Introduction
Kidins220 (kinase D-interacting substrate of 220
kDa) is a novel scaffolding protein with an important role acting
as the downstream substrate of Trks, which are neurotrophin
receptors (1). Kidins220
regulates neuronal differentiation, survival, and cytoskeleton
remodeling, by interacting with a variety of binding partners
(2). Kidins220 is a transmembrane
protein with 1715 amino acids. It elicits its function as a
platform by binding/interacting with different molecules through
the function domain/motifs at either N- or C-terminals as both
terminals face intracellularly. It has 11 ankyrin-repeats at the
N-terminal, while the C-terminal comprises PSD-95, Dlg, ZO-1
(PDZ)-binding motif, kinase light chain interacting motif (KIM), a
sterile α motif (SAM) and a proline-rich domain. There are juxta
membrane Walker A/B motifs located at both terminals (3). Kidins220 acts as a platform to
coordinate signal transduction, cytoskeleton arrangement, molecule
transport and cellular functions via these intracellular domains at
both N- and C-terminals (3). In
addition to this, it has been implicated in malignancies. For
example, Kidins220 contributes to melanoma progression by sustained
MAPK signalling and inhibiting stress-induced apoptosis (4). In neuroblastoma, Kidins220
stabilizes NGF-induced survival signaling and is associated with
morphological transition of cells from N- to S-type (5). Moreover, Kidins220 is a direct
target gene of miR-4638-5p, a microRNA with decreased expression in
castration-resistant prostate cancer (CRPC). Previous findings
indicated that miR-4638-5p, through regulating Kidins220 and the
downstream activity of VEGF and PI3K/AKT signalling pathways,
influences prostate cancer progression via angiogenesis (6).
At present, the role played by Kidins220 in
pancreatic cancer and other intestinal malignancies remains
unknown. Our preliminary investigation of Kidins220 revealed an
altered expression of Kidins220 in pancreatic cancer which provoked
the current study of Kidins220 in that cancer type. The aim of the
present study was to examine the involvement of Kidins220 in the
disease progression of pancreatic cancer and how it affects
cellular functions of pancreatic cancer cells and corresponding
molecular mechanisms.
Materials and methods
Cell lines and cell culture
PANC-1 (RRID:CVCL_0480) and MIA PaCa-2
(RRID:CVCL_0428) cancer cell lines (ATCC) were cultured in
Dulbecco's modified Eagle's medium/nutrient F-12 Ham (DMEM-F12;
Sigma-Aldrich, UK) with 10% FBS and antibiotics. Cells were treated
with rhEGF (200 ng/ml), gefitinib (ZD1839; Selleck Chem), ERK
inhibitor (GDC-0994; Selleck Chem), AKT inhibitor (MK-2206; Selleck
Chem), MMP2 (cat. no. 2621; Tocris Bioscience), and MMPs broad
spectrum inhibitor (Marimastat, cat. no. 2631; Tocris Bioscience).
The cell lines used in the study were mycoplasma-free and they were
authenticated using STR profiling.
Generation of Kidins220 lentivirus shRNA
transgenes
Lentiviral shRNA (GGC CTG CAA GAT CCA ATT ATA)
targeting Kidins220 was obtained from Cyagen Biosciences. After
amplification and purification, plasmids containing lentiviral
shRNA or scramble shRNA (CCT AAG GTT AAG TCG CCC TCG), together
with lentiviral packaging plasmids (psPAX2) and envelope plasmid
(pMD2.G) were transfected into 293 cells respectively, to generate
lentiviral particles. The lentiviral particles carrying either
Kidins220 shRNA or scramble shRNA were then used to infect target
cells, respectively. The scramble shRNA was employed as a control
for the subsequent experiments. The stable PANC-1 and MIA PaCa-2
sublines and corresponding scramble control cells were maintained
in DMEM medium and supplemented with 100 µg/ml G418.
Immunohistochemistry for pancreatic
tissue microarray
Immunohistochemical staining was conducted on a
pancreatic adenocarcinoma tissue microarray (TMA) comprising 10
normal pancreatic tissues derived from autopsy, 21 adjacent normal
pancreatic tissues, 11 pancreatic inflammation, 10 benign tumors
(pancreatic islet cell tumour), 52 malignant tumors (42 pancreatic
duct adenocarcinoma, 3 pancreatic adenosquamous carcinoma, 1
pancreatic islet cell carcinoma and 6 pancreatic metastatic
carcinoma) (PA2081a, Biomax). The primary antibody used was an
anti-Kidins220 rabbit monoclonal antibody (SC-48738) at 1:50
concentration (Santa Cruz Biotechnology, UK). The secondary
antibody solution consisted of 100 µl biotinylated antibody
stock at 5 ml dilution (Vectastain Universal Elite ABC Kit,
PK-6200, Vector Laboratories). The presence of cancerous cells was
verified by a pathologist. Assessment of the staining was performed
by determining the intensity of Kidins220 staining using ImageJ
(https://imagej.nih.gov/ij/). Briefly,
the IHC intensity was determined in 10-20 cancerous cells by a
subtraction of background of empty area on the slide for each core
on the TMA. Average intensity was calculated for each sample from
the duplicate cores of each sample, followed by statistical
analyses.
Collection of clinical cohort
Clinical cohort includes pancreatic tumors (n=149)
together with paired adjacent background tissues (n=145), collected
immediately after surgery over a period from February, 2002 to
August, 2012. Samples were stored at -80°C until use. Informed
consent was signed by the patients at Peking University Cancer
Hospital. All protocol and procedures were approved by the Peking
University Cancer Hospital Research Ethics Committee. TNM staging
was evaluated by pathologists and clinicians according to the 7th
edition of TNM Classification of Malignant Tumours from the
International Union Against Cancer (UICC) (7).
RNA extraction cDNA synthesis and
RT-PCR
Total RNA was isolated from 1-3 million cancer cells
or pancreatic tissues (300-500 mg) using TRIzol Reagent®
(Sigma-Aldrich), and first-strand cDNA was then produced using the
GoScript™ Reverse Transcription System kit. The concentration and
purity of the resulting single-stranded RNA was quantified by
measuring its absorbance at a wavelength of 260 nm using a UV 1101
Biotech spectrophotometer (WPA). Reverse transcription was
performed to convert 500 ng of RNA into cDNA using the GoScript™
Reverse Transcription System kit (Promega Corporation). PCR was
performed in PCR reaction mix with initial denaturation at 94°C for
5 min, followed by 30-35 cycles of amplification at 95°C for 30
sec, 55°C for 30 sec and 72°C for 30 sec, with a final extension at
72°C for 5 min, while GAPDH was determined as a
house-keeping control.
Quantitative polymerase chain reaction
(qPCR)
qPCR for Kidins220, EGFR, NF-κB and GAPDH was
performed using the Ampliflour™ system (Intergen
Company®) with the following thermocycling conditions:
94°C for 10 min, 90 cycles of 94°C for 10 sec, 55°C for 35 sec, and
72°C for 20 sec. MMP1 transcripts and a housekeeping gene
(GAPDH) were determined using the SYBR-Green system and
change of MMP1 in folds was calculated using the 2−∆∆Ct
method (8). The primers used for
qPCR are listed in Table SI.
Western blot analysis
Proteins were extracted using lysis buffer and then
quantified using the Bio-Rad DC Protein Assay kit (Bio-Rad
Laboratories, UK). Proteins were transferred onto PVDF membranes
after a separation in the 8 or 10% SDS-PAGE gel depending on the
molecular weight of target proteins, and subsequently blocked and
probed with primary antibody and a corresponding
peroxidise-conjugated secondary antibody. Antibody against actin
(Santa Cruz Biotechnology; sc-47778), Kidins220 (Santa Cruz
Biotechnology; sc-48738), AKT (Santa Cruz Biotechnology, sc-5298),
P-AKT1,2,3 (Santa Cruz, sc-81433), ERK (Santa Cruz Biotechnology,
sc-514302), P-ERK (Santa Cruz Biotechnology, sc-7383), EGFR (Santa
Cruz Biotechnology, sc-71034), p-Tyr antibody (PY99; Santa Cruz
Biotechnology, sc-7020), E-cadherin (Santa Cruz Biotechnology,
sc-1500), Snail (Santa Cruz Biotechnology, sc-166476), and MMP1
(Santa Cruz Biotechnology, sc-21731) were purchased from Santa Cruz
Biotechnology. The Snail (Abcam, ab167609) and NF-κB (Abcam,
ab16502) PCNA (Santa Cruz Biotechnology, sc-25280) antibodies were
purchased from Abcam (Cambridge, UK). The nuclear proteins were
prepared using a nuclear isolation buffer (1.58 M sucrose, 40 mM
Tris-HCl pH 7.5, 20 mM MgCl2, 4% Triton X-100). The
protein bands were eventually visualised using a chemiluminescence
detection kit (Luminata). The process of immunoprecipitation
involves cell lysis, followed by incubation with a specific
antibody against target protein or proteins (PY99, an antibody
targeting proteins with phosphorylated tyrosine) presenting within
the tested protein samples. The resultant antigen-antibody
complexes are then precipitated using agarose beads conjugated with
staphylococcal protein A and protein G followed by SDS-PAGE (8 or
10%) and probing with antibodies.
In vitro cell growth assay
Cells (3,000) were seeded into 200 µl medium
in three 96-well plates, and cultured at 37°C for 1, 3 and 5 days,
respectively. Following incubation, the cells were fixed and
stained with crystal violet. The absorbance was measured after
dissolving the crystal violet with acetic acid (10% v/v) and the
absorbance was determined at a wavelength of 540 nm using a
spectrophotometer (Bio-Tek, Elx800).
In vitro tumor spheroid assay
Then, 1,000 cells were seeded into 200 µl
DMEM medium into 96-well non-coated U-shape bottom 3D culture plate
(Greiner Bio-One, Ltd.). The cells were cultured at 37°C for a
period up to 14 days. Images were captured every three days to
monitor tumor growth. Culture medium was topped up every two or
three days. Size of the spheroids was measured using ImageJ
software.
Colony formation assay
One hundred cells were plated into 6-well plates and
cultured for 14 days to allow colonies to form. Colonies were fixed
with 4% formaldehyde and stained with crystal violet. The colony
numbers were counted.
In vitro cell invasion assay
Transwell inserts (Greiner Bio-One Ltd.) with an 8.0
µm pore size were coated with 50 µg Matrigel and
placed into a 24-well plate. After air drying and rehydration for
the coating with Matrigel, 20,000 cells were seeded and incubated
for 72 h at 37°C. Cells that had invaded through the matrix and
attached to the bottom side of the insert were fixed with 4%
formalin and stained with 1% crystal violet at room temperature for
15 min. The invaded cells were measured by reading the
absorbance.
In vitro Transwell migration assay
A total of 20,000 cells were seeded into Transwell
inserts (pore size, 8 µm) in a 24-well plate. After 24-h
incubation, the cells that had migrated through and moved onto the
other side of the insert were fixed with 4% formaldehyde and
stained with 1% crystal violet at room temperature for 15 min. The
migrated cells were then measured by reading the absorbance.
In vitro cell migration assay (wound
healing assay)
EVOS® FL Auto Imaging System (Life
Technologies) equipped with EVOS® Onstage Incubator
(Life Technologies) were used for the in vitro cell
migration assay. Cells in 1 ml of normal medium were pre-seeded
with an appropriate density (400,000 cells/well) in a 24-well plate
and incubated until the formation of a monolayer on the next day.
The cell monolayer was wounded with a 200 µl pipette tip.
Closure of the wound was monitored for 16 h, and images were
captured at the same positions of the wound.
In vivo subcutaneous xenograft mouse
model
Athymic nude mice (CD1, female, 3-5 weeks, 18-22 g)
were purchased from Charles River Laboratories (Charles River
Laboratories International, Inc.). The mice were kept in sterile
cages equipped with filter, at 24°C with a humidity of 50%.
Sterilised food and water were provided. After the mice were
settled for a week in the designated laboratory, PANC-1 scramble
and Kidins220 knockdown cells were subcutaneously injected into the
nude mice at a total of 5 million cells in Matrigel (2.5 mg/ml in
PBS), two inoculations per mouse and six mice per group. The mice
were terminated after an inhalation of CO2 with a flow
rate of 20% chamber volume displaced per minute and tumors were
removed 4 weeks after the inoculation. Volume of the tumors was
calculated using the formula: Tumor volume (mm3)=0.5 ×
width2 × length. The xenograft experimental procedures
and maintenance were performed in accordance with the Animals Act
1986 (Scientific procedures) and approved by the UK Home Office
(PPL PE9445FC2). This xenograft model experiment and the following
peritoneal metastatic experiment were conducted over a period from
20 March to 30 April, 2018.
In vivo peritoneal metastasis assay
Female CD1 mice, aged 3-5 weeks (18-22 g) were
purchased from Charles River Laboratories. The mice were kept under
the routine conditions. PANC-1 scramble and Kidins220 knockdown
cells were injected into the peritoneal cavity of the mouse with 3
million cells in 100 µl of PBS per mouse. Mice were
carefully monitored twice a week by measuring body weight.
Peritoneal metastasis was examined after 4 weeks monitoring. The
mice were terminated with CO2 inhalation. Metastatic
nodules were photographed using a stereo-microscrope (Olympus) and
the volume of metastatic tumors were calculated using the formula:
Tumor volume (mm3)=0.5 × width2 × length. The
peritoneal metastatic model and maintenance of the mice were
carried out by complying with the regulations of the Animals Act
1986 (Scientific procedures) under the same project licence (PPL
PE9445FC2) approved by the UK Home Office.
Statistical analysis
Data were analysed as mean ± SEM. Following a
normality check, unpaired two sample t-test was employed for
normally distributed data while non-normally distributed data was
analysed using a Mann-Whitney test. One-way ANOVA (Bonferroni
t-test) was employed for statistical analysis of multiple groups.
Differences were considered statistically significant when
P<0.05. Correlation between the predicted miRNAs and Kidins220
in the TCGA PAAD cohort was determined using Spearman test.
Kaplan-Meier survival analysis with log rank pairwise comparison
and Spearman correlation tests were carried out using SPSS software
(SPSS Standard version 13.0; SPSS, Inc.).
Results
Reduced expression of Kidins220 in
pancreatic cancer and the clinical relevance
The expression of Kidins220 in pancreatic cancer was
first evaluated by determining the transcript levels of Kidins220
in a clinical cohort comprising pancreatic tumors (n=149) and the
paired adjacent normal pancreatic tissues (n=145) using qPCR.
Clinical and pathological information together with average
Kidins220 transcript levels are shown in the Table SII. Kidins220 transcript was
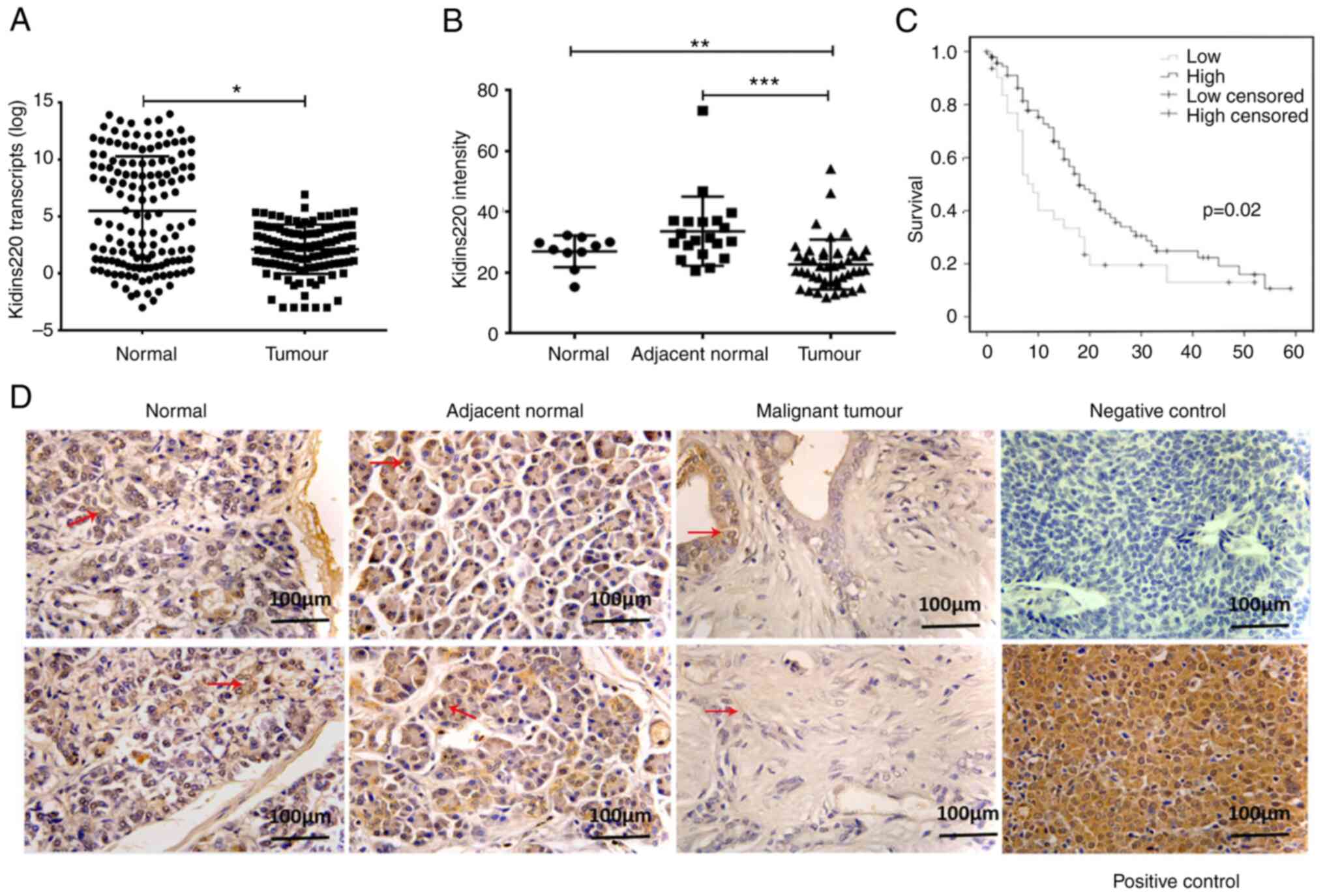
significantly reduced in pancreatic tumors in comparison with
adjacent normal tissues (P<0.05) (Fig. 1A). To examine the protein
expression of Kidins220, immunohistochemical staining was performed
on a pancreatic adenocarcinoma tissue microarray. Cytoplasmic
staining of Kidins220 was seen in both positive control (gastric
cancer tissue) and pancreatic epithelial cells in both normal and
adjacent normal tissues. Malignant tumours exhibited weaker
staining of Kidins220 in comparison with adjacent normal pancreatic
tissues (P<0.001) and normal pancreas (P<0.01) (Fig. 1B and D). Regarding patient
prognosis, Kaplan-Meier analysis of a separate clinical cohort from
a publicly available microarray database (GSE71729) showed that low
expression of Kidins220 in primary pancreatic tumors was associated
with poorer overall survival (Fig.
1C).
Kidins220 and tumorigenesis of pancreatic
cancer
In order to examine how Kidins220 may be involved in
pancreatic tumorigenesis, a comparative analysis of Kidins220
expression in benign lesions, non-invasive cancerous lesions, and
invasive adenocarcinomas was performed, using gene expression array
data (GDS3836) (9). This included
normal pancreatic tissues (n=7), intraductal papillary-mucinous
adenoma (IPMA, n=6), intraductal papillary-mucinous carcinoma
(IPMC, n=6) and invasive cancer originating in intraductal
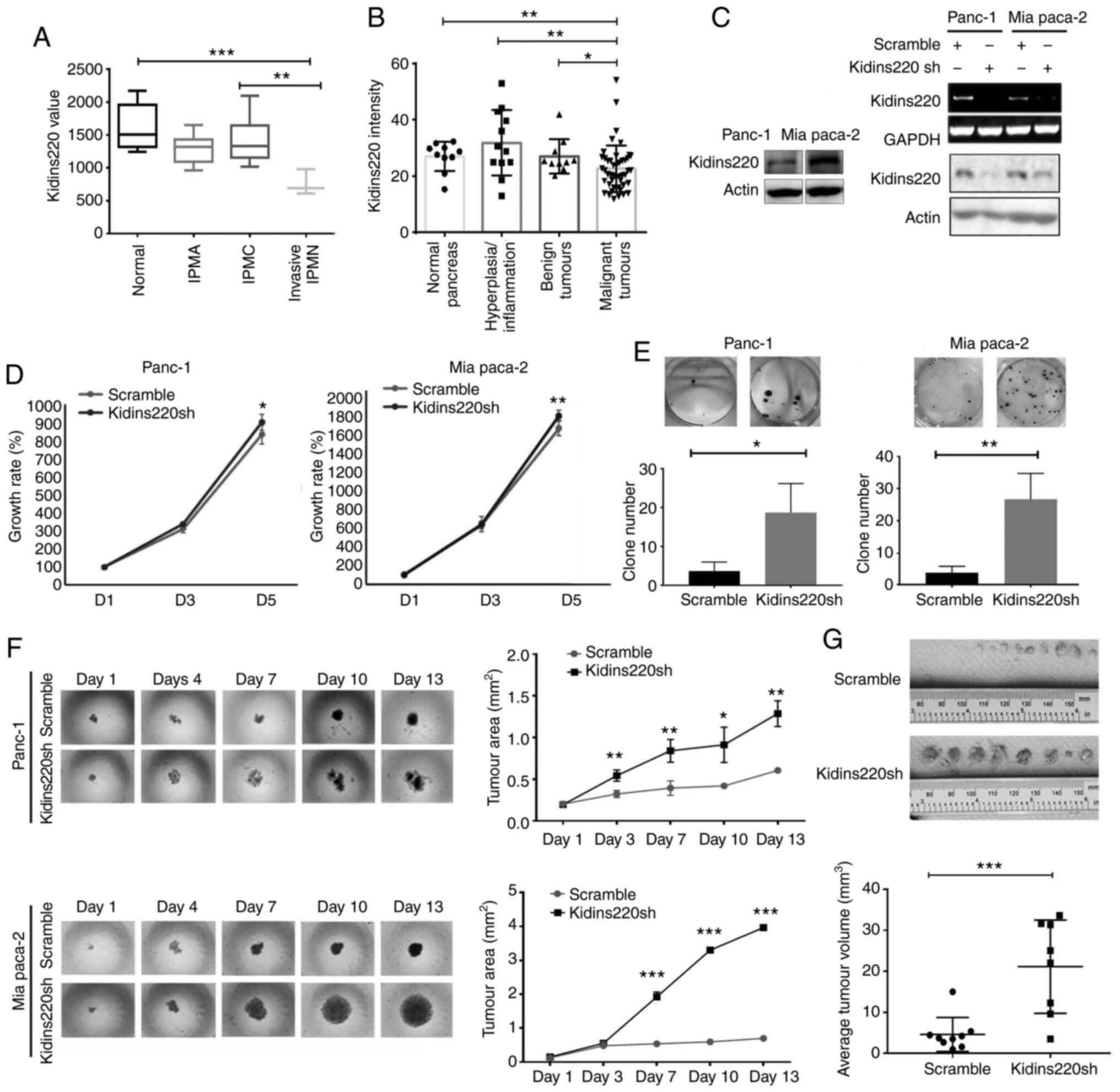
papillary-mucinous neoplasm (IPMN, n=3). As shown in Fig. 2A, a trend of reduced expression of
Kidins220 was observed in lesions which occurred during the
tumorigenesis of pancreatic cancer from IPMA, IPMC, and invasive
cancer originating in IPMN compared with normal pancreas. A
decreased Kidins220 expression was observed in the invasive cancers
compared with normal pancreatic tissues (P<0.001) and IPMC
(P<0.01). Furthermore, the semi-quantification of Kidins220 IHC
staining on the TMA showed that malignant tumors had the lowest
expression of Kidins220 compared with normal pancreas, pancreas
with hyperplasia, and benign tumors (Fig. 2B). In order to investigate the
role of Kidins220 in regulating cellular function, knockdown of
Kidins220 using lentiviral Kidins220 shRNA was conducted in PANC-1
and MIA PaCa-2 pancreatic cancer cell lines. The knockdown of
Kidins220 in both cell lines was then verified using both RT-PCR
and western blot analysis (Fig.
2C). De-regulated and uncontrolled cell proliferation is an
important trait of cancer cells. The impact of Kidins220 knockdown
on the proliferation of these two pancreatic cell lines was first
evaluated using the in vitro growth assay. The knockdown of
Kidins220 resulted in an increasing proliferation in the two cell
lines but to variable levels. A marginal increase of proliferation
was observed in the PANC-1 cells following the knockdown of
Kidins220 at Day 5 compared with the scramble control (P<0.05).
Similarly, in the MIA PaCa-2 cells, the cells with Kidins220
knockdown exhibited an increase of cell proliferation compared to
control (Fig. 2D). We also
performed colony formation assay and found that knockdown of
Kidins220 promoted the colony formation in both PANC-1 and MIA
PaCa-2 cell lines (Fig. 2E). In
the 3D spheroid model, tumor spheroids formed by
PANC-1Kidins220sh cells with knockdown of kidins220
presented bigger spheroids compared with the scramble control cells
(PANC-1scramble) at the fourth day. At the final stage,
the spheroids formed by Kidins220 knockdown cells became irregular
in comparison with the scramble cells (days 10-13). In a similar
manner, the single and suspended MIA PaCa-2 cancer cells started to
assemble themselves and form cell aggregates (day 1). The
difference of spheroid solidity was observed from Day 4. MIA
PaCa-2Kidins220sh cells also presented bigger spheroids
in comparison with the scramble control. From Day 7, the spheroids
became circular in the Kidins220 knockdown cells. At the end of the
2-week experiment, the Kidins220 knockdown MIA PaCa-2 cells grew
into much larger spheroids compared with the scramble control
(P<0.001) (Fig. 2F).
Tumorigenic capacity of the cells was also determined using a
murine xenograft model. As shown in Fig. 2G, knockdown of Kidins220 promoted
tumour growth of Panc1 cells in vivo (Fig. 2G).
Kidins220 and disease progression of
pancreatic cancer
More advanced tumours with lymph node and/or distant
metastases (which includes stages TNM3 and TNM4) (n=19), exhibited
lower transcript levels of Kidins220 compared with early stage
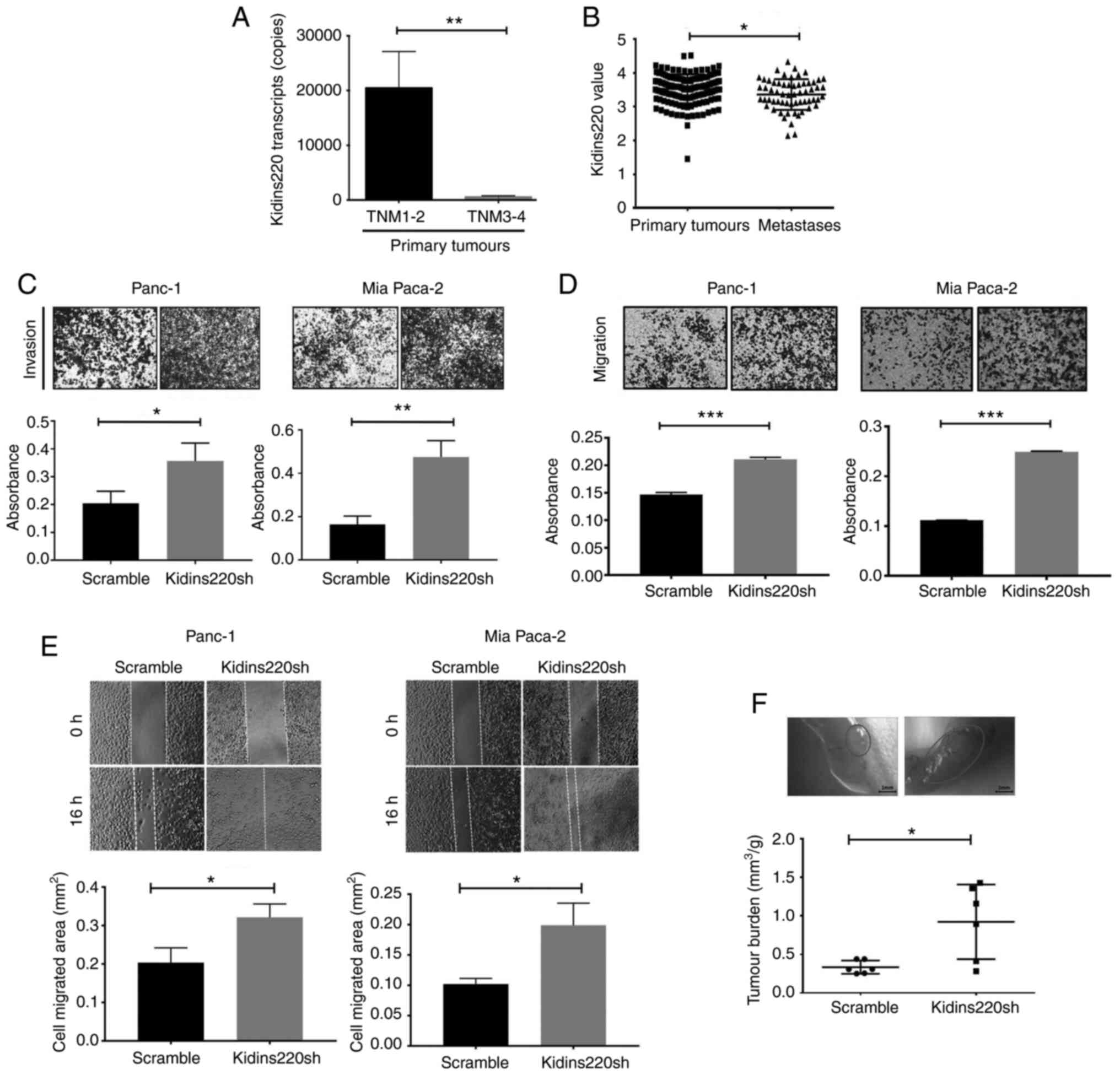
tumors (TNM1 and TNM2) (n=114) (P<0.01) (Fig. 3A). It indicates a connection
between reduced expression of Kidins220 and distant metastasis.
After a search of gene expression array databases, a dataset
comprising primary tumours (n=146) and distant metastases (n=62)
was chosen for a corresponding analysis. The distant metastasis
showed a decreased expression of Kidins220 compared with the
primary tumors (P<0.05) (GSE71729) (Fig. 3B). The IHC analysis revealed that
primary tumors with distant metastasis exhibited a lower expression
of Kidins220 protein compared to those without metastasis. Since
only two primary tumors with distant metastasis were available on
the TMA, statistical comparison did not show a significant
connection (data not shown). No obvious difference was identified
in the Kidins220 staining in the pancreatic metastatic samples from
liver, peritoneum, omentum and lymph node (data not shown) which
may be due to the limited number of samples available on the
TMA.
Knockdown of kidins220 resulted in a marked increase
of invasiveness in both PANC-1 and MIA PaCa-2 cells in comparison
with the controls (Fig. 3C).
Transwell migration assay showed that there was significantly
enhanced cell migration in PANC-1 and MIA PaCa-2 pancreatic cancer
cells as a result of the Kidins220 knockdown (Fig. 3D). This result was also in
accordance with a measurement of cell migration using wound
(scratch) assay in PANC-1 and MIA PaCa-2 cells using the wound
healing assay. Cell migration was monitored over a period of 16 h
following the wounding. It was shown that PANC-1 and MIA PaCa-2
cells with knockdown of Kidins220 migrated faster compared with the
scramble control cells (Fig. 3E).
Furthermore, PANC-1 pancreatic cancer cells (scramble and
shKidins220) were injected into the peritoneal cavity of 6 athymic
nude mice in each group. The mice were terminated after 4 weeks and
intraperitoneal exploration was conducted to detect the metastatic
tumors in the liver, stomach, pancreas, and duodenum to rectum.
Interestingly, tumor burden of the mice injected with Kidins220
knockdown PANC-1 cells was significantly increased in comparison
with scramble control (Fig. 3F).
Furthermore, there is a significant increase in average tumor
volume in the mice injected with PANC-1Kidins220sh cells
compared with the control group.
Kidins220 regulates invasion of
pancreatic cancer cells through MMP1
Our in vitro and in vivo experimental
data have shown that the knockdown of Kidins220 leads to a more
aggressive and invasive trait in pancreatic cell lines. Matrix
metalloproteinases (MMPs) are known for their role in modulating
the tumor microenvironment and enabling enhanced tumor cell
invasion. An analysis was performed for the expression profile of
MMPs in pancreatic cancer using publically available gene
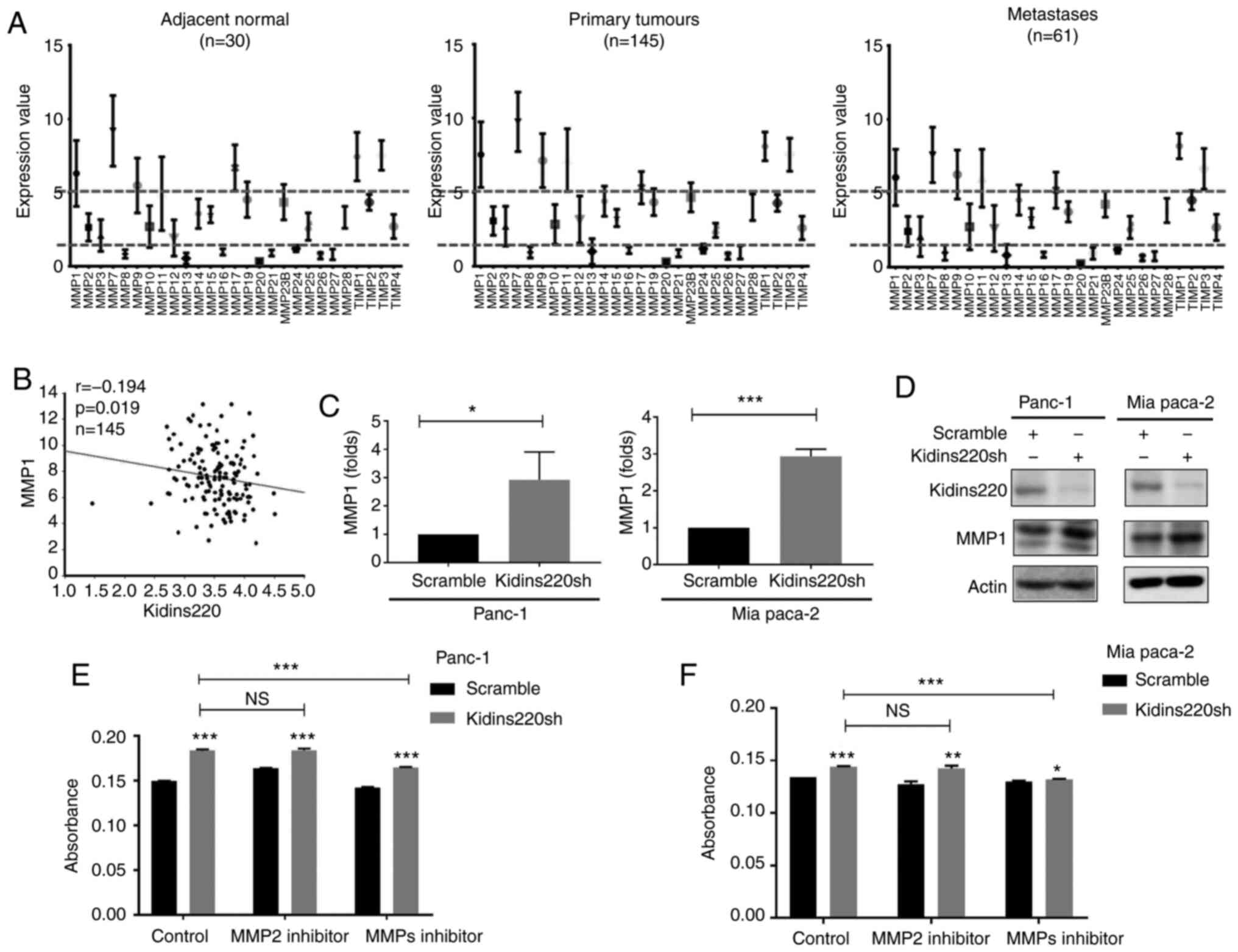
expression data (GSE71729). MMP1, MMP7, MMP9, MMP11 and MMP17 are
expressed at relatively higher levels than other MMPs in normal
pancreas which are further upregulated in primary tumors (Fig. 4A). The expression pattern of MMPs
appears to be similar from adjacent normal to primary tumors and
metastases. Spearman's correlation test revealed an inverse
correlation existing between Kidins220 and MMP1 (Fig. 4B). Subsequent quantification of
MMP1 transcripts in the Kidins220 knockdown cell lines showed an
increased expression of MMP1 (Fig.
4C). Increased protein expression of MMP1 was also identified
in these cells following the knockdown of Kidins220 (Fig. 4D). Targeting MMPs using small
inhibitors, no obvious impact on the invasiveness was observed when
the cell lines were treated with an MMP2 inhibitor. However, the
other MMP inhibitor with a concentration of 5 nM being specific to
MMP1, reduced the Kidins220 knockdown-promoted invasion in MIA
PaCa-2 cells and to a lesser extent also in the PANC-1 cells
(Fig. 4E and F).
Knockdown of Kidins220 promoted cell
migration through upregulation of EGFR/ERK/AKT signaling
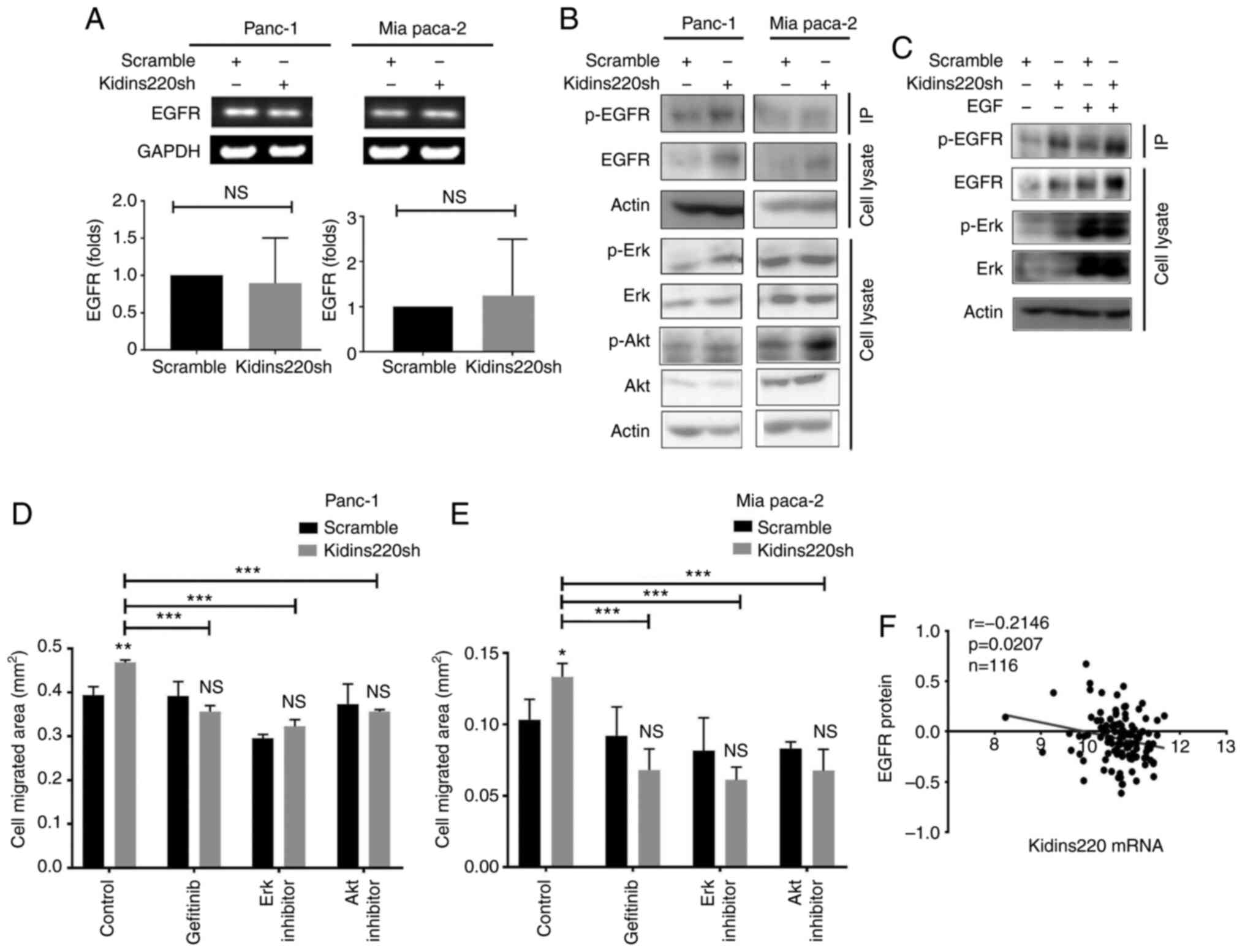
An increased protein level of EGFR was seen in both
PANC-1 and MIA PaCa-2 cancer cell lines following the knockdown of
Kidins220 without notable change of EGFR mRNA in those cell lines
(Fig. 5A and B). Increased p-EGFR
(Tyr) was also observed in the Kidins220 knockdown cells in
comparison with the scramble controls, suggesting that knockdown of
Kidins220 affected EGFR protein level and signalling. Corresponding
activation of downstream ERK and AKT were seen in both PANC-1 and
MIA PaCa-2 Kidins220 knockdown cell lines. Elevated expression and
activation of EGFR and ERK was observed in the cells exposed to
recombinant human EGF (Fig. 5C)
with further enhanced phosphorylation of EGFR and ERK seen in the
PANC-1kidins220sh cells compared to scramble control.
The involvement of the enhanced EGFR/ERK signalling was further
evaluated using small inhibitors targeting these molecules.
Blockage of EGFR using gefitinib reduced the Kindins220
knockdown-promoted migration in both PANC-1 and MIA PaCa-2 cells. A
esimilar effect was observed in the cells when they were treated
with small inhibitors targeting ERK and AKT (Fig. 5D and E). More interestingly, a
further analysis showed an inverse correlation between Kidins220
mRNA levels and EGFR protein expression in the pancreatic tumors
(Fig. 5F).
Loss of Kidins220 is accompanied with an
enhanced epithelial mesenchymal transition (EMT) through EGFR
To determine whether Kidins220 affects EGFR-induced
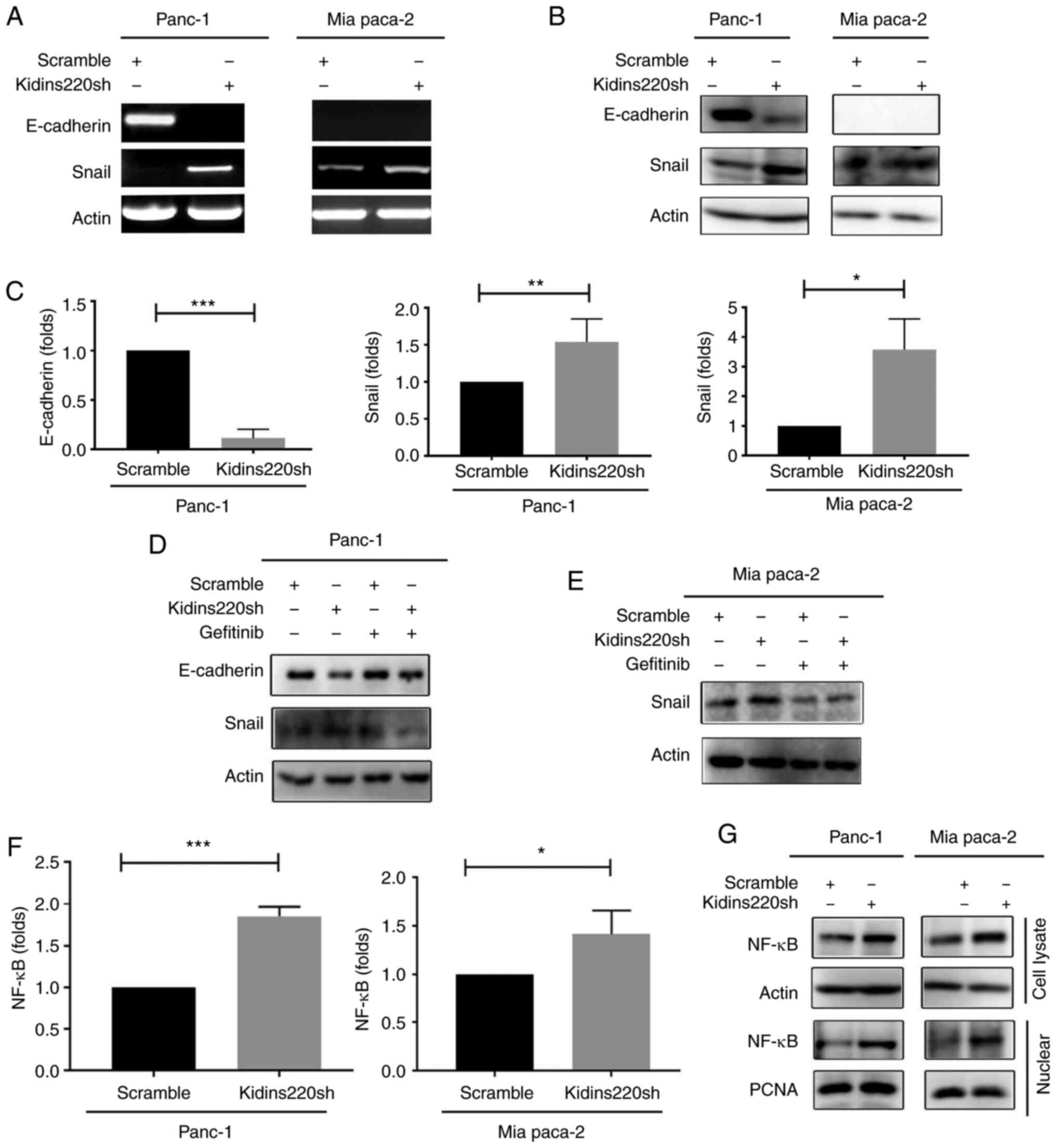
EMT in pancreatic cancer cells, the expression of EMT markers
E-cadherin and Snail were detected using RT-PCR. Downregulation of
E-cadherin and upregulation of Snail were observed in the
PANC-1kidins220sh cells. E-cadherin was undetectable in
the MIA PaCa-2 cells, but the expression of snail was increased in
the MIA PaCa-2Kidins220sh cells (Fig. 6A), and the corresponding qPCR
result for both cell lines is presented in Fig. 6C. Consistent changes were also
observed in the protein levels of these EMT markers in those cell
lines as a result of Kidins220 knockdown (Fig. 6B). Targeting EGFR using gefitinib
reversed the changes of E-cadherin and Snail in the
PANC-1Kidins220sh cells, while a similar impact was
observed for gefitinib in the MIA PaCa-2 cells on their expression
of Snail (Fig. 6D and E). Another
Kidins220 shRNA (CGAGTATTCAAGACTGAAGAT) exhibited similar impact on
the EGFR and EMT in PANC-1 cells following the knockdown of
Kidins220 (Fig. S1).
Constitutive activity of both EGFR and NFκB are
frequently observed in a variety of solid tumors as detailed in a
previous review (10). Given that
EGFR is known to be involved in the activation of NFκB (and the
subsequent potential cellular effects in solid tumours) we
determined the expression of NFκB in the pancreatic cancer cell
lines with Kidins220 knockdown. Elevated levels of both NFκB
transcripts and protein were seen in both
PANC-1Kidins220sh and MIA PaCa-2kidins220sh
cell lines compared to control. Corresponding increased nuclear
NFκB was also seen in these cell lines (Fig. 6F and G).
Discussion
Kidins220 is overexpressed in both melanoma and
neuroblastoma, and associated with disease progression (4,11,12). NGF can promote invasiveness of
pancreatic cancer cells through upregulation of MMP2 (13). This is supported by a later study
of NGF in pancreatic cancer cell lines in which NGF promoted
proliferation and invasion of pancreatic cancer cells to various
levels being associated with their differential expression of TrKA
(14). BNDF and NT3 can promote
invasiveness of pancreatic cancer cells at a low concentration
while an inhibitory effect was evident at a higher concentration
(100 ng/ml) (15). Although
subsequent in vivo studies demonstrated a therapeutic
potential of targeting Trks with a tyrosine kinase inhibitor
CEP-701 for pancreatic cancer (16), little anticancer efficacy was
evident in the relevant clinical trials (17). A cocktail of neutralising
antibodies against NTs (NGF, BDNF, NT3 and NT4/5) exhibited an
inhibitory effect on the in vivo growth of prostate cancer
cells (PC-3) and pancreatic cancer cells (ASPC) (18). Trk receptors have been shown as
differentially expressed genes in pancreatic cancer upon
chemoradiotherapy, but no obvious effect was evident for targeting
Trks using an inhibitor (AstraZeneca 1332) in the in vivo
experiment (19). Findings of a
previous study suggest that TrkA-expressing neuroendocrine tumors
of stomach and pancreas may benefit from Trk target therapy
(20). The NGF/Trk pathway is
also involved in the stress-accelerated development of
KrasG12D driven PDAC, as indicated in a murine model
(G12D) (21). However, TrkB is
also considered a protective factor in anoikis for cancer cells
particularly during their spread (22) which is yet to be fully
investigated in the pancreatic cancer. Protein kinase D is a group
of serine/threonine protein kinases comprising of three isoforms,
PKD1, PKD2 and PKD3 in mammals. In comparison with PKD2 and PKD3,
PKD1 is more actively engaged with the tumorigenesis associated
with TGFα-induced acinar-to-ductal metaplasia (ADM) and Kras
mutation (23). PKD1 is involved
in the regulation of proliferation, survival and invasion of
pancreatic cancer cells particularly when they lose their anchorage
(24). Moreover, PDK1 can promote
tumor-associated angiogenesis through upregulation of VEGF
(23). PKD2 can also enhance the
invasion of pancreatic cancer cells via the regulation of MMP7 and
MMP9 (25). PKD inhibitors have
been extensively tested for their anti-cancer potential in
vitro and in vivo (26-28) which are yet to be examined in
clinical trials. However, the development of PKD target therapy
encounters a great challenge due to the low bioavailability and
off-target effect. In contrast to the positive role played by the
NTs, Trks and PKDs, our study found a significantly reduced
expression of Kidins220 in pancreatic cancer, which is associated
with poorer overall survival. It suggests that Kidins220 as a
downstream substrate of these molecules plays a different role in
pancreatic cancers. This should be considered for the personalised
disease management when these molecules are targeted. At present,
little is known concerning the regulation of Kidins220 compared
with the understanding of its biological functions. Previous
findings suggest that reduced miR-4638-5p led to an increased
expression of Kidins220 in prostate cancer (6). Seven miRs were predicted be able to
target Kidins220 using the miRTarbase at the Enrichr platform
(29). miR-4638-5p is the second
among the seven miRs according to the combined score (Table SIII). However, miR-4638-5p was
undetectable in the TCGA pancreatic cancer cohort. We analysed the
correlation between the predicted miRs and Kidins220 in the TCGA
cohort. Spearman tests showed that miR-16-5p was inversely
correlated with Kidins220 while a positive correlation revealed
between miR-7b-5p and Kidins220 (Table SIII). The possible regulation of
Kidins220 by these putative miRs is yet to be fully investigated by
examining their expression in pancreatic cancer and specificity of
targeting Kidins220.
The formation and metastasis of pancreatic cancer
undergoes a multistep process from pancreatic intraepithelial
neoplasia (PanIN) lesions to invasive carcinomas (30,31). Based on the analysis of a gene
expression array data (GSE71729), the reduction of Kidins220
expression in invasive cancer originating in IPMN appeared to be
significant compared with normal pancreas. Furthermore, according
to IHC staining, there was a significant decrease of Kidins220
expression in the malignant tumors compared with normal pancreas,
hyperplasia or inflammation of the pancreas, and the benign tumors.
Overexpression of Kidins220 was able to protect cells from
stress-induced apoptosis, while melanoma cells with Kidins220
knockdown had a decrease in anchorage-independent growth in soft
agar and an extended cell death following UVB-induced apoptosis
(4). Similar to the findings in
melanoma, a study of Kidins220 in neuroblastoma also showed a
positive role played by this molecule in the regulation of cell
proliferation. Knockdown of Kidins220 in neuroblastoma cells
induced a decrease of proliferation through inhibition of the cell
cycle in which an arrest at G1 phase was observed. The inhibitory
effect on cell cycle was accompanied with decreased expression of
cyclin D1 and cyclin-dependent kinase 4 (CDK4) and inhibition of
hyperphosphorylated pRb to which an upregulation of p21 may
contribute (11). In the present
study, we found an enhanced tumorigenic capacity in the pancreatic
cancer cells following the knockdown of Kidins220 in an in
vitro 3D tumor spheroid experimental model although its
influence on the proliferation of pancreatic cancer cells appeared
to be much less in the 2D proliferation tests. Knockdown of
Kidins220 also increased the colony numbers of pancreatic cancer
cells and facilitated tumor growth in vivo. The impact on
in vivo tumor growth observed in the xenograft model was
more likely as a result from its regulation of both proliferation
and motility of the pancreatic cancer cells in which invasion
appeared to be predominately affected. This suggests that the
downregulation of Kidins220 may occur early during the
tumorigenesis of pancreatic cancer. Furthermore, the reduced
expression of Kidins220 in pancreatic cancer was associated with
shorter overall survival, suggesting Kidins220 as a potential
biomarker for the evaluation of prognosis of pancreatic cancer.
Kidins220 may also play an important role in
regulating the metastases of pancreatic cancer. The analysis of
Kidins220 transcript levels in the cohort of pancreatic cancer
tissue samples showed more advanced pancreatic tumors (TNM3 and
TNM4) had lower expression of Kidins220 compared with those of
early stages (TNM1 and TNM2). In melanoma, Kidins220 knockdown
reduced migratory and invasive abilities of melanoma cells in
vitro and in vivo (12). Our experiments showed that
knockdown of Kidins220 in pancreatic cell lines resulted in an
increase of cell migration and invasion. This is consistent with a
reduced expression of Kidins220 observed in more advanced diseases,
including both local invasion and spread to distant sites.
Furthermore, knockdown of Kidins220 also promoted peritoneal
metastasis of pancreatic cancer cells in a murine peritoneal
metastatic model. It suggests that the reduced expression of
Kidins220 in primary tumors conceives that pancreatic cancer cells
are a more invasive phenotype for local invasion, dissemination and
subsequent colonization at metastatic sites.
In pancreatic cancer, the expression pattern of EGF
and its receptor has been studied for several years. Overexpression
of EGFR has been indicated in pancreatic cancer and may be related
to disease progression and poor survival of pancreatic cancer
patients (32). Positive
co-expression of EGF and EGFR was significantly associated with the
poor prognosis of invasive ductal carcinoma of the pancreas.
Patients with a negative expression of EGF and its receptor also
had a 17.2 month median survival compared with the 9.7 month median
survival in patients with positive expression of EGF and EGFR
(33). In PDAC, EGFR expression
of 30.4 to 61.8% has been reported (33). EGFR expression was related to
increased invasiveness and poor prognosis. Park et al
identified increased EGFR expression, rising from PanIN to PDAC,
which indicated its potential role in the development of PDAC at an
early stage (34). These EGFR
aberrations contribute to an overactivation of pro-oncogenic
signalling pathways such as the RAS-RAF-MEK-ERK MAPK and
AKT-PI3K-mTOR pathways, which activate many cellular functions
required by cancer cells, including proliferation, migration and
invasion. The RAS-RAF-MEK-ERK MAPK pathway in particular may be the
most important pathway in mediating the biological response of EGFR
(35). These pathways have both
been implicated in the development of pancreatic cancer and are
also being evaluated as therapeutic targets (36,37).
In the present study, we found knockdown of
Kidins220 increased the phosphorylation of EGFR and total EGFR in
pancreatic cancer cell lines, without a notable change of Kidins220
transcripts. An inverse correlation between the Kidins220
transcripts and EGFR protein was also evident in the TCGA
pancreatic cancer cohort. It suggests that either a
post-transcriptional or post-translational regulation of EGFR
occurred in the pancreatic cancer cells when Kidins220 was knocked
down albeit the exact mechanism is yet to be investigated. Previous
findings showed that Kidins220 regulates the tumor formation of
melanoma through MEK/ERK signalling pathway (12). Downregulation of Kidins220
resulted in the attenuation of NGF-induced, but not BDNF-induced
MAPK signalling in neuroblastoma cells (38). Kidins220 was also involved in the
angiogenesis of castration-resistant prostate cancer through the
activity of VEGF and the PI3K/AKT pathway, which is regulated by
miR-4638-5p (6). In the present
study, corresponding changes of ERK and AKT were identified in the
Kidins220 knockdown cell lines, by which knockdown of Kidins220
increased phosphorylation of ERK and AKT. The altered expression
pattern of ERK was also further enhanced by treatment with EGF.
Furthermore, the involvement of EGFR/ERK and/or AKT in the
promotion of cell migration observed in Kidins220 knockdown cells
was elucidated using ERK, AKT, and EGFR small inhibitors. Moreover,
EGFR and Kras mutations may confer enhanced EGFR signalling and a
consequent challenge to EGFR target therapy (39,40). The role played by Kidins220 in the
EGFR and Kras mutation-related activation of EGFR signalling is yet
to be fully elucidated.
Cellular migration is an important part of the
multistep process required for cancer metastasis that also includes
proliferation, adhesion and invasion. Epithelial-mesenchymal
transition (EMT) and MMPs are two important factors in the
regulation of cancer cell migration and invasion. EMT enables
cancer cells to disseminate from a primary tumor to a distant site
and finally develop a secondary tumor (41). It occurs when tumor cells lose
their epithelial features such as loss of polarity, and gain
mesenchymal phenotype, acquiring the capability of motility and
invasion (42,43). E-cadherin is considered a
determinant molecule that maintains cell-cell adhesion and cell
polarity (43), as such
downregulation of E-cadherin is a critical event in EMT, found to
be caused by the overexpression of several different EMT-inducing
factors, such as Snail, a zinc-finger transcription repressor, and
transcriptional repressor of E-cadherin expression. In pancreatic
cancer cells, Snail exhibited a higher level of expression together
with a reduced expression of E-cadherin in poorly differentiated
cell lines compared with their expression in moderately
differentiated cell lines (44).
The present findings indicated that knockdown of Kidins220
decreased the expression of E-cadherin in PANC-1 cells and
increased the protein expression of Snail in PANC-1 and MIA PaCa-2
cells. When treating PANC-1 cells with gefitinib, the Kidins220
knockdown cells started to recover the expression of E-cadherin,
and the expression of Snail was completely inhibited with gefitinib
in PANC-1 cells. Our study also indicated an increased expression
of NF-κB at the transcription and protein level in Kidins220
knockdown pancreatic cells. Inhibition of the NF-κB pathway leads
to deregulation of epithelial-mesenchymal transition and neural
invasion in pancreatic cancer (45). However, its role in the regulation
of EMT underlying the expression of Kidins220 needs to be further
investigated.
The proteolytic activity of MMPs is required for a
cancer cell to degrade extracellular matrix during local expansion
and intravasation at nearby blood vessels, and additionally
extravasation and invasion at a distant location. High MMP1
expression is associated poor prognosis in patients with breast
cancer including disease-free and overall survival (20), but its role in pancreatic cancer
and any functional relation to Kidins220 is unknown.
Since knockdown of Kidins220 resulted in the
increased invasive capability of pancreatic cancer cells, we
examined whether MMPs, which are able to degrade the extracellular
matrix contributed to the observed tumor cell invasion (46). MMPs, particularly MMP-2 and, to a
lesser extent, MMP-9, modulate the pathogenesis of pancreatic
cancer (47,48). In the current study, we found a
positive correlation of Kidins220 and MMP1. Knockdown of Kidins220
promoted the transcript level of MMP1 in PANC-1 and MIA PaCa-2
cells. Moreover, an increased expression of MMP1 was also detected
at the protein level. In invasive melanoma, overexpression of MMP1
is regulated by the ERK signalling pathway (49). The increased expression of MMP1 in
MIA PaCa-2 Kidins220 knockdown cells can be inhibited when cells
were treated with ERK Inhibitor for 2 h. Interestingly, MMP2
inhibitor cannot prevent the increased cell invasion seen with
knockdown of Kidins220; however, an MMP broad spectrum inhibitor at
a specific MMP1-inhibiting concentration was able to reduce the
invasiveness of Kidins220 knockdown cells but to different levels.
Kidins220 knockdown-promoted invasion of MIA PaCa-2 appears to be
more dependent on the MMP1 while PANC1 appears to less responsive
to the inhibitor (Marimastat). It suggests that other MMPs or
pathways may be involved which is yet to be elucidated.
In summary, a reduced expression of Kidins220 was
observed in pancreatic cancer, and this reduced expression is
associated with disease progression, distant metastases and poor
prognosis. The reduced expression of Kidins220 in pancreatic cancer
cells is associated with enhanced tumorigenic and metastatic
traits, through upregulation of EGFR and MMP1, and promotion of
EMT. It suggests that Kidins220 is a putative marker for a more
effective and specific therapy in the personalised disease
management by targeting EGFR and its downstream signalling. This
requires further evaluation by employing both in vivo models
and a specifically designed clinical study.
There remain questions of further interest that
require additional studies for instance, the molecular mechanism
utilised by the Kidins220 to regulate the protein level of EGFR, in
which protein degradation cascades may be affected. More
importantly, the exact implication of the reduced expression of
Kidins220 in EGFR-positive pancreatic cancer should be elucidated
by considering other HER family members, K-Ras and EGFR mutation,
as this may have significant implications in selecting more
personalised therapeutic regimes for pancreatic cancer
patients.
Supplementary Data
Availability of data and materials
All data presented within the article are available
upon request from the corresponding author.
Authors' contributions
LY designed the study. SC, ZS, PHS, XG and LY
performed the in vitro experiments. SC, ZS, CL, FS and LY
conducted the in vivo experiments. XG, KJ, XT, JJ and CH
collected the pancreatic tissue samples and carried out RNA
isolation at the Beijing Cancer Hospital. PG and BA conducted the
evaluation of TMA and data analysis. SC, ZS, FS, BAS, SH, WGJ and
LY were involved in data analysis and preparation of the
manuscript. All authors read and approved the manuscript.
Ethics approval and consent to
participate
The pancreatic cancer tumors together with paired
adjacent normal pancreatic tissues were collected at the Peking
University Cancer Hospital by complying with the regulations and
approved procedures. All protocols and procedures were reviewed and
approved Peking University Cancer Hospital Research Ethics
Committee.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Acknowledgments
The authors would like to thank both Peking
University and Cardiff University for their kind support to this
collaborative research. We also extend thanks to Dr Andrew Sanders,
Ms. Catherine Zabkiewicz, Dr Jun Cai and Dr Lee Campbell for their
help on the proof reading, constructive comments and
discussions.
Funding
Not applicable.
References
|
1
|
Scholz-Starke J and Cesca F: Stepping out
of the shade: Control of neuronal activity by the scaffold protein
Kidins220/ARMS. Front Cell Neurosci. 10:682016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Neubrand VE, Cesca F, Benfenati F and
Schiavo G: Kidins220/ARMS as a functional mediator of multiple
receptor signalling pathways. J Cell Sci. 125:1845–1854. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cai S, Cai J, Jiang WG and Ye L: Kidins220
and tumour development: Insights into a complexity of cross-talk
among signalling pathways (review). Int J Mol Med. 40:965–971.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liao YH, Hsu SM and Huang PH: ARMS
depletion facilitates UV irradiation induced apoptotic cell death
in melanoma. Cancer Res. 67:11547–11556. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rogers DA and Schor NF: Kidins220/ARMS
depletion is associated with the neural-to Schwann-like transition
in a human neuroblastoma cell line model. Exp Cell Res.
319:660–669. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang Y, Shao N, Mao X, Zhu M, Fan W, Shen
Z, Xiao R, Wang C, Bao W, Xu X, et al: MiR-4638-5p inhibits
castration resistance of prostate cancer through repressing
Kidins220 expression and PI3K/AKT pathway activity. Oncotarget.
7:47444–47464. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sobin LH, Gospodarowicz MK and Wittekind
C: TNM classification of malignant tumours. 7th edition.
Wiley-Blackwell; Chichester, West Sussex: pp. 132–135. 2009
|
|
8
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
9
|
Hiraoka N, Yamazaki-Itoh R, Ino Y,
Mizuguchi Y, Yamada T, Hirohashi S and Kanai Y: CXCL17 and ICAM2
are associated with a potential anti-tumor immune response in early
intraepithelial stages of human pancreatic carcinogenesis.
Gastroenterology. 140:310–321. 2011. View Article : Google Scholar
|
|
10
|
Shostak K and Chariot A: EGFR and NF-κB:
Partners in cancer. Trends Mol Med. 21:385–393. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jung H, Shin JH, Park YS and Chang MS:
Ankyrin repeat-rich membrane spanning (ARMS)/Kidins220 scaffold
protein regulates neuroblastoma cell proliferation through p21. Mol
Cells. 37:881–887. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liao YH, Hsu SM, Yang HL, Tsai MS and
Huang PH: Upregulated ankyrin repeat-rich membrane spanning protein
contributes to tumour progression in cutaneous melanoma. Br J
Cancer. 104:982–988. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Okada Y, Eibl G, Guha S, Duffy JP, Reber
HA and Hines OJ: Nerve growth factor stimulates MMP-2 expression
and activity and increases invasion by human pancreatic cancer
cells. Clin Exp Metastasis. 21:285–292. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Diao DM, Song YC, Hou N, Xu HF, Wang JG
and Dang CX: Responses of pancreatic cancer cells to stimulations
by nerve growth factor and the role of Trk-A expression. Nan Fang
Yi Ke Da Xue Xue Bao. 32:296–300. 2012.In Chinese. PubMed/NCBI
|
|
15
|
Miknyoczki SJ, Lang D, Huang L,
Klein-Szanto AJ, Dionne CA and Ruggeri BA: Neurotrophins and Trk
receptors in human pancreatic ductal adenocarcinoma: Expression
patterns and effects on in vitro invasive behavior. Int J Cancer.
81:417–427. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Miknyoczki SJ, Chang H, Klein-Szanto A,
Dionne CA and Ruggeri BA: The Trk tyrosine kinase inhibitor CEP-701
(KT-5555) exhibits significant antitumor efficacy in preclinical
xenograft models of human pancreatic ductal adenocarcinoma. Clin
Cancer Res. 5:2205–2212. 1999.PubMed/NCBI
|
|
17
|
Shabbir M and Stuart R: Lestaurtinib, a
multitargeted tyrosine kinase inhibitor: From bench to bedside.
Expert Opin Investig Drugs. 19:427–436. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Miknyoczki SJ, Wan W, Chang H, Dobrzanski
P, Ruggeri BA, Dionne CA and Buchkovich K: The neurotrophin-trk
receptor axes are critical for the growth and progression of human
prostatic carcinoma and pancreatic ductal adenocarcinoma xenografts
in nude mice. Clin Cancer Res. 8:1924–1931. 2002.PubMed/NCBI
|
|
19
|
Johnson MD, Stone B, Thibodeau BJ,
Baschnagel AM, Galoforo S, Fortier LE, Ketelsen B, Ahmed S, Kelley
Z, Hana A, et al: The significance of Trk receptors in pancreatic
cancer. Tumour Biol. 39:10104283176922562017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Aristizabal Prada ET, Heinzle V, Knösel T,
Nölting S, Spöttl G, Maurer J, Spitzweg C, Angele M, Schmidt N,
Beuschlein F, et al: Tropomyosin receptor kinase: A novel target in
screened neuroendocrine tumors. Endocr Relat Cancer. 25:547–560.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Renz BW, Takahashi R, Tanaka T, Macchini
M, Hayakawa Y, Dantes Z, Maurer HC, Chen X, Jiang Z, Westphalen CB,
et al: β2 adrenergic-neurotrophin feedforward loop promotes
pancreatic cancer. Cancer Cell. 33:75–90.e7. 2018. View Article : Google Scholar
|
|
22
|
Desmet CJ and Peeper DS: The neurotrophic
receptor TrkB: A drug target in anti-cancer therapy? Cell Mol Life
Sci. 63:755–759. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liou GY, Döppler H, Braun UB, Panayiotou
R, Scotti Buzhardt M, Radisky DC, Crawford HC, Fields AP, Murray
NR, Wang QJ, et al: Protein kinase D1 drives pancreatic acinar cell
reprogramming and progression to intraepithelial neoplasia. Nat
Commun. 6:62002015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ochi N, Tanasanvimon S, Matsuo Y, Tong Z,
Sung B, Aggarwal BB, Sinnett-Smith J, Rozengurt E and Guha S:
Protein kinase D1 promotes anchorage-independent growth, invasion,
and angiogenesis by human pancreatic cancer cells. J Cell Physiol.
226:1074–1081. 2011. View Article : Google Scholar
|
|
25
|
Wille C, Köhler C, Armacki M, Jamali A,
Gössele U, Pfizenmaier K, Seufferlein T and Eiseler T: Protein
kinase D2 induces invasion of pancreatic cancer cells by regulating
matrix metalloproteinases. Mol Biol Cell. 25:324–336. 2014.
View Article : Google Scholar :
|
|
26
|
Evans IM, Bagherzadeh A, Charles M,
Raynham T, Ireson C, Boakes A, Kelland L and Zachary IC:
Characterization of the biological effects of a novel protein
kinase D inhibitor in endothelial cells. Biochem J. 429:565–572.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Harikumar KB, Kunnumakkara AB, Ochi N,
Tong Z, Deorukhkar A, Sung B, Kelland L, Jamieson S, Sutherland R,
Raynham T, et al: A novel small-molecule inhibitor of protein
kinase D blocks pancreatic cancer growth in vitro and in vivo. Mol
Cancer Ther. 9:1136–1146. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li QQ, Hsu I, Sanford T, Railkar R, Balaji
N, Sourbier C, Vocke C, Balaji KC and Agarwal PK: Protein kinase D
inhibitor CRT0066101 suppresses bladder cancer growth in vitro and
xenografts via blockade of the cell cycle at G2/M. Cell Mol Life
Sci. 75:939–963. 2018. View Article : Google Scholar
|
|
29
|
Kuleshov MV, Jones MR, Rouillard AD,
Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM,
Lachmann A, et al: Enrichr: A comprehensive gene set enrichment
analysis web server 2016 update. Nucleic Acids Res. 44:W90–W97.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hidalgo M: Pancreatic cancer. N Engl J
Med. 362:1605–1617. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Makohon-Moore A and Iacobuzio-Donahue CA:
Pancreatic cancer biology and genetics from an evolutionary
perspective. Nat Rev Cancer. 16:553–565. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Oliveira-Cunha M, Newman WG and
Siriwardena AK: Epidermal growth factor receptor in pancreatic
cancer. Cancers (Basel). 3:1513–1526. 2011. View Article : Google Scholar
|
|
33
|
Dong M, Nio Y, Guo KJ, Tamura K, Tian YL
and Dong YT: Epidermal growth factor and its receptor as prognostic
indicators in Chinese patients with pancreatic cancer. Anticancer
Res. 18:4613–4619. 1998.
|
|
34
|
Park SJ, Gu MJ, Lee DS, Yun SS, Kim HJ and
Choi JH: EGFR expression in pancreatic intraepithelial neoplasia
and ductal adenocarcinoma. Int J Clin Exp Pathol. 8:8298–8304.
2015.PubMed/NCBI
|
|
35
|
Wee P and Wang Z: Epidermal growth factor
receptor cell proliferation signaling pathways. Cancers (Basel).
9:522017. View Article : Google Scholar
|
|
36
|
Roy SK, Srivastava RK and Shankar S:
Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of
FOXO transcription factor, leading to cell cycle arrest and
apoptosis in pancreatic cancer. J Mol Signal. 5:102010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Williams TM, Flecha AR, Keller P, Ram A,
Karnak D, Galbán S, Galbán CJ, Ross BD, Lawrence TS, Rehemtulla A
and Sebolt-Leopold J: Cotargeting MAPK and PI3K signaling with
concurrent radiotherapy as a strategy for the treatment of
pancreatic cancer. Mol Cancer Ther. 11:1193–1202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rogers DA and Schor NF: Kidins220/ARMS is
expressed in neuroblastoma tumors and stabilizes neurotrophic
signaling in a human neuroblastoma cell line. Pediatr Res.
74:517–524. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim ST, Lim DH, Jang KT, Lim T, Lee J,
Choi YL, Jang HL, Yi JH, Baek KK, Park SH, et al: Impact of KRAS
mutations on clinical outcomes in pancreatic cancer patients
treated with first-line gemcitabine-based chemotherapy. Mol Cancer
Ther. 10:1993–1999. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee J, Jang KT, Ki CS, Lim T, Park YS, Lim
HY, Choi DW, Kang WK, Park K and Park JO: Impact of epidermal
growth factor receptor (EGFR) kinase mutations, EGFR gene
amplifications, and KRAS mutations on survival of pancreatic
adenocarcinoma. Cancer. 109:1561–1569. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yeung KT and Yang J:
Epithelial-mesenchymal transition in tumor metastasis. Mol Oncol.
11:28–39. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hotz B, Arndt M, Dullat S, Bhargava S,
Buhr HJ and Hotz HG: Epithelial to mesenchymal transition:
Expression of the regulators snail, slug, and twist in pancreatic
cancer. Clin Cancer Res. 13:4769–4776. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nomura A, Majumder K, Giri B, Dauer P,
Dudeja V, Roy S, Banerjee S and Saluja AK: Inhibition of NF-kappa B
pathway leads to deregulation of epithelial-mesenchymal transition
and neural invasion in pancreatic cancer. Lab Invest. 96:1268–1278.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gialeli C, Theocharis AD and Karamanos NK:
Roles of matrix metalloproteinases in cancer progression and their
pharmacological targeting. FEBS J. 278:16–27. 2011. View Article : Google Scholar
|
|
47
|
Koshiba T, Hosotani R, Wada M, Miyamoto Y,
Fujimoto K, Lee JU, Doi R, Arii S and Imamura M: Involvement of
matrix metalloproteinase-2 activity in invasion and metastasis of
pancreatic carcinoma. Cancer. 82:642–650. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bloomston M, Zervos EE and Rosemurgy AS
II: Matrix metalloproteinases and their role in pancreatic cancer:
A review of preclinical studies and clinical trials. Ann Surg
Oncol. 9:668–674. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Huntington JT, Shields JM, Der CJ, Wyatt
CA, Benbow U, Slingluff CL Jr and Brinckerhoff CE: Overexpression
of collagenase 1 (MMP-1) is mediated by the ERK pathway in invasive
melanoma cells: role of BRAF mutation and fibroblast growth factor
signaling. J Biol Chem. 279:33168–33176. 2004. View Article : Google Scholar : PubMed/NCBI
|