Introduction
Chordoma, characterized by having high recurrence
rate after surgery and locally invasive, as well as potentially
metastatic ability, is a rare, chronic, low-grade axial skeleton
carcinoma derived from remnants of the notochord from embryonic
development (1). The annual
incidence rate of chordoma is ~0.8 per million people, accounting
for 1-4% of all primary bone carcinoma and ~20% of spinal tumors
(2). Chordoma can occur in any
part of the spine; the sacrococcygeal region and skull base are the
most common locations, accounting for ~50 and 30% of total cases,
respectively (3,4). Surgical excision currently remains
the first-line therapy, and there is no consensus concerning the
effect of adjuvant radiotherapy after surgical treatment; the
efficacy of chemotherapy remains controversial due to reported
resistance to radiation and chemotherapy (5). Over previous decades, an increasing
number of studies have revealed potential genes associated with
chordoma, indicating that gene targeted therapy may be an avenue
for treating chordoma. For examples, T-box transcription factor T
(also known as brachyury) is considered to be the most important
gene for chordoma; brachyury is upregulated in chordoma, whilst its
expression in normal tissues or other cancers is relatively low
(6). Additionally, it has been
reported that, to a certain extent, treatments targeting EGFR may
therapeutic effects in patients with chordoma (7,8).
However, due to a lack of specific drugs for chordoma, further
specific and effective potential targets are under
investigation.
Transmembrane Emp24 protein transport domain
containing 3 (TMED3) is a member of the p24 protein family that
serves an important role in the vesicular trafficking of proteins
at the secretory endoplasmic reticulum (ER)-Golgi interface, and is
involved in different signaling pathways in eukaryotic cells
(9,10). It has been determined that the p24
protein family contains 10 TMED proteins, of which some serve roles
as regulators in the progression of different carcinomas (11). For example, TMED3 plays a
prognostic role in clear cell renal cell carcinoma, and is proposed
to be a target gene to inhibit the progression of breast cancer,
hepatocellular carcinoma, prostate cancer and gastric cancer
(12-16). Notably, Mishra et al
(17) reported that TMED3
knockdown promoted the lung metastasis of colon cancer in mice,
accompanied by the upregulation of TMED9 but downregulation of
other TMED proteins, particularly TMED7. Duquet et al
(18) demonstrated that knockdown
of TMED3 induced lung metastases from HT29 colon cancer cells in
vivo, suggesting that TMED3 may serve as a suppressor of colon
cancer metastasis. However, the function of TMED3 remains poorly
understood, and there is so far no evidence concerning TMED3 in
relation to chordoma.
The present study investigated for the first time,
to the best of the authors' knowledge, the expression of TMED3 in
chordoma cells and the effects of TMED3 knockdown on chordoma
development both in vitro and in vivo. Moreover,
differentially expressed genes associated with TMED3 in chordoma
cells were explored, revealing the potential of TMED3 as a novel
target of chordoma therapy (Fig.
S1).
Materials and methods
Cell lines and cell culture
Two human chordoma cell lines were used: MUG-Chor1
(cat. no. CRL-3219) and U-CH1 (cat. no. CRL-3217; both American
Type Culture Collection). Cells were cultured in high-glucose DMEM
(cat. no. 10-013-CVR; Corning, Inc.) supplemented with 10% fetal
bovine serum (FBS; cat. no. VS500T; Ausbian; Beijing Vian-Saga
Biological Technology, Ltd.) and 1% penicillin-streptomycin stock
solution in a humidified cell incubator with 5% CO2 at
37°C. Complete growth medium was renewed every 2-3 days and
subculture were performed when cells were 80-90% confluent. All
cell function experiments were performed in three parallel
biological replicates per group.
Vector construction and lentivirus
transduction
For the construction of plasmid vectors, the TMED3
gene sequence (gene accession no. NM_007364) was found in the
GenBank database (https://www.ncbi.nlm.nih.gov/nuccore/NM_007364.3).
Based on the principles of RNA interference, a target sequence was
designed (5′-CTC TCA CAA GAC CGT CTA CTT-3′). Restriction enzyme
sites at both ends were added. Additionally, a transcription
termination signal TTTTT sequence was added to the 3′ end of the
sense strand and a complementary termination signal sequence was
added to the 5′ end of the antisense strand. Then, the
single-stranded DNA oligonucleotides were synthesized (Sangon
Biotech Co., Ltd.). According to the manufacturer's protocol, the
inverted single-strand DNA oligonucleotide was converted into
double DNA strands with cohesive ends after annealing. Then, the
plasmid vector BR-V108 with green fluorescent protein (Shanghai
Biosciences Co., Ltd.) was linearized under the action of
AgeI (cat. no. R3552L) and EcoRI (cat. no. R3101L;
New England Biolabs, Inc.) restriction endonucleases. A
BR-V108-short hairpin (sh)RNA vector was constructed after
connecting the linearized vector to shRNA using T4 DNA Ligase (cat.
no. EL0016; Fermentas; Thermo Fisher Scientific, Inc.), and was
transformed and cloned into TOP10 competent E. coli cells
(cat. no. CB104-03; Tiangen Biotech Co., Ltd.). A vector containing
a non-specific target sequence (5′-TTC TCC GAA CGT GTC ACG T-3′)
was used as the negative control (shCtrl). Moreover, PCR was
performed to identify the vector. The primer sequences used for PCR
amplification were as follows: shTMED3 forward, 5′-CCT ATT TCC CAT
GAT TCC TTC ATA-3′ and reverse, 5′-GTA ATA CGG TTA TCC ACG CG-3′;
and shCtrl forward, 5′-CCA TGA TTC CTT CAT ATT TGC-3′ and reverse,
5′-GTA ATA CGG TTA TCC ACG CG-3′. Taq Plus DNA Polymerase (cat. no.
P201-03; Vazyme Biotech Co., Ltd.) was used and PCR was conducted
as follows: 94°C for 3 min, followed by 22 cycles of denaturation
at 94°C for 30 sec, annealing at 55°C for 30 sec and extension at
72°C for 30 sec; and a final extension at 72°C for 5 min.
The recombinant vectors were extracted according to
the instructions of the EndoFree Maxi Plasmid kit (cat. no. DP117;
Tiangen Biotech Co., Ltd.). 293T cells were seeded on a 100-mm
plate (~5×106 cells/well; Shanghai Biosciences Co.,
Ltd.) until 80% confluent and then co-transfected with recombinant
vectors (20 µg), pHelper 1.0 vector (15 µg) and pHelper 2.0 vector
(10 µg; all Shanghai Biosciences Co., Ltd.) using transfection
reagent (Lipofectamine™ 3000; Invitrogen; Thermo Fisher Scientific,
Inc.). After transfecting for 48 h, lentiviral vectors (LV-shTMED3
and LV-shCtrl) were separated and purified. Chordoma cells were
seeded on 6-well plates (~1×105 cells/well) and
transfected with lentivirus using polybrene (6 µg/ml; cat. no.
TR-1003-G; Sigma-Aldrich; Merck KGaA) according to the specific
multiplicity of infection (MOI=10). After lentiviral transduction
for 72 h, reverse transcription-quantitative (RT-q) PCR and western
blot analyses were performed in order to evaluate the expression of
TMED3, and the fluorescence of cells were detected using an
inverted fluorescence microscope.
RT-qPCR
A two-step RT-qPCR protocol was performed to
quantify the expression of TMED3 in lentivirus-transfected cells
and normal chordoma cells. Chordoma cells were seeded on 6-well
plates until 80-90% confluent. Total RNA from cells was collected
using TRIzol® reagent (cat. no. 15596018; Invitrogen;
Thermo Fisher Scientific, Inc.) and a spectrophotometer (NanoDrop™
2000; Thermo Fisher Scientific, Inc.) was used to measure and
calculate the RNA concentration of samples. RT was performed
according to the protocols of the HiScript Reverse Transcriptase
kit (cat. no. R123-01; Vazyme Biotech Co., Ltd.): A reaction
mixture that contained chordoma cell RNA (2 µg) was prepared and
first incubated at 42°C for 2 min before reacting at 50°C for 15
min and 85°C for 2 min. qPCR was performed as recommended by the
AceQ SYBR Green kit protocols (cat. no. Q111-02; Vazyme Biotech
Co., Ltd.). The primer sequences were as follows: TMED3 forward,
5′-GGC GTG AAG TTC TCC CTG GAT T-3′ and reverse, 5′-GCT GTC GTA CT
GCT TCT TCG TTT C-3′; and GAPDH forward, 5′-CGG ATT TGG TCG TAT TGG
G-3′ and reverse, 5′-GAT TTT GGA GGG ATC TCG C-3′. qPCR was
conducted as follows: 95°C for 5 min, followed by 40 cycles of
denaturation at 95°C for 30 sec, annealing at 58°C for 30 sec and
extension at 72°C for 45 sec; and a final extension at 72°C for 7
min. qPCR data were analyzed using the 2−ΔΔCq method
(19,20); after normalization to the
reference gene, relative gene expression levels were calculated by
comparing with the control group.
Western blot analysis
Western blotting was performed to examine the
expression of proteins in U-CH1 and MUG-Chor1 chordoma cells under
different conditions. Cells were seeded on 6-well plates. When
cells were 80-90% confluent, cells were lysed using RIPA buffer
(cat. no. P0013B; Beyotime Institute of Biotechnology) containing
1% PMSF (cat. no. ST506; Beyotime Institute of Biotechnology) and
deacetylase inhibitor cocktail (cat. no. P1112; Beyotime Institute
of Biotechnology) and the supernatant was collected after
centrifugation at 13,200 x g for 10 min at 4°C. Using a Pierce™ BCA
Protein Assay kit (cat. no. 23235; Thermo Fisher Scientific, Inc.),
protein samples were diluted and the absorbance was measured at 562
nm. A standard curve was prepared and the concentration of each
lysate sample was determined. Equal quantities of protein samples
(20 µg) and PageRuler™ protein marker (cat. no. 26617; Thermo
Fisher Scientific, Inc.) were loaded and separated via 12%
SDS-PAGE. Protein blots were transferred to PVDF membrane (cat. no.
88585; Thermo Fisher Scientific, Inc.) and blocked in TBS-0.05%
Tween 20 (TBST) containing 5% skim milk at room temperature for 1
h. The membranes were incubated with the following primary
antibodies overnight at 4°C: Rabbit anti-TMED3 (1:2,000; cat. no.
ab223175; Abcam); rabbit anti-GAPDH (1:3,000; cat. no. AP0063;
Bioworld Technology, Inc.); rabbit anti-Akt (1:1,000; cat. no.
4685; Cell Signaling Technology, Inc.); rabbit anti-phosphorylated
(p)-Akt antibody (1:1,000; cat. no. bs-5193r; BIOSS); rabbit
anti-cyclin D1 (1:2,000; cat. no. 2978; Cell Signaling Technology,
Inc.); rabbit anti-CDK6 (1:1,000; cat. no. ab151247; Abcam); and
rabbit anti-MAPK9 (1:1,000; cat. no. ab76125; Abcam). The membranes
were washed, followed by incubation with horseradish
peroxidase-conjugated goat anti-rabbit secondary antibody (1:3,000;
cat. no. A0208; Beyotime Institute of Biotechnology) for 1 h at
room temperature. Chemiluminescent analysis was performed using
Immobilon Western Chemiluminescent HRP substrate (cat. no.
WBKLS0050; EMD Millipore). GAPDH protein was used as the internal
reference.
Cell apoptosis analysis
Cell apoptosis was analyzed using an Annexin
V-allophycocyanin (APC) Apoptosis Detection kit (cat. no.
88-8007-74; eBioscience; Thermo Fisher Scientific, Inc.), chordoma
cells were seeded on 6-well plates until 80% confluent. The
supernatant was discarded and cells were washed using PBS buffer.
After digestion, the cell suspension was collected, followed by
centrifugation at 1,050 x g for 5 min at 4°C and washing using 1X
binding buffer. Cells were resuspended in 1X binding buffer (200
µl) at 1-5×106/ml. Then, 10 µl Annexin V-APC was mixed
with the cell suspension, which was incubated in dark for 10 min at
room temperature. Fluorescence-activated cell sorting (FACS)
analysis was performed using a flow cytometer (Guava easyCyte HT;
EMD Millipore) and InCyte 3.1 software (EMD Millipore) to detect
the fluorescence of GFP and Annexin V-APC of the stained cells. The
apoptosis rate was calculated according to the scatter diagram of
apoptosis. Apoptosis rate=rate of upper right quadrant + rate of
lower right quadrant.
MTT assay
To detect the viability of U-CH1 and MUG-Chor1
chordoma cells, an MTT assay was performed. Cells were seeded on a
96-well plate (~2,000 cells/well) and cultured for 1-5 days at
37°C. Then, MTT (5 mg/ml; cat. no. JT343; Gen-View Scientific,
Inc.) was added to each well and cells were cultured for 4 h.
Subsequently, medium was discarded and 100 µl DMSO were added. The
optical density of each well at 490/570 nm was measured; results
were analyzed after data were collected for 5 days.
Wound healing assay
Cells in the exponential growth phase were cultured
until >90% confluent on a 96-well plate (~3×104
cells/well) in DMEM containing 10% FBS. The medium was then
discarded and a 96 Wounding Replicator (V&P Scientific) was
used to make a straight scratch across the cells, after which the
cells were washed and subsequently cultured with low-serum medium
(0.5% FBS). Using the width of the scratch after culturing at 37°C
with 5% CO2 for 0, 24 and 48 h, the migration rate of
each group was calculated: Migration rate=[width (0 h)-width (24 or
48 h)]/width (0 h).
Transwell assay
Chambers were put in a 24-well plate. Serum-free
medium (100 µl) was prepared and added to the chamber for
incubation for 1-2 h at 37°C, after which the medium was replaced
and cells were added (1-2×105/ml in serum-free medium),
while 600 µl DMEM (with 30% FBS) was added to the lower chamber.
The chambers were cultured for 24 h at 37°C. Migrated cells were
then fixed using 4% paraformaldehyde for 20 min at room temperature
and were stained using Giemsa for 15 min at room temperature while
non-migrated cells were scraped off. A high-power light microscope
was used and the number of migrated cells in five fields/well was
counted (magnification, x200).
Cell cycle assay
Cells in the exponential growth phase were cultured
on a 6-cm dish in DMEM containing 10% FBS at 37°C for 24 h. Cells
at 80% confluence were resuspended in PBS buffer and fixed in 70%
precooled ethanol overnight at 4°C. After centrifugation at 1,400 x
g for 5 min at 4°C and resuspension, cells (1-6×106/ml)
were collected again and incubated with RNase (100 µg/ml; cat. no.
2158-1; Takara Bio, Inc.) and PI (50 µg/ml; cat. no. P4170;
Sigma-Aldrich; Merck KGaA) at room temperature for 30 min in the
dark. Finally, FACS analysis was performed using a flow cytometer
(Guava easyCyte HT) and ModFit LT 3.3 software (Verity Software
House) to determine the DNA content of cells.
Tumor xenograft model
A total of 10 female BALB/c nude mice (age, 4 weeks;
weight, 15.5-17.5 g) were obtained from Shanghai Laboratory Animal
Center and maintained in a standard 12:12-h light/dark cycle under
temperature (20-22°C) and humidity (40-60%)-controlled conditions
with ad libitum access to food and water. MUG-Chor1 chordoma
cells were infected with the shRNA lentivirus as described above,
digested with trypsin and resuspended. Chordoma cell suspensions
(200 µl; 1×107 cells/ml) from the shTMED3 and shCtrl
groups were injected subcutaneously into the armpits of the right
forelimbs of mice. At 5-7 days later, the dimensions of tumors were
measured using a Vernier caliper and the tumor volume in
mm3 was calculated according to the following formula:
V=π/6 x L x W x W, where V is the tumor volume, L is the long tumor
diameter and W is the short tumor diameter. At 33 days after
subcutaneous injection, mice with tumor xenografts were
anesthetized via intraperitoneal injection of 0.7% pentobarbital
(70 mg/kg) and tumors were detected using an in vivo
fluorescence imaging scanner. Fluorescence intensity was detected
by the system due to green fluorescent protein expression in the
cells. Mice were sacrificed and tumors were obtained for weight
measurements and subsequent H&E analysis. Humane endpoints were
reached when the xenograft tumor reached >10% of the animal's
body weight, the tumor diameter was >20 mm, tumors metastasized
or grew such that it led to rapid body weight loss (>20%), or
signs of immobility, a huddled posture, the inability to eat,
ruffled fur, self-mutilation, ulceration, infection or necrosis
were observed. Animals that reached study endpoints were euthanized
via cervical dislocation under anesthesia following intraperitoneal
injection of pentobarbital (70 mg/kg) (21). Death was verified by the cessation
of a heartbeat and dilated pupils.
The present study was conducted according to the
Guide for the Care and Use of Laboratory Animals of the National
Institutes of Health (22) and
approved by the Animal Ethics Committee of Zhujiang Hospital of
Southern Medical University (approval no. IACUC-2019010).
H&E and immunohistochemistry (IHC)
staining
To gain further insight into the effects of TMED3 on
tumor progression, isolated tumor masses underwent fixation in 10%
formaldehyde at room temperature for 24 h and tissue dehydration,
and were subsequently embedded in paraffin, followed by cutting
into 4-µm sections and histological analysis. Tissue was stained by
hematoxylin (cat. no. BA4041; BaSO Biotech) and eosin (cat. no.
BA4022; BaSO Biotech) for 3 min and 8 sec, respectively, at room
temperature. Light microscope was used to visualize cell patterns.
IHC analysis was performed to verify the expression of specific
gene. Briefly, after dewaxing and rehydration, sections were
incubated in citrate buffer (pH 6.0) and microwaved for 10 min.
Sections were then cooled down to room temperature and incubated in
peroxidase block (3% H2O2) for 5 min at room
temperature. Sections were then incubated in 5% normal goat serum
(cat. no. C01-03001; BIOSS) for 5 min at room temperature. Animal
tissue was stained using rabbit anti-Ki67 antibody (1:200; cat. no.
ab16667; Abcam) overnight at 4°C and horseradish
peroxidase-conjugated goat anti-rabbit antibody (1:400; cat. no.
ab6721; Abcam) for 30 min at 37°C in order to determine differences
between tumor cells in shTMED3 and shCtrl tissues. Expression was
visualized using 3,3′-diaminobenzidine (cat. no. ab64238; Abcam),
and nuclei were counterstained using hematoxylin for 10 sec at room
temperature. A light microscope was used to observe sections
(magnification, x100 and x200) and five fields of each section were
analyzed.
Clinical specimens of chordoma and para-carcinoma
tissue were collected. and preliminary validation was performed to
identify the expression of TMED3 via IHC. A total of 5 patients
with histologically confirmed chordoma were included in the study.
Specimens from patients that received surgical resection for
chordoma between April 2018 and June 2020 were stored in the
Pathology Department of Zhujiang Hospital of Southern Medical
University and used for IHC. All patients were male and ranged in
age (37-57 years). Of the 5 chordoma cases, 2 originated from the
sacrum and 3 from the skull base. Human experiments were approved
by the Ethics Review Committee of the Zhujiang Hospital of Southern
Medical University (approval no. 2020-KY-030-01), and written
consent was provided by patients. Sections were stained using
anti-TMED3 antibody (1:50; cat. no. ab151056; Abcam), an
UltraSensitive™ SP (Mouse/Rabbit) IHC kit (cat. no. KIT-9710; MXB
Biotechnologies) and hematoxylin. IHC analysis was conducted
according to the procedure mentioned above.
Human apoptosis antibody array
According to the manufacturer's protocols for a
Human Apoptosis Antibody Array kit (cat. no. ab134001; Abcam), 43
human apoptosis markers were detected in cells simultaneously.
Analysis was performed using ImageJ 1.5 software (National
Institutes of Health) to detect the signal density of each spot,
followed by normalization to the positive control spot signals.
Statistical analysis
All statistical analysis was performed using
GraphPad Prism 8 software (GraphPad Software, Inc.). Data are
presented as the mean ± SD. Data were analyzed using Student's
t-test when comparing two groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
TMED3 is highly expressed in chordoma and
downregulated by lentiviral infection
Chordoma tissues from patients were used in the
present study for preliminary analysis of TMED3 expression. All
chordoma specimens exhibited similar microscopic characteristics
(chordal-arranged, vacuolated and eosinophilic cells with intra-
and extracellular mucus). Moreover, postoperative analysis revealed
positive staining for cytokeratin, mucin-1, S-100 and vimentin;
Ki67 staining was 1-10% positive. IHC staining revealed that TMED3
was primarily distributed in the cytoplasm of cells in chordoma
tissues (Fig. S2). In U-CH1 and
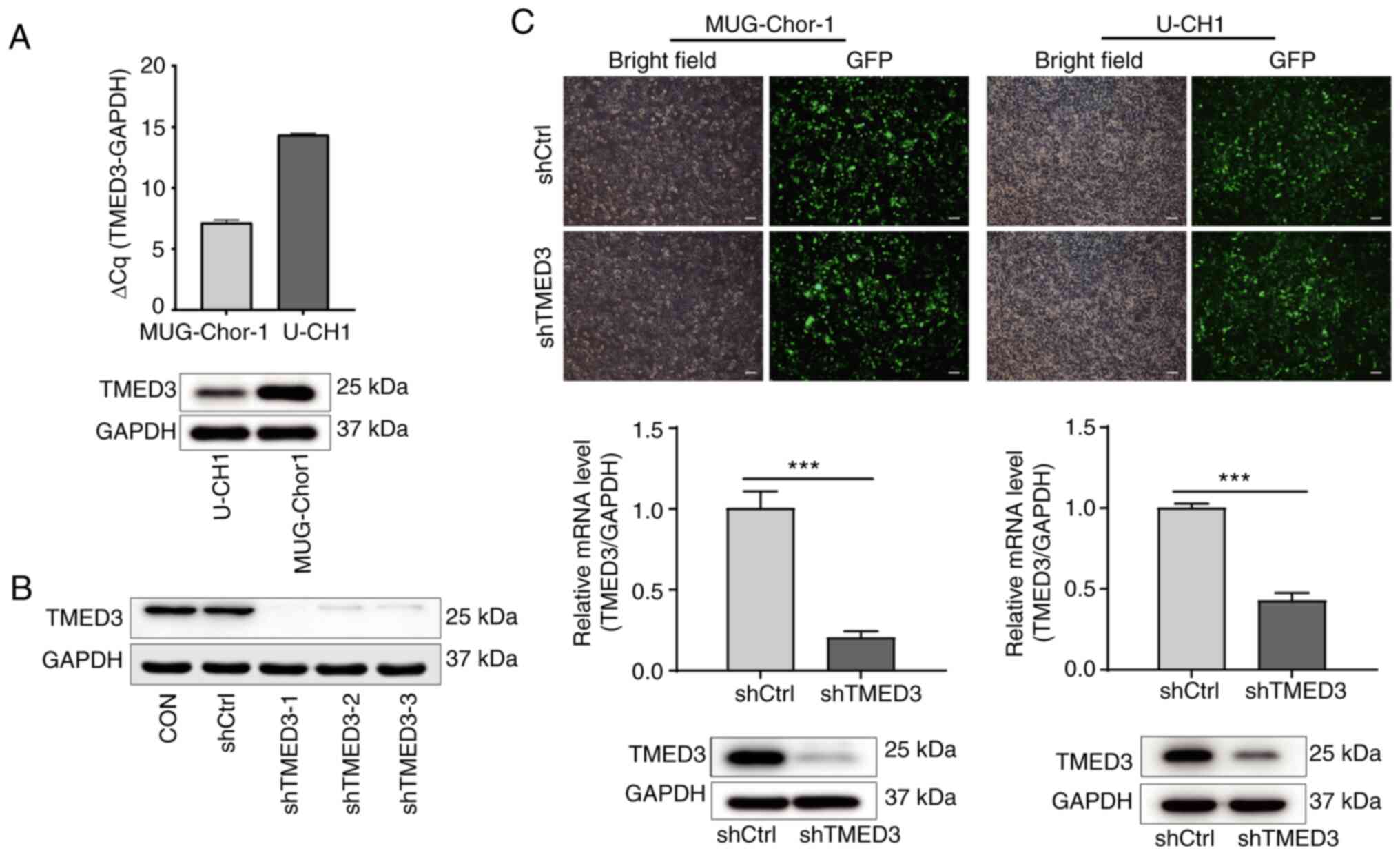
MUG-Chor1 chordoma cell lines, RT-qPCR analysis suggested that the
expression of TMED3 was high in MUG-Chor1 cells (ΔCq=7.21), while
there was moderate expression in U-CH1 cells (ΔCq=14.4; Fig. 1A). Western blot analysis revealed
that, compared with the shCtrl group, the protein levels of TMED3
in cells infected with shTMED3-1, shTMED3-2 and shTMED3-3 were
downregulated; as shTMED3-1 exhibited the strongest efficacy, this
was selected as the shRNA for subsequent experiments (Fig. 1B). After lentiviral transduction
for 72 h, the fluorescence of cells infected with LV-shCtrl or
LV-shTMED3 indicated >80% efficiency of infection. Additionally,
it was demonstrated via RT-qPCR and western blot analyses that
TMED3 was significantly downregulated in the shTMED3 group compared
with the shCtrl group (Fig. 1C).
Collectively, it was indicated that TMED3 was positively expressed
in MUG-Chor1 and U-CH1 chordoma cell lines, and that the lentiviral
knockdown was successful.
Knockdown of TMED3 inhibits chordoma cell
viability and migration
In order to obtain insight into the function of
TMED3 in chordoma cells in vitro, the effects of TMED3
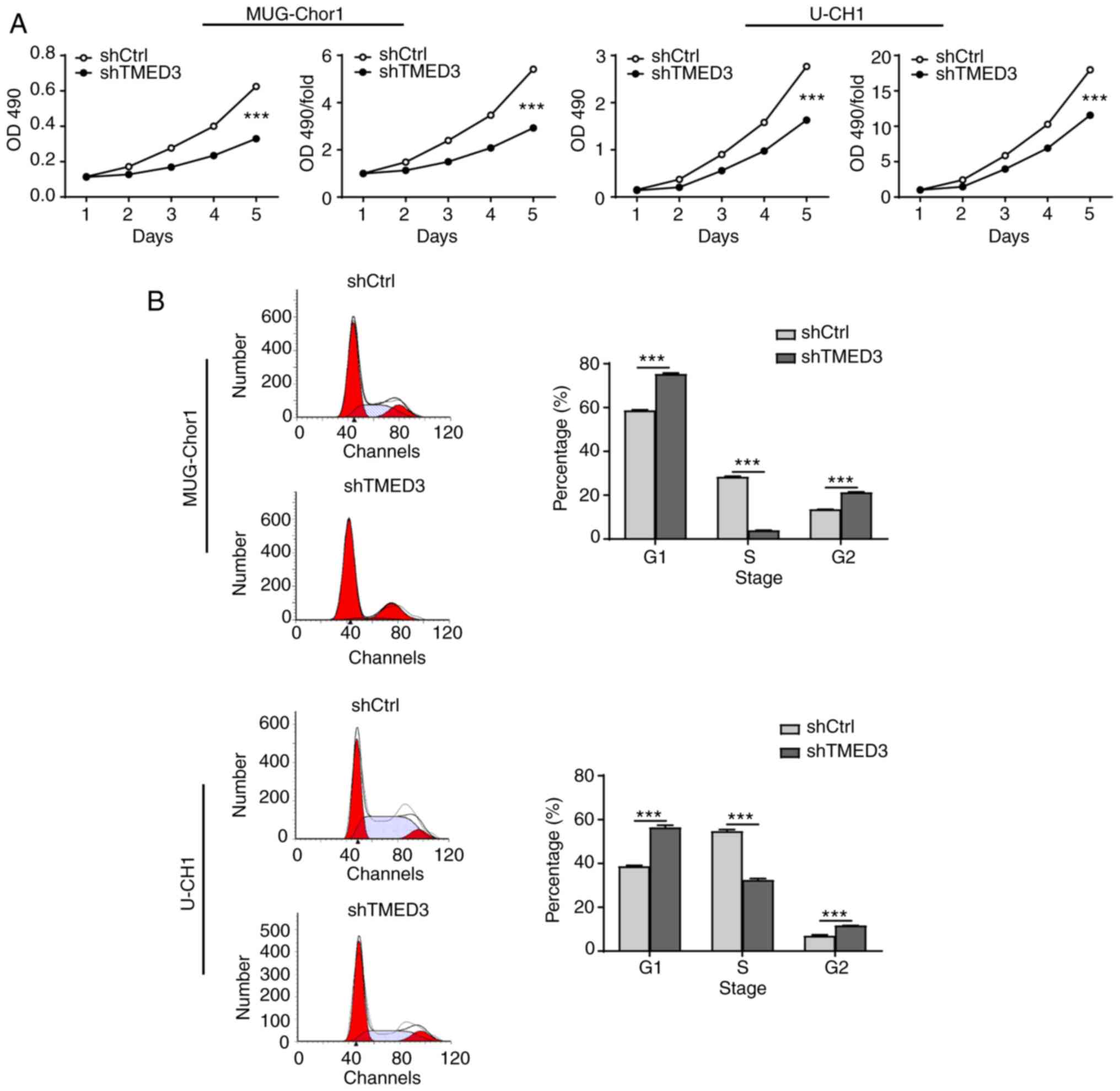
knockdown on cell viability and migration were detected. MTT assays
showed that, compared with the shCtrl group, cells in the shTMED3
group exhibited significantly reduced viability in the MUG-Chor1
[fold change (FC)=-1.85, P<0.001] and U-CH1 cell lines
(FC=-1.56, P<0.001; Fig. 2A).
Furthermore, FACS analysis was performed to investigate the cell
cycle in chordoma. Compared with shCtrl, the percentage of cells in
G1 and G2 phase was increased in the shTMED3 group, whereas the
percentage of cells in S phase decreased in the MUG-Chor1 (G1,
75.12±0.66 vs. 58.52±0.46, P<0.001; G2, 21.10±0.38 vs.
13.38±0.20, P<0.001; S, 3.78±0.28 vs. 28.14±0.40, P<0.001)
and U-CH1 cell lines (G1, 56.26±1.16 vs. 38.48±0.63, P<0.001;
G2, 11.47±0.24 vs. 7.01±0.33, P<0.001; S, 32.27±0.93 vs.
54.51±0.93, P<0.001; Fig. 2B).
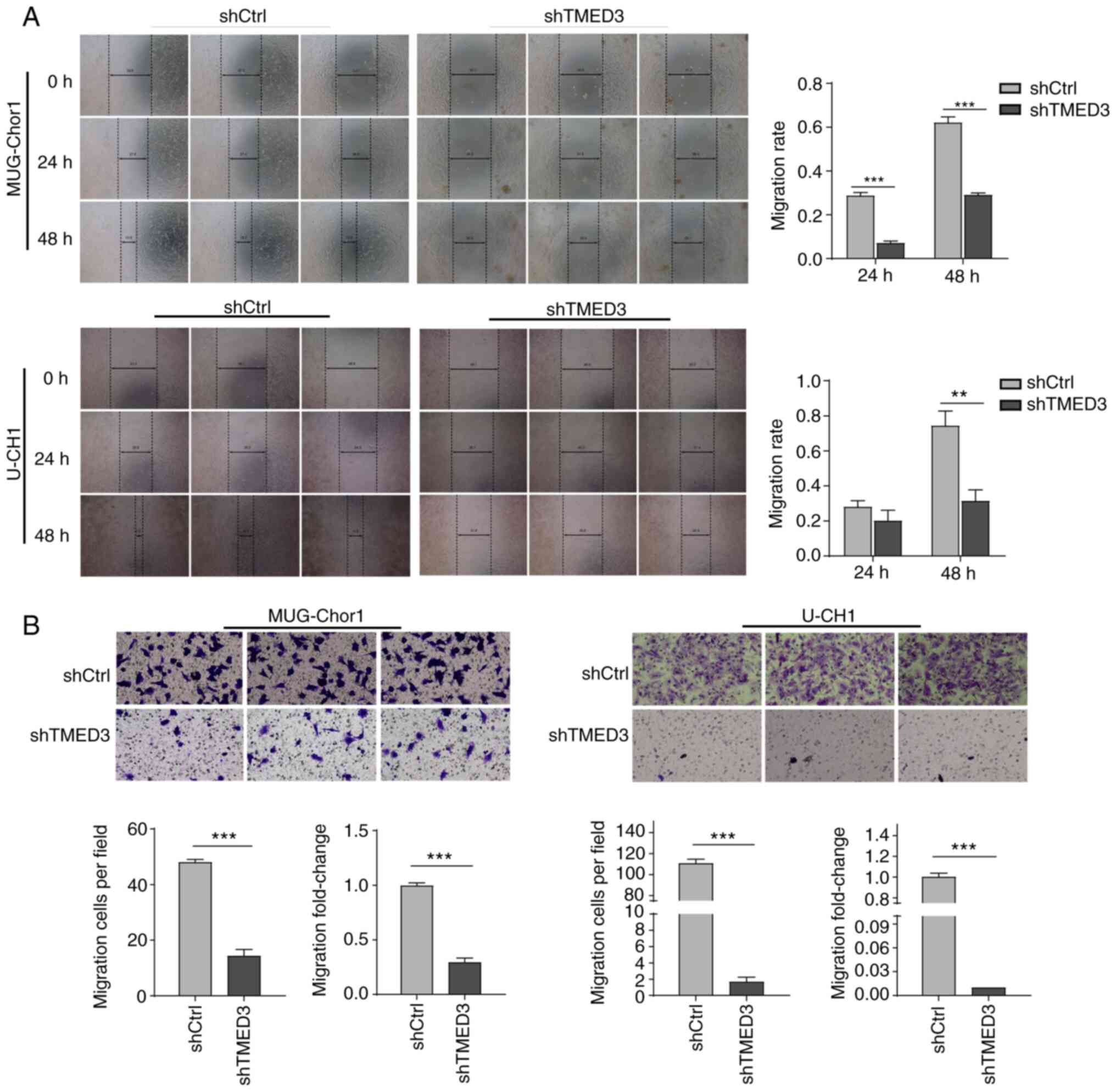
Moreover, wound healing assays revealed that the average cell
migration rate in the shTMED3 group compared with the shCtrl group
was decreased both in MUG-Chor1 (24 h, 0.07±0.01 vs. 0.29±0.02,
P<0.001; 48 h, 0.29±0.01 vs. 0.62±0.02, P<0.001) and U-CH1
cells (24 h, 0.20±0.06 vs. 0.28±0.03, P>0.05; 48 h, 0.31±0.06
vs. 0.74±0.08, P<0.01; Fig.
3A), indicating that cell migration was suppressed after
knockdown of TMED3. Similarly, Transwell assays revealed that the
migration of cells in the shTMED3 group compared with shCtrl group
was significantly inhibited by 71% in MUG-Chor1 cells (14±2.14 vs.
48±1.31, P<0.001) and 99% in U-CH1 cells (1±0.23 vs. 111±4.02,
P<0.001; Fig. 3B). The
aforementioned analysis suggested that TMED3 is tumor-associated,
and that downregulating TMED3 inhibited cell viability and
migration in chordoma.
Knockdown of TMED3 increases chordoma
cell apoptosis
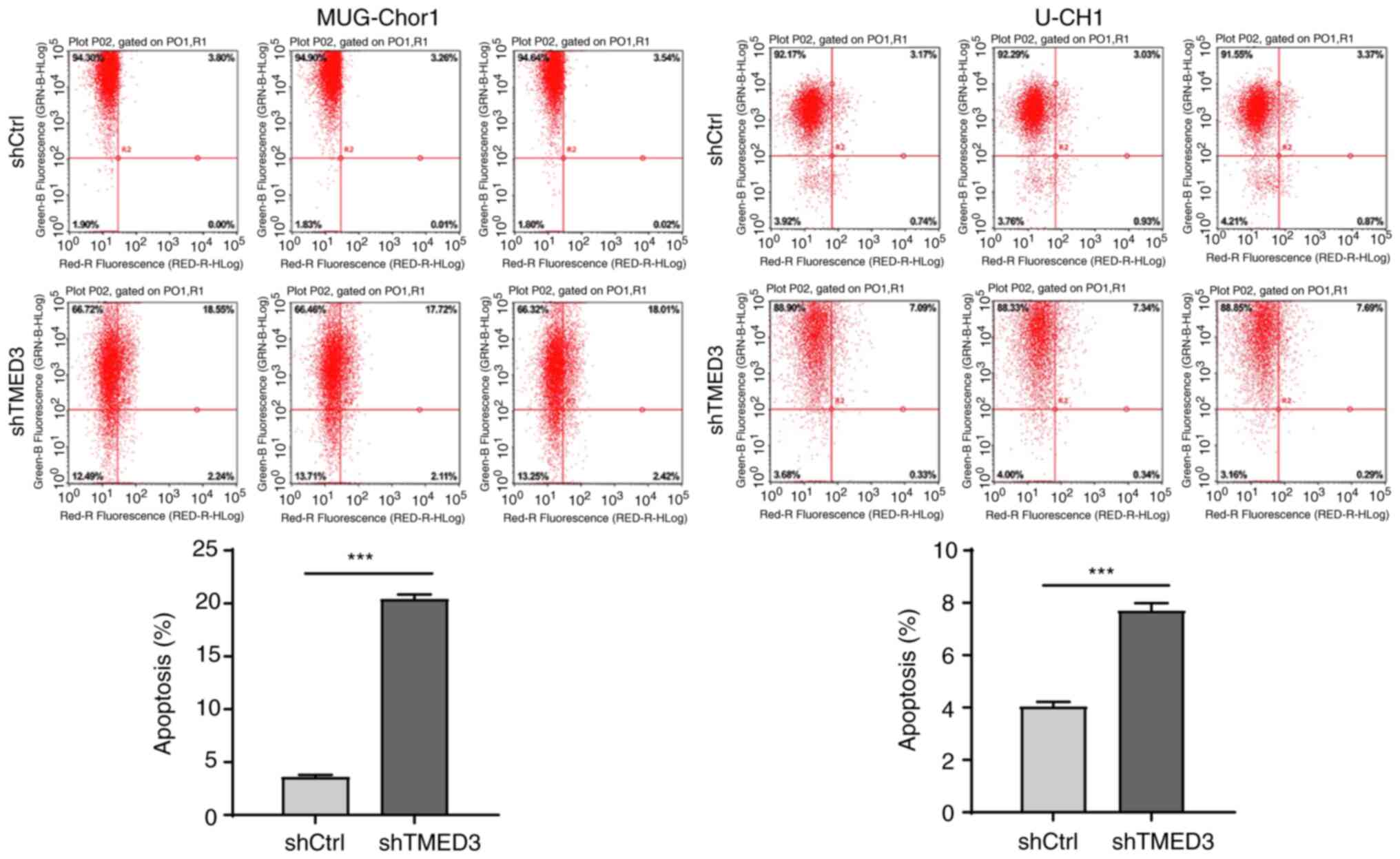
Next, in order to explore the effects of TMED3 on
chordoma cell apoptosis, Annexin V-APC was used to stain chordoma
cells; FACS analysis revealed that, compared with the shCtrl group,
the apoptosis rate was significantly increased in the shTMED3 group
in MUG-Chor1 (20.35±0.48% vs. 3.54±0.27%, P<0.001) and U-CH1
cell lines (7.69±0.29% vs. 4.04±0.18%, P<0.001; Fig. 4). These results indicated that
knockdown of TMED3 promoted chordoma cell apoptosis, and that TMED3
may be associated with chordoma progression via apoptosis signaling
pathways.
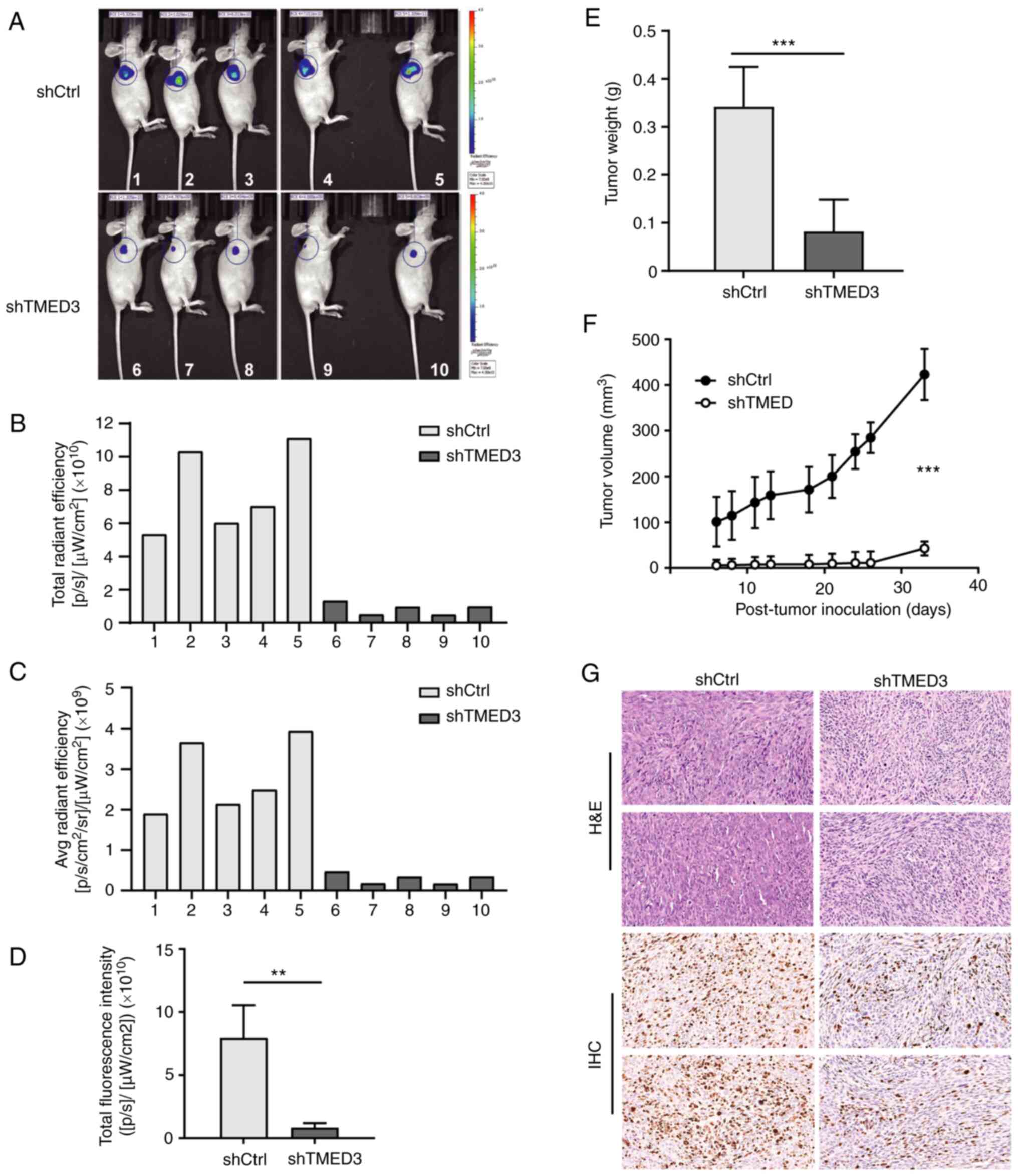
Knockdown of TMED3 inhibits chordoma
growth in vivo
To verify the role of TMED3 in chordoma in
vivo, a tumor xenograft model was established by injecting
shCtrl- or shTMED3-infected MUG-Chor1 chordoma cells subcutaneously
into nude mice, followed by measurements of tumor volume and
weight. Reductions in average tumor weight were observed in the
shTMED3 group compared with the shCtrl group (0.082±0.066 g vs.
0.342±0.083 g; P<0.001), and the average tumor volume in the
shTMED3 group was significantly smaller compared with the shCtrl
group (Fig. 5A-F). Furthermore,
IHC staining revealed that, compared with the shCtrl group, the
shTMED3 group exhibited downregulation of Ki67 expression, which is
considered to be a biomarker for tumor growth, further suggesting
that TMED3 may be an important contributor to chordoma growth
(Fig. 5G).
TMED3 regulates signaling pathways
involved in cell cycle, apoptosis and proliferation
To further explore the mechanisms underlying the
regulation of chordoma by TMED3, differentially expressed proteins
were screened using a human apoptosis marker array and western blot
analysis. The results showed that, compared with the shCtrl group,
the protein levels of Bcl-2, heat shock protein 27 (HSP27),
insulin-like growth factor (IGF)-I, IGF-II, IGF binding protein-2
(IGFBP-2) and Livin were significantly downregulated (reductions in
gray value of 42.63, 28.48, 23.87, 43.98, 25.81 and 28.91%,
respectively, P<0.05; Fig. 6A and
B) among 43 proteins involved in cell apoptosis. In addition,
western blot analysis revealed that knockdown of TMED3 resulted in
downregulation of Akt, p-Akt, CDK6 and cyclin D1, and upregulation
of MAPK9 (Fig. 6C). These results
indicated that TMED3 promoted chordoma progression by regulating
cell apoptosis-related proteins and Akt signaling (Fig. S3).
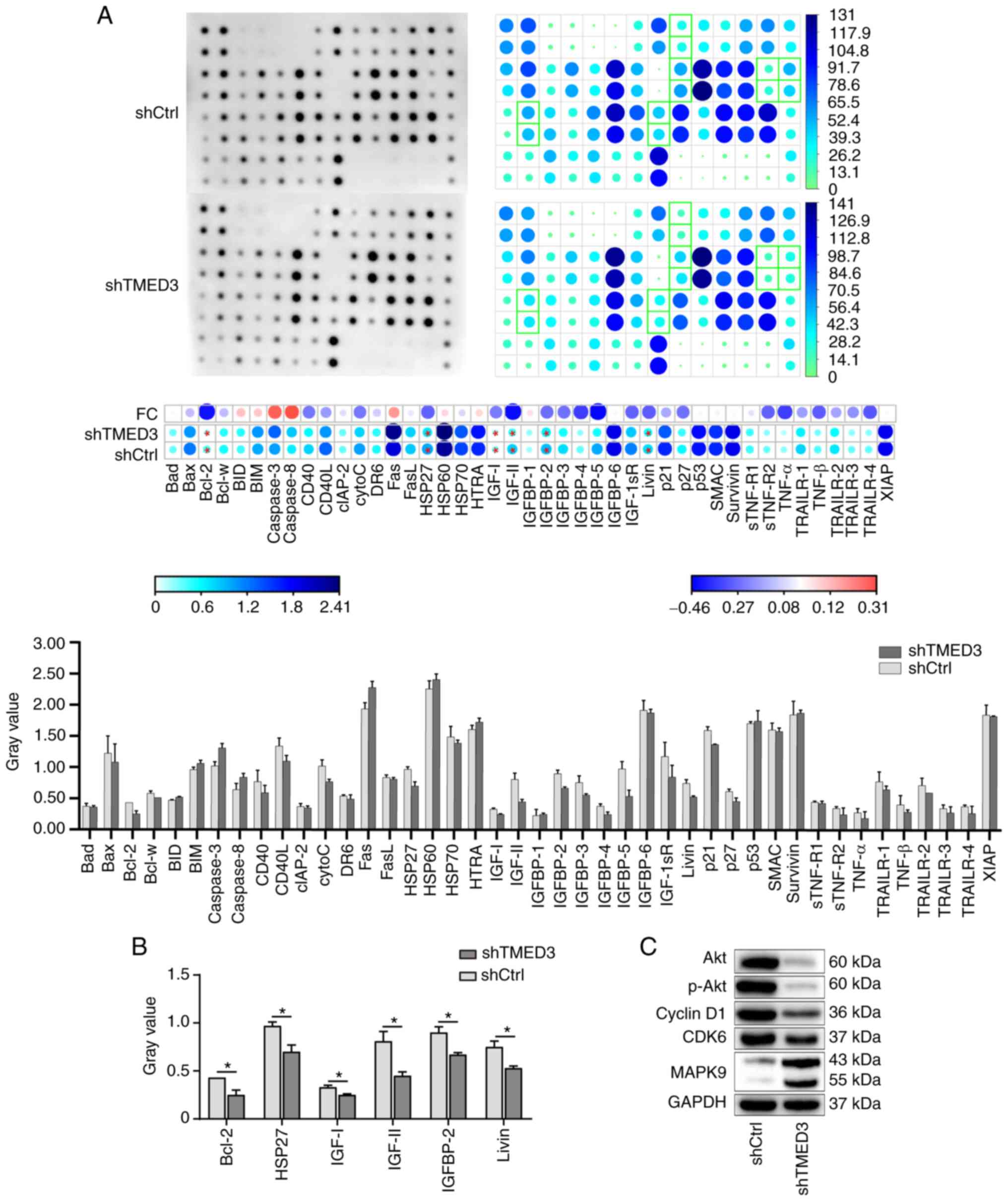 | Figure 6TMED3 regulates the expression of
genes involved in the cell cycle, apoptosis and proliferation. (A)
Human apoptosis antibody array was performed to identify the effect
of TMED3 on downstream genes associated with apoptosis. Proteins
encoded by 43 different genes were detected. Dark blue denotes high
relative expression and light green denotes low relative
expression. FC, red denotes significant upregulation and dark blue
denotes downregulation (P<0.05, FC >20% or <-20%). (B)
Proteins associated with apoptosis, including Bcl-2, HSP27, IGF-I,
IGF-II, IGFBP-2 and Livin, were detected in the human apoptosis
antibody array and showed significant differential expression.
*P<0.05. (C) Western blot analysis was performed to
determine the effect of TMED3 on the expression of proteins related
to cancer progression. TMED3, transmembrane Emp24 protein transport
domain containing 3; sh, short hairpin (RNA); shCtrl, negative
control shRNA; HSP27, heat shock protein 27; IGF, insulin-like
growth factor, IGFBP2, IGF binding protein 2; p, phosphorylated;
FC, fold change. |
Discussion
Chordoma is a rare bone cancer with limited
treatment options; currently, surgical operation is the first-line
therapy, but it is difficult to radically excise chordoma tumors
(5). Additionally, there is no
consensus concerning the effects of radiotherapy and chemotherapy
following surgical resection. Due to resistance to traditional
therapies, targeted therapy has been considered to be a more
effective way to overcome the challenges involved in chordoma
treatment. TMED3, a member of the p24 protein family that has been
shown to be critically involved in the transportation of secretory
cargo from the ER to the Golgi complex (9,10),
has been demonstrated to promote cancer progression in recent
years, including clear cell renal cell carcinoma, breast cancer and
gastric cancer (12-14).
The present study aimed to explore the role served
by TMED3 in chordoma. It was found that TMED3 protein was
distributed mainly in the cytoplasm of surgical specimens.
According to data in the Human Protein Atlas (23), TMED3 is located in the Golgi
apparatus (data not shown). Given that TMED3 plays a central role
in protein trafficking, TMED3 is predicted to be concentrated in
the ER and other cytoplasmic regions. In contrast to other cancer
types that exhibited positive staining in the nucleus, such as
colorectal, lung and breast cancers, IHC staining was weak or
negative in the nuclei of surgical chordoma specimens,
demonstrating why cancer specificity should be considered.
Additionally, the expression of TMED3 was moderately high in
chordoma cells. As TMED3 is one of the TMED family members that are
important regulators of protein transport, upregulation of TMED3
may promote anterograde and retrograde protein transport via
interactions with transmembrane and secreted proteins. It has been
reported that TMEDs are upregulated in various cancer types and
play different roles in cancer progression (24). As the function of TMED3 in
chordoma is unclear, genetic knockdown was performed in order to
gain insight into the function of TMED3. Following infection with
lentiviral vectors targeting TMED3, the viability, migration and
cell cycle progression of cells were significantly inhibited,
whereas apoptosis was enhanced; additionally, tumor growth in a
mouse xenograft model was also inhibited. Evidence obtained from
both in vivo and in vitro experiments pointed towards
TMED3 serving a role as a positive regulator in chordoma.
Consistent with these results, previous studies showed that TMED3
downregulation suppressed cell proliferation and migration in
breast cancer, gastric cancer, prostate cancer and hepatocellular
carcinoma, and that TMED3 expression is a potential biomarker of
poor prognosis in clear cell renal cell carcinoma (12-16). Conversely, Duquet et al
(18) showed that TMED3 knockdown
induced lung metastasis of HT29 colon cancer cells in vivo,
suggesting that TMED3 may serve as a suppressor of colon cancer
metastasis. Thus, the complex roles of TMED3 merit further
exploration.
In order to investigate the mechanisms underlying
the effects of TMED3 in chordoma, after TMED3 silencing, an
apoptosis array was used to detect the expression of genes involved
in human apoptosis pathways. Apoptosis is the process of programmed
cell death, and insufficient apoptosis contributes to the
progression of various cancer types (25). According to the protein array,
IGF-I, IGF-II and IGFBP-2, which are involved in apoptosis
signaling pathways (26), were
identified to be downregulated in MUG-Chor1 cells. IGF-I and
IGF-II, widely distributed in multiple tissues in humans, bind
their receptors with high affinity and subsequently activate a
cascade of downstream events, leading to cancer progression by
promoting cell proliferation and survival, and tumor growth and
metastasis (27). Furthermore,
IGFBP2, one of the six members of the IGFBP family that can react
with ligands either extracellularly or intracellularly to regulate
cell survival and viability, has been reported to serve as an
oncogene (26-28). It was reported that overexpression
or exogenous IGFBP-2 promoted cell proliferation in metastatic
cancer, including glioblastoma and ovarian, prostate and bladder
cancers, which was blocked by IGFBP-2 knockdown (29). The present study reported findings
consistent with previous studies, suggesting that TMED3 modulates
cancer-related genes to inhibit cell apoptosis, leading to chordoma
progression. Additionally, Bcl-2, Livin and HSP27, members of the
inhibitor of apoptosis protein family, were also observed to be
downregulated after knockdown of TMED3 in MUG-Chor1 cells, which
was consistent with previous studies and supported the idea that
targeting the expression of TMED3 results in apoptosis in chordoma
(30-32).
Furthermore, the expression of critical genes
associated with signal transduction was analyzed. Western blot
analysis revealed that Akt, p-Akt, CDK6 and cyclin D1 were
downregulated following TMED3 knockdown, whereas MAPK9 was
upregulated, suggesting at a potential relationship with TMED3.
Akt, also known as protein kinase B, is a serine-threonine protein
kinase that functions as an essential regulator in multiple
biological processes, such as metabolism, cell proliferation, cell
survival, metastasis and angiogenesis (33,34). It has been demonstrated that Akt
reacts with >100 substrates by modulating the phosphatase
activity of specific groups, and abnormal activation of Akt has
been widely observed in different cancer types including prostate,
gastric, pancreatic, ovarian and breast cancers (35). A previous study indicated that
both PI3K/Akt and RAS/MAPK pathways were activated in chordoma,
which was consistent with the present study (36-38).
Additionally, cyclin D1 and CDK6 are associated with
the cell cycle, and were downregulated in TMED3-silenced chordoma
cells in the present study (39).
PI3K and Akt mediate suppression of cyclin D1 threonine residue
phosphorylation, inhibiting subsequent ubiquitination to prevent
degradation via the RAS signaling pathway, leading to cell cycle
progression (39,40). In the present study, chordoma
cells infected with shTMED3 were arrested in G2 phase, indicating
that TMED3 may regulate the cell cycle via the PI3K/Akt pathway in
which CDK6 and cyclin D1 are involved. Conversely, compared with
chordoma cells in the shCtrl group, MAPK9 was found to be
upregulated in TMED3-silenced cells. MAPK9 (also known as JNK2) is
a member of the MAPK family that serves a key role in response to a
variety of signals, leading to regulation of multiple signaling
pathways in mammalian cells (41). The effects of MAPK9 in cancers
depend on the tissue type. MAPK9 was found to be abnormally
activated in multiple caner types, including glioma, lung
carcinoma, lymphoblastic leukemia and prostate carcinoma, which
indicated that MAPK9 critically contributes to cancer progression
and development (42).
Conversely, in certain cancers, it has reported that MAPK9 plays a
role as a negative regulator in cell proliferation via activation
of the JNK pathway (43,44). Fan et al (45), for example, showed that
specifically inhibiting JNK1/2 expression suppressed the
vinblastine-induced phosphorylation of the anti-apoptotic proteins
Bcl-2 and Bcl-XL, leading to inactivation of Bcl-2 and
Bcl-XL, which may explain why Bcl-2 protein was
significantly downregulated in shTMED3 cells in the present study.
Based on the findings of the present study, knockdown of TMED3 may
inhibit phosphorylation and thus lead to the inhibition of
biological processes that contribute to chordoma progression.
In conclusion, it was demonstrated that TMED3 was
highly expressed in chordoma, and that knockdown of TMED3 inhibited
chordoma progression both in vivo and in vitro by
suppressing cell proliferation and migration, and promoting
apoptosis. Furthermore, proteins associated with apoptosis
signaling pathways, including Bcl-2, HSP27, IGF-I, IGF-II, IGFBP-2
and Livin, were significantly downregulated. Additionally, TMED3
may regulate chordoma via PI3K/Akt or MAPK signaling pathways, as
Akt, CDK6, Cyclin D1 and MAPK9 exhibited altered expression
following TMED3 silencing. To further determine the potential
mechanisms of TMED3, there were plans to perform a bioinformatics
analysis using public databases; however, sufficient data was not
obtained from the database. The present study doesn't fully explain
the underlying upstream mechanisms involved in the effects of
TMED3, nor the interactions of the downstream proteins; these
issues will be the focus of future studies. Nevertheless, these
findings highlighted a potentially important role for TMED3 in
chordoma progression and indicated that TMED3 may be a potential
target for chordoma therapy.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JY, YZ and ZC made substantial contributions to the
study conception and design. JY, HH, DX and YD were involved in
performing the experiments and analyzing the data. HH, ZC and YZ
edited and revised the manuscript. All authors read and approved
the final manuscript. JY, HH, DX, YD, YZ and ZC confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
The animal study was conducted according to the
Guide for the Care and Use of Laboratory Animals of the National
Institutes of Health and approved by the Animal Ethics Committee of
Zhujiang Hospital of Southern Medical University (approval no.
IACUC-2019010). The human study was approved by the Ethics
Committee of Zhujiang Hospital of Southern Medical University
(approval no. 2020-KY-030-01). Written informed consent was
obtained from all patients enrolled in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
We gratefully thank Professor Qingling Zhang
(Zhujiang Hospital of Southern Medical University) for her academic
advice. Additionally, we thank Dr Chenyu Zhang (Shanghai
Biosciences Co., Ltd.) and Dr Shengjie Zhu (Fudan University) for
their help as technical consultants.
Funding
The present study was supported by the Spine Research Foundation
of Zhujiang Hospital (grant no. 20190102).
References
|
1
|
Sahyouni R, Goshtasbi K, Mahmoodi A and
Chen JW: A historical recount of chordoma. J Neurosurg Spine.
28:422–428. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thanindratarn P, Dean DC, Nelson SD,
Hornicek FJ and Duan Z: Advances in immune checkpoint inhibitors
for bone sarcoma therapy. J Bone Oncol. 15:1002212019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stacchiotti S and Sommer J; Chordoma
Global Consensus Group: Building a global consensus approach to
chordoma: A position paper from the medical and patient community.
Lancet Oncol. 16:e71–e83. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guan JY, He XF, Chen Y, Zeng QL, Mei QL
and Li YH: Percutaneous intratumoral injection with pingyangmycin
lipiodol emulsion for the treatment of recurrent sacrococcygeal
chordomas. J Vasc Interv Radiol. 22:1216–1220. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kayani B, Hanna SA, Sewell MD, Saifuddin
A, Molloy S and Briggs TW: A review of the surgical management of
sacral chordoma. Eur J Surg Oncol. 40:1412–1420. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang XR, Ng D, Alcorta DA, Liebsch NJ,
Sheridan E, Li S, Goldstein AM, Parry DM and Kelley MJ: T
(brachyury) gene duplication confers major susceptibility to
familial chordoma. Nat Genet. 41:1176–1178. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stacchiotti S, Tamborini E, Lo Vullo S,
Bozzi F, Messina A, Morosi C, Casale A, Crippa F, Conca E, Negri T,
et al: Phase II study on lapatinib in advanced EGFR-positive
chordoma. Ann Oncol. 24:1931–1936. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Magnaghi P, Salom B, Cozzi L, Amboldi N,
Ballinari D, Tamborini E, Gasparri F, Montagnoli A, Raddrizzani L,
Somaschini A, et al: Afatinib is a new therapeutic approach in
chordoma with a unique ability to target EGFR and brachyury. Mol
Cancer Ther. 17:603–613. 2018. View Article : Google Scholar
|
|
9
|
Strating JR and Martens GJ: The p24 family
and selective transport processes at the ER-Golgi interface. Biol
Cell. 101:495–509. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Barr FA, Preisinger C, Kopajtich R and
Korner R: Golgi matrix proteins interact with p24 cargo receptors
and aid their efficient retention in the Golgi apparatus. J Cell
Biol. 155:885–891. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Connolly DJ, O'Neill LA and McGettrick AF:
The GOLD domain-containing protein TMED1 is involved in
interleukin-33 signaling. J Biol Chem. 288:5616–5623. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Peng C, Huang K, Liu G, Li Y and Yu C:
MiR-876-3p regulates cisplatin resistance and stem cell-like
properties of gastric cancer cells by targeting TMED3. J
Gastroenterol Hepatol. 34:1711–1719. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pei J, Zhang J, Yang X, Wu Z, Sun C, Wang
Z and Wang B: TMED3 promotes cell proliferation and motility in
breast cancer and is negatively modulated by miR-188-3p. Cancer
Cell Int. 19:752019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ha M, Moon H, Choi D, Kang W, Kim JH, Lee
KJ, Park D, Kang CD, Oh SO, Han ME, et al: Prognostic role of TMED3
in clear cell renal cell carcinoma: A retrospective Multi-cohort
analysis. Front Genet. 10:3552019. View Article : Google Scholar :
|
|
15
|
Zheng H, Yang Y, Han J, Jiang WH, Chen C,
Wang MC, Gao R, Li S, Tian T, Wang J, et al: TMED3 promotes
hepatocellular carcinoma progression via IL-11/STAT3 signaling. Sci
Rep. 6:370702016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vainio P, Mpindi JP, Kohonen P, Fey V,
Mirtti T, Alanen KA, Perälä M, Kallioniemi O and Iljin K:
High-throughput transcriptomic and RNAi analysis identifies AIM1,
ERGIC1, TMED3 and TPX2 as potential drug targets in prostate
cancer. PLoS One. 7:e398012012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mishra S, Bernal C, Silvano M, Anand S and
Ruiz IAA: The protein secretion modulator TMED9 drives
CNIH4/TGFalpha/GLI signaling opposing TMED3-WNT-TCF to promote
colon cancer metastases. Oncogene. 38:5817–5837. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Duquet A, Melotti A, Mishra S, Malerba M,
Seth C, Conod A and Altaba AR: A novel genome-wide in vivo screen
for metastatic suppressors in human colon cancer identifies the
positive WNT-TCF pathway modulators TMED3 and SOX12. EMBO Mol Med.
6:882–901. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pfaffl MW: A new mathematical model for
relative quantification in real-time RT-PCR. Nucleic Acids Res.
29:e452001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Laferriere CA and Pang DS: Review of
intraperitoneal injection of sodium pentobarbital as a method of
euthanasia in laboratory rodents. J Am Assoc Lab Anim Sci.
59:254–263. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
National Research Council (US) Committee
for the Update of the Guide for the Care and se of Laboratory
Animals: Guide for the Care and Use of Laboratory Animals. 8th
edition. National Academies Press (US); Washington, DC: 2011
|
|
23
|
Thul PJ, Akesson L, Wiking M, Mahdessian
D, Geladaki A, Blal HA, Alm T, Asplund A, Björk L, Breckels LM, et
al: A subcellular map of the human proteome. Science.
356:eaal33212017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Aber R, Chan W, Mugisha S and
Jerome-Majewska LA: Transmembrane emp24 domain proteins in
development and disease. Genet Res (Camb). 101:e142019. View Article : Google Scholar
|
|
25
|
Wong RS: Apoptosis in cancer: From
pathogenesis to treatment. J Exp Clin Cancer Res. 30:872011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brahmkhatri VP, Prasanna C and Atreya HS:
Insulin-like growth factor system in cancer: Novel targeted
therapies. Biomed Res Int. 2015:5380192015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Simpson A, Petnga W, Macaulay VM,
Weyer-Czernilofsky U and Bogenrieder T: Insulin-like growth factor
(IGF) pathway targeting in cancer: Role of the IGF axis and
opportunities for future combination studies. Target Oncol.
12:571–597. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bach LA: IGF-binding proteins. J Mol
Endocrinol. 61:T11–T28. 2018. View Article : Google Scholar
|
|
29
|
Baxter RC: IGF binding proteins in cancer:
Mechanistic and clinical insights. Nat Rev Cancer. 14:329–341.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kasof GM and Gomes BC: Livin, a novel
inhibitor of apoptosis protein family member. J Biol Chem.
276:3238–3246. 2001. View Article : Google Scholar
|
|
31
|
Garrido C, Schmitt E, Cande C, Vahsen N,
Parcellier A and Kroemer G: HSP27 and HSP70: Potentially oncogenic
apoptosis inhibitors. Cell Cycle. 2:579–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Deveraux QL, Schendel SL and Reed JC:
Antiapoptotic proteins. The bcl-2 and inhibitor of apoptosis
protein families. Cardiol Clin. 19:57–74. 2001. View Article : Google Scholar
|
|
33
|
Kandel ES and Hay N: The regulation and
activities of the multifunctional serine/threonine kinase Akt/PKB.
Exp Cell Res. 253:210–229. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fresno Vara JA, Casado E, de Castro J,
Cejas P, Belda-Iniesta C and Gonzalez-Baron M: PI3K/Akt signalling
pathway and cancer. Cancer Treat Rev. 30:193–204. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hoxhaj G and Manning BD: The PI3K-AKT
network at the interface of oncogenic signalling and cancer
metabolism. Nat Rev Cancer. 20:74–88. 2020. View Article : Google Scholar :
|
|
36
|
Tamborini E, Virdis E, Negri T, Orsenigo
M, Brich S, Conca E, Gronchi A, Stacchiotti S, Manenti G, Casali
PG, et al: Analysis of receptor tyrosine kinases (RTKs) and
downstream pathways in chordomas. Neuro Oncol. 12:776–789. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fry MJ: Phosphoinositide 3-kinase
signalling in breast cancer: How big a role might it play? Breast
Cancer Res. 3:304–312. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ippen FM, Grosch JK, Subramanian M, Kuter
BM, Liederer BM, Plise EG, Mora JL, Nayyar N, Schmidt SP,
Giobbie-Hurder A, et al: Targeting the PI3K/Akt/mTOR pathway with
the pan-Akt inhibitor GDC-0068 in PIK3CA-mutant breast cancer brain
metastases. Neuro Oncol. 21:1401–1411. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sherr CJ, Beach D and Shapiro GI:
Targeting CDK4 and CDK6: From discovery to therapy. Cancer Discov.
6:353–367. 2016. View Article : Google Scholar :
|
|
40
|
Diehl JA, Zindy F and Sherr CJ: Inhibition
of cyclin D1 phosphorylation on threonine-286 prevents its rapid
degradation via the ubiquitin-proteasome pathway. Genes Dev.
11:957–972. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yoon CH, Kim MJ, Kim RK, Lim EJ, Choi KS,
An S, Hwang SG, Kang SG, Suh Y, Park MJ and Lee SJ: c-Jun
N-terminal kinase has a pivotal role in the maintenance of
self-renewal and tumorigenicity in glioma stem-like cells.
Oncogene. 31:4655–4666. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bubici C and Papa S: JNK signalling in
cancer: In need of new, smarter therapeutic targets. Br J
Pharmacol. 171:24–37. 2014. View Article : Google Scholar :
|
|
43
|
Wu Q, Wu W, Fu B, Shi L, Wang X and Kuca
K: JNK signaling in cancer cell survival. Med Res Rev.
39:2082–2104. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sabapathy K and Wagner EF: JNK2: A
negative regulator of cellular proliferation. Cell Cycle.
3:1520–1523. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fan M, Goodwin M, Vu T, Brantley-Finley C,
Gaarde WA and Chambers TC: Vinblastine-induced phosphorylation of
Bcl-2 and Bcl-XL is mediated by JNK and occurs in parallel with
inactivation of the Raf-1/MEK/ERK cascade. J Biol Chem.
275:29980–29985. 2000. View Article : Google Scholar : PubMed/NCBI
|