Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of
the most common types of primary carcinoma of the pancreas, with
~56,770 new cases and ~45,750 mortalities annually in the USA in
2019 (1). The 5-year survival rate
remains at 6% due to late diagnosis and therapy resistance
(2). Therefore, it is necessary to
investigate appropriate methods for the diagnosis and treatment of
PDAC.
Vitamin C (VC), a strong reducing agent and
antioxidant, can be oxidized into dehydroascorbic acid (DHA) when
in solution (3). Human cells
uptake VC and DHA through sodium-dependent VC transporters (SVCTs)
and glucose transporters (GLUTs) (4,5). The
high‑affinity SVCT2 transporter is ubiquitously expressed and more
commonly expressed in cancer cells compared with SVCT1 (6). Based on its chemical properties, VC
functions as a cofactor of Fe(II) and is part of the
α-ketoglutarate-dependent dioxygenase family, participating in
biological processes, including collagen synthesis, regulation of
hypoxia-inducible factor stability, and methylation of DNA and
histones (7,8).
Epigenetics, such as DNA methylation, refers to
heritable alterations in gene expression without changes in the DNA
sequence (9). The ten-eleven
translocation (TET) protein family members are considered key
enzymes for DNA demethylation, which can oxidize 5-methylcytosine
(5mC) to 5-hydroxymethylcytosine (5hmC) in a
Fe2+/α-ketoglutarate dependent reaction (10,11).
There are three TET proteins, TET1, 2 and 3, among which TET2 is
the most commonly mentioned member and has been investigated in
previous tumor studies (12,13).
Previous studies have shown that VC is indispensable for TET
enzymes to maintain catalytic activity (14-16).
VC can promote DNA demethylation through TETs, particularly TET2,
to upregulate the expression levels of tumor suppressor genes in
tumor cells and achieve an antitumor effect in leukemia and solid
tumors, including liver, ovarian and kidney cancer (17-20).
Considering the involvement of VC in epigenetic
modifications and the key role of epigenetic dysregulation in the
development of PDAC, VC treatment may be a promising strategy for
PDAC therapy. Although numerous studies have reported the antitumor
activity of VC treatment both in vitro and in vivo
(21-25), previous clinical trials and case
studies concerning PDAC have failed to confirm a clinical efficacy
of VC treatment (26,27). Furthermore, VC treatment has failed
to demonstrate an improvement in patient quality of life and
chemosensitivity (28-31). This may indicate that VC treatment
does not conform to all patients and that biomarkers are required
to identify the sensitivity of VC treatment in individual patients.
Therefore, it is essential to understand the role of VC treatment
in PDAC and to precisely investigate the underlying mechanisms of
action, which would be beneficial to the development of alternative
therapies or to allow for the proper selection of patients for
effective VC treatment in PDAC.
The present study provided the first evidence that
SVCT2 predicted good prognoses in patients with PDAC. VC directly
inhibited the viability of human pancreatic cancer cells in
vitro. Furthermore, VC promoted DNA demethylation of the PH
domain leucine-rich repeat protein phosphatase 2 (PHLPP2) promoter
to upregulate the expression levels of PHLPP2 and exert a tumor
inhibitory effect. As such, the present study illustrated a new
mechanism for VC in the suppression of pancreatic cancer, and
indicated that PHLPP2 may be a biomarker and an epigenetic target
for the clinical application of VC in PDAC.
Materials and methods
Cell culture and reagents
Capan-1 (a poorly-differentiated primary PDAC cell
line) and PANC-1 (a well-differentiated metastasis PDAC cell line)
were purchased from The Cell Bank of Type Culture Collection of
Chinese Academy of Sciences. The PDAC cell lines were cultured in
DMEM (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10%
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and
50 IU/ml penicillin/streptomycin (Gibco; Thermo Fisher Scientific,
Inc.) at 37˚C. VC (Sigma‑Aldrich; Merck KGaA) was diluted in
PBS.
Cell viability assay
Capan-1 and PANC-1 cells were seeded in a 96-well
plate at a density of 1x104 cells/well. Following 24 h
of incubation, the cells were treated with 0, 0.5, 1, 2, 4 or 8 mM
VC for 48 h at 37˚C. Subsequently, cell viability was evaluated
using Cell Counting Kit‑8 (CCK‑8; Dojindo Molecular Technologies,
Inc.), according to the manufacturer's instructions. Absorbance was
measured at 450 nm using an Infinite M200 PRO microplate reader
(Tecan Group, Ltd.).
Colony formation assay
Capan-1 and PANC-1 cells were seeded in a 6-well
plate (1x103/well) and incubated for 24 h, followed by
treatment with 0.5 mM VC at 37˚C for a further 48 h. Following
incubation in fresh medium for 10 days, the cells were washed with
PBS, fixed using 4% paraformalde-hyde for 15 min at room
temperature and stained using 0.01% crystal violet for 20 min at
room temperature. Subsequently, the cell colonies were imaged and
counted in the whole view using an inverted light microscope
(magnification, x40).
Isolation of peripheral blood mononuclear
cells (PBMCs)
Blood samples from 5 patients with PDAC, including 3
males and 2 females (median age, 61.40 years; age range, 55 to 61
years), were obtained in November 2019 from Huashan Hospital, Fudan
University (Shanghai, China). Human blood (~8 ml) was added slowly
into 4 ml Percoll (GE Healthcare). The cells were separated by
gradient centrifugation at 4˚C for 20 min at 850 x g. The upper
phase (buffer and plasma) was discarded and the PBMCs were
collected. The cells were washed using PBS and centrifuged for 5
min at 500 x g at 4˚C. The remaining red blood cells were lysed
using ACK Lysing Buffer (Gibco; Thermo Fisher Scientific, Inc.),
washed in PBS and centrifuged for 5 min at 500 x g at 4˚C. The
cells were finally cultured in DMEM supplemented with 10% FBS and
50 IU/ml penicillin/streptomycin and treated with or without 4 mM
VC at 37˚C for 48 h. The present study was approved by the Huashan
Hospital Institutional Review Board of Fudan University (permit no.
005/13) and written informed consent was obtained from each
participant.
Annexin V/7‑aminoactinomycin D (7‑AAD)
assay
Capan-1, PANC-1 cells and PBMCs were seeded in
6-well plates at 1x106 cells/well and treated with 4 mM
VC at 37˚C for 48 h. The cells were then stained using Annexin V
(BD Biosciences) at 4˚C for 30 min, followed by staining with 7‑AAD
(Invitrogen; Thermo Fisher Scientific, Inc.) for 5 min at room
temperature. The percentage of apoptotic cells and necrotic cells
was measured using a BD FACSCalibur flow cytometer (BD Biosciences)
and analyses using FlowJo software (version 10; FlowJo LLC).
Wound healing assay
For cell migration assays, 1x106
cells/well were seeded in 6-well plates and incubated for 24 h. A
wound was created using a plastic 200-µl tip and rinsed by PBS. The
cells were incubated in DMEM containing 1% FBS with or without 4 mM
VC. Images were obtained at 0 or 24 h post-wounding using an
inverted light microscope (magnification, x100), and the scratch
area was analyzed using the ImageJ wound healing tool (version
1.52a; National Institutes of Health). The cell recovered area
after 24 h was divided by the cell-free area at 0 h of the same
well to obtain the percentage of wound closure.
Genomic DNA isolation and dot blot
Genomic DNA from Capan-1 and PANC-1 cells was
extracted using TIANamp Genomic DNA kit (Tiangen Biotech Co.,
Ltd.), according to the manufacturer's instructions. The
concentration of DNA was quantified using a NanoDrop (Thermo Fisher
Scientific, Inc.). DNA samples were denatured and spotted onto a
nitrocellulose membrane (GE Healthcare) and air‑dried. DNA was
fixed to the membrane by UV crosslinking for 15 min. The membrane
was then blocked with 5% fat-free milk in 0.1% TBS-Tween-20 (TBST)
for 1 h at room temperature and incubated with the primary
anti-5hmC antibody (cat. no. 40000; 1:500; Active Motif, Inc.) or
anti-5mC antibody (cat. no. ab10805; 1:1,000; Abcam) at 4˚C
overnight. The membrane was incubated with a horseradish
peroxidase-conjugated secondary antibody, anti-mouse IgG (cat. no.
7076S; 1:3,000; CST Biological Reagents Co., Ltd.) for 1 h at room
temperature, washed with TBST, then detected using the ImageQuant
LAS 4000 mini scanner (GE Healthcare) with Immobilon-ECL-Ultra-West
ern-HRP-Substrate (EMD Millipore). The 5hmC and 5mC intensity was
quantified using Image J software (version 1.52a; National
Institutes of Health).
RNA extraction and reverse
transcription‑quantitative PCR (RT‑qPCR)
Total RNA from Capan-1 and PANC-1 cells was
extracted using TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. RNA was
reverse-transcribed using a Hifair II 1st Strand cDNA Synthesis
SuperMix kit (Yeasen). qPCR was performed using the SYBR Green mix
(Yeasen) on a 7500 Real-Time PCR system (Applied Biosystems; Thermo
Fisher Scientific, Inc.) according to manufacturer's instructions.
Quantitative gene amplification was performed using the following
ther-mocycling conditions: 40 cycles of pre‑denaturation at 95˚C
for 10 sec, annealing/extension at 60˚C for 34 sec. Relative
expression levels were normalized using the 2-ΔΔCq
(32) method, and β-actin was used
as an internal control. The primers used for RT-qPCR were as
follows: β-actin forward, 5′-AGC GAG CAT CCC CCA AAG TT-3′ and
reverse, 5′-GGG CAC GAA GGC TCA TCA TT-3′; PHLPP2 forward, 5′-AGT
CTT TAC CAT CCG CCT GC-3′ and reverse, 5′-TGG AGT GTG CAA CAA GGG
T-3′; adenomatous polyposis coli (APC) forward, 5′-AAG CAT GAA ACC
GGC TCA CAT-3′ and reverse, 5′-CATTCGTGTAGTTGAACCCTGA-3′;
mitogen-activated protein kinase kinase 4 (MAP2K4) forward, 5′‑CGG
CTC TTC ACT CCC AAC AA-3′ and reverse, 5′-GCT TTG CGT TTA CTT TGT
GCC-3′; F-Box and WD repeat domain containing 7 forward, 5′-TGC AAA
AGA GCC TCT ACC ACA-3′ and reverse, 5′-CAA GCC CAG TGG TAC TTG
TAT-3′; helicase-like transcription factor (HLTF) forward, 5′-GAA
TTG TCT AGC TCC CGC CC-3′ and reverse, 5′-TAG AAG ATC CTT TCG CCC
TGC-3′; HECT domain and ankyrin repeat containing E3 ubiquitin
protein ligase 1 forward, 5′-GGA GTT GCC CGA GGA TAA TGA-3′ and
reverse, 5′-TGC TGC AAT GTG AAG CAA GC‑3′; PLAG1 like zinc finger 1
forward (PLAGL1), 5′‑TCC AGA ACT TTC CAA GCG GG-3′ and reverse,
5′-TCA GAT GTG ACA CGA GGC AG-3′; SNF2 histone linker PHD RING
heli-case (SHPRH) forward, 5′-ACA GGC TGC ATC ATT CGT GA-3′ and
reverse, 5′-TCC CTC GGG AAG AGT GAG AG-3′; TOP1 binding
arginine/serine rich protein forward, 5′-GTC GAG GTG AGG GAG TGA
AA-3′ and reverse, 5′-AAG CCA GTA AGT CGT CGC AC-3′; AT-rich
interactive domain-containing protein 1A (ARID1A) forward, 5′-GCC
GAA TCT CAT GCC TTC CA-3′ and reverse, 5′-GGC CGC TTG TAA TTC TGC
TGT-3′; and SWI/SNF related, matrix associated, actin dependent
regulator of chromatin, subfamily a, member 2 (SMARCA2) forward,
5′-CTG TTT TGA CCG GTT GCC TG-3′ and reverse, 5′-CCA GTC AGT AGA
GTA ATG CTG C-3′.
5hmC and 5mC assay
EpiMark® 5hmC and 5mC Analysis kit (New
England Biolabs, Inc.) was used to quantify 5hmC and 5mC expression
levels, according to the manufacturer′s protocol. Genomic DNA was
treated with T4 β-glucosyltransferase (New England Biolabs, Inc.)
with or without UDP-Glucose substrate (New England Biolabs, Inc.)
at 37˚C overnight. Converted DNA was then digested with or without
MspI and HpaII at 37˚C overnight. 5hmC and 5mC
expression levels were quantitatively analyzed using RT-qPCR with
primers designed at peak regions containing the GGCC sequence that
span one CCGG MspI/HpaII recognition site on PHLPP2.
Primers used for RT-qPCR were as follows: PHLPP2 forward, 5′-GCT
TGC CTG CCC TTG TTA AA3′ and reverse, 5′-CAG TCT GTG TCC CCC ATC
TG-3′.
Small interfering RNA (siRNA)
transfection
siRNA was designed by Suzhou Genepharma Co., Ltd. to
downregulate PHLPP2 and TET2 expression levels. Capan-1 and PANC-1
cells were transfected with siRNAs using Lipofectamine™ 2000
(Invitrogen; Thermo Fisher Scientific, Inc.). A mixture of 100 nM
siRNA and Lipofectamine® were mixed for 20 min at room
temperature, then added to the cells and incubated for 6 h. The
siRNA sequences were as follows: Control siRNA, 5′-UUC UCC GAA CGU
GUC ACG UTT-3′; PHLPP2 siRNA, 5′-GCA ACU UUC UGA CUA CUU UTT-3′;
and TET2 siRNA, 5′-GGA UGU AAG UUU GCC AGA ATT-3′. Subsequent
experiments were performed 48 h post-transfection.
Western blot assays
Capan-1 and PANC-1 cells were serum starved
overnight and treated with epidermal growth factor (10 ng/ml;
PeproTech, Inc.) for 15 min. Cells were lysed using RIPA lysis
buffer (Beyotime Institute of Biotechnology). Following protein
concentration analysis using a BCA protein assay kit (Beyotime
Institute of Biotechnology), SDS-PAGE sample loading buffer
(Beyotime Institute of Biotechnology) was added to the protein
sample. Then, 50 µg proteins were separated via 10% SDS-PAGE
(Epizyme) and transferred to nitrocellulose filter membranes (EMD
Millipore). Subsequently, the membrane was blocked using 5%
fat-free milk in TBST for 1 h at room temperature and incubated
with the primary anti-phosphorylated-c-Raf (Ser338) antibody (cat.
no. 9427S; 1:1,000; CST Biological Reagents Co., Ltd.), anti-c-Raf
antibody (cat. no. 9422S; 1:1,000; CST) or anti-GAPDH antibody
(cat. no. 5174S; 1:1,000; CST Biological Reagents Co., Ltd.) at 4˚C
overnight. The membrane was incubated with a horseradish
peroxidase-conjugated secondary antibody, anti-Rabbit IgG (cat. no.
7074S; 1:5,000; CST Biological Reagents Co., Ltd.) for 1 h at room
temperature, washed with TBST and then bands were detected using
the ImageQuant LAS 4000mini scanner (GE Healthcare). The images
were analyzed using Image J software (version 1.52a; National
Institutes of Health).
Bioinformatics analysis
The RNA-Seq data and clinical information including
overall survival data, disease free time, TNM stage and status of
178 patients of The Cancer Genome Atlas (TCGA) PAAD dataset were
download the from UCSC Xena platform (https://xena.ucsc.edu/). Patient characteristics were
listed in Table SI. The RPPA data
of 116 PAAD patients were accessed and processed using the
linkedomics (http://linkedo-mics.org/login.php) (33). For survival analysis, the median of
SVCT1/2, GLUT1 and PHLPP2 expression was used to separate PAAD
patients from TCGA database into the 'high' and 'low' groups.
Survival analysis for overall survival (OS) and disease‑free
survival (DFS) was performed using the Kaplan Meier method and the
log rank test was carried out using the R software 'survival'
package (https://github.com/ther-neau/survival). The
correlation between the expression of genes of TCGA PAAD dataset
was analyzed by Pearson's correlation and curves were added
according to the TIMER database algorithm (34). To identify he potential tumor
suppressor target genes of VC, genes included in the TSGene 2.0
database (35) and positively
correlated with TET2 and SVCT2 (Pearson's correlation ≥0.5) were
screened to generate a heat map using R software 'pheatmap' package
(https://CRAN.R-project.org/package=pheatmap) and to
validate experimentally.
Statistical analysis
Statistical significance was evaluated by an
unpaired Student's t-tests or one-way ANOVA followed by Tukey's
test for multiple comparisons using GraphPad Prism 7.0 software
(GraphPad Software, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
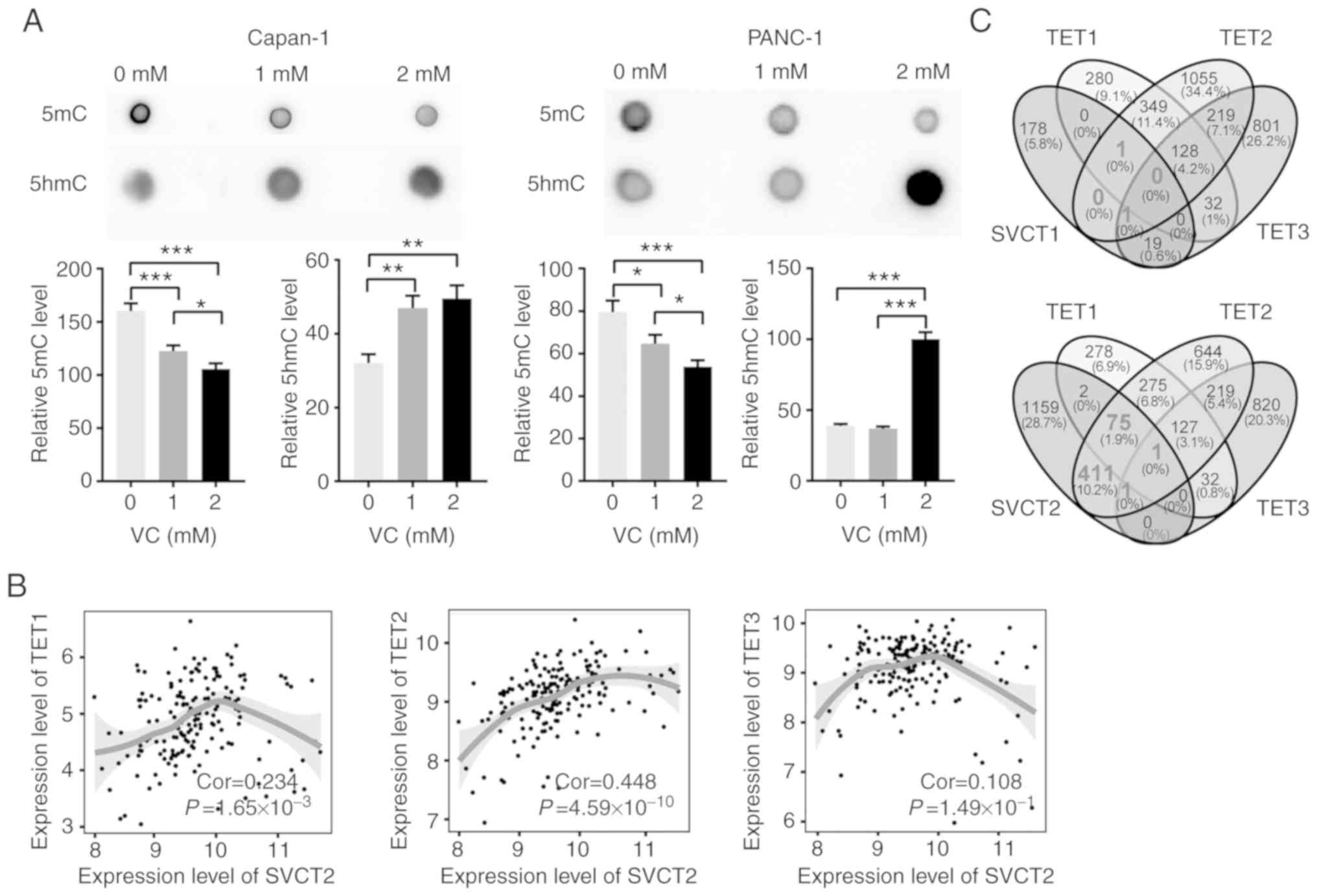
VC exhibits antitumor effects in
pancreatic cancer
To investigate the role of VC in the progression of
pancreatic cancer, the associations between the expression levels
of VC/DHA transporters (SVCT1, SVCT2 and GLUT1) and survival rates
of patients with PDAC were identified. According to the
Kaplan‑Meier survival analysis of TCGA data (Fig. 1A), high expression levels of SVCT2
predicted good overall survival (P=0.039), while high expression of
GLUT1 was associated with poor overall survival (P=0.028) of
patients with PDAC. Patients with high or low expression of SVCT1
had no significant difference in the survival rate, which may due
to the fact that SVCT2 is the predominant transporter of VC in the
pancreas (36). The level of SVCT2
in patients with stage I disease was higher than that in patients
with stage III or IV disease, and significantly higher than that in
patients with stage II. However, there was no significant
difference in DFS rate between patients with high or low expression
levels of SVCT2 (Fig. S1). Since
SVCT2 is an important VC transporter, these results indicated that
VC may exhibit an antitumor effect in pancreatic cancer. In order
to confirm the aforementioned hypothesis, human pancreatic cancer
cells Capan-1 and PANC-1 were treated with various concentrations
of VC for 48 h in vitro. CCK‑8 analyses demonstrated that VC
treatment inhibited the proliferation of Capan-1 and PANC-1 cells
in a concentration-dependent manner (Fig. 1B). Subsequently, colony formation
assays were performed to further evaluate the role of VC on tumor
cell growth. The ability of both Capan-1 and PANC-1 cells to form
colonies was significantly impaired in response to VC treatment
(Fig. 1C). Annexin V/7-AAD
staining assays were utilized to determine whether VC treatment
contributed to cell apoptosis. VC significantly induced cell
apoptosis in both cell lines (Fig.
1D). Meanwhile, the effect of VC on cell migration was
evaluated using wound healing assays. Since both Capan-1 and PANC-1
cells couldn't survive in serum-starved medium for 24 h, 1% FBS was
used for the wound healing assays, which may be a limitation of the
study. Following VC treatment, Capan-1 and PANC-1 cells had a
significantly reduced migratory ability compared with those in the
control group (Fig. 1E). To
evaluate the safety of VC, the apoptosis of PBMCs, which were
isolated from patients with pancreatic cancer and treated with or
without VC, were analyzed using flow cytometry. The percentage of
apoptotic PBMCs was not significantly increased following VC
supplementation (Fig. S2), while
VC could significantly induce apoptosis in PDAC cells (Fig. 1D). Therefore, the aforementioned
data suggest that VC could inhibit the proliferation, growth and
migration of tumor cells, while inducing tumor cell apoptosis.
Collectively, the current findings suggested that VC may exhibit an
antitumor effect in pancreatic cancer.
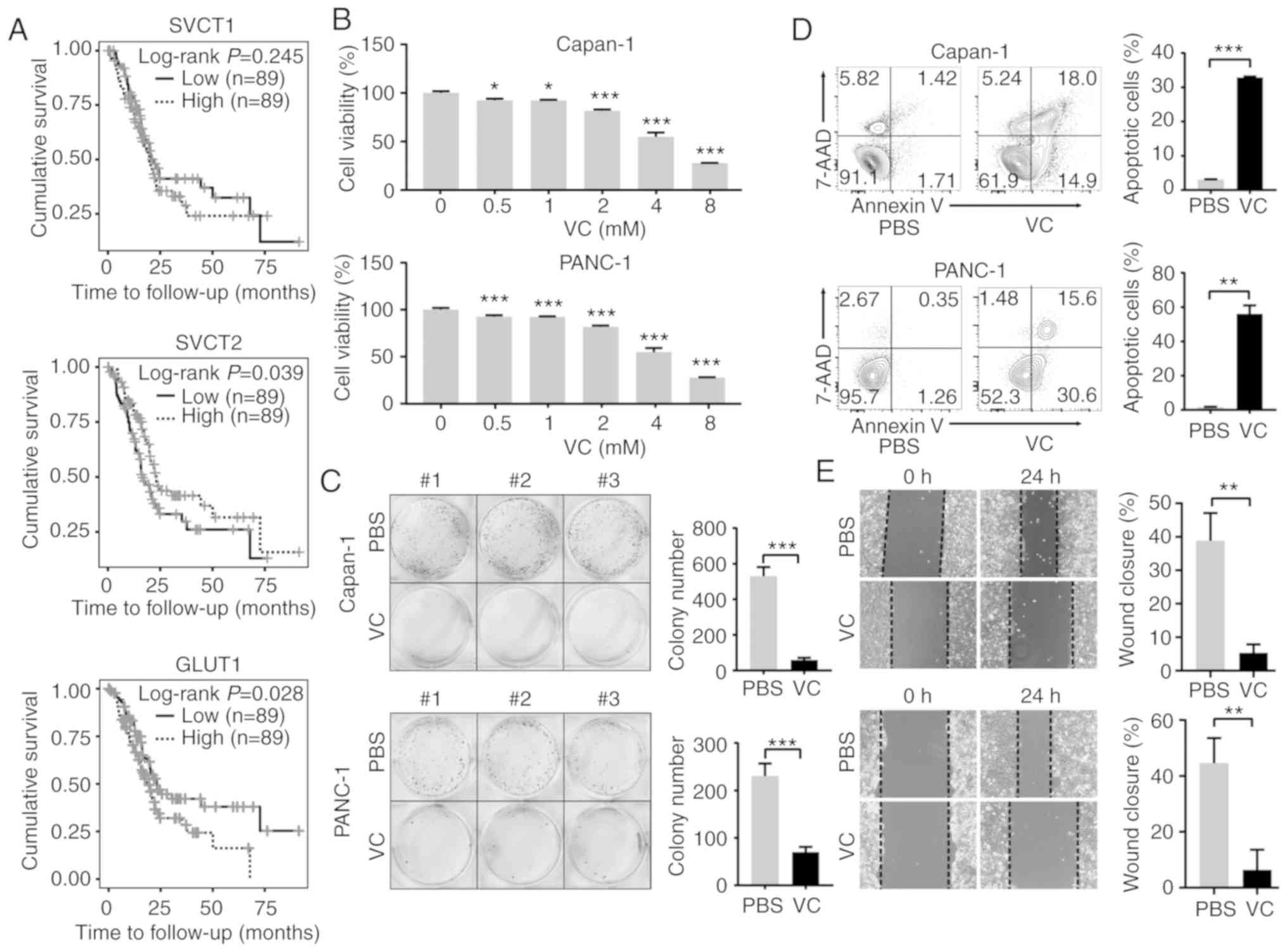 | Figure 1VC exhibits an antitumor effect in
pancreatic cancer. (A) Kaplan‑Meier analysis was used to analyze
the associations between SVCT1, SVCT2 and GLUT1 expression levels
and the overall survival rate of patients with pancreatic cancer.
(B) Capan-1 or PANC-1 cells were exposed to various concentrations
of VC for 48 h, and cell viability was assessed using Cell Counting
Kit‑8 assays. Data are presented as the percentage of the untreated
control. *P<0.05, ***P<0.001 vs. 0 mM
VC. (C) Colony formation assay was performed to measure cell
proliferation. Capan-1 or PANC-1 cells were seeded into plates at a
density of 1,000/well and grown at 37˚C for 10 days after exposure
to 0.5 mM VC for 48 h. Cell colonies were stained with 0.1% crystal
violet (left), and colony numbers were quantified using ImageJ
software (right). ***P<0.001. (D) Capan-1 or PANC-1
cells were treated with 4 mM VC for 48 h, and then examined by flow
cytometry to assess the levels of apoptosis.
**P<0.01, ***P<0.001. (E) Following
incubation for 24 h, a scratch was made at the 0 h time point, then
Capan-1 or PANC-1 cells were treated with 4 mM VC for a further 24
h before images (magnification, x100) were obtained and data were
quantified. **P<0.01. Data are presented as the mean
± standard error of three independent experiments. SVCT,
sodium-dependent VC transporter; VC, vitamin C; GLUT1, glucose
transporter 1; 7-AAD, 7-aminoactinomycin D. |
VC increases demethylation of pancreatic
cancer cells relying on TET2
Epigenetic regulation plays a crucial role in the
development of PDAC; VC takes part in the epigenetic regulation,
particularly in the conversion of 5mC to 5hmC by enhancing the
catalytic activity of TET dioxygenases (5). Thus, the effect of VC on the global
levels of 5mC/5hmC was examined in both Capan-1 and PANC-1 cell
lines using dot blot assays. A significant increase in global 5hmC
levels was observed following VC treatment compared with PBS
treatment, accompanied by a significant reduction in 5mC levels in
both cell lines (Fig. 2A). The
mRNA level of SVCT2 was positively correlated with that of TET1,
TET2 and TET3 in tumor tissues from PDAC patients from TCGA
database, with TET2 demonstrating the strongest correlation among
the TET family (Fig. 2B). To
further determine the downstream genes that may be regulated by
TET2 and VC, Pearson's correlation analysis was performed based on
the gene expression levels from the PDAC dataset. A total of 488
genes were significantly correlated with the expression levels of
TET2 and SVCT2, while only 78 and 2 genes were positively
correlated with SVCT2 for TET1 and TET3, respectively (Fig. 2C). This suggests VC may increase
the expression level of 5hmC in pancreatic cancer cells, which
mainly relies on TET2.
VC specifically upregulates PHLPP2
through DNA demethylation
To seek the potential target genes of VC, the TSGene
2.0 database, an updated gene set of tumor suppressor genes, was
used to screen the genes that may be involved in the antitumor
activity of VC. Initially, the present study screened for genes
that positively correlated with TET2 and SVCT2 (Pearson's
correlation ≥0.5). Then, tumor suppressor genes identified by the
TSGene database within these genes were selected, resulting in 36
genes. Among the 36 genes, 11 genes were selected based on previous
reports associated with the progression of PDAC (37-41)
(Fig. 3A). RT-qPCR was used to
validate the expression levels of these 11 genes after VC treatment
in both Capan-1 and PANC-1 cell lines. Following VC treatment,
SHPRH, ARID1A, SMARCA2, PLAGL1 and HLTF were significantly
decreased, and only PHLPP2 was significantly increased in Capan-1.
Following treatment of PANC-1 cells with VC, MAP2K4 was
significantly downregulated, and PHLPP2 and APC were significantly
upregulated. Notably, only PHLPP2 exhibited a significant increase
in both cell lines, with a 2.4-fold change in Capan-1 and 5.3-fold
change in PANC-1 following exposure to VC (Fig. 3B). To further investigate whether
the upregulation of PHLPP2 resulted from a VC-stimulated increase
in TET2-dependent DNA demethylation, a promoter methylation assay
was utilized to measure the expression levels of 5mC/5hmC at the
PHLPP2 promoter. VC supplementation significantly increased the
percentage of 5hmC, accompanied with a decrease in 5mC at the
PHLPP2 promoter region. Knocking down the expression levels of TET2
by siRNA (Fig. S3A), led to a
decrease in 5hmC levels and increase in 5mC levels at the PHLPP2
promoter, while VC supplementation could not reverse the reduction
(Fig. 3C). Thus, the current
findings suggested that VC could upregulate the PHLPP2 expression
levels by enhancing PHLPP2 promoter demethylation, depending on
TET2.
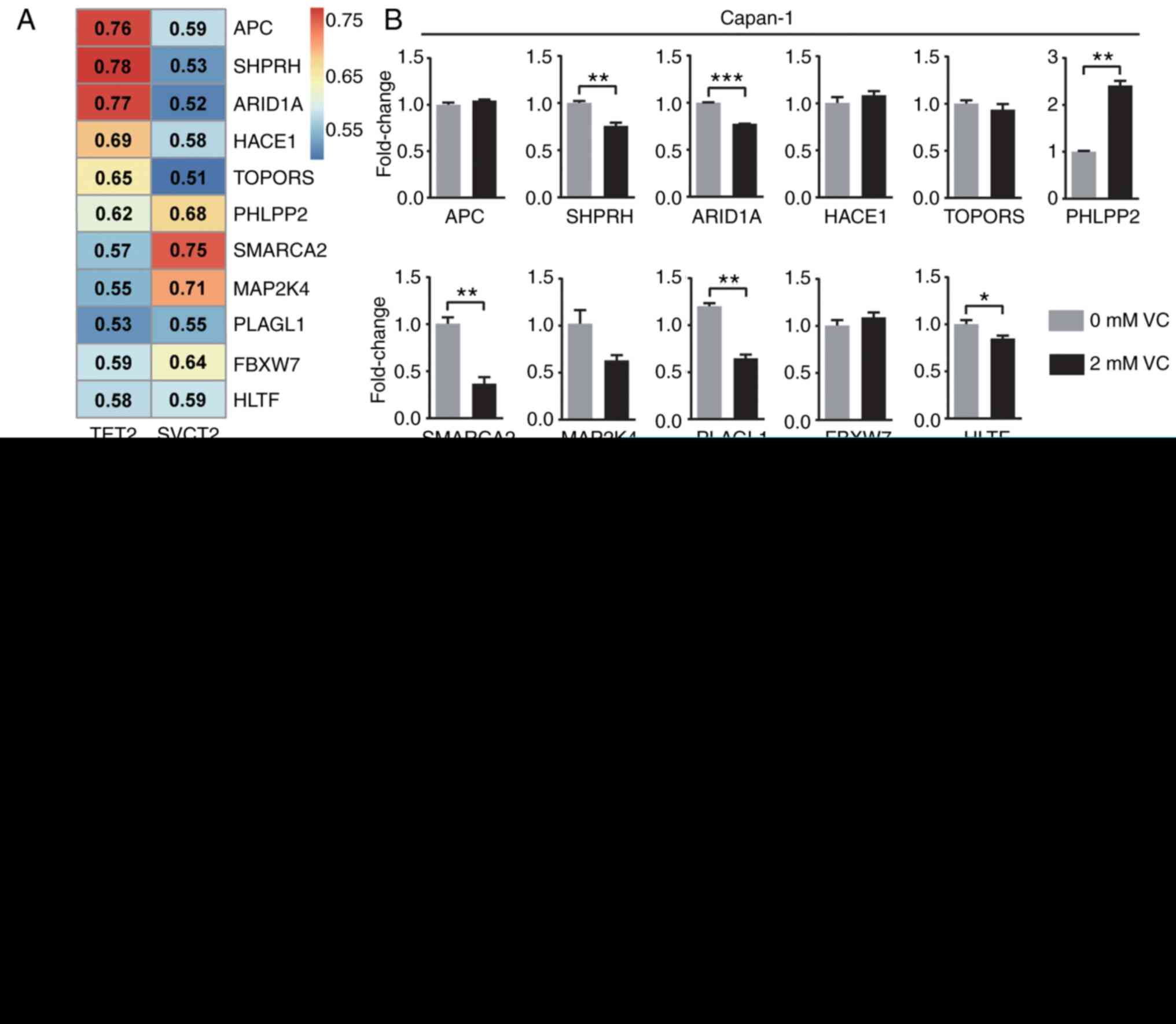 | Figure 3VC specifically upregulates PHLPP2
through DNA demethylation. (A) Heatmap of the correlation between
tumor suppressor genes and SVCT2/TET2. (B) Quantitative changes in
the mRNA expression levels of tumor suppressor genes in Capan-1 or
PANC-1 cells with VC treatment. (C) The levels of 5mC/5hmC in the
PHLPP2 promoter of siNC/siPHLPP2-treated PDAC cells were detected
following VC supplementation. Data are presented as the mean ±
standard error of the mean of three independent experiments.
*P<0.05, **P<0.01,
***P<0.001. PHLPP2, PH domain leucine-rich repeat
protein phosphatase 2; si, small interfering RNA; VC, vitamin C;
NC, negative control; 5mC, 5-methylcytosine; 5hmC,
5-hydroxymethylcytosine; SVCT, sodium-dependent VC transporter;
TET2, ten-eleven translocation 2; APC, adenomatous polyposis coli;
SHPRH, SNF2 histone linker PHD RING helicase; ARID1A, AT-rich
interactive domain-containing protein 1A; HACE1, HECT domain and
ankyrin repeat containing E3 ubiquitin protein ligase 1; TOPORS,
TOP1 binding arginine/serine rich protein; SMARCA2, SWI/SNF
related, matrix associated, actin dependent regulator of chromatin,
subfamily a, member 2; MAP2K4, mitogen‑activated protein kinase
kinase 4; PLAGL1, PLAG1 like zinc finger 1; FBXW7, F‑Box and WD
repeat domain containing 7; HLTF, helicase‑like transcription
factor. |
Antitumor effect of VC partially depends
on PHLPP2
PHLPP2 is a tumor suppressor gene that has been
reported to inhibit progression of various cancer types, including
breast, prostate, bladder and lung cancer (42). Similarly, as shown in the
Kaplan‑Meier analysis, patients with high PHLPP2 expression levels
had a significantly higher overall survival rate compared with
those with low PHLPP2 levels (P=0.003; Fig. 4A). To confirm whether PHLPP2 plays
a major role in the inhibitory effect of VC on PDAC, PHLPP2
expression in Capan-1 and PANC-1 cells was knocked down using
siRNAs (Fig. S3B), which were
then subjected to VC treatment. VC-induced cell apoptosis was
significantly reduced in response to PHLPP2 silencing; however,
VC-induced cell necrosis was reduced by knockdown of the expression
of PHLPP2 only in Capan-1 cells (Figs.
4B and S4). Meanwhile,
VC-mediated reduction of the migration in both cell lines was
partially restored through silencing of PHLPP2 (Fig. 4C). Furthermore, western blot assays
were used to detect the phosphorylation levels of Raf1, which has
been reported to be dephosphorylated by PHLPP2 at S338 (43). PHLPP2‑knockdown resulted in a
significant increase in Raf1 phosphorylation, with VC able to
attenuate the increase (Fig. 4D).
The RPPA data of patients with PDAC from TCGA database also
demonstrated that PHLLP2 exhibited a significant negative
correlation with the levels of phosphory-lated Raf1 (Pearson's
correlation, -0.249; P=6.91x10-3), while SVCT2 was also
negatively correlated with the levels of C-Raf pS338 (Pearson's
correlation, -0.162; P=8.16x10-2; Fig. S5). Collectively, the current data
demonstrated that the antitumor effect of VC was partially
dependent on PHLPP2.
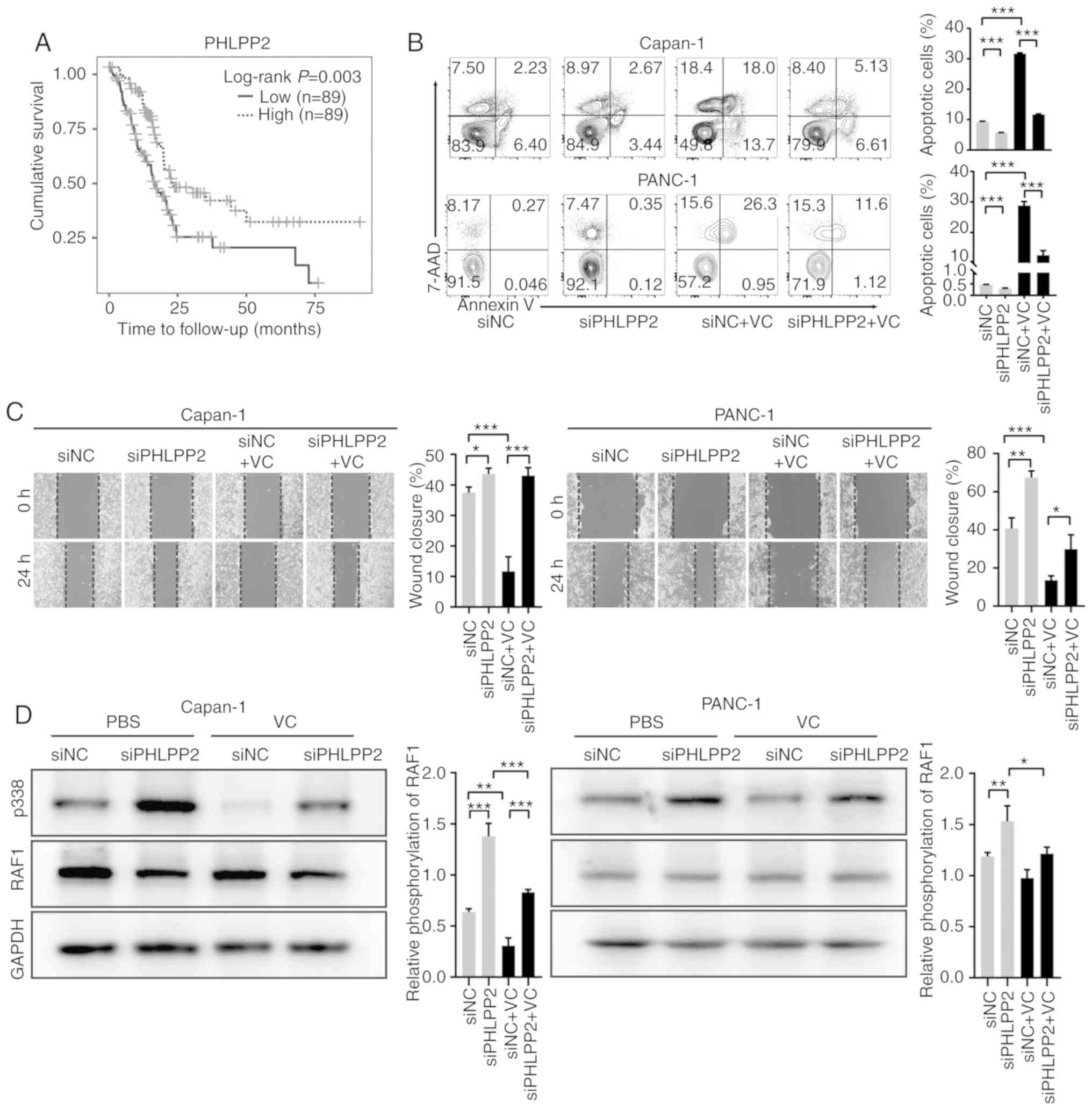 | Figure 4Antitumor effect of VC partially
depends on PHLPP2. (A) Survival analysis of PHLPP2 in patients with
PDAC. (B) Following PHLPP2-knockdown, Capan-1 or PANC-1 cells were
treated with 4 mM VC, and apoptosis was assessed using Annexin
V/7AAD staining. (C) The migratory ability of the cells was
assessed using wound healing assays. (D) siNC and siPHLPP2-treated
PDAC cells were serum starved overnight and treated with epidermal
growth factor (10 ng/ml) for 15 min, and cell lysates were analyzed
for phosphorylated and total RAF1 expression levels. Data are
presented as the mean ± standard error of the mean of three
independent experiments. *P<0.05,
**P<0.01, ***P<0.001. PHLPP2, PH domain
leucine-rich repeat protein phosphatase 2; si, small interfering
RNA; VC, vitamin C; NC, negative control; 7-AAD, 7-aminoactinomycin
D. |
Discussion
VC treatment has been reported to exhibit an
antitumor effect in cancer treatment; however, there is a lack of
convincing clinical evidence, particularly in pancreatic cancer
(19,23,44).
The present study investigated the association between the
epigenetic regulation of VC and pancreatic cancer, which
demonstrated that VC exerted its antitumor activity partially by
upregulating PHLPP2 expression levels through DNA demethylation.
The 5hmC levels of PHLPP2 may act as a biomarker and an epigenetic
target of VC for pancreatic cancer therapy.
The mechanisms of action underlying the antitumor
effect of VC include oxidative stress and epigenetic regulation,
with most previous studies predominantly focusing on the former
(24,25). However, Ge et al (45) have found that oxidation-resistant
VC derivatives, which lack the ability to induce oxidative stress
in tumor cells, still exhibit antitumor efficacy. Previously,
epigenetic regulation has gained increasing attention as a
supplementary mechanism for the action of VC against tumors
(16,17,45,46).
It has been well established that epigenetic dysregulation is
recognized as a hallmark in the development of multiple tumor
types, such as breast, liver, lung and prostate cancer (11,47,48).
For example, the levels of 5hmC were found to be significantly
decreased, accompanied with a 5mC increase in various cancer types,
including PDAC, which may lead to the silencing of tumor suppressor
genes (47,48). Since VC serves as a co-factor of
TET2 and is indispensable for its optimal catalytic activity, high
dose VC treatment may enhance the activity of TET2 leading to an
increase in the levels of 5hmC in tumors (45). Consistent with the aforementioned
findings, the present study demonstrated that VC could reverse the
loss of 5hmC through TET2, and identified an increase in 5hmC
levels in the promoter region of the tumor suppressor gene
PHLPP2.
The PHLPP family consists of PHLPP1 and PHLPP2.
PHLPP2 can dephosphorylate AKT and Raf1, resulting in the induction
of apoptosis and the inhibition of migration, which is consistent
with the current results (49,50).
PHLPP2 has been reported to be downregulated in various tumor
types, including PDAC (51), and
in vitro studies have demonstrated that overexpression of
PHLPP2 can inhibit tumor growth in prostate, bladder and
endometrial cancer (50,52). The present study confirmed that
PHLPP2 re-expression could inhibit pancreatic cancer cells.
Furthermore, the participation of VC‑induced epigenetic
reprogramming was also identified in PHLPP2 re-expression.
Due to the paradoxical conclusions in clinical
trials of VC treatment, biomarkers are required for the proper
selection of patients that are sensitive to VC-based therapies, to
substantially improve the clinical efficacy of VC (44,46).
According to the current results, several conclusions regarding
biomarkers could be made. For example, patients with low expression
levels of SVCT2 may not benefit from VC treatment because VC cannot
be effectively transported into tumor cells (53). In addition, patients with low
levels of 5hmC in tumor tissues may be more responsive to VC
treatment. Specifically, in PDAC, low expression levels of 5hmC in
the promotor region of PHLPP2 in tumor tissues may be an ideal
indicator for VC treatment.
In conclusion, the present study identified that
SVCT2 is positively correlated with the prognosis of patients with
PDAC. VC treatment can directly inhibit human pancreatic tumor
cells, which is partially dependent on the upregulation of PHLPP2
through DNA demethylation. The current results suggest a novel
epigenetic regulatory mechanism for the anti-tumor effect of VC and
provide a theoretical basis for using PHLPP2 as a novel biomarker
and epigenetic target in VC treatment of pancreatic cancer.
Supplementary Data
Funding
This work was supported by the National Natural
Science Foundation of China (grant nos. 81830080, 81870375,
81870456 and 81600438) and the Shanghai Rising-Star Program (grant
no. 18QA1401000).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
FL and JL designed the research. LC, HS, ZL, HC, WZ,
YL and WL performed experiments and analyzed the data. FL, JL, LC
and HS wrote manuscript. LC, HS, ZL and HC contributed equally to
the work. FL and JL revised the manuscript and jointly supervised
the work. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Huashan
Hospital Institutional Review Board of Fudan University (Shanghai,
China; permit no. 005/13) and written informed consent was obtained
from each participant.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors wish to thank to Ms. Diana Tseng (Fudan
University, Shanghai, China) for critically reading the
manuscript.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2019. CA Cancer J Clin. 69:7–34. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lee S, Kim J, Jung S, Li C, Yang Y, Kim
KI, Lim JS, Kim Y, Cheon CI and Lee MS: SIAH1-induced p34SEI-1
polyubiq-uitination/degradation mediates p53 preferential vitamin C
cytotoxicity. Int J Oncol. 46:1377–1384. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vera JC, Rivas CI, Fischbarg J and Golde
DW: Mammalian facilitative hexose transporters mediate the
transport of dehydro-ascorbic acid. Nature. 364:79–82. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tsukaguchi H, Tokui T, Mackenzie B, Berger
UV, Chen XZ, Wang Y, Brubaker RF and Hediger MA: A family of
mammalian Na+-dependent L-ascorbic acid transporters. Nature.
399:70–75. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
McCarty MF: Expression and/or activity of
the SVCT2 ascorbate transporter may be decreased in many aggressive
cancers, suggesting potential utility for sodium bicarbonate and
dehydro-ascorbic acid in cancer therapy. Med Hypotheses.
81:664–670. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kuiper C and Vissers MC: Ascorbate as a
co‑factor for fe‑ and 2-oxoglutarate dependent dioxygenases:
Physiological activity in tumor growth and progression. Front
Oncol. 4:3592014. View Article : Google Scholar
|
|
8
|
Mastrangelo D, Pelosi E, Castelli G,
Lo-Coco F and Testa U: Mechanisms of anti-cancer effects of
ascorbate: Cytotoxic activity and epigenetic modulation. Blood
Cells Mol Dis. 69:57–64. 2018. View Article : Google Scholar
|
|
9
|
Navada SC, Steinmann J, Lübbert M and
Silverman LR: Clinical development of demethylating agents in
hematology. J Clin Invest. 124:40–46. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tahiliani M, Koh KP, Shen Y, Pastor WA,
Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L and
Rao A: Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in
mammalian DNA by MLL partner TET1. Science. 324:930–935. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ushijima T: Cancer epigenetics: Now
harvesting fruit and seeding for common diseases. Biochem Biophys
Res Commun. 455:1–2. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rasmussen KD and Helin K: Role of TET
enzymes in DNA meth-ylation, development, and cancer. Genes Dev.
30:733–750. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Woods BA and Levine RL: The role of
mutations in epigenetic regulators in myeloid malignancies. Immunol
Rev. 263:22–35. 2015. View Article : Google Scholar
|
|
14
|
Minor EA, Court BL, Young JI and Wang G:
Ascorbate induces ten-eleven translocation (Tet) methylcytosine
dioxygenase-mediated generation of 5-hydroxymethylcytosine. J Biol
Chem. 288:13669–13674. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yin R, Mao SQ, Zhao B, Chong Z, Yang Y,
Zhao C, Zhang D, Huang H, Gao J, Li Z, et al: Ascorbic acid
enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA
demethylation in mammals. J Am Chem Soc. 135:10396–10403. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Blaschke K, Ebata KT, Karimi MM,
Zepeda‑Martinez JA, Goyal P, Mahapatra S, Tam A, Laird DJ, Hirst M,
Rao A, et al: Vitamin C induces Tet-dependent DNA demethylation and
a blastocyst-like state in ES cells. Nature. 500:222–226. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cimmino L, Dolgalev I, Wang Y, Yoshimi A,
Martin GH, Wang J, Ng V, Xia B, Witkowski MT, Mitchell-Flack M, et
al: Restoration of TET2 function blocks aberrant self-renewal and
leukemia progression. Cell. 170:1079–1095.e20. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin JR, Qin HH, Wu WY, He SJ and Xu JH:
Vitamin C protects against UV irradiation-induced apoptosis through
reactivating silenced tumor suppressor genes p21 and p16 in a
Tet-dependent DNA demethylation manner in human skin cancer cells.
Cancer Biother Radiopharm. 29:257–264. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gillberg L, Ø rskov AD, Liu M, Harsløf
LBS, Jones PA and Grønbaek K: Vitamin C‑A new player in regulation
of the cancer epigenome. Semin Cancer Biol. 51:59–67. 2018.
View Article : Google Scholar
|
|
20
|
Ngo B, Van Riper JM, Cantley LC and Yun J:
Targeting cancer vulnerabilities with high-dose vitamin C. Nat Rev
Cancer. 19:271–282. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Takemura Y, Satoh M, Satoh K, Hamada H,
Sekido Y and Kubota S: High dose of ascorbic acid induces cell
death in mesothelioma cells. Biochem Biophys Res Commun.
394:249–253. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Reddy VG, Khanna N and Singh N: Vitamin C
augments chemotherapeutic response of cervical carcinoma HeLa cells
by stabilizing P53. Biochem Biophys Res Commun. 282:409–415. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cha J, Roomi MW, Ivanov V, Kalinovsky T,
Niedzwiecki A and Rath M: Ascorbate supplementation inhibits growth
and metastasis of B16FO melanoma and 4T1 breast cancer cells in
vitamin C‑deficient mice. Int J Oncol. 42:55642013. View Article : Google Scholar
|
|
24
|
Baek MW, Cho HS, Kim SH, Kim WJ and Jung
JY: Ascorbic acid induces necrosis in human laryngeal squamous cell
carcinoma via ROS, PKC, and calcium signaling. J Cell Physiol.
232:417–425. 2017. View Article : Google Scholar
|
|
25
|
Su X, Shen Z, Yang Q, Sui F, Pu J, Ma J,
Ma S, Yao D, Ji M and Hou P: Vitamin C kills thyroid cancer cells
through ROS‑dependent inhibition of MAPK/ERK and PI3K/AKT pathways
via distinct mechanisms. Theranostics. 9:4461–4473. 2019.
View Article : Google Scholar :
|
|
26
|
Polireddy K, Dong R, Reed G, Yu J, Chen P,
Williamson S, Violet PC, Pessetto Z, Godwin AK, Fan F, et al: High
dose parenteral ascorbate inhibited pancreatic cancer growth and
metastasis: Mechanisms and a phase I/IIa study. Sci Rep.
7:171882017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Welsh JL, Wagner BA, van't Erve TJ, Zehr
PS, Berg DJ, Halfdanarson TR, Yee NS, Bodeker KL, Du J, Roberts LJ
II, et al: Pharmacological ascorbate with gemcitabine for the
control of metastatic and node-positive pancreatic cancer (PACMAN):
Results from a phase I clinical trial. Cancer Chemother Pharmacol.
71:765–775. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hoffer LJ, Levine M, Assouline S,
Melnychuk D, Padayatty SJ, Rosadiuk K, Rousseau C, Robitaille L and
Miller WH Jr: Phase I clinical trial of i.v. ascorbic acid in
advanced malignancy. Ann Oncol. 19:1969–1974. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Carr AC, Vissers MC and Cook JS: The
effect of intravenous vitamin C on cancer- and chemotherapy-related
fatigue and quality of life. Front Oncol. 4:2832014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Espey MG, Chen P, Chalmers B, Drisko J,
Sun AY, Levine M and Chen Q: Pharmacologic ascorbate synergizes
with gemcitabine in preclinical models of pancreatic cancer. Free
Radic Biol Med. 50:1610–1619. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Drisko JA, Serrano OK, Spruce LR, Chen Q
and Levine M: Treatment of pancreatic cancer with intravenous
vitamin C: A case report. Anticancer Drugs. 29:373–379. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
33
|
Vasaikar SV, Straub P, Wang J and Zhang B:
LinkedOmics: Analyzing multi-omics data within and across 32 cancer
types. Nucleic Acids Res. 46(D1): D956–D963. 2018. View Article : Google Scholar :
|
|
34
|
Li T, Fan J, Wang B, Traugh N, Chen Q, Liu
JS, Li B and Liu XS: TIMER: A web server for comprehensive analysis
of tumor‑infiltrating immune cells. Cancer Res. 77:e108–e110. 2017.
View Article : Google Scholar
|
|
35
|
Zhao M, Kim P, Mitra R, Zhao J and Zhao Z:
TSGene 2.0: An updated literature-based knowledgebase for tumor
suppressor genes. Nucleic Acids Res. 44(D1): D1023–D1031. 2016.
View Article : Google Scholar :
|
|
36
|
Subramanian VS, Srinivasan P and Said HM:
Uptake of ascorbic acid by pancreatic acinar cells is negatively
impacted by chronic alcohol exposure. Am J Physiol Cell Physiol.
311:C129–C135. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Waddell N, Pajic M, Patch AM, Chang DK,
Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al:
Whole genomes redefine the mutational landscape of pancreatic
cancer. Nature. 518:495–501. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Smith AJ, Wen YA, Stevens PD, Liu J, Wang
C and Gao T: PHLPP negatively regulates cell motility through
inhibition of Akt activity and integrin expression in pancreatic
cancer cells. Oncotarget. 7:7801–7815. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang Z, Wang F, Du C, Guo H, Ma L, Liu X,
Kornmann M, Tian X and Yang Y: BRM/SMARCA2 promotes the
proliferation and chemoresistance of pancreatic cancer cells by
targeting JAK2/STAT3 signaling. Cancer Lett. 402:2132242017.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Niu N, Lu P, Yang Y, He R, Zhang L, Shi J,
Wu J, Yang M, Zhang ZG, Wang LW, et al: Loss of Setd2 promotes
Kras-induced acinar-to-ductal metaplasia and epithelia-mesenchymal
transition during pancreatic carcinogenesis. Gut. Jul 11–2019.Epub
ahead of print. PubMed/NCBI
|
|
41
|
Grant RC, Selander I, Connor AA,
Selvarajah S, Borgida A, Briollais L, Petersen GM, Lerner-Ellis J,
Holter S and Gallinger S: Prevalence of germline mutations in
cancer predisposition genes in patients with pancreatic cancer.
Gastroenterology. 148:556–564. 2015. View Article : Google Scholar :
|
|
42
|
Newton AC and Trotman LC: Turning off AKT:
PHLPP as a drug target. Annu Rev Pharmacol Toxicol. 54:537–558.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li X, Stevens PD, Liu J, Yang H, Wang W,
Wang C, Zeng Z, Schmidt MD, Yang M, Lee EY and Gao T: PHLPP is a
negative regulator of RAF1, which reduces colorectal cancer cell
motility and prevents tumor progression in mice. Gastroenterology.
146:1301–1312. e1–e10. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shenoy N, Creagan E, Witzig T and Levine
M: Ascorbic acid in cancer treatment: Let the phoenix fly. Cancer
Cell. 34:7007062018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ge G, Peng D, Xu Z, Guan B, Xin Z, He Q,
Zhou Y, Li X, Zhou L and Ci W: Restoration of
5-hydroxymethylcytosine by ascorbate blocks kidney tumour growth.
EMBO Rep. 19:pii: e45401. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cimmino L, Neel BG and Aifantis I: Vitamin
C in stem cell reprogramming and cancer. Trends Cell Biol.
28:698–708. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yang H, Liu Y, Bai F, Zhang JY, Ma SH, Liu
J, Xu ZD, Zhu HG, Ling ZQ, Ye D, et al: Tumor development is
associated with decrease of TET gene expression and
5-methylcytosine hydrox-ylation. Oncogene. 32:663–669. 2013.
View Article : Google Scholar
|
|
48
|
Huang Y and Rao A: Connections between TET
proteins and aberrant DNA modification in cancer. Trends Genet.
30:4644742014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Huang C, Liao X, Jin H, Xie F, Zheng F, Li
J, Zhou C, Jiang G, Wu XR and Huang C: MEG3, as a Competing
endogenous RNA, binds with miR-27a to promote PHLPP2 protein
translation and impairs bladder cancer invasion. Mol Ther Nucleic
Acids. 16:51–62. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Liu J, Eckert MA, Harada BT, Liu SM, Lu Z,
Yu K, Tienda SM, Chryplewicz A, Zhu AC, Yang Y, et al:
m6A mRNA meth-ylation regulates AKT activity to promote
the proliferation and tumorigenicity of endometrial cancer. Nat
Cell Biol. 20:1074–1083. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Nitsche C, Edderkaoui M, Moore RM, Eibl G,
Kasahara N, Treger J, Grippo PJ, Mayerle J, Lerch MM and Gukovskaya
AS: The phosphatase PHLPP1 regulates Akt2, promotes pancreatic
cancer cell death, and inhibits tumor formation. Gastroenterology.
142:377–387. e1–e5. 2012. View Article : Google Scholar
|
|
52
|
Nowak DG, Katsenelson KC, Watrud KE, Chen
M, Mathew G, D'Andrea VD, Lee MF, Swamynathan MM, Casanova-Salas I,
Jibilian MC, et al: The PHLPP2 phosphatase is a druggable driver of
prostate cancer progression. J Cell Biol. 218:1943–1957. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kuiper C, Vissers MC and Hicks KO:
Pharmacokinetic modeling of ascorbate diffusion through normal and
tumor tissue. Free Radic Biol Med. 77:340–352. 2014. View Article : Google Scholar : PubMed/NCBI
|