Introduction
According to current research, 15-20% of all cases
of cancer can be attributed to infectious agents, including
Helicobacter pylori and human papillomavirus (HPV), followed
by hepatitis B virus (HBV), hepatitis C virus (HCV) and the
Epstein‑Barr virus (EBV) (1‑3). In HPV, specifically types 16, 18,
31, 33, 35, 39, 45, 51, 52, 56, 58 and 59 that are commonly
referred to as high-risk (HR), are not only associated with
cervical cancer (CC), the third most prevalent type of cancer
worldwide in women in 2017, but also with other tumor types,
including anal, penile, vulvar, vaginal and head and neck cancer
(1,4).
Persistent infections with HR-HPVs are necessary,
but not sufficient to cause cancer, indicating the existence of
multistep actions in viral carcinogenesis that contribute to the
characteristic hallmarks underlying the phenotype of tumors
(3,5). Thus, it has been of great interest to
study mechanisms by which persistent infections with these viruses
contribute to cancer development. HPV has been demonstrated to
induce a series of mechanisms that contribute to the evasion of the
immune response and apoptosis‑activated cell death, and finally the
transformation, proliferation and cellular immortalization of the
host cell (6,7).
HPV life cycle disruption due to integration of the
viral genome into the cellular genome (8,9) and
activation of the cell methylation machinery have been demonstrated
to be involved in the carcinogenic process of CC among other
factors (10-12). Specifically, the HR‑HPV E7
oncoprotein has been reported to serve a crucial role in oncogenic
transformation due to its ability to form complexes with members of
the retinoblastoma protein (pRB) family and destabilize them
(13,14), in addition to the ability to
interact with other proteins, including histone deacetylase 1
(HDAC1) (15) and DNA
methyltransferase 1 (Dnmt1) (16).
E7 binds to HDAC1 via its zinc finger‑like motif
through an intermediary protein Mi2β, which is a component of the
nucleosome remodeling and histone deacetylation (NURD) complex that
has the ability to modify chromatin structure trough both
deacetylation of histones and ATP-dependent nucleosome
repositioning (17,18). The formation of this complex is
necessary for the maintenance of viral episomes, controlling cell
proliferation and extending cell life (15,17).
Consistent with this, a chromatin immunoprecipitation assay in Ca
Ski cells demonstrated that E7 and HDACs are associated with the
major histocompatibility complex (MHC) class I promoter and histone
deacetylation (19), as well as
chromatin repression and the downregulation of MHC class I
genes and MHC class I heavy chain, and repression of the genes
encoding the transporter associated with antigen processing subunit
1 (TAP1) and low molecular weight protein 2 (LMP2) (20). Furthermore, an association with
Dnmt1 is directed and mediated by the conserved region 3 (CR3)
zinc‑finger region of E7, which is known to contribute to E7
transformation functions and stimulate the methyltransferase
activity of Dnmt1, which may lead to aberrant genome methylation
and cellular transformation as a consequence of the silencing of
tumor-suppressor genes (16).
Another study has demonstrated that in samples of normal cervix and
cervical cancer, HPV types 16 and 18 activate the cell methylation
machinery to methylate viral DNA, as well as the promoter regions
of cellular genes, including cyclin A1, Rubicon-like autophagy
enhancer, retinoic acid receptor β2, cadherin 1 (CDH1),
death-associated protein kinase 1, human telomerase reverse
transcriptase 1 (hTERT1), hTERT2, hypermethylated in
cancer 1 and Twist Family BHLH Transcription Factor 1 (21). Therefore, previous evidence
suggests that HR-HPV E7 serves an important role in the
activation of the cellular methylation machinery, which regulates
the transcription of viral and cellular genes either during their
productive infection during its life cycle or during the
carcinogenic process.
The mechanisms by which E7 is involved in the
regulation of gene expression at the chromatin level are not well
understood. It has been observed that a common characteristic of
several cancer types associated with viruses is the decreased
expression of CDH1, which encodes E-cadherin, through
epigenetic mechanisms (22-24).
In the case of cancer types associated with HPV infections, it has
been demonstrated that HPV16 E7 suppresses the transcription
of CDH1, which reduces protein expression of E-cadherin
(25,26). In addition, HPV16 E7 has
been reported to increase the amount and activity of Dnmt1 in NIKS
cells, which are derived from foreskin keratinocytes transfected
with HPV16 E7 or NIKS bearing episomal HPV16 DNA (26); however, NIKS cells not infected
with HPV16 and NIKS bearing episomal HPV16 DNA did not exhibit any
differences in the CpG methylation status of the CDH1
promoter regions, as all CpG sites were unmethylated (26). It is evident that HPV activates the
methylation machinery via E7/Dnmt1; however, it is not clear how
HPV induces CDH1 repression by epigenetic mechanisms.
Since HR-HPVs E7 has been demonstrated to interact
with Dnmt1 and HDAC1, the aim of the current study was to determine
the methylation pattern of the CDH1 promoter region in HeLa,
SiHa and Ca Ski cell lines positive for HPV16 and HPV18.
Additionally, associations with transcription factors Snai1 and
Snai2, which are negative regulators of CDH1 expression and
inducers of the epithelial-mesenchymal transition (EMT) process
(27), were evaluated. HeLa, SiHa
and Ca Ski cell lines were selected for the present study, as they
are representative of the most frequent cancer types of the uterine
cervix with positive HR-HPV infection with different viral loads
and epithelial origins, including cervical adenocarcinoma, cervical
squamous cell carcinoma and cervical epidermoid carcinoma (28-34).
Materials and methods
Cell lines
As reported by the American type culture collection,
HeLa cells are derived from a female African-American patient with
uterine cervical adenocar-cinoma and are reported to contain 10-50
integrated copies of HPV18 per cell (30-36).
SiHa cells are derived from a female Asian patient with grade II
cervical squamous cell carcinoma and are reported to contain 1-2
integrated copies of HPV16 per cell (29,30,33,36).
Ca Ski cells are derived from a female Caucasian patient with
cervical epidermoid carcinoma and are reported to contain 500-600
integrated copies of HPV16 per cell (28,30,36).
HaCaT is a non-tumorigenic immortalized human epidermal cell line
derived from skin keratinocytes. All cell lines were authenticated
through STR DNA profiling (ID no. DP0297) by the University of
Colorado DNA Sequencing & Analysis Core. HeLa, SiHa and HaCaT
cell lines (ATCC) were cultured in DMEM (cat. no. 12800-058; Gibco;
Thermo Fisher Scientific, Inc.); Ca Ski cells were cultured in RPMI
medium (cat. no. 31800‑014; Gibco; Thermo Fisher Scientific, Inc.).
All cell lines were supplemented with 10% fetal bovine serum (FBS;
cat. no. 16000‑044; Gibco; Thermo Fisher Scientific, Inc.) and 1X
penicillin-streptomycin (cat. no. 15140122; Gibco; Thermo Fisher
Scientific, Inc.) and were incubated in a humidified chamber at
37°C with 5% CO2.
Untransfected MCF-7 cells and MCF-7 cell stable
clones transfected with a pcDNA 3.1 expression vector (Invitrogen;
Thermo Fisher Scientific, Inc) with the bicistronic E6/E7 region
from HPV18 (MCF-7 pE6/E7) were kindly provided by Dr Erick de la
Cruz Hernández (Juarez Autonomous University of Tabasco,
Villahermosa, Mexico) and were cultured with 800 µg/ml
Geneticin in DMEM/F12 for 3 weeks as previously described (37-39).
Untransfected C33-A cells and C33-A cell stable clones transfected
with a pcDNAE7 plasmid (C33-A pE7/HPV16) were kindly provided by Dr
Patricio Gariglio (CINVESTAV-IPN, Mexico City, Mexico) and were
cultured with 800 µg/ml Geneticin in DMEM for 2 weeks as
previously described (40).
Treatments with 5‑aza‑2'‑deoxycytidine
(5‑AzadC) and trichostatin A (TSA)
5-AzadC causes DNA demethylation or
hemi-demethylation that results in gene activation by inhibiting
Dnmt activity (41). TSA has been
used as a histone deacetylase inhibitor, which causes histone
hyperacetylation that leads to chromatin relaxation and modulation
of gene expression (42). 5-AzadC
and TSA were obtained from Sigma-Aldrich; Merck KGaA (cat. nos.
A3656 and T8552, respectively) and resuspended in DMSO
(Sigma-Aldrich; Merck KGaA; cat. no. D8418) to obtain working stock
solutions (10.0 mM 5-AzadC and 1.0 mM TSA). The 5-AzadC and TSA
stock solutions were aliquoted, protected from light and stored at
‑80°C for later use. A total of 4.5×105 HeLa and
5×105 SiHa cells were seeded in p60 boxes in triplicate
and were treated with 5 and 10 µM 5-AzadC and 200 and 500 nM
TSA. Untreated cells or cells treated with DMSO were used as
controls. The total volume of culture medium was 3 ml, which was
supplemented with 10% FBS and did not contain any antibiotics.
Assays were performed protected from light and the cells were
incubated for 48 h at 37°C with 5% CO2. The culture
medium was replaced after 24 h due to the half-life of 5-AzadC and
TSA.
Transfection with short interfering RNA
(siRNA)
siRNAs targeting HPV16 and HPV18 E7 were designed as
previously described (43,44) and obtained from Ambion (Thermo
Fisher Scientific, Inc.; cat. nos. s237642 and s237640).
Silencer® Select GAPDH siRNA (Hs, Mm, Rn) (cat.
no. 4390849; Ambion; Thermo Fisher Scientific, Inc.) was used as a
positive transfection control to select the transfection agent
(Lipofectamine® 2000 or siPORT™ NeoFX™) and to optimize
gene silencing without affecting cell viability according to the
manufacturer's protocol and as previously described (45-50).
siRNAs were resuspended in UltraPure™ DNase/RNasefree distilled
water (cat. no. 10977‑015; Thermo Fisher Scientific, Inc.) to
obtain a working stock of 10 µM. siRNAs were then aliquoted
and stored at ‑80°C for later use.
Transfection with siRNA was performed in triplicate
using the siPORT™ NeoFX™ kit (cat. no. AM4511; Ambion; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
A total of 7.5×104 HeLa and 8.0×104 SiHa
cells/well were seeded in a 12-well plate. HeLa cells were
transfected with 30 nM siRNA against HPV18 E7, whereas SiHa cells
were transfected with 30 nM siRNA against HPV16 E7. In addition, 30
nM siRNA targeting GAPDH was transfected as a positive
control in both cell lines. Cells subjected to treatment with
siPORT™ NeoFX™ Transfection Agent with Opti-MEM® I (cat.
no. 31985070; Thermo Fisher Scientific, Inc.) were used as a
reference control to obtain the relative values, and untransfected
cells were used as a reference control for statistical comparisons.
Culture medium supplemented with 12% FBS and no antibiotics was
added to make up a final transfection volume of 1.2 ml. Cells were
incubated for 48 h at 37°C with 5% CO2 and subsequently
the extraction of nucleic acids and proteins was performed.
Transfection efficiency was determined by measuring the expression
of E7 and GAPDH at the mRNA level.
Bisulfite conversion and DNA methylation
analysis
Total genomic DNA was isolated from the treated and
transfected cell lines using the Wizard® Genomic DNA
Purification kit (cat. no. A1120; Promega Corporation) and 1.5
µg genomic DNA was treated with bisulfite according to the
manufacturer's protocol of the EZ DNA Methylation-Gold™ kit (cat.
no. D5006; Zymo Research Corp.). Methylation of CpG sites at the
CDH1 promoter region was analyzed by the bisulfite
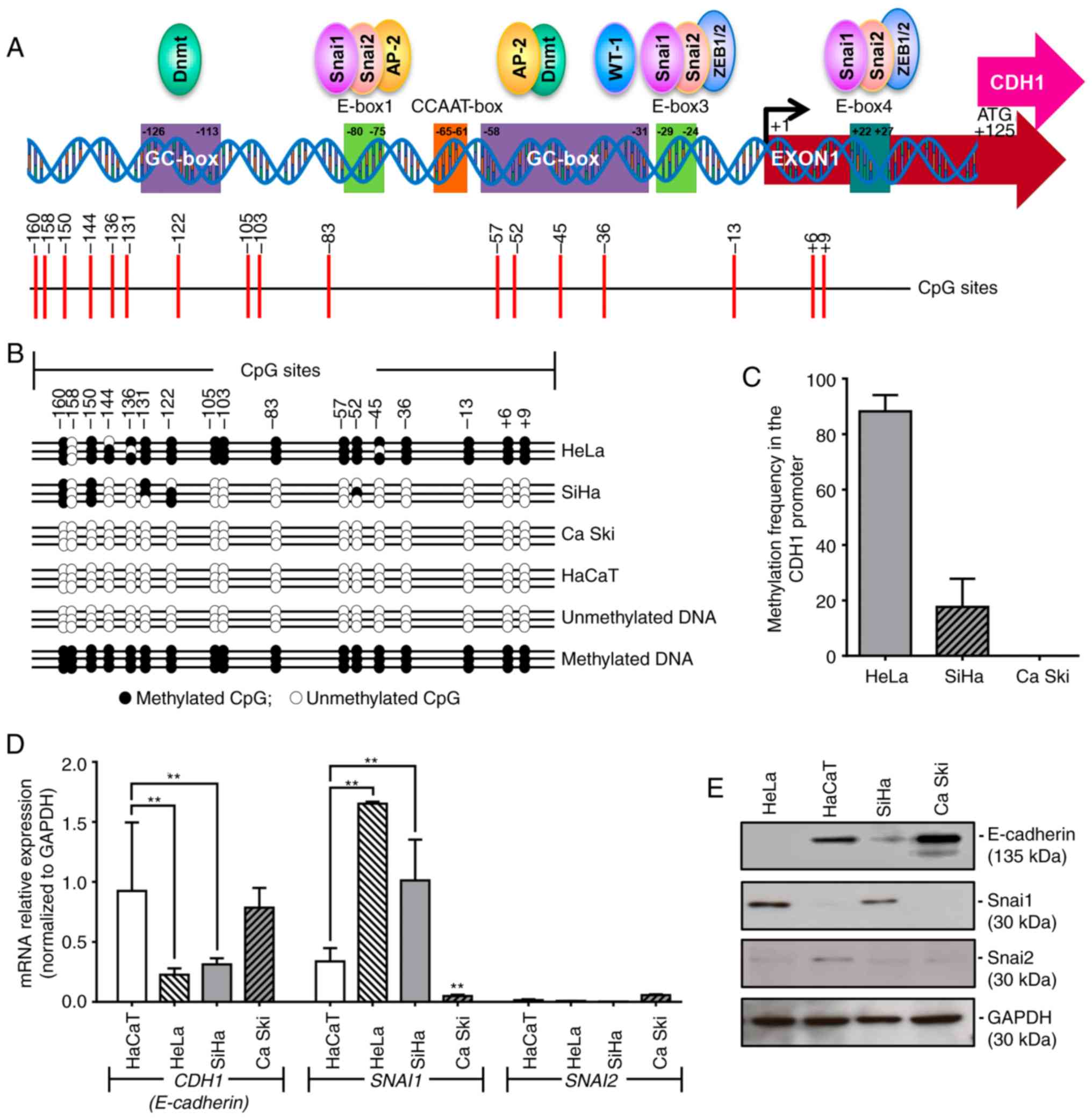
sequencing PCR (BSP) protocol (Fig.
1A) (10-12,51,52)
or using oligonucleotides for MSP-protocol provided by Dr Alfonso
Dueñas-Gonzalez (INCan-UNAM, Mexico City, Mexico); the PCR
conditions for MSP-protocol were the same as those used for the BSP
protocol. PCR was performed with a total volume of 25 µl,
containing 1X PCR Gold Buffer, 1 mM dNTP, 2 mM MgCl2, 10
pMol forward CDH1-BSP (5'-TTT TAG TAA TTT TAG GTT AGA GGG
TTA T-3') and reverse CDH1-BSP (5'-AAA CTC ACA AAT ACT TTA
CAA TTC C-3') oligonucleotides, 1 U AmpliTaq Gold® DNA
Polymerase (cat. no. 4338856; Applied Biosystems; Thermo Fisher
Scientific, Inc.) and 300 ng bisulfite‑treated DNA. The
thermocycling conditions were as follows: 95°C for 7 min, followed
by 35 cycles of 95°C for 35 sec, 57°C for 35 sec and 72°C for 60
sec, and a final extension at 72°C for 7 min. PCR products were
treated using ExoI (cat. no. EN0581; Thermo Fisher
Scientific, Inc.) and SAP (cat. no. EF0651; Thermo Fisher
Scientific, Inc.) enzymes. The treated PCR products were sequenced
using the BigDye® v3.1 Cycle Sequencing kit (cat. no.
4337455; Applied Biosystems; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol in an ABI PRISM™
3100-Avant Genetic Analyzer (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The BSP oligonucleotides were designed by
MethPrimer v2.0 software (The Li Lab; PUMCH; Chinese Academy of
Medical Sciences) using GenBank sequence DQ335132.1 for the
CDH1 gene (53). The
sequencing data obtained from BSP were analyzed using Chromas
v2.6.4 software (Technelysium Pty., Ltd.) and Lasergene v7 package
(DNASTAR, Inc.).
RNA extraction and reverse
transcription‑quantitative (RT‑q) PCR
Total RNA was extracted from the treated and
transfected cells using TRIzol® reagent (cat. no.
15596026; Invitrogen; Thermo Fisher Scientific, Inc.). The RNA was
treated with DNase I (cat. no. EN0521; Invitrogen; Thermo
Fisher Scientific, Inc.), and purified with Direct‑zol™ RNA
MicroPrep (cat. no. R2060; Zymo Research Corp.) according to the
manufacturer's protocol. Complementary DNA (cDNA) was obtained from
the purified RNA using the SuperScript™ IV First-Strand Synthesis
system (cat. no. 18091050; Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. The RNA‑primer mix
was incubated at 65°C for 5 min and 4°C for 1 min, and the RT
reaction mix was incubated at 55°C for 15 min and 80°C for 10 min
to inactivate the reaction and placed on ice for subsequent use.
Subsequently, 60 ng cDNA was subjected to qPCR to determine the
expression levels of the genes of interest using the primers listed
in Table I. qPCR conditions were
as follows: 95°C for 10 min, followed by 40 cycles of 95°C for 20
sec, 60°C for 30 sec and 72°C for 35 sec, and a final extension at
72°C for 7 min. This was followed by a melting curve analysis at
65‑95°C. All qPCR assays were analyzed using Rotor-Gene Q Series
v2.1.0 software (Qiagen, Inc.).
 | Table IPrimer sequences used for the BSP and
qPCR assays. |
Table I
Primer sequences used for the BSP and
qPCR assays.
| Gene | Assay Primer
sequences (5'→3') |
|---|
| CDH1 | BSP F:
TTTTAGTAATTTTAGGTTAGAGGGTTAT |
| R:
AAACTCACAAATACTTTACAATTCC |
| CDH1 | qPCR F:
GTCAGTTCAGACTCCAGCCC |
| R:
AAATTCACTCTGCCCAGGACG |
| SNAI1 | qPCR F:
ACCACTATGCCGCGCTCTT |
| R:
GGTCGTAGGGCTGCTGGAA |
| SNAI2 | qPCR F:
GACCCTGGTTGCTTCAAGGA |
| R:
TGTTGCAGTGAGGGCAAGAA |
| E7
HPV16 | qPCR F:
CAGCTCAGAGGAGGAGGATG |
| R:
TGCCCATTAACAGGTCTTCC |
| E7
HPV18 | qPCR F:
TGAAATTCCGGTTGACCTTC |
| R:
CACGGACACACAAAGGACAG |
| GAPDH | qPCR F:
AAGGTCGGAGTCAACGGATTTG |
| R:
CCATGGGTGGAATCATATTGGAA |
| HPRT | qPCR F:
GGACTAATTATGGACAGGACTG |
| R:
GCTCTTCAGTCTGATAAAATCTAC |
To obtain expression levels of CDH1,
SNAI1, SNAI2, HPV16 E7 and HPV18 E7 in the
HeLa, SiHa, Ca Ski and HaCaT cells, a commercial sample of RNA
extracted from normal cervix negative for HPV (Human Cervix Total
RNA; cat. no. AM6992; Ambion; Thermo Fisher Scientific, Inc.) was
used as a reference. The ΔCq values for each gene were normalized
to the reference gene GAPDH using the 2−ΔΔCq
method (54). The commercial
sample of normal cervix negative for HPV was set as 1, and the
results are not presented. For statistical analysis, the HaCaT cell
line was used for comparison. For experiments involving the
treatment of HeLa and SiHa cells with 5-AzadC and TSA, cells
treated with DMSO were used as a reference, but data were not
included in the graphs. For experiments involving the transfection
of HeLa and SiHa cells with siRNAs, the cells treated with siPORT™
NeoFX™ Transfection Agent with Opti-MEM® I were used as
a reference, but results were not included in the graphs. ΔCq
values of CDH1, SNAI1, SNAI2, HPV16 E7 and
HPV18 E7 were normalized using the 2−ΔΔCq method
(39) with GAPDH as
reference for 5-AzadC and TSA treatments, and HPRT as
reference for experiments involving siRNA transfections. For
statistical analysis, untreated (Unt) HeLa and SiHa cells were used
for comparisons.
Protein extraction and western blot
analysis
Proteins were obtained using a lysis buffer
containing 5 mM EDTA, 150 mM NaCl, 5 mM Tris-HCl pH 9.0, 1%
Nonidet-P40 and 1.2 mg/ml cOmplete™ protease inhibitor cocktail
(Roche Applied Science). Protein extracts were forced through a
22-gauge needle 10 times and centrifuged for 10 min at 17,000 × g
at 4°C. Protein concentration was determined using the Bradford
method. Subsequently, 30 mg protein was loaded and separated on 12%
SDS-PAGE gels followed by transfer to nitrocellulose membranes.
Membranes were blocked with 5% nonfat dry milk in 1X TBS with 0.1%
Tween‑20 (TBST) at 4°C for 1 h with gentle agitation and incubated
overnight at 4°C with antibodies against E-cadherin (cat. no.
sc-8426; 1:1,000), GAPDH (cat. no. sc-48167; 1:1,000), β-actin
(cat. no. sc-1616; 1:1,000; all from Santa Cruz Biotechnology,
Inc.), Snai1 (cat. no. L70G2; 1:1,000; Cell Signaling Technology,
Inc.) and Snai2 (cat. no. C19G7; 1:1,000; Cell Signaling
Technology, Inc.) diluted in TBST with 5% BSA (cat. no. 9998; Cell
Signaling Technology, Inc.). Subsequently, membranes were incubated
with secondary antibodies for 2 h at room temperature, including
goat anti-mouse IgG-horseradish peroxidase (HRP; cat. no. sc-2005;
1;10,000), donkey anti-rabbit IgG-HRP (cat. no. sc-2313; 10,000)
and donkey anti-goat IgG-HRP (cat. no. sc-2020; 1:5,000; all from
Santa Cruz Biotechnology, Inc.) diluted in TBST with 5% non-fat dry
milk. Immobilon Western Chemiluminescent HRP substrate (EMD
Millipore) was used for protein detection, and images were acquired
using C-DiGit Blot scanner equipment (Li-Cor Biosciences) and
processed in the Image Studio™ Lite version 5.2 software (Li-Cor
Biosciences).
Nitrocellulose membranes were incubated with two
primary antibodies in the following manner: i) Incubation was
performed as described against a primary antibody, including
E-cadherin, Snai1 or Snai2 with their respective secondary
antibody; ii) images were acquired; iii) membranes were washed 3×5
min with TBST at room temperature; iv) membranes were re-probed
with a second primary antibody, including GAPDH or β-actin with
their respective secondary antibody; and v) images were
acquired.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism v4.0 (GraphPad Software, Inc.). One-way ANOVA with Turkey's
post hoc test were used to evaluate significant differences in gene
expression and methylation levels, and results were presented as
the mean ± SD. P<0.05 was considered to indicate a statistically
significant difference.
Results
Different methylation patterns in the
CDH1 promoter region are present in HPV16‑ and HPV18‑positive
cancer cell lines
A common site‑specific methylation pattern in
certain CpG islands (−160, −150, −131 and −122) of the CDH1
promoter region was detected in HeLa and SiHa cells, whereas other
CpG islands (−45, −136, −105, −103, −83, −57, −52, −45, −36, −13,
+6 and +9) were identified to be methylated in HeLa only (Fig. 1B). Of note, Ca Ski cells did not
exhibit methylation of any of the 17 CpG sites of the CDH1
promoter that were analyzed (Fig.
1B). Quantification of the methylation levels in the
CDH1 promoter region indicated that HeLa presented a
methylation frequency of 88.24%, SiHa cells exhibited a methylation
frequency of 17.65% and Ca Ski cells demonstrated no methylation
(frequency, 0%; Fig. 1C).
Based on previous studies (25-28),
initial experiments were conducted using the C33-A, C33-A
transfected with pE7/HPV16 (C33-A pE7/HPV16), MCF-7 and MCF-7
transfected with pE6/E7 from HPV18 (MCF-7 pE6/E7) cell lines as
HPV-negative cancer models. Validation of C33-A and MCF-7 cell
stable clone selection with pE7/HPV16 and pE6/E7, respectively, was
performed by evaluating E7 mRNA expression by RT-PCR (Fig. S1B). However, C33-A vs. C33-A
pE7/HPV16 and MCF-7 vs. MCF-7 pE6/E7 did not exhibit any
differences in the methylation of CDH1 promoter regions
(Fig. S1A) or in the expression
of CDH1 at mRNA and protein levels (Fig. S1B and C). As the MCF-7 cell line
is an adenocarcinoma that derives from the mammary gland and
exhibits an epithelial phenotype with a high expression level of
CDH1, similar to that observed in Ca Ski cells, and since the C33-A
cell line originally does no express CDH1, C33-A and MCF7 cells
were eliminated from the study; neither the effect of oncoprotein
E7 on the suppression of CDH1 expression, nor the
methylation patterns in the CDH1 promoter could be evaluated
in these cells.
Methylation levels in the CDH1 promoter
are associated with the mRNA and protein expression levels of
CDH1
Analysis of CDH1 expression in the different
cell lines demonstrated a significant decrease of the CDH1
mRNA level in the HeLa (P<0.001) and SiHa (P<0.01) cell lines
compared with the HaCaT cell line (Fig. 1D). A similar decrease was observed
at the protein level (Fig. 1E). By
contrast, the Ca Ski cell line exhibited high CDH1
expression, similar to the control HaCaT cell line, at the mRNA and
protein level (Fig. 1D and E).
CDH1 expression level is associated with
the expression level of SNAI1
The present study measured the expression of
SNAI1 and SNAI2 to test the hypothesis that the
expression of CDH1 is regulated by the transcription factors
Snai1 and Snai2, which mediate EMT by negatively regulating the
expression of CDH1 (27,55-57).
The results revealed that the mRNA expression level of SNAI1
is significantly increased in HeLa (P<0.0001) and SiHa cells
(P<0.001), but significantly reduced in Ca Ski cells (P<0.01)
compared with HaCaT cells (Fig.
1D). This result was also reflected at the protein level
(Fig. 1E). No significant
differences were observed in the expression of SNAI2 at the
mRNA (Fig. 1D) and protein
(Fig. 1E) level among all cell
lines. Since the Ca Ski cell line exhibits a non-mesenchymal
phenotype, a high expression level of E-cadherin and low expression
levels of Snai1 and Snai2, this cell line was excluded from further
analysis.
Treatment with 5‑AzadC and TSA affect
methylation and re‑expression of CDH1
HeLa and SiHa cells were treated with different
concentrations of 5-AzadC and TSA, followed by analysis of the
expression levels of CDH1, SNAI1, and SNAI2.
HeLa and SiHa cells treated with the vehicle (DMSO) were used for
normalizing the expression values, as well for performing
comparisons in statistical analysis.
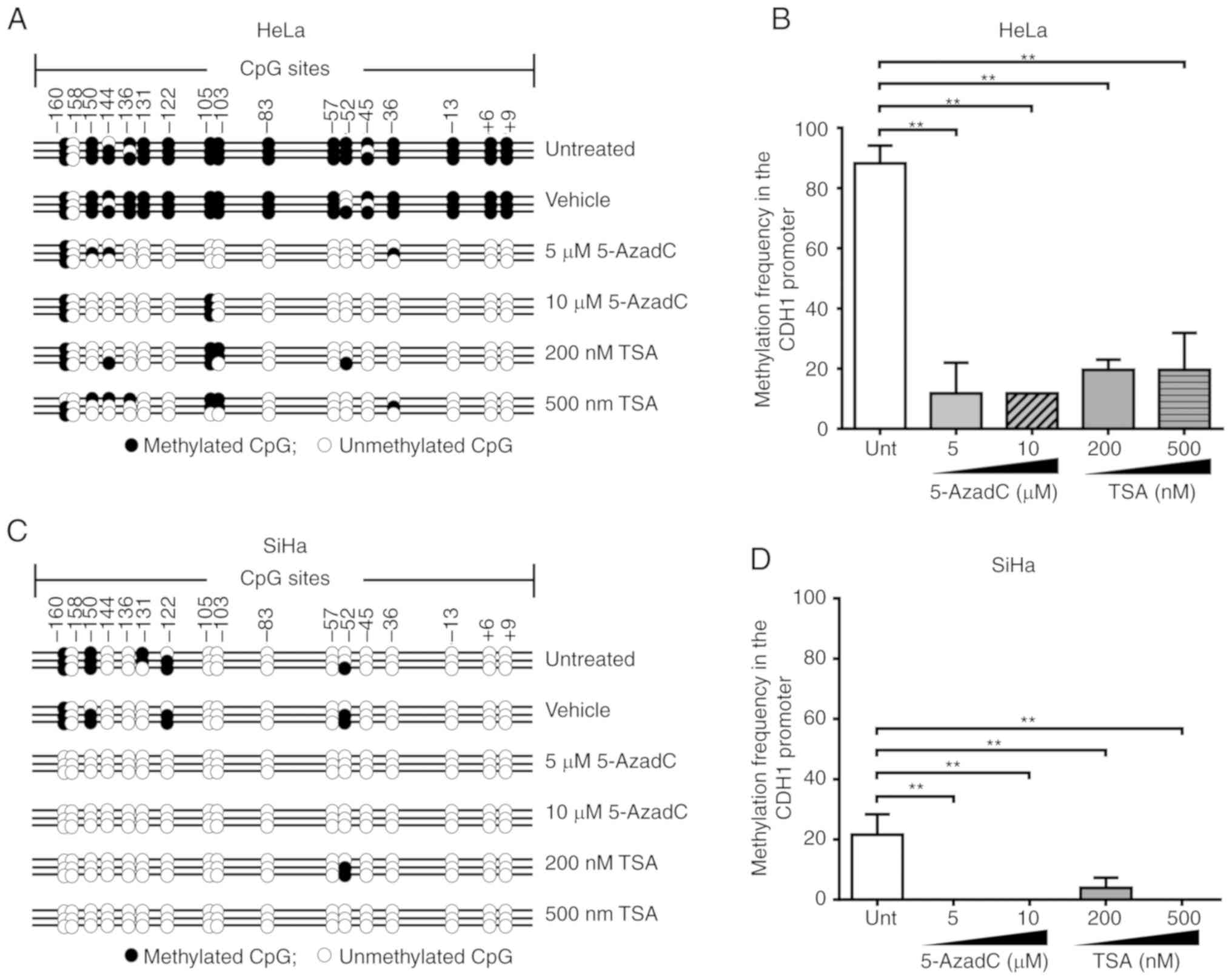
The results demonstrated that in the HeLa cell line,
the methylation pattern was maintained at CpG sites -103, -105, and
-160 of the CDH1 promoter region following treatment with
5-AzadC and TSA. By contrast, in the SiHa cell line, this region
was completely demethylated following treatment with 5 or 10
µM 5-AzadC or 500 nM TSA, and only CpG site -52 was
methylated following treatment with 200 nM TSA (Fig. 2A and C). Compared with untreated
HeLa and SiHa cells, a decrease in methylation of 76.48% was
observed following 5 or 10 µM 5-AzadC treatment, whereas
following treatment with 200 or 500 nM TSA, a decrease in
methylation of 68.63% was observed. By contrast, in SiHa cells, a
decrease of 21.57% was observed following treatment with 5 or 10
µM 5-AzadC or 500 nM TSA, whereas following treatment with
200 nM TSA, a 17.65% decrease in methylation was observed.
Therefore, it was demonstrated in the two cell lines that treatment
with 5‑AzadC or TSA significantly diminished the level of
CDH1 methylation (P<0.001; Fig. 2B and D). However, no significant
differences were observed between the two treatments in diminishing
the methylation levels of CDH1.
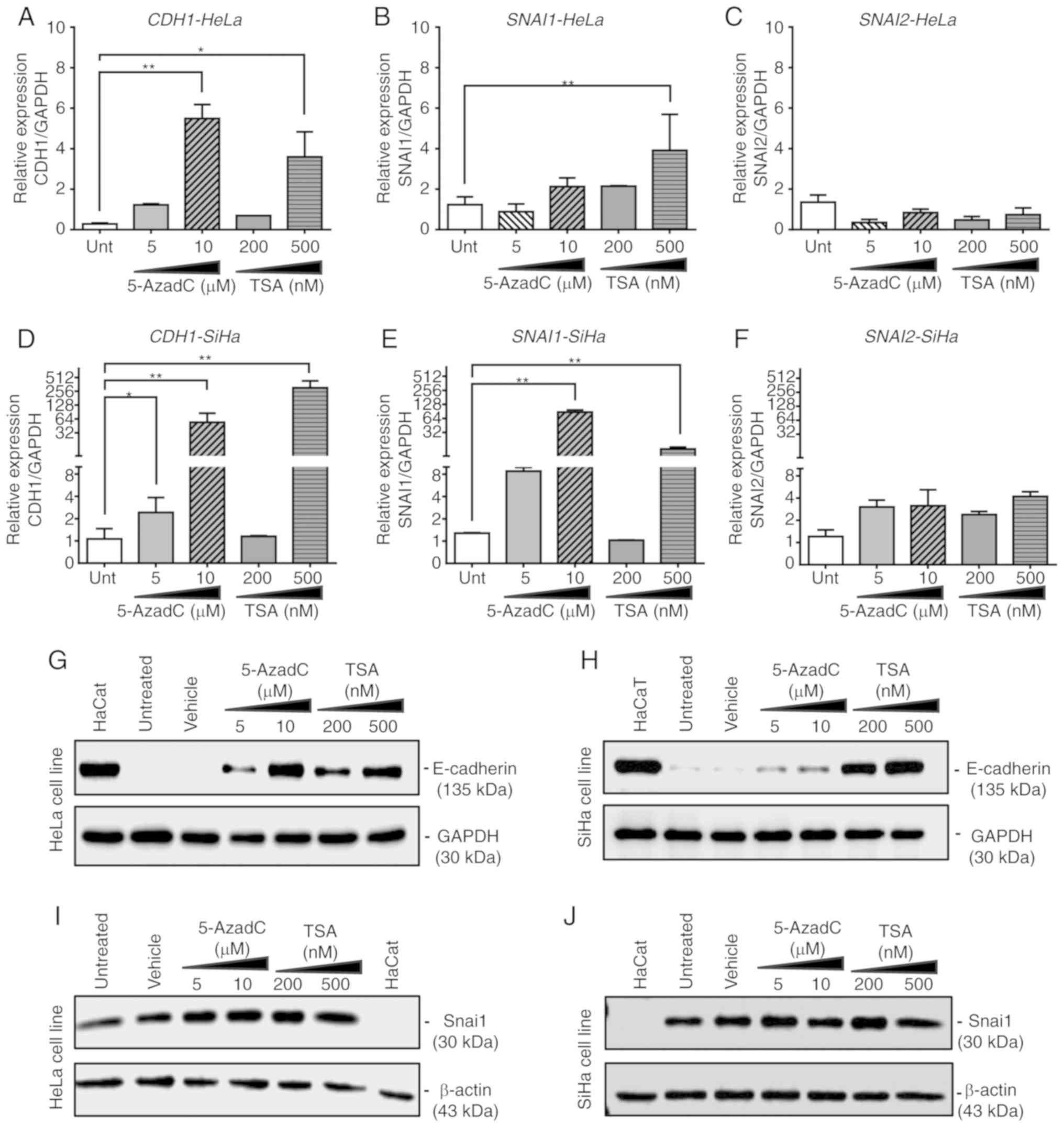
In the HeLa cell line, treatments with 10 µM
5-AzadC (P=0.004) and 500 nM TSA (P=0.03) significantly increased
the expression of CDH1 mRNA and protein (Fig. 3A and G). In addition, 500 nM TSA
significantly increased the mRNA expression of SNAI1
(P=0.01, Fig. 3B) in HeLa cells
without notable changes in protein expression (Fig. 3I). In the SiHa cell line, the mRNA
expression of CDH1 significantly increased following
treatment with 5-AzadC at 5 and 10 µM (P<0.05 and
P<0.01, respectively) or 500 nM TSA (P=0.004; Fig. 3D); a similar effect was observed at
the protein level (Fig. 3H). In
addition, the expression of SNAI1 mRNA was significantly
increased following treatment with 10 µM 5-AzadC (P=0.004)
or 500 nM TSA (P=0.01) (Fig. 3E)
in SiHa cells without notable changes at the protein level
(Fig. 3J), similar to that
observed in HeLa.
5-AzadC and TSA also increased the mRNA expression
level of SNAI1 in the HeLa (Fig. 3B) and SiHa cell lines (Fig. 3E); however, no changes were
observed in SNAI1 protein levels in the two cell lines (Fig. 3I and J). The expression of SNAI2
was not significantly modified at the mRNA level under any
treatment condition in the tested cell lines (Fig. 3C and F).
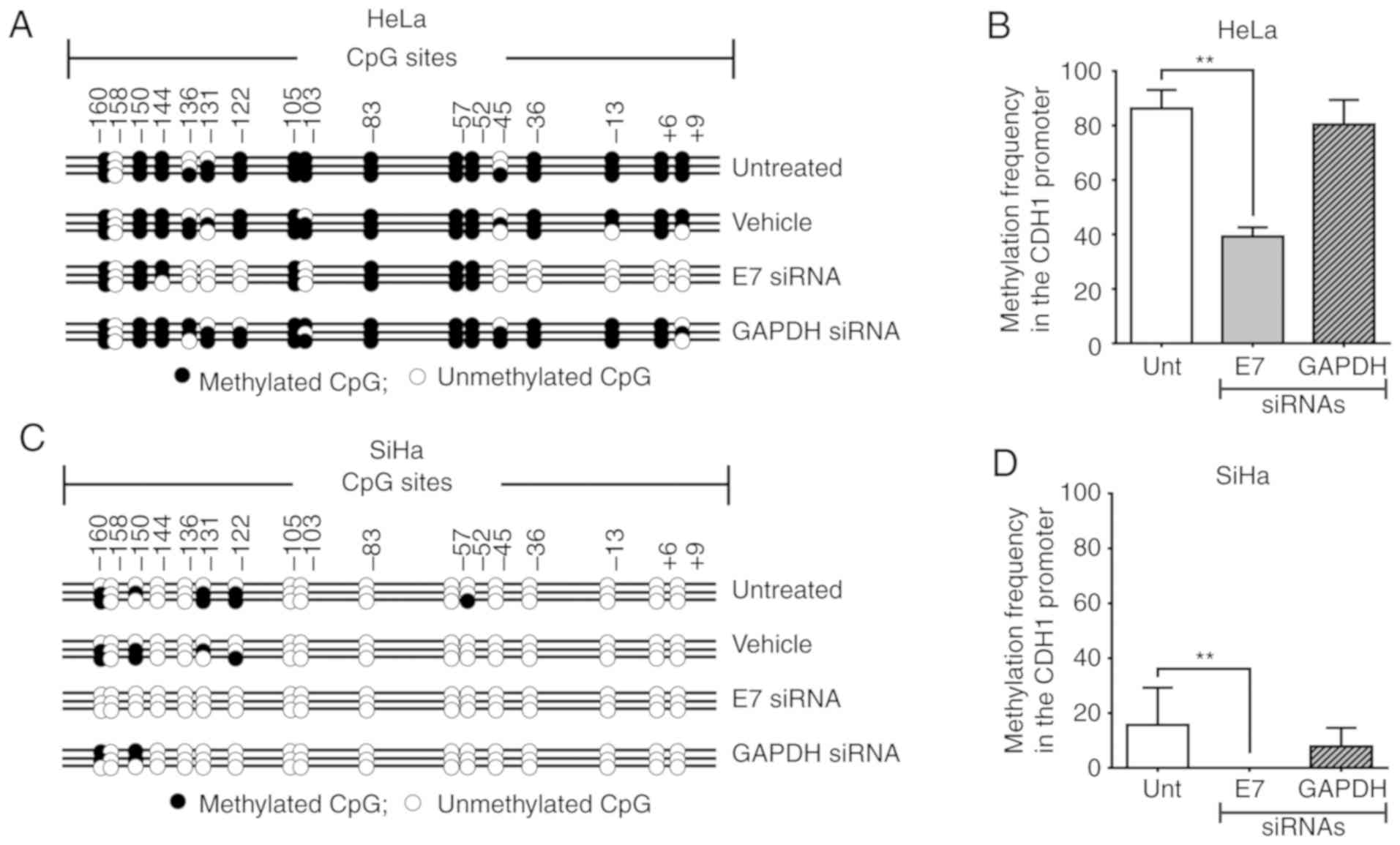
Suppression of E7 by siRNA modifies the
methylation patterns of the CDH1 promoter and induces CDH1
expression in HeLa and SiHa cell lines
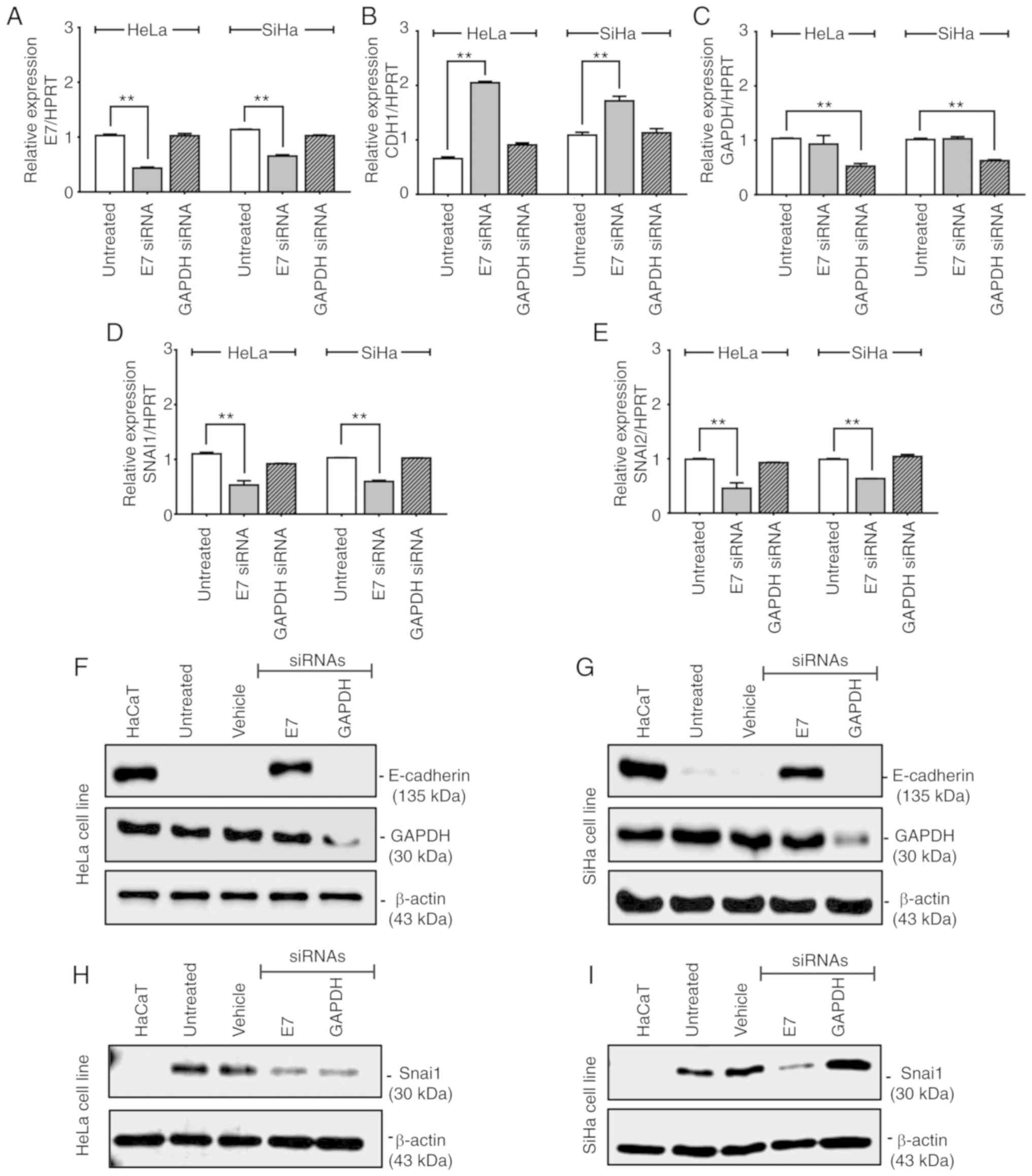
To examine the involvement of E7 in the
CDH1 methylation and expression patterns, HeLa and SiHa cell
lines were transfected with siRNA against E7, which resulted
in 57.9 and 42.5% reduction in the E7 mRNA level,
respectively (Fig. 5A and C). In
the HeLa cell line, E7 silencing led to demethylation of
CDH1 CpG promoter sites located between +9 and -45, as well
as at -103 and between -122 and -136 (Fig. 4A). In addition, in the SiHa cell
line, total demethylation of the CDH1 promoter was observed
following E7 silencing (Fig.
4C). Therefore, partial silencing of E7 in HeLa and SiHa
cells yielded a significant decrease in CDH1 promoter
methylation compared with untreated control cells (P<0.001;
Fig. 4B and D). Increased
CDH1 mRNA and protein levels were observed following
E7 silencing in HeLa (Fig. 5B
and F) and SiHa (Fig. 5B and
G) cells. The use of Silencer® Select GAPDH
siRNA as a positive control siRNA excluded the possibility that
changes in the methylation pattern of the CDH1 promoter were
due to the siRNA transfection conditions as GAPDH silencing
did not induce significant changes in the methylation pattern of
the CDH1 promoter in HeLa (P=0.7140; Fig. 4B) and SiHa (P=0.3248; Fig. 4D) cells; therefore, the changes in
the methylation pattern were likely due to E7 silencing.
Suppression of E7 inhibits SNAI1 and
SNAI2 expression in HeLa and SiHa cells
Silencing of E7 not only induced the
expression of CDH1, but also significantly decreased the
expression of SNAI1 and SNAI2 (P<0.001) in HeLa
and SiHa cells (Fig. 5D, E, H and
I). This suggested that E7 may be not only involved in
suppressing the expression of CDH1, but may also regulate
the expression of SNAI1 and SNAI2, which negatively
regulate CDH1. No significant changes were observed in the mRNA
expression of E7, CDH1, SNAI1 and SNAI2
following GAPDH silencing in HeLa and SiHa cells (P>0.5;
Fig. 5A, B, D and E). However,
silencing of GAPDH with siRNA resulted in a decrease in the
protein expression of Snai1 in HeLa cells (Fig. 5H).
As housekeeping genes GAPDH and HPRT
were used to normalize the expression levels of the genes studied,
no significant differences were noted in the reduction of
SNAI1 expression at the mRNA level (Fig. 5D). Further analysis of the
expression levels of GAPDH, CDH1 and SNAI1
genes normalized against β-actin demonstrated that GAPDH
expression was 2.6-fold higher in HaCaT, 1.6-fold higher in HeLa,
3.3-fold higher in SiHa and 3.8-fold higher in Ca Ski cells
compared with a commercial sample of RNA extracted from normal
cervical tissue (Fig. S2).
Discussion
HR-HPV activates the cell methylation machinery,
which not only methylates its own genome, but also the promoter
regions of cellular genes (21).
Laurson et al (26) have
demonstrated that E7 induces the expression of Dnmt1 and suppresses
the expression of CDH1. Reduction of E-cadherin has been
reported to contribute to the persistence of HPV, which is in
agreement with reports that E7 interacts with and induces the
expression of Dnmt1 and triggers its de novo methylation
activity (16) (Fig. 6A). The results of the present study
agree with previous findings by Laurson et al (26), in which E7 from HPV16 suppressed
the expression of CDH1. However, in this previous study, no
significant differences were observed in the methylation pattern of
the CDH1 promoter of NIKS cells overexpressing E7, as the 17
CpG sites analyzed were not methylated (23). In addition, treatment with 5-AzadC
re-established the expression of CDH1 at the mRNA and
protein levels (26); however, the
concentration of 5-AzadC used was not stated and the changes in the
expression of other cellular genes were not discussed.
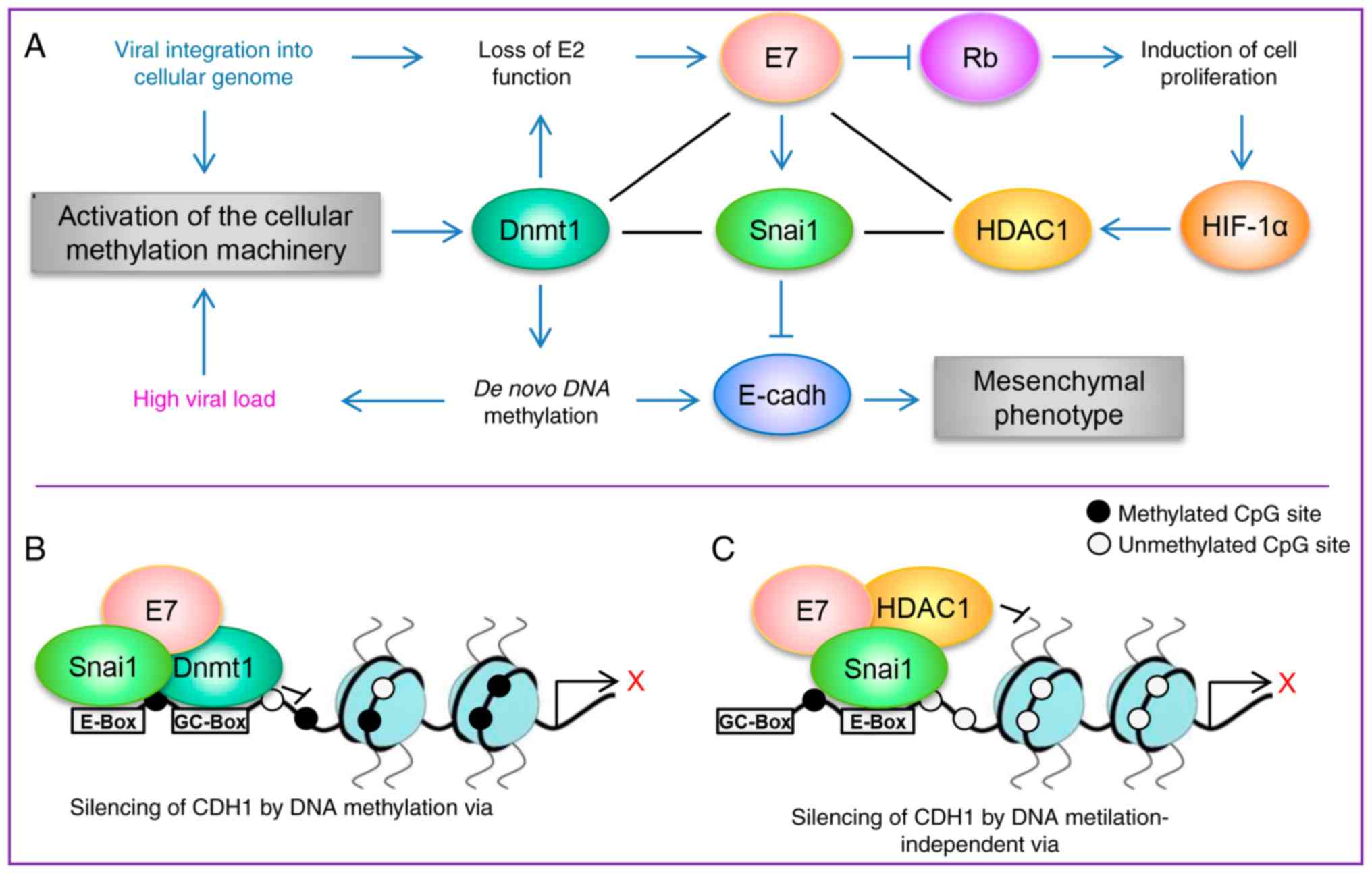 | Figure 6A schematic model demonstrating how
high-risk HPV E7 may induce epithelial-mesenchymal transition via
Snai1. (A) A schematic model of proteins that interact with or are
induced by E7. The loss of E2 function, either from the integration
or methylation of the E2 binding sites in the HPV long control
region, leads to the dysregulated expression of the oncoproteins E6
and E7. E7 serves an important role in regulating several pathways;
E7 induces cellular proliferation via pRB and E7 not only induces
the expression of Dnmt1 and HDAC1, but also physically interacts
with them, which promotes epigenetic regulation of HPV and cellular
genes. The results of the present study indicated that E7 may not
only suppresses the expression of CDH1 through the
methylation of its promoter region, but also induce the expression
of Snai1, which is a negative regulator of CDH1 expression.
(B and C) Proposed models of how E7 may regulate the expression of
CDH1. HPV, human papilloma virus; CDH1, cadherin 1;
Dmnt1, DNA methyltransferase 1; HDAC1, histone deacetylase type 1;
HIF-1α, hypoxia-inducible factor 1α; pRB, retinoblastoma
protein. |
The present study evaluated the re-expression of
CDH1 at the mRNA and protein levels in HeLa and SiHa cell
lines treated with 5-AzadC and TSA, as well as changes in the
methylation pattern of the promoter region of CDH1.
Additionally, although TSA is not a demethylating agent,
demethylation in the promoter of CDH1 following treatment
with TSA was observed. This result was an agreement with previous
studies that have reported that TSA induces DNA demethylation and
proposed that a change in chromatin modification, including the
deacetylation of histones induced by an HDAC inhibitor, such as
TSA, may render a gene susceptible to DNA demethylation (58,59).
In addition, an increase in SNAI1 mRNA level
was observed after treatment with 500 nM TSA in HeLa cells, whereas
in SiHa cells an increase in SNAI1 mRNA level was observed
after treatment with 10 µM 5-AzadC and 500 nM TSA; however,
no apparent changes were observed in SNAI1 protein level in the two
cell lines. This result was in agreement with studies in which such
treatments not only re-established the expression of CDH1,
but also induced upregulation of SNAI1 and SNAI2 at
the mRNA level (60,61), which was likely due to
modifications in the methylation pattern in the promoter regions of
SNAI1 and SNAI2 (62). The role of SNAI1 regulation
by epigenetic mechanisms is largely unknown. A previous study has
demonstrated that the treatment with 5‑AzadC in fibroblast cell
IMR90, induced pluripotent stem cells from IMR90, BeWo and
HTR8/SVneo cell lines induces a greater expression of SNAI1
and SNAI2 at the mRNA level and that the regulation of the
two genes is mediated by DNA methylation of their first intron and
not due to DNA methylation of their promoter region; however, this
previous study did not determine the expression of these genes at
the protein level (62). The
present study did not determine the methylation status of the
SNAI1 promoter region as it is transcriptionally active in
HeLa and SiHa cells. The differences observed in the effect on
SNAI1 expression at the mRNA and protein levels by treatment with
5-AzadC and TSA indicated that other factors may regulate the
expression of SNAI1 in the two cell lines.
On the other hand, the results published by Laurson
et al (26) suggested that
in the NIKS-cell model, suppression of CDH1 expression by E7
was independent of the methylation status of the CDH1
promoter region, which was observed in the SiHa cell line in the
current study. The previous study also suggested that repression of
CDH1 may be regulated via SNAI2 (SLUG);
however, it was reported that the expression of SNAI2 was
not altered by the presence of E7 (26). The results of the present study
revealed that while SNAI1 mRNA and protein was expressed in
HeLa and SiHa cells, SNAI2 mRNA expression was barely
detectable in HaCaT and Ca Ski cells. SNAI2 has been
demonstrated to be upregulated in HaCaT cells during the process of
cell motility and wound-healing (63).
The results of the present study demonstrated that
following silencing of E7 from HPV16 and HPV18, CDH1
expression was recovered in HeLa and SiHa cells, which is in
agreement with a previous study by Caberg et al (25). This previous study reported that
following 24-h transfection of SiHa cells with siRNA against HPV16
E7, CDH1 was upregulated and an increase of the
Retinoblastoma protein (pRB), which is responsible for a major G1
check point, blocking S-phase entry and cell growth, and activating
protein 2α were detected without changes in the mRNA expression
levels of SNAI1 and SNAI2 (25). By contrast, the present study
demonstrated that following 48-h transfection with siRNA against
E7, changes in the methylation pattern of the CDH1
promoter region were observed in HeLa and SiHa cells. In addition,
an increase in the mRNA and protein expression levels of
CDH1 were identified, as well as a decrease in the mRNA and
protein expression levels of SNAI1 and SNAI2.
Therefore, the current results suggested that E7 not only
suppressed the expression of CDH1 via the methylation of its
promoter, but also regulated the expression of SNAI1, a
negative regulator of CDH1 involved in EMT and associated
with metastasis (27,55-57).
These observations are also concordant with the mechanism of action
reported for other oncogenic viruses, where the X protein of HBV
(HBx), core protein of HCV and latent membrane protein 1 (LMP1) of
EBV promote EMT and metastasis by inducing the expression
SNAI1 and suppressing CDH1 expression (64-67).
Following silencing GAPDH with siRNA,
SNAI1 expression was partially suppressed in HeLa cells at
the protein level, but not at mRNA level. This was consistent with
a previous study that demonstrated that the interaction of GAPDH
with Sp1 resulted in increased expression of Snai1, which promoted
the proliferation and metastasis of cancer cells, and that
suppression of GAPDH with shRNA resulted in a significant
decrease of Snai1 in the HCT116 and LoVo cell lines (68). This suggested that GAPDH may serve
a role in the metastasis of cervical adenocarcinoma (HeLa) by
affecting EMT through the upregulation of Snai1 expression mediated
by Sp1, similar to its role reported in colon cancer (68), but further studies are required to
verify this.
It is currently unknown how HPV may activate the
expression of SNAI1; however, it has been reported that in
other cancer types associated with virus, HBx, core and LMPI
proteins increased SNAI1 expression through the activation
of the PI3K/Akt and MAPK pathways by transforming growth factor-β
(TGF-β) action (64,69-72),
which is in agreement with a previous study by Peinado et al
(73) suggesting that TGF-β
induces SNAI1 transcription through MAPK and PI3K.
In the present study, the signaling pathways
involved in regulating SNAI1 expression were not determined;
however, previous studies have reported that HR-HPV infection
activates the PI3K/Akt/mTOR pathway (74), E7 from HPV upregulates Akt activity
though the pRB protein (75) and
TGF-β stimulates EMT and tumor invasion in SiHa cells (76).
In summary, the results of the present demonstrated
that HR-HPV E7 may regulate the expression of CDH1 by two
different pathways, in which Snai1 is involved. The first pathway
involves hypermethylation of the CDH1 promoter region and
the expression of Snai1, as observed in the HeLa cell line. The
second pathway involves hypomethylation of the CDH1 promoter
region with expression of Snai1 as observed in SiHa cell line,
suggesting that CDH1 and SNAI1 may be considered as
biomarkers of metastasis in uterine cervical cancer. Therefore,
based on the present results and previous evidence that E7
interacts with Dnmt1 and HDAC1 (15-17),
it would be beneficial to determine if E7 from HR‑HPV may interact
with Snai1 to form a co-repressor complex with either Dnmt1 or
HDAC1 in the CDH1 promoter (Fig. 6B and C), which may explain the
suppression of CDH1 expression during the EMT process.
Supplementary Data
Funding
The present study received additional support of
the Fundación Miguel Alemán, A.C. to AGC. PRC received a fellowship
from Programa de Movilidad Internacional de Estudiantes de la
Coordinación de Estudios de Posgrado (CEP-UNAM) during his doctoral
stay at the Universidad Autónoma de Madrid (UAM).
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
PRC performed all experiments, interpreted the data
and wrote the manuscript. VAV, GDBO and CCPM performed western blot
analysis and interpreted the data. AC and AGC conceived and
designed the study, interpreted the data and edited the manuscript.
All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
This study is part of the doctoral dissertation
project of Pedro Rosendo Chalma, a Doctoral student from Programa
de Doctorado en Ciencias Biomédicas (PDCB), Instituto de
Investigaciones Biomédicas, Universidad Nacional Autónoma de México
(UNAM) and received a fellowship from Consejo Nacional de Ciencia y
Tecnología (CONACyT; grant no. 203376). The present study was
supported by CONACyT (grant no. 253804 to AG-C) and MINECO (grant
no. SAF2013-44739-R to AC). The authors would like to thank Ms.
Miriam C. Guido Jiménez (UNAM) and Ms. Raquél López Paniagua
(INCan) for their technical support, Dr Erick de la Cruz Hernández
(Juarez Autonomous University of Tabasco) and Dr Patricio Gariglio
(CINVESTAV-IPN) for providing the cell lines and Dr Alfonso Dueñas
Gonzalez (INCan-UNAM) for providing the oligonucleotides for
CDH1 used in the MSP-Protocol.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
2
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
zur Hausen H: Papillomaviruses in the
causation of human cancers-a brief historical account. Virology.
384:260–265. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bansal A, Singh MP and Rai B: Human
papillomavirus-associated cancers: A growing global problem. Int J
Appl Basic Med Res. 6:84–89. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Doorbar J: Latent papillomavirus
infections and their regulation. Curr Opin Virol. 3:416–421. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schiffman M, Doorbar J, Wentzensen N, de
Sanjosé S, Fakhry C, Monk BJ, Stanley MA and Franceschi S:
Carcinogenic human papillomavirus infection. Nat Rev Dis Primers.
2:160862016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Durst M, Kleinheinz A, Hotz M and Gissmann
L: The physical state of human papillomavirus type 16 DNA in benign
and malignant genital tumours. J Gen Virol. 66:1515–1522. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lehn H, Villa LL, Marziona F, Hilgarth M,
Hillemans HG and Sauer G: Physical state and biological activity of
human papillomavirus genomes in precancerous lesions of the female
genital tract. J Gen Virol. 69:187–196. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Badal S, Badal V, Calleja-Macias IE,
Kalantari M, Chuang LS, Li BF and Bernard HU: The human
papillomavirus-18 genome is efficiently targeted by cellular DNA
methylation. Virology. 324:483–492. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fernandez AF, Rosales C, Lopez-Nieva P,
Graña O, Ballestar E, Ropero S, Espada J, Melo SA, Lujambio A,
Fraga MF, et al: The dynamic DNA methylomes of double-stranded DNA
viruses associated with human cancer. Genome Res. 19:438–451. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kalantari M, Lee D, Calleja-Macias IE,
Lambert PF and Bernard HU: Effects of cellular differentiation,
chromosomal integration and 5-aza-2'-deoxycytidine treatment on
human papillomavirus-16 DNA methylation in cultured cell lines.
Virology. 374:292–303. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Boyer SN, Wazer DE and Band V: E7 protein
of human papilloma virus-16 induces degradation of retinoblastoma
protein through the ubiquitin-proteasome pathway. Cancer Res.
56:4620–4624. 1996.PubMed/NCBI
|
|
14
|
Munger K, Phelps WC, Bubb V, Howley PM and
Schlegel R: The E6 and E7 genes of the human papillomavirus type 16
together are necessary and sufficient for transformation of primary
human keratinocytes. J Virol. 63:4417–4421. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brehm A, Nielsen SJ, Miska EA, McCance DJ,
Reid JL, Bannister AJ and Kouzarides T: The E7 oncoprotein
associates with Mi2 and histone deacetylase activity to promote
cell growth. EMBO J. 18:2449–2458. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Burgers WA, Blanchon L, Pradhan S, de
Launoit Y, Kouzarides T and Fuks F: Viral oncoproteins target the
DNA methyltransfer-ases. Oncogene. 26:1650–1655. 2007. View Article : Google Scholar
|
|
17
|
Longworth MS and Laimins LA: The binding
of histone deacety-lases and the integrity of zinc finger‑like
motifs of the E7 protein are essential for the life cycle of human
papillomavirus type 31. J Virol. 78:3533–3541. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang Y, LeRoy G, Seelig HP, Lane WS and
Reinberg D: The dermatomyositis‑specific autoantigen Mi2 is a
component of a complex containing histone deacetylase and
nucleosome remod-eling activities. Cell. 95:279–289. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li H, Ou X, Xiong J and Wang T: HPV16E7
mediates HADC chromatin repression and downregulation of MHC class
I genes in HPV16 tumorigenic cells through interaction with an MHC
class I promoter. Biochem Biophys Res Commun. 349:1315–1321. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Georgopoulos NT, Proffitt JL and Blair GE:
Transcriptional regulation of the major histocompatibility complex
(MHC) class I heavy chain, TAP1 and LMP2 genes by the human
papilloma-virus (HPV) type 6b, 16 and 18 E7 oncoproteins. Oncogene.
19:4930–4935. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Milutin Gasperov N, Sabol I, Planinić P,
Grubišić G, Fistonić I, Ćorušić A and Grce M: Methylated host cell
gene promoters and human papillomavirus type 16 and 18 predicting
cervical lesions and cancer. PLoS One. 10:e01294522015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu J, Lian Z, Han S, Waye MM, Wang H, Wu
MC, Wu K, Ding J, Arbuthnot P, Kew M, et al: Downregulation of
E-cadherin by hepatitis B virus X antigen in hepatocellullar
carcinoma. Oncogene. 25:1008–1017. 2006. View Article : Google Scholar
|
|
23
|
McLaughlin-Drubin ME and Munger K: Viruses
associated with human cancer. Biochim Biophys Acta. 1782:127–150.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tsai CN, Tsai CL, Tse KP, Chang HY and
Chang YS: The Epstein-Barr virus oncogene product, latent membrane
protein 1, induces the downregulation of E-cadherin gene expression
via activation of DNA methyltransferases. Proc Natl Acad Sci USA.
99:10084–10089. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Caberg JH, Hubert PM, Begon DY, Herfs MF,
Roncarati PJ, Boniver JJ and Delvenne PO: Silencing of E7 oncogene
restores functional E-cadherin expression in human papillomavirus
16-transformed keratinocytes. Carcinogenesis. 29:1441–1447. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Laurson J, Khan S, Chung R, Cross K and
Raj K: Epigenetic repression of E-cadherin by human papillomavirus
16 E7 protein. Carcinogenesis. 31:918–926. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cano A, Perez-Moreno MA, Rodrigo I,
Locascio A, Blanco MJ, del Barrio MG, Portillo F and Nieto MA: The
transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pattillo RA, Hussa RO, Story MT, Ruckert
AC, Shalaby MR and Mattingly RF: Tumor antigen and human chorionic
gonadotropin in CaSki cells: A new epidermoid cervical cancer cell
line. Science. 196:1456–1458. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Friedl F, Kimura I, Osato T and Ito Y:
Studies on a new human cell line (SiHa) derived from carcinoma of
uterus. I Its establishment and morphology. Proc Soc Exp Biol Med.
135:543–545. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Diao MK, Liu CY, Liu HW, Li JT, Li F,
Mehryar MM, Wang YJ, Zhan SB, Zhou YB, Zhong RG and Zeng Y:
Integrated HPV genomes tend to integrate in gene desert areas in
the CaSki, HeLa, and SiHa cervical cancer cell lines. Life Sci.
127:46–52. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yee C, Krishnan-Hewlett I, Baker CC,
Schlegel R and Howley PM: Presence and expression of human
papillomavirus sequences in human cervical carcinoma cell lines. Am
J Pathol. 119:361–366. 1985.PubMed/NCBI
|
|
32
|
Schneider-Gädicke A and Schwarz E:
Different human cervical carcinoma cell lines show similar
transcription patterns of human papillomavirus type 18 early genes.
EMBO J. 5:2285–2292. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pater MM and Pater A: Human papillomavirus
types 16 and 18 sequences in carcinoma cell lines of the cervix.
Virology. 145:313–318. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pater MM and Pater A: Expression of human
papillomavirus types 16 and 18 DNA sequences in cervical carcinoma
cell lines. J Med Virol. 26:185–195. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Spence RP, Murray A, Banks L, Kelland LR
and Crawford L: Analysis of human papillomavirus sequences in cell
lines recently derived from cervical cancers. Cancer Res.
48:324–328. 1988.PubMed/NCBI
|
|
36
|
Meissner JD: Nucleotide sequences and
further characterization of human papillomavirus DNA present in the
CaSki, SiHa and HeLa cervical carcinoma cell lines. J Gen Virol.
80:1725–1733. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
De la Cruz-Hernandez E, Garcia-Carranca A,
Mohar-Betancourt A, Dueñas-González A, Contreras-Paredes A,
Pérez-Cardenas E, Herrera-Goepfert R and Lizano-Soberón M:
Differential splicing of E6 within human papillomavirus type 18
variants and functional consequences. J Gen Virol. 86:2459–2468.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vazquez-Vega S, Sanchez-Suarez LP,
Andrade-Cruz R, Castellanos-Juarez E, Contreras-Paredes A,
Lizano-Soberon M, Garcia-Carranca A and Benitez Bribiesca L:
Regulation of p14ARF expression by HPV-18 E6 variants. J Med Virol.
85:1215–1221. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vazquez-Vega S, Sanchez-Suarez LP,
Contreras-Paredes A, Castellanos-Juárez E, Peñarroja-Flores R,
Lizano-Soberón M, Andrade-Cruz R, García-Carrancá A and
Benítez-Bribiesca L: Nuclear co-expression of p14ARF and p16INK4A
in uterine cervical cancer-derived cell lines containing HPV.
Cancer Biomark. 8:341–350. 2011. View Article : Google Scholar
|
|
40
|
Gutier rez J, Ga rcia-Villa E,
Ocadiz-Delgado R, Cortés-Malagón EM, Vázquez J, Roman-Rosales A,
Alvarez-Rios E, Celik H, Romano MC, Üren A, et al: Human
papillomavirus type 16 E7 oncoprotein upregulates the retinoic acid
receptor-beta expression in cervical cancer cell lines and K14E7
transgenic mice. Mol Cell Biochem. 408:261–272. 2015. View Article : Google Scholar
|
|
41
|
Momparler RL: Epigenetic therapy of cancer
with 5-aza-2'-deox-ycytidine (decitabine). Semin Oncol. 32:443–451.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Vigushin DM, Ali S, Pace PE, Mirsaidi N,
Ito K, Adcock I and Coombes RC: Trichostatin A is a histone
deacetylase inhibitor with potent antitumor activity against breast
cancer in vivo. Clin Cancer Res. 7:971–976. 2001.PubMed/NCBI
|
|
43
|
Jiang M and Milner J: Selective silencing
of viral gene expression in HPV-positive human cervical carcinoma
cells treated with siRNA, a primer of RNA interference. Oncogene.
21:6041–6048. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lea JS, Sunaga N, Sato M, Kalahasti G,
Miller DS, Minna JD and Muller CY: Silencing of HPV 18 oncoproteins
with RNA interference causes growth inhibition of cervical cancer
cells. Reprod Sci. 14:20–28. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sledz CA, Holko M, de Veer MJ, Silverman
RH and Williams BR: Activation of the interferon system by
short-interfering RNAs. Nat Cell Biol. 5:834–839. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Makpol S, Zainuddin A and Chua KH: GAPDH
expression as a measurement of transfection efficiency for p16
INK4a gene silencing (siRNA) in senescent human diploid
fibroblasts. Am J Mol Biol. 2:390–397. 2012. View Article : Google Scholar
|
|
47
|
Han H: RNA interference to knock down gene
expression. Methods Mol Biol. 1706:293–302. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Borawski J, Lindeman A, Buxton F, Labow M
and Gaither LA: Optimization procedure for small interfering RNA
transfection in a 384-well format. J Biomol Screen. 12:546–559.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Peter Hahn JD, Wolfgang Bielke and Kang
Jie: Patent: EP2240582 B1-Positive controls for expression
modulating experiments. European Patent Office. October
23–2013.
|
|
50
|
Cheng A, Magdaleno S and Vlassov AV:
Optimization of trans-fection conditions and analysis of siRNA
potency using real-time PCR. Methods Mol Biol. 764:199–213. 2011.
View Article : Google Scholar
|
|
51
|
Badal V, Chuang LS, Tan EH, Badal S, Villa
LL, Wheeler CM, Li BF and Bernard HU: CpG methylation of human
papilloma-virus type 16 DNA in cervical cancer cell lines and in
clinical specimens: Genomic hypomethylation correlates with
carcinogenic progression. J Virol. 77:6227–6234. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kalantari M, Calleja-Macias IE, Tewari D,
Hagmar B, Lie K, Barrera-Saldana HA, Wiley DJ and Bernard HU:
Conserved methylation patterns of human papillomavirus type 16 DNA
in asymptomatic infection and cervical neoplasia. J Virol.
78:12762–12772. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hoffmann I, Hilger M and Mueller O: Homo
sapiens promoter of E-cadherin from HEK293 cells.
Max-Planck-Institut fuer Molekulare Physiologie; Dortmund: 2006
|
|
54
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
55
|
Peinado H, Portillo F and Cano A:
Transcriptional regulation of cadherins during development and
carcinogenesis. Int J Dev Biol. 48:365–375. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Moreno-Bueno G, Cubillo E, Sarrio D,
Peinado H, Rodríguez-Pinilla SM, Villa S, Bolós V, Jordá M, Fabra
A, Portillo F, et al: Genetic profiling of epithelial cells
expressing E-cadherin repressors reveals a distinct role for Snail,
Slug, and E47 factors in epithelial-mesenchymal transition. Cancer
Res. 66:9543–9556. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Peinado H, Olmeda D and Cano A: Snail, Zeb
and bHLH factors in tumour progression: An alliance against the
epithelial phenotype? Nat Rev Cancer. 7:415–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Cameron EE, Bachman KE, Myöhänen S, Herman
JG and Baylin SB: Synergy of demethylation and histone deacetylase
inhibition in the re-expression of genes silenced in cancer. Nat
Genet. 21:103–107. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ou JN, Torrisani J, Unterberger A,
Provençal N, Shikimi K, Karimi M, Ekström TJ and Szyf M: Histone
deacetylase inhibitor Trichostatin A induces global and
gene‑specific DNA demeth-ylation in human cancer cell lines.
Biochem Pharmacol. 73:1297–1307. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Meng F, Sun G, Zhong M, Yu Y and Brewer
MA: Anticancer efficacy of cisplatin and trichostatin A or
5‑aza‑2'‑deoxycytidine on ovarian cancer. Br J Cancer. 108:579–586.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Liu YN, Lee WW, Wang CY, Chao TH, Chen Y
and Chen JH: Regulatory mechanisms controlling human E-cadherin
gene expression. Oncogene. 24:8277–8290. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Chen Y, Wang K, Qian CN and Leach R: DNA
methylation is associated with transcription of Snail and Slug
genes. Biochem Biophys Res Commun. 430:1083–1090. 2013. View Article : Google Scholar :
|
|
63
|
Savagner P, Kusewitt DF, Carver EA,
Magnino F, Choi C, Gridley T and Hudson LG: Developmental
transcription factor slug is required for effective
re-epithelialization by adult kerati-nocytes. J Cell Physiol.
202:858–866. 2005. View Article : Google Scholar
|
|
64
|
Arzumanyan A, Friedman T, Kotei E, Ng IO,
Lian Z and Feitelson MA: Epigenetic repression of E-cadherin
expression by hepatitis B virus x antigen in liver cancer.
Oncogene. 31:563–572. 2012. View Article : Google Scholar
|
|
65
|
Horikawa T, Yoshizaki T, Kondo S, Furukawa
M, Kaizaki Y and Pagano JS: Epstein-Barr Virus latent membrane
protein 1 induces Snail and epithelial-mesenchymal transition in
metastatic nasopharyngeal carcinoma. Br J Cancer. 104:1160–1167.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Liu H, Xu L, He H, Zhu Y, Liu J, Wang S,
Chen L, Wu Q, Xu J and Gu J: Hepatitis B virus X protein promotes
hepatoma cell invasion and metastasis by stabilizing Snail protein.
Cancer Sci. 103:2072–2081. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Nie D, Shan X, Nie L, Duan Y, Chen Z, Yang
Y, Li Z, Tian L, Gao Q, Shan Y and Tang N: Hepatitis C virus core
protein interacts with Snail and histone deacetylases to promote
the metastasis of hepatocellular carcinoma. Oncogene. 35:3626–3635.
2016. View Article : Google Scholar
|
|
68
|
Liu K, Tang Z, Huang A, Chen P, Liu P,
Yang J, Lu W, Liao J, Sun Y, Wen S, et al:
Glyceraldehyde-3-phosphate dehydrogenase promotes cancer growth and
metastasis through upregulation of SNAIL expression. Int J Oncol.
50:252–262. 2017. View Article : Google Scholar
|
|
69
|
Liu Y, Xu Y, Ma H, Wang B, Xu L, Zhang H,
Song X, Gao L, Liang X and Ma C: Hepatitis B virus X protein
amplifies TGF‑β promotion on HCC motility through down-regulating
PPM1a. Oncotarget. 7:33125–33135. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Park GB, Kim D, Kim YS, Kim S, Lee HK,
Yang JW and Hur DY: The Epstein-Barr virus causes
epithelial-mesenchymal transition in human corneal epithelial cells
via Syk/src and Akt/Erk signaling pathways. Invest Ophthalmol Vis
Sci. 55:1770–1779. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Sides MD, Klingsberg RC, Shan B, Gordon
KA, Nguyen HT, Lin Z, Takahashi T, Flemington EK and Lasky JA: The
Epstein-Barr virus latent membrane protein 1 and transforming
growth factor-β1 synergistically induce epithelial–mesenchymal
transition in lung epithelial cells. Am J Respir Cell Mol Biol.
44:852–862. 2011. View Article : Google Scholar
|
|
72
|
Taniguchi H, Kato N, Otsuka M, Goto T,
Yoshida H, Shiratori Y and Omata M: Hepatitis C virus core protein
upregulates transforming growth factor-beta 1 transcription. J Med
Virol. 72:52–59. 2004. View Article : Google Scholar
|
|
73
|
Peinado H, Quintanilla M and Cano A:
Transforming growth factor beta-1 induces snail transcription
factor in epithelial cell lines: Mechanisms for epithelial
mesenchymal transitions. J Biol Chem. 278:21113–21123. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Surviladze Z, Sterk RT, DeHaro SA and
Ozbun MA: Cellular entry of human papillomavirus type 16 involves
activation of the phosphatidylinositol 3-kinase/Akt/mTOR pathway
and inhibition of autophagy. J Virol. 87:2508–2517. 2013.
View Article : Google Scholar :
|
|
75
|
Menges CW, Baglia LA, Lapoint R and
McCance DJ: Human papillomavirus type 16 E7 up-regulates AKT
activity through the retinoblastoma protein. Cancer Res.
66:5555–5559. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Yi JY, Hur KC, Lee E, Jin YJ, Arteaga CL
and Son YS: TGFbeta1-mediated epithelial to mesenchymal transition
is accompanied by invasion in the SiHa cell line. Eur J Cell Biol.
81:457–468. 2002. View Article : Google Scholar : PubMed/NCBI
|