Introduction
Acquired resistance of tumor cells to cell death is
crucial in tumorigenesis and a major obstacle to tumor therapeutic
strategies (1). Resistance to cell
death may be attributed to several abnormally expressed genes. One
such gene is Heat Shock Protein Family A Member 9 (HSPA9) coding
for mortalin, the mitochondrial stress protein 70 also known as
glucose regulated protein 75 (GRP75), which is an essential member
of the heat shock proteins 70 family (2,3).
Mortalin expression is upregulated in tumors and its expression
level is positively correlated with tumor aggressiveness and
metastasis (4-7). Furthermore, soluble mortalin has been
detected in the serum of patients with colorectal cancer and its
level is positively correlated with patients' poor prognosis
(8,9). Mortalin possesses numerous crucial
functions, including in intracellular trafficking and protein
quality control (10). Being
located in the mitochondria, mortalin is involved in mitochondrial
biogenesis and regulation of mitochondrial membrane potential,
export and import of mitochondrial proteins and energy generation
(10,11). Furthermore, mortalin protects tumor
cells from apoptosis by chelation of p53 (3). A previous study from our laboratory
demonstrated that mortalin protects tumor cells from
antibody-dependent complement-mediated cytotoxicity (CDC) by
promoting the removal of the membrane attack complex (MAC; C5b-9)
from complement-attacked cells via released vesicles (12). In addition, by directly binding to
the complement proteins C8 and C9, mortalin can inhibit MAC
assembly and incorporation into membranes, which is a pivotal step
in CDC (12,13). Knockdown or inhibition of mortalin
by small interfering (si)RNA or MKT-077, respectively, increases
cell sensitivity to CDC (13,14).
Accumulating evidence reported that the expression level and
function of mortalin are altered in tumor cells, resulting in
immune resistance and tumor progression. Subsequently, mortalin has
been considered as a potential target for cancer therapy. The
present study hypothesized that synthetic peptides which have amino
acid sequences derived from mortalin's sequence could block the
protective interactions of mortalin with its client proteins and
might trigger either spontaneous and/or elevated immune cell death.
In the present study, mimetic peptides derived from mortalin
sequence were generated and their efficacy to induce cancer cell
death was tested, as single agents and as adjuvants to CDC. To
promote peptide entry into cells, a transactivator of transcription
(TAT) sequence (15,16)
was added to their C-terminal domain. This study evaluated the
anti-cancer toxic activity and underlying mechanism of several
mimetic mortalin peptides.
Materials and methods
Cell culture
The human erythroleukemia cells K562 and the Raji
and Ramos lymphoma cells were grown in RPMI-1640. The mantle cell
lymphoma cells Z-138 were grown in Iscove's modified Dulbecco's
medium. The human ovarian carcinoma cells SKOV3, the human breast
carcinoma cells T47D and the human prostate carcinoma cells PC3
were cultured in Dulbecco's Modified Eagle Medium. All growth media
(Sigma-Aldrich; Merck KGaA) were supplemented with 10% fetal calf
serum (Gibco; Thermo Fisher Scientific, Inc.), glutamine and
pyruvate (Bio-Rad Laboratories, Inc). Cells were purchased from the
American Type Culture Collection and were mycoplasma-free.
Blood samples (5 ml) were collected from patients
with B-cell chronic lymphocytic leukemia (B-CLL) and from healthy
volunteers and placed in EDTA-containing plastic tubes. This study
was approved by the Helsinki Committee of the Rabin Medical Center,
Petach Tikva, Israel and informed consent was provided by all
participants. Blood was diluted 2-fold with Hanks' Balanced Salt
Solution (Sigma-Aldrich; Merck KGaA) and placed on top of 15 ml
Lymphoprep (Stemcell Technologies, Inc.) in conical plastic tube
and centrifuged at 400 × g for 30 min at room temperature. The
upper layer was aspirated and the peripheral blood mononuclear cell
(PBMC) layer was collected. PBMC were washed, resuspended in HBSS,
counted and kept on ice until used. Most of the cells isolated from
patients were B-CLL cells.
Reagents
Peptides were purchased from either Mimotopes,
Genemed Synthesis, Inc. or Blavatnik Research Center (Tel-Aviv
University). The sequences of the peptides were chosen following
computational modelling of human mortalin structure (Compugen
Ltd.), and predicting protein interaction sites in the N-terminal
nucleotide-binding domain (NBD, 52-433) or the C-terminal
substrate-binding domain (SBD, 434-679) of mortalin. A TAT sequence
corresponding to the human immunodeficiency virus type 1 entry
sequence RKKRRQRRR was added at the C-terminus of each peptide. A
biotin was added at the N-terminus domain of the peptides. Peptides
were purified by HPLC to >90% purity, tested by mass
spectrometry and kept lyophilized at -20°C. Fresh stock peptide
solutions (2-10 mM) were prepared in DMSO. The sequences of the
peptides were as follows: Mot-P2, PSQIGAFVLMKMKETAENYL (mortalin
no. 163-182, NBD); Mot-P7, GEDFDQALLRHIVKEFKRET (mortalin no.
275-294, NBD); Mot-P8, NMALQRVREAAEKAKSEL (mortalin no. 302-319,
NBD); Mot-P10, RAQFEGIVTDLIRRTIA (mortalin no. 348-364, NBD);
Mot-P14, MVKNAEKYAEEDR (mortalin no. 561-573, SBD); Mot-P16,
FKDQLPADECN KLKEEISKMRELLA (mortalin no. 599-623, SBD); and
scram-bled peptide, KERYNEAKEDMVA.
Normal human serum (NHS; 3H biomedical AB) was
stored at −70°C. A polyclonal anti-human C3 antiserum was prepared
in goats by subcutaneous injection of human C3. First inoculation
was done with Complete Freund's adjuvant, followed by three more
subcutaneous injections with Incomplete Freund's adjuvant. Mouse
anti-mortalin antibody was purchased from StressMarq Biosciences,
Inc. (cat. no. SMC-133) or Abcam (cat. no. ab94668), and mouse
anti-actin antibody was provided by EMD Millipore (cat. no.
MAB1501). Peroxidase-conjugated goat anti-mouse IgG (cat. no.
115-035-003), FITC-conjugated rabbit anti-goat IgG (cat. no.
305-095-003), AlexaFluor 488-conjugated anti-human IgG (cat. no.
109-545-003) and FITC-conjugated goat anti-mouse IgG (cat. no.
115-095-003) were purchased from Jackson ImmunoResearch
Laboratories, Inc. Mouse monoclonal antibody directed to a
neoepitope in human C5b-9 (clone aE11) was from Hycult Biotech
(cat. no. HM2167). Mouse anti-human CD46 (cat. no. BLG-352404),
anti-human CD55 (cat. no. BLG-311302) and anti-human CD59 (cat. no.
BLG-304702) monoclonal antibodies were from BioLegend, Inc.
Monoclonal anti-CD20 antibody (Rituximab) was from Roche
Pharmaceutical Ltd., (cat. no. RO 45-2294).
JC-1 mitochondrial staining kit, staurosporine
(STS), methyl-β-cyclodextrin (MβCD), filipin III and bovine serum
albumin (BSA) were purchased from Sigma-Aldrich; Merck KGaA. The
caspase inhibitor Q-VD-OPh (QVD) was from R&D Systems and
Annexin V Apoptosis Detection Kit was from Thermo Fisher
Scientific, Inc. DCFDA- Cellular ROS Detection Assay kit was from
Abcam. Cell Cycle Kit was from Merck KGaA. Fluorescein
(DTAF)-conjugated streptavidin was from Jackson ImmunoResearch
Laboratories, Inc. TMB substrate and TMB Stop solution were from
SouthernBiotech. The CytoTox-ONE lactate dehydrogenase (LDH)
release kit and CellTiter-Glo luminescent cell viability assay were
from Promega Corporation. N-acetyl cysteine (NAC) was from Enzo
Life Sciences.
SMARTpool siRNA for RNA interference was from GE
Healthcare Dharmacon, Inc. The human HSPA9 gene (mortalin)
targeting sequences were a mixture of GAGGUGAAAUCCACAAAUG,
GACUAUCGC UCCAUGCCAA, CCUAUGGUCUAGACAAAUC and AAACGCAAGUGGAAAUUAA.
The non-specific siRNA targeting sequences were
UAAGGCUAUGAAGAGAUAC, AUGUAUUGGCCUGUAUUAG, AUGAACGUGAAUUGC UCAA and
UGGUUUACAUGUCGACUAA.
Evaluation of cell death, ATP synthesis
and cell cycle analysis
Cancer cells were incubated with mortalin peptides
(5-100 µM) for 30 min (0.25×106 cells/100
µl in 12×75-mm glass test tubes) or overnight
(0.25×106 cells/200 µl in 24-well plates) at
37°C. Adherent carcinoma cells were first detached from plate by
trypsinization and washed with HBSS before treatment with peptides.
Next, cells were labeled with propidium iodide (PI) or DAPI, both
at 1 µg/ml, and immediately analyzed by Cytoflex Flow
Cytometer (Beckman Coulter). Cell death was calculated after
quantifying PI/DAPI-negative, viable cells in a 30 sec flow. Viable
cell counts in negative control samples were defined as Total
cells. The quantification of cell death was performed as follows:
Cell death (%) = [(total cells− the number of viable cells in
experimental) / total cells] × 100. Alternatively, following
peptide treatment, cells were washed, mixed with 0.02% trypan blue
in isotonic buffer and percent cell death was determined after
counting live/dead cells in a hemocytometer under a light
microscope. Percentage of cell death for control cells incubated
without a peptide (C) was subtracted from that of experimental
peptide-treated cells (E) to calculated the final percentage of
dead cells as follows: Cell Death = [(E-C) / (100-C) × 100]. After
treatment, cells were also labelled with annexin-V-FITC (Thermo
Fisher Scientific, Inc.) to detect changes in plasma membrane
asymmetry and permeability.
Cell death was also evaluated according to LDH
release following the manufacturers' instructions. Briefly, cells
were seeded at a density of 5×104 cells/well in 50
µl of culture medium in a 96-well plate and were treated
with mortalin peptides at various concentrations for 30-60 min at
37°C. A negative control included solvent (DMSO) treatment. For
total LDH release (representing death of all cells, i.e., 100% cell
death), cells were treated with the kits' lysis solution. Cells at
room temperature were mixed with CytoTox-ONE reagent for 10 min at
22°C. Next, a stop solution was added and fluorescence emission at
590 nm (excitation at 560 nm) was measured in a SpectraMAX M5
microplate reader and corresponded to the LDH release. The
percentage of cell death was calculated according to the following
equation: (LDH released by peptide - LDH released by DMSO) / (total
LDH Released - LDH released by DMSO) × 100.
To determine the peptide's effect on cellular ATP
levels, cells were treated with the peptides at various
concentrations for 30 min at 37°C. ATP level was quantified with
the CellTiter-Glo kit according to the manufacturers' instructions.
Briefly, luminescence level, representing ATP level, was recorded
in a microplate reader in peptide-treated and DMSO-treated cells,
and the percentage of ATP level in peptide-treated relative to
DMSO-treated cells was calculated.
To identify changes in cell cycle, cells were
treated with peptides at various concentrations for 36 h at 37°C.
Cells were washed with PBS, mixed with 70% ice-cold ethanol and
fixed for at least 2 h at −20°C. Subsequently, cells were labeled
with Muse Cell Cycle Reagent for 30 min at room temperature and
analyzed in a Cytoflex Flow Cytometer. The percentage of cells in
stages G0/G1 (2N), S (2N-4N) and G2/M (4N) was determined as
previously described (17).
Measurement of ROS level and
mitochondrial membrane potential
To measure the ROS level, cells were stained for 30
min at 37°C with the probe 2′,7′-dichlorofluorescin diacetate
(DCFDA; 20 µM) according to the manufacturers' instructions
and the fluorescence was quantified in a Cytoflex Flow Cytometer.
For maximal ROS inhibition, cells were treated for 30 min at 37°C
with 5 mM NAC prior to staining with DCFDA.
To detect changes in the mitochondrial membrane
potential upon treatment with peptides, cells were stained with
JC-1 (1 mg/ml) for 20 min at 37°C. Fluorescence was measured in a
Cytoflex flow cytometer. In normal cells, JC-1 emits red
fluorescence (590 nm) and upon dissipation of the mitochondrial
membrane potential, its emission changes to green fluorescence (529
nm) (18). The percentage of
depolarized cells is indicated by the decrease in red
fluorescence.
Peptide imaging by confocal
microscopy
PC3, SKOV3 and T47D cells (1-2×105
cells/ml) were seeded into 4-well chamber slides (Sigma-Aldrich;
Merck KGaA) and kept in culture for 24 h in CO2
incubator. Then, cells were treated with biotinylated peptides at
the indicated concentrations, for 5-10 min at 37°C. Subsequently,
cells were fixed with methanol for 10 min at -20°C, blocked with 5
mg/ml BSA at room temperature for 1 h, and permeabilized (0.2%
Triton X-100) for 60 min at room temperature. For nucleus staining,
300 nM DAPI solution was added for 5 min at room temperature. For
peptide staining, cells were incubated with 5 µg/ml
fluorescein-conjugated streptavidin for 30 min at room temperature.
After washing with PBS containing 0.2% Triton X-100, the chambers
were drained, coverslips were glued onto them with Mowiol
(Sigma-Aldrich; Merck KGaA) and cells were kept in the dark for 24
h. Cell imaging was performed using Leica-SP5 confocal microscope
(Leica Microsystems, Inc.).
Measurement of peptide binding by
ELISA
Microtiter plate wells were coated overnight at 4°C
with BSA, C3, C9 or mortalin (all at 5 µg/ml) in PBS. Wells
were washed 3 times with PBS-T (0.05% Twin) and blocked with 1% BSA
for 1 h at room temperature. Subsequently, biotinylated peptides
were added at 0.5 or 2.5 µg/ml to the wells for 1 h at room
temperature. After washing, peroxidase-conjugated streptavidin was
diluted in PBS according to the manufacturers' instructions and was
added to the wells and incubated for 1 h at room temperature in the
dark. Then, TMB One Component HRP Microwell Substrate was added and
absorbance was read at 605 nm (450 nm if a stop solution was added)
in a microplate reader.
CDC assay
Malignant B-cells in HBSS (5×105/100
µl) were treated with Rituximab antibody against CD20 (RTX;
2 µg/ml) for 30 min on ice. Next, 50% NHS was added for 60
min at 37°C in a shaking water bath. Cells treated with NHS without
antibody served as negative controls. Cell death was measured
either by trypan blue inclusion under a light microscope or by
determining PI staining using flow cytometry. Percentage of cell
death was calculated as described above.
Quantification of C3, C5b-9, CD20 and
membrane complement regulators
Ramos cells were treated with RTX (1.5 µg/ml)
for 30 min at 4°C. Cells were then mixed with 50% NHS and further
incubated for 10 min at 37°C. To quantify bound C3, cells were
treated with a goat anti-C3 antibody (1:1,000), followed by
FITC-conjugated rabbit anti-goat IgG antibody (1:200). To quantify
deposited C5b-9 (MAC), cells were labeled with a mouse anti-C5b-9
aE11 antibody (1:50), followed by FITC-conjugated goat anti-mouse
IgG antibody (1:50). Cells were incubated with each antibody for 30
min on ice.
To measure CD20 expression, Ramos cells labeled with
2 µg/ml RTX for 30 min at 4°C were stained with Alexa-fluor
488-conjugated anti-human IgG antibody for 1 h on ice (1:500). To
quantify the membrane complement regulators, cells were treated
with mouse anti-CD46, anti-CD55 or anti-CD59 anti-bodies for 30 min
at 4°C (1:50) and then with FITC-conjugated secondary antibodies
for 30 min at 4°C (1:100). The labeled cells were analyzed by flow
cytometry and the mean fluorescence intensity (MFI) was determined
with CytExpert software v2.0 (Beckman Coulter).
Transient transfection by
electroporation
K562 cells (2.5×106) were mixed in an
electroporation cuvette (Cell Projects) with mortalin siRNA (500
pmol) or a nonspecific siRNA as control for 10 min at room
temperature. Next, cells were electroporated with a BTX ECM 830
Electro Square Porator (225V, 14 msec), mixed with culture medium
and cultured for 48 h at 37°C. To validate siRNA insertion and
transfection efficiency, transfected cells (0.5×106)
were lysed with SDS-PAGE sample buffer, loaded on the gels and
proteins were separated by 4-10% gradient SDS-PAGE. Proteins were
transferred onto nitrocellulose membranes. Membranes were blocked
with 5% skimmed milk and were incubated with primary antibodies
against actin and mortalin for 1 h at room temperature (1:500).
Membranes were incubated with peroxidase-conjugated secondary
antibody for 1 h at room temperature (1:10,000; Jackson
ImmunoResearch Laboratories, Inc.). Bands were detected using
Super-Signal West Pico Chemiluminescent Substrate (Pierce; Thermo
Fisher Scientific, Inc.) and exposed to Super RX film (Fujifilm
Wako Pure Chemical Corporation). Relative expression level of
mortalin was normalized to actin using ImageJ v1.4 software
(National Institutes of Health).
Statistical analysis
Student's paired t-test was used to compare two
groups. Two-way ANOVA was performed to compare two groups: a tested
reagent and the interaction between them. GraphPad Prism software
v8 (GraphPad Software, Inc.) was used for statistical analysis.
Data were expressed as the means ± standard deviation. P<0.05
was considered to indicate a statistically significant
difference.
Results
Mortalin-derived peptides are
cytotoxic
Mortalin is an essential housekeeping gene and cell
protector from many toxic moieties (3,12).
Its concentration is elevated in cancer cells, suggesting that
blocking mortalin protective activities may be an effective
anti-cancer strategy. In the present study, the efficacy of
mortalin mimetic peptides as inducers of cancer cell death was
evaluated. Six mortalin mimetic peptides predicted by computational
analysis to be exposed on the surface of human mortalin and a
scrambled control peptide were synthesized. Initial analysis with
Ramos cells clearly indicated that adding a TAT sequence at the
C-terminal of the peptides significantly increased peptide
cytotoxicity (Fig. S1).
Therefore, all peptides described in the present study possessed a
C-terminal TAT. A dose-response analysis of the cytotoxicity of the
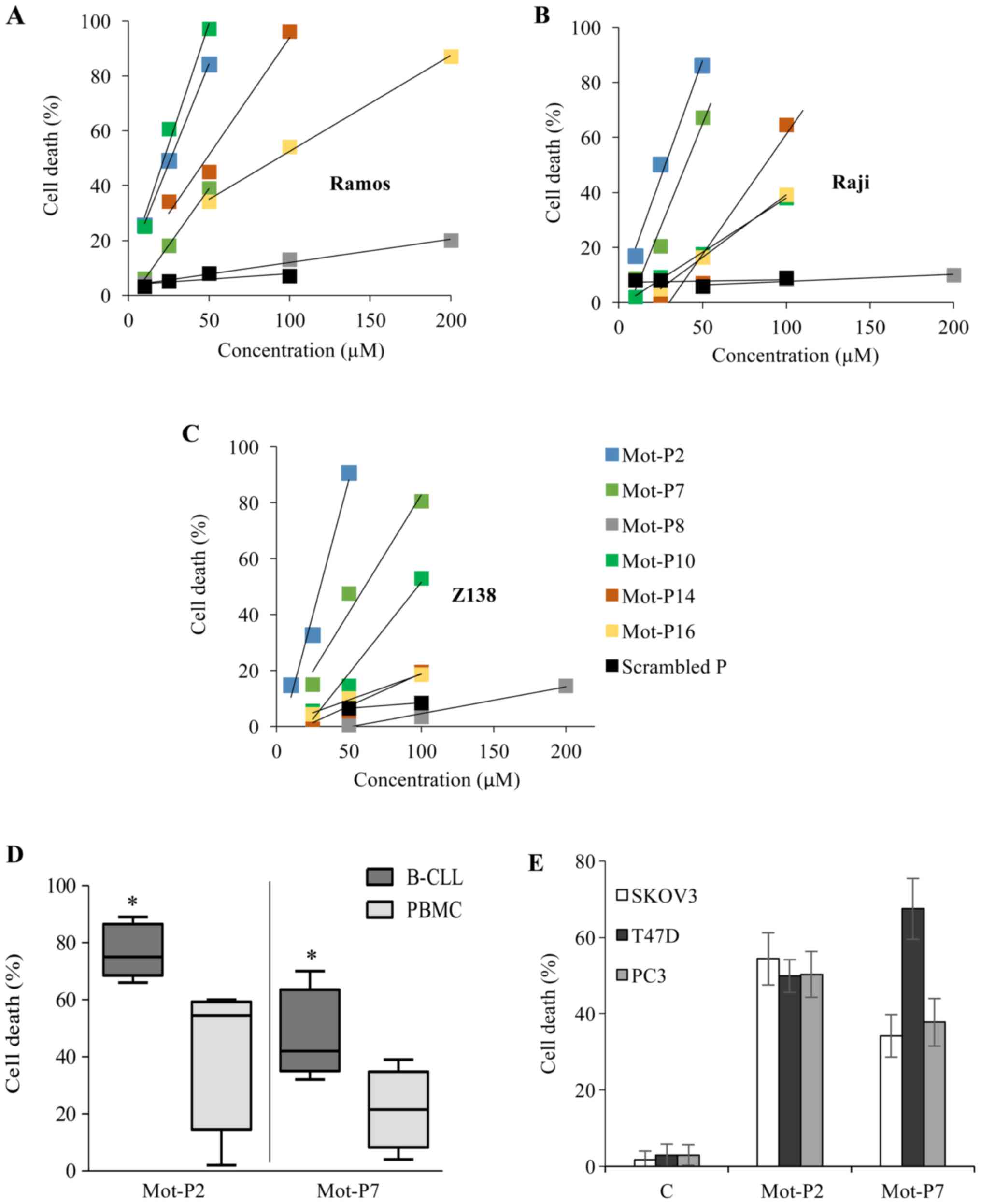
six peptides over 24 h in Ramos, Raji and Z-138 lymphoma cells is
presented in Fig. 1A-C. Based on
these titration curves, the lethal dose (LD50) of the peptides was
calculated (Table SI). The two
most potent peptides, Mot-P2 (LD50, 26-30 µM) and Mot-P7
(LD50, 40-63 µM), were chosen for further experiments.
Subsequently, the toxicity of the peptides was tested on B-CLL
cells from patients and on PBMC from healthy donors. Mot-P2 and
Mot-P7 (both at 50 µM) were demonstrated to be toxic for
both B-CLL cells and PBMC; however, PBMC were less sensitive
(Fig. 1D). Mot-P2 caused 77.0±9.4%
and 42.7±27.4% cell death in B-CLL and PBMC cells, respectively
(P=0.03). Furthermore, Mot-P7 caused 47.8±15.4% and 21.5±14.3% cell
death in B-CLL and PBMC cells, respectively (P=0.03). Primary B-CLL
cells and B-cell leukemia/lymphoma established cell lines could
express a similar sensitivity to toxicity from Mot-P2 and Mot-P7.
At the concentration of 50 µM, cell death of Ramos, Raji and
Z138 cells by Mot-P2 was 90-100%, and by Mot-P7, 50-85% (Fig. 1A-C). Under the same conditions,
~75% of primary CLL cells were lysed by Mot-P2 and ~45% by Mot-P7
(Fig. 1D). The peptides were also
tested on the three human carcinoma cell lines PC3 (human prostate
cancer); T47D (breast cancer) and SKOV3 (ovarian carcinoma). The
results demonstrated that cell death was almost maximal after 4 h
treatment. As presented in Fig.
1E, Mot-P2 (50 µM) and Mot-P7 (100 µM) were toxic
for all carcinoma cell lines.
Peptide uptake and intracellular
distribution in carcinoma cells
The intracellular distribution of biotin-labeled
Mot-P2 and Mot-P7 was studied in carcinoma cells by confocal
micros-copy. This could not be performed on lymphoma cells because
of their size and morphology. Cells were incubated with the
peptides (5 min at 37°C) and labeled with Streptavidin-FITC as
aforementioned. Mot-P2 and Mot-P7 were rapidly taken up by PC-3,
SKOV-3 and T47D cells (Fig. S2).
The results demonstrated that Mot-P2 was distributed throughout the
cytoplasm and nucleus, which was similar to a previous study
reporting nuclear accumulation of the TAT protein basic domain
(19). The distribution of Mot-P7
was slightly different, with more peptides localized at the plasma
membrane and less in the cytoplasm and nucleus. The kinetics of the
peptides' internalization process was followed up to 15 min.
However, with increasing time, the data became less informative, as
an increasing percentage of the cells started undergoing cell death
and a nuclear/pancytoplasmic/membrane distribution of the peptides
became apparent.
Effects of Mot-P2 and Mot-P7 on plasma
membrane
Since Mot-P2 and Mot-P7 are highly cytotoxic,
analysis was performed before cell death got too extensive. A
kinetic analysis of Mot-P2 and Mot-P7 cytotoxicity on Ramos cells
was performed with a relatively higher concentration of Mot-P7 (100
µM P7 and 25 µM P2, in order to reach a similar level
of cell death). Almost maximal cell death was achieved by both
peptides within 30 min, although Mot-P2 acted slightly faster than
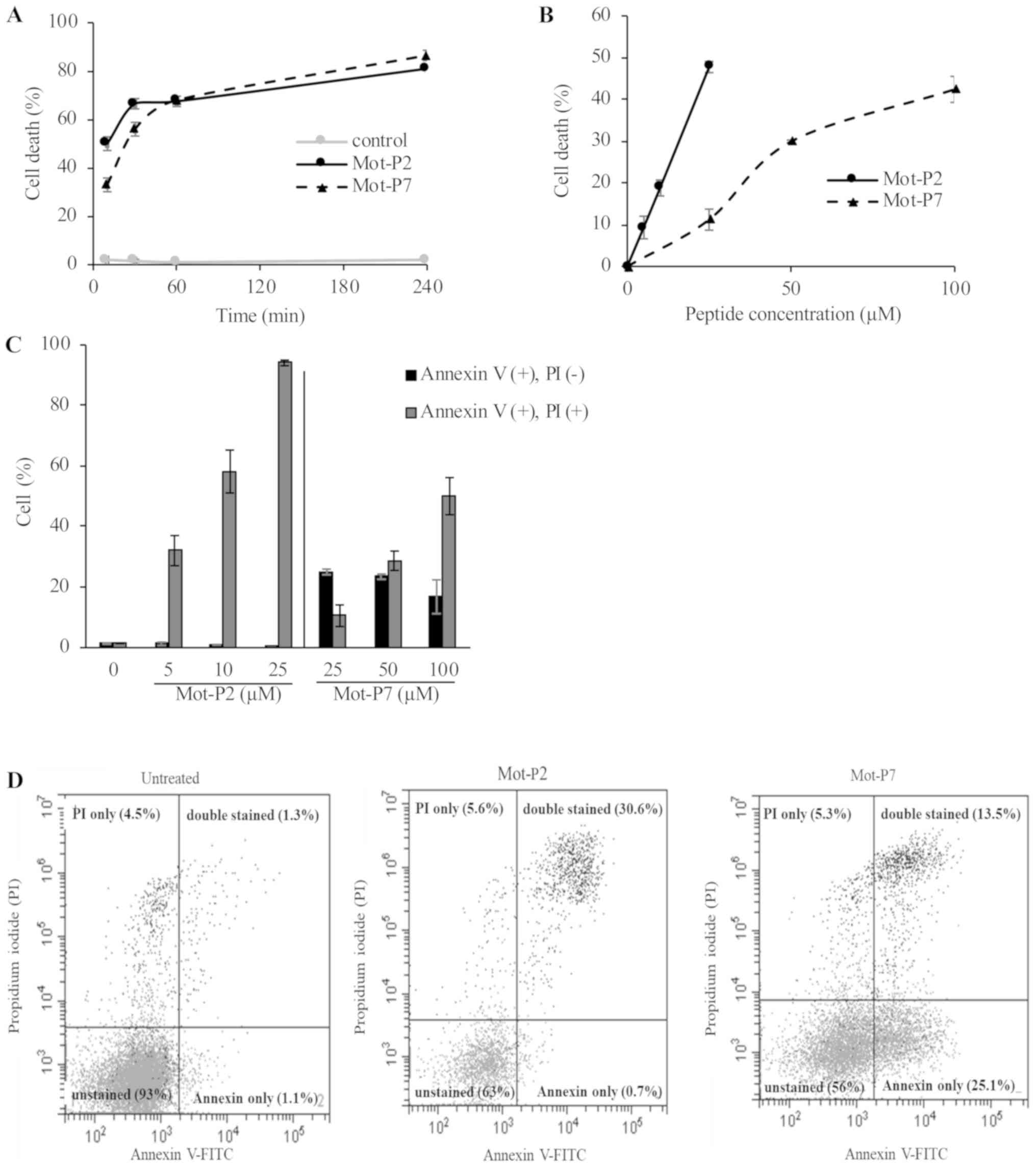
Mot-P7 (Fig. 2A). Plasma membrane
perforation was also confirmed by LDH leakage, demonstrating a
higher toxicity for Mot-P2 (Fig.
2B). Various cell death processes are known to disturb plasma
membrane asymmetry and cause flip-flopping of phosphatidylserine
(PS) from the inner to the outer leaflet of the plasma membrane
(20,21). Externalized PS was therefore
quantified by binding with Annexin V. Ramos cells were treated with
Mot-P2 or Mot-P7 and labeled with annexin-V-FITC and PI to detect
changes in plasma membrane asymmetry and permeability. Following
Mot-P2 treatment, all damaged cells were positive for Annexin V and
PI (Fig. 2C and D). PS
externalization and PI entry occurred therefore in all damaged
cells. Conversely, Mot-P7-treated cells exhibited two distinct
damaged cell populations: one with intact plasma membrane
(PI-negative) and labeled with annexin-V-FITC and one labeled both
by Annexin V and PI (Fig. 2C and
D). Interestingly, at a lower Mot-P7 concentration, more cells
were single Annexin V-positive, whereas, at a higher Mot-P7
concentration, more cells were double Annexin V- and PI-positive.
These results suggested that the mechanisms of action of Mot-P2 and
Mot-P7 are distinct. Caspases that mediate apoptotic cell death
activate PS externalization (20).
QVD is a pan-caspase inhibitor (22). Prior to exposure to peptides, Ramos
cells were treated with QVD or with DMSO as control. Subsequently,
cells were treated with Mot-P2 or Mot-P7. In addition, cells were
also treated with staurosporine, which is an apoptosis inducer
(21). As presented in Fig. 3A, pre-treatment with QVD inhibited
the death (i.e., double labeling) of staurosporine-treated cells.
However, QVD had no effect on cell death following Mot-P2 or
Mot-P7. Therefore, unlike staurosporine, the peptides activate a
caspase-independent, probably necrotic cell death.
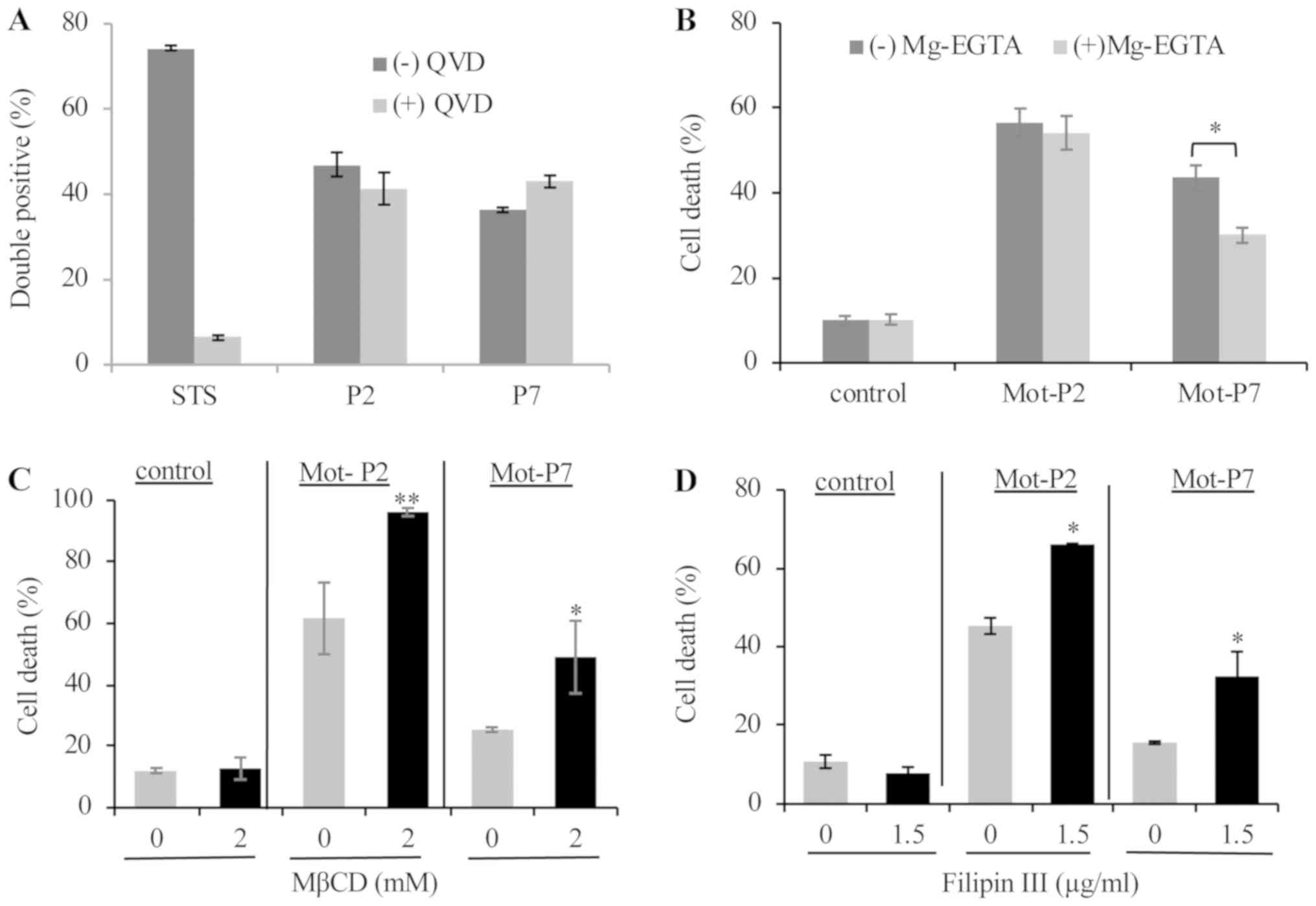 | Figure 3Role of calcium and cholesterol in
peptide cytotoxicity. (A) Ramos cells were pretreated with 50
µM QVD or DMSO as control for 1 h at 37°C. Cells were then
treated with 20 µM Mot-P2 or 75 µM Mot-P7 or 2
µM of STS as a positive control for 4 h at 37°C. PS
externalization and cell membrane integrity were determined by flow
cytometry after staining with Annexin V-FITC and PI, respectively.
Percentage of double-positive cells is presented. (B) Ramos cells
were pretreated for 30 min at 37°C with Mg-EGTA for calcium
chelation. Toxic doses of Mot-P2 or Mot-P7 (25 µM or 50
µM, respectively) were added for additional 30 min. Cells
were washed, labeled with PI and cell death was analyzed by flow
cytometry. Peptide-treated. n=3. *P<0.05 vs.
untreated cells, n=4. (C and D) Ramos cells were pretreated for 30
min at 37°C with (C) 0 or 2 mM MβCD or (D) with 0 or 1.5
µg/ml Filipin III (C) for membrane cholesterol depletion or
inhibition, respectively. Next, cells were treated for 30 min at
37°C with 25 µM Mot-P2 or 50 µM Mot-P7. Cells were
washed, labeled with PI and analyzed by flow cytometry. n=3,
*P<0.05 and **P<0.01 vs. untreated
cells. MβCD, methyl-β-cyclodextrin; STS, staurosporine; PS,
phosphatidylserine. |
Due to the much higher calcium ion concentration
outside the cells compared with inside cells, pore formation can
lead to a rapid influx of calcium ions into the cytoplasm, calcium
toxicity, and cell death (23). To
examine the impact of calcium influx on peptide cytotoxicity,
extracellular calcium was chelated with EGTA (10 mM) and
supplemented with MgCl2 (2.5 mM; Mg-EGTA) prior to
peptide treatment. The results demonstrated that the toxic effect
of Mot-P7, but not that of Mot-P2, was partially inhibited by
extracellular calcium ablation (Fig.
3B).
Membrane cholesterol is known to modulate the
interaction of some membranolytic peptides and proteins with the
plasma membrane and affect therefore their cytotoxicity (24-26).
To determine whether cholesterol serve a role in Mot-P2 and Mot-P7
cytotoxicity, the effect of two cholesterol membrane blocking or
depleting agents, methyl-β-cyclodextrin (MβCD) and filipin III,
respectively, on cell death was tested. As demonstrated in Fig. 3C and D, both cholesterol depletion
and inhibition led to a significant increase in Mot-P2 and Mot-P7
cytotoxicity in Ramos cells, suggesting that plasma membrane
cholesterol may limit Mot-P2 and Mot-P7 cytotoxicity.
Effects of Mot-P2 and Mot-P7 on the
mitochondria
The observation of a membrane damaging effect by
Mot-P2 and Mot-P7 suggested that both peptides may partially induce
cell death by causing mitochondrial toxicity. The effect of
peptides on the mitochondrial membrane potential was examined by
cell staining with JC-1, which is a cationic dye that accumulates
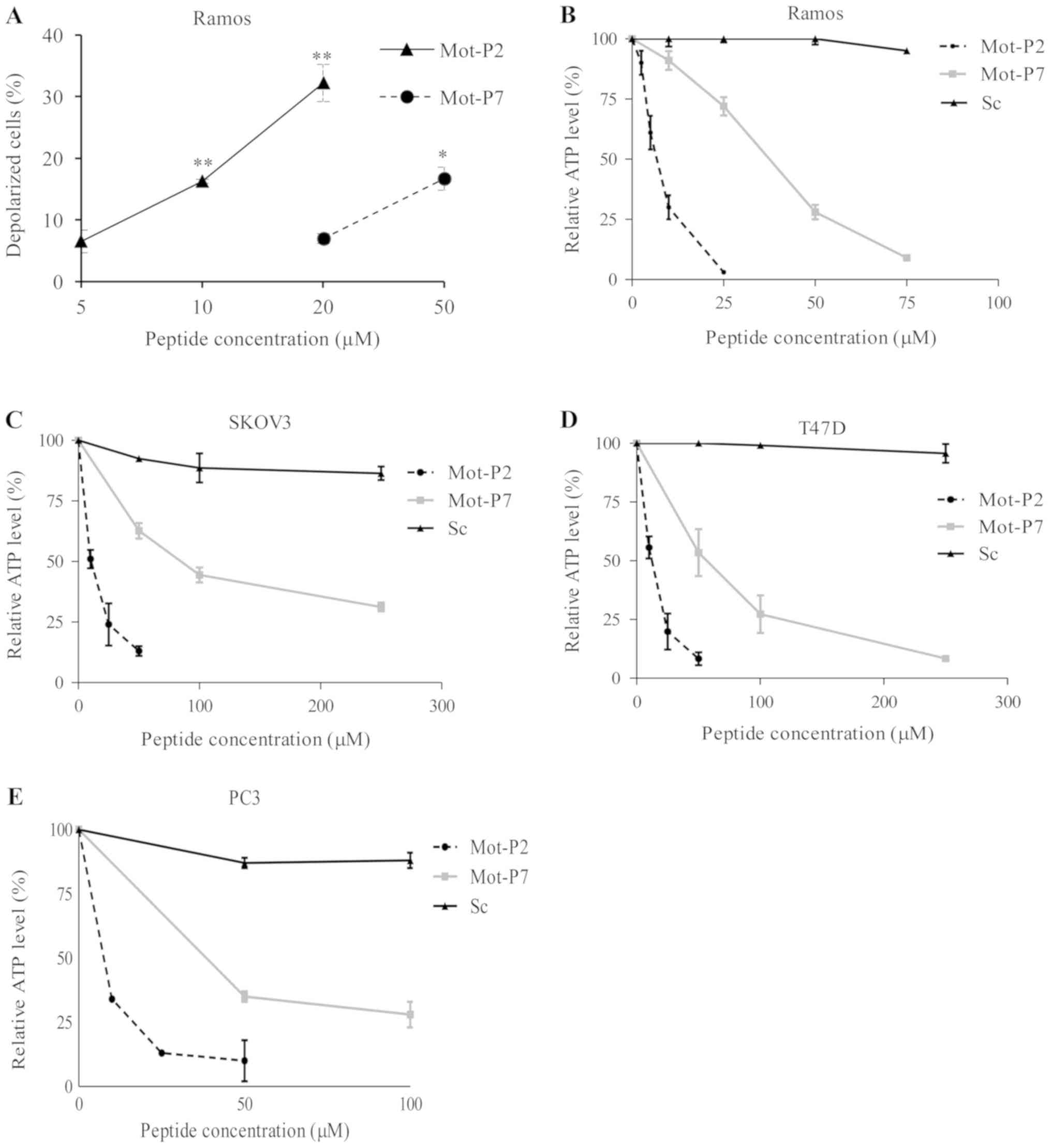
in the mitochondria of healthy cells. As presented in Fig. 4A, Mot-P2 and Mot-P7 both dissipated
the mitochondrial membrane potential in Ramos cells; however, the
effect was higher with Mot-P2.
After mitochondrial damage, ATP production may be
decreased to a level causing or amplifying cell death. The effect
of Mot-P2 and Mot-P7 on the ATP level in Ramos, SKOV3, T47D and PC3
cells was subsequently evaluated. Compared with the scrambled
peptide, both Mot-P2 and Mot-P7 caused, within 30 min, a dramatic
dose-dependent drop in the cellular ATP level, in the four cell
lines tested (Fig. 4B-E).
Furthermore, a Mot-P7 concentration 4-8 times higher than Mot-P2
was required to cause a 50% drop in ATP level. In addition, Mot-P2
and Mot-P7 caused, within 10 min at 37°C, concomitantly plasma
membrane pore formation (LDH release) and mitochondrial damage (ATP
drop; Fig. S3).
Besides inhibition of ATP synthesis, peptide-induced
mitochondrial stress may result in overproduction of ROS. Mortalin
is a mitochondrial chaperone involved in cell protection from ROS
(27). The effect of Mot-P2 and
Mot-P7 on ROS production was therefore evaluated using the
ROS-sensitive fluorescent dye DCFDA. The results demonstrated that
Ramos cell treatment with Mot-P2 or Mot-P7 significantly increased
ROS level (Fig. 5A). To
investigate whether ROS contributes to cell death, cells were
pre-incubated with NAC, which is a ROS scavenger (28), prior to peptide treatment. The
results demonstrated that Mot-P2-induced cell death was not
affected by ROS inhibition (Fig.
5B). Conversely, Mot-P7-induced cell death was inhibited by NAC
(Fig. 5B), suggesting a difference
in the mode of action of the two peptides.
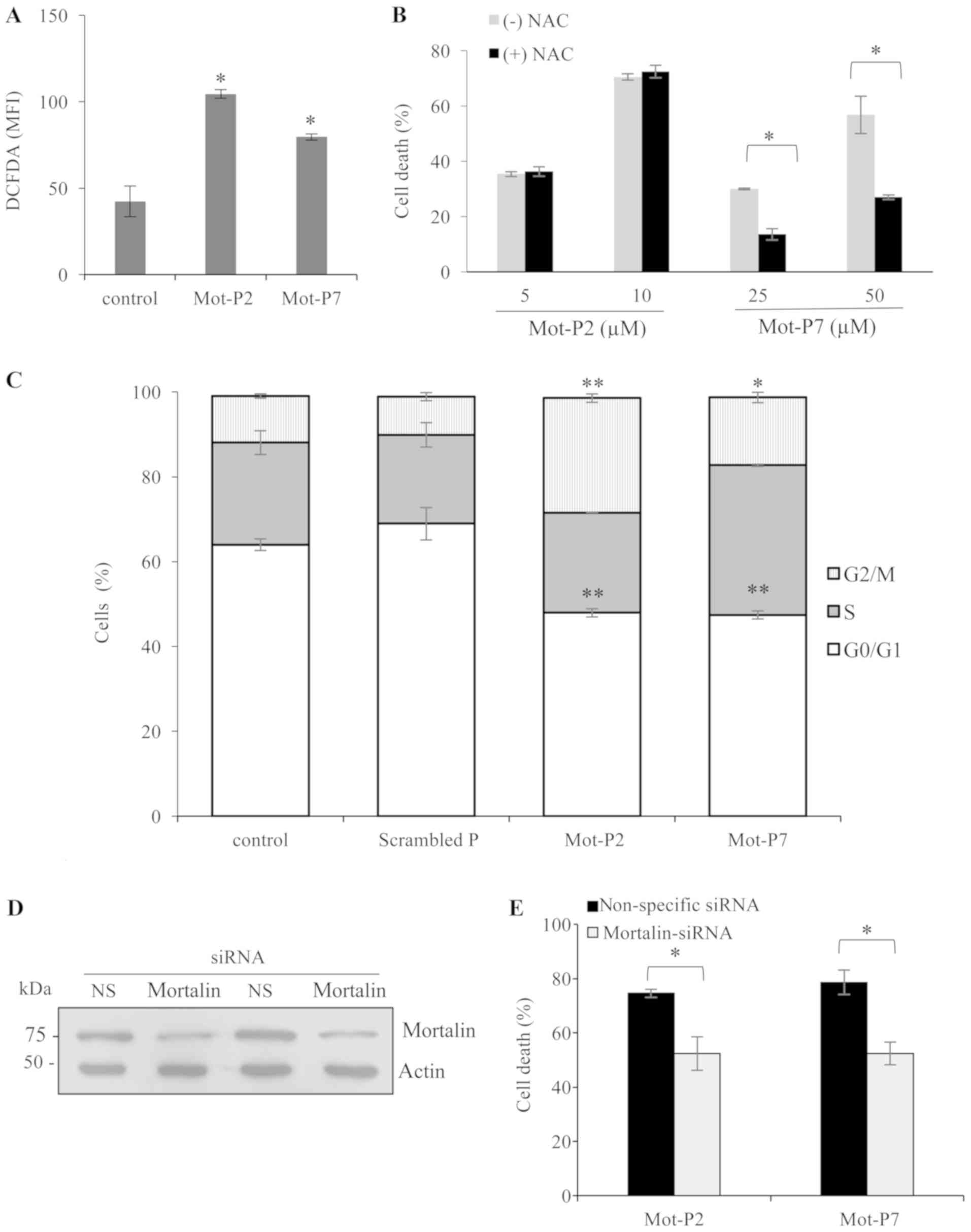 | Figure 5Mot-P2 and Mot-P7 activities. (A) ROS
are involved in Mot-P7- but not Mot-P2-induced cell death. (A)
Ramos cells were stained with 20 µM DCFDA for 30 min at 37°C
in the dark. Next, cells were treated with 10 µM Mot-P2 or
50 µM Mot-P7 for 1 h at 37°C. Mean fluorescence intensity of
DCFDA was analyzed by flow cytometry. n=3. *P<0.05
vs. DMSO-treated cells. (B) Ramos cells were pretreated with 5 mM
NAC for 30 min at 37°C. Next, cells were treated with the indicated
concentrations of Mot-P2 or Mot-P7 for 1 h at 37°C. Cells were
washed, labeled with PI and analyzed by flow cytometry.
*P<0.05 vs. cells not treated with NAC. n=4. (C)
Sub-toxic Mot-P2 and Mot-P7 induced cell cycle arrest. Ramos cells
were treated with a sub-toxic dose of Mot-P2, Mot-P7 or a scrambled
peptide (5, 15 or 15 µM, respectively) or DMSO (control) for
36 h at 37°C. Next, cells were fixed with 70% cold ethanol for 2 h
and stained with PI. Distribution of cells among the various cell
cycle stages was analyzed by flow cytometry. n=3,
*P<0.05 and **P<0.01 vs. control cells.
(D and E) Silencing of mortalin lowers peptide cytotoxicity. (D)
K562 cells were transfected with mortalin siRNA or NS siRNA as a
control. At 48 h post transfection, mortalin knockdown was verified
by western blotting. (E) Transfected cells were seeded in wells of
96-well plates and treated with 10 µM Mot-P2 or 75 µM
Mot-P7 for 1 h at 37°C. The extent of cell death was measured by
LDH release. n=3. *P<0.05 vs. NS siRNA transfected
cells. NAC, N-acetyl cysteine; DCFDA, 2′,7′-dichlorofluorescin
diacetate; PI, propidium iodide; NS, non specific; si, small
interfering. |
Effect of Mot-P2 and Mot-P7 at non-toxic
concentration on cell proliferation
The effect of the peptides at low, non-toxic,
concentrations on cell proliferation was investigated. Treatment of
Ramos cells with 5 µM Mot-P2 or 25 µM Mot-P7,
moderately, but significantly, reduced the rate of cell
proliferation in the absence of cell death (Fig. S4). The ability of Mot-P2 and
Mot-P7 to alter cell cycle progression was therefore examined.
Ramos cells were grown with a subtoxic dose of Mot-P2 or Mot-P7 for
36 h; then the distribution of the cells among the cell cycle
division phases was analyzed. Treatment with Mot-P2 and Mot-P7, but
not scrambled peptide, reduced the percentage of cells in G0/G1 and
increased the percentage of cells in G2/M (Fig. 5C). This suggests that the peptides
cause a cell cycle arrest in the G2/M phase.
Intracellular mortalin is a target of
Mot-P2 and Mot-P7
Mortalin protects cells from various insults by
binding to its numerous target proteins. The mimetic mortalin
peptides were hypothesized to interfere with some of these
interactions and to potentially reverse cell protection by
mortalin. Mortalin downregulation was performed in K562 cells.
Mortalin was downregulated in K562 cells with a specific siRNA as
previously described (13). Cells
were transfected with mortalin siRNA or a control non-specific
siRNA and grown for 48 h prior to peptide treatment. The results
demonstrated that transfection induced a decrease in mortalin
expression (Fig. 5D). In addition,
cell death following treatment with Mot-P2 and Mot-P7 was
significantly decreased (Fig. 5E).
These findings were confirmed in PC3 and T47D cells. The results
demonstrated that mortalin downregulation in PC3 and T47D cells
also partially decreased cell sensitivity to Mot-P2 and Mot-P7
(Fig. S5). These results
suggested that Mot-P2 and Mot-P7 may partially induce cytotoxicity
by binding either to mortalin or to a mortalin binder. Still, both
peptides may also have an additional cytotoxic activity that could
be independent of mortalin; however, this remains to be
elucidated.
Combined effect of Mot-P2 and Mot-P7 with
CDC
As previously described, mortalin protects cells
from anti-body-mediated CDC (12,14),
and mortalin inhibitors potentiate the cytotoxic action of
complement (13). Since Mot-P2 and
Mot-P7 can partially target their action to mortalin or
mortalin-binders (Fig. 5E), it was
anticipated that Mot-P2 and Mot-P7 would affect CDC. CDC was tested
on Ramos cells coated with RTX antibodies and then treated with
human complement. Based on an antibody titration assay (Fig. S6), the RTX concentration that
activates complement on lymphoma cells and yields a low percentage
cell death (~30-40%) was determined. Next, Ramos cells were treated
with Mot-P2 or Mot-P7 concentrations causing low-toxicity (25-35%
cell death) and with RTX and complement (NHS), also at a low-toxic
concentration. The combined treatments with peptide and complement
led to an additive cytotoxic effect (Fig. 6A). Thus, whereas Mot-P2 alone,
Mot-P7 alone and NHS alone caused 28, 35 and 34% cell death,
respectively, Mot-P2 + NHS and Mot-P7 + NHS induced 52 and 49% cell
death, respectively. Combining either Mot-P2 or Mot-P7 with CDC may
therefore result in increased cytotoxicity.
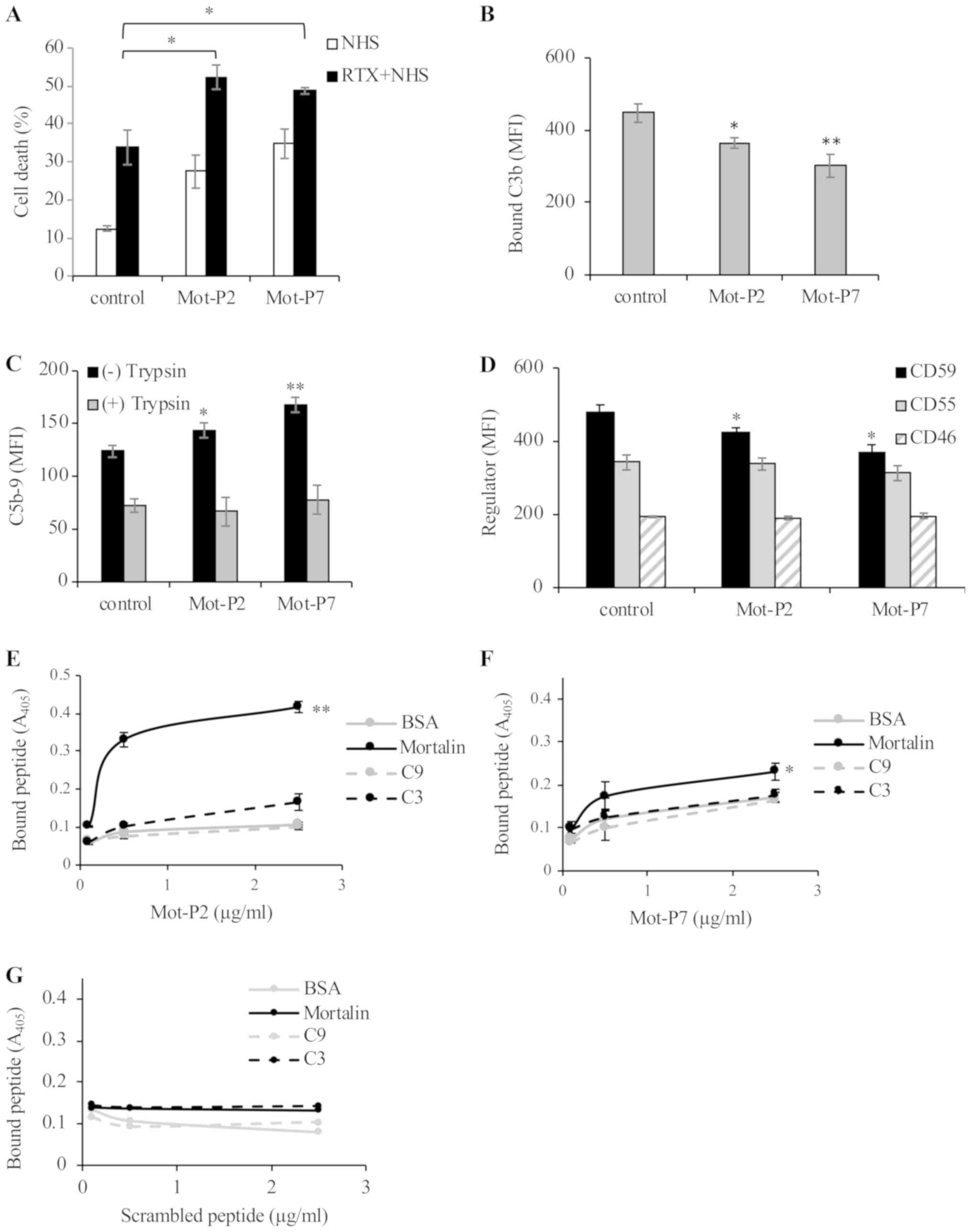 | Figure 6Combination of peptide- and
complement-dependent cytotoxicity. (A) Ramos cells were treated
with a sub-toxic dose of Mot-P2 or Mot-P7 (5 and 50 µM,
respectively) or with DMSO for 30 min at 37°C. Next, cells were
treated with or without Rituximab (RTX, 2 µg/ml) for 30 min
at 4°C and with 50% NHS for 60 min at 37°C. Cells were washed,
labeled with PI and cell death was analyzed by flow cytometry.
*P<0.05 vs. DMSO-treated control cells. (B) Ramos
cells were treated with 5 µM Mot-P2 or 50 µM Mot-P7
or with DMSO for 30 min at 37°C. To measure C3 deposition, cells
were incubated with RTX (1.5 µg/ml) for 30 min at 4°C and
further incubated with 50% NHS for 10 min at 37°C. Cells were
washed, treated with goat anti-C3 antibodies followed by
FITC-conjugated rabbit anti-goat IgG antibodies and analyzed by
flow cytometry. *P<0.05 and **P<0.01
vs. DMSO-treated control cells. (C) Ramos cells were treated 5
µM Mot-P2 or 50 µM Mot-P7 or with DMSO (control) for
30 min at 37°C. To measure C5b-9 (MAC) deposition level, cells were
incubated with 2 µg/ml RTX for 30 min at 4°C, followed by
treatment with 50% NHS, for 10 min at 37°C. Next, cells were washed
and treated or not with trypsin (100 µg/ml) for 20 min at
room temperature. Then, cells were labeled with mouse anti-C5b-9
neo-epitope antibody aE11 followed by FITC-conjugated goat
anti-mouse IgG antibodies. Cells were analyzed by flow cytometry.
*P<0.05 and **P<0.01 vs. control cells.
(D) Ramos cells were treated with 5 µM Mot-P2 or 50
µM Mot-P7 or with DMSO (control) for 30 min at 37°C. To
determine the expression level of the membrane complement
regulatory proteins, cells were labeled with mouse anti-CD46,
anti-CD55 or anti-CD59 antibodies, followed by FITC-conjugated goat
anti-mouse IgG antibodies and analyzed by flow cytometry.
*P<0.05 vs. DMSO-treated control cells. (E-G)
Purified h uman mortalin, C9, C3 or BSA (5 µg/ml each) was
attached overnight to wells of a 96-well plate at 4°C. Next, wells
were washed and blocked with 1% BSA for 1 h at room temperature
followed by adding to the wells biotinylated peptides (E) Mot-P2,
(F) Mot-P7 or a (G) scrambled peptide (0.5 or 2.5 µg/ml) for
1 h at room temperature. Wells were washed, treated with
peroxidase-conjugated streptavidin for 1 h at room temperature.
Bound peptides were quantified using TMB substrate and absorbance
was read at 405 nm in a microplate reader. *P<0.05
and **P<0.01 vs. BSA. MFI, mean fluorescence
intensity; PI, propidium iodide; NHS, normal human serum; BSA,
bovine serum albumin |
These peptides may enhance RTX-mediated CDC by
enhancing the cell CD20 level, by enhancing the cell C3 and/or
C5b-9 deposition levels and/or C5b-9 deposition by suppressing the
expression level of the membrane complement regulators CD46, CD55
and CD59 and/or by lowering the protective capacity of mortalin.
The results demonstrated that Mot-P2 and Mot-P7 had no effect on
the amount of bound RTX, suggesting that the peptides did not
modify CD20 expression level on the cells (Fig. S7). Subsequently, the impact of the
peptides on C3 and C5b-9 deposition was analyzed. Ramos cells were
treated with Mot-P2 or Mot-P7 and then with RTX and NHS. Treatment
time with NHS was 10 min, which corresponded to the peak time for
C3 and C5b-9 depositions (29).
Both Mot-P2 and Mot-P7 slightly decreased the extent of C3
deposition (Fig. 6B), and slightly
enhanced the C5b-9 deposition level (Fig. 6C). Expression level analysis of the
membrane complement regulatory proteins on peptide-treated and
control cells demonstrated that Mot-P2 and Mot-P7 had no effect on
CD46 and CD55, but slightly decreased the CD59 expression level
(Fig. 6D). Consequently, the
reduced expression of CD59 in peptide-treated cells may account for
the elevated deposition of C5b-9. However, more deposited C5b-9
does not necessarily indicate higher cytotoxic activity. A
significant proportion of the deposited C5b-9 is only peripherally
attached to the cells and does not contribute to cytotoxicity
(30). Unlike the fully inserted
cytotoxic C5b-9, the peripherally attached C5b-9 is sensitive to
trypsinization (29). To quantify
the amount of the fully inserted C5b-9 on peptide-treated and
control cells, Ramos cells were treated with Mot-P2 or Mot-P7
first, followed by RTX and NHS (10 min) and then with trypsin. As
presented in Fig. 6C,
trypsinization decreased the amount of deposited C5b-9, with no
significant difference in the amount of trypsin-resistant C5b-9
between peptide-treated and control cells. These findings suggested
that the Mot-P2 and Mot-P7-enhancing effect on CDC may not result
from the observed reduced CD59 expression and enhanced C5b-9
deposition. To determine whether peptides could bind directly to
intracellular mortalin and therefore affect its protective
activity, the direct binding of biotin-labeled Mot-P2 or Mot-P7 to
mortalin was tested by ELISA. Peptide binding to mortalin was
compared with their binding to C3, C9 and BSA. The results
demonstrated that, unlike a scrambled peptide, Mot-P2 bound
strongly to mortalin (Fig. 6E and
G). In addition, Mot-P7 demonstrated only a moderate yet
significant binding to mortalin (Fig.
6F). Mot-P2 and Mot-P7 did not bind to C3 or C9.
Discussion
Because of its ubiquitous elevated expression in
cancer cells and its suggested role in cell protection from toxic
insults (10), mortalin has been
considered as a potential target in cancer therapy. This hypothesis
was supported by previous studies using the mortalin inhibitor
MKT-077 (13,31). The present study hypothesized that
mortalin mimetic peptides could interfere with the protective
mechanisms of mortalin and might therefore be used as tumor
therapeutic agents. Several peptides from the nucleotide-binding or
the substrate-binding domains of mortalin were designed and a TAT
sequence was added to their C-terminal to promote their cell
penetration ability. Two peptides, Mot-P2 and Mot-P7, from the
nucleotide-binding domain of mortalin, were demonstrated to be
highly toxic to lymphoma and carcinoma cell lines and to primary
CLL cells. However, these two peptides were significantly less
toxic for healthy PBMC. The peptide cytotoxic efficiency at low
micro-molar concentrations was also attributed to the linked TAT.
However, the lack of cytotoxicity of a TAT-linked scrambled
peptide, as well as the lower toxicity of Mot-P8, suggested that
cytotoxicity is peptide-dependent. This was further confirmed
following mortalin knockdown, when cells became less sensitive to
the peptides. In addition, at a non-toxic concentration, the two
peptides interfered with cell cycle progression and induced a G2/M
arrest.
The low LD50 for Mot-P2 and Mot-P7 encouraged us to
investigate their mode of action. The results identified several
similarities and differences in their modes of action (Table I). The two peptides were highly
cytotoxic and induced cell death within minutes after cell binding.
Cell death was necrosis, as PI entered into and LDH leaked out of
the affected cells, indicating plasma membrane damage. Within
minutes, phosphatidylserine flipped to the extracellular surface of
the plasma membrane, possibly after activating scramblase or by
another mechanism (20). Loss of
plasma membrane asymmetry was described first for apoptosis;
however, it has also been demonstrated in cell development, injury,
senescence and necrosis (21).
 | Table ISimilarities and differences in the
mode of action of Mot-P2 and Mot-P7. |
Table I
Similarities and differences in the
mode of action of Mot-P2 and Mot-P7.
| Peptide
characteristics | Mot-P2 | Mot-P7 |
|---|
| LD50,
µMa | 26-30 | 40-63 |
| LDH release | + | + |
| PS
externalization | + | +b |
| Ca2+
entry dependencec | − | + |
| Cholesterol
protectiond | + | + |
| MIMDe | ++ | + |
| ATP level
drops | + | + |
| ROS generated | + | + |
| ROS involved in
cell deathf | − | + |
| Cell growth
inhibitiong | + | + |
| Cell cycle
inhibitiong | + | + |
| Mortalin
dependence | + | + |
| Direct binding to
mortalin | ++ | + |
| Additive effect
with CDCh | + | + |
The present study demonstrated that Mot-P2 and
Mot-P7 had different effects on plasma membrane. Whereas Mot-P2
activated concomitant PS externalization and PI entry, Mot-P7
activated PS externalization only in some cells and both PS
externalization and PI entry in other cells. These findings
suggested that Mot-P7 may activate PS externalization independent
of plasma membrane perforation in some cells. This was supported by
the observation that in the first 10-15 min, less LDH was released
from Mot-P7-treated cells compared with Mot-P2-treated cells.
Furthermore, cell death induced by Mot-P7 was partially inhibited
by chelation of extracellular calcium, which was not the case for
Mot-P2. Calcium ions play a crucial role in cell death regulation
and numerous pharmaceuticals target Ca2+-mediated
processes (23). An increase in
intracellular calcium is also essential to trigger plasma membrane
remodeling and PS externalization (32). Mot-P7 may be able to use calcium
ion influx for both PS externalization and further perforation of
the plasma membrane. Subsequently, in absence of extracellular
calcium ions, Mot-P7 may become less effective.
Plasma membrane is the initial cellular compartment
with which the peptides interact. Cholesterol is known to influence
plasma membrane damage by membrane-disrupting peptides or proteins
in two ways. It may either serve as a receptor for them and
facilitate membrane damage (33)
or it may promote removal or blockade of the toxic molecules and
protect the cells (25,26). Membranes with higher cholesterol
levels have a decreased rate of peptide insertion (34). In the present study, both depletion
and blocking of cholesterol induced enhanced cell sensitivity to
Mot-P2 and Mot-P7. Therefore, plasma membrane cholesterol may have
a protective role for both peptides. However, whether cholesterol
could facilitate peptide removal and/or membrane repair remains to
be determined.
The second organelle affected by the peptides was
the mitochondrion. This effect could be secondary to calcium influx
or to another, yet undefined, toxic moiety or could be directly
activated by peptides binding to the mitochondrial outer membrane.
The results from confocal analysis revealed peptides accumulation
inside the cytoplasm within 5 min following cell binding. This was
more distinct with Mot-P2 than with Mot-P7. Furthermore, the
mitochondrial membrane potential depolarization induced by Mot-P2
was higher than that of Mot-P7. The TAT sequence has been
demonstrated to allow peptides passage through mitochondrial
membranes via mechanisms that do not involve the regular import
pathway (35). In addition, the
ATP-depleting activity of Mot-P2 was more pronounced than that of
Mot-P7. Both peptides induced ROS overproduction; however, ROS
scavenging by NAC decreased Mot-P7-induced, but not Mot-P2-induced,
cell death. Mot-P7 may be less effective without extracellular
calcium ions. This may account for the dependence of Mot-P7 on ROS
for cell death. Compared with Mot-P7, Mot-P2 induced a more
rigorous and faster damage to the plasma membrane and the
mitochondria and activated rapid cell death independent of calcium
entry and ROS generation. Evaluating the combined action of Mot-P2
and Mot-P7 may therefore be of interest. It is possible that the
effect of the two peptides may be additive or synergistic. However,
because both peptides target mortalin in the cell, their
combination might lead to reduction in their cytotoxic activity.
Binding of one peptide to mortalin could delay the binding of the
second peptide due to the possible competition or allosteric
effect.
Since mortalin protects cells from CDC, the impact
of Mot-P2 and Mot-P7 on CDC was also investigated in the present
study. The results demonstrated that the peptides and CDC acted
additively but not synergistically. Under the conditions used, the
peptides had no effect on CD20 expression; however, they slightly
decreased C3 deposition but had no effect on C5b-9 membrane
insertion. The elevated cell death may therefore not result from
peptide-induced enhanced complement activation. The additive
cytotoxicity of the peptides and C5b-9 could result from one or
more of the following effects. Firstly, the peptide could target
mortalin in the cells and block its protective activity against
CDC, enhancing therefore CDC. In this respect, direct binding of
Mot-P2 and Mot-P7 to mortalin is shown. In addition, the peptides
are both derived from the nucleotide-binding domain of mortalin,
known to bind complement C8 and C9 and inhibit CDC (14). Secondly, the peptides could bind to
essential mortalin binders and therefore sensitize cells to CDC.
Thirdly, the lytic signals of C5b-9 could amplify the toxic effects
of the peptides. Fourthly, the two cell death pathways could act
independently, and the resulting additive cell death could reflect
the cumulative damage they inflicted on the target cells. Further
investigation is required to determine whether the actions of the
peptides and the anti-body/complement are dependent or independent.
However, when considering cancer therapy, even as combined
mono-therapies, they may have an advantage over each monotherapy
given alone (36,37).
In conclusion, the present study demonstrated that
the two mortalin-TAT peptides were highly cytotoxic to numerous
cancer cell types. The peptides acted as single agents but also
exhibited additive cytotoxicity when combined with
antibody-mediated complement-dependent cytotoxicity. They may
therefore be considered as valuable anti-cancer biotherapies.
Peptide-based therapeutics for cancer have their pros and cons, and
numerous attempts have been made to leverage their cytotoxicity and
to overcome some of these challenges (38,39).
The use of Mot-P2 or Mot-P7 as monotherapy for cancer may be even
more effective when combined with anti-cancer antibodies. It may
improve the clinical efficacy while maintaining acceptable clinical
toxicity. Combination therapy also leads to reduced side effects,
since lower dosage of each drug is required. Subsequently, these
two peptides may stop cell cycle, when sub-toxic doses are applied.
Furthermore, the high efficacy of combination therapy may produce a
more effective response, in fewer cycles of treatment; this option
may therefore reduce drug resistance incidence (40).
Supplementary Data
Acknowledgments
Not applicable.
Funding
This study was supported by the Israel Cancer
Association (grant no. 20150110) and The Varda and Boaz Dotan
Research Center in Hemato-Oncology (Tel Aviv University; grant no.
060141520).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZF designed the study. RJ and ZF wrote the article.
RJ performed most of the experiments. AW performed the experiments
with the carcinoma cell lines. MS, LZ and ND contributed to some of
the experiments. OB collected the clinical samples. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Helsinki Committee of
the Rabin Medical Center, Petach Tikva, Israel and informed consent
was provided by all participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests
References
|
1
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ran Q, Wadhwa R, Kawai R, Kaul SC, Sifers
RN, Bick RJ, Smith JR and Pereira-Smith OM: Extramitochondrial
localization of mortalin/mthsp70/PBP74/GRP75. Biochem Biophys Res
Commun. 275:174–179. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wadhwa R, Yaguchi T, Hasan MK, Mitsui Y,
Reddel RR and Kaul SC: Hsp70 family member, mot-2/mthsp70/GRP75,
binds to the cytoplasmic sequestration domain of the p53 protein.
Exp Cell Res. 274:246–253. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dundas SR, Lawrie LC, Rooney PH and Murray
GI: Mortalin is over-expressed by colorectal adenocarcinomas and
correlates with poor survival. J Pathol. 205:74–81. 2005.
View Article : Google Scholar
|
|
5
|
Takano S, Wadhwa R, Yoshii Y, Nose T, Kaul
SC and Mitsui Y: Elevated levels of mortalin expression in human
brain tumors. Exp Cell Res. 237:38–45. 1997. View Article : Google Scholar
|
|
6
|
Ando K, Oki E, Zhao Y, Ikawa-Yoshida A,
Kitao H, Saeki H, Kimura Y, Ida S, Morita M, Kusumoto T, et al:
Mortalin is a prognostic factor of gastric cancer with normal p53
function. Gastric Cancer. 17:255–262. 2014. View Article : Google Scholar
|
|
7
|
Wadhwa R, Takano S, Kaur K, Deocaris CC,
Pereira-Smith OM, Reddel RR and Kaul SC: Upregulation of
mortalin/mthsp70/Grp75 contributes to human carcinogenesis. Int J
Cancer. 118:2973–2980. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rozenberg P, Kocsis J, Saar M, Prohászka
Z, Füst G and Fishelson Z: Elevated levels of mitochondrial
mortalin and cytosolic HSP70 in blood as risk factors in patients
with colorectal cancer. Int J Cancer. 133:514–518. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jubran R, Kocsis J, Garam N, Maláti É,
Gombos T, Barabás L, Gráf L, Prohászka Z and Fishelson Z:
Circulating mitochondrial stress 70 protein/mortalin and cytosolic
Hsp70 in blood: Risk indicators in colorectal cancer. Int J Cancer.
141:2329–2335. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kaul SC, Deocaris CC and Wadhwa R: Three
faces of mortalin: A housekeeper, guardian and killer. Exp
Gerontol. 42:263–274. 2007. View Article : Google Scholar
|
|
11
|
Voisine C, Craig EA, Zufall N, von Ahsen
O, Pfanner N and Voos W: The protein import motor of mitochondria:
Unfolding and trapping of preproteins are distinct and separable
functions of matrix Hsp70. Cell. 97:565–574. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pilzer D and Fishelson Z: Mortalin/GRP75
promotes release of membrane vesicles from immune attacked cells
and protection from complement-mediated lysis. Int Immunol.
17:1239–1248. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pilzer D, Saar M, Koya K and Fishelson Z:
Mortalin inhibitors sensitize K562 leukemia cells to
complement-dependent cytotoxicity. Int J Cancer. 126:1428–1435.
2010.
|
|
14
|
Saar Ray M, Moskovich O, Iosefson O and
Fishelson Z: Mortalin/GRP75 binds to complement C9 and plays a role
in resistance to complement-dependent cytotoxicity. J Biol Chem.
289:15014–15022. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Frankel AD and Pabo CO: Cellular uptake of
the tat protein from human immunodeficiency virus. Cell.
55:1189–1193. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Raucher D and Ryu JS: Cell-penetrating
peptides: Strategies for anticancer treatment. Trends Mol Med.
21:560–570. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Darzynkiewicz Z, Juan G and Bedner E:
Determining cell cycle stages by flow cytometry. Curr Protoc Cell
Biol Chapter. 8(Unit 8): 42001.
|
|
18
|
Smiley ST, Reers M, Mottola-Hartshorn C,
Lin M, Chen A, Smith TW, Steele GD Jr and Chen LB: Intracellular
heterogeneity in mito-chondrial membrane potentials revealed by a
J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci USA.
88:3671–3675. 1991. View Article : Google Scholar
|
|
19
|
Vivès E, Brodin P and Lebleu B: A
truncated HIV-1 Tat protein basic domain rapidly translocates
through the plasma membrane and accumulates in the cell nucleus. J
Biol Chem. 272:16010–16017. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nagata S: Apoptosis and Clearance of
Apoptotic Cells. Annu Rev Immunol. 36:489–517. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hirt UA and Leist M: Rapid,
noninflammatory and PS-dependent phagocytic clearance of necrotic
cells. Cell Death Differ. 10:1156–1164. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Caserta TM, Smith AN, Gultice AD, Reedy MA
and Brown TL: Q-VD-OPh, a broad spectrum caspase inhibitor with
potent antiapoptotic properties. Apoptosis. 8:345–352. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhivotovsky B and Orrenius S: Calcium and
cell death mechanisms: A perspective from the cell death community.
Cell Calcium. 50:211–221. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mason AJ, Marquette A and Bechinger B:
Zwitterionic phospholipids and sterols modulate antimicrobial
peptide-induced membrane destabilization. Biophys J. 93:4289–4299.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Prenner EJ, Lewis RNAH, Jelokhani-Niaraki
M, Hodges RS and McElhaney RN: Cholesterol attenuates the
interaction of the antimicrobial peptide gramicidin S with
phospholipid bilayer membranes. Biochim Biophys Acta. 1510:83–92.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Moskovich O, Herzog LO, Ehrlich M and
Fishelson Z: Caveolin-1 and dynamin-2 are essential for removal of
the complement C5b-9 complex via endocytosis. J Biol Chem.
287:19904–19915. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu Y, Liu W, Song XD and Zuo J: Effect of
GRP75/mthsp70/PBP74/mortalin overexpression on intracellular ATP
level, mitochondrial membrane potential and ROS accumulation
following glucose deprivation in PC12 cells. Mol Cell Biochem.
268:45–51. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun SY: N-acetylcysteine, reactive oxygen
species and beyond. Cancer Biol Ther. 9:109–110. 2010. View Article : Google Scholar
|
|
29
|
Moskovich O and Fishelson Z:
Quantification of complement C5b-9 binding to cells by flow
cytometry. The Complement System: Methods and Protocols. 1100th
edition. Gadjeva M: Humana Press; Totowa, NJ: pp. 103–108. 2014,
View Article : Google Scholar
|
|
30
|
Bhakdi S, Tranum-Jensen J and Klump O: The
terminal membrane C5b-9 complex of human complement. Evidence for
the existence of multiple protease-resistant polypeptides that form
the trans-membrane complement channel. J Immunol. 124:2451–2457.
1980.PubMed/NCBI
|
|
31
|
Koya K, Li Y, Wang H, Ukai T, Tatsuta N,
Kawakami M, Shishido and Chen LB: MKT-077, a novel rhodacyanine dye
in clinical trials, exhibits anticarcinoma activity in preclinical
studies based on selective mitochondrial accumulation. Cancer Res.
56:538–543. 1996.PubMed/NCBI
|
|
32
|
Kunzelmann-Marche C, Freyssinet JM and
Martínez MC: Regulation of phosphatidylserine transbilayer
redistribution by store-operated Ca2+ entry: Role of actin
cytoskeleton. J Biol Chem. 276:5134–5139. 2001. View Article : Google Scholar
|
|
33
|
Heuck AP, Moe PC and Johnson BB: The
cholesterol-dependent cytolysin family of gram-positive bacterial
toxins. Subcell Biochem. 51:551–577. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Won A, Ruscito A and Ianoul A: Imaging the
membrane lytic activity of bioactive peptide latarcin 2a. Biochim
Biophys Acta. 1818:3072–3080. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Del Gaizo V, MacKenzie JA and Payne RM:
Targeting proteins to mitochondria using TAT. Mol Genet Metab.
80:170–180. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Palmer AC and Sorger PK: Combination
cancer therapy can confer benefit via patient-topatient variability
without drug additivity or synergy. Cell. 171:1678–1691.e13. 2017.
View Article : Google Scholar
|
|
37
|
Foucquier J and Guedj M: Analysis of drug
combinations: Current methodological landscape. Pharmacol Res
Perspect. 3:e001492015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kurrikoff K, Aphkhazava D and Langel Ü:
The future of peptides in cancer treatment. Curr Opin Pharmacol.
47:27–32. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Guidotti G, Brambilla L and Rossi D:
Peptides in clinical development for the treatment of brain tumors.
Curr Opin Pharmacol. 47:102–109. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bayat Mokhtari R, Homayouni TS, Baluch N,
Morgatskaya E, Kumar S, Das B and Yeger H: Combination therapy in
combating cancer. Oncotarget. 8:38022–38043. 2017. View Article : Google Scholar : PubMed/NCBI
|