Introduction
Although China in part of the low prostate cancer
(PCa) incidence area, the incidence of PCa increased by 4.7%
annually between 2005 and 2011, and the mortality increased by 5.5%
annually between 2000 and 2011, making it a burden on the
healthcare system (1). The early
stages of PCa are typically androgen receptor (AR)-dependent, and
the disease can be controlled by active monitoring, androgen
deprivation/hormone therapy (ADT) and radical prostatectomy.
However, ADT causes weight gain, lean muscle loss, diabetes and
osteopenia (2,3). ADT has also been demonstrated to
induce apoptosis resistance and may lead to the development of
AR-independent PCa (4).
Non-hormonal, non-toxic treatments that inhibit AR-dependent PCa
are highly desirable.
Previous studies have reported that certain natural
products, including lycopene, soy isoflavones, resveratrol and
green tea extract can inhibit the expression of the AR and
prostate-specific antigen (PSA) in androgen-dependent PCa LNCaP
cells (5-7). These cells represent the most
commonly used cellular model of AR-dependent PCa. The combination
of ADR with a herbal supplement including cholecalciferol,
α-tocopherol, L-selenomethionine and epigallocatechin was
beneficial in a phase II clinical trial, leading to a decrease or
stabilization of the level of PSA (6).
Ophiopogonin D' (OPD') and Ophiopogonin D (OPD) are
two triterpene saponins derived from Ophiopogon japonicus,
which is a plant used in traditional Chinese medicine. OPD and OPD´
have the same molecular weight (854.47 kDa), central structure
(platycodigenin) and glycosylradical, but different
glycosideradicals. Previous studies have demonstrated that OPD
exhibits antitumor effects (8,9).
However, little is known about the effects of OPD'. Our previous
study revealed that OPD' induced apoptosis in androgen-independent
PCa cells through a receptor-interacting serine/threonine-protein
kinase 1 (RIPK1)-mediated mechanism (10).
RIPK1 serves an important role in cell death and
inflammation. Activation of RIPK1 promotes cell death in radio
resistant breast cancer and enhances the antitumor activity of
docetaxel in patient-derived breast cancer xenografts (11,12).
Abnormal expression of RIPK1 is associated with a range of human
degenerative and inflammatory diseases, including central nervous
system pathologies, amyotrophic lateral sclerosis, Alzheimer's
disease, Parkinson's disease, traumatic brain injury, stroke and
lysosomal storage diseases (13).
In prostate cancer, RIPK1 interacts directly with the AR to
suppress PCa cell proliferation in vitro and tumor growth
in vivo (14). Inhibitor of
apoptosis protein antagonists and sorafenib (a RIPK1 inhibitor)
induce apoptosis or necroptosis in castration-resistant
(AR-independent) or autophagy-deficient PCa cells (15-17).
Necroptosis is different from apoptosis in terms of
the underlying mechanisms and the characteristics of cell death and
is important to eliminate apoptosis-resistant tumor cells (18). Apoptosis involves the formation of
apoptotic bodies and requires the activation of endogenous and/or
exogenous caspase-dependent apoptosis pathways (19). Necroptosis is characterized by
increased membrane permeability, swelling organelles and cleavage
of the cell nucleus (20).
Necroptosis is regulated by multiple pathways, including the
RIPK1/RIPK3/mixed lineage kinase domain-like protein (MLKL)
pathway. Recent studies suggest that splenomegaly is largely
dependent on RIPK3-MLKL-mediated necroptosis, but is independent of
RIPK1 kinase activity (21). By
contrast, monocytosis is dependent on RIPK1 kinase activity, but
not RIPK3-MLKL (21).
Our previous study demonstrated that OPD' inhibited
the in vitro and in vivo growth of AR-independent PCa
via RIPK1 with no significant effects on the body weight of nude
mice (10). The aim of the present
study was to explore the effects and mechanisms of action of OPD'
in an AR-dependent PCa cell line LNCaP.
Materials and methods
Test compound, chemicals and
reagents
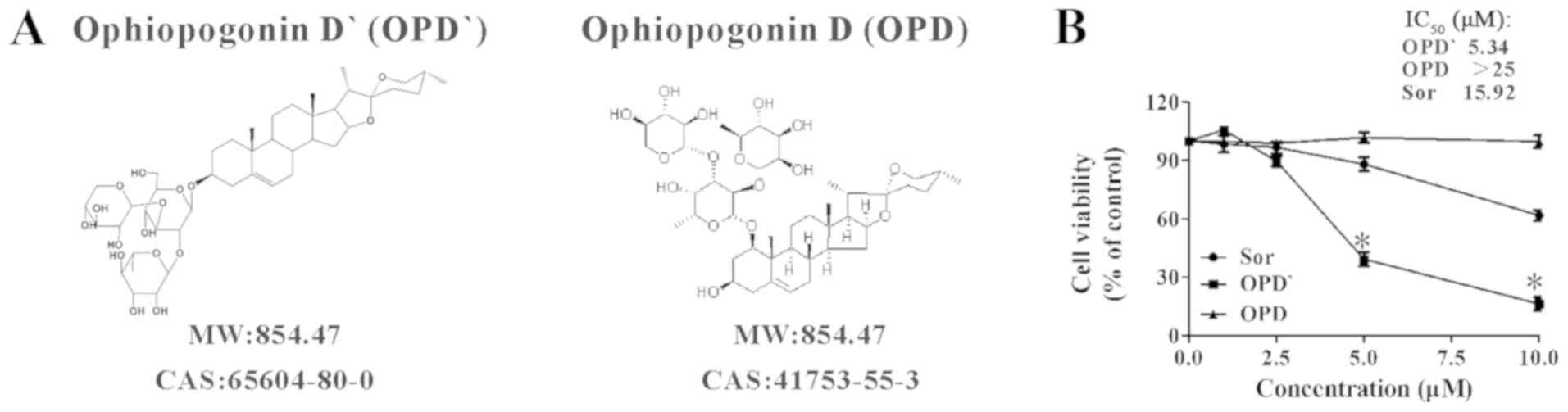
The OPD and OPD' used in the present study (Fig. 1A) were obtained from Chengdu Must
Bio-Technology and had a purity of >96%. The anti-human RIPK1
(1:1,000; cat. no. 3493), RIPK3 (1:1,000; cat. no. 13526), caspase
8 (1:1,000; cat. no. 9746), Fas-associated death domain (FADD;
1:500; cat. no. 2782) and mouse anti-rabbit IgG (light-chain
specific; 1:1,000; cat. no. 45262) antibodies were obtained from
Cell Signaling Technology, Inc. The RIPK3 (1:50; cat. no. ab56164),
anti-MLKL (1:1,000; cat. no. ab184718) and anti-p-MLKL antibodies
(1:1,000; cat. no. ab187091) were purchased from Abcam. The
anti-β-actin (1:1,000; cat. no. TA-09), horseradish
peroxidase-conjugated anti-rabbit IgG (1:2,000, cat. no. ZB-2306)
and horseradish peroxidase-conjugated anti-mouse IgG (1:2,000; cat.
no. ZB-2305) antibodies were obtained from OriGene Technologies,
Inc. Sorafenib (positive control), Necrostatin-1 (Nec-1, a RIPK1
inhibitor) and Z-VAD-FMK (a caspase inhibitor) were purchased from
Selleck Chemicals. Necrosulfonamide (NSA) was purchased from Santa
Cruz. N-acetylcysteine (NAC) was purchased from Beyotime Institute
of Biotechnology.
Cell lines and cell culture
The LNCaP, PC3 and DU145 cell lines were purchased
from the American Type Culture Collection. Cells were incubated in
a stable, humidified environment at 37°C with 5% CO2 and
were passaged every 2-3 days when they became confluent. The three
cell lines were cultured in their own special medium supplemented
with 10% FBS as previously described (22).
Cell survival assay
The Cell Counting Kit-8 (CCK-8; Beyotime Institute
of Biotechnology) assay was used to examine the effects of OPD' on
the survival of human LNCaP cells. LNCaP cells (8,000 cells/well)
were treated with 0-25 µM OPD' or sorafenib (used as a
positive control) for 24 h, and 10 µl CCK8 was added in per
well for 3 h at 37°C. The absorbance of the sample at 450 nm was
measured using a Tecan Infinite M200 microplate reader (Tecan Group
Ltd.). The proportion of viable cells was calculated based on the
following formula: Viable cells = [OD (OPD')-OD(blank)] /
[OD(DMSO)-OD(blank)] × 100%. IC50 was calculated by the
LOGIT method (23).
Apoptosis assay
The effects of OPD' or sorafenib on the proportion
of LNCaP cells undergoing apoptosis and necroptosis were examined
using an Annexin V-FITC/propidium iodide (PI) Apoptosis Detection
kit (BestBio, Ltd.). LNCaP cells (2×105 cells/well) were
treated with 2.5-10 µM OPD' or sorafenib for 18 h. The
samples were collected for FITC/PI staining for 15 min according to
the manufacturer's instructions and analyzed by a FACSCalibur flow
cytometer (BD Biosciences) and FlowJo 7.6.1 software (BD
Biosciences).
Ultrastructural study
LNCaP cells (1×106) were treated with 5
µM OPD' for 24 h. The cultured cells were collected, fixed
with 2% glutaraldehyde in 0.1 M PBS overnight, post-fixed with 1%
osmium tetroxide for 2 h at 4°C and observed under a FEITecnai 10
electron microscope (Thermo Fisher Scientific, Inc.) at ×12,000
magnification.
Co-immunoprecipitation of RIPK3- and
MLKL-bound complexes
LNCaP cells were treated with OPD' for 6 h, and
protein lysates were collected using ice-cold NP-40 cell lysis
buffer (Beyotime Institute of Biotechnology) with 1 mM PMSF. A 50%
protein A/G agarose mixture (cat. no. P2055; Beyotime Institute of
Biotechnology) was added (100 µl per 1 ml sample solution),
and the sample was agitated on a horizontal shaker for 60 min at
4°C. The sample was centrifuged at 825 × g for 2 min at 4°C, and
the supernatant was divided into two parts. Subsequently, 10
µl anti-RIPK3 antibody (cat. no. ab56164; Abcam) or
rabbit-IgG (cat. no. A0716; Beyotime Institute of Biotechnology)
was added to yield ~500 µl total volume, and the sample was
incubated at 4°C overnight to allow antibody binding. New 50%
protein A/G agarose was added to the sample solution, which was
incubated at 4°C for 1 h. The sample was washed with NETN lysis
buffer (20 mM Tris, pH 8.0; 150 mM NaCl; 0.5% NP-40; 1 mM DTT; 0.5
mM EDTA; protease inhibitor mixture) 5 times at 4°C. The resulting
complexes were collected and used for western blotting
analysis.
Drug treatments
The cells were pre-treated with inhibitors (10
µM Nec-1, 10 µM NSA, 20 µM Z-VAD-FMK or 5 mM
NAC) for 2 h and treated with OPD' for 24 or 18 h (total inhibitor
treatment duration was 26 or 20 h). Cell survival rate and flow
cytometry analyses were performed using CCK-8 and Annexin V-FITC/PI
staining, respectively.
Small interfering (si)RNA
transfection
siRNA sequences against FADD were purchased from
Guangzhou RiboBio Co., Ltd. The siRNA sequences were as follows:
siRNA-FADD#1, GACCGAGCTCAAGTTCCTA; and siRNA-FADD#2,
CTGAGAATCTGGAAGAACA. A total of 2×105 LNCaP cells were
transfected with 50 nM siRNA-FADD with Lipofectamine®
2000 (Invitrogen; Thermo Fisher Scientific, Inc.) and Opti-MEM I
reduced serum medium (Invitrogen; Thermo Fisher Scientific, Inc.)
for 6 h at 37°C. Subsequently, the cells were incubated in 5
µM OPD' for another 6 or 24 h. Western blotting and flow
cytometry analyses were performed using anti-FADD antibodies and
Annexin V-FITC/PI staining, respectively.
Western blotting assays
LNCaP, DU145 and PC3 cells were treated with OPD',
and the total protein was extracted with a protein extraction kit
including RIPA, protease inhibitor mixture and phosphorylase
inhibitor mixture (BestBio, Ltd.). The samples were centrifuge at
13,200 × g for 15 min at 4°C, and the supernatant was transferred
to the new Eppendorf tube. Protein concentration was determined by
the bicinchoninic assay method, and the samples were balanced with
RIPA, protease inhibitor mixture, phosphorylase inhibitor mixture
(250:1:1) and 5X sample loading buffer (cat. no. P0015; Beyotime
Institute of Biotechnology) and denatured for 10 min at 100°C. A
total of 50 µg protein sample per lane was separated by
8-12% SDS-PAGE at 80 V for 30 min and 120 V for 60 min, transferred
to a PVDF membrane at 200 mA and 4°C for 150 min and blocked with
5% skimmed milk for 120 min at room temperature. The membrane was
incubated with the primary antibodies overnight at 4°C with gentle
agitation and washed with TBS + 0.1% Tween-20. The secondary
antibodies were added and incubated for 2 h at room temperature.
The membrane was visualized using a chemiluminescence kit (EMD
Millipore), detected using a Fusion FX5 Spectra instrument (Vilber
Lourmat Sté) and analyzed by Image Lab (Bio-Rad Laboratories,
Inc.).
Statistical analysis
Continuous variables are presented as the means ±
SEM. Statistical analysis was performed by one-way ANOVA and
Tukey's test. All tests were performed using SPSS 13.0 (SPSS, Inc.)
and were two-sided. P<0.05 was considered to indicate a
statistically significant difference.
Results
OPD' inhibits LNCaP cell
proliferation
The inhibitory effects of OPD' on LNCaP cell
survival were assessed using the CCK-8 assay. Sorafenib has been
recommended for the treatment of refractory PCa, and is also a
RIPK1 inhibitor (16,20,24);
thus, it was used as a positive control in the present study. OPD'
exhibited stronger proliferation inhibitory effects compared with
OPD and sorafenib in LNCaP cells at 24 h, with IC50
values of 5.34 µM for OPD', 15.92 µM for sorafenib
and >25 µM for OPD (Figs.
1B and S1).
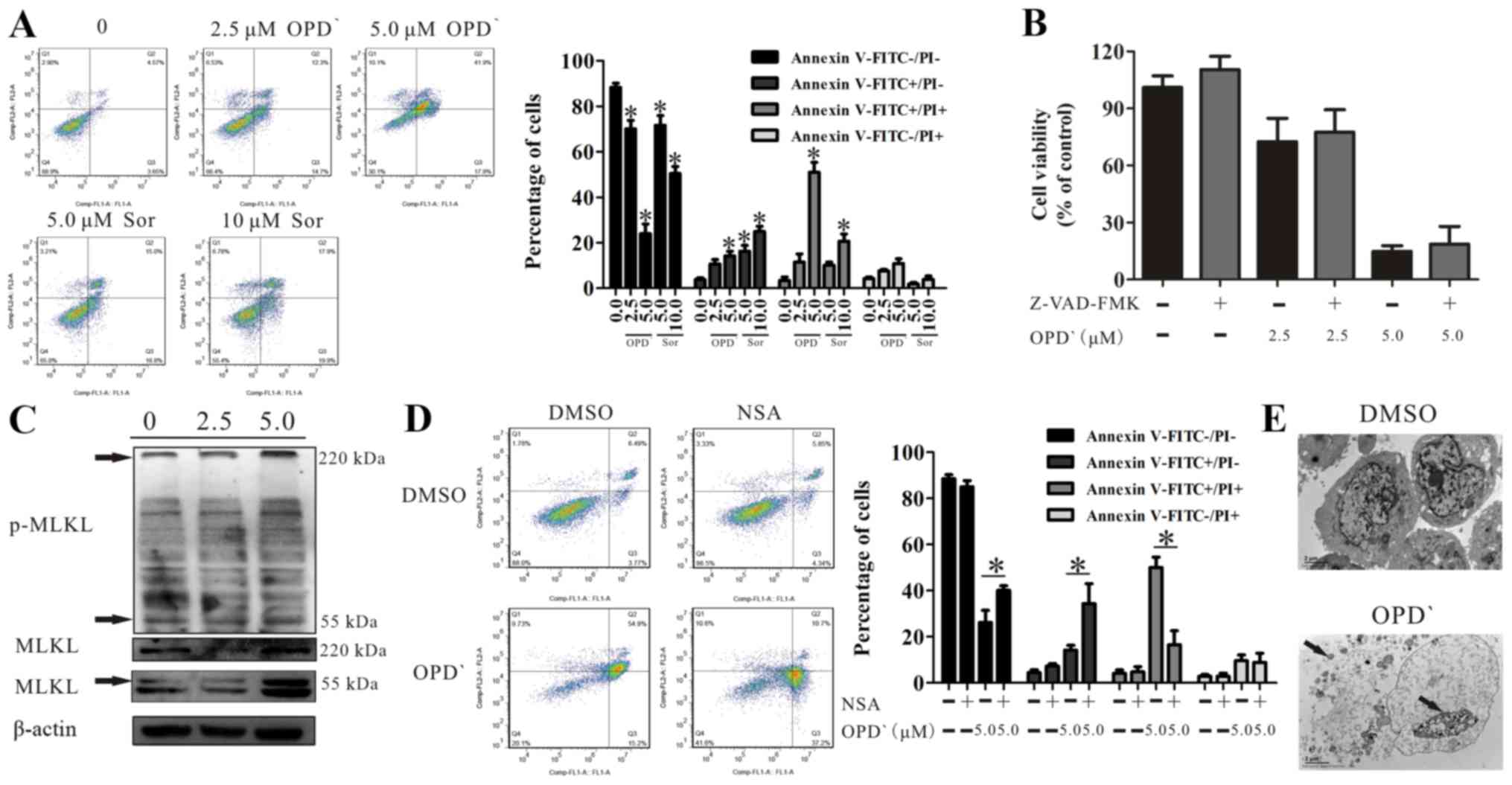
OPD' induces necrosis in LNCaP cells
As indicated in Fig.
2A, 24-h treatment with OPD' led to an increase in the
proportion of FITC-positive and FITC/PI dual-positive cells (0 vs.
5 µM OPD', 3.9±1.3 vs. 14.2±3.6 and 3.5±2.6 vs. 51.0±7.5,
respectively). Sorafenib treatment (10 μM) resulted in a greater
increase in the proportion of FITC-positive cells and increased the
proportion of FITC/PI dual-positive cells, but this effect was less
potent compared with that of OPD' (Fig. 2A). The proportion of FITC/PI
dual-positive cells is considered to reflect late apoptosis or
necrosis (25), and apoptosis is
mainly mediated by caspase pathways (19); the effects of OPD' were not
reversed following pre-treatment with 20 µM Z-VAD-FMK (a
pan-caspase inhibitor) for 2 h (Fig.
2B), suggesting that these cells were not undergoing
apoptosis.
MLKL is a marker of necrosis (26). The results of western blot analysis
demonstrated that OPD' exposure increased the protein expression
levels of MLKL (55 and 220 kDa) and p-MLKL (220 kDa) (Figs. 2C and S2). Treatment of cells with OPD' and
NSA, a MLKL inhibitor, reversed the impact of OPD' on the
proportion of FITC−/PI− and
FITC+/PI+ cells (5 µM OPD' vs. NSA + 5
µM OPD', 26.2±9.5 vs. 40.1±3.5 and 50.1±7.8 vs. 16.6±10.5,
respectively; Fig. 2D). In
addition, transmission electron microscopy images demonstrated
changes in the cell morphology following treatment with 5 µM
OPD' for 24 h, with leakage of cell contents, increased
permeability of the cell membranes and cleavage of the cell
nucleus, but no apoptotic bodies, which suggested that the cells
were undergoing necrosis (Fig. 2E)
(20).
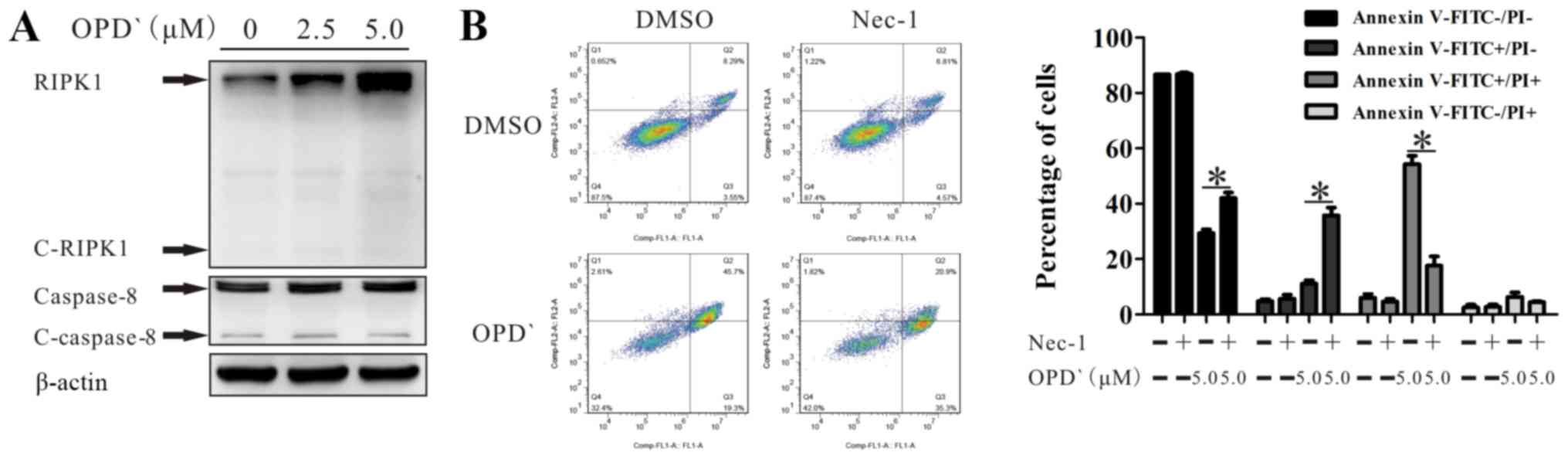
OPD'-induced necroptosis is dependent on
RIPK1
RIPK1 is involved in programmed cell death (27), and is cleaved by cleaved-caspase 8
(C-caspase 8). Western blot analysis indicated that treatment with
5 µM OPD' for 6 h increased the protein expression levels of
RIPK1 and caspase 8, without any effect on the levels of
cleaved-RIPK1 (C-RIPK1) or C-caspase 8 in LNCaP cells (Fig. 3A). Treatment with 2.5 and 5
µM OPD' increased RIPK1 without any effects on c-caspase 8
and possible slight increases in C-RIPK1 and caspase 8 at 2.5
µM (Fig. 3A). When the
cells were co-treated with OPD' and Nec-1, the effects of OPD' on
the proportion of FITC+/PI+ cells(late
apoptosis and necrosis) were reversed (Fig. 3B), suggesting that RIPK1 was
necessary for OPD'-induced necrosis. Compared with cells treated
with only OPD', the proportion of FITC+/PI-
cells was increased following co-treatment with OPD' and Nec-1
(Fig. 3B). Thus, early apoptosis
was still induced, but late apoptosis/necrosis was not induced when
cells were treated with Nec-1.
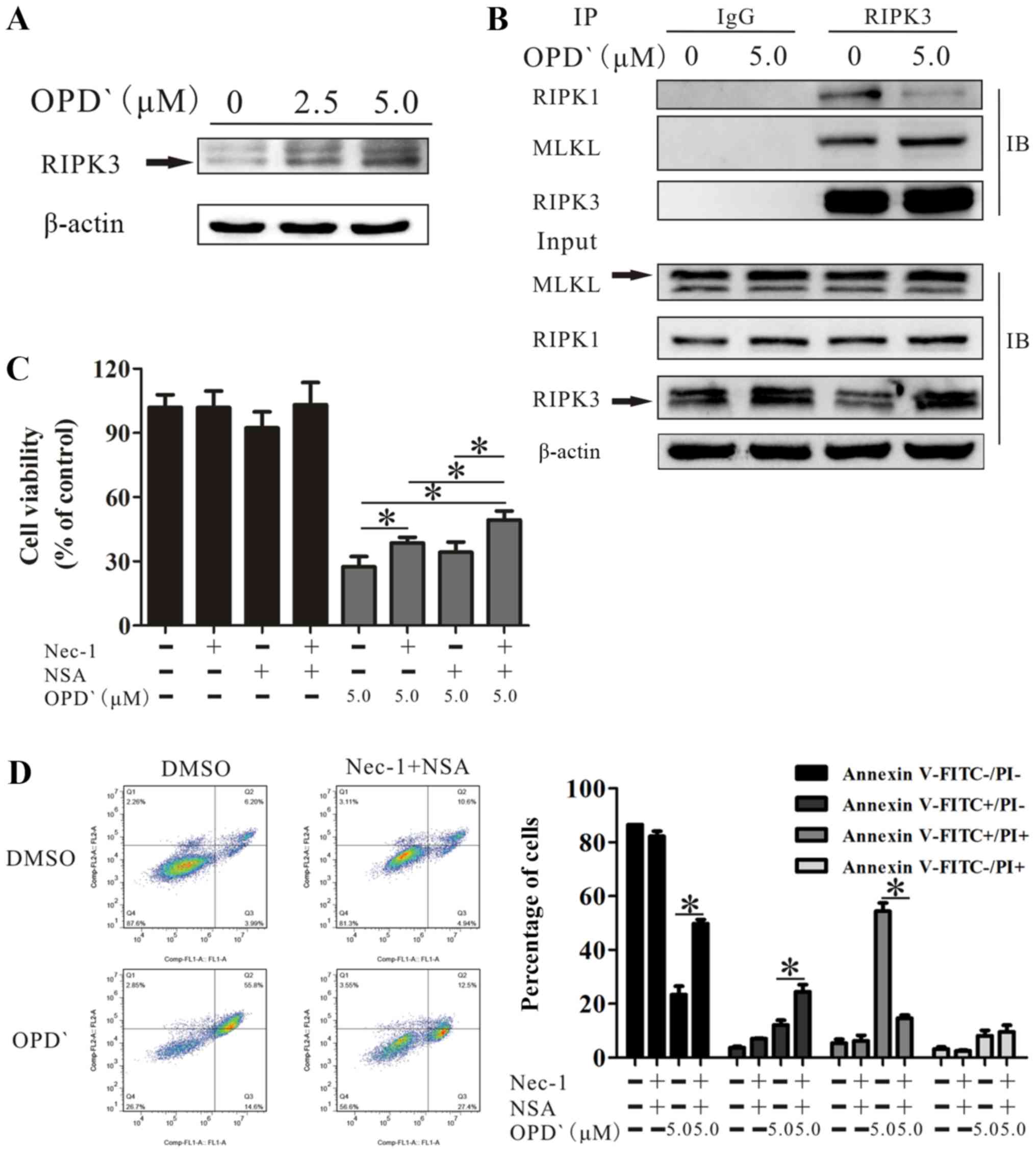
OPD' induces necroptosis by multiple
pathways involving RIPK1 and RIPK3/MLKL
The RIPK1/RIPK3/MLKL pathway is a classical
necroptosis pathway (28).
Therefore, the effects of OPD' on the expression levels of these
three proteins were examined. OPD' exposure increased the protein
expression level of RIPK3 (Fig.
4A). Co-immunoprecipitation analysis results revealed that
RIPK3, but not RIPK1, interacted with MLKL. This indicated that
RIPK3 interacted with RIPK1 and MLKL, but RIPK3 did not require
RIPK1 for binding MLKL (Fig.
4B).
Based on the above results, cells treated with only
OPD', OPD' plus Nec-1, OPD' plus NSA or OPD' plus Nec-1 and NSA
were compared; the results demonstrated that the effects of OPD' on
cell viability were decreased by co-treatment with Nec-1 or Nec-1
and NSA (Fig. 4C). The effect was
not reversed by co-treatment with NSA (P=0.109; Fig. 4C). However, the combination of
Nec-1 and NSA was significantly more effective compared with the
Nec-1 alone at inhibiting the effects of OPD' (Fig. 4C). The FICT/PI double staining
analysis results demonstrated that the co-treatment of cells with
OPD', Nec-1 and NSA inhibited the effects of OPD' on the
proportions of FITC−/PI− and
FITC+/PI+ cells (5 µM OPD' vs.
Nec-1+NSA + 5 µM OPD', 23.3±7.1 vs. 49.8±3.4 and 54.3±7.0
vs. 14.6±2.7, respectively; Fig.
4D), resulting in an increase in the proportion of
FITC+/PI- cells (12.1±3.9 vs. 24.4±6.1;
Fig. 4D). Thus, Nec-1 and NSA
attenuated the effects of OPD' on LNCaP cells.
OPD' regulates Fas ligand (FasL), AR and
PSA through RIPK1
In addition to the RIPK1/RIPK3/MLKL pathway, RIPK1
also activates mitogen-activated protein kinases, such as JNK
(29), which upregulate the
expression of Fas, FasL and Bim (30) and interact with the AR to inhibit
the development of PCa (14). As
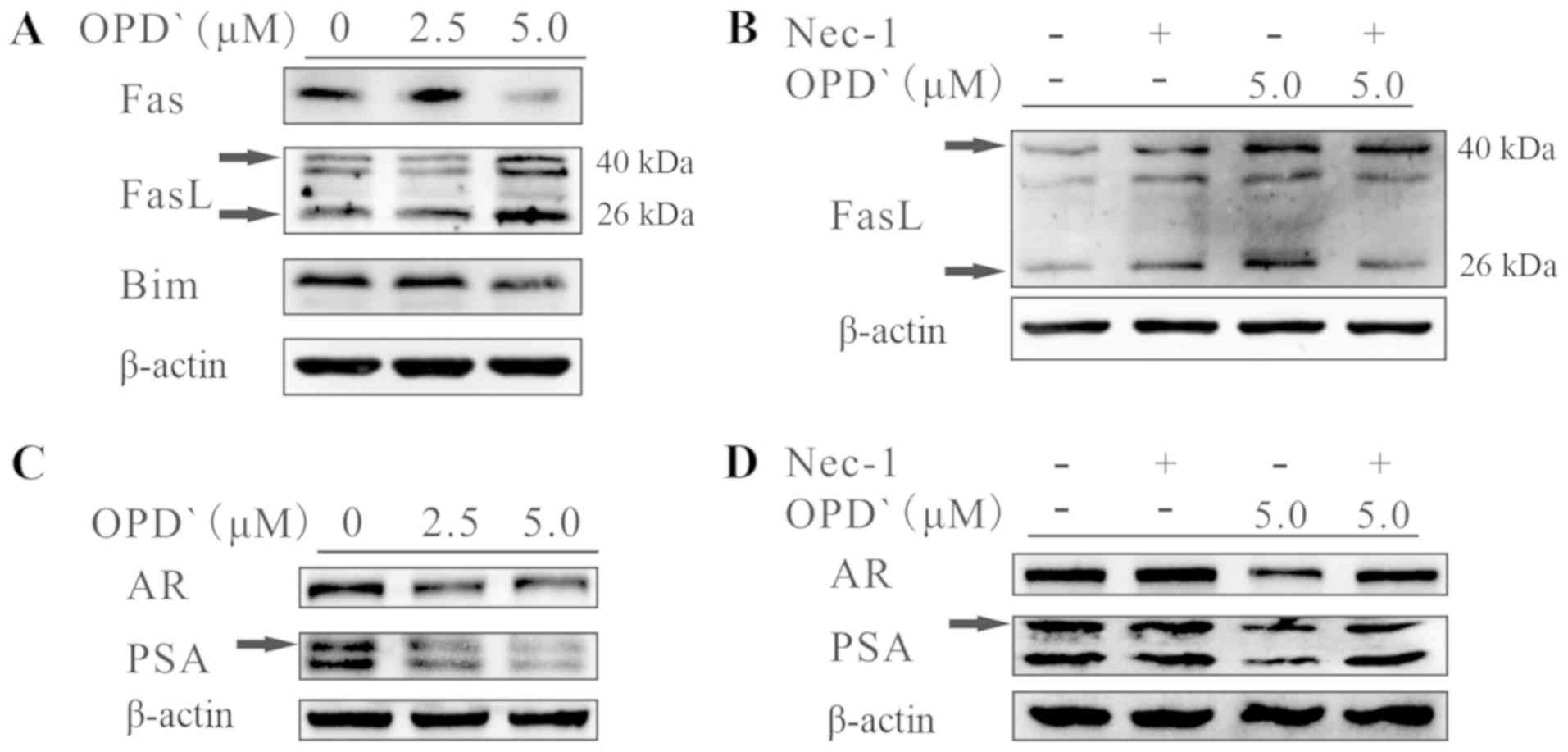
presented in Fig. 5A, exposure of
LNCaP cells to 5 µM OPD' for 6 h increased the protein
expression levels of FasL (40 kDa) and soluble FasL (26 kDa),
whereas the protein expression levels of Fas and Bim were
decreased. In addition, the effects of OPD' on soluble FasL were
reversed by pre-treatment with Nec-1 for 2 h prior to OPD'
treatment (Fig. 5B). Following
treatment with OPD', the protein expression levels of the AR and
PSA were decreased (Fig. 5C). This
effect was reversed by pre-treatment with Nec-1 for 2 h (Fig. 5D). These results suggested that
OPD' may affect FasL via RIPK1, thus inhibiting the AR and
inhibiting PCa cell proliferation, while also inducing
necroptosis.
OPD' determines the pattern of cell death
through FADD
The type II tumor necrosis factor receptor
1-interacting protein complex (comprising RIPK1, caspase 8, FADD
and tumor necrosis factor receptor type 1-associated death domain
protein) requires FADD to induce apoptosis or necroptosis (31,32).
As presented in Fig. 6A, the
baseline FADD protein expression level in LNCaP cells was higher
compared with that in PC3 and DU145 cell lines. Although OPD'
treatment increased the protein expression of FADD in LNCaP
cells(Fig. 6B), only a slight
increase was observed in the protein expression level of FADD
following exposure of DU145 cells to 1 µM and 2.5 µM
OPD' for 6 h. Of note, the FADD protein level exhibited a decrease
in PC3 cells (Fig. 6B). The
effects of OPD' on the proportion of
FITC−/PI− and FITC+/PI+
cells were reversed in LNCaP cells by pre-treatment with siRNA-FADD
(5 µM OPD' vs. siRNA-F1 + 5 µM OPD' and siRNA-F2 + 5
µM OPD', 42.6±6.7 vs. 58.6±7.6 and 59.8±8.6, and 30.9±9.8
vs. 18.5±3.2 and 19.3±4.0, respectively; Fig. 6C).
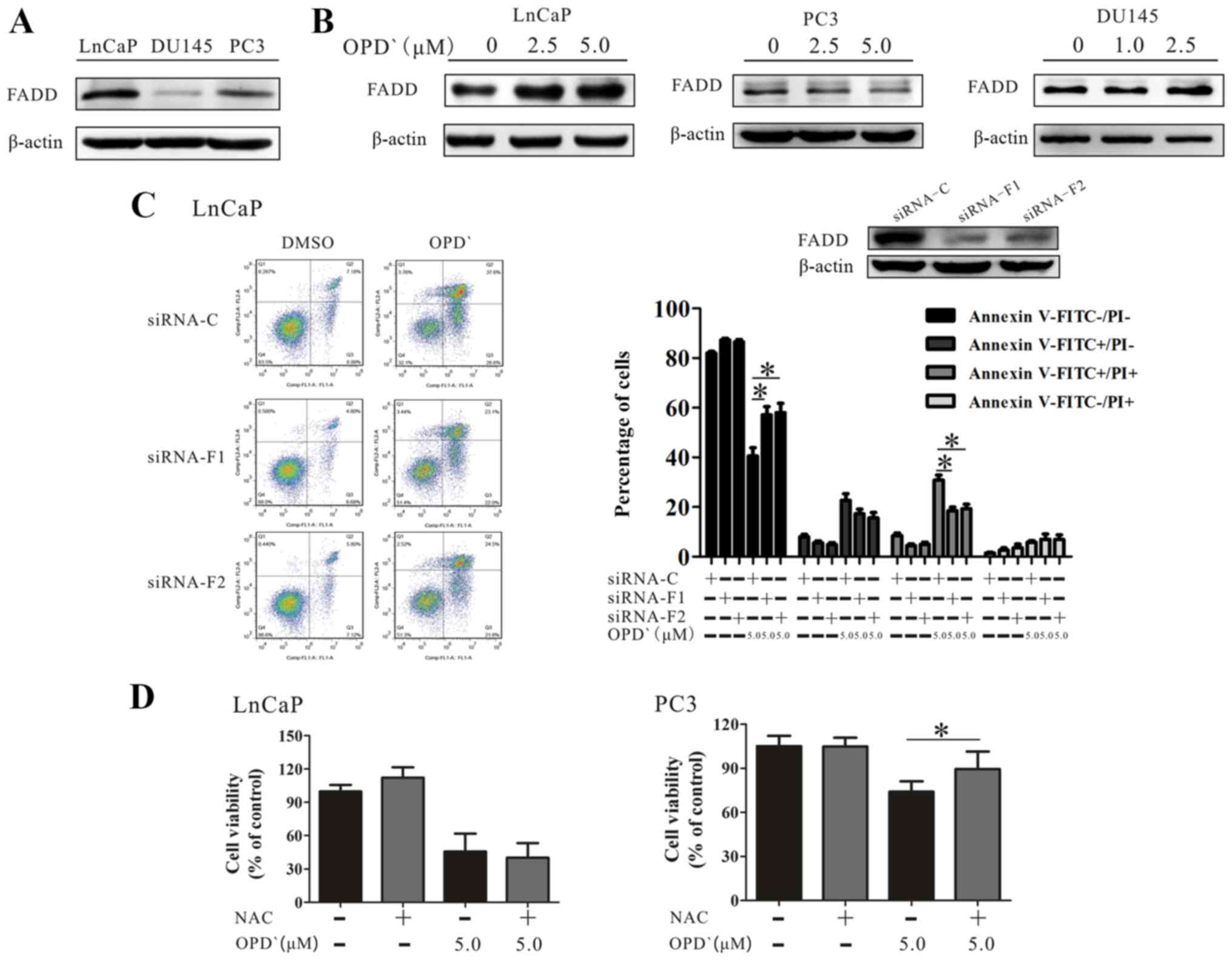 | Figure 6OPD' determines the pattern of cell
death mediated by FADD. (A) The baseline FADD protein expression in
LNCaP, PC3 and DU145 cells was examined by western blotting. (B)
LNCaP, PC3 and DU145 cells were treated with OPD' for 6 h, and FADD
protein expression was examined by western blotting. (C) Annexin
V-FITC/PI staining was used to analyze the apoptotic rates of LNCaP
cells following treatment with OPD' and siRNA-FADD (n=3). (D) The
Cell Counting Kit-8 assay was used to analyze the viability of
LNCaP and PC3 cells following treatment with OPD' and NAC (n=3).
*P<0.05. OPD', Ophiopogonin D'; FADD, Fas-associated
death domain; siRNA, small interfering RNA; C, siRNA-control;
siRNA-F1, siRNA-FADD#1; siRNA-F2, siRNA-FADD#2; PI, propidium
iodide; NAC, N-acetylcysteine. |
RIPK1 induces apoptosis by generating reactive
oxygen species (ROS) in the absence of FADD (33). The effects of OPD' were reversed by
pretreatment of PC3 cells with an antioxidant NAC, but this was not
observed in LNCaP cells (Fig. 6D).
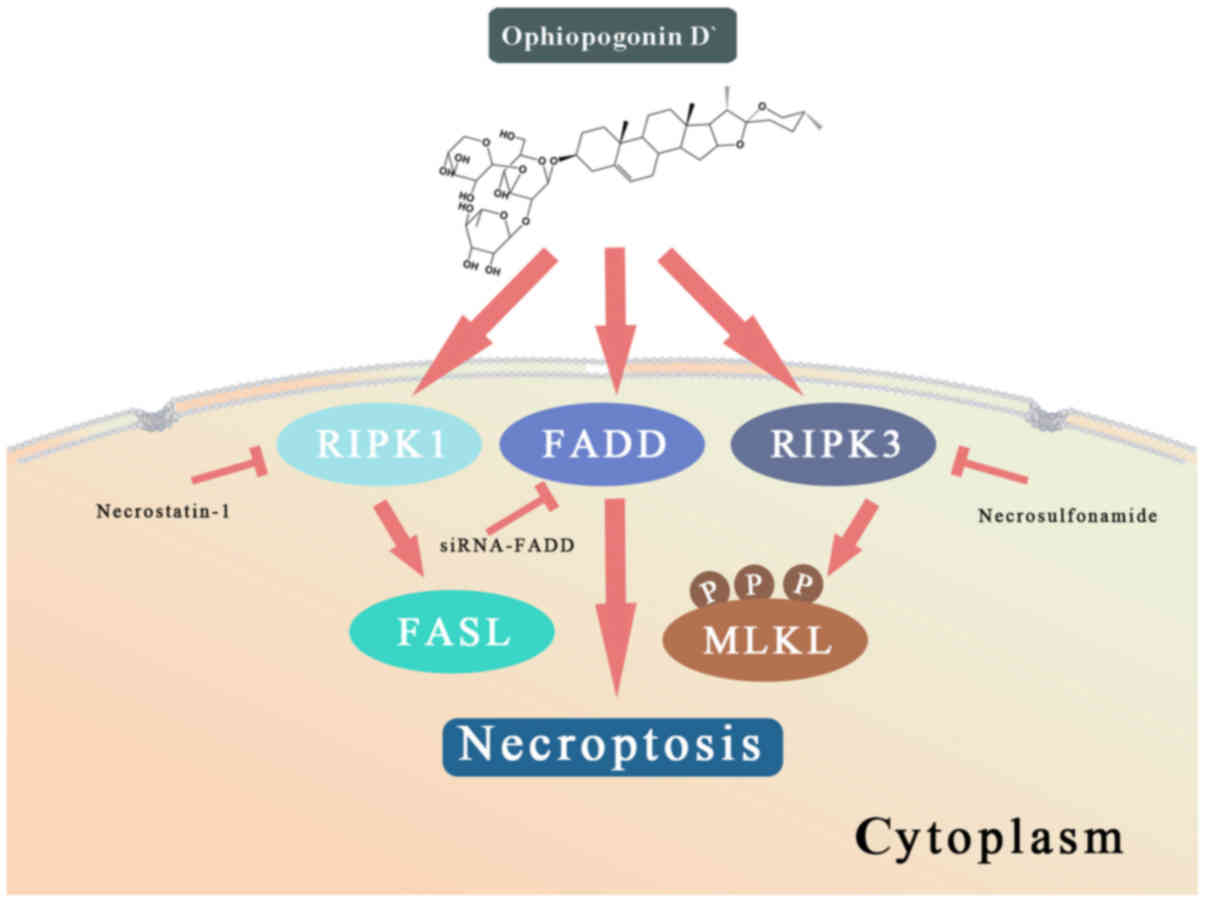
These results suggested that OPD' induced necrop-tosis through
multiple pathways, and that FADD served a key role in determining
the form of cell death (Fig.
7).
Discussion
A previous study has suggested that promoting
apoptosis may cure PCa (34).
Apoptosis resistance frequently develops after ADT, leading to
treatment-resistant PCa (4). The
occurrence of apoptosis resistance is associated with the induction
of autophagy by androgen deprivation, which reduces the efficacy of
ADT (35,36). More specifically, upregulation of
autophagy-associated proteins has been demonstrated to promote
resistance to ADT (35,37,38).
Another study has indicated that the transformation of cell death
patterns between apoptosis and necroptosis is controlled by
autophagy machinery (39). In
prostate cells defective in autophagy, toxic agents induced
necroptosis rather than apoptosis (16). Inhibition of autophagy has also
been demonstrated to improve the efficacy of abiraterone (40). Thus, suppressing autophagy and/or
inducing necroptosis may make ADT more effective (34). The results of the present study
suggested that OPD' induced necroptosis in LNCaP cells.
RIPK1 is a crucial regulator of necroptosis
(41). The results of the present
study suggested that OPD' induced necroptosis via several
RIPK1-dependent mechanisms. RIPK3 is known to interact with RIPK1
to induce necroptosis (19). The
present study demonstrated that OPD' activated MLKL through RIPK3
to induce necroptosis. Of note, when MLKL was inhibited by NSA, the
OPD'-induced necroptosis and apoptosis was increased. The results
of the present study also revealed that RIPK1 did not directly
interact with RIPK3, but was able to induce necroptosis in a
RIPK3/MLKL-independent manner. Thus, although MLKL and RIPK3 maybe
involved in the necroptotic effects of OPD', other regulators
appear to be involved. RIPK3 may induce apoptosis by disrupting
FUN14 domain-containing 1 activation (42) when MLKL is inhibited by NSA. RIPK1
also regulates FasL, which is required for the execution of
necroptosis in red blood cells (43), as well as mitogen-activated protein
kinases such as JNK (29), which
promotes the protein expression of Fas, FasL and Bim (30). In the present study, OPD' treatment
increased the protein expression levels of FasL (but not Bim or
Fas) in LNCaP cells in a RIPK1-dependent manner, which supported
the involvement of FasL.
The AR is a transcription factor that activates
genes that promote PCa cell proliferation and survival (44). AR antagonists or surgery are
typically used to treat patients with early-stage PCa. The results
of the present study demonstrated that OPD' inhibited the AR
protein expression via RIPK1. PSA is an important index used for
the diagnosis and evaluation of PCa progression and is related to
the expression of the AR (45,46).
In the present study, OPD' decreased the protein expression of PSA
via a mechanism dependent on RIPK1. Other saponin products, such
asginsenosides, have also been demonstrated to inhibit the AR
protein expression during testosterone stimulation (47). In addition to saponins, an ethanol
extract of Algerian propolis was demonstrated to decrease androgen
receptor transcriptional activity and the secretion of PSA
(48). Thus, a body of evidence
supports the use of plant-derived compounds to reduce the AR
expression and activity.
Tumors are composed of genetically heterogeneous
cellular clones, which undergo further genetic changes during
disease progression and in response to clinical treatment (49). Prostate tumors contain functionally
and phenotypically distinct cells (50). The androgen receptor status of
prostate tumor cells is one of the major indicators used for the
staging of PCa (51). Our previous
study demonstrated that OPD' exhibited activity against
androgen-independent PC3 and DU145 prostate cancer cells, which was
mediated through RIPK1 (10).
Although OPD' increased RIPK1 protein expression in these cells and
androgen-dependent LNCaP cells, the treatment resulted in different
effects. Interestingly, OPD' induced apoptosis in PC3 and DU145
cells, which are more sensitive compared with PC3 cells (10), but led to necroptosis in LNCaP
cells. This may be due to the different baseline expression and
activity of FADD in the different cell lines.
FADD is not only a crucial adaptor protein in death
receptor-mediated apoptosis, but is also important for necroptosis
(52). FADD, caspase 8 and RIPK1
form a death-inducing signaling complex that has the potential to
initiate either apoptosis or necroptosis depending on the
conditions and cell type (53). In
the absence of FADD, RIPK1 induces apoptosis by generating ROS
(33). The results of the present
study demonstrated that OPD' increased the protein expression of
FADD and induced necroptosis in LNCaP cells, whereas in our
previous study, it reduced the protein expression of FADD and
induced apoptosis in PC3 cells (10). Further experiments in the present
study revealed that the effects of OPD' were reversed by
pre-treatment of PC3 cells with NAC, which was not observed in
LNCaP cells. The level of OPD'-induced apoptosis did not change
following pre-treatment of cells with siRNA-FADD. This may be
attributed to PC3 cells having a high mitochondrial oxygen
concentration and generating more O2- compared with
LNCaP cells (54). Although normal
ROS levels maintain cell proliferation, excessive ROS induces cell
death, and PC3 cells were already being exposed to high levels of
ROS (55). FADD induces apoptosis
in the absence of RIPK1 (56), and
when RIPK1 was inhibited by Nec-1, the level of OPD'-induced
apoptosis in LNCaP cells increased.
In summary, the results of the present study
demonstrated that OPD' inhibited the proliferation of LNCaP cells,
and that these effects were at least partially RIPK1-dependent.
OPD' also affected the expression levels of FasL, AR and PSA in a
RIPK1-dependent manner. OPD' may inhibit the viability of diverse
types of prostate cancer cell and exert its effects via multiple
mechanisms. Further studies are needed to replicate these results
and mechanisms in vivo. However, the current results are
novel, suggesting that OPD' may have potential as an anti-tumor
agent in the treatment of PCa.
Supplementary Data
Funding
This work was supported by the National Natural
Science Foundation of China (grant nos. 81603347, 81673167 and
81171991).
Availability of data and materials
All data used or analyzed in this study are
available from the corresponding author upon reasonable
request.
Authors' contributions
ZL and HX organized, conceived, designed and
supervised the study. CW, MZ, WS and JW designed and conducted the
experiments and drafted the manuscript. YL, HW, JG, NL and JL
participated in the study design and data interpretation. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Keating NL, O'Malley AJ and Smith MR:
Diabetes and cardiovascular disease during androgen deprivation
therapy for prostate cancer. J Clin Oncol. 24:4448–4456. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ross RW and Small EJ: Osteoporosis in men
treated with androgen deprivation therapy for prostate cancer. J
Urol. 167:1952–1956. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mohler JL, Wu W and Fiandalo MV: The role
of intracrine androgen metabolism, androgen receptor and apoptosis
in the survival and recurrence of prostate cancer during androgen
deprivation therapy. Curr Drug Targets. 14:420–440. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Siler U, Barella L, Spitzer V, Schnorr J,
Lein M, Goralczyk R and Wertz K: Lycopene and vitamin E interfere
with autocrine/paracrine loops in the Dunning prostate cancer
model. FASEB J. 18:1019–1021. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dorff TB, Groshen S, Tsao-Wei DD, Xiong S,
Gross ME, Vogelzang N, Quinn DI and Pinski JK: A Phase II trial of
a combination herbal supplement for men with biochemically
recurrent prostate cancer. Prostate Cancer Prostatic Dis.
17:359–365. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McLarty J, Bigelow RLH, Smith M, Elmajian
D, Ankem M and Cardelli JA: Tea polyphenols decrease serum levels
of prostate-specific antigen, hepatocyte growth factor, and
vascular endothelial growth factor in prostate cancer patients and
inhibit production of hepatocyte growth factor and vascular
endothelial growth factor in vitro. Cancer Prev Res (Phila).
2:673–682. 2009. View Article : Google Scholar
|
|
8
|
Lee JH, Kim C, Lee SG, Sethi G and Ahn KS:
Ophiopogonin D, a Steroidal Glycoside Abrogates STAT3 Signaling
Cascade and Exhibits Anti-Cancer Activity by Causing GSH/GSSG
Imbalance in Lung Carcinoma. Cancers (Basel). 10:E4272018.
View Article : Google Scholar
|
|
9
|
Lee JH, Kim C, Lee SG, Yang WM, Um JY,
Sethi G and Ahn KS: Ophiopogonin D modulates multiple oncogenic
signaling pathways, leading to suppression of proliferation and
chemosensitization of human lung cancer cells. Phytomedicine.
40:165–175. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lu Z, Wang H, Zhu M, Song W, Wang J, Wu C,
Kong Y, Guo J, Li N, Liu J, et al: Ophiopogonin D', a Natural
Product From Radix Ophiopogonis, Induces in Vitro and in Vivo
RIPK1-Dependent and Caspase-Independent Apoptotic Death in
Androgen-Independent Human Prostate Cancer Cells. Front Pharmacol.
9:4322018. View Article : Google Scholar
|
|
11
|
Perez-Añorve IX, Gonzalez-De la Rosa CH,
Soto-Reyes E, Beltran-Anaya FO, Del Moral-Hernandez O,
Salgado-Albarran M, Angeles-Zaragoza O, Gonzalez-Barrios JA,
Landero-Huerta DA, Chavez-Saldaña M, et al: New insights into
radioresistance in breast cancer identify a dual function of
miR-122 as a tumor suppressor and oncomiR. Mol Oncol. 13:1249–1267.
2019.PubMed/NCBI
|
|
12
|
Newell M, Goruk S, Mazurak V, Postovit L
and Field CJ: Role of docosahexaenoic acid in enhancement of
docetaxel action in patient-derived breast cancer xenografts.
Breast Cancer Res Treat. 177:357–367. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Degterev A, Ofengeim D and Yuan J:
Targeting RIPK1 for the treatment of human diseases. Proc Natl Acad
Sci USA. 116:9714–9722. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hsu CL, Liu JS, Lin TW, Chang YH, Kuo YC,
Lin AC, Ting HJ, Pang ST, Lee LY, Ma WL, et al: Characterization of
a novel androgen receptor (AR) coregulator RIPK1 and related
chemicals that suppress AR-mediated prostate cancer growth via
peptide and chemical screening. Oncotarget. 8:69508–69519. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Martens S, Jeong M, Tonnus W, Feldmann F,
Hofmans S, Goossens V, Takahashi N, Bräsen JH, Lee EW, Van der
Veken P, et al: Sorafenib tosylate inhibits directly necrosome
complex formation and protects in mouse models of inflammation and
tissue injury. Cell Death Dis. 8:pp. e29042017, View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kharaziha P, Chioureas D, Baltatzis G,
Fonseca P, Rodriguez P, Gogvadze V, Lennartsson L, Björklund AC,
Zhivotovsky B, Grandér D, et al: Sorafenib-induced defective
autophagy promotes cell death by necroptosis. Oncotarget.
6:37066–37082. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
McCann C, Crawford N, Majkut J, Holohan C,
Armstrong CWD, Maxwell PJ, Ong CW, LaBonte MJ, McDade SS, Waugh DJ
and Longley DB: Cytoplasmic FLIP(S) and nuclear FLIP(L) mediate
resistance of castrate-resistant prostate cancer to apoptosis
induced by IAP antagonists. Cell Death Dis. 9:10812018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mohammad RM, Muqbil I, Lowe L, Yedjou C,
Hsu HY, Lin LT, Siegelin MD, Fimognari C, Kumar NB, Dou QP, et al:
Broad targeting of resistance to apoptosis in cancer. Semin Cancer
Biol. 35(Suppl): S78–S103. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bock FJ and Tait SWG: Mitochondria as
multifaceted regulators of cell death. Nat Rev Mol Cell Biol.
October 21–2019.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou W and Yuan J: SnapShot: Necroptosis.
Cell. 158:464–464.e1. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Polykratis A, Martens A, Eren RO,
Shirasaki Y, Yamagishi M, Yamaguchi Y, Uemura S, Miura M, Holzmann
B, Kollias G, et al: A20 prevents inflammasome-dependent arthritis
by inhibiting macrophage necroptosis through its ZnF7
ubiquitin-binding domain. Nat Cell Biol. 21:731–742. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lu Z, Zhou R, Kong Y, Wang J, Xia W, Guo
J, Liu J, Sun H, Liu K, Yang J, et al: S-equol, a Secondary
Metabolite of Natural Anticancer Isoflavone Daidzein, Inhibits
Prostate Cancer Growth In Vitro and In Vivo, Though Activating the
Akt/FOXO3a Pathway. Curr Cancer Drug Targets. 16:455–465. 2016.
View Article : Google Scholar
|
|
23
|
Sebaugh JL: Guidelines for accurate
EC50/IC50 estimation. Pharm Stat. 10:128–134. 2011. View Article : Google Scholar
|
|
24
|
Mohler JL, Kantoff PW, Armstrong AJ,
Bahnson RR, Cohen M, D'Amico AV, Eastham JA, Enke CA, Farrington
TA, Higano CS, et al: National comprehensive cancer network:
Prostate cancer, version 1.2014. J Natl Compr Canc Netw.
11:1471–1479. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen S, Cheng AC, Wang MS and Peng X:
Detection of apoptosis induced by new type gosling viral enteritis
virus in vitro through fluorescein annexin V-FITC/PI double
labeling. World J Gastroenterol. 14:2174–2178. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sun L, Wang H, Wang Z, He S, Chen S, Liao
D, Wang L, Yan J, Liu W, Lei X, et al: Mixed lineage kinase
domain-like protein mediates necrosis signaling downstream of RIP3
kinase. Cell. 148:213–227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Luan Q, Jin L, Jiang CC, Tay KH, Lai F,
Liu XY, Liu YL, Guo ST, Li CY, Yan XG, et al: RIPK1 regulates
survival of human melanoma cells upon endoplasmic reticulum stress
through autophagy. Autophagy. 11:975–994. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Moriwaki K, Bertin J, Gough PJ, Orlowski
GM and Chan FK: Differential roles of RIPK1 and RIPK3 in
TNF-induced necroptosis and chemotherapeutic agent-induced cell
death. Cell Death Dis. 6:e16362015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Festjens N, Vanden Berghe T, Cornelis S
and Vandenabeele P: RIP1, a kinase on the crossroads of a cell's
decision to live or die. Cell Death Differ. 14:400–410. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cohen M, Pierredon S, Wuillemin C, Delie F
and Petignat P: Acellular fraction of ovarian cancer ascites induce
apoptosis by activating JNK and inducing BRCA1, Fas and FasL
expression in ovarian cancer cells. Oncoscience. 1:262–271. 2014.
View Article : Google Scholar
|
|
31
|
Holler N, Zaru R, Micheau O, Thome M,
Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B and Tschopp
J: Fas triggers an alternative, caspase-8-independent cell death
pathway using the kinase RIP as effector molecule. Nat Immunol.
1:489–495. 2000. View
Article : Google Scholar
|
|
32
|
Schwabe RF and Luedde T: Apoptosis and
necroptosis in the liver: A matter of life and death. Nat Rev
Gastroenterol Hepatol. 15:738–752. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Antunovic M, Matic I, Nagy B, Caput
Mihalic K, Skelin J, Stambuk J, Josipovic P, Dzinic T, Paradzik M
and Marijanovic I: FADD-deficient mouse embryonic fibroblasts
undergo RIPK1-dependent apoptosis and autophagy after NB-UVB
irradiation. J Photochem Photobiol B. 194:32–45. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang KX, Firus J, Prieur B, Jia W and
Rennie PS: To die or to survive, a fatal question for the destiny
of prostate cancer cells after androgen deprivation therapy.
Cancers (Basel). 3:1498–1512. 2011. View Article : Google Scholar
|
|
35
|
Zhang HY, Ma YD, Zhang Y, Cui J and Wang
ZM: Elevated levels of autophagy-related marker ULK1 and
mitochondrion-associated autophagy inhibitor LRPPRC are associated
with biochemical progression and overall survival after androgen
deprivation therapy in patients with metastatic prostate cancer. J
Clin Pathol. 70:383–389. 2017. View Article : Google Scholar
|
|
36
|
Ziparo E, Petrungaro S, Marini ES, Starace
D, Conti S, Facchiano A, Filippini A and Giampietri C: Autophagy in
prostate cancer and androgen suppression therapy. Int J Mol Sci.
14:12090–12106. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Berchem GJ, Bosseler M, Sugars LY, Voeller
HJ, Zeitlin S and Gelmann EP: Androgens induce resistance to
bcl-2-mediated apoptosis in LNCaP prostate cancer cells. Cancer
Res. 55:735–738. 1995.PubMed/NCBI
|
|
38
|
Bonkhoff H, Fixemer T and Remberger K:
Relation between Bcl-2, cell proliferation, and the androgen
receptor status in prostate tissue and precursors of prostate
cancer. Prostate. 34:251–258. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Goodall ML, Fitzwalter BE, Zahedi S, Wu M,
Rodriguez D, Mulcahy-Levy JM, Green DR, Morgan M, Cramer SD and
Thorburn A: The Autophagy Machinery Controls Cell Death Switching
between Apoptosis and Necroptosis. Dev Cell. 37:337–349. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ma X, Zou L, Li X, Chen Z, Lin Z and Wu X:
Inhibition of Autophagy Improves the Efficacy of Abiraterone for
the Treatment of Prostate Cancer. Cancer Biother Radiopharm.
34:181–188. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Newton K, Wickliffe KE, Dugger DL,
Maltzman A, Roose-Girma M, Dohse M, Kőműves L, Webster JD and Dixit
VM: Cleavage of RIPK1 by caspase-8 is crucial for limiting
apoptosis and necroptosis. Nature. 574:428–431. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhou H, Zhu P, Guo J, Hu N, Wang S, Li D,
Hu S, Ren J, Cao F and Chen Y: Ripk3 induces mitochondrial
apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury.
Redox Biol. 13:498–507. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
LaRocca TJ, Stivison EA, Mal-Sarkar T,
Hooven TA, Hod EA, Spitalnik SL and Ratner AJ: CD59 signaling and
membrane pores drive Syk-dependent erythrocyte necroptosis. Cell
Death Dis. 6:e17732015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dehm SM and Tindall DJ: Molecular
regulation of androgen action in prostate cancer. J Cell Biochem.
99:333–344. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tosoian JJ, Trock BJ, Landis P, Feng Z,
Epstein JI, Partin AW, Walsh PC and Carter HB: Active surveillance
program for prostate cancer: An update of the Johns Hopkins
experience. J Clin Oncol. 29:2185–2190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lev A, Lulla AR, Ross BC, Ralff MD, Makhov
PB, Dicker DT and Eldeiry WS: ONC201 Targets AR and AR-V7
Signaling, Reduces PSA, and Synergizes with Everolimus in Prostate
Cancer. Mol Cancer Res. 16:754–766. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bae JS, Park HS, Park JW, Li SH and Chun
YS: Red ginseng and 20(S)-Rg3 control testosterone-induced prostate
hyperplasia by deregulating androgen receptor signaling. J Nat Med.
66:476–485. 2012. View Article : Google Scholar
|
|
48
|
Zabaiou N, Fouache A, Trousson A,
Buñay-Noboa J, Marceau G, Sapin V, Zellagui A, Baron S, Lahouel M
and Lobaccaro JA: Ethanolic extract of Algerian propolis decreases
androgen receptor transcriptional activity in cultured LNCaP cells.
J Steroid Biochem Mol Biol. 189:108–115. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Paschalis A, Sheehan B, Riisnaes R,
Rodrigues DN, Gurel B, Bertan C, Ferreira A, Lambros MBK, Seed G,
Yuan W, et al: Prostate-specific Membrane Antigen Heterogeneity and
DNA Repair Defects in Prostate Cancer. Eur Urol. 76:469–478. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Deng Q and Tang DG: Androgen receptor and
prostate cancer stem cells: Biological mechanisms and clinical
implications. Endocr Relat Cancer. 22:T209–T220. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yadav N and Heemers HV: Androgen action in
the prostate gland. Minerva Urol Nefrol. 64:35–49. 2012.PubMed/NCBI
|
|
52
|
Tourneur L and Chiocchia G: FADD: A
regulator of life and death. Trends Immunol. 31:260–269. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ang RL and Ting AT: Detection of RIPK1 in
the FADD-Containing Death Inducing Signaling Complex (DISC) During
Necroptosis. Methods Mol Biol. 1857:101–107. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wiese AK, Prior S, Chambers TC and Higuchi
M: Intracellular Oxygen Concentration Determined By Mitochondrial
Respiration Regulates Production of Reactive Oxygen Species. Integr
Cancer Biol Res. 1:0062017.PubMed/NCBI
|
|
55
|
Rasul A, Di J, Millimouno FM, Malhi M,
Tsuji I, Ali M, Li J and Li X: Reactive oxygen species mediate
isoalantolactone-induced apoptosis in human prostate cancer cells.
Molecules. 18:9382–9396. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Anderton H, Bandala-Sanchez E, Simpson DS,
Rickard JA, Ng AP, Di Rago L, Hall C, Vince JE, Silke J, Liccardi
G, et al: RIPK1 prevents TRADD-driven, but TNFR1 independent,
apoptosis during development. Cell Death Differ. 26:877–889. 2019.
View Article : Google Scholar
|