Introduction
Breast cancer is one of the most common malignancies
among women (1), and is the
leading cause of female cancer-related mortality worldwide; it
currently accounts for >40,000 deaths annually (2). The conventional therapies for breast
cancer are surgery and chemotherapy (3,4);
however, these treatments have severe side-effects and cause
patients physical and psychological distress. Therefore, the
identification of a key therapeutic target molecule that can be
utilized to eliminate the side-effects of current treatments is
required.
A number of researchers have investigated various
means of treating breast cancer, and the phosphoinositide 3-kinase
(PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway is one of
the most frequent targets (5,6).
This pathway is known to regulate several cellular functions, such
as cell proliferation, apoptosis, cell cycle and angiogenesis, and
various metabolic functions, and has been demonstrated to play a
key role in cancer progression (2). The PI3K pathway is commonly mutated
in breast cancer via different mechanisms, which include i) the
enhanced PI3K activity by mutation and/or amplification of PI3K
encoding genes (e.g., PIK3CA, PIK3CB, or PIK3R1); ii) the excessive
activation of receptor tyrosine kinases, such as human epidermal
growth factor receptor 2 (HER2), epidermal growth factor receptor
(EGFR), or insulin-like growth factor 1 receptor (IGF-1R); iii) the
overexpression of the downstream effectors [e.g., Akt1, Akt2, or
3-phosphoinositide-dependent protein kinase-1 (PDK1)]; or iv) loss
of function of tumor suppressor genes, such as phosphatase and
tensin homolog (PTEN) and inositol polyphosphate 4-phosphatase type
II (INPP4B) (7,8). In particular, the increased activity
of the PI3K pathway has been linked with breast cancer
tumori-genesis, drug resistance and clinical outcomes (8); thus, the inhibition of excessive PI3K
pathway activation is viewed as a promising anticancer treatment
strategy in breast cancer. The therapeutic efficacies of PI3K
inhibitors, Akt inhibitors, mTOR inhibitors and dual PI3K/mTOR
inhibitors are currently being evaluated in clinical studies for
the treatment of breast cancer and other types of cancer, including
lung cancer, colorectal cancer and leukemia (9-12).
Despite several clinical trial failures, everolimus (a selective
mTOR inhibitor and rapamycin analogue) has been approved by the
European Medicines Agency (EMA) and the United States Food and Drug
Administration (US FDA) for the treatment of renal cancer, breast
cancer and neuroendocrine tumors of gastrointestinal or lung origin
(13). In addition, alpelisib
(Piqray™, an orally available PI3K inhibitor) was recently approved
for the treatment breast cancer by the FDA (14).
Thus, the authors focused on the development of
potent PI3Kα inhibitors for the treatment of cancer, and identified
a novel imidazopyridine scaffold and synthesized a novel
imidazo[1,2-a]pyridine derivative,
2,4-difluoro-N-(5-(3-(5-methyl-1,2,4-oxadiazol-3-yl)
imidazo[1,2-a]pyridin-6-yl) pyridin-3-yl)benzenesulfonamide
(HS-146) (15). The present study
evaluated whether HS-146 exerts anticancer effects on breast cancer
and investigated the molecular mechanisms of action of HS-146 using
the MCF-7 human breast cancer cell line.
Materials and methods
Preparation of HS-146
The synthetic route for the preparation of HS-146 is
presented in Fig. 1. The pyridyl
sulfonamide group at the C6 position was installed by Suzuki
coupling using the known intermediate oxadiazole 'A2' to
provide 'B'. Treatment of 'B' with 2,4-difluorobenzenesulfonyl
chloride and pyridine then afforded HS-146.
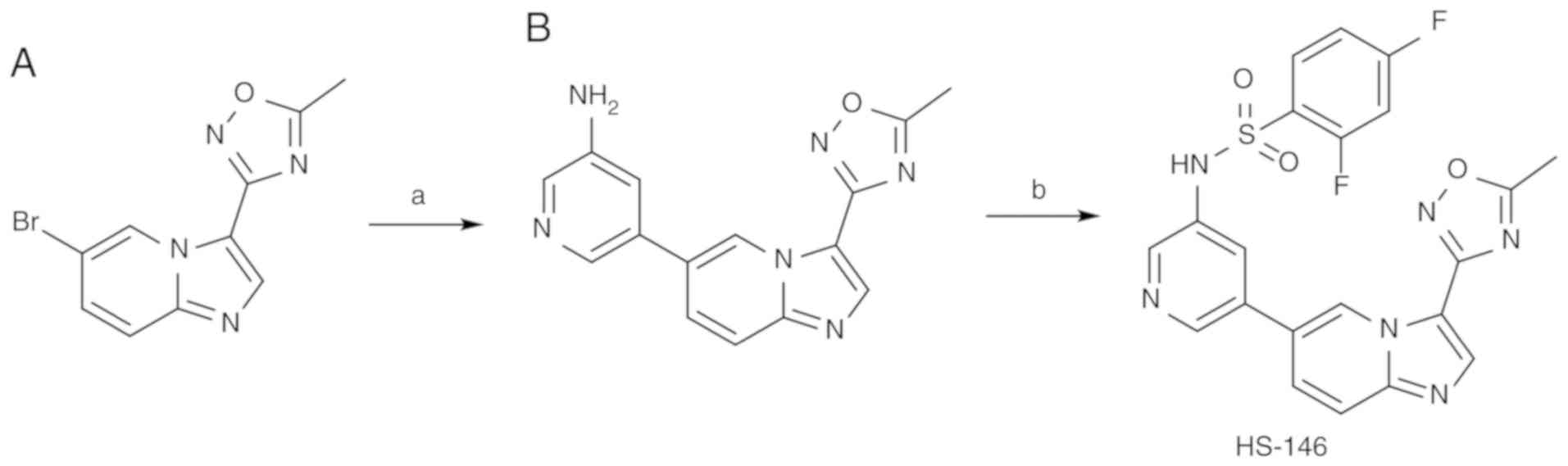 | Figure 1Synthetic route for HS-146. Reagents
and conditions: (A)
5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-3-amine,
K2CO3, Pd(dppf)Cl2.
CH2Cl2, 1,4-dioxane, H2O, 100°C, 6
h; (B) 2,4-difluorobenzenesulfonyl chloride, pyridine, DCM, room
temperature, 24 h. |
Step 1: Synthesis of
5-(3-(5-methyl-1,2,4-oxadiazol-3-yl) imidazo[1,2-
a]pyridin-6-yl)pyridin-3-amine
To a mixture of
3-[6-bromoimidazo[1,2-a]pyridin-3-yl)-5-methyl-1,2,4-oxadiazole
(450 mg, 1.8 m mol) were added 5-(4,4,5,5- tetramethyl-1,3,2-
dioxaborolan-2-yl)pyridin-3-amine (470 mg, 2.1 mmol),
K2CO3 (980 mg, 7.1 mmol) and Pd(dppf)
Cl2 CH2Cl2 (73 mg, 5 mol%).
1,4-Dioxane:H2O (=3:1, 4 ml) was added as a solvent, and
the mixture was then heated to 100°C for 6 h. The reaction mixture
was cooled to room temperature and concentrated. Purification by
flash column chromatography (DCM/MeOH=10:1) afforded
5-(3-(5-methyl-1,2,4-oxadiazol-3-yl)imidazo[1,2-a]pyridin-6-
yl)pyridin-3-amine (360 mg, 69%).
Step 2: Synthesis of
2,4-difluoro-N-(5-(3-(5-methyl-1,2,4-oxa-
diazol-3-yl)imidazo[1,2-a]pyridin-6-yl)pyridin-3-yl)benzenes
ulfonamide
To a solution of 5-(3-(5-methyl-1,2,4-oxadiazol-3-
yl)imidazo[1,2-a]pyridin- 6- yl)pyridin-3- amine (24 mg,
0.082 mmol) and pyridine (20 μl, 0.24 mmol) in DCM (5 ml)
was added 2,4-difluorobenzenesulfonyl chloride (21 μl, 0.099
mmol). The resulting reaction mixture was stirred at room
temperature for 24 h and then concentrated in vacuo. The
residue was purified by flash column chromatography (DCM/MeOH=20:1)
to yield
2,4-difluoro-N-(5-(3-(5-methyl-1,2,4-oxadiazol-3-yl)imidazo
[1,2-a]pyridin- 6-yl)pyridin- 3-yl) benzenesulfonamide (34
mg, 87% yield). 1H NMR(DMSO-d6, 300
MHz) δ 2.73 (s, 3H), 7.29 (t, 1H, J=3.3Hz), 7.55 (t, 1H, J=2.9 Hz),
7.82 (m, 2H), 7.96 (d, 1H, J=3.1 Hz), 8.05 (dd, 1H, J=2.9, 4.9 Hz),
8.38 (d, 2H, J=1.2 Hz), 8.65 (s, 1H), 9.21 (s, 1H), 11.25 (s, 1H).
HRMS (EI+) m/z calcd for
C21H14F2N6O3S
[M+H]+, 469.0895; found, 469.0907, HPLC purity > 99%.
For the in vitro experiments, HS-146 was prepared as a 10 mM
stock solution in dimethyl sulfoxide (DMSO, AppliChem), aliquoted
and then stored at -20°C.
Docking simulation
The binding mode of HS-146 was determined using
Discovery Studio 4.5 (www.3dsbiovia.com), and docking simulation was
performed using the DS CHARMm-based CDOCKER docking algorithm.
Ligand preparation was conducted with HS-146 under standard
conditions (pH 6.5-8.5). The X-ray structure (PDB 4YKN) was used as
a protein target (16). For
clarity, P-loop and a part of α-helix are presented in the carbon
alpha wire style.
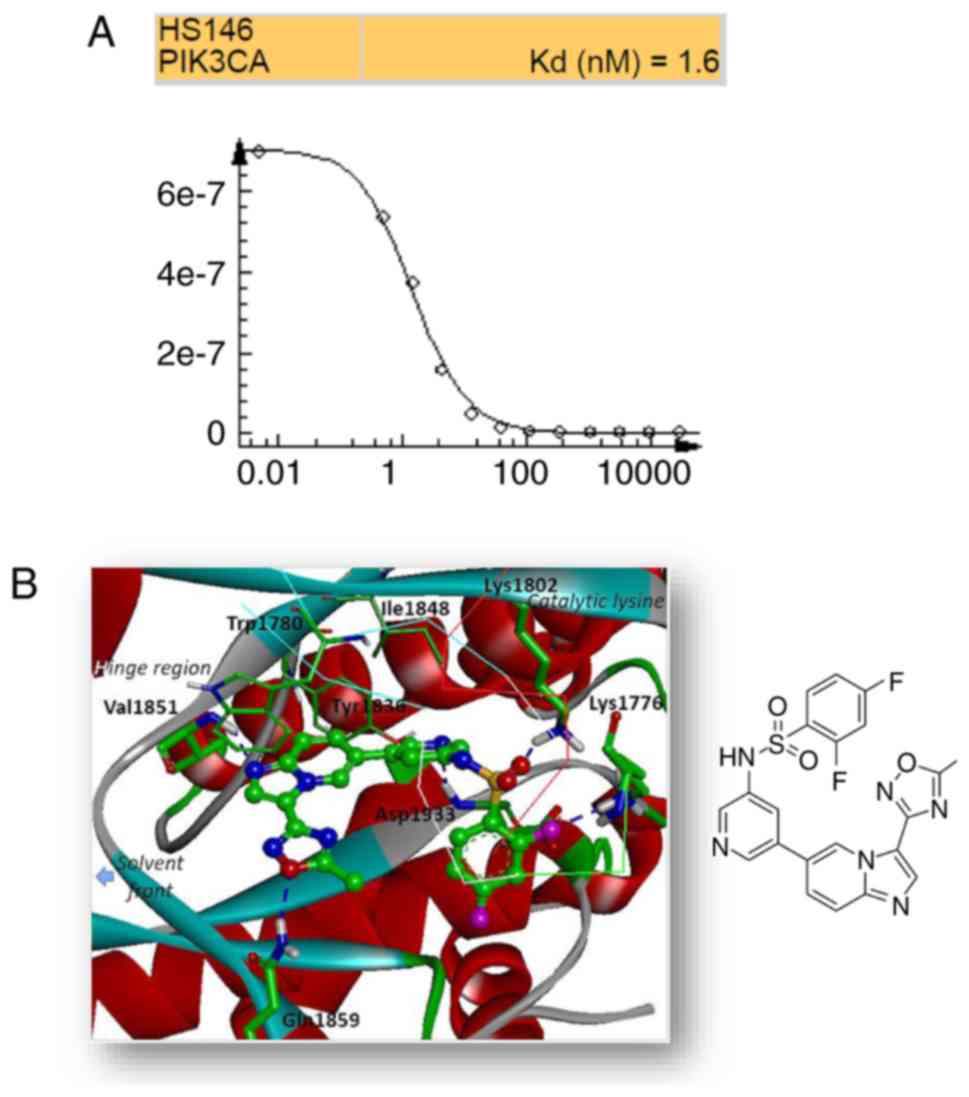
High-throughput binding assay
To identify the selectivity of HS-146, the binding
affinity of the compound against a panel of 70 kinases was tested
in a high-throughput binding assay conducted by Eurofins DiscoverX
(Fremont, CA 94538, www.discoverx.com) at a single 10 μM
concentration. In the assay, the compound exhibited a strong and
competitive binding to PI3Kα with a Kd value of
1.6 nM (Fig. 2A and Table SI).
Cell lines and cell culture
The human breast cancer cell lines MCF-7,
MDA-MB-231, SKBR3, and BT-474 and the human breast epithelial cell
line, MCF-10A, were used in the present study. MCF-7 and MDA-MB-231
cells were cultured in RPMI-1640 (Welgene). SKBR3 cells were
cultured in DMEM medium (Welgene) supplemented with 10% fetal
bovine serum (FBS, Welgene) and 1% penicillin/streptomycin (Gibco;
Thermo Fisher Scientific, Inc.), and BT-474 cells were maintained
in Hybri-Care medium (American Type Culture Collection)
supplemented with 10% FBS, and 1% penicillin/streptomycin. MCF-10A
cells were grown in DMEM/F12 medium (Invitrogen; Thermo Fisher
Scientific, Inc.) supplemented with EGF 20 ng/ml (PeproTech, Inc.),
insulin 10 μg/ml (Sigma-Aldrich; Merck KGaA),
hydrocorti-sone 500 ng/ml (Sigma-Aldrich; Merck KGaA), cholera
toxin 100 ng/ml (Sigma-Aldrich; Merck KGaA), 5% horse serum
(Invitrogen; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin. All cells were cultured in a humidified 5%
CO2 atmosphere at 37°C.
Cell viability assay
An MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)
assay was performed to evaluate the inhibitory effect of HS-146 on
cell proliferation. In brief, the MCF-7, BT-474, SKBR3, MDA-MB-231,
or MCF-10A cells were seeded at 1-10×103 cells per well
in 96-well culture plates and allowed to adhere overnight. The
media were then replaced with fresh medium containing the vehicle
(0.1% DMSO as a control) or various concentrations (0.1, 0.5, 1, 5,
10, or 50 μM) of HS-146 and incubated for 24, 48, or 72 h at
37°C, when 10 μl MTT (0.2 mg/ml final concentration,
Sigma-Aldrich; Merck KGaA) was added to each well and incubated for
3 h at 37°C. Media containing MTT were then carefully removed and
100 μl of DMSO (Junsei Chemical Co.) were added to each well
and gently mixed for 30 min. Well optical densities (OD) were then
measured at 540 nm using a microplate reader (Tecan Group, Ltd.).
The half maximal inhibitory concentration (IC50) was
calculated from the concentration-response curve obtained by
plotting the cell survival vs. the drug concentration.
Cell cycle analysis
The MCF-7 cells were treated with vehicle or various
concentrations (0.1, 1, or 10 μM) of HS-146 at 37°C for 24
h, washed with phosphate-buffered saline (PBS), fixed in 70%
ethanol overnight at -20°C, and stained with propidium iodide
(PI)/RNase staining buffer (BD Biosciences) for 30 min at 37°C in
the dark. Cell cycle distributions were determined using a
FACSCalibur flow cytometer equipped with CellQuest software (BD
Biosciences).
TUNEL assay
The apoptosis of MCF-7 cells was determined by
terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end
labeling (TUNEL) assay using a DeadEnd™ Colorimetric TUNEL System
(Promega Corp.) according to the manufacturer's instructions.
Briefly, the cells were fixed with 4% paraformaldehyde in PBS for
25 min at room temperature and washed with PBS for 5 min. The cells
were permeabilized with 0.2% Triton X-100 solution in PBS for 5 min
at room temperature. The cells were then incubated with
equilibration buffer for 10 min at room temperature and incubated
with rTdT reaction mixture for 1 h at 37°C. After washing with 2X
saline sodium citrate (2X SSC) solution for 15 min and PBS for 5
min at room temperature, the cells were incubated with DAB solution
for 10 min in the dark. TUNEL-positive cells were observed under a
microscope (Olympus Corp.).
Flow cytometric analysis of cell
apoptosis
The apoptotic rate of the MCF-7 cells was determined
by flow cytometry using a FITC Annexin V Apoptosis Detection kit
(BD Biosciences). Briefly, collected cells were washed with cold
PBS and incubated with binding buffer containing 5 μl FITC
Annexin V and 5 μl PI for 15 min at room temperature in dark
conditions. The stained cells were analyzed using a CytoFLEX flow
cytometer equipped with CytExpert software (Beckman Coulter,
Inc.).
Assessment of mitochondrial membrane
potential
The MCF-7 cells were pre-treated with or without 5
μM SC79 (Sigma-Aldrich; Merck KGaA, an Akt activator) for 1
h, and then treated 10 μM HS-146 for 24 h. Cells were
stained with 0.05 μg/ml of rhodamine 123 (a fluorescent
mitochondrial dye) for 1 h at 37°C, collected, and resuspended in
PBS containing 1% FBS. Changes in mitochondrial membrane potential
were monitored by measuring green fluorescence intensities using a
CytoFLEX flow cytometer equipped with CytExpert software (Beckman
Coulter, Inc.).
Western blot analysis
Cells were rinsed with ice-cold PBS and lysed using
RIPA buffer (Thermo Fisher Scientific, Inc.) containing protease
inhibitor cocktail (GenDEPOT) and phosphatase inhibitor cocktail
(GenDEPOT). Total protein concentrations were measured using a BCA
protein assay kit (Thermo Fisher Scientific, Inc.). Equal amounts
of total protein (30-50 μg) were then boiled in sample
buffer (Bio-Rad Laboratories, Inc.), separated by 8-12% SDS-PAGE,
and transferred onto PVDF membranes (EMD Millipore). Membranes were
blocked with PBS containing Tween-20 in 5% non-fat milk for 1 h at
room temperature, incubated with primary antibodies [1:1,000 v/v;
cyclin D1 (sc-8396), cyclin E (sc-247), cyclin-dependent kinase
(Cdk)2 (sc-6248), Cdk4 (sc-56277), p21Waf1/Cip1
(sc-6246) (all from Santa Cruz Biotechnology, Inc.),
poly(ADP-ribose) polymerase (PARP; #9542, Cell Signaling
Technology, Inc.), Mcl-1 (sc-12756, Santa Cruz Biotechnology,
Inc.), caspase-7 (#12827), phospho-Akt (#9271), total Akt (#9272),
phospho-mTOR (#2971), total mTOR (#2972), phospho-glycogen synthase
kinase (GSK)3β (#9323, Cell Signaling Technology), total GSK3β
(#9315, Cell Signaling Technology), phospho-p70S6K1 (#9234, Cell
Signaling Technology), phospho-eukaryotic translation initiation
factor 4E-binding protein 1 (4E-BP1; #2855) (all from Cell
Signaling Technology, Inc.), hypoxia-inducible factor (HIF)-1α
(610958, BD Biosciences), vascular endothelial growth factor (VEGF,
sc-152, Santa Cruz Biotechnology, Inc.), vimentin (V2258,
Sigma-Aldrich Merck KGaA), E-cadherin (#3195, Cell Signaling
Technology, Inc.) and β-actin (sc-47778, Santa Cruz Biotechnology,
Inc.)] overnight at 4°C and then incubated with horseradish
peroxidase (HRP)-conjugated goat anti-rabbit and goat anti-mouse
IgG secondary antibodies (1:5,000 v/v; #7074, #7076, Cell Signaling
Technology, Inc.) for 1 h at room temperature. Signals were
detected using Amersham™ ECL™ Prime Western Blotting Detection
Reagent (GE Healthcare), and densitometric analysis of the bands
was performed using ImageJ 1.48v software (NIH).
4,6-Diamidio-2-phenylinodole (DAPI)
staining
MCF-7 cells were plated onto cover glasses in
RPMI-1640 medium and allowed to reach 70% confluence (~24 h), fixed
in ice-cold 4% paraformaldehyde, washed with PBS, and stained with
2 μg/ml DAPI (Sigma-Aldrich; Merck KGaA) for 20 min at 37°C.
Stained cells were observed under a fluorescence microscope (Nikon
Corp.).
Immunofluorescence assay
MCF-7 cells grown on cover glasses were treated with
the vehicle or with HS-146 (10 μM) for 6 h, washed with PBS
(3×5 min), fixed with 4% para-formaldehyde for 10 min at 4°C,
permeabilized with 0.2% Triton X-100, and blocked with a 5% BSA
solution for 1 h at room temperature. Cells were then incubated
with phospho-Akt antibody (rabbit polyclonal, 1:200, #9271, Cell
Signaling Technology) overnight at 4°C and then with Alexa Fluor
488-conjugated goat anti-rabbit IgG secondary antibody (1:500,
#A-11008, Thermo Fisher Scientific, Inc.) for 1 h at room
temperature. After washing, the cover glasses were mounted on slide
glasses using mounting medium containing DAPI (Vector Laboratories,
Inc.). Images were acquired using a fluorescence microscope (Nikon
ECLIPSE Ts2-FL; Nikon Corp.).
Enzyme-linked immunosorbent assay
(ELISA)
MCF-7 cells were seeded into 6-well plates,
pre-treated with the vehicle or various concentrations of HS-146
(0.1-10 μM) for 1 h and then treated with 100 μM
cobalt(II) chloride (CoCl2) for 24 h to generate hypoxic
conditions. Culture media were then harvested and centrifuged at
1,593 × g for 5 min. VEGF levels in supernatants were measured
using a human VEGF ELISA kit (AbFrontier).
Wound healing migration assay
MCF-7 cells were seeded into 6-well culture plates
and cultured until 90% confluent. The cells were wounded with a
razor blade, and injury lines were marked. After wounding, the
detached cells were removed by washing with serum-free medium, and
the plates were further incubated for 48 h in serum-free RPMI-1640
medium containing the vehicle or various concentrations of HS-146
(0.1, 1 or 10 μM). The cells were then rinsed with
serum-free medium and fixed in absolute methanol. The cell
migratory behavior was observed under a phase contrast microscope
(Olympus Corp.) and documented.
Transwell invasion assay
The membranes of the upper Transwell chamber were
coated with 20 μl Matrigel (Matrigel:RPMI medium, 1:2; BD
Biosciences) and incubated for 2 h at 37°C. 5×104 cells
resuspended in serum-free medium were then seeded in the upper
chambers containing the vehicle or various concentration of HS-146
(0.1, 1, or 10 μM), and bottom chambers were filled with 750
μl RPMI-1640 containing 10% FBS. After 72 h, non-invasive
cells on the upper membrane surfaces were removed by wiping with
cotton swabs, and cells that invaded the membranes were fixed with
methanol and stained with crystal violet solution (Sigma-Aldrich;
Merck KGaA) for 10 min at room temperature. The membranes were then
dried in air and the invading cells were observed under an optical
microscope (CKX53; Olympus Corp.).
Statistical analysis
The results are presented as the means ± standard
deviations (SD). Statistical significances of group differences
were analyzed by one-way ANOVA followed by Tukey's multiple
comparison test in GraphPad PRISM 5.03 (GraphPad Software).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Binding mode
To obtain insight of the inhibitory mechanisms of
HS-146, its binding mode in the ATP binding site of PI3Kα was
investigated. The lowest energy conformation of HS-146-PI3Kα
complex was calculated using Discovery Studio 4.5 (Fig. 2B). The imidazopyridine core was
located in the adenine binding site in the ATP binding pocket and
formed a hydrogen bond with the backbone amide proton of Val1851.
In addition, the oxadiazole moiety attached at the C3 position of
imiazo[1,2-a]pyridine appeared to form a hydrogen bond with
the side-chain amide proton of Gln1859. The pyridine moiety was
located at the ribose binding pocket and interacted with the
aspartate backbone in DFG motif. The binding of HS-146 was further
stabilized by hydrogen bond formation with the two lysine side
chains: Lys1802 interacted with oxygen atom of sulfonamide linkage,
and Lys1776 in the P-loop interacted with the fluorobenzene group
of HS-146. HS-146 was bound with moderate strength by van der Waals
interactions with non-polar residues (e.g., Trp1780, Tyr1836 and
Ile1848).
HS-146 is cytotoxic to human breast
cancer cell lines
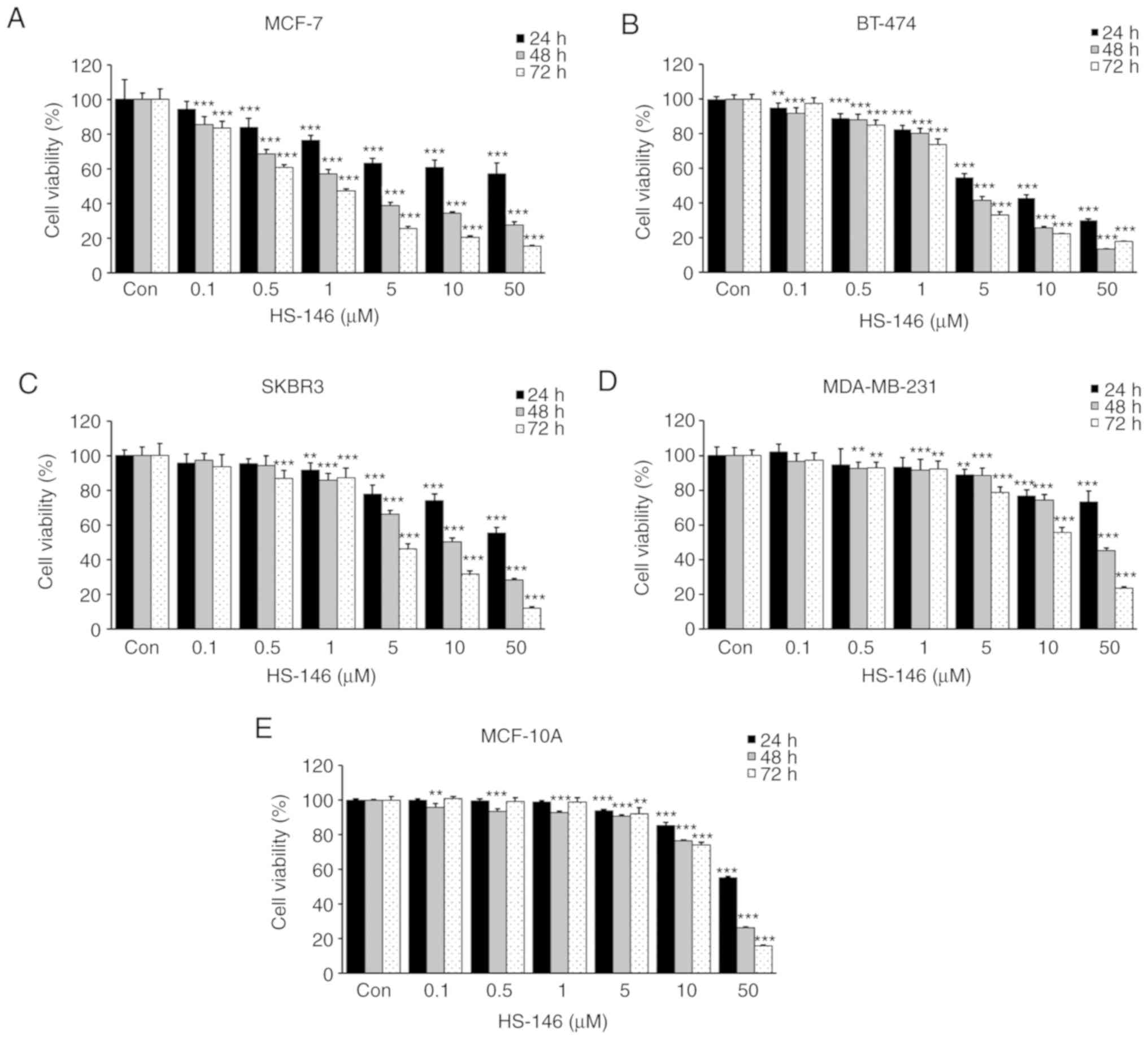
To evaluate the anticancer effect of HS-146,
cytotoxicity tests were conducted on the MCF-7, BT-474, SKBR3 and
MDA-MB-231 human breast cancer cell lines. The cells were treated
with HS-146 at 0, 0.1, 0.5, 1, 5, 10, or 50 μM for 24, 48,
or 72 h, and cell viabilities were assessed by an MTT assay. As a
result, HS-146 decreased the viability of all 4 cell lines in a
time- and concentration-dependent manner (Fig. 3). The IC50 values for
HS-146 in the MCF-7, BT-474, SKBR3 and MDA-MB-231 cells were 2.5,
4.1, 10.6 and 43.4 μM at 48 h, respectively. In addition, to
determine whether HS-146 was cytotoxic to non-cancerous cells, the
viability of the MCF-10A cells treated with HS-146 was measured.
The result revealed that HS-146 was almost non-toxic up to 5
μM, and exerted minimal toxic effects on non-cancerous cells
at 10 μM (Fig. 3E). These
results indicated that HS-146 was most effective at inhibiting the
proliferation of MCF-7 cells; thus, the MCF-7 cells were used in
subsequent experiments.
HS-146 induces cell cycle arrest in the
G0/G1 phase
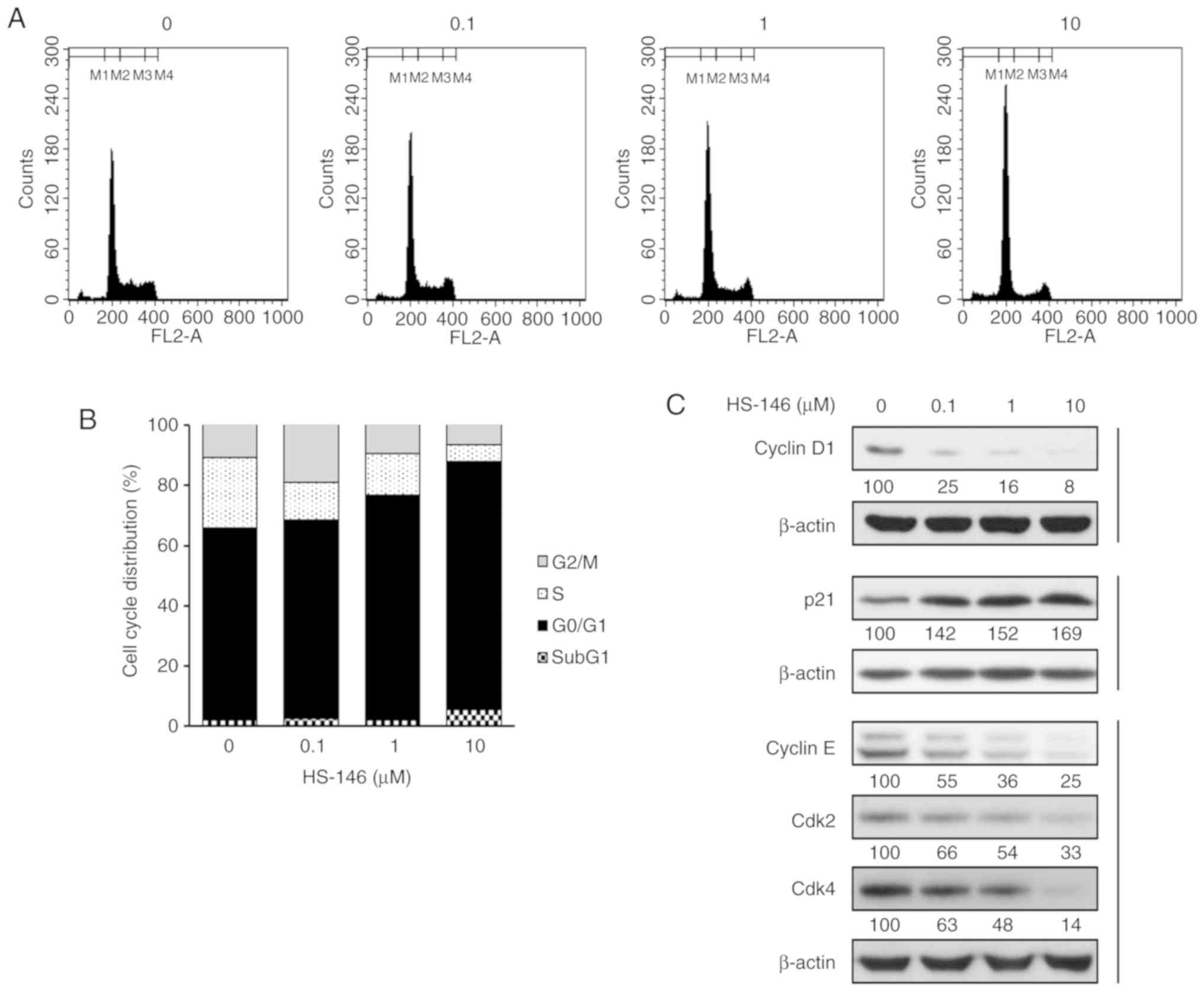
To confirm whether the inhibition of cell
proliferation by HS-146 was associated with cell cycle arrest, cell
cycle distributions were evaluated by flow cytometry. The MCF-7
cells were treated with the vehicle or various concentrations of
HS-146 (0, 0.1, 1, or 10 μM) for 24 h and then fixed and
PI-stained. The DNA contents were then assessed by measuring the
fluorescence intensities. As shown in Fig. 4, HS-146 increased the proportion of
cells in the G0/G1 phase in a concentration-dependent manner by
almost 20% from 63.93% (untreated controls) to 82.29% (10
μM), and decreased the percentages of cells in the S and
G2/M phases from 23.33% (control) to 5.64% (10 μM) and from
11.06% (control) to 6.87% (10 μM), respectively. These
results indicated that HS-146 induced G0/G1 arrest and
concomitantly reduced the percentages of cells in the S and G2/M
phases (Fig. 4A and B). In
addition, the expression levels of the regulatory subunits, cyclin
D1 and cyclin E, the cyclin-dependent kinases (Cdks) partners of
these cyclins, Cdk2 and Cdk4, and p21Waf1/Cip1, which
are markers of cell cycle arrest (G1/S checkpoint effectors) were
investigated by western blot analysis. Cyclin D1 has been reported
to be a key requirement for cell proliferation in a number of types
of cancers (17). HS-146 was found
to downregulate cyclin D1, cyclin E, Cdk2 and Cdk4 expression in a
concentration-dependent manner (Fig.
4C). p21Waf1/Cip1 is involved in the inhibition of
cell cycle progression and in the induction of apoptosis (18). In the present study, HS-146
upregulated its expression in a concentration-dependent manner
(Fig. 4C). These results indicated
that the inhibition of cell proliferation by HS-146 was due to
G0/G1 cell cycle arrest.
HS-146 induces the apoptosis of MCF-7
cells
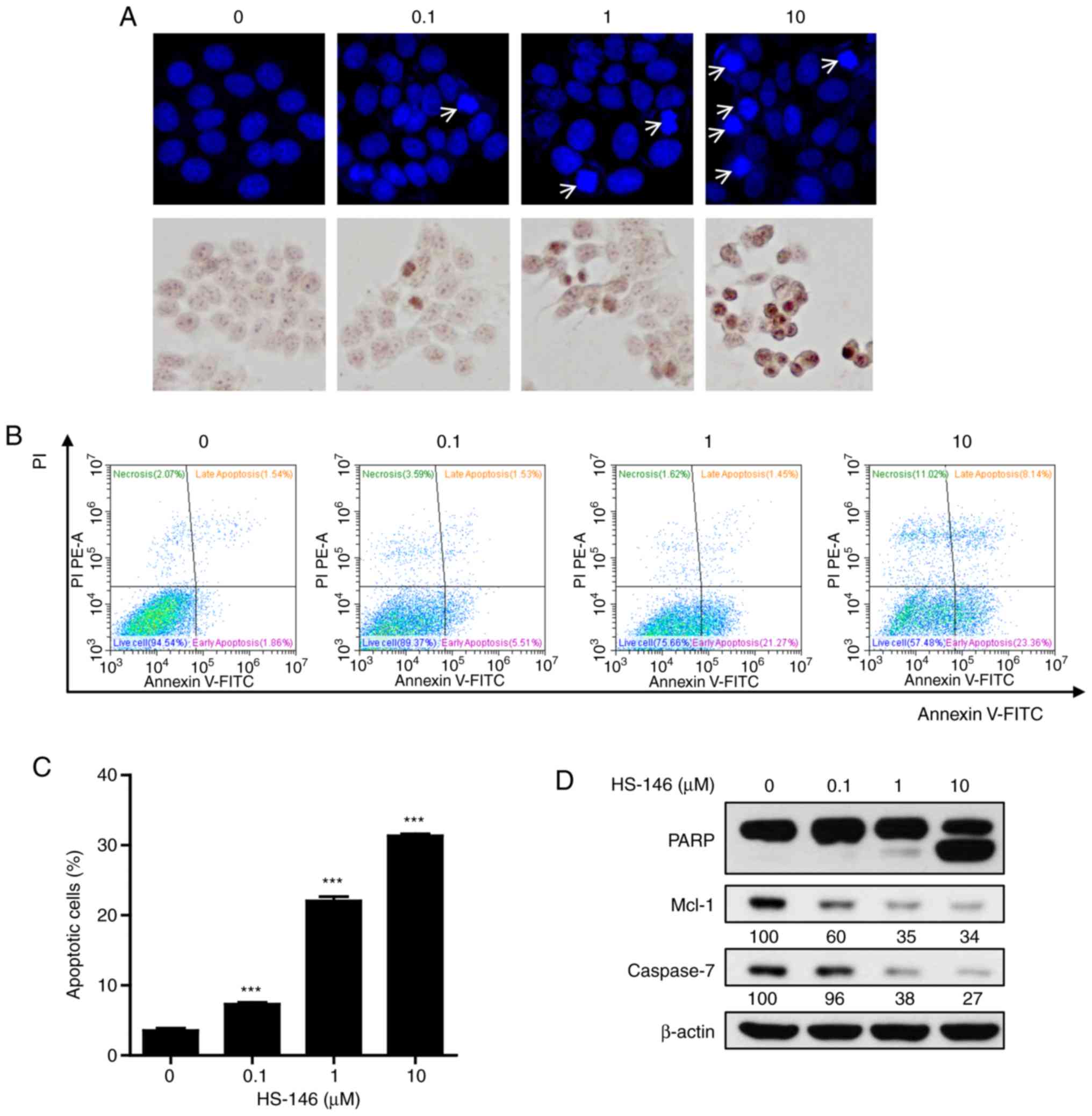
To explore the anticancer mechanisms of HS-146, the
morphological changes of nuclei were investigated by DAPI and TUNEL
staining. MCF-7 cells were treated with various concentrations of
HS-146 (0.1, 1, or 10 μM) and then compared with the
untreated controls. As shown in Fig.
5A, HS-146 increased nuclear condensation and fragmentation in
the MCF-7 cells in a concentration-dependent manner. These results
were supported by the quantitative data of Annexin V-FITC/PI
apoptosis assay (Fig. 5B and C).
HS-146 significantly increased the proportion of the Annexin
V-positive cells (early and late apoptotic cells) in a
concentration-dependent manner (P<0.001 vs. the vehicle-treated
control). Furthermore, the expression levels of the
apoptosis-related proteins, PARP, Mcl-1 and caspase-7, were
examined by western blot analysis in the HS-146-treated cells. As
shown in Fig. 5D, HS-146 increased
the level of cleaved PARP (a pro-apoptotic marker) and suppressed
the levels of Mcl-1 and caspase-7 in a concentration-dependent
manner. These results suggested that the anticancer effects of
HS-146 were due to the induction of apoptosis.
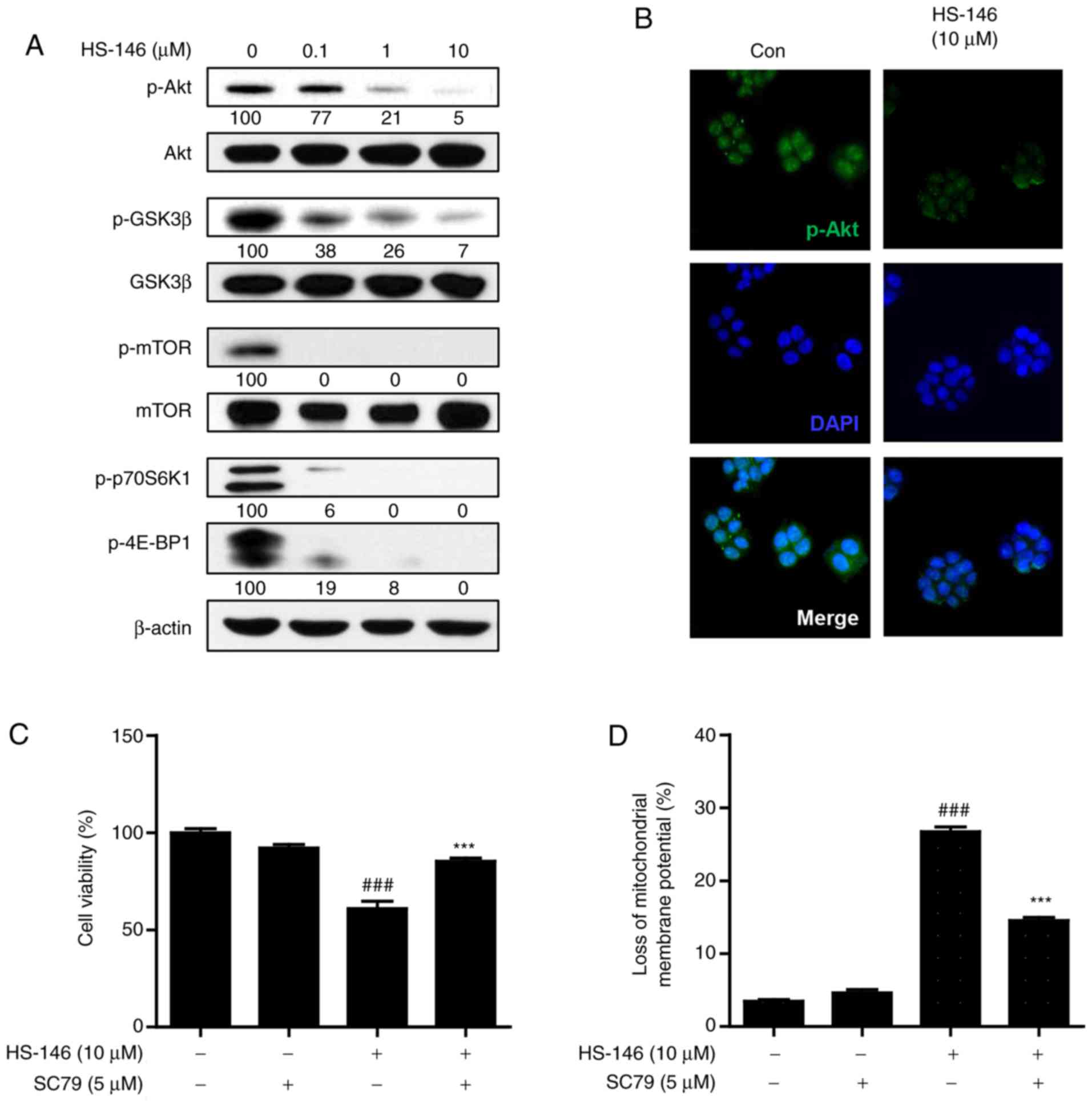
HS-146 inhibits the PI3K/Akt/mTOR
pathway
To investigate the molecular mechanisms responsible
for the anticancer activity of HS-146, the effects of HS-146 on the
PI3K/Akt/mTOR signaling pathway were evaluated. MCF-7 cells were
treated with various concentrations of HS-146 (0.1, 1, or 10
μM) for 6 h and compared with the untreated controls. The
expression levels of phosphorylated and total forms of Akt and of
its downstream effectors, GSK3β, mTOR, p70S6K1 and 4E-BP1, were
assessed by western blot analysis. As shown in Fig. 6A, the protein expression levels of
phospho-Akt, phospho-GSK3β, phospho-p70S6K1 and phospho-4E-BP1 were
effectively inhibited by HS-146 in a concentration-dependent
manner. Treatment with HS-146 almost abrogated phosphorylated mTOR
even at 0.1 μM. To determine whether HS-146 effectively
inhibits Akt activity, the MCF-7 cells were treated with 10
μM HS-146 and the expression of p-Akt was assessed by
fluorescence staining. The expression of p-Akt was markedly
decreased by HS-146 as compared with the untreated controls
(Fig. 6B). Furthermore, to verify
whether the growth inhibitory effects of HS-146 are associated with
the suppression of Akt, the cells were pre-treated with 5 μM
SC79 (a selective Akt activator) for 1 h and then treated with 10
μM HS-146 for 24 h. After staining with MTT or rhodamine 123
(a mitochondrial dye), the cell viabilities and mitochondrial
membrane potentials were analyzed, respectively. As expected, SC79
significantly reversed the reduction in cell viability and the loss
of mitochondrial membrane potential induced by HS-146 treatment
(Fig. 6C and D). Collectively,
these results suggest that the inhibition of PI3K/Akt/mTOR
signaling by HS-146 in MCF-cells is associated with the induction
of apoptosis and the inhibition of proliferation.
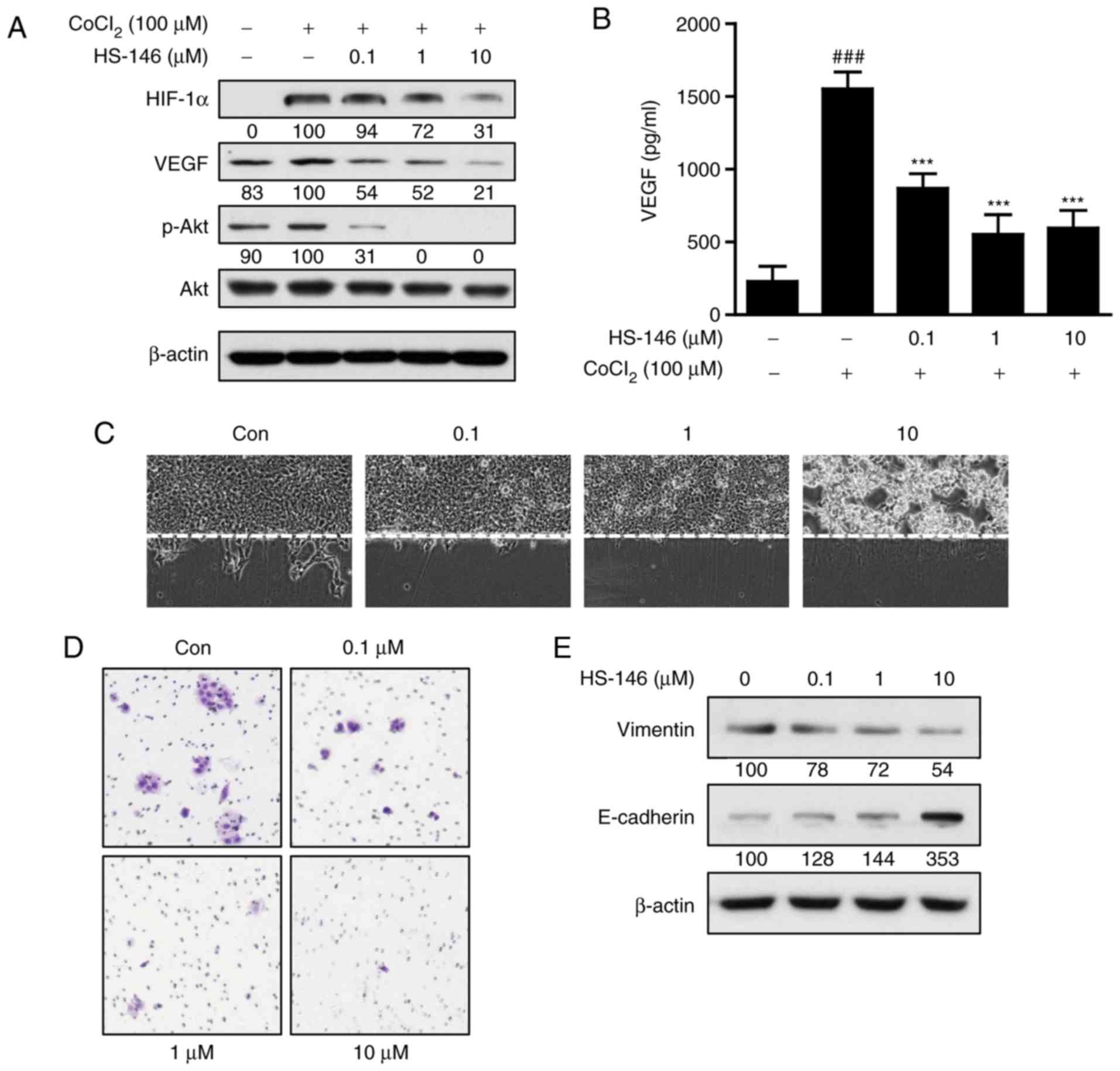
HS-146 inhibits breast cancer cell
metastatic ability
The ability to inhibit metastasis is a crucial
aspect of breast cancer treatment. Thus, to investigate whether
HS-146 has the potential to inhibit metastasis, MCF-7 cells were
treated with various concentrations of HS-146 (0.1, 1, or 10
μM) under hypoxic conditions induced by treatment of the
cells with 100 μM CoCl2 for 24 h. As shown in
Fig. 7A, HIF-1α expression was
increased under hypoxic conditions in the untreated controls, and
HS-146 inhibited this upregulation of HIF-1α and phosphorylated Akt
expression in a concentration-dependent manner. The effects of
HS-146 on the expression of VEGF (an immediate downstream target of
HIF-1α) were also examined by ELISA and western blot analysis. Both
techniques revealed that VEGF expression was increased by hypoxia
and that HS-146 inhibited this increase in a
concentration-dependent manner (Fig.
7A and B).
In addition, the effects of HS-146 on the migratory
and invasive activities of the MCF-7 cells were examined by a wound
healing and Transwell invasion assay. Treatment of the cells with
HS-146 inhibited MCF-7 cell migration in the wound healing assay in
a concentration-dependent manner, and cyto-toxicity was observed at
the concentration of 10 μM due to the serum-free conditions
(Fig. 7C). Furthermore, Transwell
invasion assays revealed that HS-146 reduced the invading cell
numbers in a concentration-dependent manner (Fig. 7D). To investigate mechanisms
responsible for the inhibitory effects of HS-146 on VEGF-induced
cell migration and invasion, the expression levels of
epithelial-mesenchymal transition (EMT)-related molecules, such as
vimentin and E-cadherin, were examined by western blot analysis.
The results revealed that HS-146 treatment decreased the level of
vimentin, while it increased the level of E-cadherin in a
concentration-dependent manner (Fig.
7E).
Discussion
A number of research groups continue to identify
novel strategies for the treatment of cancer. Over the past two
decades, one of the most promising novel therapies involves
treatments based on small molecule inhibitors that target
substances critical for the development and progression of cancer.
Developed small molecule inhibitors for the treatment of breast
cancer include Cdk4/6 inhibitors (palbociclib, riboci-clib and
abemaciclib), PARP inhibitors (olaparib, veliparib and talazoparib)
and PI3K inhibitors (buparlisib and alpelisib) (19). Some of these inhibitors have been
approved by the FDA for the treatment of breast cancer or have
progressed to late-stage clinical trials (19,20)
(https://clinicaltrials.gov/ct2/show/NCT03820830?term=NCT03820830&draw=2&rank=1,
https://clinicaltrials.gov/ct2/show/NCT03286842?term=NCT03286842&draw=2&rank=1,
https://clinical-trials.gov/ct2/show/NCT01945775?term=NCT01945775&draw=2&rank=1).
The PI3K/Akt/mTOR signaling pathway is one of the most frequently
dysregulated pathways in human cancer and the activation of this
pathway has been estimated to occur in as many as 70% of breast
cancer cases (20-23). Activating mutations in PIK3CA have
been shown to drive the initiation and progression of breast
cancer, and thus, the selective inhibition of PI3Kα is viewed as an
important therapeutic strategy (24). Recently, several investigators have
reported that PIK3CA mutation is significantly associated with
estrogen receptor (ER)-positive breast cancer (25-27).
In the present in vitro study, it was found
that HS-146 (a novel PI3Kα inhibitor) exerts anticancer effects and
that the underlying mechanisms of its effects are associated with
the regulation of the PI3K/Akt/mTOR pathway, and the consequent
suppression of cancer cell proliferation by cell cycle arrest,
increased apoptosis and the inhibition of metastasis. The effects
of potential anticancer agents are often determined by
investigating their effects on the proliferation, apoptosis and the
induction of cancer cell cycle arrest (28,29).
Therefore, the present study first investigated the ability of
HS-146 to suppress the proliferation of 4 different breast cancer
cell lines, namely the MCF-7 (ER+/PR+),
BT-474 (ER+/PR+/HER2+), SKBR3
(HER2+) and MDA-MB-231 (triple-negative) breast cancer
cell lines. HS-146 was found to inhibit the proliferation of all 4
cell lines and to most effectively inhibit that of MCF-7 cells. It
was also found that ER-positive cells (MCF-7 and BT-474) were more
sensitive to HS-146 compared with ER-negative cells (SKBR3 and
MDA-MB-231).
Cancer development is a result of a number of
cellular events, one of which is the abnormal regulation of the
cell cycle (30). Cyclin D1
positively regulates the transition from the G1 phase to the S
phase, and the Cdk inhibitor, p21Waf1/Cip1, negatively
regulates cell cycle progression (31,32).
Since cyclin D1 over-expression has been linked to the development
and progression of cancer, it is viewed as an attractive
developmental target for anticancer agents, and to date, several
compounds have been demonstrated to induce cyclin D1 degradation
(31). The formation of complexes
of cyclin D-Cdk4 and cyclin E-Cdk2 are crucial for the G1-S
progression in mammalian cells and (33,34).
Cdk inhibitors such as p27Kip1 and
p21Waf1/Cip1 is known to regulate negatively the
activity of these complexes (35).
In the present study, HS-146 increased the numbers of cells in the
G0/G1 phase and upregulated the protein expression of
p21Waf1/Cip1; by contrast, it downregulated the protein
expression of cyclin D1, cyclin E, Cdk2 and Cdk4. Thus, it appears
that HS-146 inhibits MCF-7 proliferation by inducing G0/G1
arrest.
Apoptosis is an essential normal cellular activity
and is required for tissue remodeling and for the removal of
damaged or aging cells (36). The
induction of apoptosis is a possible outcome of DNA damage, as has
been widely reported (28,37), and several studies have been
performed to identify means with which to induce apoptosis
(36,38,39).
In the present study, it was found that HS-146 induced apoptosis,
as evidenced by observations of DNA fragmentation and nuclear
condensation, as well as quantitative data using Annexin V-FITC/PI
apop-tosis assay. The anti-apoptotic Bcl-2 family protein, Mcl-1,
is known to play an important role in breast cancer cell survival
and chemoresistance (40), whereas
caspase-7 is an effector of apoptosis and activated caspase-7 can
cleave PARP during apoptosis (41). The present study demonstrated that
HS-146 inhibited the activation of Mcl-1 and caspase-7, and
increased PARP cleavage. These findings confirm that the anticancer
effects of HS-146 are mediated via the intrinsic apoptotic pathway
in MCF-7 cells.
The PI3K/Akt/mTOR signaling pathway has been well
documented to play a major role in carcinogenesis in breast cancer
cells. Activated PI3K converts PIP2 (phosphatidylino-sitol
bisphosphate) to PIP3 (phosphatidylinositol triphosphate), which
results in the phosphorylation of Akt. In addition, Akt is
activated by PDK1 and mTORC2 (mTOR complex 2), of which activation
regulates GSK3β and cyclin D1, and promotes cell cycle progression.
Furthermore, Akt also activates mTORC1 (mTOR complex 1), which
leads to the phosphorylation of p70S6K and 4E-BP1 (5,6).
Based on this mechanism, the present study evaluated the inhibitory
effect of HS-146 on PI3K/Akt/mTOR signaling. The results of western
blot analysis revealed that HS-146 markedly inhibited the
activation (phosphorylation) of Akt, mTOR, GSK3β, p70S6K1 and
4E-BP1. In addition, it was confirmed that Akt activation was
markedly suppressed in HS-146-treated MCF-7 cells by
immunofluorescence staining. It was further demonstrated that the
pharmacological activation of Akt by SC79 antagonized the growth
inhibitory effects of HS-146 against MCF-7 cells. Thus, the
anticancer effects of HS-146 may be partly mediated via the
inhibition of the Akt signaling pathway.
The activation of the PI3K/Akt pathway in tumor
cells can increase the expression of HIF-1α protein, which
activates the transcription of VEGF promoter, a key factor of tumor
angiogenesis that regulates key features, such as angiogenesis and
invasion. Numerous researchers have suggested that uncontrolled
angiogenesis is central to tumor growth and metastasis (42-44).
Thus, the present study investigated the anti-angiogenic and
anti-metastatic effects of HS-146 on MCF-7 cells. HS-146 suppressed
the hypoxia-induced increase in the expression of HIF-1α, VEGF and
p-Akt, the amount of secreted VEGF, and MCF-7 migration and
invasion. EMT is known to play a central role in acquiring
mesenchymal features from epithelial cells, and enhances the cell
migratory and invasive potential, resulting in cancer progression
(45,46). Therefore, the suppressive effects
of HS-146 against the migration and invasion of MCF-7 cells were
further supported by western blot analysis that confirmed the
inhibition of EMT by the decrease in the expression of vimentin and
the increase in the expression of E-cadherin by HS-146
treatment.
In conclusion, the present study synthesized HS-146
and found that it functions as a PI3Kα inhibitor with potent
anticancer effects on human breast cancer cells. In MCF-7 cells,
HS-146 effectively inhibited proliferation, induced G0/G1 cell
cycle arrest and induced apoptosis by inhibiting the PI3K/Akt/mTOR
signaling pathway. These findings suggest that HS-146 has potential
for use as a therapeutic candidate for breast cancer, which offers
a developmental starting point for the treatment for other human
cancers.
Supplementary Data
Acknowledgments
Not applicable.
Funding
The present study was supported by the National
Research Foundation of Korea (grant nos. 2018R1A2A1A05077263,
2019M3E5D1A02069621, 2014M3C1A3051476 and 2014009392) and the
Institute for Basic Science (IBS-R010-A2).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SSH and SH (the principal director and study
supervisor) were responsible for the design of the study and for
obtaining funding. OHK, JHL and SM participated in the study design
and experiments, and wrote the manuscript. SYP carried out
experiments and the statistical analysis. All authors participated
in the preparation of the manuscript, and read and approved the
final version.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Harbeck N and Gnant M: Breast cancer.
Lancet. 389:1134–1150. 2017. View Article : Google Scholar
|
|
2
|
Pierobon M, Ramos C, Wong S, Hodge KA,
Aldrich J, Byron S, Anthony SP, Robert NJ, Northfelt DW, Jahanzeb
M, et al: Enrichment of PI3K-AKT-mTOR pathway activation in hepatic
metastases from breast cancer. Clin Cancer Res. 23:4919–4928. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Peng Y, Wang Y, Tang N, Sun D, Lan Y, Yu
Z, Zhao X, Feng L, Zhang B, Jin L, et al: Andrographolide inhibits
breast cancer through suppressing COX-2 expression and angiogenesis
via inactivation of p300 signaling and VEGF pathway. J Exp Clin
Cancer Res. 37:2482018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cai F, Zhang L, Xiao X, Duan C, Huang Q,
Fan C, Li J, Liu X, Li S and Liu Y: Cucurbitacin B reverses
multidrug resistance by targeting CIP2A to reactivate protein
phosphatase 2A in MCF-7/adriamycin cells. Oncol Rep. 36:1180–1186.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li GY, Jung KH, Lee H, Son MK, Seo J, Hong
SW, Jeong Y, Hong S and Hong SS: A novel imidazopyridine
derivative, HS-106, induces apoptosis of breast cancer cells and
represses angiogenesis by targeting the PI3K/mTOR pathway. Cancer
Lett. 329:59–67. 2013. View Article : Google Scholar
|
|
6
|
Popolo A, Pinto A, Daglia M, Nabavi SF,
Farooqi AA and Rastrelli L: Two likely targets for the anti-cancer
effect of indole derivatives from cruciferous vegetables:
PI3K/Akt/mTOR signalling pathway and the aryl hydrocarbon receptor.
Semin Cancer Biol. 46:132–137. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Miller TW, Rexer BN, Garrett JT and
Arteaga CL: Mutations in the phosphatidylinositol 3-kinase pathway:
Role in tumor progression and therapeutic implications in breast
cancer. Breast Cancer Res. 13:2242011. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Qin H, Liu L, Sun S, Zhang D, Sheng J, Li
B and Yang W: The impact of PI3K inhibitors on breast cancer cell
and its tumor microenvironment. PeerJ. 6:e50922018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xing Y, Lin NU, Maurer MA, Chen H, Mahvash
A, Sahin A, Akcakanat A, Li Y, Abramson V, Litton J, et al: Phase
II trial of AKT inhibitor MK-2206 in patients with advanced breast
cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN
loss/PTEN mutation. Breast Cancer Res. 21:782019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Frustaci AM, Tedeschi A, Deodato M,
Zamprogna G, Cairoli R and Montillo M: Duvelisib: A new
phosphoinositide-3-kinase inhibitor in chronic lymphocytic
leukemia. Future Oncol. 15:2227–2239. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Massacesi C, Di Tomaso E, Urban P, Germa
C, Quadt C, Trandafir L, Aimone P, Fretault N, Dharan B, Tavorath R
and Hirawat S: PI3K inhibitors as new cancer therapeutics:
Implications for clinical trial design. Onco Targets Ther.
9:203–210. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bendell JC, Varghese AM, Hyman DM, Bauer
TM, Pant S, Callies S, Lin J, Martinez R, Wickremsinhe E, Fink A,
et al: A first-in-human phase 1 study of LY3023414, an oral
PI3K/mTOR dual inhibitor, in patients with advanced cancer. Clin
Cancer Res. 24:3253–3262. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lin T, Leung C, Nguyen KT and Figlin RA:
Mammalian target of rapamycin (mTOR) inhibitors in solid tumours.
Clin Pharm. 8:2016.
|
|
14
|
Markham A: Alpelisib: First global
approval. Drugs. 79:1249–1253. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim O, Jeong Y, Lee H, Hong SS and Hong S:
Design and synthesis of imidazopyridine analogues as inhibitors of
phosphoinositide 3-kinase signaling and angiogenesis. J Med Chem.
54:2455–2466. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang H, Medeiros PF, Raha K, Elkins P,
Lind KE, Lehr R, Adams ND, Burgess JL, Schmidt SJ, Knight SD, et
al: Discovery of a potent class of PI3Kα inhibitors with unique
binding mode via encoded library technology (ELT). ACS Med Chem
Lett. 6:531–536. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
John RR, Malathi N, Ravindran C and
Anandan S: Mini review: Multifaceted role played by cyclin D1 in
tumor behavior. Indian J Dent Res. 28:187–192. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu S, Bishop WR and Liu M: Differential
effects of cell cycle regulatory protein p21(WAF1/Cip1) on
apoptosis and sensitivity to cancer chemotherapy. Drug Resist
Updat. 6:183–195. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nur Husna SM, Tan HT, Mohamud R, Dyhl-Polk
A and Wong KK: Inhibitors targeting CDK4/6, PARP and PI3K in breast
cancer: A review. Ther Adv Med Oncol. 10:17588359188085092018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee JJ, Loh K and Yap YS: PI3K/Akt/mTOR
inhibitors in breast cancer. Cancer Biol Med. 12:342–354. 2015.
|
|
21
|
Castaneda CA, Cortes-Funes H, Gomez HL and
Ciruelos EM: The phosphatidyl inositol 3-kinase/AKT signaling
pathway in breast cancer. Cancer Metastasis Rev. 29:751–759. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fruman DA, Chiu H, Hopkins BD, Bagrodia S,
Cantley LC and Abraham RT: The PI3K pathway in human disease. Cell.
170:605–635. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Janku F, Yap TA and Meric-Bernstam F:
Targeting the PI3K pathway in cancer: Are we making headway? Nat
Rev Clin Oncol. 15:273–291. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu XL, Xu YC, Wang YX, Chen Y, Wang BB,
Wang Y, Chen YH, Tan C, Hu LD, Ma QY, et al: Decrease in
phosphorylated ERK indicates the therapeutic efficacy of a clinical
PI3Kα-selective inhibitor CYH33 in breast cancer. Cancer Lett.
433:273–282. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sabine VS, Crozier C, Brookes CL, Drake C,
Piper T, van de Velde CJ, Hasenburg A, Kieback DG, Markopoulos C,
Dirix L, et al: Mutational analysis of PI3K/AKT signaling pathway
in tamoxifen exemestane adjuvant multinational pathology study. J
Clin Oncol. 32:2951–2958. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zardavas D, Marvelde LT, Milne R, Joensuu
H, Moynahan ME, Hennessy B, Bieche I, Saal LH, Stal O, Iacopetta B,
et al: Tumor PIK3CA genotype and prognosis: A pooled analysis of
4,241 patients (pts) with early-stage breast cancer (BC). J Clin
Oncol. 33 (Suppl. 15): S5162015. View Article : Google Scholar
|
|
27
|
Yang SX, Polley E and Lipkowitz S: New
insights on PI3K/AKT pathway alterations and clinical outcomes in
breast cancer. Cancer Treat Rev. 45:87–96. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dhatchana Moorthy N, Muthu Ramalingam B,
Iqbal S, Mohanakrishnan AK, Gunasekaran K and Vellaichamy E: Novel
isothiacalothrixin B analogues exhibit cytotoxic activity on human
colon cancer cells in vitro by inducing irreversible DNA damage.
PLoS One. 13:e02029032018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee JH, Lee H, Yun SM, Jung KH, Jeong Y,
Yan HH, Hong S and Hong SS: IPD-196, a novel phosphatidylinositol
3-kinase inhibitor with potent anticancer activity against
hepatocellular carcinoma. Cancer Lett. 329:99–108. 2013. View Article : Google Scholar
|
|
30
|
Vermeulen K, Van Bockstaele DR and
Berneman ZN: The cell cycle: A review of regulation, deregulation
and therapeutic targets in cancer. Cell Prolif. 36:131–149. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Alao JP: The regulation of cyclin D1
degradation: Roles in cancer development and the potential for
therapeutic invention. Mol Cancer. 6:242007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Deng T, Yan G, Song X, Xie L, Zhou Y, Li
J, Hu X, Li Z, Hu J, Zhang Y, et al: Deubiquitylation and
stabilization of p21 by USP11 is critical for cell-cycle
progression and DNA damage responses. Proc Natl Acad Sci USA.
115:4678–4683. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Morgan DO: Principles of CDK regulation.
Nature. 374:131–134. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Reed SI, Bailly E, Dulic V, Hengst L,
Resnitzky D and Slingerland J: G1 control in mammalian cells. J
Cell Sci Suppl. 18:69–73. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sherr CJ and Roberts JM: CDK inhibitors:
Positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lim JH, Lee YM, Park SR, Kim DH and Lim
BO: Anticancer activity of hispidin via reactive oxygen
species-mediated apoptosis in colon cancer cells. Anticancer Res.
34:4087–4093. 2014.PubMed/NCBI
|
|
37
|
Norbury CJ and Zhivotovsky B: DNA
damage-induced apop-tosis. Oncogene. 23:2797–2808. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang L, Liu Y, Wang M, Qian Y, Dai X, Zhu
Y, Chen J, Guo S and Hisamitsu T: Celastrus orbiculatus extract
triggers apoptosis and autophagy via PI3K/Akt/mTOR inhibition in
human colorectal cancer cells. Oncol Lett. 12:3771–3778. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kumar D, Das B, Sen R, Kundu P, Manna A,
Sarkar A, Chowdhury C, Chatterjee M and Das P: Andrographolide
analogue induces apoptosis and autophagy mediated cell death in
U937 cells by inhibition of PI3K/Akt/mTOR pathway. PLoS One.
10:e01396572015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Basu A and Sridharan S: Regulation of
anti-apoptotic Bcl-2 family protein Mcl-1 by S6 kinase 2. PLoS One.
12:e01738542017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Walsh JG, Cullen SP, Sheridan C, Luthi AU,
Gerner C and Martin SJ: Executioner caspase-3 and caspase-7 are
functionally distinct proteases. Proc Natl Acad Sci USA.
105:12815–12819. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Karar J and Maity A: PI3K/AKT/mTOR pathway
in angiogen-esis. Front Mol Neurosci. 4:512011. View Article : Google Scholar
|
|
43
|
Choi MJ, Lee H, Lee JH, Jung KH, Kim D,
Hong S and Hong SS: The effect of HS-111, a novel thiazolamine
derivative, on apop-tosis and angiogenesis of hepatocellular
carcinoma cells. Arch Pharm Res. 35:747–754. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kim YW, Jang EJ, Kim CH and Lee JH:
Sauchinone exerts anticancer effects by targeting AMPK signaling in
hepatocellular carcinoma cells. Chem Biol Interact. 261:108–117.
2017. View Article : Google Scholar
|
|
45
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Singh S and Chakrabarti R: Consequences of
EMT-driven changes in the immune microenvironment of breast cancer
and therapeutic response of cancer cells. J Clin Med. 8:pii: E642.
2019. View Article : Google Scholar
|