in PBS containing 30% glycerol.
 | Table ISummary of parental and engineered
cell lines. |
Table I
Summary of parental and engineered
cell lines.
| NSCLC cell
lines | EGFR mutation | Expression
construct | Notes |
|---|
| Parental | | | |
| H1299 | Wild-type | NA | - |
| A549 | Wild-type | NA | - |
| PC9 | Del E746-A750 | NA | EMT status:
Epithelial |
| | |
Gefitinib-sensitive |
| H1975 | L858R/T790M | NA | - |
| HCC827 | Del E746-A750 | NA |
Gefitinib-sensitive |
| | | The lncRNA
expression |
| | | levels were
obtained from CCLE |
| | | Gene Expression
profile dataset (46) |
| HCC827 GR5 | Del E746-A750 | NA |
Gefitinib-resistant |
| | | The lncRNA
expression |
| | | levels were
obtained from CCLE |
| | | Gene Expression
profile dataset (46) |
| Engineered | | | |
| H1299 EGFR WT | Wild-type | pIRESpuro: EGFR
WT | - |
| H1299 EGFR
19del | Del E746-A750 | pIRESpuro: EGFR Del
E746-A750 | - |
| H1299 EGFR
L858R | L858R | pIRESpuro: EGFR
L858R | EMT status:
Mesenchymal |
| PC9-GR | Del E746-A750 | NA | EMT status:
Mesenchymal |
|
Gefitinib-resistant |
| H1299 EGFR
L858R | L858R | pIRESpuro: EGFR
L858R | Endogenous
LINC00460 |
| LINC00460 KO | | | knockdown
(Cas9) |
| H1299 EGFR
L858R | L858R | pIRESpuro: EGFR
L858R | Endogenous |
| LINC00460 KO
EV | | pcDNA3-Hyg (Empty
Vector) | LINC00460 |
| | | knockdown
(Cas9) |
| H1299 EGFR
L858R | L858R | pIRESpuro: | Endogenous |
| LINC00460 KO
OE | | EGFR L858R
pcDNA3-Hyg: | LINC00460 knockdown
(Cas9) |
| LINC00460 | LINC00460
overexpression |
Clinical samples
A total of 62 patients with recurrent post-operative
EGFR-mutant lung adenocarcinoma treated with gefitinib during the
period from January, 2008 to January, 2019 were enrolled in this
study. LINC00460 expression was analyzed in 62 samples (FFPE slides
of surgical specimens in 23 cases and cell-free RNA in 39 cases).
Written informed consent was obtained from all patients prior to
their participation in this study. RNA was extracted from the FFPE
slides using an RNeasy FFPE kit (cat. no. 73504, Qiagen) and
separated serum was stored at −80°C until use. Cell-free RNA in
peripheral blood was extracted and purified using a QIAamp
Circulating Nucleic Acid kit (cat. no. 55114, Qiagen) in accordance
with the manufacturer's protocol and stored at −20°C until use.
This single-center study was conducted at the Toho University Omori
Medical Center and was approved by the Human Genome/Gene Analysis
Research Ethics Committee (authorization no. A17117).
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted using an RNeasy Mini kit, a
miRNeasy Mini kit or an RNeasy FFPE kit (Qiagen). To synthesize
cDNA, total RNA (500 ng) was reacted with PrimeScript RT Master Mix
(Takara Bio Inc.). Real-time PCR with specific primers (10
µM) and SYBR Premix Ex Taq (Takara Bio Inc.) was used to
measure all RNA expression levels. All reactions were performed in
a total volume of 25 µl. The PCR amplification program
consisted of initial denaturation at 95°C for 30 sec, followed by
40 cycles of PCR at 95°C for 5 sec and 60°C for 30 sec. The
comparative ΔΔCq method was used to determine the relative
expression levels of RNAs (47).
The relative amount of target mRNA and lncRNA was normalized to
that of GAPDH mRNA. Melting curve analysis validated the
specificity of the primers. The sequences of the primers used for
qPCR were as follows: LINC00460 forward,
5′-GTGGATGAGAACGAAGGTTACG-3′ and reverse,
5′-CTTTCCCACGCTCAGTCTTTC-3′; human GAPDH forward,
5′-GCACCGTCAAGGCTGAGAAC-3′ and reverse, 5′-TGGTGAAGACGCCAGTGGA-3′;
human IL-6 forward, 5′-GGCACTGGCAGAAAACAACC-3′ and reverse,
5′-GCAAGTCTCCTCATTGAATCC-3′; human zinc finger E-box-binding
homeobox 1 (ZEB1) forward, 5′-TT CAAACCCATAGTGGTTGCT-3′ and
reverse, 5′-TGGGAG ACACCAAACCAACTG-3′; human vimentin (VIM)
forward, 5′-AGGCGAGGAGAGCAGGATTTC-3′ and reverse, 5′-AGT
GGGTATCAACCAGAGGGAG-3′); human E-cadherin (CDH1) forward,
5′-TCGCTTACACCATCCTCAGC-3′ and reverse, 5′-GGAAACTCTCTCGGTCCAGC-3′;
and human MALAT1 forward, 5′-AAAGCAAGGTCTCCCCACAAG-3′ and reverse,
5′-GGTCTGTGCTAGATCAAAAGGC-3′.
Western blot analysis
The cells were lysed with RIPA buffer (Santa Cruz
Biotechnology). The total protein concentration was determined by
BCA Protein Assay (Takara Bio Inc.). Gels for SDS-PAGE were cast
using a TGX Fast Cast Acrylamide kit 10% (cat. no. 1610173,
Bio-Rad). The lysates (30 µg of protein in total volume of
20 µl per lane) were run at a constant voltage of 200 V in
Tris-Glycine SDS running buffer. The gels were then directly
transferred to PVDF membranes (0.2 nm) using a Trans-Blot Turbo
Blotting System (Bio-Rad). Following transfer, the membranes were
incubated in blocking buffer [Tris-buffered saline with 0.1%
Tween-20 (TBS-T)] containing 10% skim milk) at room temperature for
1 h. Each protein of interest was stored with a specific primary
antibody in a Dilution Buffer (TBS-T with 2% BSA) at 4°C overnight,
washed 3 times with TBS-T, and then reacted with an HRP secondary
antibody in a Dilution Buffer at room temperature for 1 h.
Chemiluminescence was developed using the Clarity Western ECL
substrate, and the blots were scanned with an Amersham Imager 600
device (GE Healthcare Biosciences). The antibodies used in this
experiment are listed in Table
II.
| Table IIList of antibodies used in this
study. |
Table II
List of antibodies used in this
study.
| Antibody | Manufacturer | Species | Dilution |
|---|
| p-EGFR (Y1068) | Abcam (ab5644) | Rabbit
monoclonal | 1:1,000 for WB |
| EGFR | Abcam
(ab52894) | Rabbit
monoclonal | 1:1,000 for WB |
| p-STAT3 (Y705) | Abcam
(ab76315) | Rabbit
monoclonal | 1:2,000 for WB |
| STAT3 | Abcam
(ab68153) | Rabbit
monoclonal | 1:2,000 for WB |
| p-AKT (S473) | Cell Signaling
Technology (#4060) | Rabbit
monoclonal | 1:1,000 for WB |
| AKT | Cell Signaling
Technology (#9272) | Rabbit
monoclonal | 1:1,000 for WB |
| p-ERK1/2
(T202/Y204) | Cell Signaling
Technology (#4370) | Rabbit
monoclonal | 1:2,000 for WB |
| ERK1/2 | Cell Signaling
Technology (#9102) | Rabbit
monoclonal | 1:1,000 for WB |
| GAPDH | Abcam (ab9485) | Rabbit
monoclonal | 1:1,000 for WB |
| Lamin B1 | Abcam
(ab65986) | Rabbit
monoclonal | 1:1,000 for WB |
| Normal Rabbit
IgG | Sigma-Aldrich
(12-370) | Rabbit | 1:50 for RIP |
FLAG (M2)
WB | Sigma-Aldrich
(F1804) | Mouse
monoclonal | 1:50 for RIP,
1:1,000 for |
| Vimentin (V9) | Santa Cruz
Biotechnology (sc-6260) | Mouse
monoclonal | 1:1,000 for WB,
1:100 for IF |
| E-cadherin
(G-10) | Santa Cruz
Biotechnology (sc-8426) | Mouse
monoclonal | 1:1,000 for WB,
1:100 for IF |
| Cleaved Caspase-3
(Asp175) (5A1E) | Cell Signaling
Technology (#9664) | Rabbit
monoclonal | 1:1,000 for WB |
| Cleaved PARP
(Asp214) | Cell Signaling
Technology (#9541) | Rabbit
monoclonal | 1:2,000 for WB |
| B-actin | Proteintech
(60008-1-Ig) | Mouse
monoclonal | 1:5,000 for WB |
| HRP-linked
anti-rabbit IgG | Cell Signaling
Technology (#7074) | | 1:3,000 for WB |
| HRP-linked
anti-mouse IgG | Cell Signaling
Technology (#7076) | | 1:3,000 for WB |
Plasmid construction
The pIRESpuro-EGFR (wild-type, 19del, L858R)
expression vector was a gift from Dr Y. Kanno, Faculty of
Pharmaceutical Sciences, Toho University. The KOD plus neo (Toyobo)
was used for PCR to amplify the full-length LINC00460 sequence from
a cDNA library derived from the total RNA from the H1299 cells. The
PCR products were then used to clone and express LINC00460 in a
pcDNA 3.1/Hygro (+) mammalian expression vector (Invitrogen; Thermo
Fisher Scientific). All PCR products were verified by DNA
sequencing.
Subcellular fractionation
The PC9 cell pellet was suspended in hypotonic
buffer [10 mM Hepes (pH 7.5), 10 mM KCl, and 1.5 mM
MgCl2]. Following incubation at 4°C for 10 min, the
cells were centrifuged for 20 min at 1,000 x g to collect the
cytoplasmic and nuclear fractions. Western blot analysis was used
to validate the separation of each fraction. GAPDH was used as a
cytoplasm marker. Lamin B1 were used as a nucleus marker. RNA
enrichment in each fraction was performed by RT-qPCR.
RNA immunoprecipitation (RIP) assays
based on the miTRAP method
miTRAP (an MS2-based RIP assay) was used to pull
down endogenous miRNAs that bound to LINC00460. Cell lysates were
suspended in RIP buffer containing 20 mM Tris (pH 7.5), 150 mM
NaCl, 1 mM MgCl2, 5% glycerol, 0.5% NP-40 and RNase
inhibitor. Subsequently, anti-FLAG antibody was added followed by
incubation for 2 h at 4°C. Complexes of the MS2 stem-loop RNAs and
FLAG-MS2 proteins were purified by using protein G-conjugated
magnetic beads (Dynabeads Protein G, Life Technologies; Thermo
Fisher Scientific). Precipitated RNA was used for cDNA synthesis
and measured by RT-qPCR. The sequences of the primers were as
follows: miR-149-5p forward, 5′-TCTGGCTCCGTGTCTTCACTCCC-3′. The
reverse primer was supplied with the Mir-X™ miRNA RT-qPCR TB
Green® kit (cat. no. 638314, Takara Bio Inc.). The
antibodies used in this experiment are listed in Table II.
Luciferase reporter assays
The siCHECK™ vector (Promega) was used to determine
the quantity of the RNA interference (RNAi) of miR-149-5p in
accordance with the manufacturer's instructions. The cells were
co-transfected with a psiCHECK2 dual-luciferase reporter
(containing LINC00460, LINC00460 Mut, IL-6, or IL-6 Mut) and miRNA
mimics using Lipofectamine™ 2000 (Invitrogen; Thermo Fisher
Scientific). Each group was plated in triplicate using 48-well
plates. Renilla luciferase activity was normalized to
Firefly luciferase activity.
Small interfering RNA (siRNA)-mediated
silencing
PC9-GR cells (1×105) were cultured in
medium containing 10% FBS (antibiotic-free). siRNAs (100 nM;
si-LINC00460#1 and≈#2) were used for transfection using
TransIT-siQUEST Transfection Reagent (Mirus Bio) for 24 h after
seeding the cells. Following 48 h of incubation, the cells were
used to carry out subsequent experimentations (RT-qPCR and WST-8
assay). The target sequences for LINC00460 were as follows:
si-LINC00460#1, 5′-UAGCAAUUGCUGGAAUC-3′; and si-LINC00460#2,
5′-CACACUUCTCGGCUAAG-3′. Luciferase siRNA and scrambled siRNA were
used as negative controls.
LINC00460 knockout by CRISPR-Cas9
A vector expressing LINC00460 dual guide RNAs
(gRNAs) and Cas9 was transfected into the H1299 cells in 6-well
plates using Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific). After 18 h, the cells were re-seeded in a 10-cm
dish. The following day, puromycin (1.5 µg/ml, Thermo Fisher
Scientific) was added to the cell culture for selection. Individual
surviving colonies were removed and expanded in 35-mm dishes.
Positive clones were identified by RT-qPCR and further verified by
genomic PCR and DNA sequencing. Genomic DNA was extracted from the
cell lines with a PureLink™ Genomic DNA Mini kit (cat. no. 1820-00,
Thermo Fisher Scientific). The cells subjected to knockout
experiments were rescued by transfection with the LINC00460
expression vector (pcDNA3.1-Hyg: LINC00460) and selected on a
medium containing hygromycin B (200 µg/ml, Wako). The empty
vector (i.e., pcDNA3.1-Hyg) was used as a control. The target
sequences of the gRNAs for the LINC00460 coding region were as
follows: gRNA1, 5′-TCCCTGTGCGCGCATAGCAG-3′; and gRNA2,
5′-CCATGCAGGGCCCCCCTCAT-3′. The primer sequences of genomic PCR
were as follows: Primer-F (forward),
5′-GTGGCAGGGCAGGCCAGCTTTCCCAAAA TG-3′; and Primer-R (reverse),
5′-GGGGCTAAGCTCACC CTTTTATACCAGCACCAATC-3′.
Enzyme-linked immunosorbent assay
(ELISA)
The concentrations of human IL-6 in the cell culture
supernatants were measured using the Human IL-6 Quantikine ELISA
kit (cat. no. D6050, R&D Systems) in accordance with the
manufacturer's instructions. The density of each well in a 96-well
plate was determined with a microplate reader set to 450 nm. The
concentrations of each sample were determined using a standard
curve to calculate the values as pg/ml.
Wound healing assays
Scratch wounds of the cell mono-layer were created
using a p200 pipet tip, after which the initial images were
captured. Second images were captured following 48 h of incubation
at 37°C. This assay was performed in the presence of mitomycin C
(0.2 mg/ml) to confirm that the result was not due to cell
proliferation. The wounded areas were quantified using ImageJ
software (https://imagej.nih.gov/ij/).
Immunofluorescence assays
The cells were fixed with 4% paraformaldehyde for 30
min and permeabilized with 0.2% Triton X-100 in PBS for 10 min.
Following 1 h of exposure to blocking buffer, coverslips were
incubated at 4°C with primary antibodies a Dilution Buffer (PBS
with 0.1% Tween-20 and 2% BSA), washed with PBS-T for 3 times, and
incubated with secondary antibodies for 30 min. Finally, coverslips
were mounted in SlowFade™ Gold Antifade Mountant with DAPI
(Invitrogen; Thermo Fisher Scientific) and imaged with a BX61
microscope (Olympus). The antibodies used in this experiment are
shown in Table II.
Cell viability assay
The cells were seeded in a 96-well plate
(5×103 cells/well). Cell viability was performed using a
Cell Counting Kit-8 containing WST-8 (CCK-8; Dojindo Laboratories)
according to the manufacturer's protocol. The number of viable
cells was quantified by determining the absorbance at 450 nm with a
microplate reader.
Caspase-3/7 assays
Each sample was evaluated with a caspase-3/7
activation assay performed on a Caspase-Glo 3/7® Assay
System (cat. no. G8090, Promega), according to the manufacturer's
instructions.
Cell treatments
293 cells were pre-treated with gefitinib (1
µM) or the solvent control (DMSO). After 1 h, the cells were
treated with EGF (200 ng/ml). Following 24 h of incubation at 37°C,
the expression level of LINC00460 was measured by RT-qPCR. PC9
cells were treated with rh IL-6 (50 ng/ml) for 24 h, followed by
western blot analysis. PC9-GR cells were treated with gefitinib,
erlotinib and osimertinib (1 µM) or the solvent control
(DMSO) for 24 h following siRNA transfection. After a further 24 h,
the cells were used to perform subsequent experiments (RT-qPCR and
WST-8 assay).
Determination of clinical outcomes
The progression-free survival (PFS) and overall
survival (OS) of patients with a high or low LINC00460 expression
was estimated. The PFS of patients treated with EGFR-TKIs was
assessed from the date that EGFR-TKI therapy commenced to the first
sign of disease progression, as determined by findings from
computed tomography or magnetic resonance imaging and the Response
Evaluation Criteria In Solid Tumors (RECIST) criteria. OS was
defined as the period from the date of diagnosis until death from
any cause.
Statistical analyses
Statistical analyses were conducted using SPSS
software for Windows, version 12.0 (SPSS, Inc.). Samples were
classified as 'high' or 'low' according to whether LINC00460
expression was higher or lower than the median for all samples.
Survival curves were drawn with the Kaplan-Meier method and
statistical analysis was performed using the log-rank test. The
t-test was used to compare data between 2 groups. One-way ANOVA
with Tukey's post-hoc test was used for comparisons among multiple
groups.
Results
Bioinformatics analysis of RNA-seq and
clinical data
The following method was used to screen candidate
lncRNAs. Compared with the normal tissues, lncRNA expression was
significantly dysregulated in the TCGA database (TCGA-LUAD),
indicating the differential expression of lncRNAs in specimens of
lung adenocarcinoma with EGFR-activating mutations (exon 19
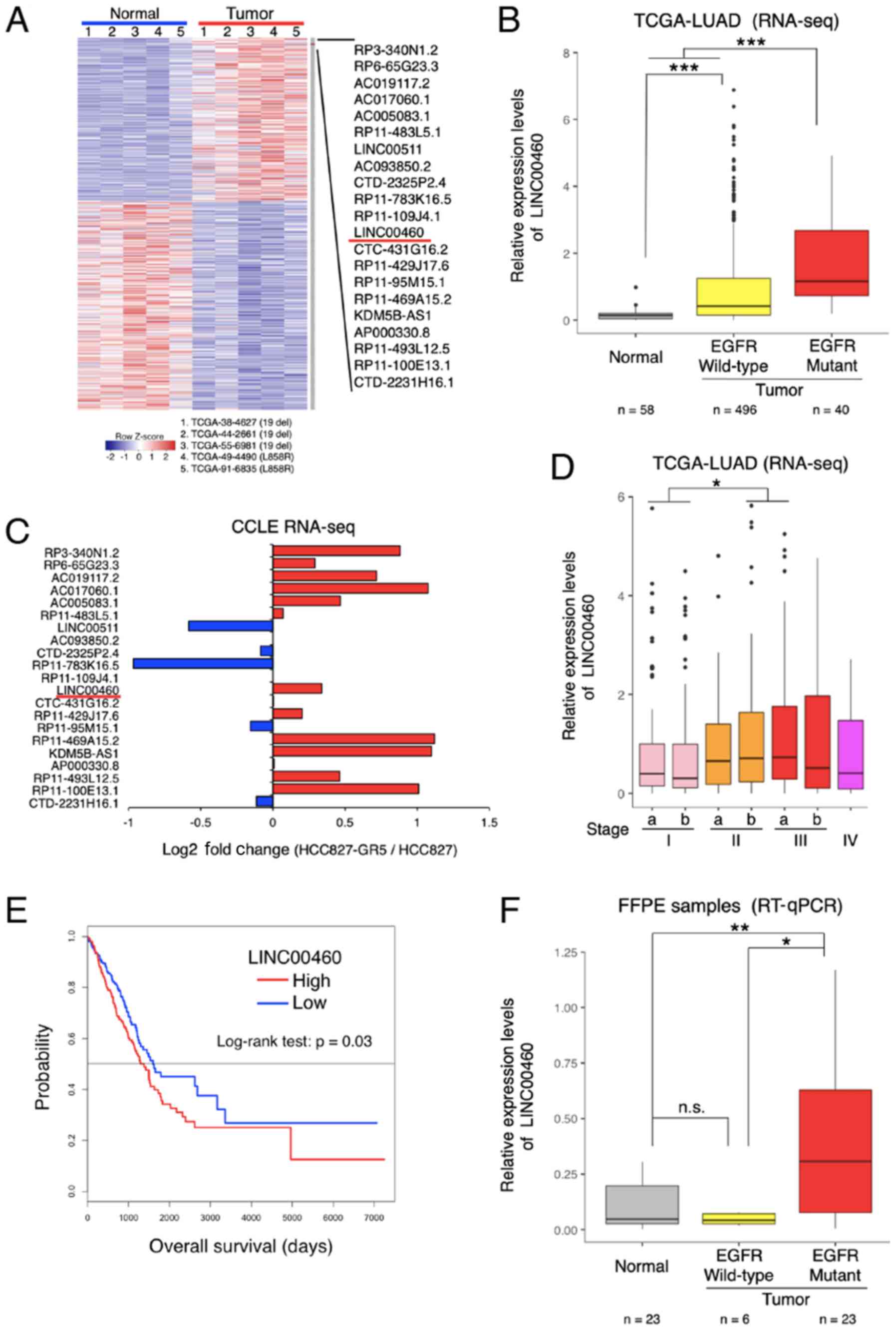
deletion and L858R) (Fig. 1A).
Among the most differentially overexpressed lncRNAs (21 lncRNAs,
P<0.01, Z value >2), lncRNA-LINC00460 was selected for
further analysis (NCBI Reference Sequence: NR_034119). The
LINC00460 expression levels were significantly higher in wild-type
EGFR and in specimens with activation mutations (exon 19 deletion
and L858R) compared with normal tissues (P<0.0001; Fig. 1B). According to the RNA-seq dataset
of the CCLE database, LINC00460 expression was higher in a
gefitinib-resistant NSCLC cell line (HCC827 GR5) compared with a
gefitinib-sensitive cell line (HCC827) (Fig. 1C). Furthermore, the analysis of the
clinical TCGA dataset revealed that LINC00460 expression was
significantly associated with an advanced tumor stage (Fig. 1D) and a poor prognosis (Fig. 1E). Consistent with the results of
TCGA RNA-seq, LINC00460 expression was found to be significantly
higher in lung adenocarcinoma tissues with EGFR-activating
mutations than in normal tissues (P=0.0099) by RT-qPCR assay
(Fig. 1F).
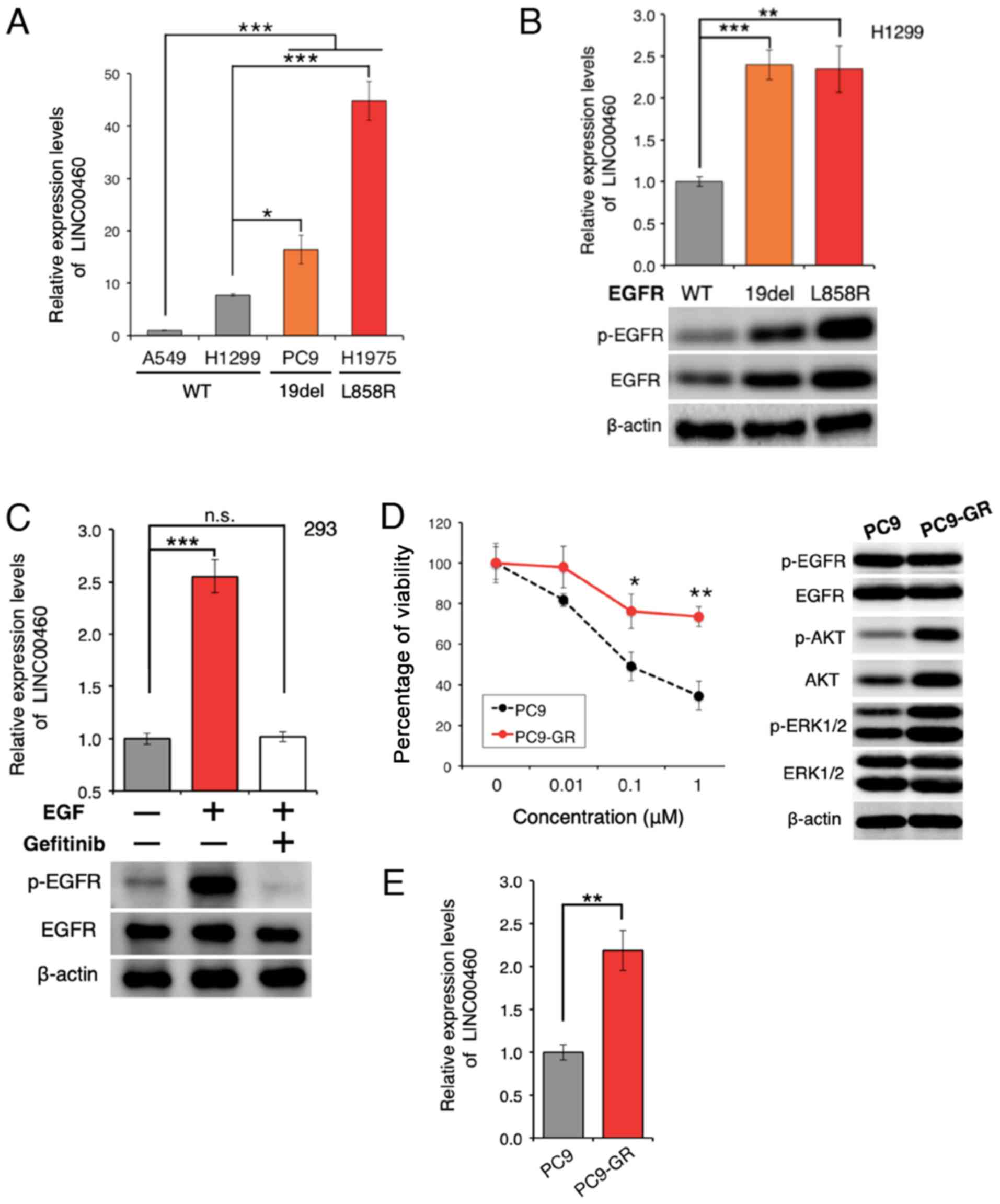
lncRNA expression in cell lines
The expression of LINC00460 was analyzed in 4 lung
cancer cell lines. LINC00460 expression was found to be
significantly higher in lung cancer cell lines with
EGFR-activating mutations than in wild-type cell lines
(Fig. 2A). Furthermore,
transfection with active EGFR mutations resulted in a higher
LINC00460 expression, as compared with the expression in H1299
cells expressing wild-type EGFR (Fig.
2B). EGFR activation induced by treatment with EGF yielded
similar results; however, EGFR inactivation by pre-treatment
with gefitinib significantly attenuated the EGFR-induced increase
in LINC00460 expression (Fig. 2C).
These results suggest that the abnormal activation of EGFR is
associated with the overexpression of LINC00460. To determine
whether LINC00460 is overexpressed in gefitinib-resistant cells,
gefitinib-resistant PC9 cells (PC9-GR cells) were generated
(Fig. 2D). The expression level of
LINC00460 was found to be significantly higher in the PC9-GR cells
than in the PC9 cells (Fig. 2E),
indicating that LINC00460 is involved in resistance against
EGFR-TKI.
Function of LINC00460 as a ceRNA for
miR-149-5p facilitating IL-6 production
To investigate whether LINC00460 transcripts induce
EGFR-TKI resistance, the potential modes of action of LINC00460
were determined in experimental studies. To predict the biological
function of LINC00460 transcripts in EGFR-activating mutated
lung cancer cells, LINC00460 localization patterns were
investigated in PC9 cells. As LINC00460 is mainly localized in the
cytoplasm (Fig. 3A), it was
hypothesized that it functions as a ceRNA and acts as a decoy for
miRNA from the corresponding miRNA-targeted transcripts (43). To examine this hypothesis, we used
bioinformatics analysis (miRDB and TargetScan) to identify
candidate miRNAs that interact with LINC00460. To pull down
endogenous miRNAs associated with LINC00460 in lung cancer cells,
an MS2-based RIP was performed based on the previously described
miTRAP method (44) (Fig. 3B). Subsequently, the
LINC00460-specific enrichment of the candidate miRNAs was validated
by RT-qPCR. miR-149-5p enrichment was found to be significantly
greater in RNAs retrieved from MS2-LINC00460 than in those from
controls (MS2-Luciferase mRNA) and MS2-LINC00460-Mut (MRE-deleted
MS2-LINC00460) (Fig. 3C). However,
we could not pull down other candidate-targeted miRNAs (data not
shown). These data suggest the presence of a specific interaction
between LINC00460 and miR-149-5p in NSCLC cells and demonstrate
that LINC00460 has two putative miRNA response elements (MREs) of
miR-149-5p (Fig. 3B). Of note,
bioinformatics analysis revealed that miR-149-5p binds to the MRE
in the 3′UTR of IL-6 mRNA. However, in lung adenocarcinoma, the
association between LINC00460, miR-149-5p and IL-6 is unknown.
Therefore, subsequent experiments focused on the ceRNA function of
LINC00460 to target miR-149-5p.
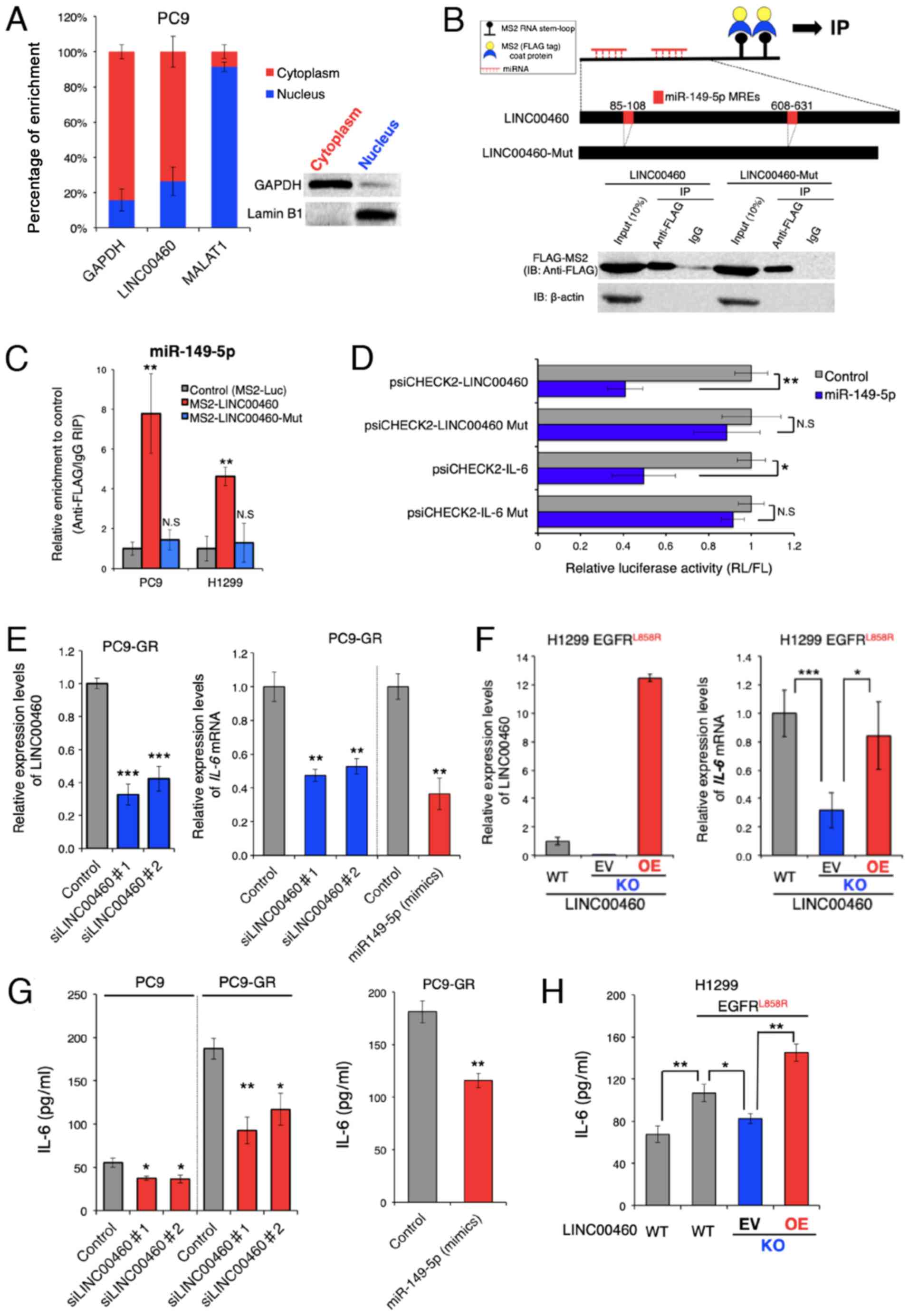 | Figure 3LINC00460 transcripts function as a
ceRNA for miR-149-5p in regulating IL-6 expression. (A) Cell
nucleus/cytoplasm fractionation and RT-qPCR indicating the cellular
distribution of LINC00460 in PC9 cells. GAPDH was used as a
cytoplasm marker. MALAT1 and Lamin B1 were used as nucleus markers.
Data are presented as the means ± SD (n=3). (B) Upper panel:
Schematic of the binding 2X MS2 stem-loop fused bait RNA to MS2
coat proteins used for the RIP assay based on the miTRAP method and
schematic outline of predicted binding sites for miR-149-5p on the
LINC00460 sequence. Lower panel: Western blot analysis of FLAG-tag
fused MS2 proteins isolated from PC9 input fractions or co-purified
with (MS2-)LINC00460 and (MS2-)LINC00460 Mut (deletion mutant of
the MREs), respectively. IgG served as a negative control for
nonspecific pull downs. β-actin was used as an internal control.
(C) MS2-based RIP assay with anti-FLAG antibody in PC9 or H1299
cells. The cells were incubated for 48 h following transfection
with a MS2-FLAG plasmid, along with MS2-LINC00460, MS2-LINC00460
Mut, or MS2bs-Luc (control vectors). The results of RT-qPCR are
presented as the means ± SD (n=3). (D) Relative luciferase activity
of psiCHECK2-LINC00460, LINC00460 Mut, IL-6, and IL-6 Mut upon
transfection with miR-149-5p mimics in H1299 cells.
psiCHECK2-LINC00460 and psiCHECK2-IL-6 are psiCHECK2 vectors that
contain LINC00460 and the 3'UTR of IL-6 mRNA, respectively.
Mut stands for deletion mutant of miR-149-5p MREs. Data are
presented as the ratio of Renilla luciferase activity (RL)
to Firefly luciferase activity (FL). Error bars represent the means
± SD (n=3). (E) Relative expression levels of LINC00460 (left
panel) and IL-6 mRNA (right panel) following transfection
with control (si-Luc), si-LINC00460#1, and si-LINC00460#2. The
results of RT-qPCR are presented as the means ± SD (n=3). (F) Left
panel: Detection of LINC00460 in knockout (KO) clones by RT-qPCR.
H1299 cells expressing EGFR L858R were transfected with the Cas9
plasmid vector for LINC00460 knockout. The KO cells were rescued by
the LINC00460 expression vector. Right panel: Relative expression
of IL-6 mRNA in the engineered H1299 cells. The results of
RT-qPCR are presented as the means ± SD (n=3). (G and H)
Concentrations of IL-6 (pg/ml) in cell culture supernatants were
measured by ELISA. (G) Concentration of IL-6 in PC9 or PC9-GR
following transfection with control (si-Luc), si-LINC00460#1,
si-LINC00460#2 (left), and miR-149-5p mimics (right panel). (H)
Concentration of IL-6 in engineered H1299 cells. WT, wild type; KO,
knockout; EV, empty vector; OE, overexpression. The results of
ELISA are presented as the means ± SD (n=3). The level of
statistical significance was P-value set at <0.05
(*P<0.05, **P<0.01,
***P<0.001). n.s., not significant. |
To further investigate the RNAi effect, the coding
regions of LINC00460 or the 3'UTR of IL-6 were cloned into the
psiCHECK2™ vector, which transcribes the fusion of the reporter
gene (R. luciferase) and the sequence of interest.
Co-transfection of these vectors and a vector expressing miR-149-5p
mimics significantly inhibited the luciferase activity of
psiCHECK2-LINC00460 and psiCHECK2-IL-6, while each of the MRE
deletion mutants (psiCHECK2-LINC00460 Mut or IL-6 Mut) did not
exhibit a significant response to the miR-149-5p mimics (Fig. 3D). These results indicate that
LINC00460 and IL-6 mRNA contained functional miR-149-5p binding
sites.
Subsequently, the effect of LINC00460 expression on
IL-6 production was investigated, as IL-6 mRNA expression is
regulated by miR-149-5p. The expression of IL-6 mRNA was
significantly decreased by the siRNA knockdown of LINC00460
transcripts (Fig. 3E, right
panel). This result was consistent with the outcome of the enforced
expression of miR-149-5p in PC9-GR cells (Fig. 3E). To accurately determine the
potential effect of LINC00460 on the responses to EGFR-TKIs, a
Cas9-mediated LINC00460 knockout cell line was generated from H1299
cells, which harbored EGFR L858R rather than EGFR T790M and
exhibited intrinsic resistance to EGFR-TKIs (48-50)
(Figs. 3F, right panel and S1).
The knockdown of LINC00460 significantly deceased IL-6 mRNA
expression, whereas the overexpression of LINC00460 in LINC00460
knockout cells (performing a rescue experiment) increased IL-6 mRNA
expression (Fig. 3F, left panel).
ELISA revealed that IL-6 expression was attenuated at the protein
level when LINC00460 was knocked down, and when the expression of
miR-149-5p mimics was enforced in PC9-GR cells (Fig. 3G). Although the acquisition of an
EGFR-activating mutation (L858R) by the H1299 cells induced
the significant production of IL-6 and LINC00460 expression,
LINC00460 knockdown decreased the IL-6 protein levels in parental
cells (Figs. 2B and 3H). Conversely, the enforced
overexpression of LINC00460 significantly promoted IL-6 production
in the cells with LINC00460 knockdown (Fig. 3H). Taken together, these findings
suggested that LINC00460 expression, at least in part, promoted
IL-6 production by directly acting as a decoy for miR-149-5p
associated with the degradation of IL-6 mRNA.
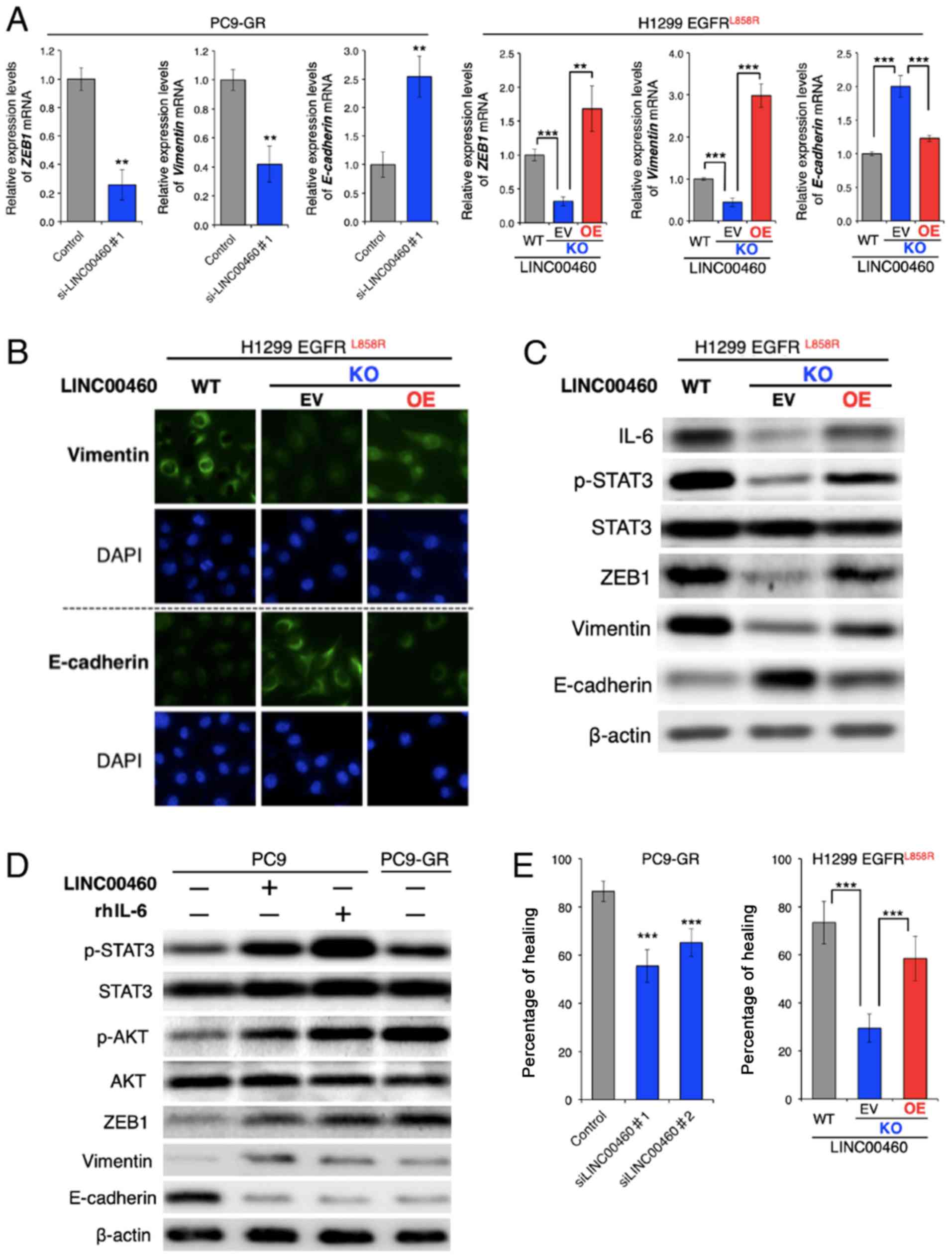
Effects of LINC00460 on phenotypes
associated with EGFR-TKI resistance in EGFR-mutated lung
cancer
The overexpression of IL-6 in tumors triggers
inflammation and primary resistance or a low response to EGFR-TKIs
in preclinical models (22).
Research on the mechanisms involved in EGFR-TKI resistance has
reported that IL-6 induces an EMT phenotype by activating the
JAK/STAT3 or AKT signaling pathway (51), which suggests the loss of
E-cadherin and increased vimentin and ZEB1 expression (21).
Although the parental PC9-GR and H1299 cells had a
mesenchymal phenotype, RT-qPCR analysis of the ZEB1,
vimentin (VIM) and E-cadherin (CDH1) mRNA expression
levels revealed that LINC00460 knockdown significantly decreased
ZEB1 and VIM, and induced CDH1 expression. The
overexpression of LINC00460 exerted the opposite effect on the
expression levels of these EMT-related mRNAs in the cells in which
H1299 LINC00460 had been knocked out (Fig. 4A). In addition, an
immunofluorescence assay revealed that LINC00460 knockdown
increased the E-cadherin and decreased the vimentin protein levels.
Consistently, LINC00460 overexpression exerted the opposite effect
(Fig. 4B). Furthermore, western
blot analysis clearly indicated that LINC00460 knockdown suppressed
the phosphorylation of STAT3 and altered the expression levels of
EMT-related proteins, including ZEB1, vimentin and E-cadherin, and
that these changes were reversed by LINC00460 overexpression
(Fig. 4C). In PC9 cells, which are
an epithelial phenotype, transfection with LINC00460 and treatment
with human recombinant IL-6 (rhIL-6) induced a similar expression
pattern of EMT-related genes to that of gefitinib-resistant PC9-GR
cells. These stimuli activated the STAT3 and AKT pathways, promoted
the expression levels of ZEB1 and vimentin, and inhibited
E-cadherin expression (Fig. 4D).
In the context of EMT, silencing LINC00460 expression inhibited the
migration of PC9-GR cells. The migration of the cells subjected to
LINC00460 knockout was significantly lower than of the parental
H1299 cells. Moreover, cells subjected to LINC00460 knockout and
subsequently to the overexpression of LINC00460 exhibited a
phenotype similar to the original (Fig. 4E). Taken together, these results
indicate that LINC00460 overexpression promoted an EMT-like
phenotype by upregulating IL-6 production.
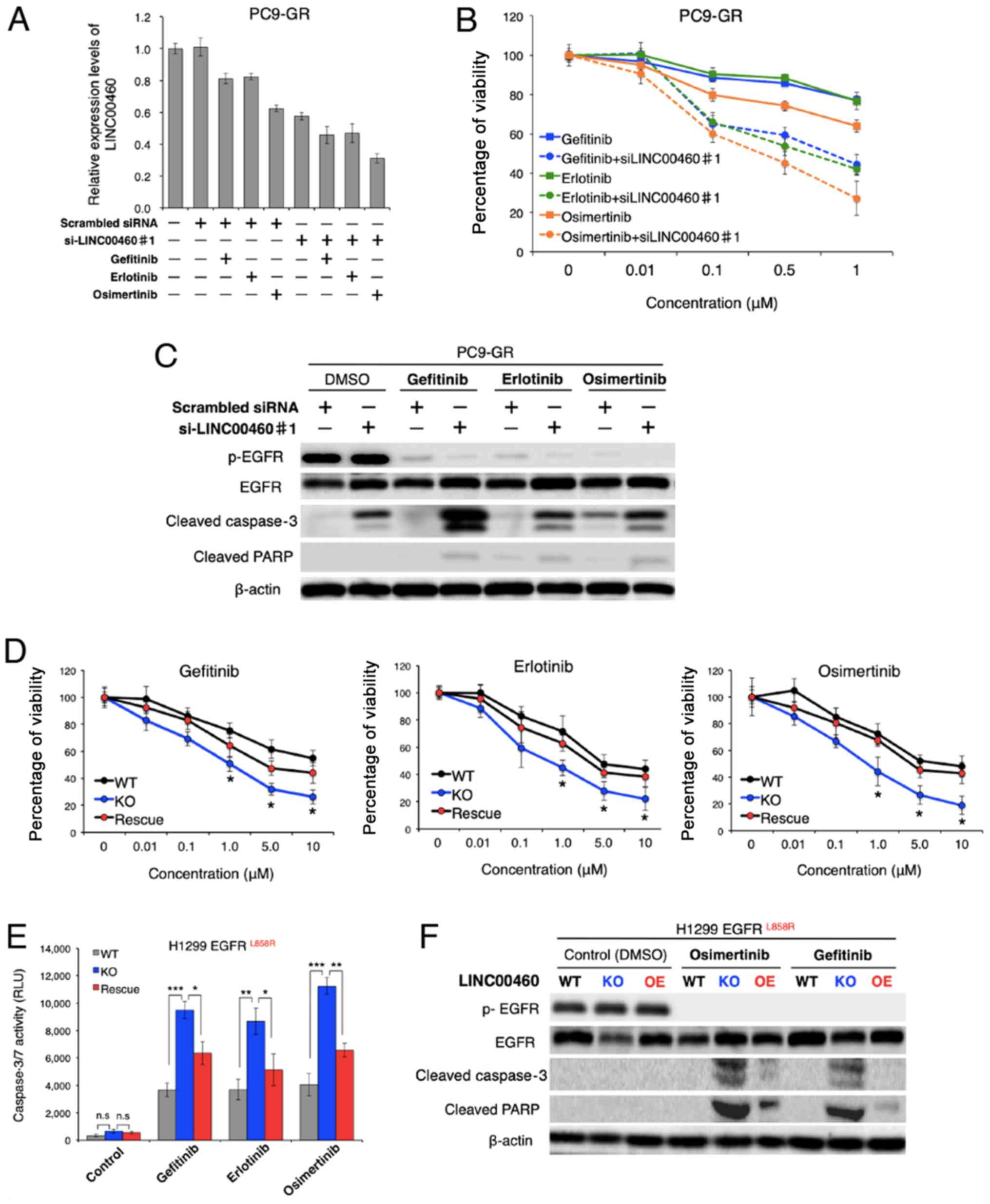
Effect of LINC00460 on responses to
EGFR-TKIs
To evaluate the therapeutic potential of LINC00460
for gefitinib-resistant NSCLC, the issue of whether LINC00460
silencing restores responses to EGFR-TKIs (gefitinib, erlotinib and
osimertinib) was investigated in NSCLC harboring activating
EGFR mutations in vitro. Of note, while treatment
with each EGFR-TKIs alone exerted a minimal inhibitory effect on
the viability of PC9-GR cells, the combined application of single
EGFR-TKIs and LINC00460 siRNA markedly decreased the LINC00460
expression level and cell viability of PC9-GR (Fig. 5A and B). Moreover, the results of
western blot analysis revealed that all combinations of a single
EGFR-TKI and LINC00460 siRNA promoted the cleavage of caspase-3 and
PARP in PC9-GR cells, indicating that apoptosis was induced in
these cells (Fig. 5C).
Subsequently, the question of whether the expression levels of
LINC00460 are associated with cell viability in response to
EGFR-TKIs in NSCLC harboring EGFR-activating mutations
without T790M was investigated. As shown in Fig. 5D, viability following treatment
with various EGFR-TKIs was significantly decreased by LINC00460
knockout in H1299 cells harboring L858R. Conversely, the
overexpression of LINC00460 in the cells which had been subjected
to LINC00460 knockout cells increased the number of surviving cells
in all experiments. In addition, caspase 3/7 activity in these
cells following treatment with EGFR-TKIs was investigated.
Caspase-Glo 3/7 assay and western blot analysis indicated that the
effects of LINC00460 expression on cell viability were, at a
minimum, involved in significantly inducing or inhibiting the
apoptotic pathway (Fig. 5E and F).
Taken together, these findings suggested that the combined use of
siRNA targeting LINC00460 and EGFR-TKIs induced the apoptosis of
EGFR-mutated, LINC00460-overexpressing, NSCLC cells
resistant to EGFR-TKI monotherapy.
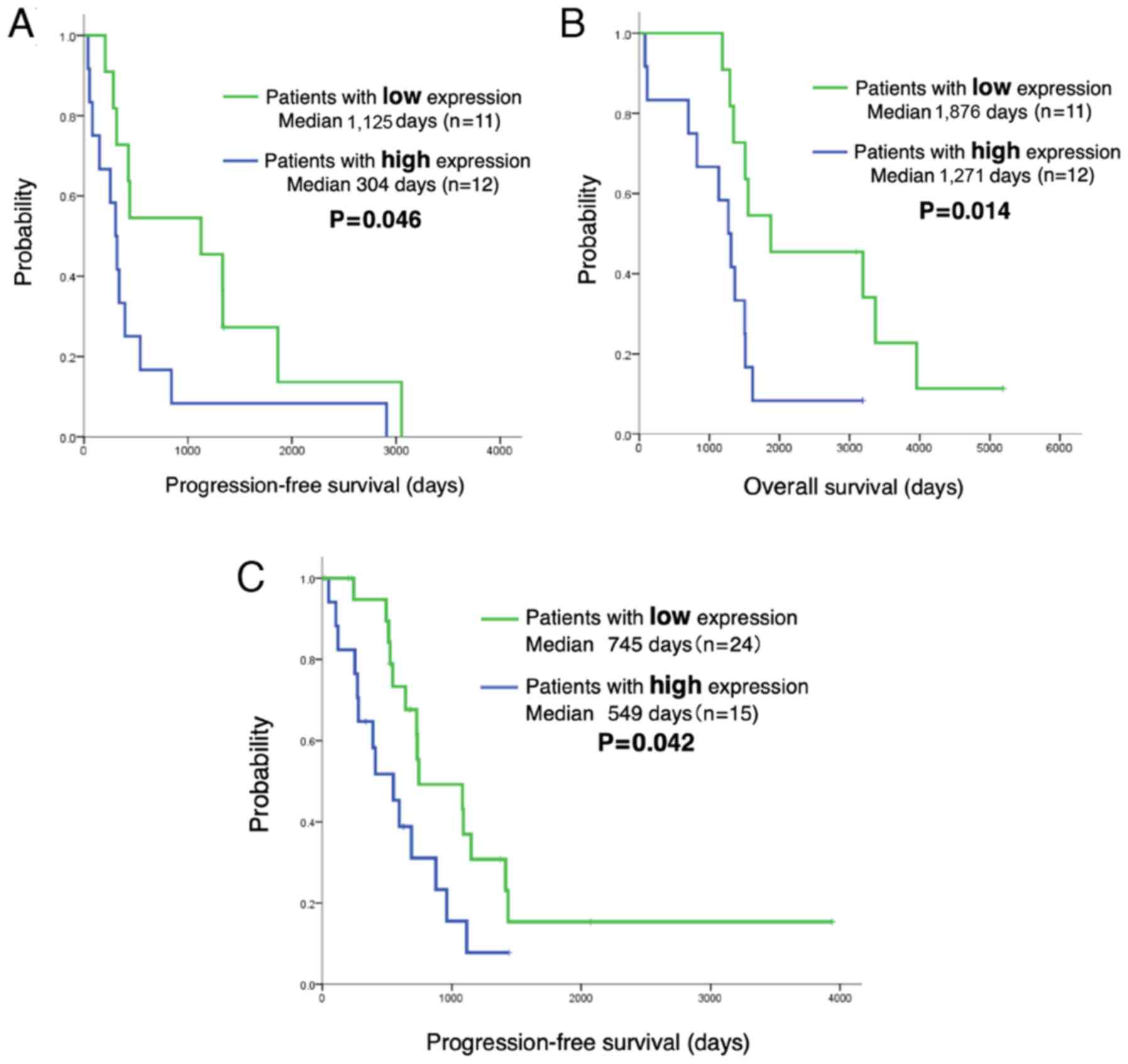
LINC00460 expression predicts a shorter
PFS and OS of patients with EGFR-mutant NSCLC treated with
EGFR-TKIs
The analysis of the LINC00460 status in 62 patients
with EGFR-mutant lung adenocarcinoma treated with EGFR-TKI
revealed no significant differences in the clinical characteristics
(age, sex, performance status, smoking history, exon 19 deletion or
L858R, EGFR-TKI and metastatic site) between those with a high
LINC00460 expression (n=27) and those with a low LINC00460
expression (n=35). The PFS and OS of patients in relation to
LINC00460 expression was then estimated. Notably, Kaplan-Meier
survival analysis indicated that patients with a high LINC00460
expression in their tumors (n=12) had a significantly shorter PFS
and OS following gefitinib therapy than those with a low expression
(n=11) (median PFS: 304 vs. 1,125 days, P=0.046, respectively; OS:
1,271 vs. 1,876 days, P=0.014; Fig. 6A
and B). Moreover, PFS was significantly shorter for patients
with a high LINC00460 expression in cell-free RNA (n=15) following
EGFR-TKI therapy than for those with a low LINC00460 expression
(n=24) (median PFS: 549 vs. 745 days, P=0.042, respectively)
(Fig. 6C).
Discussion
The present study on the role of lncRNAs in lung
adenocarcinoma with EGFR mutation revealed that LINC00460
overexpression in lung cancer cells was caused by the abnormal
activation of EGFR signaling attributable to the excessive
stimulation of EGF and acquisition of a cancer driver mutation in
EGFR. EGFR expression further increased when these
cells acquired gefitinib resistance. The assessment of the
biological significance of LINC00460 in lung cancer cells suggested
that this RNA functioned as a competing endogenous RNA decoy for
the tumor suppressive miRNA-miR-149-5p, which then significantly
promoted IL-6 production and EMT-like traits. In addition, with
respect to the novel clinical importance of LINC00460 in
EGFR mutation-positive lung cancer, it was found that the
silencing LINC00460 in combination with treatment with EGFR-TKIs
(gefitinib, erlotinib and osimertinib) exerted a more potent
inhibitory effect on the viability of EGFR-mutant NSCLC
cells. These findings suggest that a high LINC00460 expression
renders EGFR-TKI treatment ineffective and may prevent the
development of intrinsic resistance to EGFR-TKIs and the emergence
of drug-tolerant cells in EGFR-mutated lung cancers that
overexpress LINC00460.
LINC00460 has recently been reported to be an
oncogenic lncRNA in types of various cancer (29). These findings suggest that
LINC00460 is overexpressed in various types of cancer, including
nasopharyngeal, colorectal, ovarian and lung cancers, and that
LINC00460 expression promotes cell growth and migration and
increases tumor volume and weight in vivo (52-55).
These studies also indicate that the binding of LINC00460 to
various miRNAs is the molecular mechanism through which LINC00460
affects cell function.
Subsequently, this study whether LINC00460
overexpression disrupts the miRNA regulatory machinery, which may
affect EGFR-TKI resistance in lung cancer cells and worsens the
outcomes of lung cancer patients. It was found that LINC00460, the
oncogenic function of which was attributed to the inhibition of
miR-149-5p, promoted its target gene, IL-6, in EGFR-mutated
lung cancer with limited responsiveness to EGFR-TKIs. The tumor
suppressor, miR-149-5p, has been shown to inhibit fibroblast
activation by reducing IL-6 expression (56). In addition, cancer-associated
fibroblasts enhance EMT and the stem-like traits of human gastric
cancer cells by inducing IL-6 expression by means of an miR-149-5p
dependent mechanism (56).
Moreover, a previous study on nasopharyngeal cancer indicated that
LINC00460 facilitated carcinogenesis by acting as a decoy for
miR-149-5p to upregulate IL-6 expression (52). The discovery of the function of
this LINC00460 supports the current findings regarding
EGFR-mutated lung cancer. IL-6 is a cytokine associated with
a number of chronic inflammatory diseases. Furthermore, IL-6
expression is involved in the regulation of tumor growth and
metastatic spread, including that of lung cancers. Activated mutant
EGFRs activates the gp130/JAK/STAT3 pathway by upregulating IL-6 in
primary human lung adenocarcinoma cells (57). The IL-6-inducible activation of
JAK1/STAT3 signaling also induces de novo resistance to
EGFR-TKIs in NSCLC with a T790M mutation (58). These results do not contradict the
current findings that LINC00460 overexpression activates STAT3 by
promoting IL-6 production in EGFR-activating mutant lung
cancer cells with EGFR-TKI resistance (Fig. 4C and D).
In addition, the acquisition of mesenchymal-like
traits by the activation of IL-6 inducible STAT3 and AKT signaling
contributes to EGFR-TKI resistance (59). EMT also functions as a potential
mechanism of EGFR-TKI resistance in NSCLC (18,23).
This property has also been observed in a subset of NSCLC patients
who developed EGFR-TKI resistance (59). EMT was originally reported to be an
essential process in the developmental stage and cell migration
during early embryogenesis. When EMT occurs in cancer cells, they
lose their epithelial properties and gain mesenchymal
characteristics. The epithelial cell marker, E-cadherin, is a cell
adhesion molecule that suppresses cancer cell motility, whereas the
mesenchymal cell markers, VIM and ZEB1 enhance cancer cell
migration and invasion (23).
Consistent with these results, a previous study indicated that
LINC00460 promoted EMT, as well as cell migration and invasion
(53). Taken together, these
previous findings support those of the current study, indicating
that the upregulation of LINC00460 is involved in the mechanism of
cancer malignancy and EGFR-TKI resistance induced by IL-6-inducible
EMT. However, the effects of LINC000460 on cell phenotypes and drug
sensitivity suggest that LINC00460 interacts with various other
miRNAs and proteins to control cell signaling, in addition to the
molecular mechanism described in the present study. Furthermore,
LINC00460 may code for a small peptide under certain conditions
(60), although we were unable to
identify this peptide in the present study (data not shown).
Therefore, further studies of the molecular mechanisms underlying
LINC00460 efficacy are warranted.
A high LINC00460 expression has recently been
reported to be associated with a poor prognosis in certain types of
cancer, including nasopharyngeal carcinoma (52) and colorectal carcinoma (55). A similar association was observed
in the present analysis of EGFR-mutated lung adenocarcinoma.
In this study, patients with a high LINC00460 expression in tumors
had a significantly shorter PFS following gefitinib therapy and OS
compared with those with a low LINC00460 expression. A recent
clinical study on patients harboring EGFR-mutant lung cancer
suggested that patients with high IL-6 levels in tumors had a
significantly shorter PFS following gefitinib therapy (61), which is in accordance with the
current findings that LINC00460 regulated IL-6 levels in lung
cancer. Moreover, it was found that the sensitivity to EGFR-TKIs
(gefitinib, erlotinib and osimertinib) was reduced by an increased
LINC00460 expression. Osimertinib is a third-generation EGFR-TKI
and is approved for the treatment of EGFR-T790M-positive NSCLC in
patients with acquired resistance to first- or second-generation
EGFR-TKIs. Currently, first-line treatment with osimertinib is
considered a standard therapy for patients with EGFR-mutant NSCLC.
However, resistance to osimertinib in these patients is similar to
that observed with other EGFR-TKIs. The current study deepens our
understanding of the molecular mechanisms underlying resistance to
osimertinib in EGFR-mutated NSCLC. It is expected that the
LINC00460 expression level may be a novel predictor of the effects
of EGFR-TKI and a prognostic indicator of EGFR mutation-positive
NSCLC.
This study had some limitations. First, it was a
retrospective single-center study with a small sample size.
Although differences in clinical outcome in relation to LINC00460
expression were identified, the number of patients enrolled was too
small to consider the association of LINC00460 expression with the
PFS and OS. Thus, a large-scale multicenter study is required to
confirm the validity of these results.
In conclusion, the findings of this study suggest
that LINC00460 overexpression is associated with a poor response to
the latest EGFR-TKI (i.e., osimertinib), as well as to
first-generation EGFR-TKIs (i.e., gefitinib and erlotinib), and
with poor outcomes following gefitinib treatment. A novel
therapeutic strategy targeting LINC00460 may overcome EGFR-TKI
resistance and improve the prognosis of patients with
EGFR-mutant lung adenocarcinoma. Advances in the preclinical
and clinical development of RNAi drugs and their drug delivery
systems, and in technologies for RNA detection, have increased the
sophistication of drug and diagnostic development processes
(62,63). The discovery of the importance of
LINC00460 may lead to its use as a prognostic predictor, diagnostic
indicator of EGFR-TKIs, and as a molecular target of
pharmaceuticals. Moreover, the present findings may lead to
development of individualized treatments for lung cancer.
Therefore, the clinical importance of research on LINC00460 is
likely to increase.
Supplementary Data
Funding
This study was supported by JSPS KAKENHI (grant nos.
JP15K09195 and JP18K08189).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article or are available from the
corresponding author on reasonable request.
Authors' contributions
KI, YN, TM and SH conceived and designed the study.
KKa, HK, TI and YN performed the experiments. KI and YN wrote the
manuscript. SS, YT, NT, AI, SH and KKi were involved in the
conception and design of the study and reviewed and edited the
manuscript. All authors have read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
This single-center study was conducted at the Toho
University Omori Medical Center and was approved by the Human
Genome/Gene Analysis Research Ethics Committee (autho-rization no.
A17117). Written informed consent was obtained from all patients
prior to participation.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Mr. Atsushi Kakimoto
of Konica Minolta Inc., as well as Mr. Yuichiro Kanno and Mr.
Tomohiro Ariyama, of the Faculty of Pharmaceutical Sciences, Toho
University for providing assistance with this study. The authors
would also like to thank Mr. David Kipler for his editorial review
of the article. The abstract for this article was presented at the
2019 Annual Meeting of the American Society of Clinical Oncology
(May, 31 to June 4, 2019) in Chicago, IL, USA and was published as
abstract no. e20529 in the Journal of Clinical Oncology 37,
2019.
Glossary
Abbreviations:
|
lncRNAs
|
long non-coding RNAs
|
|
NSCLC
|
non-small cell lung cancer
|
|
EGFR
|
epidermal growth factor receptor
|
|
EGFR-TKI
|
epidermal growth factor
receptor-tyrosine kinase inhibitor
|
|
IL-6
|
interleukin-6
|
|
PFS
|
progression-free survival
|
|
OS
|
overall survival
|
|
PCR
|
polymerase chain reaction
|
|
HGF
|
hepatocyte growth factor
|
|
EMT
|
epithelial-mesenchymal transition
|
|
TCGA
|
The Cancer Genome Atlas
|
|
GDC
|
Genomic Data Commons
|
|
CCLE
|
Cancer Cell Line Encyclopedia
|
|
RECIST
|
response evaluation criteria in solid
tumors
|
|
EGF
|
epidermal growth factor
|
|
KO
|
knockout
|
|
OE
|
overexpression
|
References
|
1
|
Hoffman PC, Mauer AM and Vokes EE: Lung
cancer. Lancet. 355:479–485. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Paez JG, Jänne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et
al: EGFR mutations in lung cancer: Correlation with clinical
response to gefitinib therapy. Science. 304:1497–1500. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, et al: Activating mutations in the
epidermal growth factor receptor underlying responsiveness of
non-small-cell lung cancer to gefitinib. N Engl J Med.
350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maemondo M, Inoue A, Kobayashi K, Sugawara
S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I,
et al North-East Japan Study Group: Gefitinib or chemotherapy for
non-small-cell lung cancer with mutated EGFR. N Engl J Med.
362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rosell R, Carcereny E, Gervais R,
Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R,
Pallares C, Sanchez JM, et al: Spanish Lung Cancer Group in
collaboration with Groupe Français de Pneumo-Cancérologie and
Associazione Italiana Oncologia Toracica: Erlotinib versus standard
chemotherapy as first-line treatment for European patients with
advanced EGFR mutation-positive non-small-cell lung cancer
(EURTAC): A multicentre, open-label, randomised phase 3 trial.
Lancet Oncol. 13:239–246. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang JC, Wu YL, Schuler M, Sebastian M,
Popat S, Yamamoto N, Zhou C, Hu CP, O'Byrne K, Feng J, et al:
Afatinib versus cisplatin-based chemotherapy for EGFR
mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6):
Analysis of overall survival data from two randomised, phase 3
trials. Lancet Oncol. 16:141–151. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mok TS, Wu YL, Ahn MJ, Garassino MC, Kim
HR, Ramalingam SS, Shepherd FA, He Y, Akamatsu H, Theelen WS, et al
AURA3 Investigators: Osimertinib or Platinum-Pemetrexed in EGFR
T790M-Positive Lung Cancer. N Engl J Med. 376:629–640. 2017.
View Article : Google Scholar
|
|
8
|
Mitsudomi T, Morita S, Yatabe Y, Negoro S,
Okamoto I, Tsurutani J, Seto T, Satouchi M, Tada H, Hirashima T, et
al West Japan Oncology Group: Gefitinib versus cisplatin plus
docetaxel in patients with non-small-cell lung cancer harbouring
mutations of the epidermal growth factor receptor (WJTOG3405): An
open label, randomised phase 3 trial. Lancet Oncol. 11:121–128.
2010. View Article : Google Scholar
|
|
9
|
Zhou C, Wu YL, Chen G, Feng J, Liu XQ,
Wang C, Zhang S, Wang J, Zhou S, Ren S, et al: Erlotinib versus
chemotherapy as first-line treatment for patients with advanced
EGFR mutation-positive non-small-cell lung cancer (OPTIMAL,
CTONG-0802): A multicentre, open-label, randomised, phase 3 study.
Lancet Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kuan FC, Kuo LT, Chen MC, Yang CT, Shi CS,
Teng D and Lee KD: Overall survival benefits of first-line EGFR
tyrosine kinase inhibitors in EGFR-mutated non-small-cell lung
cancers: A systematic review and meta-analysis. Br J Cancer.
113:1519–1528. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Soria JC, Ohe Y, Vansteenkiste J,
Reungwetwattana T, Chewaskulyong B, Lee KH, Dechaphunkul A, Imamura
F, Nogami N, Kurata T, et al FLAURA Investigators: Osimertinib in
Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl
J Med. 378:113–125. 2018. View Article : Google Scholar
|
|
12
|
Kobayashi S, Boggon TJ, Dayaram T, Jänne
PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG and Halmos
B: EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tong JH, Yeung SF, Chan AW, Chung LY, Chau
SL, Lung RW, Tong CY, Chow C, Tin EK, Yu YH, et al: MET
Amplification and Exon 14 Splice Site Mutation Define Unique
Molecular Subgroups of Non-Small Cell Lung Carcinoma with Poor
Prognosis. Clin Cancer Res. 22:3048–3056. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yano S, Yamada T, Takeuchi S, Tachibana K,
Minami Y, Yatabe Y, Mitsudomi T, Tanaka H, Kimura T, Kudoh S, et
al: Hepatocyte growth factor expression in EGFR mutant lung cancer
with intrinsic and acquired resistance to tyrosine kinase
inhibitors in a Japanese cohort. J Thorac Oncol. 6:2011–2017. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Marcoux N, Gettinger SN, O'Kane G, Arbour
KC, Neal JW, Husain H, Evans TL, Brahmer JR, Muzikansky A, Bonomi
PD, et al: EGFR-Mutant Adenocarcinomas That Transform to Small-Cell
Lung Cancer and Other Neuroendocrine Carcinomas: Clinical Outcomes.
J Clin Oncol. 37:278–285. 2019. View Article : Google Scholar
|
|
17
|
Suda K, Tomizawa K, Fujii M, Murakami H,
Osada H, Maehara Y, Yatabe Y, Sekido Y and Mitsudomi T: Epithelial
to mesenchymal transition in an epidermal growth factor
receptor-mutant lung cancer cell line with acquired resistance to
erlotinib. J Thorac Oncol. 6:1152–1161. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Weng CH, Chen LY, Lin YC, Shih JY, Lin YC,
Tseng RY, Chiu AC, Yeh YH, Liu C, Lin YT, et al:
Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per
se is a common mechanism for acquired resistance to EGFR TKI.
Oncogene. 38:455–468. 2019. View Article : Google Scholar
|
|
19
|
Chung JH, Rho JK, Xu X, Lee JS, Yoon HI,
Lee CT, Choi YJ, Kim HR, Kim CH and Lee JC: Clinical and molecular
evidences of epithelial to mesenchymal transition in acquired
resistance to EGFR-TKIs. Lung Cancer. 73:176–182. 2011. View Article : Google Scholar
|
|
20
|
Li L, Gu X, Yue J, Zhao Q, Lv D, Chen H
and Xu L: Acquisition of EGFR TKI resistance and EMT phenotype is
linked with activation of IGF1R/NF-κB pathway in EGFR-mutant NSCLC.
Oncotarget. 8:92240–92253. 2017.PubMed/NCBI
|
|
21
|
Yao Z, Fenoglio S, Gao DC, Camiolo M,
Stiles B, Lindsted T, Schlederer M, Johns C, Altorki N, Mittal V,
et al: TGF-β IL-6 axis mediates selective and adaptive mechanisms
of resistance to molecular targeted therapy in lung cancer. Proc
Natl Acad Sci USA. 107:15535–15540. 2010. View Article : Google Scholar
|
|
22
|
Shibue T and Weinberg RA: EMT, CSCs, and
drug resistance: The mechanistic link and clinical implications.
Nat Rev Clin Oncol. 14:611–629. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Djebali S, Davis CA, Merkel A, Dobin A,
Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F,
et al: Landscape of transcription in human cells. Nature.
489:101–108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kuramochi-Miyagawa S, Watanabe T, Gotoh K,
Takamatsu K, Chuma S, Kojima-Kita K, Shiromoto Y, Asada N, Toyoda
A, Fujiyama A, et al: MVH in piRNA processing and gene silencing of
retrotransposons. Genes Dev. 24:887–892. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Santosh B, Varshney A and Yadava PK:
Non-coding RNAs: Biological functions and applications. Cell
Biochem Funct. 33:14–22. 2015. View Article : Google Scholar
|
|
27
|
Schmitt AM and Chang HY: Long Noncoding
RNAs in Cancer Pathways. Cancer Cell. 29:452–463. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yarmishyn AA and Kurochkin IV: Long
noncoding RNAs: A potential novel class of cancer biomarkers. Front
Genet. 6:1452015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tao H, Yang JJ, Zhou X, Deng ZY, Shi KH
and Li J: Emerging role of long noncoding RNAs in lung cancer:
Current status and future prospects. Respir Med. 110:12–19. 2016.
View Article : Google Scholar
|
|
30
|
Wang Q, Cheng N, Li X, Pan H, Li C, Ren S,
Su C, Cai W, Zhao C, Zhang L, et al: Correlation of long non-coding
RNA H19 expression with cisplatin-resistance and clinical outcome
in lung adenocarcinoma. Oncotarget. 8:2558–2567. 2017.
|
|
31
|
Wang B, Jiang H, Wang L, Chen X, Wu K,
Zhang S, Ma S and Xia B: Increased MIR31HG lncRNA expression
increases gefitinib resistance in non-small cell lung cancer cell
lines through the EGFR/PI3K/AKT signaling pathway. Oncol Lett.
13:3494–3500. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cheng N, Cai W, Ren S, Li X, Wang Q, Pan
H, Zhao M, Li J, Zhang Y, Zhao C, et al: Long non-coding RNA UCA1
induces non-T790M acquired resistance to EGFR-TKIs by activating
the AKT/mTOR pathway in EGFR-mutant non-small cell lung cancer.
Oncotarget. 6:23582–23593. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen LL: Linking Long Noncoding RNA
Localization and Function. Trends Biochem Sci. 41:761–772. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Quinn JJ and Chang HY: Unique features of
long non-coding RNA biogenesis and function. Nat Rev Genet.
17:47–62. 2016. View Article : Google Scholar
|
|
35
|
Goff LA and Rinn JL: Linking RNA biology
to lncRNAs. Genome Res. 25:1456–1465. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang KC, Yang YW, Liu B, Sanyal A,
Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta
RA, et al: A long noncoding RNA maintains active chromatin to
coordinate homeotic gene expression. Nature. 472:120–124. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Flynn RA and Chang HY: Long noncoding RNAs
in cell-fate programming and reprogramming. Cell Stem Cell.
14:752–761. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pandey RR, Mondal T, Mohammad F, Enroth S,
Redrup L, Komorowski J, Nagano T, Mancini-Dinardo D and Kanduri C:
Kcnq1ot1 antisense noncoding RNA mediates lineage-specific
transcriptional silencing through chromatin-level regulation. Mol
Cell. 32:232–246. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang L, Froberg JE and Lee JT: Long
noncoding RNAs: Fresh perspectives into the RNA world. Trends
Biochem Sci. 39:35–43. 2014. View Article : Google Scholar :
|
|
40
|
Good MC, Zalatan JG and Lim WA: Scaffold
proteins: Hubs for controlling the flow of cellular information.
Science. 332:680–686. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hung T and Chang HY: Long noncoding RNA in
genome regulation: Prospects and mechanisms. RNA Biol. 7:582–585.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kallen AN, Zhou XB, Xu J, Qiao C, Ma J,
Yan L, Lu L, Liu C, Yi JS, Zhang H, et al: The imprinted H19 lncRNA
antagonizes let-7 microRNAs. Mol Cell. 52:101–112. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu B, Sun L, Liu Q, Gong C, Yao Y, Lv X,
Lin L, Yao H, Su F, Li D, et al: A cytoplasmic NF-κB interacting
long noncoding RNA blocks IκB phosphorylation and suppresses breast
cancer metastasis. Cancer Cell. 27:370–381. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tay Y, Rinn J and Pandolfi PP: The
multilayered complexity of ceRNA crosstalk and competition. Nature.
505:344–352. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu J, Lichtenberg T, Hoadley KA, et al:
An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive
High-Quality Survival Outcome Analytics. Cell. 173:400–416.e411.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Barretina J, Caponigro G, Stransky N,
Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV,
Sonkin D, et al: The Cancer Cell Line Encyclopedia enables
predictive modelling of anticancer drug sensitivity. Nature.
483:603–607. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Livak KJ and Schmittgen TD: Analysis of
Relative Gene Expression Data Using Real-Time Quantitative PCR and
the 2(-Delta Delta C(T)). Method Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
48
|
Ran FA, Hsu PD, Wright J, Agarwala V,
Scott DA and Zhang F: Genome engineering using the CRISPR-Cas9
system. Nat Protoc. 8:2281–2308. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Aparicio-Prat E, Arnan C, Sala I, Bosch N,
Guigó R and Johnson R: DECKO: Single-oligo, dual-CRISPR deletion of
genomic elements including long non-coding RNAs. BMC Genomics.
16:8462015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chen YR, Fu YN, Lin CH, Yang ST, Hu SF,
Chen YT, Tsai SF and Huang SF: Distinctive activation patterns in
constitutively active and gefitinib-sensitive EGFR mutants.
Oncogene. 25:1205–1215. 2006. View Article : Google Scholar
|
|
51
|
Yadav A, Kumar B, Datta J, Teknos TN and
Kumar P: IL-6 promotes head and neck tumor metastasis by inducing
epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling
pathway. Mol Cancer Res. 9:1658–1667. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kong YG, Cui M, Chen SM, Xu Y, Xu Y and
Tao ZZ: LncRNA-LINC00460 facilitates nasopharyngeal carcinoma
tumorigenesis through sponging miR-149-5p to up-regulate IL6. Gene.
639:77–84. 2018. View Article : Google Scholar
|
|
53
|
Li K, Sun D, Gou Q, Ke X, Gong Y, Zuo Y,
Zhou JK, Guo C, Xia Z, Liu L, et al: Long non-coding RNA linc00460
promotes epithelial-mesenchymal transition and cell migration in
lung cancer cells. Cancer Lett. 420:80–90. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lian Y, Yan C, Xu H, Yang J, Yu Y, Zhou J,
Shi Y, Ren J, Ji G and Wang K: A Novel lncRNA, LINC00460, Affects
Cell Proliferation and Apoptosis by Regulating KLF2 and CUL4A
Expression in Colorectal Cancer. Mol Ther Nucleic Acids.
12:684–697. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Liu X, Wen J, Wang H and Wang Y: Long
non-coding RNA LINC00460 promotes epithelial ovarian cancer
progression by regulating microRNA-338-3p. Biomed Pharmacother.
108:1022–1028. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Li P, Shan JX, Chen XH, Zhang D, Su LP,
Huang XY, Yu BQ, Zhi QM, Li CL, Wang YQ, et al: Epigenetic
silencing of microRNA-149 in cancer-associated fibroblasts mediates
prostaglandin E2/interleukin-6 signaling in the tumor
microenvironment. Cell Res. 25:588–603. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Gao SP, Mark KG, Leslie K, Pao W, Motoi N,
Gerald WL, Travis WD, Bornmann W, Veach D, Clarkson B, et al:
Mutations in the EGFR kinase domain mediate STAT3 activation via
IL-6 production in human lung adenocarcinomas. J Clin Invest.
117:3846–3856. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kim SM, Kwon OJ, Hong YK, Kim JH, Solca F,
Ha SJ, Soo RA, Christensen JG, Lee JH and Cho BC: Activation of
IL-6R/JAK1/STAT3 signaling induces de novo resistance to
irreversible EGFR inhibitors in non-small cell lung cancer with
T790M resistance mutation. Mol Cancer Ther. 11:2254–2264. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Sequist LV, Waltman BA, Dias-Santagata D,
Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger
S, Cosper AK, et al: Genotypic and histological evolution of lung
cancers acquiring resistance to EGFR inhibitors. Sci Transl Med.
3:75ra262011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Matsumoto A, Pasut A, Matsumoto M,
Yamashita R, Fung J, Monteleone E, Saghatelian A, Nakayama KI,
Clohessy JG and Pandolfi PP: mTORC1 and muscle regeneration are
regulated by the LINC00961-encoded SPAR polypeptide. Nature.
541:228–232. 2017. View Article : Google Scholar
|
|
61
|
Tamura T, Kato Y, Ohashi K, Ninomiya K,
Makimoto G, Gotoda H, Kubo T, Ichihara E, Tanaka T, Ichimura K, et
al: Potential influence of interleukin-6 on the therapeutic effect
of gefitinib in patients with advanced non-small cell lung cancer
harbouring EGFR mutations. Biochem Biophys Res Commun. 495:360–367.
2018. View Article : Google Scholar
|
|
62
|
Matsui M and Corey DR: Non-coding RNAs as
drug targets. Nat Rev Drug Discov. 16:167–179. 2017. View Article : Google Scholar
|
|
63
|
Setten RL, Rossi JJ and Han SP: The
current state and future directions of RNAi-based therapeutics. Nat
Rev Drug Discov. 18:421–446. 2019. View Article : Google Scholar : PubMed/NCBI
|