Introduction
Hepatocellular carcinoma (HCC) is the second most
common malignant tumor with the highest morbidity and mortality
(1). Oxaliplatin (OXA), as a
platinum-based chemotherapeutic agent (2), has exhibited efficacy in the
treatment of HCC; however, the majority of hepatobiliary cancer
guidelines do not recommend the use of platinum drugs as first-line
treatment due to the low sensitivity of HCC these drugs (3-6). A
number of studies performed using established cancer cell lines
have demonstrated that resistant cells are characterized by aerobic
glycolysis and lactate levels, a by-product of glycolysis; these
levels are enhanced in drug-resistant or metastatic cancers,
suggesting that the Warburg effect in these cancers may reflect
metabolic adaptations associated with the development of resistance
to chemotherapy (7). The study by
Fekir et al indicated that the upregulation of pyruvate
dehydrogenase kinase (PDK)4 was associated with chemoresistance
that could be successfully reversed by the PDK4 inhibitor,
dichloroacetate (DCA), in 4 HCC cell lines (8). The study by Choiniere et al
demonstrated that tumor cells which have a more vigorous metabolism
were more sensitive to changes in the metabolic mode; in addition,
the metabolism of HCC cells derived from liver cells is
particularly strong (9).
Therefore, chemotherapy combined with therapies that target tumor
cell metabolism may hold great potential for the treatment of
HCC.
Tumor cells, including HCC cells, exhibit a unique
form of metabolism, known as the Warburg effect (10). Although the Warburg effect (aerobic
glycolysis) of tumor cells was discovered a hundred years ago, the
mechanisms of its occurrence have not yet been fully elucidated
(11). PDK1, as a key regulator in
the aerobic glycolysis of tumor cells, phosphorylates the E1
subunit of pyruvate dehydrogenase (PDH) at Ser232, leading to its
inactivation (12). Inactivated
PDH fails to catalyze the conversion of pyruvate into acetyl-CoA,
thus preventing pyruvate from entering tricarboxylic acid cycle
(TCA) (12,13). In other words, PDK1 may regulate
cellular glucose metabolism by controlling the conversion of
pyruvate. Wang et al confirmed that the expression of PDK1
in HCC tissues was significantly higher than that of adjacent
normal tissues by immunohistochemical staining (14). The results of the study by Battello
et al revealed that HCC cells exhibited an enhanced
transcription and expression of hypoxia-inducible factor (HIF)-1α
under normal oxygen conditions through the inflammatory cytokine,
oncostatin M (OSM), resulting in the HIF-1-regulated PDK1
expression in HCC cells (15),
indicating that PDK1 plays an important role in the process of HCC.
In a previous study by the authors, it was demonstrated that
dicoumarol (DIC) can bind to the lipoamide binding pocket of
PDK1's, exerting a more effective selective inhibitory activity
compared to the classic inhibitor of PDK1, sodium dichloroacetate
(DCA), and inhibits glycolysis in human ovarian cancer cells
(16).
Therefore, the present study aimed to target PDK1 to
explore its significance for the metabolic transformation of HCC
cells and its potential for enhancing the sensitivity of
chemotherapeutic drugs. Furthermore, the present study proposes a
possible mechanism of the Warburg effect, providing an effective
strategy for determining the role of oxidative phosphorylation and
glycolysis in tumors, such as HCC.
Materials and methods
Reagents and antibodies
DIC (Selleck Chemicals) was dissolved in 2% dimethyl
sulfoxide (DMSO).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
N-acetyl-L-cysteine (NAC), Hoechst 33342 and anti-p-PDHE1A
(s232) antibody (SAB1305601) were purchased from Sigma-Aldrich;
Merck KGaA. Anti-β-actin antibody (sc-81760) was from Santa Cruz
Biotechnology, Inc. Anti-PDH (ab67592) and anti-PDK1 (ab202468)
were from Abcam.
Analysis of combined drug effects
Drug synergy was determined by the combination index
method derived from the median-effect principle of Chou and
Talalay. Data obtained from the growth inhibitory experiments of
DIC and OXA were used to perform these analyses. The resulting
combination index (CI) theorem of Chou-Talalay offers quantitative
definition for additive effect (CI=1), synergism (CI >1), and
antagonism (CI >1) in drug combinations (17,18).
Inhibition of PDK1 by shRNA
Short hairpin (sh)RNA targeting PDK1 and
scrambled-shRNA (scr-shRNA) were purchased from GenePharma. The
PDK1 shRNA sequence was as follows:
5′-CTT-CGG-ATC-AGT-GAA-TGC-TTG-3′, the scrambled-shRNA (scr-shRNA)
sequence, used as a negative control, was
5′-GTT-CTC-CGA-ACG-TGT-CAC-GT-3′. Following adherence, the cells
were transfected with the shRNA plasmid using TurboFect™
transfection reagent (Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. Cells were plated in 6-well plates or
96-well plates and transfected the following day with 4 or 0.2
µg of PDK1-shRNA or scr-shRNA using 6 or 0.4 µl of
transfection reagent, respectively. After 48 h, cells were
collected for use in the indicated assays.
Mice
Male BALB/c-nu mice aged 6 weeks old and weighing
approximately 15 g each were purchased from Beijing Vital River
Laboratory Animal Technology Co., Ltd. They were kept at
temperature (22-24°C) with a stable humidity (55±15%) with free
access to food/water in a 12 h/12 h light/dark cycle and housed in
specific pathogen-free conditions at Jilin University (19). A total of 5×106 SNU-449
cells were subcutaneously injected into the backs of the mice.
After 7 days, when the tumor volume reached approximately 100
mm3, the nude mice were randomly divided into 4 groups
and were administered treatment intraperitoneally (i.p.) every day
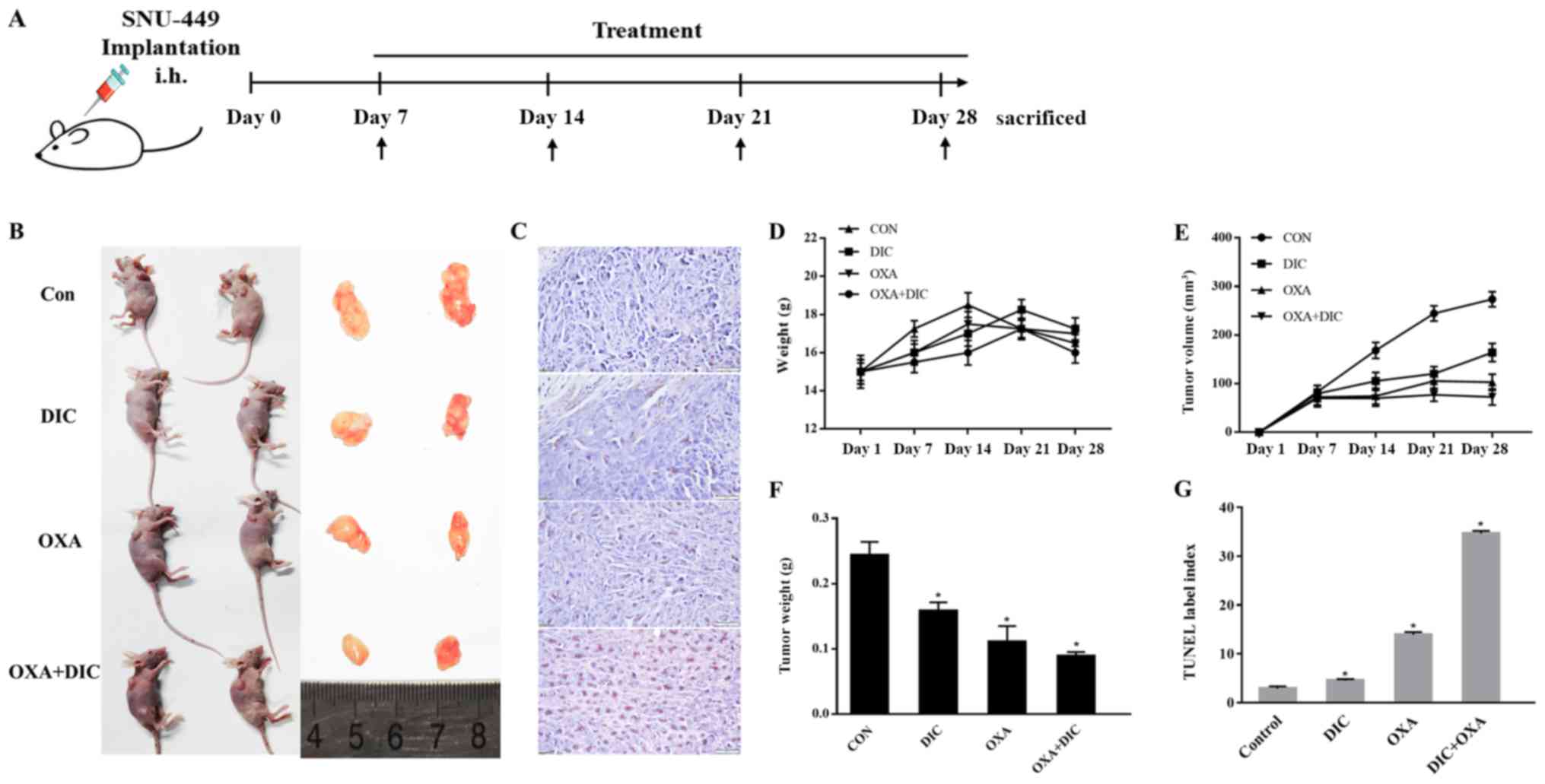
(Fig. 3A). The control group,
administered 200 µl saline; the DIC group, administered 25
mg/kg DIC; the OXA group, administered 200 mg/kg OXA (Shandong New
Time Pharmaceutical Co., Ltd.); and the OXA + DIC group,
administered the corresponding drugs indicated above. All animal
experiments were performed in accordance with the National
Guidelines for Experimental Animal Welfare and with approval of the
Animal Welfare and Research Ethics Committee at Jilin University
(Changchun, China).
Cells and cell culture
The human HCC cell lines, SNU-387 and SNU-449, were
purchased from the Chinese Academy of Medical Sciences. The two
cell lines were cultured in RPMI-1640 medium containing 10% fetal
bovine serum and 1% antibiotics (HyClone; GE Healthcare Life
Sciences), and were grown in a humidified cell culture incubator
containing 5% CO2 and 95% air at 37°C.
Cell viability assay
The in vitro cell viability was examined
using the standard MTT assay, as previously described (20). Briefly, the SNU-387 and SNU-449
cells were seeded in 96-well plates at 8,000 cells/well. The
following day, increasing concentrations (0, 4, 16, 64 and 256
µM) of OXA with or without DIC were added to each well, and
the plates were then incubated at 37°C for 24 h. For the SNU-449
cells, the following day, increasing concentrations of OXA and DIC
with or without pre-treatment with 5 mM NAC for 1 h were added to
each well, and the plates were incubated at 37°C for 24 h.
Subsequently, 10 µl of 5 mg/ml MTT reagent (Sigma-Aldrich;
Merck KGaA) in phosphate-buffered saline (PBS) were added to each
well, and the plate was incubated at 37°C for an additional 4 h.
The formazan crystals were dissolved in 150 µl of DMSO, and
after the plate was shaken for 10 min, the optical density at 570
nm was recorded using a multifunctional microplate reader (BMG
Labtech).
Western blot analysis
SNU-449 and SNU-387 cells were treated with DIC (25
µM), OXA (64 and 256 µM), OXA (64 and 256 µM)
and DIC (25 µM) for 24 h. The cells were lysed in cold RIPA
buffer containing 1% PMSF and 1% β-mercaptoethanol, and the lysate
was collected by centrifugation at 12,000 × g for 10 min at 4°C.
Total protein in each sample was quantified using the Bio-Rad
protein reagent (Bio-Rad Laboratories, Inc.). Approximately 50
µg of total protein from each sample was denatured at 95°C
for 10 min, separated by 12-15% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis, and transferred onto
Immune-Blot poly-vinylidene fluoride membranes (Bio-Rad
Laboratories, Inc.). After blocking in 5% (w/v) non-fat milk in
Tris-buffered saline for 2 h, the membranes were incubated with
specific primary antibodies (1:1,000) (indicated above in 'Reagents
and antibodies') overnight at 4°C. Following incubation of the
membranes with secondary antibodies [peroxidase-conjugated
AffiniPure goat anti-mouse IgG (H+L; cat. no. SA00001-1), and
peroxidase-conjugated AffiniPure goat anti-rabbit IgG (H+L; cat.
no. SA00001-2) from ProteinTech Group, Inc. at room temperature for
1.5-2 h, the signals were detected using enhanced chemiluminescence
reagents followed by Syngene Bio Imaging (Synoptics). The band
densities were measured using Syngene Bio Imaging tools (the
software was used for densitometry).
Hoechst 33342 staining assay
Morphological alterations of apoptotic cells were
detected by staining the nuclear chromatin of the SNU-449 cells
with Hoechst 33342. In brief, 2×104 SNU-449 cells were
cultured in 24-well plates per well at 37°C and treated as
indicated for an additional 24 h. The cells were washed with cold
PBS and fixed using 4% (w/v) paraformaldehyde for 15 min. The
plates were then incubated with 1 µg/ml Hoechst 33342 for 10
min and observed under a fluorescence microscope (IX-71; Olympus
Corp.).
Apoptosis assay
A total of 3×104 SNU-387 or SNU-449 cells
per well were seeded into 6-well plates and divided into 7 groups
according to the treatments they received: The control (unstained),
control (stained), DIC (25 µM), OXA (64 µM or 256
µM), co-treated with DIC (25 µM) and OXA (64 or 256
µM). Following the 24-h treatment, apoptosis was measured by
staining the cells with Annexin V and propidium iodide (PI) using
the FITC Annexin V Apoptosis Detection kit from BD Pharmingen (BD
Biosciences). The cells were analyzed on a C6 Flow Cytometer, and
the signal was quantified using C6 Software and a Workstation
Computer (BD Accuri™).
Gene Expression Profiling Interactive
Analysis (GEPIA)
GEPIA is a newly developed interactive web server
for analyzing the RNA sequencing expression data of 9,736 tumor and
8,587 normal samples from TCGA and the GTEx projects, using a
standard processing pipeline (21,22).
GEPIA performs survival analysis based on gene expression
levels.
Integrative Molecular Database of
Hepatocellular Carcinoma (NCCDB)
NCCDB is a database of 15 curated public HCC
expression datasets that cover approximately 4,000 clinical samples
and develop to serve as a one-stop online resource for exploring
HCC gene expression with user-friendly interfaces. Among these 15
datasets, HCCDB15 is the dataset with the largest number of
samples, including 356 HCC samples and 49 adjacent tissue samples
(23).
Determination of glucose uptake and
lactate production
The SNU-449 cells were treated with indicated drugs
[DIC (25 µM), OXA (64 and 256 µM), OXA (64 and 256
µM) and DIC (25 µM)] for 24 h, washed with PBS, and
cultured in RPMI-1640 culture medium to achieve a confluency of
80%. The culture medium was then collected, and the glucose and
lactate concentrations were measured with a glucose assay kit and a
lactate assay kit (Beyotime Institute of Biotechnology, Inc.),
respectively. The data were normalized by the corresponding total
protein amounts from each sample.
Oxygen consumption rate (OCR) and
extracellular acidification rate (ECAR) analysis
A total of 8×104 SNU-449 and SNU-387
cells were seeded into 96-well plates and incubated at 37°C
overnight to allow adherence. The following day, various
concentrations of DIC (25 and 50 µM) were added into the
indicated wells. Each treatment was repeated in 3 wells. The OCR
and ECAR were measured using oxygen-sensitive (Mito-Xpress) and
pH-sensitive (pH-Xtra) fluorescent probes (Luxcel Bioscience).
Measurement of reactive oxygen species
(ROS) levels
The SNU-449 cells were treated with the control
(unstained), control (stained with Mito SOX™ Red Mitochondrial
Superoxide Indicator), DIC (25 µM), OXA (64 or 256
µM), co-treated with DIC (25 µM) and OXA (64 or 256
µM) for 24 h, and the ROS level in each sample was then
detected by cell staining with Mito SOX™ Red Mitochondrial
Superoxide Indicator (Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's instructions. Positive cells
containing a high level of ROS were detected by the BD Accuri™ C6
Plus personal flow cytometry (BD Biosciences).
Measurement of mitochondrial membrane
potential (MMP)
MMP was determined by using JC-1 dye, contained
within the Mitochondrial Membrane Potential Assay kit (Beyotime
Institute of Biotechnology, Inc.). Following treatment with DIC (25
µM), OXA (64 or 256 µM), co-treated with DIC (25
µM) and OXA (64 or 256 µM) for 24 h, the cells were
incubated with 1 ml of 1X JC-1 for 30 min at 37°C in the dark, and
the ratio of cells positive for red fluorescence (JC-1 polymer
indicating intact MMP) to those positive for green fluorescence
(monomeric form of JC-1, an indicator for loss of MMP) was
determined by flow cytometry using BD Accuri C6.
Immunohistochemistry
The mouse tissues were fixed in 4% (w/v)
paraformaldehyde, dehydrated in graded ethanol and embedded in
paraffin. Samples were cut into 3-µm-thick sections using a
Leica microtome (16). TUNEL
staining was carried out with an In Situ Cell Death Detection kit,
POD (Roche Diagnostics GmbH).
Statistical analysis
Data are expressed as the means ± standard error
(SE). Differences were analyzed using one-way analysis of variance
(ANOVA) followed by Tukey's multiple comparisons test. All
experiments were repeated 3 times. A value of P<0.05 was
considered to indicate a statistically significant difference.
Statistical analysis was performed using SPSS 12.0 statistical
software (SPSS, Inc.).
Results
PDK1 is highly expressed in HCC, and DIC
inhibits PDK1 activity in SNU-449 and SNU-387 cells
In order to determine the effects of metabolic
reprogramming on drug resistance in HCC cells, the present study
first focused on PDK1, a 'switch' that controls pyruvate in glucose
metabolism and plays an important role in the metabolic pattern of
aerobic glycolysis of tumor cells (known as the Warburg effect).
The expression of PDK1 in liver HCC (LIHC) was higher than that in
normal liver issue (Fig. 1A) based
on the data from GEPIA. In addition, patients with a high PDK1
expression exhibited a lower overall survival than patients with a
low expression (Fig. 1B).
Consistent results were also obtained with the HCCDB15 dataset from
the HCCDB database in HCC (Fig.
1C) and in adjacent tissues (Fig.
1D). The above-mentioned results indicated that PDK1 may play
an important role in the occurrence and development of HCC.
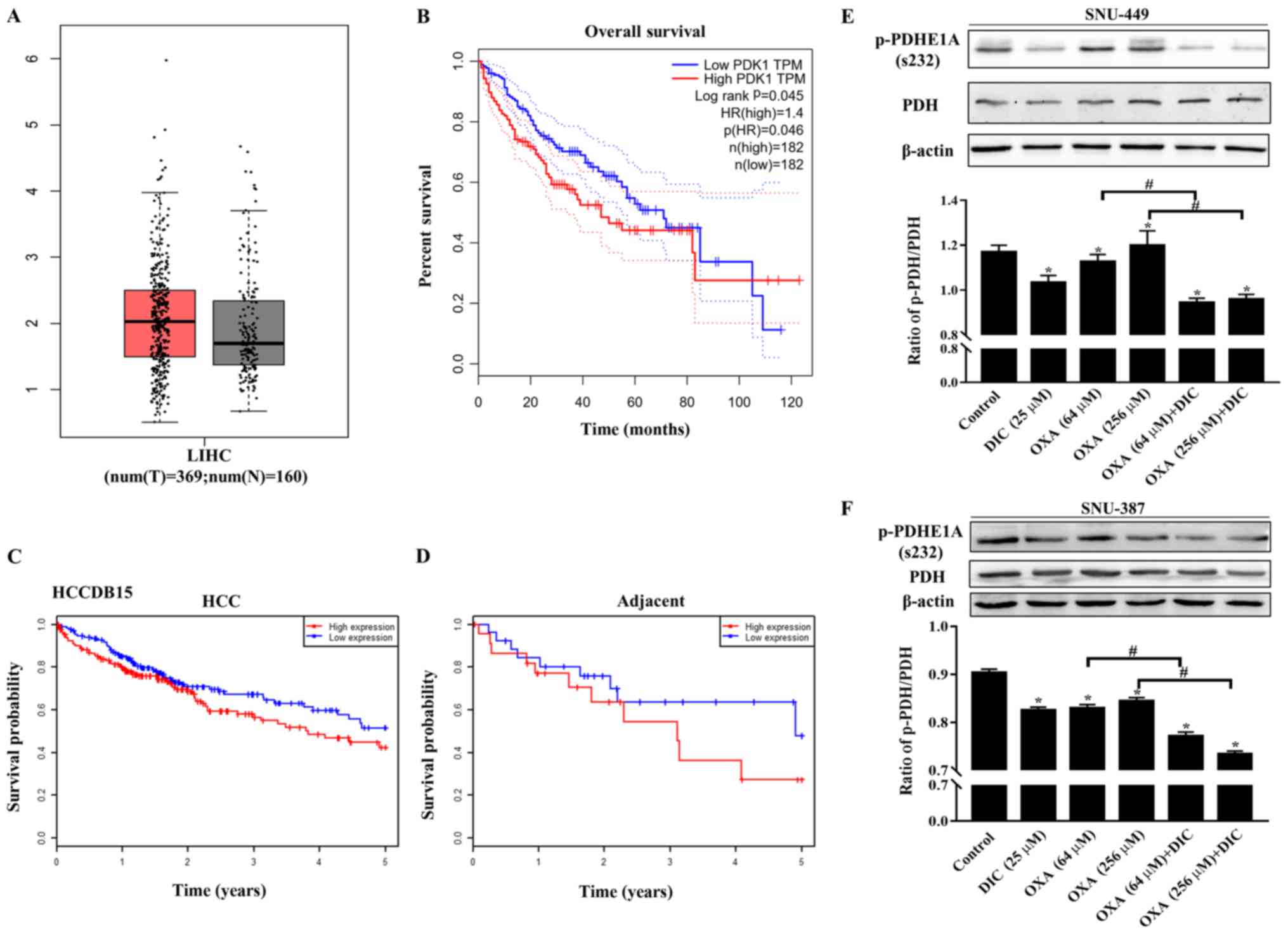 | Figure 1PDK1 is highly expressed in HCC, and
DIC inhibits PDK1 activity in SNU-449 cells and SNU-387 cells. (A)
Bioinformatics analysis results of the expression of PDK1 in liver
hepatocellular carcinoma (LIHC) acquired from GEPIA. (B) Overall
survival curve showing the significant association between PDK1 and
survival in LIHC (P<0.05) acquired from GEPIA. The dotted lines
represent the hazard ratio. (C and D) Survival probability curves
showing the association between PDK1 and survival in HCC and
adjacent tissues. (E) SNU-449 cells were treated with DIC (25
µM), OXA (64 and 256 µM), OXA (64 and 256 µM)
and DIC (25 µM) for 24 h. The levels of PDH and p-PDHE1A
(Ser232) were detected by western blot analysis. β-actin was used
as an internal control. The lower panel represents the quantified
graph of the ratio of p-PDH/PDH of the upper panel.
*P<0.05 vs. control; #P<0.05 DIC
treatment vs. no DIC treatment at the same concentration of OXA.
(F) SNU-387 cells were treated as described in (E). The levels of
PDH and p-PDHE1A (Ser232) were detected by western blot analysis.
β-actin was used as an internal control. The lower panel represents
the quantified graph of the ratio of p-PDH/PDH of the upper panel.
*P<0.05 vs. control; #P<0.05 DIC
treatment vs. no DIC treatment at the same concentration of OXA.
Data are presented as the means ± SE from 3 independent
experiments. HCC, hepatocellular carcinoma; GEPIA, Gene Expression
Profiling Interactive Analysis; PDK1, pyruvate dehydrogenase kinase
1; LIHC, liver hepatocellular carcinoma; DIC, dicoumarol; OXA,
oxaliplatin; PDH, pyruvate dehydrogenase. |
In addition, the phosphorylation of the
PDK1-specific phosphorylation site (Ser232) on PDH was evaluated to
assess the inhibitory effects of DIC on PDK1. It was found that the
levels of phosphorylated PDH (Ser 232) in the SNU-449 (Fig. 1E) and SNU-387 cells (Fig. 1F) treated with DIC exhibited a
significant decreased compared with those of the control group,
consistent with previous findings by the authors obtained with
human ovarian cancer cells SKOV3 and A2780 (16). Moreover, the groups treated with
both OXA and DIC exhibited a significant inhibition of PDK1
activity compared with the corresponding OXA treatment group
(Fig. 1E and F).
DIC and OXA synergistically inhibit tumor
growth in vitro and in vivo
Subsequently, the present study verified whether
combined treatment with DIC and OXA would affect tumor growth. MTT
assay revealed that SNU-449 cell viability decreased following
treatment with OXA (0-256 µM) for 24 h, and further
decreased following combined treatment with OXA and DIC (25
µM) (Fig. 2A). The trend
observed in the viability of the SNU-387 cells was consistent with
that of the SNU-449 cells (Fig.
2B). To further confirm this combined drug effect, the
combination index (CI) of both drugs was calculated. The calculated
CI was <1, indicating that DIC and OXA exerted a combined drug
effect (Fig. 2C). However, when
shRNA was used to inhibit PDK1, DIC did not enhance the inhibitory
effects of OXA on the viability of 2 cell lines (Fig. 2F and G). The above-mentioned
results indicate that the synergistic effects of DIC and OXA may be
the result of the inhibitory effect of DIC on PDK1. An interesting
point was noted: The SNU-449 cells seemed to be more sensitive to
the effects of DIC (Fig. 2A and
B); thus, the expression of PDK1 at the basal level was
examined in both cell lines. It was found that the SNU-449 cells
exhibited a higher PDK1 expression level than the SNU-387 cells
(Fig. 2D and E), indicating that
the difference in PDK1 levels may be the reason for the difference
in the inhibition of the viability of the 2 cell lines by DIC.
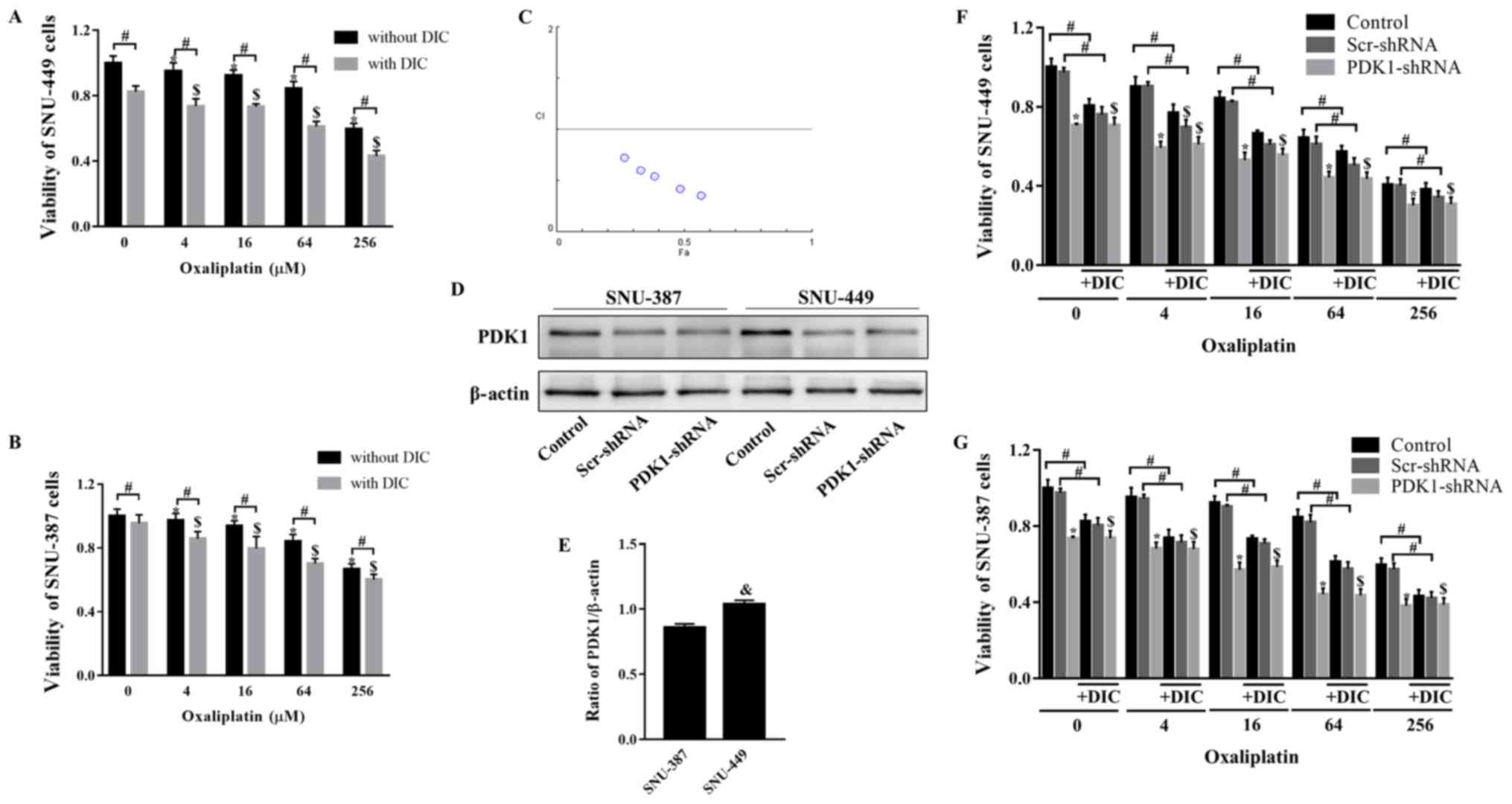 | Figure 2DIC and OXA synergistically inhibit
tumor growth in vitro. (A) SNU-449 cells and (B) SNU-387
cells were treated with OXA (4, 16, 64, or 256 µM) for 24 h
with or without DIC (25 µM) and cell viability was assayed
by MTT assay. *,$P<0.05 vs. control;
#P<0.05 DIC treatment vs. no DIC treatment at the
same concentration of OXA. (C) The combination index (CI) of DIC
and OXA obtained from the growth inhibitory experiments, additive
effect (CI=1), synergism (CI <1) and antagonism (CI >1) in
drug combinations. (D) SNU-387 and SNU-449 cells were transiently
transfected for 48 h with the shPDK1 expression vector or empty
vector, and western blot analysis was performed to examine the
levels of PDK1. (E) Quantified results of PDK1 in SNU-387 and
SNU-449 cells. &P<0.05 SNU-449 vs. SNU-387 cells.
(F) SNU-449 cells and (G) SNU-387 cells were treated with OXA (4,
16, 64 or 256 µM) for 24 h with or without DIC (25
µM) and treated as described in (D), and cell viability was
assayed by MTT assay. *,$P<0.05 vs. control.
#P<0.05 DIC treatment vs. no DIC treatment at the
same concentration of OXA. Data are presented as the means ± SE
from 3 independent experiments. HCC, hepatocellular carcinoma;
PDK1, pyruvate dehydrogenase kinase 1; DIC, dicoumarol; OXA,
oxaliplatin. |
This effect was further investigated in vivo.
SNU-449 tumor xenografts were established in nude mice and the mice
were treated with normal saline (control), DIC (25 mg/kg), OXA (200
mg/kg), or DIC (25 mg/kg) and OXA (200 mg/kg) after 7 days
(Fig. 3A). During the 21-day
treatment period, the body weights of the mice from all groups did
not alter significantly, suggesting that all treatments were safe
for the animals (Fig. 3D).
However, the tumor weight (Fig.
3E) and volume (Fig. 3F)
decreased to a certain extent in the DIC and OXA groups compared
with the control group, and a more pronounced decrease was observed
in the group treated with both DIC and OXA (Fig. 3B, E and F). Finally, apoptosis in
SNU-A449 xenografts was detected using a TUNEL assay. The results
revealed significantly higher TUNEL+ signals in the DIC
and OXA combination group consistent with the above-mentioned
results (Fig. 3C and G). These
results demonstrated DIC and OXA synergistically inhibited tumor
growth in vitro and in vivo.
DIC enhances the sensitivity of HCC cells
to OXA-induced apoptosis
In order to clarify the mechanisms responsible for
the inhibition of tumor growth observed following combined
treatment with DIC and OXA, indicators of apoptosis were examined
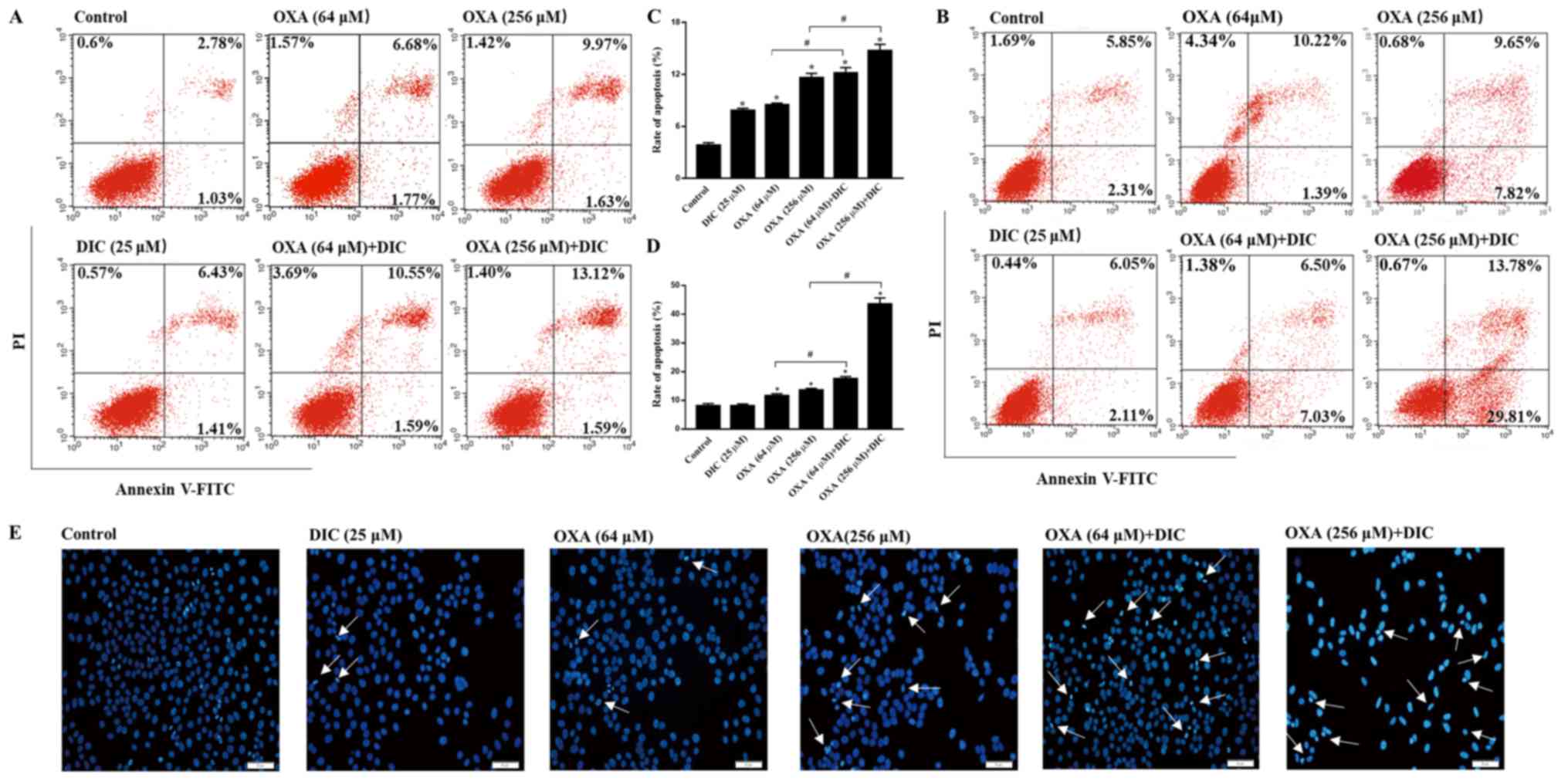
in vitro. Consistent with the results obtained in
vivo, the groups treatmed with DIC and OXA alone exhibited a
certain degree of apoptosis compared to the control group, as shown
by Annexin V-PI staining. However, marked apoptosis was observed in
the combined treatment groups of SNU-449 (Fig. 4A) and SNU-387 (Fig. 4B) HCC cells in a
concentration-dependent manner, respectively. The sum of the early
and late apoptotic rates of the 2 cell lines are also shown
(Fig. 4C and D). In the
morphological analysis, the SNU-449 cells exhibited nuclear
shrinking, become round in shape and were slightly smaller than the
normal cells following combined treatment with DIC and OXA for 24 h
compared with each individual treatment, as shown by Hoechst 33342
staining (Fig. 4E). The
above-mentioned results indicated that combined treatment with OXA
and DIC induced the apoptosis of HCC cells.
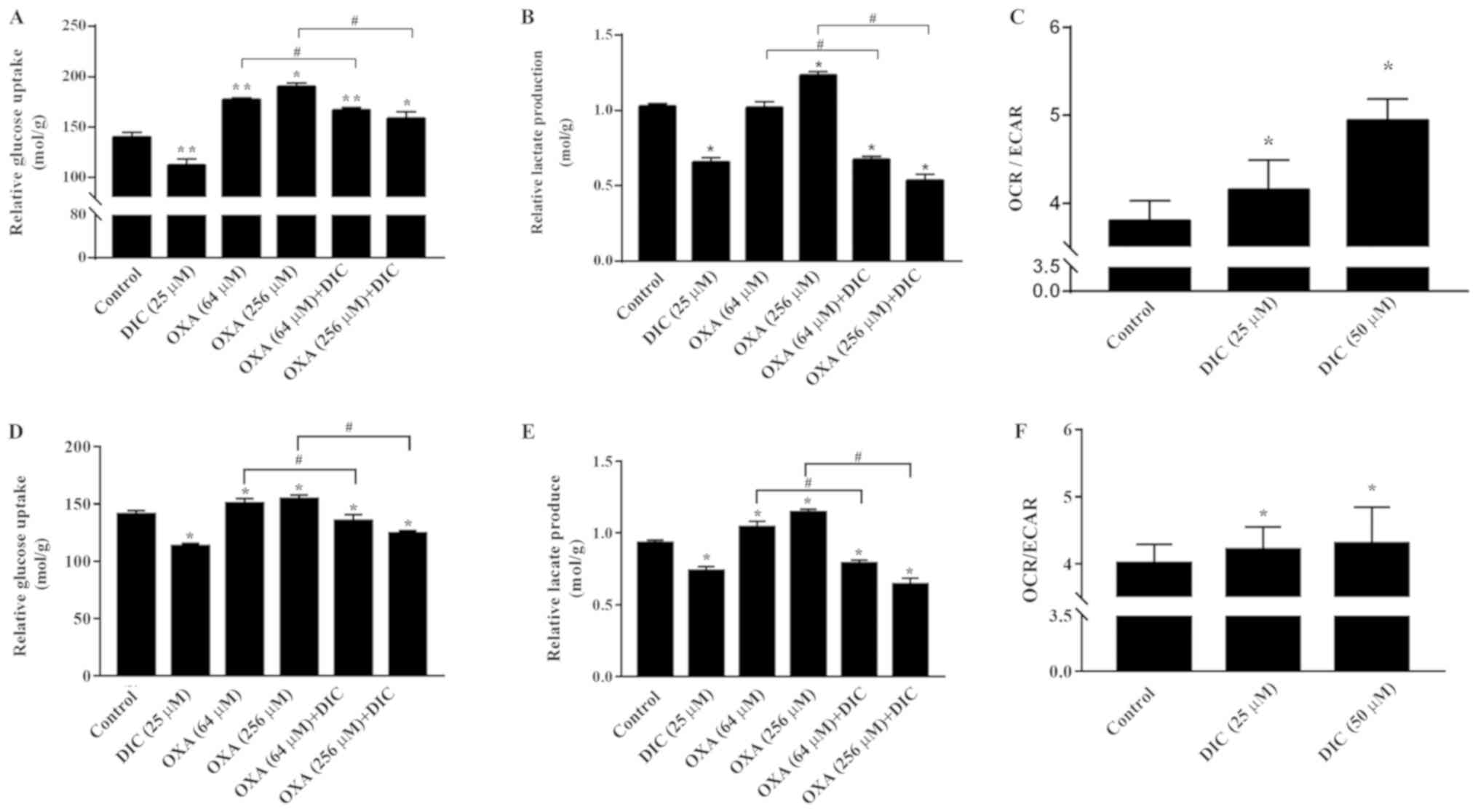
DIC alters metabolism of HCC cells and
enhances oxidative phosphorylation
Subsequently, the mechanisms through which DIC
treatment increases the apoptosis of HCC cells induced by OXA were
investigated. Based on the inhibition of PDK1 by DIC (Fig. 1E and F) and the important role of
PDK1 in glucose metabolism of tumor cells (24), some glucose metabolism indicators
of HCC cells in SNU-449 and SNU-387 were examined. Relative glucose
uptake (Fig. 5A) and lactate
production (Fig. 5B) were
decreased in the DIC-treated group compared to the control group.
Consistently, the addition of DIC (25 µM) in combination
with OXA also decreased relative glucose uptake (Fig. 5A) and lactic acid production
(Fig. 5B) compared with the OXA
group (64 and 256 µM), although OXA treatment alone exerted
no significant effects and even increased lactic acid production
and glucose uptake. To further examine the trend of glucose
metabolism following DIC treatment, the OCR and ECAR of SNU-449
cells were examined; OCR is commonly used to indicate the
occurrence of oxidative phosphorylation and ECAR is used as an
indicator of glycolysis during glucose metabolism. It was found
that the increase in OCR was more significant than ECAR, which was
almost 10 and 30% higher in the DIC-treated cells, respectively
(Fig. 5C). In addition, the
glucose uptake (Fig. 5D), lactate
production (Fig. 5E) and the ratio
of OCR to ECAR (Fig. 5F) in the
SNU-387 cells were basically consistent with those in the SNU449
cells. The above-mentioned results indicate that DIC can alter the
metabolism of HCC cells and enhance oxidative phosphorylation.
DIC promotes a decrease in MMP and
increases the production of mitochondrial ROS (mtROS) in HCC
cells
Subsequently, the present study wished to determine
the effects on the mitochondria due to the change in the metabolic
mode in HCC cells, and the mechanisms through which DIC enhances
the sensitivity of cells to apoptosis induced by OXA. It was found
that the mitochondrial inner membrane potential decreased upon
treatment with DIC compared to OXA alone treatment (Fig. 6A and B). An important byproduct of
oxidative phosphorylation, mtROS, increased significantly following
treatment with both DIC and OXA compared to OXA alone treatment
(Fig. 6C and D). Notably, the
viability of the SNU-449 cells treated with DIC (25 µM) or
OXA (64 and 256 µM) alone was restored when the cells were
also treated with NAC (5 mM), an antioxidant that increases ROS
scavenging (Fig. 6E). The
above-mentioned results indicate that DIC increases HCC cell
apoptosis by promoting the reduction of MMP and increasing the
production of mtROS in HCC cells.
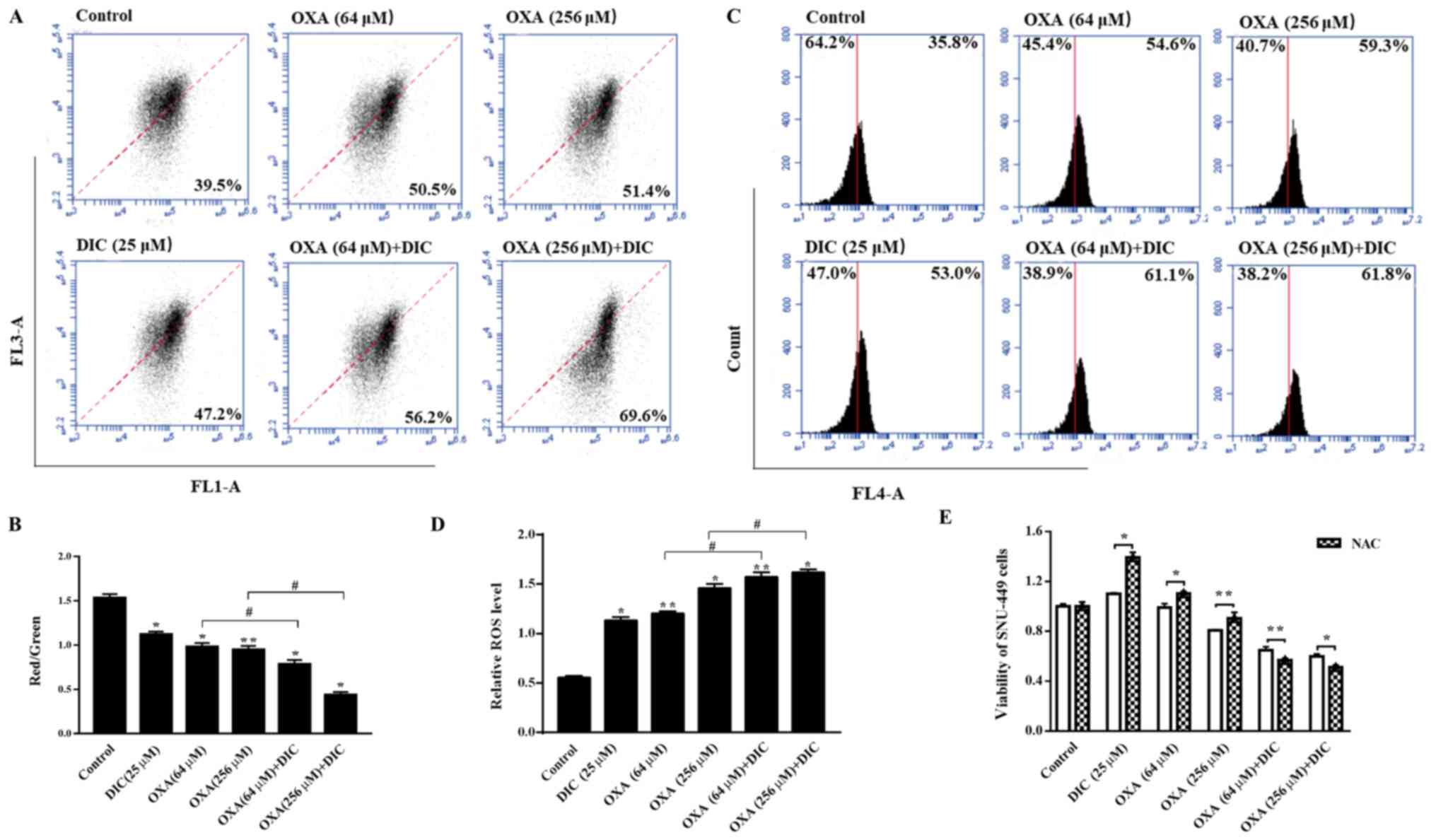 | Figure 6DIC promotes decline in MMP and
increases the production of mtROS in HCC cells. SNU-449 cells were
treated with DIC (25 µM), OXA (64 and 256 µM), OXA
(64 and 256 µM) and DIC (25 µM) for 24 h, and the (A)
mitochondrial membrane potential and (C) mitochondrial ROS
production were measured by flow cytometry and quantified. (B) The
graph shows the change in mitochondrial membrane potential after
DIC (25 µM), OXA (64 and 256 µM), OXA (64 and 256
µM) and DIC (25 µM) treated for 24 h, the increased
ratio of red fluorescence with green fluorescence represents the
depolarization of the mitochondrial membrane.
*P<0.05, **P<0.01 vs. control;
#P<0.05 DIC treatment vs. no DIC treatment at the
same concentration of OXA. (D) The graph shows the quantification
of mitochondrial ROS production after above treatments.
*P<0.05, **P<0.01 vs. control;
#P<0.05 DIC treatment vs. no DIC treatment at the
same concentration of OXA. (E) Upon treatment of the SNU-449 cells
with DIC (25 µM), OXA (64 and 256 µM), OXA (64 and
256 µM) and DIC (25 µM) for 24 h, the cell viability
after NAC addition was assayed by MTT assay. *P<0.05,
**P<0.01 NAC treatment vs. no NAC treatment at the
same concentration of DIC and OXA. Data are presented as the means
± SE from 3 independent experiments. HCC, hepatocellular carcinoma;
DIC, dicoumarol; OXA, oxaliplatin; MMP, mitochondrial membrane
potential; ROS, reactive oxygen species; mt, mitochondrial. |
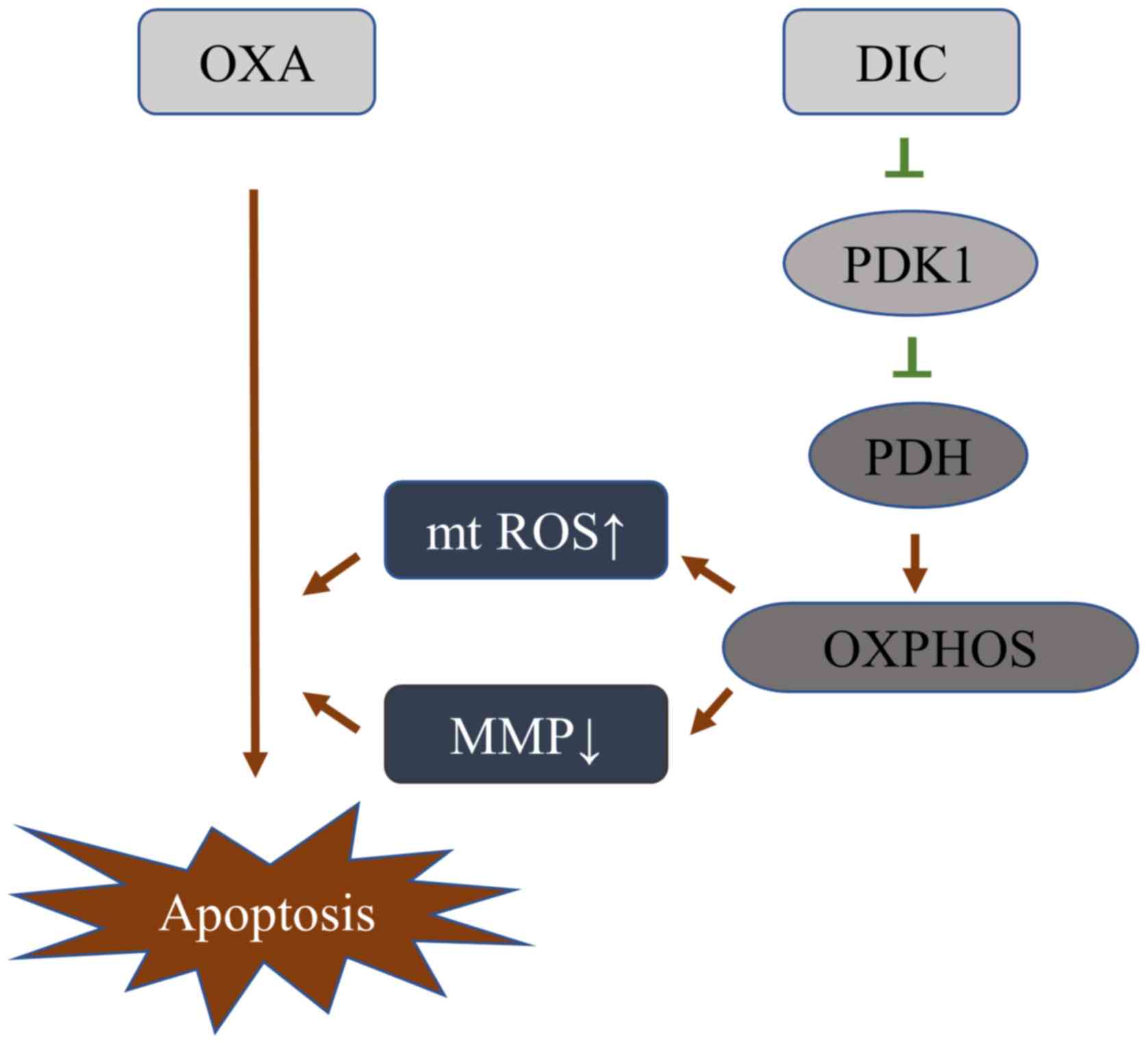
DIC alters the metabolic pattern of HCC
cells by inhibiting PDK1 activity, thereby increasing its
sensitivity to OXA
Based on the above-mentioned results, it was
initially clarified that DIC can inhibit the activity of PDK1, and
promote the metabolism of HCC cells towards oxidative
phosphorylation, resulting in a decrease in mitochondrial membrane
potential and an increase in ROS, thereby enhancing the sensitivity
of HCC cells to OXA-induced apoptosis (Fig. 7).
Discussion
Consistent with previous findings by the authors in
SKOV3 cells (16), the present
study verified that DIC can inhibit PDK1 activity in HCC cells
(Fig. 2E and 1F). However, it was also found that when
DIC (25 µM) was used in combination with OXA, it enhanced
the sensitivity of HCC cells to OXA-induced apoptosis (Fig. 2A and B), rather than the
cytotoxicity directly caused by high-dose DIC (16). In addition, it was found that the
higher the content of PDK1 in the cell, the more sensitive it seems
to be the inhibitory effect of DIC (Fig. 2A, B and E). In the present study,
DIC reduced the phosphorylation of PDH by inhibiting the activity
of PDK1, which converted the metabolism of HCC cells to oxidative
phosphorylation, leading to an increase in mtROS and a decrease in
MMP, thereby increasing the sensitivity to apoptosis induced by the
chemotherapeutic drug, OXA (Fig.
7).
HCC cells exhibit aerobic glycolysis, which can
rapidly supply the basic molecules required for biosynthesis
including the de novo generation of nucleotides, lipids and
proteins, and can be diverted into multiple branching pathways that
emanate from glycolysis (25), the
necessary reducing equivalents and the acidic microenvironment to
promote tumor growth and proliferation (26). In addition, this metabolic
characteristic of tumor cells is also closely related to
chemotherapeutic resistance (27).
Tumor cells which lack functional AMPK or LKB1 demonstrate an
enhanced susceptibility to OXPHOS inhibitors, as they fail to
decrease energy consumption in response to the energetic stress
induced by these agents, which then leads to an energetic crisis
and cytotoxicity (28). Therefore,
understanding the underlying mechanisms of aerobic glycolysis in
tumor cells is necessary for the effectiveness of chemotherapeutic
drugs (25,27,29).
In addition to the tumor cell's own metabolism which will affect
its sensitivity to chemotherapeutic drugs, the macroenvironment
includes the different tissues of the organism, capable of
exchanging signals and fueling the tumor at 'a distance' (30). Clinically, an association between
PKM2 expression levels and drug resistance has been reported
(27,31-33).
As a gatekeeper for aerobic glycolysis, PDK1 is highly expressed in
HCC (34) (Fig. 1A). Therefore, the HCC cell lines,
SNU-449 and SNU-387, were selected in the present study to examine
the role of PDK1 in aerobic glycolysis and chemotherapeutic
resistance.
PDK1 inactivates mitochondrial respiration through
the phosphorylation of PDH (35).
PDH catalyzes the oxidative decarboxylation of pyruvate to
acetyl-CoA, one of the mechanisms of the entry into the
tricarboxylic acid cycle of pyruvate in the mitochondria (36). Therefore, the inhibition of PDK1
activity by DIC promotes the entry of pyruvate into the
mitochondria and the metabolic changes caused by it. The study by
Sun et al demonstrated that increased PDH activity shifted
metabolism from glycolysis to oxidative phosphorylation and
decreased the hyperpolarization of the mitochondrial membrane
potential, resulting in mitochondrial pore opening, allowing ROS
and cytochrome c transport from the mitochondria to the cytoplasm,
thereby inducing apoptosis through caspase activation (37). The present study wished to
determine whether the inhibition of PDK1 activity could induce
apoptosis or enhance sensitivity to chemotherapeutic drugs through
this mechanism. It has been previously demonstrated that certain
concentrations of DIC can induce the apoptosis of SKOV3 cells
(16). Thus, the present study
wished to determine whether the combined application of DIC for
chemotherapy-insensitive and metabolically robust HCC cells would
increase their apoptotic sensitivity. Therefore, the present study
targeted PDK1, which is highly expressed in HCC cells, and verified
its key regulatory role in metabolic processes, providing a
theoretical basis for coping with the role of metabolic processes
in the adaptive changes of tumors under stress, such as
chemotherapeutic drugs.
The classic inhibitor of PDK1 is sodium
dichloroacetate (DCA); however, its clinical trials has been
terminated due to its severe side-effects (38-40).
However, when used in co-treatment with the chemotherapeutic drug,
paclitaxel (PTX), DCA markedly decreased cell autophagy, enhanced
apoptosis and inhibited the proliferation of A549 and H1975 cells
(41). Thus, the development of
novel medications, which can improve the demerits of DCA, have been
examined for the past 2 decades due to poor pharmacokinetics, and
low potency and selectivity (34).
It was previously found DIC that bound to the PDK1
lipoamide-binding pocket, suggesting it had the potential to
inhibit PDK1 activity (16). In
addition, DIC, as a clinically used drug for the prevention and
treatment of thromboembolic diseases, has a guaranteed safety. The
new use of 'old drugs' can also greatly reduce the cost of drug
development.
In conclusion, the findings of the present study
indicate that the inhibition of PDK1 may be a potential strategy
for targeting metabolism in HCC with chemotherapy. DIC already has
demonstrated clinical applications and its novel application in
cancer treatment, as an 'old drug' that shows novel efficacy,
brings new hope for antitumor therapy. Combined treatment with
traditional chemotherapeutic drugs may also provide new insight
into the treatment of HCC.
Acknowledgments
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (nos. 81672948 and
81772794), the Jilin Provincial Research Foundation for Health
Technology Innovation (no. 2019J009), the Jilin Provincial Research
Foundation for the Development of Science and Technology Projects
(no. 20191004004TC), the Jilin Provincial Industrial Innovation
Project (no. 2018C052-7) and 'the Fundamental Research Funds for
the Central Universities, Jilin University (JLU)'.
Availability of data and materials
The data that support the findings of this research
are available from the corresponding author upon reasonable
request.
Authors' contributions
HX performed most of the experiments and was
responsible for writing the manuscript. YH conducted the data
analysis. JM and YZ performed the animal experiments. YL provided
materials, analyzed the data and revised the manuscript. LS and JS
conducted the research, analyzed data and provided funding. All
authors have read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
All patient data were obtained from online datasets,
and thus no ethics approval was required. All animal experiments
were performed in accordance with the National Guidelines for
Experimental Animal Welfare and with approval of the Animal Welfare
and Research Ethics Committee at Jilin University (Changchun,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors have declared that no competing interest
exists.
References
|
1
|
Wallace MC, Preen D, Jeffrey GP and Adams
LA: The evolving epidemiology of hepatocellular carcinoma: A global
perspective. Expert Rev Gastroenterol Hepatol. 9:765–779. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ngan CY, Yamamoto H, Takagi A, Fujie Y,
Takemasa I, Ikeda M, Takahashi-Yanaga F, Sasaguri T, Sekimoto M,
Matsuura N, et al: Oxaliplatin induces mitotic catastrophe and
apoptosis in esophageal cancer cells. Cancer Sci. 99:129–139.
2008.
|
|
3
|
Abdel-Rahman O: Revisiting
oxaliplatin-based regimens for advanced hepatocellular carcinoma.
Curr Oncol Rep. 16:3942014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ma J, Zeng S, Zhang Y, Deng G, Qu Y, Guo
C, Yin L, Han Y, Cai C, Li Y, et al: BMP4 promotes oxaliplatin
resistance by an induction of epithelial-mesenchymal transition via
MEK1/ERK/ELK1 signaling in hepatocellular carcinoma. Cancer Lett.
411:117–129. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yin X, Tang B, Li JH, Wang Y, Zhang L, Xie
XY, Zhang BH, Qiu SJ, Wu WZ and Ren ZG: ID1 promotes hepatocellular
carcinoma proliferation and confers chemoresistance to oxaliplatin
by activating pentose phosphate pathway. J Exp Clin Cancer Res.
36:1662017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sheng J, Shen L, Sun L, Zhang X, Cui R and
Wang L: Inhibition of PI3K/mTOR increased the sensitivity of
hepatocellular carcinoma cells to cisplatin via interference with
mitochondriallysosomal crosstalk. Cell Prolif. 52:e126092019.
View Article : Google Scholar
|
|
7
|
Morandi A and Indraccolo S: Linking
metabolic reprogramming to therapy resistance in cancer. Biochim
Biophys Acta Rev Cancer. 1868:1–6. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fekir K, Dubois-Pot-Schneider H, Désert R,
Daniel Y, Glaise D, Rauch C, Morel F, Fromenty B, Musso O, Cabillic
F, et al: Retrodifferentiation of human tumor hepatocytes to stem
cells leads to metabolic reprogramming and chemoresistance. Cancer
Res. 79:1869–1883. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Choiniere J, Wu J and Wang L: Pyruvate
dehydrogenase kinase 4 deficiency results in expedited cellular
proliferation through E2F1-mediated increase of cyclins. Mol
Pharmacol. 91:189–196. 2017. View Article : Google Scholar :
|
|
10
|
Dai Y, Xiong X, Huang G, Liu J, Sheng S,
Wang H and Qin W: Dichloroacetate enhances adriamycin-induced
hepatoma cell toxicity in vitro and in vivo by increasing reactive
oxygen species levels. PLoS One. 9:e929622014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu Y, Gao W, Zhang Y, Wu S, Liu Y, Deng X,
Xie L, Yang J, Yu H, Su J, et al: ABT737 reverses cisplatin
resistance by targeting glucose metabolism of human ovarian cancer
cells. Int J Oncol. 53:1055–1068. 2018.PubMed/NCBI
|
|
12
|
Saunier E, Benelli C and Bortoli S: The
pyruvate dehydrogenase complex in cancer: An old metabolic
gatekeeper regulated by new pathways and pharmacological agents.
Int J Cancer. 138:809–817. 2016. View Article : Google Scholar
|
|
13
|
Koukourakis MI, Giatromanolaki A, Sivridis
E, Gatter KC and Harris AL; Tumor and Angiogenesis Research Group:
Pyruvate dehydrogenase and pyruvate dehydrogenase kinase expression
in non small cell lung cancer and tumor-associated stroma.
Neoplasia. 7:1–6. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang J, Liu F, Ao P, Li X, Zheng H, Wu D,
Zhang N, She J, Yuan J and Wu X: Correlation of PDK1 expression
with clinico-pathologic features and prognosis of hepatocellular
carcinoma. OncoTargets Ther. 9:5597–5602. 2016. View Article : Google Scholar
|
|
15
|
Battello N, Zimmer AD, Goebel C, Dong X,
Behrmann I, Haan C, Hiller K and Wegner A: The role of HIF-1 in
oncostatin M-dependent metabolic reprogramming of hepatic cells.
Cancer Metab. 4:32016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang W, Su J, Xu H, Yu S, Liu Y, Zhang Y,
Sun L, Yue Y and Zhou X: Dicumarol inhibits PDK1 and targets
multiple malignant behaviors of ovarian cancer cells. PLoS One.
12:e01796722017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chou TC: Drug combination studies and
their synergy quantifi-cation using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rodea-Palomares I, Petre AL, Boltes K,
Leganés F, Perdigón-Melón JA, Rosal R and Fernández-Piñas F:
Application of the combination index (CI)-isobologram equation to
study the toxicological interactions of lipid regulators in two
aquatic bioluminescent organisms. Water Res. 44:427–438. 2010.
View Article : Google Scholar
|
|
19
|
Xu H, Sun L, He Y, Yuan X, Niu J, Su J and
Li D: Deficiency in IL-33/ST2 Axis Reshapes Mitochondrial
Metabolism in Lipopolysaccharide-Stimulated Macrophages. Front
Immunol. 10:1272019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
He Y, Meng H, Xu H, Fan L, Zhou Z, Xu B,
Sun L and Gao Y: Regulation of integrated stress response
sensitizes U87MG glioblastoma cells to temozolomide through the
mitochondrial apoptosis pathway. Anat Rec (Hoboken). 301:1390–1397.
2018. View Article : Google Scholar
|
|
21
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45(W1):
W98–W102. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sun CC, Zhou Q, Hu W, Li SJ, Zhang F, Chen
ZL, Li G, Bi ZY, Bi YY, Gong FY, et al: Transcriptional E2F1/2/5/8
as potential targets and transcriptional E2F3/6/7 as new biomarkers
for the prognosis of human lung carcinoma. Aging (Albany NY).
10:973–987. 2018. View Article : Google Scholar
|
|
23
|
Lian Q, Wang S, Zhang G, Wang D, Luo G,
Tang J, Chen L and Gu J: HCCDB: A Database of Hepatocellular
Carcinoma Expression Atlas. Genomics Proteomics Bioinformatics.
16:269–275. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Emmanouilidi A and Falasca M: Targeting
PDK1 for Chemosensitization of Cancer Cells. Cancers (Basel).
9:92017. View Article : Google Scholar
|
|
25
|
Liberti MV and Locasale JW: The warburg
effect: How does it benefit cancer cells? Trends Biochem Sci.
41:211–218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang W, Zhang SL, Hu X and Tam KY:
Targeting tumor metabolism for cancer treatment: is pyruvate
dehydrogenase kinases (PDKs) a viable anticancer target? Int J Biol
Sci. 11:1390–1400. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bhattacharya B, Mohd Omar MF and Soong R:
The Warburg effect and drug resistance. Br J Pharmacol.
173:970–979. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ngoi NYL, Eu JQ, Hirpara J, Wang L, Lim
JSJ, Lee SC, Lim YC, Pervaiz S, Goh BC and Wong ALA: Targeting cell
metabolism as cancer therapy. Antioxid Redox Signal. 32:285–308.
2020. View Article : Google Scholar
|
|
29
|
Icard P, Shulman S, Farhat D, Steyaert JM,
Alifano M and Lincet H: How the Warburg effect supports
aggressiveness and drug resistance of cancer cells? Drug Resist
Updat. 38:1–11. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Amoedo ND, Obre E and Rossignol R: Drug
discovery strategies in the field of tumor energy metabolism:
Limitations by metabolic flexibility and metabolic resistance to
chemotherapy. Biochim Biophys Acta Bioenerg. 1858:674–685. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shin YK, Yoo BC, Hong YS, Chang HJ, Jung
KH, Jeong SY and Park JG: Upregulation of glycolytic enzymes in
proteins secreted from human colon cancer cells with 5-fluorouracil
resistance. Electrophoresis. 30:2182–2192. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guo W, Zhang Y, Chen T, Wang Y, Xue J,
Zhang Y, Xiao W, Mo X and Lu Y: Efficacy of RNAi targeting of
pyruvate kinase M2 combined with cisplatin in a lung cancer model.
J Cancer Res Clin Oncol. 137:65–72. 2011. View Article : Google Scholar
|
|
33
|
Shi HS, Li D, Zhang J, Wang YS, Yang L,
Zhang HL, Wang XH, Mu B, Wang W, Ma Y, et al: Silencing of pkm2
increases the efficacy of docetaxel in human lung cancer xenografts
in mice. Cancer Sci. 101:1447–1453. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jeoung NH: Pyruvate Dehydrogenase Kinases:
Therapeutic Targets for Diabetes and Cancers. Diabetes Metab J.
39:188–197. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Qin L, Tian Y, Yu Z, Shi D, Wang J, Zhang
C, Peng R, Chen X, Liu C, Chen Y, et al: Targeting PDK1 with
dichloroacetophenone to inhibit acute myeloid leukemia (AML) cell
growth. Oncotarget. 7:1395–1407. 2016. View Article : Google Scholar :
|
|
36
|
Wada M, Horinaka M, Yasuda S, Masuzawa M,
Sakai T and Katoh N: PDK1 is a potential therapeutic target against
angiosarcoma cells. J Dermatol Sci. 78:44–50. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sun W, Xie Z, Liu Y, Zhao D, Wu Z, Zhang
D, Lv H, Tang S, Jin N, Jiang H, et al: JX06 Selectively inhibits
pyruvate dehydrogenase kinase PDK1 by a covalent cysteine
modification. Cancer Res. 75:4923–4936. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tataranni T and Piccoli C: Dichloroacetate
(DCA) and cancer: An overview towards clinical applications. Oxid
Med Cell Longev. 2019:82010792019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Stacpoole PW, Martyniuk CJ, James MO and
Calcutt NA: Dichloroacetate-induced peripheral neuropathy. Int Rev
Neurobiol. 145:211–238. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Stacpoole PW: The pharmacology of
dichloroacetate. Metabolism. 38:1124–1144. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lu X, Zhou D, Hou B, Liu QX, Chen Q, Deng
XF, Yu ZB, Dai JG and Zheng H: Dichloroacetate enhances the
antitumor efficacy of chemotherapeutic agents via inhibiting
autophagy in non-small-cell lung cancer. Cancer Manag Res.
10:1231–1241. 2018. View Article : Google Scholar : PubMed/NCBI
|