1. Introduction
SQSTM1/P62, an adapter and scaffolding protein
described cursorily by Park (1)
in 1995, is a 62-kD protein that binds to the Src homology 2 domain
of p56lck and is involved in the degradation and destruction of
ubiquitinated proteins. Over time, an increasing number of studies
have extensively shown that SQSTM1/p62 is a multifunctional protein
that impinges on a variety of tumor biological behaviors, such as
cell growth, proliferation, migration, invasion, metastasis,
autophagy, apoptosis and ubiquitination (2-5).
SQSTM1/p62 has important and versatile roles in cancer owing to its
distinct and important structures, including a ubiquitin-associated
(UBA) region and a Phox and Bem1p (PB1) domain, which mediate the
ubiquitin-proteasome degradation pathway, a LC3-interacting region
(LIR), which mediates the autophagy pathway, and a Kelch-like
ECH-associated protein 1 (KEAP1)-interacting region, which is
involved in the NF-κB pathway (6). The coexistence of a UBA region and
LIR indicates that SQSTM1/p62 serves as a bridge between autophagy
and the ubiquitin-proteasome system, which is of the utmost
importance for SQSTM1/p62 regulation of various biological
behaviors (7,8). In addition, after incorporation into
a completed autophagosome, SQSTM1/p62 is quickly degraded in
autolysosomes; therefore, it is an important index useful for
monitoring autophagic degradation. Generally, increased SQSTM1/p62
expression reflects inhibition of autophagy. Analogously, decreased
SQSTM1/p62 expression indicates activation of autophagy, and thus,
to a certain degree, it is used to monitor autophagic flux in
cells; however, these differences in expression may be context
specific (9). For instance, the
SQSTM1/p62 level may not change when autophagy is induced, as
SQSTM1/p62 may be involved in other signaling pathways, interacting
with numerous molecules through other domains as described in the
aforementioned literature (9).
Additionally, the phosphorylation of SQSTM1/p62 at Ser403 may
post-transcriptionally participate in the regulation of the
autophagic clearance of ubiquitinated proteins (10). Notably, recent evidence has
confirmed that in gastric cancer, SQSTM1/p62 can directly bind and
transport the long non-coding RNA ARHGAP5-AS1 as cargo to
autophagosomes, ultimately leading to RNA recycling by
autolysosomes (11). Furthermore,
SQSTM1/p62 may directly recruit and interact with autophagy-linked
FYVE protein, a protein similar to SQSTM1/p62 that is essential to
the formation and autophagic degradation of ubiquitinated proteins,
to degrade ubiquitinated proteins via autophagy (12). In particular, SQSTM1/p62 plays a
crucial role in autophagy and the regulation of other
transcriptional regulators, including nuclear factor erythroid 2
(NRF2) and NF-κB. For example, NRF2 degradation is primarily
powered by the KEAP1, whereas, phosphorylation of SQSTM1/p62 at
S349 may tremendously enhance the adhesion to KEAP1 and
subsequently disrupts association between KEAP1 and NRF2, and
promotes NRF2 stabilization and activation to facilitate growth of
tumor cells, implying crosstalk between SQSTM1/p62-mediated
autophagy and the KEAP1-NRF2 system (13,14). Moreover, SQSTM1/p62 acts as a
direct transcriptional target of NF-κB and can in turn activate the
NF-κB pathway by stimulating inhibitor of NF-κB kinase subunit
β/IκB by TNF receptor-associated factor 6 (TRAF6)
polyubiquitination (15-17). In addition, SQSTM1/p62 p.R321C
mutation leads to autophagy inhibition by activating the NF-κB
pathway in Paget's disease of the bone (18), and as autophagy is inhibited,
positive feedback between SQSTM1/p62 and NF-κB interaction can
excessively and sustainably trigger the NF-κB pathway, leading to
epithelial-mesenchymal transition (EMT) in various RAS-mutated
cells (19).
Recently, an increasing number of studies have
confirmed that under conditions of various intracellular or
extracellular pressures, including nutrient deficiency, and
endoplasmic reticulum, oxidative and metabolic stresses, such as
hypoxia and high concentrations of insulin, SQSTM1/p62 may act as
either a proto-oncogene or tumor suppressor gene, promoting or
inhibiting malignant tumor progression, respectively (20-23). By directly binding to numerous
cancer-associated genes, such as Tribbles 3, EGFR, COX-2, MMP1/2,
membrane type-MMP, c-Myc, Snail, Twist (20), RAD51 recombinase, filamin A
(21), ring finger protein 168
(an E3 ubiquitin-protein ligase) (22) and checkpoint kinase 1 (23), SQSTM1/p62 can mitigate genetic
instability and DNA damage foci, and induce DNA repair, thereby
playing a role in cancer oncogenesis, malignant progression,
senescence and chemoradiotherapeutic sensitivity (20-23). Since SQSTM1/p62 is at the center
of a hub of various complicated signaling pathways in different
cancer types and as its functions have wide implications in
different cellular systems, the underlying regulatory mechanisms of
its role in steering tumor progression are not entirely known and
remain to be revealed in the future. The present review focuses on
the mechanisms and functions of SQSTM1/p62 in regulating diverse
biological behaviors in different types of cancer.
2. Gastrointestinal tumors
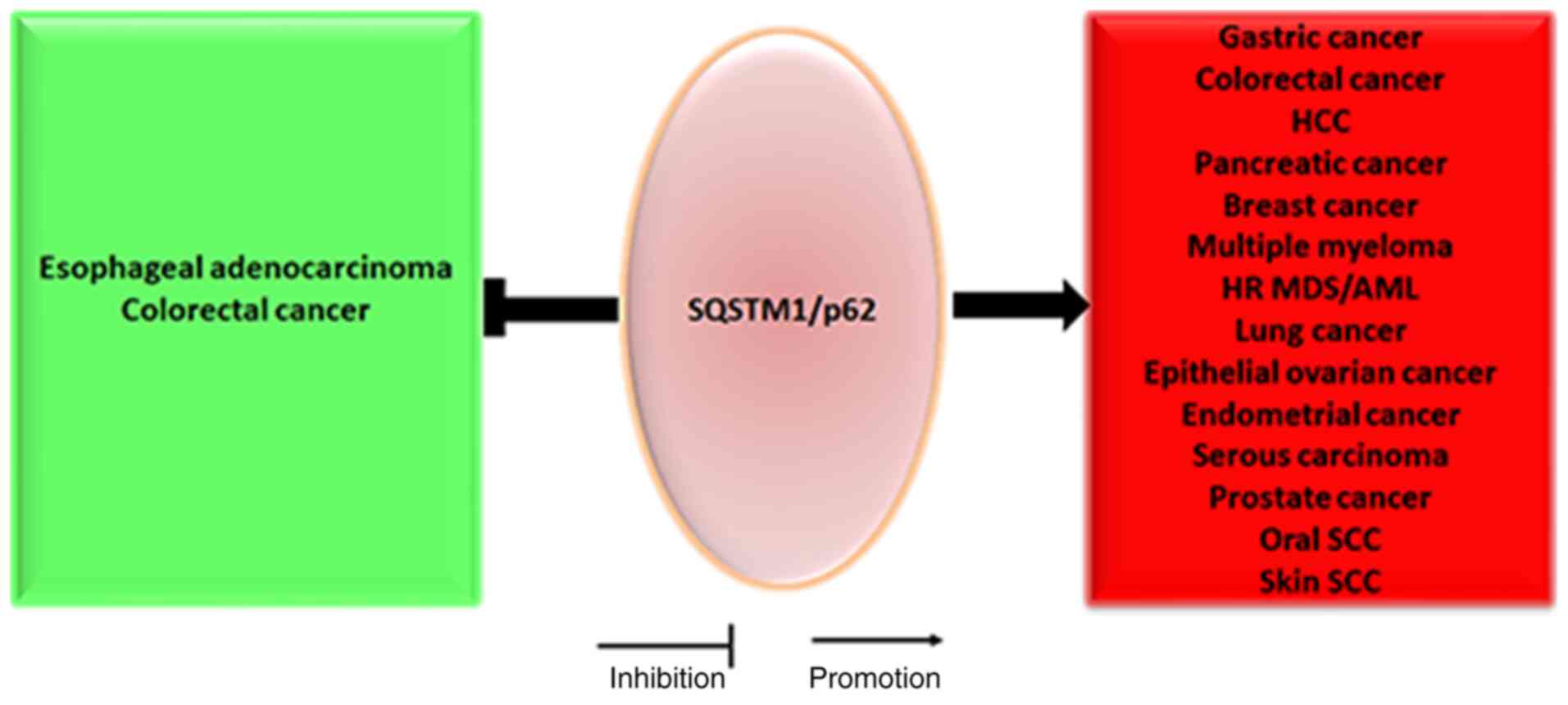
It has been established that SQSTM1/p62, as a tumor
oncogene, is frequently abnormally upregulated and involved in the
aggression of gastrointestinal tumors, including gastric,
colorectal and pancreatic cancer (24). Several lines of evidence also
suggest that specific expression patterns of SQSTM1/p62 in tumor
cells are closely related to invasion and metastasis, and can
indicate prognosis. Notably, the punctiform expression of
SQSTM1/p62 in the cytoplasm and/or cell nucleus may be an
independent prognostic factor of esophageal adenocarcinoma,
particularly early esophageal adenocarcinoma, in which the
expression of SQSTM1/p62 is usually aberrantly upregulated
(24,25). However, patients with higher
SQSTM1/p62 expression in the tumor cell cytoplasm and nucleus have
a better prognosis, while patients with lower SQSTM1/p62 or lower
combined LC3 and SQSTM1/p62 expression in tumor cells have more
aggressive esophageal adenocarcinoma (25). The overexpression of SQSTM1/p62 in
gastric cancer is related to hematogenous and hepatic metastasis,
particularly in early gastric cancer, with an unfavorable
prognosis. However, SQSTM1/p62 expression in colorectal cancer
treated with 5-fluorouracil has not been shown to be significantly
associated with prognosis (26,27). All of these data indicate that
SQSTM1/p62 dysregulation may be an early and important event in
tumorigenesis. SQSTM1/p62 expression in different gastrointestinal
tumors has different prognostic significance, possibly due to the
location of the cancer lesion and the immune response to cancer
cells that express SQSTM1/p62 and to the tumor microenvironment,
which exhibits a high density of regulatory forkhead box
P3+ T cells (28).
Furthermore, inhibition of SQSTM1/p62 may repress
autophagy, impeding tumor aggression by involving the MEK/ERK
signaling pathway in KRAS- and BRAFV600E-mutated colorectal cancer
cells. The evidence for this comes from the finding of a previous
study where positive cytoplasmic SQSTM1/p62 staining for
conspicuously present in the majority of colorectal cancer tissues.
Although there was no significant association between
SQSTM1/p62-positive staining and KRAS mutations, patients with
SQSTM1/p62-positive staining in the cytoplasm, particularly those
with KRAS mutations, may have better overall survival rates
(29). However, KRAS and
BRAFV600E mutations have been shown to activate the MEK/ERK
signaling pathway to promote autophagy and downregulate SQSTM1/p62
expression in colorectal cancer cells when the phosphoinositol-3
kinase (PI3K)/mTOR signaling pathway is inhibited (30). Therefore, the activation of
autophagy and the downregulation of SQSTM1/p62 may indirectly
mirror the effects of KRAS and BRAF mutations in cells. However,
autophagy can be repressed by SQSTM1/p62 inhibition, leading to the
cessation of cancer cell growth and tumor formation (31).
Nevertheless, SQSTM1/p62 expression is not always
decreased and has sometimes been increased when autophagy was
activated. For instance, a study showed that SQSTM1/p62 expression
in colorectal cancer cells was inhibited by the
β-catenin/transcription factor (TCF)4 complex, which inhibits
autophagosome formation (32).
However, under starvation-induced autophagy conditions, SQSTM1/p62
expression was markedly increased, as β-catenin was spontaneously
degraded by LC3 binding to the LIR of β-catenin (31). Thus, it seems that detecting
SQSTM1/p62 expression alone may not always be a reliable approach
for evaluating the autophagy process; it may be necessary to also
assess the expression of other autophagy-related molecules, such as
Beclin1 or ATG8.
In addition, recent studies have shown that
SQSTM1/p62 plays an indispensable role in regulating drug
sensitivity in colorectal cancer by inducing autophagy, apoptosis
and DNA damage. For example, high expression of SQSTM1/p62 can be
induced directly by activated heat shock factor 1 in colorectal
cancer cells treated with a heat shock protein 90 (HSP90)
inhibitor, which may accelerate the process of autophagy and
suppress cell death, ultimately resulting in the weak anticancer
effects of HSP90. By contrast, SQSTM1/p62 inhibition may markedly
enhance colorectal cancer chemosensitivity to HSP90 (33). Alternatively, it was previously
reported that SQSTM1/p62 and LC3 were overexpressed in stage III-IV
colon cancer exposed to 5-FU therapy, which indicated that high
levels of SQSTM1/p62 and activation of autophagy may represent the
self-defense of a tumor against internal and external stress to
compromise therapeutic efficacy (27). This outcome may be partially
reflected at the genetic level; for example, in escin-treated CRC
cells, activated autophagy and SQSTM1/p62 accumulation have been
shown to play a protective role against escin-induced apoptosis and
DNA damage, as SQSTM1/p62 abrogates the ataxia-telangiectasia
mutated/phosphorylated histone family member X pathway (34). However, a high level of SQSTM1/p62
may enhance the efficacy of photodynamic therapy by promoting tumor
cell death (35). Therefore, on
the one hand, SQSTM1/p62 may act as a protective factor in tumor
cell survival by promoting autophagy and inhibiting apoptosis under
drug-induced stress, while on the other hand, it may act as a
driving force to accelerate the death of tumor cells subjected to
photodynamic therapy via mechanisms that remain unknown.
In conclusion, SQSTM1/p62 is involved in the
occurrence and development of gastrointestinal tumors through
various signaling pathways (Fig.
1). SQSTM1/p62 plays a key role in the process of autophagy.
However, increased or decreased SQSTM1/p62 expression does not
necessarily represent the inactivation or activation of autophagy.
In fact, SQSTM1/p62 expression may be elevated or decreased in
association with activated or suppressed autophagy. Additionally,
patients with an elevated SQSTM1/p62 level do not necessarily tend
to have a poor prognosis, nor does a decreased SQSTM1/p62
expression level indicate a favorable prognosis. Nonetheless, it
seems certain that the inhibition of SQSTM1/p62 expression can
significantly enhance the therapeutic strategies used in the
treatment of gastrointestinal tumors. Therefore, targeting
SQSTM1/p62-mediated signaling pathways may be an ancillary strategy
used with therapies against these cancer types.
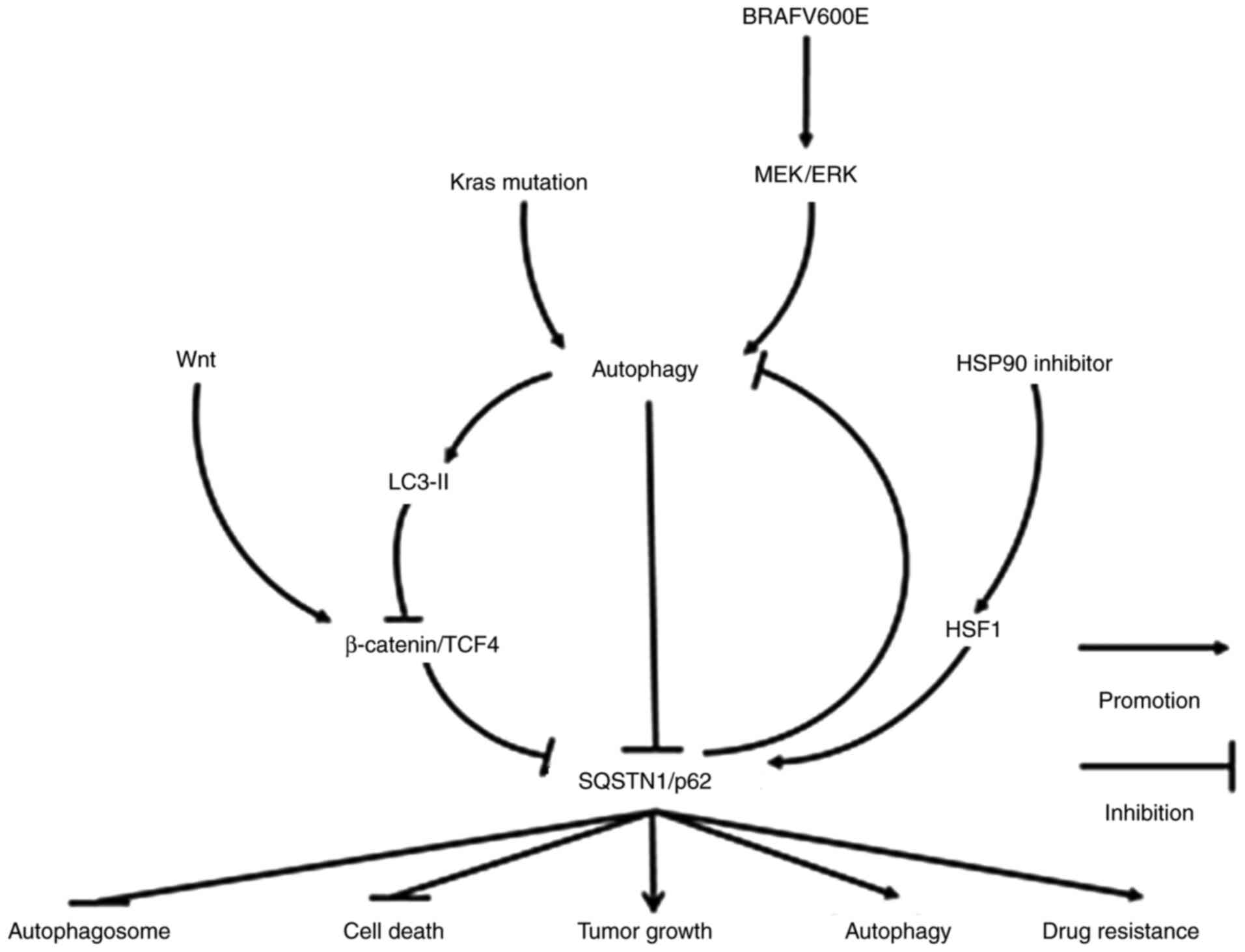 | Figure 1Molecular mechanisms of SQSTM1/p62 in
regulating gastrointestinal tract tumors. SQSTM1/p62 expression may
be decreased following the activation of Wnt/β-catenin/TCF4, a
pathway inhibiting autophagy, including autophagy caused by KRAS
mutation and BRAFV600E activation. However, under conditions of
starvation-induced autophagy or in the presence of an HSP90
inhibitor, SQSTM1/p62 expression may also increase due to the
degradation of LC-3II-bound β-catenin in autophagosomes or the
activation of heat shock factor 1, respectively. The elevated
expression of SQSTM1/p62 ultimately not only inhibits the formation
of autophagosomes and cell death, but also facilitates autophagy,
tumor growth and drug resistance. SQSTM1, sequestosome 1; TCF4,
transcription factor 4; HSP90, heat shock protein 90; HSF1, heat
shock factor protein 1. |
3. Hepatocellular carcinoma (HCC)
The pattern and significance of SQSTM1/p62
expression are different between cancer cells and peripheral
stromal cells. SQSTM1/p62 expression has been shown to be higher in
HCC cells but lower in peripheral mesenchymal astrocytes, where
SQSTM1/p62 directly promotes the interaction between the vitamin D
receptor and retinoid X receptor to inhibit the process of liver
fibrosis, inflammation and cancer (36). In this respect, SQSTM1/p62 may
function as an anti-oncogene in HCC formation by regulating steroid
hormone reactions.
In HCC, SQSTM1/p62 gene amplification, mutation and
hepatitis C virus infection can cause excessive SQSTM1/p62
aggregation and phosphorylation. Under these conditions, glucose is
carried into the glucuronic acid metabolic pathway, and glutamate
is converted into glutathione by activating nuclear factor
erythroid 2 (NRF2), which further promotes SQSTM1/p62 expression.
The positive feedback induces aggressive cancer cell proliferation
and chemoresistance (37-39). In addition, SQSTM1/p62 can bind to
the N-terminal region of TRAF6 [a ubiquitin ligase activating the
NF-κB cascade by binding to the N-terminal region of ζ-protein
kinase C (PKC)] to activate the NF-κB signaling pathway, thus
facilitating invasion, migration and distant metastasis of HCC.
Enhanced expression of SQSTM1/p62 participates in the formation of
benign adenoma by liver cells with autophagy deficiency;
furthermore, in addition to activating NRF2, enhanced SQSTM1/p62
expression in conditions of cellular reactive oxygen stress or
inflammation in HCC precancerous lesions can induce c-myc
expression by initiating the mechanistic target of rapamycin
complex 1 (m-TORC1) signaling pathway, rather than the
ubiquitination pathway, to promote the occurrence of liver cancer.
Even after HCC resection, high expression of SQSTM1/p62 in the
remainder of the liver is an indicator of HCC recurrence (40). In summary, accumulation of
SQSTM1/p62 as an oncogene in HCC may eventually promote malignant
tumor cell progression by either interacting with NRF2 or TRAF6 to
activate the NF-κB signaling pathway or to upregulate c-myc
expression to initiate the m-TORC1 signaling pathway.
The promoter region of SQSTM1/p62 contains
antioxidant response elements (AREs), making SQSTM1/p62 an NRF2
(and NRF1) response gene. Thus, the SQSTM1/p62 gene is induced in
response to NRF2 activation. Substantial analyses of a high number
of cancer biopsy samples of different forms of cancer have
indicated that the NRF2 pathway is frequently activated by somatic
mutations (41). Thus, in cancer
cells where NRF2 is constitutively activated, the transcription of
SQSTM1/p62 and a number of other protective oxidative stress
response genes is expected to be increased. Thus, elevated
SQSTM1/p62 gene expression is considered to be a consequence of
mutations that frequently occur in cancer. A study showed that
autophagy-deficient mouse livers exhibited aberrant SQSTM1/p62
accumulation and developed severe liver damage as SQSTM1/p62
accumulation disrupted the KEAP1-NRF2 association and promoted NRF2
stabilization and accumulation. However, in SQSTM1/p62-deficient
mouse livers, KEAP1 was primarily degraded via the autophagy
pathway in a SQSTM1/p62-dependent manner, and NRF2 accumulation
caused severe liver dysfunction through the autophagy pathway, not
the proteasome pathway, independent of SQSTM1/p62 expression
(42).
Additionally, SQSTM1/p62 regulates the
chemosensitivity and chemoresistance of HCC in different ways. When
dehydroepiandrosterone induces autophagic cell death, unexpectedly,
SQSTM1/p62 is not degraded through the autophagic process; it
aggregates upon upregulation via the initiation of the Jun
N-terminal kinase (JNK)-NRF2-SQSTM1/p62 signaling pathway, in which
JNK methylation triggers NRF2 to induce SQSTM1/p62 expression
(43). On the other hand, a
ferroptosis agonist promotes the expression of SQSTM1/p62 in HCC
cells, which can further inactivate the cobinding factor KEAP1 to
release and thus activate NRF2. Once released, NRF2 accumulates in
the cell nucleus and induces the expression of various downstream
oncogenes, such as ferritin heavy chain 1, NAD(P)H dehydrogenase
quinone 1 (NQO1) and heme oxygenase-1 (HO-1), which suppresses
cellular ferroptosis and causes drug resistance (38). Notably, SQSTM1/p62 also regulates
both apoptosis and pyroptosis via acetylation. Researchers have
confirmed that SQSTM1/p62 colocalizes with histone deacetylase 6
(HDAC6) during ubiquitylation in mitochondria and abolishes HDAC6
deacetylation (44). The
inhibition of SQSTM1/p62 may facilitate HDAC6-mediated
deacetylation of α-tubulin and cortactin to disrupt the stability
of microtubes and the formation of autolysosomes. Unexpectedly, the
inhibition of autolysosomes can cause the downregulation not the
upregulation of SQSTM1/p62 (45),
suggesting that the underlying mechanisms for regulating
SQSTM1/p62-related networks under autophagy conditions should be
studied further. Therefore, it seems that SQSTM1/p62 may weaken the
curative effects of some anticancer drugs primarily through the
SQSTM1/p62-NRF2 axis.
In summary, it appears that SQSTM1/p62 in HCC cells
may act primarily as an oncogene, while it may also function as a
tumor repressor gene in peripheral stromal cells; in these cells,
SQSTM1/p62 regulates chemosensitivity and chemoresistance through a
variety of processes, including autophagic cell death, apoptosis,
ferroptosis and pyroptosis, via different signaling pathways,
particularly protein deacetylation and the SQSTM1/p62-NRF2 positive
feedback loop (Fig. 2).
Therefore, targeting SQSTM1/p62 in intricate networks may be a good
adjuvant therapeutic approach to HCC treatment.
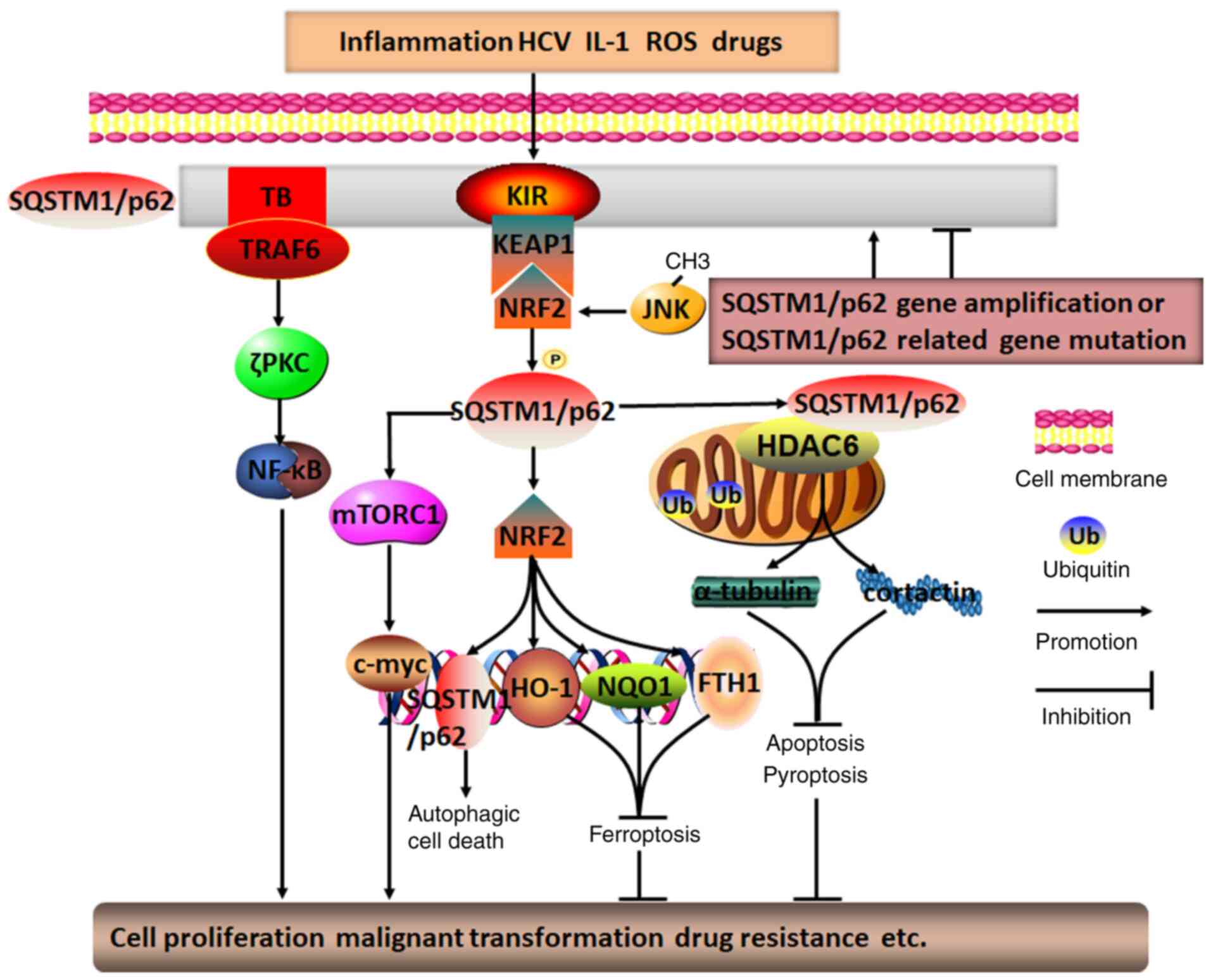 | Figure 2Molecular mechanisms of SQSTM1/p62 in
regulating HCC. Diverse intracellular and extracellular risk
factors, including SQSTM1/p62 gene amplification,
SQSTM1/p62-related gene mutation, inflammation, HCV infection,
ROS-induced stress, drugs and IL-1, may promote SQSTM1/p62
aggregation and phosphorylation. Subsequently, SQSTM1/p62 is
capable of activating the mTOR/c-myc, TRAF6/ζPKC/NF-κB, and
SQSTM1/p62/KEAP1/NRF2 pathways through its distinctive TB and KIR
domains. The positive feedback loop of SQSTM1/p62/KEAP1/NRF2,
involving JNK methylation-induced NRF2 activation, may generate
various secondary messengers, such as SQSTM1/p62, HO-1, NQO1 and
FTH1. In addition, by colocalizing with HDAC6 during ubiquitylation
in mitochondria, SQSTM1/p62 may inhibit HDAC6 deacetylation of
α-tubulin and cortactin, which may ultimately result in p62
directly or indirectly regulating HCC autophagic cell death,
apoptosis, pyroptosis, ferroptosis, cell proliferation, malignant
transformation and drug resistance, among others. TRAF6, TNF
receptor-associated factor 6; TB, TRAF6-binding domain; KIR,
KEAP1-interacting region; PKC, protein kinase C; SQSTM1,
sequestosome 1; ROS, reactive oxygen species; FTH1, ferritin heavy
chain 1; KEAP1, Kelch-like ECH-associated protein 1; NRF2, nuclear
factor erythroid 2; HO-1, heme oxygenase 1; NQO1, NAD(P)H
dehydrogenase quinone 1; HDAC6, histone deacetylase 6; HCC,
hepatocellular carcinoma; HCV, hepatitis C virus. |
4. Breast cancer
A mechanism of SQSTM1/p62 function as an oncogene
similar to that involved in HCC has been observed in breast cancer;
that is, expression of SQSTM1/p62 in the presence of NRF2 can be
decreased in the cytoplasm and increased in the cell nucleus
through the SQSTM1/p62-KEAP1-NRF2 axis, which causes NRF2 to
activate the expression of numerous downstream oncogenes.
Furthermore, the aggregation of NRF2 in the cell nucleus may also
significantly promote SQSTM1/p62 expression. In particular,
SQSTM1/p62 mediates CD44-NRF2 activation during autophagy in cancer
stem cells (CSCs), leading to aggressive cancer growth and drug
resistance (46). Thus, the
formation of the SQSTM1/p62 and the NRF2 positive feedback loop
ultimately endows CSCs with self-renewal capacity and
chemoradiotherapy resistance (47,48). Notably, SQSTM1/p62 has been shown
to interact with microRNAs to regulate breast cancer malignancy.
That is, in a previous study, SQSTM1/p62 prevented myc degradation
by directly abrogating the expression of the microRNAs let7a and
let7b, resulting in persistent overexpression of myc in the cancer
cells (49).
To some degree, it seems that SQSTM1/p62 plays a
central role in in complex networks to regulate the occurrence and
development of breast cancer. Recent data have demonstrated that
SQSTM1/p62, as a bridge between Notch1 intracellular domain
(Notch1-IC) and LC-3-II in the autophagosome, has the ability to
abolish the Notch1 signaling pathway through its role in the
autophagosome, which engulfs and degrades Notch1-IC (50,51). Moreover, HER-2-mediated activation
of SQSTM1/p62 can maintain cancer cell vitality and markedly
accelerate cell proliferation and malignant growth through various
signaling pathways, including the PI3K/AKT signaling pathway (which
is initiated through SQSTM1/p62-induced PTEN degradation), the
AKT/glycogen synthase kinase 3β/β-catenin signaling pathway, the
Wnt/β-catenin signaling pathway, the NF-κB signaling pathway and
the KEAP1-NRF2 signaling pathway (51-53). Among these pathways, the
SQSTM1/p62-activated NF-κB signaling pathway under conditions of
autophagy deficiency may lead to high rates of p65 phosphorylation
and transport into the cell nucleus, inducing the upregulation of
NF-κB target genes, such as IFN-γ, TNF-α and IL-6, which promote
cancer cell proliferation (54).
In addition, SQSTM1/p62 can recruit and phosphorylate JNK to
facilitate the development of cancer cells by directly binding to
Van Gogh-like 2 in the Wnt/PCP signaling pathway (55). In addition, SQSTM1/p62 dysfunction
may cause autophagy deficiency and initiate the mitochondrial
apoptosis pathway by inducing the accumulation of the
apoptosis-related BH3-only protein NBK/Bik on endoplasmic reticulum
membranes, thus converting autophagic cells to apoptotic cells and
suppressing cancer cell proliferation (56). Furthermore, in a previous study,
SQSTM1/p62 degradation was diminished due to the inhibition of
cathepsin in the autolysosome in cancer cells treated with
chemicals that induce autophagy, and apoptosis occurred due to the
increased reactive oxygen species (ROS) level that was induced upon
SQSTM1/p62 accumulation (57).
SQSTM1/p62 aggregation in breast cancer cells
usually acts as an indicator of chemoresistance to multiple drugs,
such as Adriamycin, PI3K/AKT inhibitors and Pseudomonas
aeruginosa mannose-sensitive hemagglutinin (46). Breast cancer with high expression
of SQSTM1/p62 has been linked to distant lymphatic and vessel
metastasis, and is correlated with an unfavorable prognosis in
these patients, particularly those with triple-negative breast
cancer (58-61). As a result, the inhibition of
SQSTM1/p62 expression in these cancer cells may lead to the
abrogation of cell growth and proliferation, enhancing the efficacy
of therapeutic treatments. Nevertheless, an exception has also been
reported in gemcitabine-treated estrogen receptor (ER)-positive
breast cancer cells, where ERK phosphorylation may promote
SQSTM1/p62 expression to accelerate the process of autophagic cell
death. By contrast, SQSTM1/p62 silencing may abrogate the
ER/ERK/SQSTM1/p62 signaling pathway to interrupt excessive
autophagy, thereby protecting cancer cells from cytotoxic effects
(62). Taken together, these
findings imply that SQSTM1/p62 may enhance either chemosensitivity
or chemoresistance depending on the type of breast cancer,
information of importance for implementing precision therapies to
treat breast cancer.
In general, SQSTM1/p62 can regulate breast cancer
cell proliferation, malignant transformation, invasion, migration
and distant metastasis through multiple apoptosis- and
autophagy-associated signaling pathways; thus, it plays a dual role
in breast cancer, acting both as an oncogene to confer cancer cell
chemoresistance and as a tumor repressor to confer chemosensitivity
(Fig. 3).
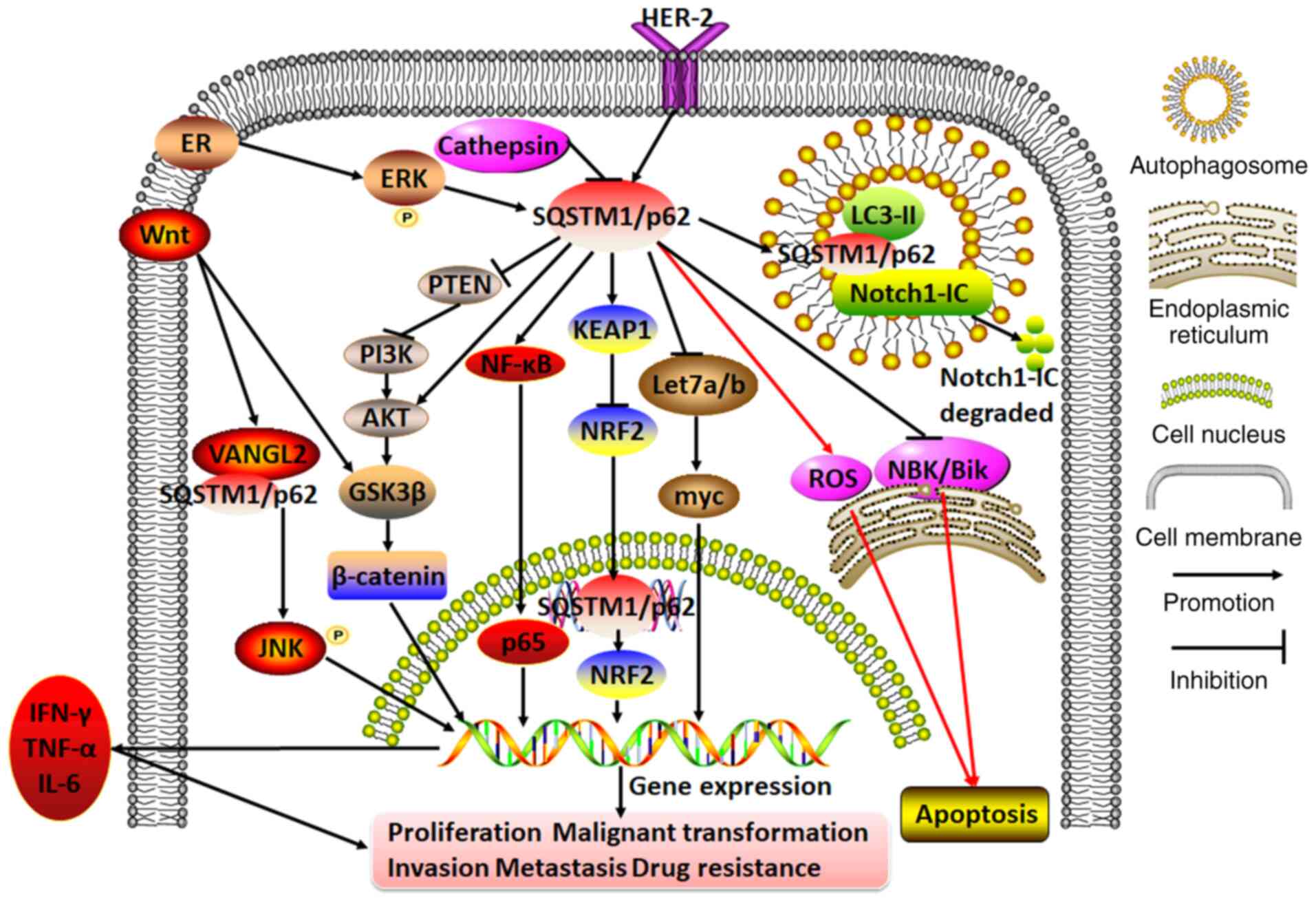 | Figure 3Molecular mechanisms of SQSTM1/p62 in
regulating breast cancer. In breast cancer cells, cathepsin on the
autolysosome may repress SQSTM1/p62 expression. By contrast, ER or
HER-2 can initiate SQSTM1/p62 expression. The hyperexpression of
SQSTM1/p62 exacerbates cell proliferation, malignant
transformation, invasion, metastasis and drug resistance by
participating in diverse cascades, including the PTEN/PI3K/AKT,
Wnt/β-catenin, AKT/glycogen synthase kinase 3β/β-catenin,
KEAP1/NRF2, NF-κB (which releases IFN-γ, TNF-α and IL-6),
Wnt/VANGL2/SQSTM1/p62/JNK and Let7a/b/myc pathways. Moreover,
servicing as a bridge between Notch1-IC and LC3-II, SQSTM1/p62
mediates Notch1-IC degradation in autophagosomes. In addition,
SQSTM1/p62 aggregation may either promote or inhibit apoptosis by
repressing NBK/Bik expression at the endoplasmic reticulum or
inducing ROS expression. VANGL2, Van Gogh-like 2; ROS, reactive
oxygen species; SQSTM1, sequestosome 1; ER, estrogen receptor;
PI3K, phosphoinositol-3 kinase; KEAP1, Kelch-like ECH-associated
protein 1; NRF2, nuclear factor erythroid 2; Notch1-IC, Notch1
intracellular domain. |
5. Hematological malignancy
In multiple myeloma cells treated with TNF-α,
high-risk myelodysplastic syndromes/acute myeloid leukemia (AML)
del(5q) cells and hematopoietic stem cells/progenitor cells with
miR-146a deletion, the sustained expression of SQSTM1/p62 has an
oncogenic effect that leads to the marked activation of NF-κB or
p38MAPK cascades through the following mechanisms: i)
Direct binding to and ubiquitination of TRAF-6 through its
TRAF-binding domain to activate the NF-κB signaling pathway
(63-65); ii) direct binding with
receptor-interacting protein 1 through its C-terminal region ZZ
domain, as well as with atypical PKCs through its N-terminal PB1
domain to activate the NF-κB signaling pathway (66,67); and iii) direct binding to
p38MAPK through its p38 domain to activate the
p38MAPK signaling pathway (68). Activated NF-κB can further enhance
SQSTM1/p62 gene expression, ultimately generating predominately
positive feedback loops to continuously amplify its deleterious
effect in exacerbating unrestrained growth, proliferation and
malignant transformation, and inhibiting osteoblast activation to
hamper bone formation (66,69). As a result, the deprivation of
SQSTM1/p62, leading to the inhibition of SQSTM1/p62 ZZ domain
activity, may attenuate the NF-κB signaling pathway by
downregulating phospho-focal adhesion kinase, p-IκBα and NF-κB
expression, promoting osteoblast differentiation and generating
myeloid progenitor cells and osteoclasts, thereby inhibiting the
growth of multiple myeloma cells (67,69).
Given that SQSTM1/p62 is closely linked to autophagy
and ubiquitylation-mediated proteasomal processes, defective
autophagy in multiple myeloma cells may lead to an increase in
undigested and toxic proteins with SQSTM1/p62 located in the
endoplasmic reticulum; thus, SQSTM1/p62 may be regarded as a
reliable biomarker for the chemosensitivity of multiple myeloma to
proteasome inhibitors (70).
Furthermore, both SQSTM1/p62 and Kruppel-like factor 4 (KLF4), an
intranuclear transcription factor, are upregulated in multiple
myeloma cells that are resistant to the proteasome inhibitor
carfilzomib. Notably, KLF4 has been found to bind to the promoter
regions encoding ubiquitin-binding domains to upregulate SQSTM1/p62
and trigger the ubiquitin-proteasome and autophagy pathways, thus
inducing cancer cell resistance to carfilzomib (71). In addition, all-trans retinoic
acid (ATRA)-induced NF-κB activation may upregulate SQSTM1/p62
expression. By contrast, SQSTM1/p62 may decrease the response to
ATRA in AML patients with a poor prognosis (72,73). However, SQSTM1/p62 may function as
a tumor suppressor in chronic myeloid leukemia, as the
proto-oncogene BCR-ABL can be translocated to autophagosomes by
SQSTM1/p62 for degradation by the cysteine protease cathepsin B in
autolysosomes (74). Notably, a
recent article reported that in mice lacking SQSTM1/p62 expression
in all hematopoietic cells, including macrophages, no changes in
NF-κB signaling were found (75).
These data query the importance of the signaling role of SQSTM1/p62
in non-transformed cells. On the other hand, another paper
emphasized the possible autophagy-independent roles of SQSTM1/p62
in cancer (76). In summary,
SQSTM1/p62 may either transmit deleterious messages to induce
resistance to chemotherapeutic drugs through the
ubiquitin-proteasome, autophagy or ATRA/NF-κB pathways in patients
with multiple myeloma or AML, or provide attenuating signals
through the autophagy pathway in patients with chronic myeloid
leukemia.
In summary, by participating in multiple signaling
pathways, particularly in the positive feedback loop cascade with
NF-κB, SQSTM1/p62 may serve as a proto-oncogene to induce
hematological malignancy with resistance to various anti-tumor
drugs. Unexpectedly, SQSTM1/p62 may, under certain circumstances,
sensitize cancer cells to some chemotherapeutics (Fig. 4). Therefore, silencing SQSTM1/p62
expression may enhance chemosensitivity, but notably, elevated
SQSTM1/p62 expression does not necessarily indicate that cancer
cells will become resistance to antitumor reagents; therefore,
these observations should be used only to guide the treatment of
specific hematological malignancies.
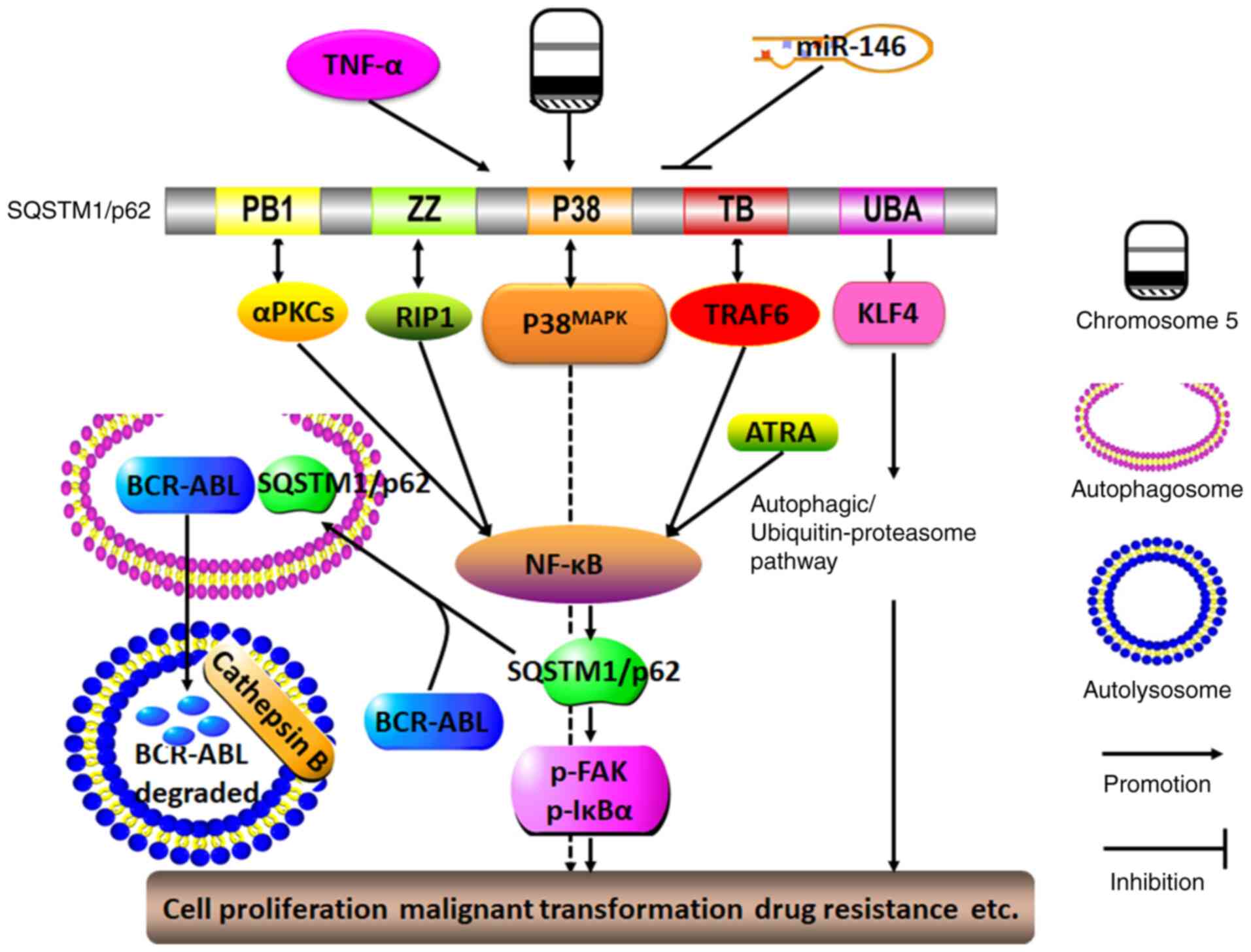 | Figure 4Molecular mechanisms of SQSTM1/p62 in
regulating hematological malignancy. The presence of TNF-α, del(5q)
and miR-146a deletions may lead to the sustained expression of
SQSTM1/p62. By interacting with TRAF-6, RIP1 and αPKCs via its TB,
ZZ and PB1 domains, respectively, SQSTM1/p62 may activate the NF-κB
signaling pathway, which in turn promotes SQSTM1/p62 expression.
ATRA may also trigger the NF-κB signaling pathway, thereby forming
a positive feedback loop with SQSTM1/p62 and NF-κB. By interacting
with p38MAPK via the P38 domain, SQSTM1/p62 can function
in conjunction with the activated autophagy/ubiquitin-proteasome
pathway as triggered by the interaction between SQSTM1/p62 and KLF4
through the SQSTM1/p62 UBA domain, and SQSTM1/p62 can ultimately
promote cell proliferation, malignant transformation and drug
resistance. By contrast, SQSTM1/p62 may function as a tumor
suppressor by driving the oncogene BCR-ABL into autophagosomes, and
it is degraded by cathepsin B in autolysosomes. aPKC, atypical
protein kinase C; PB1, Phox and Bem1p; ZZ, ZZ-type zinc finger
domain; P38, P38MAPK domain; RIP1, receptor-interacting
protein 1; TB, TRAF6-binding domain; UBA, ubiquitin-associated
domain; KLF4, Kruppel-like factor 4; SQSTM1, sequestosome 1; TRAF6,
TNF receptor-associated factor 6. |
6. Lung cancer
Recent investigations have confirmed that SQSTM1/p62
is usually upregulated in the cytoplasm of non-small cell lung
cancer (NSCLC) cells, which indicates a more progressive phenotype
for these cancer cells and a shorter survival period for patients
(77). However, when autophagy is
defective, the expression of SQSTM1/p62 is negatively associated
with Tumor-Node-Metastasis stage and lymph node metastases, which
indicates that it may be considered an independent prognostic
factor for NSCLC (78).
It has been confirmed that SQSTM1/p62 plays an
oncogenic role through its involvement with different pathways,
such as the NF-κB signaling pathway and SQSTM1/p62-KEAP1-NRF2
cascade, to regulate autophagy and apoptosis. For instance,
emerging evidence has indicated that nickel exposure may cause the
degradation of SQSTM1/p62 through the autophagy-related mTOR-Unc-51
like autophagy activating kinase1-Beclin1 cascade. However, nickel
exposure increases SQSTM1/p62 expression by stabilizing TNF mRNA,
which in turn promotes SQSTM1/p62 mRNA transcription due to the
activation of the NF-κB/REL-associated protein signaling pathway;
thus, TNF may function as a key transcription regulator binding to
the promoter regions of the SQSTM1/p62 gene (79). Similarly, in cisplatin-treated
lung adenocarcinoma cells or lung adenocarcinoma cells in
nutrient-rich environments, the binding of SQSTM1/p62 and TRAF6
through the TRAF6-binding motif may also activate the NF-κB
signaling pathway through TRAF6-mediated ubiquitination of mTORC1,
which is then transported to the lysosome by SQSTM1/p62 and thereby
inactivated; this positive feedback loop promotes the malignant
transformation of bronchial epithelial cells, the abolishment of
autophagy, the proliferation of cancer cells and the secondary
chemoresistance of cells to cisplatin (80,81). These results indicate that, in
some cases, increased SQSTM1/p62 expression may indirectly result
in the inhibition of autophagy. In addition, a number of other
factors contribute to the occurrence of lung cancer. For instance,
arsenic, cadmium and isodeoxyelephantopin may activate the
SQSTM1/p62-KEAP1-NRF2 cascade, in which SQSTM1/p62 transports KEAP1
to autophagosomes, where KEAP1 is spontaneously digested in the
cytoplasm; alternatively, exposure to these compounds might
activate the NRF2-KEAP1-SQSTM1/p62 cascade, in which NRF2 in the
nucleus directly triggers SQSTM1/p62 binding to KEAP1 to release
NRF2. NRF2 is subsequently imported into nucleus where it actively
stimulates the expression of secondary downstream molecules such as
HO-1, SQSTM1/p62 and Bcl-2/Bcl-xL by binding to ARE promoter
regions. Thus, the formation of another positive feedback loop
between SQSTM1/p62 and NRF2 leads to the increased expression of
antiapoptotic proteins and antioxidases, ultimately causing cancer
cell proliferation, growth and malignant transformation (82-84). Unexpectedly, SQSTM1/p62 is capable
of weakening the oncolytic effects of measles virus in lung cancer,
as seen through its ability to evoke mitochondrial autophagy to
degrade the Edmonton strain (85).
Since SQSTM1/p62 acts as an oncogene to facilitate
malignant cancer progression, the inhibition of excessive
SQSTM1/p62 aggregation may represent an effective anti-cancer
strategy. For instance, silencing SQSTM1/p62 may lead to the
initiation of the autophagic cell death process and the abrogation
of cell proliferation not only in lung cancer, but also in
pancreatic cancer and squamous cell carcinoma of the esophagus,
oral cavity and skin (86). The
silencing or degradation of SQSTM1/p62 apparently disrupts the
SQSTM1/p62/TRAF6/NF-κB cascade and activates the apoptosis pathway.
Moreover, as SQSTM1/p62 may directly block the formation of the
Fas/Cav-1 complex, non-functional SQSTM1/p62 may result in the
escape of the Fas/Cav-1 complex to activate caspase-8 and cleave
Beclin-1, and produce diverse mediators, including ROS, that
eventually enhance the killing effects of resveratrol or the
combined effects of the Chinese herb Yu Ping Feng San and cisplatin
(81,87). Furthermore, silencing SQSTM1/p62
expression to downregulate neuronal precursor cell expression of
developmentally downregulated gene 9 may increase the response to
cisplatin treatment in small cell lung cancer cells (88). Alternatively, SQSTM1/p62 can bind
with both the TRAF-C domain of TRAF6 and the coiled-coil domain of
Beclin1 to disrupt the interaction of the TRAF6-Beclin1 and
TRAF6-evolutionarily conserved signaling intermediate in Toll
pathways (ECSIT); thus, SQSTM1/p62 may simultaneously abolish
TRAF6-ECSIT signaling and activate the NF-κB pathway in response to
toll-like receptor 4 stimulation, thereby abrogating Beclin1 and
ECSIT ubiquitination, along with the activation of autophagy and
cancer invasion of both lung and breast cancer cells (89,90).
In conclusion, SQSTM1/p62 functions as an oncogene
to disrupt the malignant transformation of lung cancer by forming
several positive feedback loops, particularly loops involving
SQSTM1/p62 and NF-κB or NRF2 (Fig.
5). Therefore, interrupting deleterious cycles of these
amplifying feedback loops may be considered a feasible way to
obtain marked clinical benefits for patients suffering from lung
cancer.
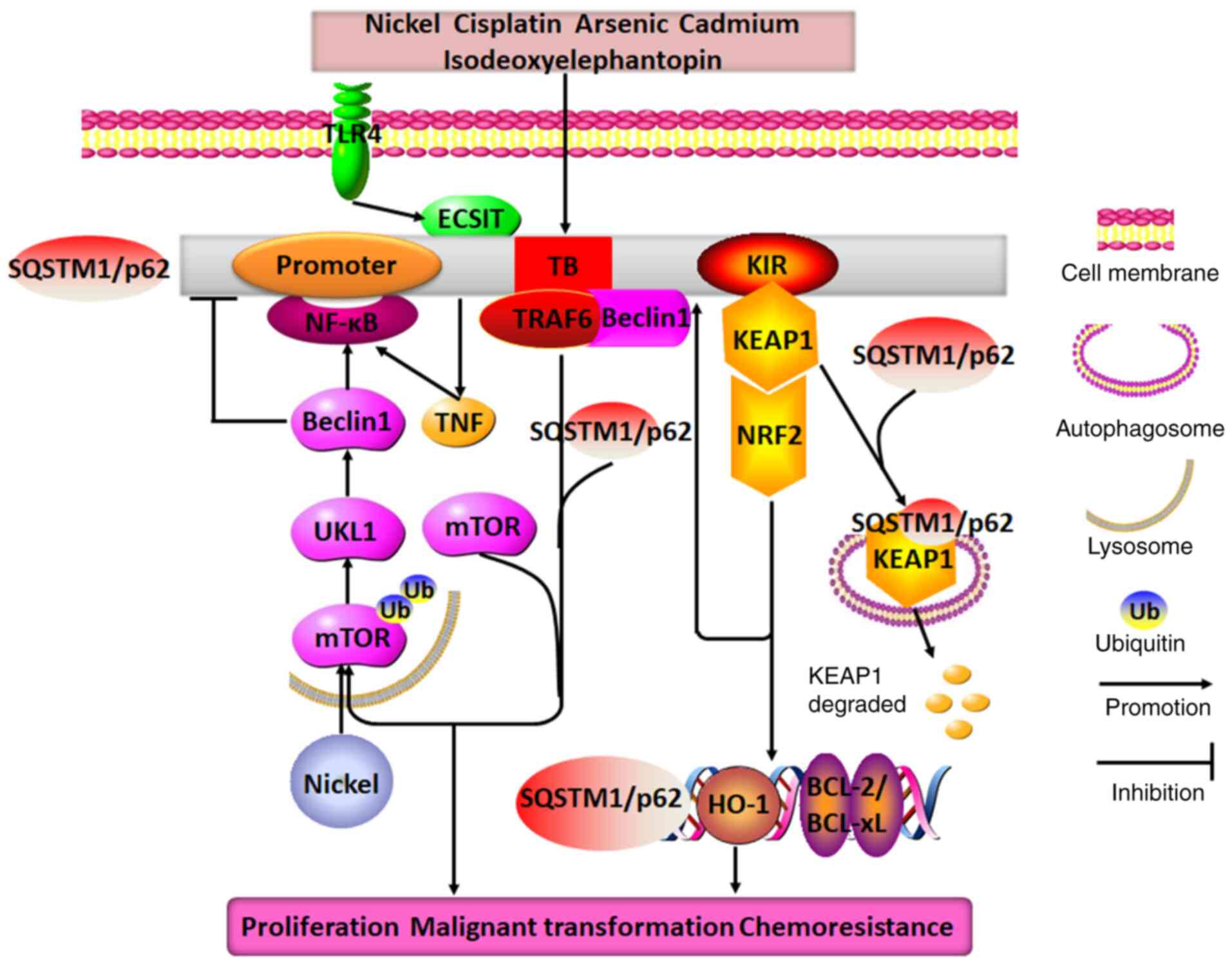 | Figure 5Molecular mechanisms of SQSTM1/p62 in
regulating lung cancer. Nickel exposure may cause SQSTM1/p62
degradation through the mTOR/ULK1/Beclin1 pathway, and it
upregulates p62 expression by stimulating the TNF/NF-κB/SQSTM1/p62
positive feedback loop. Additionally, when exposed to different
drugs, such as cisplatin, arsenic, cadmium, isodeoxyelephantopin
and TLR4, SQSTM1/p62 expression is observed to be increased in lung
cancer cells, where it may directly interact with TRAF6, KEAP1,
Beclin1 and ECSIT through its TB and KIR domains to form two
distinct positive feedback loops: SQSTM1/p62/TRAF6/mTORC1/NF-κB and
SQSTM1/p62/KEAP1/NRF2. These feedback loops promote the expression
of SQSTM1/p62, HO-1 and BCL2/BCL-xL. Therefore, the end result of
the combined effects of these positive feedback loops with
SQSTM1/p62 at their core is the promotion of lung cancer cell
proliferation, malignant transformation and chemoresistance. TRAF6,
TNF receptor-associated factor 6; TB, TRAF6-binding domain; KIR,
KEAP1-interacting region; ECSIT, evolutionarily conserved signaling
intermediate in Toll pathways; SQSTM1, sequestosome 1; ULK1, Unc-51
like autophagy activating kinase; TLR4, toll-like receptor 4;
KEAP1, Kelch-like ECH-associated protein 1; HO-1, heme oxygenase
1. |
7. Genital system cancer
Recent evidence has confirmed that SQSTM1/p62
expression is markedly higher in epithelial ovarian cancer,
particularly in cisplatin-resistant ovarian cancer, than in benign
ovarian tumors (91). The
positive rates of SQSTM1/p62 are also significantly higher in the
advanced stage than in the early stage of ovarian cancer (92). However, a study has reported that
compared with patient-matched primary tumor tissues, metastatic and
recurrent ovarian cancer samples had lower SQSTM1/p62 expression
levels, and a low level of SQSTM1/p62 was closely related to cancer
recurrence, metastasis and paclitaxel resistance; furthermore, the
positive rates of SQSTM1/p62 in cisplatin-resistant ovarian cancer
cells were also lower than those in cisplatin-sensitive cells
(93,94). In some specific cancer types, such
as seminomas and non-seminomas, SQSTM1/p62 expression is negligibly
changed (95). Therefore, the
function and mechanism of SQSTM1/p62 in ovarian cancer should be
further explored.
Moreover, high SQSTM1/p62 expression in the
cytoplasm and low expression in the nucleus is associated with an
advanced stage, a residual tumor and unfavorable survival in
patients with endometrial cancer, epithelial ovarian cancer, serous
carcinoma or cervical cancer (92,96,97). The possible underlying mechanisms
might be that by binding NRF2 and the cotranscription factor TCF20
through its binding domain containing the ARE element, activated
SQSTM1/p62 can initiate the activation of NRF2 and TCF20, which
subsequently enter the nucleus to stimulate SQSTM1/p62 expression;
this outcome leads to resistance to cisplatin or sulforaphane by
initiating cascades of antioxidant gene expression and facilitating
differentiation, proliferation and anti-apoptotic behavior in
drug-resistant cancer cells (97). Additionally, SQSTM1/p62-mediated
upregulation of the cell cycle protein Skp2 through autophagy, not
proteasomal processes, can cause the degradation of p21 and p27,
which results in abrogation of quinacrine-induced apoptosis
(98).
Upon treatment with vitamin K3, ovarian cancer cells
may escape ROS-mediated oxidative damage by activating
SQSTM1/p62/KEAP1/NRF2 signaling (99). By contrast, after treatment with
cisplatin to block autophagy, SQSTM1/p62 may function as an
anti-oncogene to accelerate the apoptosis of ovarian cancer cells
by interacting with the apoptosis-related protein caspase-8
(100).
In summary, SQSTM1/p62 expression varies in
different genital system cancer types and different cellular
locations. However, depending largely on the type of cancer,
SQSTM1/p62 can display various functions by engaging in diverse
signaling events, and it may be a new reliable prognostic biomarker
for endometrial cancer and ovarian cancer, and a drug-sensitive
biomarker for use in neoadjuvant chemotherapy.
8. Urological cancer
Similar to that in genital system cancer, high
expression of SQSTM1/p62 may be an indicator of poor prognosis for
patients with prostate cancer. SQSTM1/p62 expression is
substantially higher in prostate cancer, particularly prostate
cancer at an advanced stage, with resistance to androgen, and with
low androgen expression, compared with that in benign prostatic
hyperplasia (101,102); moreover, its expression in the
cytoplasm is highly associated with advanced Gleason grade, a
positive resection margin and the recurrence of prostate cancer.
The higher the SQSTM1/p62 expression level in the cytoplasm, the
poorer the prognosis in these patients. Thus, by targeting
SQSTM1/p62-induced constitutive NRF2 activation and Bcl-xL
expression, verteporfin, a drug used to treat macular degeneration,
can effectively block autophagy and enhance drug sensitivity in
prostate cancer (103). More
importantly, at the genetic level, elevated SQSTM1/p62 levels may
also be relevant to the gene fusion of TMPRSS2 and ERG, and gene
deletions in PTEN, 3p13, 5q21 and 6q15 (104), which suggests that crosstalk
between SQSTM1/p62 and prostate cancer-related genes may play a
pivotal role in the occurrence and development of prostate
cancer.
SQSTM1/p62 can drive the autophagy process in
response to the activation of the Raf/MEK/ERK signaling pathway in
both malignant melanoma and prostate cancer (105). In addition, SQSTM1/p62 can bind
to MEKK3 and neighbor of BRCA1 (NBR1) through its PB1 domain,
activate Cdc42-associated kinase 1 (Ack1) through its UBA domain
and then stimulate the following downstream events: i) Under
nutrient-deprived conditions, MEKK3 induces SQSTM1/p62
phosphorylation, and phosphorylated SQSTM1/p62 recruits TRAF6,
which directs mTORC1 to lysosomes (106); and ii) in the presence of EGF,
Ack1 dissociation from SQSTM1/p62 and NBR1 in autophagosome
precursors results in prolonged survival of EGFR through slow
endocytosis rather than lysosomal degradation pathways (107). These cascades markedly promote
autophagy, proliferation and malignant processes in PTEN-deficient
prostate organoids, prostate cancer and cervical cancer cells.
Furthermore, by directly simulating the KEAP1/NRF2/ARE signaling
pathway, SQSTM1/p62 may contribute to the aggressive behavior of
prostate cancer (108).
In addition, it seems that SQSTM1/p62 expression in
prostate cancer cells may have some underlying connections with the
surrounding microenvironment. SQSTM1/p62 expression is frequently
upregulated in cancer cells, but downregulated in nearby
microenvironments. For example, emerging studies have validated
that, in contrast to prostate cancer cells, in cancer-associated
fibroblasts (CAFs), SQSTM1/p62 expression is usually decreased. The
lower the expression of SQSTM1/p62, the higher the Gleason grade in
prostate cancer. Notably, SQSTM1/p62 deficiency in adipocytes near
prostate cancer cells can be beneficial to tumor nutrient
availability, the EMT and invasiveness, by increasing osteopontin
secretion and inactivating mTORC1-related energy-consuming pathways
(109). Furthermore, SQSTM1/p62
deletion stimulates the downstream
SQSTM1/p62/mTORC1/c-Myc/glutathione (GSH)/IL-6/TGFβ pathway, in
which mTORC1 and c-Myc inactivation resulting from SQSTM1/p62
deletion decreases GSH expression and stimulates the IL-6/TGFβ
pathway to generate a CAF phenotype that favors malignant
transformation, proliferation and invasion (110). Furthermore, it has been revealed
that both IL-1β and hematopoietic HS-5 derived from cancer-related
bone marrow stromal cells upregulate and phosphorylate SQSTM1/p62
by activating the AMPK pathway in androgen receptor
(AR)-independent prostate cancer, and that phosphorylation of
SQSTM1/p62 recruits and degrades AR, eventually causing repression
of apoptosis, promotion of proliferation and resistance to multiple
drugs (101,111). In summary, downregulation of
SQSTM1/p62 in cancer-related surrounding cells may assist tumor
cells in promoting malignant transformation in prostate cancer.
In renal cell carcinoma, in response to hypoxia,
SQSTM1/p62 can directly integrate and inactivate the
ubiquitin-protein ligase VHL E3 to improve the activity and
stability of hypoxia-inducible factor 1α (HIF1α), which in turn
forces SQSTM1/p62 to trigger the glycolytic pathway. Therefore, the
collaboration of SQSTM1/p62 and HIF1α may provide more energy to
facilitate renal cancer cell survival in harsh environments
(112). By contrast, when the
proteasome degradation pathway is mitigated, SQSTM1/p62 can
directly bind to and direct ubiquitylated HIF2α to autophagosomes,
where HIF2α is degraded through the SQSTM1/p62-dependent autophagic
degradation pathway. Conversely, when the autophagic degradation
pathway is inhibited, ubiquitylated HIF2α can be degraded via the
proteasome degradation pathway (113). These data indicate that
SQSTM1/p62 may function as an oncogene in manipulating the
occurrence and development of renal cell carcinoma by inducing the
action of HIF as a switch between the autophagic degradation
pathway and proteasome degradation pathway.
Moreover, in bladder cancer, overexpressed
SQSTM1/p62 may protect bladder cancer cells from oxidative stress
by stimulating the KEAP1/NRF2 signaling pathway, which upregulates
the expression of several antioxidant genes, such as
glutamate-cysteine ligase catalytic subunit, glutathione
S-transferase Mu 5 and glutathione peroxidase 2, promoting cell
proliferation and inhibiting apoptosis (114).
In summary, SQSTM1/p62 is closely linked not only to
oncogenic characteristics or tumor repressor gene deletions in
tumor cells, but also to tumor microenvironments. For example,
under nutrient-rich or nutrient-deprivation conditions,
inflammation, hypoxia and growth factors can upregulate SQSTM1/p62
expression to stimulate downstream signal transduction, which
activates a variety of oncogenes and accelerates malignant
transformation, growth, proliferation, distant metastasis and drug
resistance in urinary system cancer (Fig. 6).
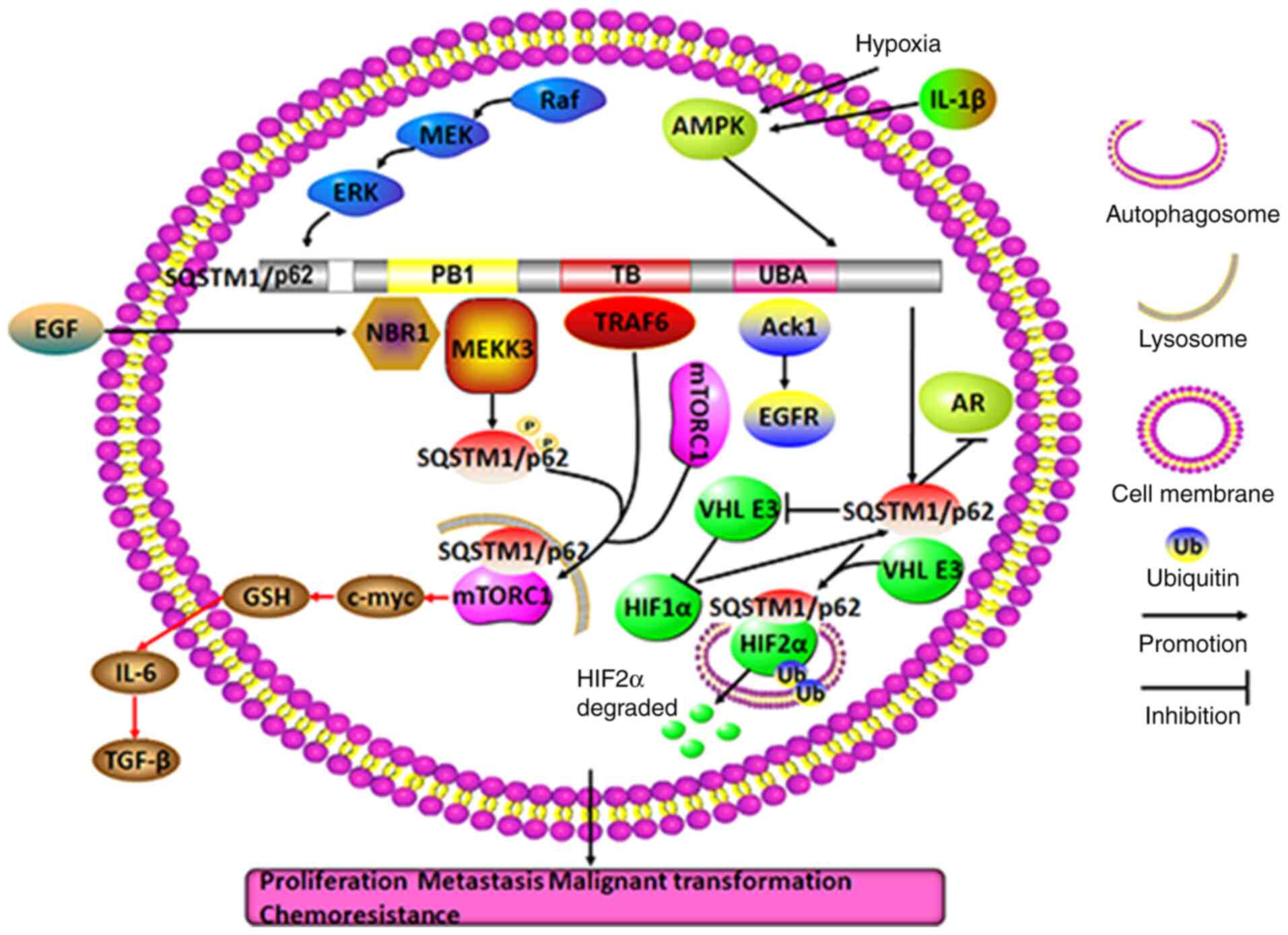 | Figure 6Molecular mechanisms of SQSTM1/p62 in
regulating urological cancer. In prostate cancer, SQSTM1/p62
aggregation and phosphorylation may facilitate autophagy and AR
degradation owing to the activation of the Raf/MEK/ERK pathway and
the IL-1β and hypoxia-activated AMPK pathway, respectively.
Moreover, in a nutrient-rich environment or in the presence of EGF,
SQSTM1/p62 can promote autophagy, proliferation and malignant
transformation by tightly binding either to MEKK3 or NBR1 to
recruit TRAF6 and direct mTORC1 to lysosomes through the SQSTM1/p62
PB1 domain or to Ack1 to prolong EGFR survival through the
SQSTM1/p62 UBA domain. In CAFs, SQSTM1/p62 deletion may cause
stimulation of the mTORC1/c-myc/GSH/IL-6/TGF-β pathway, which
facilitates the action of nearby prostate cancer cells in combating
malignant transformation, proliferation and invasion (red arrow).
In renal cell carcinoma, hypoxia-induced SQSTM1/p62 may not only
directly bind to and drive ubiquitylated HIF2α to autophagosomes
for degradation, but may also form a positive feedback loop with
HIF1α by inactivating VHL E3, facilitating renal cell survival in
unfavorable environments. TRAF6, TNF receptor-associated factor 6;
PB1, Phox and Bem1p; NBR1, neighbor of BRCA1 gene 1; TB,
TRAF6-binding domain; UBA, ubiquitin-associated domain; SQSTM1,
sequestosome 1; NBR1, neighbor of BRCA1; Ack1, activate
Cdc42-associated kinase 1; CAFs, cancer-associated fibroblasts;
mTORC1, mechanistic target of rapamycin complex 1; GSH,
glutathione; HIF1α, hypoxia-inducible factor 1α. |
9. Head and neck neoplasms and skin
cancer
Accumulating evidence has indicated that SQSTM1/p62
may play roles via positive feedback between SQSTM1/p62 and NRF2 in
squamous cell carcinoma (115-119). Highly aggressive oral squamous
carcinoma cells have high SQSTM1/p62 expression in the cytoplasm
and low SQSTM1/p62 expression in the nucleus, but not the opposite
expression trend (115). In
addition, SQSTM1/p62 aggregation in the cytoplasm has been found to
be associated with poor prognosis and drug resistance to PI3K/AKT
inhibitors in autophagy-deficient head and neck squamous cell
carcinoma (116,117). Usually, the change in SQSTM1/p62
expression, to some degree, reflects the change in LC3-II
expression. As autophagy is initiated, LC3-II is formed on
autophagosome; meanwhile SQSTM1/p62 incorporates into the
autophagosome by interacting with LC3-II and then will be degraded
in autolysosomes (9). Autophagy
is triggered in oral squamous cell carcinoma cells, but SQSTM1/p62
expression may not fluctuate with LC3-II. By contrast, in skin
squamous cell carcinoma, strong activation of autophagy and low
expression of SQSTM1/p62 have been correlated with a poor prognosis
(118). Similarly,
arsenic-mediated overexpression of SQSTM1/p62 triggers positive
feedback between SQSTM1/p62 and NRF2, which causes the expression
of numerous secondary messengers, including the glutamate-cysteine
ligase catalytic subunit, HO-1 and NQO1, and promotes keratinocyte
proliferation and malignant transformation (119). Therefore, inducing the oncolytic
virus ΔPK to degrade SQSTM1/p62 by activating the calcium protease
calpain pathway, not the autophagy pathway, may be a novel
therapeutic approach for treating tumor stem cell-rich malignant
melanomas (120). Nevertheless,
SQSTM1/p62, as a direct target of miR-372, can suppress cancer cell
migration by inducing NQO1 expression to repress ROS activation in
head and neck squamous cell carcinoma (121).
Collectively, these observations imply that
SQSTM1/p62 expression is typically higher in the cytoplasm than in
the nucleus, usually indicating that SQSTM1/p62 participates in a
series of downstream cascades involved in oncogene activation.
Thus, inhibiting SQSTM1/p62 activation in different ways or
decreasing its expression may be an effective treatment against
head and neck carcinoma and skin cancer.
10. Nerve and brain tumors
The SQSTM1/p62/NRF2 axis may play a central role in
nerve and brain tumors by participating in autophagy. Increasing
evidence shows that SQSTM1/p62 can cause neuronal degeneration and
neural stem cell differentiation through its abnormal aggregation
and the inhibition of superoxide dismutase (122-124). Additionally, SQSTM1/p62
overexpression, along with NRF2 activation, is capable of
stimulating classical macroautophagy and mitochondrial autophagy,
which allow neuroblastoma cell and glioblastoma multiforme cell
survival by antagonizing apoptosis, resulting in drug resistance to
proteasome inhibitors (125,126). As a result, deleting the
SQSTM1/p62 UBA domain to abolish the defensive function of
autophagy and promote apoptosis may be a potential and powerful way
to inhibit the proteasome in these tumors (127).
In summary, SQSTM1/p62 plays an oncogenic role in
these tumors by engaging in macroautophagy and mitochondrial
autophagy to prevent apoptosis, thereby contributing to
uncontrolled cell growth and unresponsiveness to chemotherapeutic
drugs.
11. EMT
Recently, numerous studies have shown that
SQSTM1/p62 participates in EMT-modulated malignant transformation
(128-132). Tumor cells undergoing EMT shift
from being in an epithelial state to being in a mesenchymal state,
and in the mesenchymal state, a variety of signaling pathways are
activated, including TGF-β/Smad and Wnt/β-catenin pathways, which
is followed by decreased expression of epithelial markers,
including E-cadherin and occludin, and by increased expression of
mesenchymal markers, including vimentin, Twist1, snail1/2 and
N-cadherin. The EMT renders most cancer cells aggressive (128). For instance, in squamous cell
carcinoma of the skin, malignant melanoma, hepatic carcinoma,
breast cancer and bladder cancer, TGF-β-mediated SQSTM1/p62
aggregation in the cytoplasm of autophagy-defective cells can
initiate the EMT process to exacerbate cancer cell growth,
proliferation, migration, invasion and distant metastasis. As
SQSTM1/p62 binds to Smad4 and Twist1 through its UBA domain to
prevent degradation by autophagy and the proteasome pathway, the
activation of Smad4 and Twist1 causes increased N-cadherin and
decreased E-cadherin, occludin and claudin-1 expression, ultimately
stimulating the EMT (129-131). In addition, SQSTM1/p62 can
inhibit autophagy and promote the EMT to accelerate tumor invasion
and metastasis by directly binding with HDAC6 to impair the
acetylation of α-tubulin and microtubules, which leads to disrupted
fusion of autophagosomes and lysosomes, and subsequently to the
impairment of autophagosomal degradation (132). Similarly, in breast cancer MCF-7
cells, in the presence of insulin receptor substrate 1/2,
SQSTM1/p62 releases disheveled 2 (Dvl2) to trigger the EMT by
activating the EMT-related molecules c-Myc and cyclinD1, which are
involved in the Wnt/β-catenin signaling pathway. However, when
autophagy is activated, SQSTM1/p62 transports both Dvl2 and snail2
into autophagosomes, where they are degraded, thereby inhibiting
EMT initiation (133,134).
In summary, functioning as a proto-oncogene or tumor
suppressor gene, SQSTM1/p62 can play a dual role in activating or
silencing EMT. The prerequisite condition for exerting its
biological functions depends largely on whether autophagy is
activated. When autophagy is ongoing, SQSTM1/p62 can block the EMT
process and malignant cancer progression by promoting the
degradation of EMT-related factors through the autophagy pathway.
By contrast, when autophagy is inhibited or damaged, SQSTM1/p62
activates the EMT signaling pathways and promotes cancer cell
malignant transformation by binding to and stabilizing EMT-related
factors (Fig. 7).
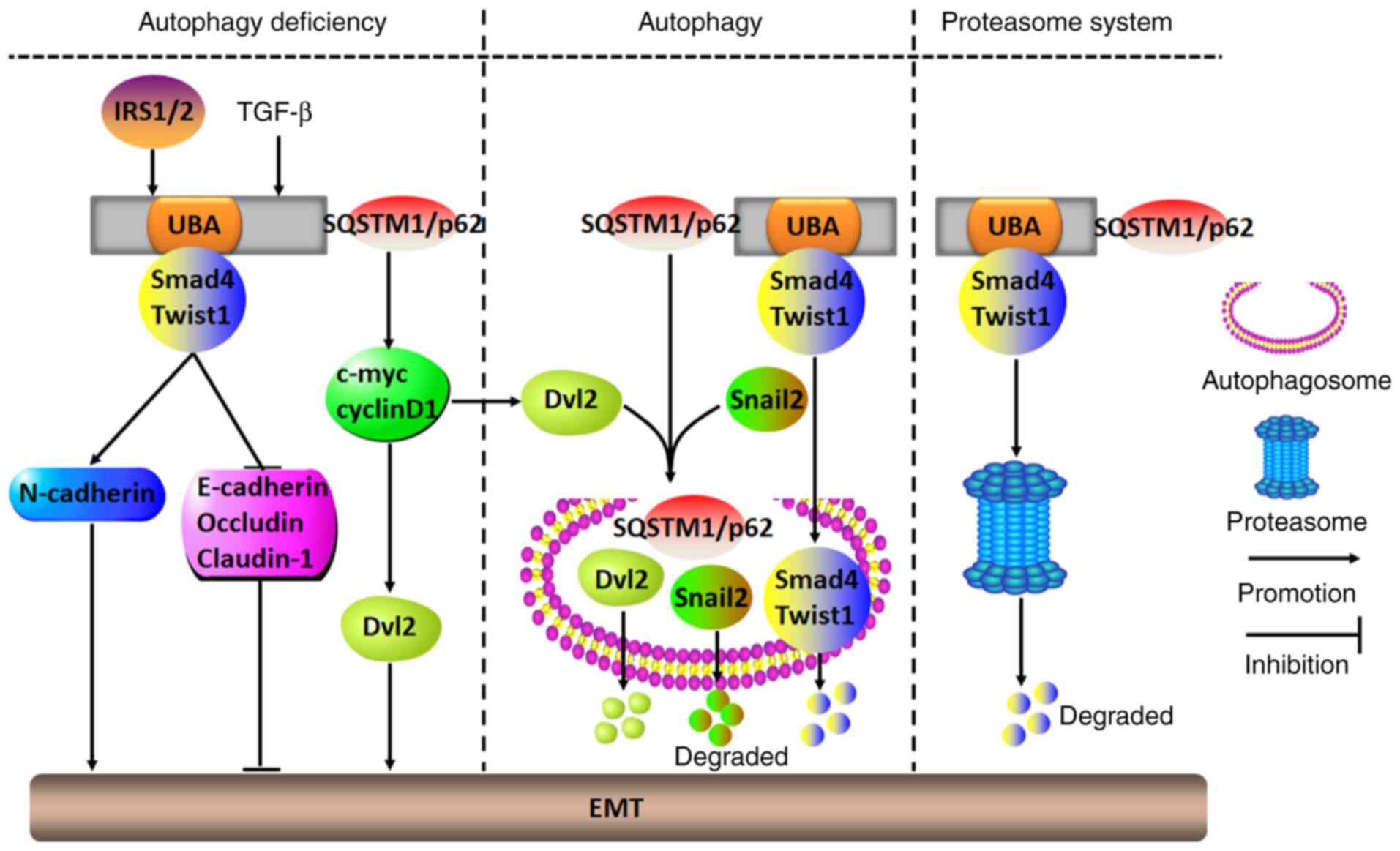 | Figure 7Molecular mechanisms of SQSTM1/p62 in
regulating the tumor EMT. In numerous tumor cells with deficient
autophagy, IRS1/2- or TGF-β-induced SQSTM1/p62 aggregation may
initiate the EMT to accelerate malignant aggression by either
binding to and stabilizing Smad4 and Twist1 through its UBA domain
or stimulating c-myc and cyclinD1 to release Dvl2. However, when
cells show autophagic ability, SQSTM1/p62 may act as a carrier to
transport EMT-related molecules, including Dvl2, snail2, Smad4 and
Twist1, to autophagosomes for their degradation, which results in
inhibition of the EMT. Alternatively, SQSTM1/p62 may also repress
the EMT by directing Smad4 and Twist1 into proteasomes for
degradation. UBA, ubiquitin-associated domain; Dvl2, Disheveled 2;
EMT, epithelial-mesenchymal transition; SQSTM1, sequestosome 1;
IRS1/2, insulin receptor substrate 1/2. |
12. Conclusions and prospects
To date, the molecular mechanisms and functions of
SQSTM1/p62 in regulating malignant progression are unclear, but
SQSTM1/p62 has been shown to participate in the occurrence and
development of various tumors in multiple ways, including by
affecting genetic stability, transcription and post-transcriptional
regulation, to modify and interact with different molecules.
For instance, at the transcriptional level, either
NRF2 or TNF may upregulate SQSTM1/p62 mRNA expression to facilitate
hepatocellular malignant transformation of hepatocytes or bronchial
epithelial cells by directly binding the promoter region of
SQSTM1/p62 (40,79). At the post-transcriptional level,
SQSTM1/p62 may be directly targeted by miR-372 to inhibit cancer
cell migration in head and neck squamous cell carcinoma (121), and it may also be targeted by
miR-487a to promote malignant progression of esophageal cancer
cells (135). Notably, high
levels of the lncRNA associated with small nucleolar RNA host gene
16 may increase SQSTM1/p62 expression by directly inhibiting its
target molecule miR-17-5p, a miRNA that directly targets
SQSTM1/p62, to facilitate HCC cell proliferation, migration and
invasion (136). Since
SQSTM1/p62 can function as an oncogene or tumor suppressor gene in
different tumors under different circumstances (Fig. 8), SQSTM1/p62 can be considered to
be like a multifunctional ship, ferry or car trafficking different
bad or good cargos to different factories for further
processing.
Under normal conditions, SQSTM1/p62 expression is
inversely associated with LC3-II expression when autophagy is
activated; however, to a certain extent, elevated SQSTM1/p62
expression not only induces autophagy and autophagic cell death,
but also inhibits autophagy, and vice versa. These results
demonstrate that, in addition to the classical autophagy pathway,
SQSTM1/p62 participates in a variety of signaling pathways to
regulate tumor progression. However, until recently, the
relationships between autophagy and SQSTM1/p62 had not been well
known. Therefore, although guidelines for monitoring autophagy have
been described, these data indicate that monitoring autophagic flux
merely by assessing the fluctuation in SQSTM1/p62 and LC3-II levels
is insufficient, and it is advisable to assess the functional
status of the ubiquitin proteasome system (137). As SQSTM1/p62 acts as a bridge or
an axis between the autophagy pathway and the ubiquitin-proteasome
degradation pathway, when either pathway is disrupted, SQSTM1/p62
activity may be diverted to the other pathway.
In an overwhelming majority of cases, cancer cells
with SQSTM1/p62 aggregation in the cytoplasm are more aggressive
than those in which SQSTM1/p62 resides only in the nucleus,
implying that excessively activated SQSTM1/p62 shuttling from the
nucleus to the cytoplasm is involved in diverse signaling pathways,
particularly the EMT-related TGF-β/Smad and Wnt/β-catenin,
SQSTM1/p62/KEAP1/NRF2 and SQSTM1/p62/TRAF6/NF-κB pathways. The
formation of positive feedback loops clearly amplifies the effects
of carcinogenesis, which results in pleiotropic effects on the
clinicopathological parameters of human tumors (Table I). As drug resistance in malignant
cancer is the most critical challenge to effective treatment
encountered thus far, it is of great significance to search for
specific molecule-targeted drugs. It is promising that recent
studies have demonstrated that XRK3F2, a novel small-molecule
inhibitor specifically targeting the SQSTM1/p62-ZZ domain, can
selectively impair leukemia-initiating cells in AML and multiple
myeloma cells to inhibit tumor aggression by inhibiting the
SQSTM1/p62-ZZ domain-dependent autophagic or TNFα plus IL17
signaling pathways, respectively; therefore, XRK3F2 may be, to some
extent, a potentially useful tool for targeting therapies
specifically to these tumors in the future (138,139). In addition, the crosstalk
between components in tumor microenvironments (SQSTM1/p62 and
various autocrine or paracrine factors originating from tumor
stromal cells, etc.) and tumor cells may markedly increase the
expression of SQSTM1/p62 in tumor cells by augmenting or inhibiting
specific signaling pathways, as well as by boosting tumor stem cell
self-renewal, malignant tumor growth and aggression, and
chemoradiotherapeutic resistance. To date, the communication
between tumor microenvironments and tumor cells has been unclear,
and further understanding may elucidate the underlying mechanisms
of SQSTM1/p62 serving as a bridge between tumor microenvironments
and tumor cells. A SQSTM1/p62 DNA vaccine or gene fusion vaccine
based on the SQSTM1/p62 gene and tumor antigens has already
exhibited complementary antitumor effects and few adverse effects
in experimental subjects, such as dogs, mice, and rats, as well as
in breast cancer, lung cancer, melanoma, ovarian cancer, renal
cancer and sarcoma; these effects were achieved by augmenting
immunoreactions among CD3-positive cells surrounding the tumor
cells and stimulating a fibrotic response around the tumor cells
(140-143). Therefore, unmasking the
underlying mechanisms of SQSTM1/p62 in regulating the tumor
microenvironment and tumor cells may be very worthwhile and lead to
an increase in the effective precision therapy treatments available
for various tumors in the future.
 | Table IClinicopathological significance and
biological pathways of SQSTM1/p62 in human tumors. |
Table I
Clinicopathological significance and
biological pathways of SQSTM1/p62 in human tumors.
| Tumors | Location and
expression | Pathways | Vessel
invasion | Lymph node and
distant metastasis | TNM stage |
Chemoresistance | Prognosis | (Refs.) |
|---|
| Esophageal
adenocarcinoma | Cytoplasm ↑,
Nucleus↑ | N.A. | N.A. | Positive | N.A. | N.A. | Good | (25) |
| Gastric cancer | Cytoplasm ↑,
Nucleus↑ | N.A. | Positive | Positive or
Negative | Positive or No | N.A. | Poor | (24,26) |
| Colorectal
cancer | Cytoplasm↑,
Nucleus↑or↓ | Autophagy, MEK/ERK
PI3K/mTOR, Wnt/β-catenin, ATM/γH2AX | N.A. | No | No | Yes | Good or No | (24,27, 29-32,34) |
| Pancreatic
adenocarcinoma | Cytoplasm ↑,
Nucleus↑ | N.A. | N.A. | No | No | N.A. | No | (24) |
| HCC | Cytoplasm↑ | Vitamin D,
SQSTM1/p62/KEAP1/NRF2, SQSTM1/p62/TRAF6/NF-κB,
SQSTM1/p62/HDAC6 | N.A. | N.A. | N.A. | Yes | Poor | (36-40, 42-44) |
| Breast cancer | Cytoplasm ↑,
Nucleus↑ |
CD44/SQSTM1/p62/NRF2,
SQSTM1/p62/KEAP1/NRF2, SQSTM1/p62/TRAF6/NF-κB, PI3K/AKT
Wnt/β-catenin, AKT/GSK3β/β-catenin, ER/ERK, Wnt/PCP,
IRS1/2/SQSTM1/P62/Dvl2, TRAF6-Beclin1 | Positive | Positive | Positive or No | Yes | Poor | (46-55, 57-61, 90,133) |
| Multiple
myeloma | Cytoplasm↑ |
SQSTM1/P62/TRAF6/NF-κB,
SQSTM1/P62/RIP1/NF-κB, SQSTM1/P62/aPKCs/NF-κB,
SQSTM1/p62/P38MAPK | N.A. | N.A. | N.A. | Yes | Poor | (62-66, 69,70) |
| HR MDS/AML | Cytoplasm↑,
Nucleus↑ |
SQSTM1/p62/TRAF6/NF-κB | N.A. | N.A. | N.A. | N.A. | Poor | (62,63) |
| Chronic myeloid
leukemia | Cytoplasm↑ | Autophagy | N.A. | N.A. | N.A. | No | N.A. | (73) |
| Lung cancer | Cytoplasm↑ | mTOR-ULK1-Beclin1,
SQSTM1/p62/TRAF6/NF-κB, SQSTM1/p62/KEAP1/NRF2, TRAF6-Beclin1 | N.A. | Negative | Negative | Yes | Poor | (77-83,90) |
| Epithelial ovarian
cancer | Cytoplasm↑ or↓ |
SQSTM1/p62/KEAP1/NRF2, SQSTM1/p62/caspase
8 | N.A. | Positive | Positivea | Yes | Poor | (91,92, 94,99, 100) |
| Seminoma | Cytoplasm/Nucleus,
NC | N.A. | N.A. | N.A. | N.A. | N.A. | N.A. | (95) |
| Non-seminoma | Cytoplasm/Nucleus,
NC | N.A. | N.A. | N.A. | N.A. | N.A. | N.A. | (95) |
| Endometrial
cancer | Cytoplasm↑ | N.A. | N.A. | Positive | No | N.A. | Poor | (89) |
| Serous
carcinoma | Cytoplasm↑ | N.A. | N.A. | Positive | No | N.A. | Poor | (96) |
| Cervical
cancer | Cytoplasm ↑,
Nucleus↑ |
SQSTM1/p62/NRF2 | N.A. | N.A. | N.A. | N.A. | N.A. | (97) |
| Prostate
cancer | Cytoplasm↑ | SQSTM1/p62/NRF2,
Raf/MEK/ERK, SQSTM1/p62/MEKK3, SQSTM1/P62/TRAF6/mTORC1,
SQSTM1/p62/mTORC1/c-myc/GSH/IL-6/TGFβ, AMPK/SQSTM1/p62/AR | N.A. | Positive | Positive | Yes | Poor | (101,103, 105-111) |
| Malignant
melanoma | Cytoplasm↑ | Raf/MEK/ERK,
EGF/TGFβ/Twist1 | N.A. | N.A. | N.A. | N.A. | N.A. | (105,128) |
| Renal cell
carcinoma | Cytoplasm↑ | Glycolytic pathway,
Autophagic pathway, Proteasome degradation pathway | N.A. | N.A. | N.A. | N.A. | N.A. | (112,113) |
| Bladder cancer | Cytoplasm↑ | TGFβ/Smad4,
SQSTM1/p62/KEAP1/NRF2 | N.A. | N.A. | N.A. | N.A. | N.A. | (115,129) |
| Oral SCC | Cytoplasm↑,
Nucleus↓ | N.A. | Positive | Positive | Positive | N.A. | Poor | (115) |
| Head and neck
SCC | Cytoplasm↑ | PI3K/AKT,
SQSTM1/p62/NRF2 | N.A. | N.A. | N.A. | Yes | N.A. | (116) |
| Skin SCC | Cytoplasm↓ or↑ | SQSTM1/P62/NRF2,
EGF/TGFβ/Twist1 | N.A. | Negative | Negative | N.A. | Poor | (117-119, 129) |
| Neuroblastoma
mitophagy | Cytoplasm↑ |
SQSTM1/P62/NRF2, | N.A. | N.A. | N.A. | Yes | N.A. | (125) |
| Glioblastoma | Cytoplasm↑ | SQSTM1/P62/NF-κB,
autophagy | N.A. | N.A. | N.A. | Yes | N.A. | (126) |
Availability of data and materials
Not applicable.
Authors' contributions
JT drafted the manuscript and YL created the
figures; all the images have been drawn using with Pathway Builder
Tool 2.0 (www.proteinlounge.com) and ScienceSlides 2016 version
(www.visiscience.com). SX and YL
participated in the literature collection, JL and QY designed and
created the tables, and HZ and KD designed the study and supervised
the creation of the manuscript. All authors have read and approved
the final manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Professor Iain
Charles Bruce (College of Medicine, Zhejiang University, Hangzhou,
China) for providing English language assistance.
Abbreviations:
|
SQSTM1
|
sequestosome 1
|
|
UBA
|
ubiquitin-associated domain
|
|
PB1
|
Phox and Bem1p
|
|
LIR
|
LC3-interacting region
|
|
HSP90
|
heat shock protein 90
|
|
HCC
|
hepatocellular carcinoma
|
|
NRF2
|
nuclear factor erythroid 2
|
|
TRAF6
|
TNF receptor-associated factor 6
|
|
ARE
|
antioxidant response element
|
|
JNK
|
Jun N-terminal kinase
|
|
KEAP1
|
Kelch-like ECH-associated protein
1
|
|
NQO1
|
NAD(P)H dehydrogenase quinone 1
|
|
HDAC6
|
histone deacetylase 6
|
|
CSC
|
cancer stem cell
|
|
ROS
|
reactive oxygen species
|
|
PI3K
|
phosphoinositol-3 kinase
|
|
KLF4
|
Kruppel-like factor 4
|
|
ATRA
|
all-trans retinoic acid
|
|
AML
|
acute myeloid leukemia
|
|
NSCLC
|
non-small cell lung cancer
|
|
NBR1
|
neighbor of BRCA1
|
|
Ack1
|
activate Cdc42-associated kinase
1
|
|
CAFs
|
cancer-associated fibroblasts
|
|
EMT
|
epithelial-mesenchymal transition
|
|
AR
|
androgen
|
|
HIF1α
|
hypoxia-inducible factor 1α
|
|
Dvl2
|
disheveled 2
|
|
ECSIT
|
evolutionarily conserved signaling
intermediate in toll pathways
|
References
|
1
|
Park I, Chung J, Walsh CT, Yun Y,
Strominger JL and Shin J: Phosphotyrosine-independent binding of a
62-kDa protein to the src homology 2 (SH2) domain of p56lck and its
regulation by phosphorylation of Ser-59 in the lck unique
N-terminal region. Proc Natl Acad Sci USA. 92:12338–12342. 1995.
View Article : Google Scholar
|
|
2
|
Moscat J and Diaz-Meco MT: p62 at the
crossroads of autophagy, apoptosis, and cancer. Cell.
137:1001–1004. 2009. View Article : Google Scholar
|
|
3
|
Ishaq M, Khan MA, Sharma K, Sharma G,
Dutta RK and Majumdar S: Gambogic acid induced oxidative stress
dependent caspase activation regulates both apoptosis and autophagy
by targeting various key molecules (NF-κB, Beclin-1, p62 and NBR1)
in human bladder cancer cells. Biochim Biophys Acta.
1840:3374–3384. 2014. View Article : Google Scholar
|
|
4
|
Li S, Yang G, Zhu X, Cheng L, Sun Y and
Zhao Z: Combination of rapamycin and garlic-derived
S-allylmercaptocysteine induces colon cancer cell apoptosis and
suppresses tumor growth in xenograft nude mice through
autophagy/p62/Nrf2 pathway. Oncol Rep. 38:1637–1644. 2017.
View Article : Google Scholar
|
|
5
|
Wang Y, Zhang N, Zhang L, Li R, Fu W, Ma
K, Li X, Wang L, Wang J, Zhang H, et al: Autophagy regulates
chromatin ubiquitination in DNA damage response through elimination
of SQSTM1/p62. Mol Cell. 63:34–48. 2016. View Article : Google Scholar
|
|
6
|
Lee Y and Weihl CC: Regulation of
SQSTM1/p62 via UBA domain ubiquitination and its role in disease.
Autophagy. 13:1615–1616. 2017. View Article : Google Scholar
|
|
7
|
Seibenhener ML, Babu JR, Geetha T, Wong
HC, Krishna NR and Wooten MW: Sequestosome 1/p62 is a polyubiquitin
chain binding protein involved in ubiquitin proteasome degradation.
Mol Cell Biol. 24:8055–8068. 2004. View Article : Google Scholar
|
|
8
|
Cohen-Kaplan V, Livneh I, Avni N, Fabre B,
Ziv T, Kwon YT and Ciechanover A: p62- and ubiquitin-dependent
stress-induced autophagy of the mammalian 26S proteasome. Proc Natl
Acad Sci USA. 113:E7490–E7499. 2016. View Article : Google Scholar
|
|
9
|
Klionsky DJ, Abdel-Aziz AK, Abdelfatah S,
Abdellatif M, Abdoli A, Abel S, Abeliovich H, Abildgaard MH, Abudu
YP, Acevedo-Arozena A, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy (4th edition) 1.
Autophagy. 17:1–382. 2021. View Article : Google Scholar
|
|
10
|
Matsumoto G, Wada K, Okuno M, Kurosawa M
and Nukina N: Serine 403 phosphorylation of p62/SQSTM1 regulates
selective autophagic clearance of ubiquitinated proteins. Mol Cell.
44:279–289. 2011. View Article : Google Scholar
|
|
11
|
Zhu L, Zhu Y, Han S, Chen M, Song P, Dai
D, Xu W, Jiang T, Feng L, Shin VY, et al: Impaired autophagic
degradation of lncRNA ARHGAP5-AS1 promotes chemoresistance in
gastric cancer. Cell Death Dis. 10:3832019. View Article : Google Scholar
|
|
12
|
Clausen TH, Lamark T, Isakson P, Finley K,
Larsen KB, Brech A, Øvervatn A, Stenmark H, Bjørkøy G, Simonsen A
and Johansen T: p62/SQSTM1 and ALFY interact to facilitate the
formation of p62 bodies/ALIS and their degradation by autophagy.
Autophagy. 6:330–344. 2010. View Article : Google Scholar
|
|
13
|
Ichimura Y, Waguri S, Sou YS, Kageyama S,
Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, et
al: Phosphorylation of p62 activates the Keap1-Nrf2 pathway during
selective autophagy. Mol Cell. 51:618–631. 2013. View Article : Google Scholar
|
|
14
|
Park JY, Sohn HY, Koh YH and Jo C:
Curcumin activates Nrf2 through PKCδ-mediated p62 phosphorylation
at Ser351. Sci Rep. 11:84302021. View Article : Google Scholar
|
|
15
|
Ling J, Kang Y, Zhao R, Xia Q, Lee DF,
Chang Z, Li J, Peng B, Fleming JB, Wang H, et al: KrasG12D-induced
IKK2/β/NF-κB activation by IL-1α and p62 feedforward loops is
required for development of pancreatic ductal adenocarcinoma.
Cancer Cell. 21:105–120. 2012. View Article : Google Scholar
|
|
16
|
Duran A, Linares JF, Galvez AS,
Wikenheiser K, Flores JM, Diaz-Meco MT and Moscat J: The signaling
adaptor p62 is an important NF-kappaB mediator in tumorigenesis.
Cancer Cell. 13:343–354. 2008. View Article : Google Scholar
|
|
17
|
Nakamura K, Kimple AJ, Siderovski DP and
Johnson GL: PB1 domain interaction of p62/sequestosome 1 and MEKK3
regulates NF-kappaB activation. J Biol Chem. 285:2077–2089. 2010.
View Article : Google Scholar
|
|
18
|
Usategui-Martin R, Gestoso-Uzal N,
Calero-Paniagua I, De Pereda JM, Del Pino-Montes J and
González-Sarmiento R: A mutation in p62 protein (R321C), associated
to Paget's disease of bone, causes a blockade of autophagy and an
activation of NF-kB pathway. Bone. 133:1152652020. View Article : Google Scholar
|
|
19
|
Wang Y, Xiong H, Liu D, Hill C, Ertay A,
Li J, Zou Y, Miller P, White E, Downward J, et al: Autophagy
inhibition specifically promotes epithelial-mesenchymal transition
and invasion in RAS-mutated cancer cells. Autophagy. 15:886–899.
2019. View Article : Google Scholar
|
|
20
|
Hua F and Hu ZW: TRIB3-P62 interaction,
diabetes and autophagy. Oncotarget. 6:34061–34062. 2015. View Article : Google Scholar
|
|
21
|
Hewitt G, Carroll B, Sarallah R,
Correia-Melo C, Ogrodnik M, Nelson G, Otten EG, Manni D, Antrobus
R, Morgan BA, et al: SQSTM1/p62 mediates crosstalk between
autophagy and the UPS in DNA repair. Autophagy. 12:1917–1930. 2016.
View Article : Google Scholar
|
|
22
|
Wang Y, Zhu WG and Zhao Y: Autophagy
substrate SQSTM1/p62 regulates chromatin ubiquitination during the
DNA damage response. Autophagy. 13:212–213. 2017. View Article : Google Scholar
|
|
23
|
Wang L, Howell MEA, Sparks-Wallace A,
Hawkins C, Nicksic CA, Kohne C, Hall KH, Moorman JP, Yao ZQ and
Ning S: p62-mediated selective autophagy endows virus-transformed
cells with insusceptibility to DNA damage under oxidative stress.
PLoS Pathog. 15:e10075412019. View Article : Google Scholar
|
|
24
|
Mohamed A, Ayman A, Deniece J, Wang T,
Kovach C, Siddiqui MT and Cohen C: P62/Ubiquitin IHC expression
correlated with clinicopathologic parameters and outcome in
gastrointestinal carcinomas. Front Oncol. 5:702015. View Article : Google Scholar
|
|
25
|
Adams O, Dislich B, Berezowska S, Schläfli
AM, Seiler CA, Kröll D, Tschan MP and Langer R: Prognostic
relevance of autophagy markers LC3B and p62 in esophageal
adenocarcinomas. Oncotarget. 7:39241–39255. 2016. View Article : Google Scholar
|
|
26
|
Masuda GO, Yashiro M, Kitayama K, Miki Y,
Kasashima H, Kinoshita H, Morisaki T, Fukuoka T, Hasegawa T,
Sakurai K, et al: Clinicopathological correlations of
autophagy-related proteins LC3, beclin 1 and p62 in gastric cancer.
Anticancer Res. 36:129–136. 2016.
|
|
27
|
Park JM, Huang S, Wu TT, Foster NR and
Sinicrope FA: Prognostic impact of beclin 1p62/sequestosome 1 and
LC3 protein expression in colon carcinomas from patients receiving
5-fluorouracil as adjuvant chemotherapy. Cancer Biol Ther.
14:100–107. 2013. View Article : Google Scholar
|
|
28
|
Kosumi K, Masugi Y, Yang J, Qian ZR, Kim
SA, Li W, Shi Y, da Silva A, Hamada T, Liu L, et al: Tumor SQSTM1
(p62) expression and T cells in colorectal cancer. Oncoimmunology.
6:e12847202017. View Article : Google Scholar
|
|
29
|
Schmitz KJ, Ademi C, Bertram S, Schmid KW
and Baba HA: Prognostic relevance of autophagy-related markers LC3,
p62/sequestosome 1, beclin-1 and ULK1 in colorectal cancer patients
with respect to KRAS mutational status. World J Surg Oncol.
14:1892016. View Article : Google Scholar
|
|
30
|
Goulielmaki M, Koustas E, Moysidou E,
Vlassi M, Sasazuki T, Shirasawa S, Zografos G, Oikonomou E and
Pintzas A: BRAF associated autophagy exploitation: BRAF and
autophagy inhibitors synergise to efficiently overcome resistance
of BRAF mutant colorectal cancer cells. Oncotarget. 7:9188–9221.
2016. View Article : Google Scholar
|
|
31
|
Ren F, Shu G, Liu G, Liu D and Zhou J,
Yuan L and Zhou J: Knockdown of p62/sequestosome 1 attenuates
autophagy and inhibits colorectal cancer cell growth. Mol Cell
Biochem. 385:95–102. 2014. View Article : Google Scholar
|
|
32
|
Petherick KJ, Williams AC, Lane JD,
Ordóñez-Morán P, Huelsken J, Collard TJ, Smartt HJ, Batson J, Malik
K, Paraskeva C and Greenhough A: Autolysosomal β-catenin
degradation regulates Wnt-autophagy-p62 crosstalk. EMBO J.
32:1903–1916. 2013. View Article : Google Scholar
|
|
33
|
Samarasinghe B, Wales CT, Taylor FR and
Jacobs AT: Heat shock factor 1 confers resistance to Hsp90
inhibitors through p62/SQSTM1 expression and promotion of
autophagic flux. Biochem Pharmacol. 87:445–455. 2014. View Article : Google Scholar
|
|
34
|
Wang Z, Chen Q, Li B, Xie JM, Yang XD,
Zhao K, Wu Y, Ye ZY, Chen ZR, Qin ZH, et al: Escin-induced DNA
damage promotes escin-induced apoptosis in human colorectal cancer
cells via p62 regulation of the ATM/γH2AX pathway. Acta Pharmacol
Sin. 39:1645–1660. 2018. View Article : Google Scholar
|
|
35
|
Kim JH and Kim IW: p62 manipulation
affects chlorin e6-mediated photodynamic therapy efficacy in
colorectal cancer cell lines. Oncol Lett. 19:3907–3916. 2020.
|
|
36
|
Duran A, Hernandez ED, Reina-Campos M,
Castilla EA, Subramaniam S, Raghunandan S, Roberts LR, Kisseleva T,
Karin M, Diaz-Meco MT and Moscat J: p62/SQSTM1 by binding to
vitamin D receptor inhibits hepatic stellate cell activity,
fibrosis, and liver cancer. Cancer Cell. 30:595–609. 2016.
View Article : Google Scholar
|
|
37
|
Shimizu T, Inoue K, Hachiya H, Shibuya N,
Aoki T and Kubota K: Accumulation of phosphorylated p62 is
associated with NF-E2-related factor 2 activation in hepatocellular
carcinoma. J Hepatobiliary Pancreat Sci. 23:467–471. 2016.
View Article : Google Scholar
|
|
38
|
Saito T, Ichimura Y, Taguchi K, Suzuki T,
Mizushima T, Takagi K, Hirose Y, Nagahashi M, Iso T, Fukutomi T, et
al: p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular
carcinoma through Nrf2-dependent metabolic reprogramming. Nat
Commun. 7:120302016. View Article : Google Scholar
|
|
39
|
Jain A, Lamark T, Sjottem E, Larsen KB,
Awuh JA, Øvervatn A, McMahon M, Hayes JD and Johansen T: p62/SQSTM1
is a target gene for transcription factor NRF2 and creates a
positive feedback loop by inducing antioxidant response
element-driven gene transcription. J Biol Chem. 285:22576–22591.
2010. View Article : Google Scholar
|
|
40
|
Umemura A, He F, Taniguchi K, Nakagawa H,
Yamachika S, Font-Burgada J, Zhong Z, Subramaniam S, Raghunandan S,
Duran A, et al: p62, upregulated during preneoplasia, induces
hepatocellular carcinogenesis by maintaining survival of stressed
HCC-initiating cells. Cancer Cell. 29:935–948. 2016. View Article : Google Scholar
|
|
41
|
Hoadley KA, Yau C, Wolf DM, Cherniack AD,
Tamborero D, Ng S, Leiserson MDM, Niu B, McLellan MD, Uzunangelov
V, et al: Multiplatform analysis of 12 cancer types reveals
molecular classification within and across tissues of origin. Cell.
158:929–944. 2014. View Article : Google Scholar
|
|
42
|
Taguchi K, Fujikawa N, Komatsu M, Ishii T,
Unno M, Akaike T, Motohashi H and Yamamoto M: Keap1 degradation by
autophagy for the maintenance of redox homeostasis. Proc Natl Acad
Sci USA. 109:13561–13566. 2012. View Article : Google Scholar
|
|
43
|
Vegliante R, Desideri E, Di Leo L and
Ciriolo MR: Dehydroepiandrosterone triggers autophagic cell death
in human hepatoma cell line HepG2 via JNK-mediated p62/SQSTM1
expression. Carcinogenesis. 37:233–244. 2016. View Article : Google Scholar
|
|
44
|
Yan J, Seibenhener ML, Calderilla-Barbosa
L, Diaz-Meco MT, Moscat J, Jiang J, Wooten MW and Wooten MC:
SQSTM1/p62 interacts with HDAC6 and regulates deacetylase activity.
PLoS One. 8:e760162013. View Article : Google Scholar
|
|
45
|
Chen Q, Yue F, Li W, Zou J, Xu T, Huang C,
Zhang Y, Song K, Huang G, Xu G, et al: Potassium
bisperoxo(1,10-phenanthroline) oxovanadate (bpV(phen)) induces
apoptosis and pyroptosis and disrupts the P62-HDAC6 protein
interaction to suppress the acetylated microtubule-dependent
degradation of autophagosomes. J Biol Chem. 290:26051–26058. 2015.
View Article : Google Scholar
|
|
46
|
Ryoo IG, Choi BH, Ku SK and Kwak MK: High
CD44 expression mediates p62-associated NFE2L2/NRF2 activation in
breast cancer stem cell-like cells: Implications for cancer stem
cell resistance. Redox Biol. 17:246–258. 2018. View Article : Google Scholar
|
|
47
|
Ryoo IG, Choi BH and Kwak MK: Activation
of NRF2 by p62 and proteasome reduction in sphere-forming breast
carcinoma cells. Oncotarget. 6:8167–8184. 2015. View Article : Google Scholar
|
|
48
|
Jain A, Rusten TE, Katheder N, Elvenes J,
Bruun JA, Sjøttem E, Lamark T and Johansen T: p62/sequestosome-1,
autophagy-related gene 8, and autophagy in drosophila are regulated
by nuclear factor erythroid 2-related factor 2 (NRF2), independent
of transcription factor TFEB. J Biol Chem. 290:14945–14962. 2015.
View Article : Google Scholar
|
|
49
|
Xu LZ, Li SS, Zhou W, Kang ZJ, Zhang QX,
Kamran M, Xu J, Liang DP, Wang CL, Hou ZJ, et al: p62/SQSTM1
enhances breast cancer stem-like properties by stabilizing MYC
mRNA. Oncogene. 36:304–317. 2017. View Article : Google Scholar
|
|
50
|
Gao C, Cao W, Bao L, Zuo W, Xie G, Cai T,
Fu W, Zhang J, Wu W, Zhang X and Chen YG: Autophagy negatively
regulates Wnt signalling by promoting dishevelled degradation. Nat
Cell Biol. 12:781–790. 2010. View Article : Google Scholar
|
|
51
|
Ahn JS, Ann EJ, Kim MY, Yoon JH, Lee HJ,
Jo EH, Lee K, Lee JS and Park HS: Autophagy negatively regulates
tumor cell proliferation through phosphorylation dependent
degradation of the Notch1 intracellular domain. Oncotarget.
7:79047–79063. 2016. View Article : Google Scholar
|
|
52
|
Cai-McRae X and Karantza V: p62: A hub of
multiple signaling pathways in HER2-induced mammary tumorigenesis.
Mol Cell Oncol. 2:e9750352015. View Article : Google Scholar
|
|
53
|
Cai-McRae X, Zhong H and Karantza V:
Sequestosome 1/p62 facilitates HER2-induced mammary tumorigenesis
through multiple signaling pathways. Oncogene. 34:2968–2977. 2015.
View Article : Google Scholar
|
|
54
|
Wei H, Wang C, Croce CM and Guan JL:
p62/SQSTM1 synergizes with autophagy for tumor growth in vivo.
Genes Dev. 28:1204–1216. 2014. View Article : Google Scholar
|
|
55
|
Puvirajesinghe TM, Bertucci F, Jain A,
Scerbo P, Belotti E, Audebert S, Sebbagh M, Lopez M, Brech A,
Finetti P, et al: Identification of p62/SQSTM1 as a component of
non-canonical Wnt VANGL2-JNK signalling in breast cancer. Nat
Commun. 7:103182016. View Article : Google Scholar
|
|
56
|
Chen S, Zhou L, Zhang Y, Leng Y, Pei XY,
Lin H, Jones R, Orlowski RZ, Dai Y and Grant S: Targeting
SQSTM1/p62 induces cargo loading failure and converts autophagy to
apoptosis via NBK/Bik. Mol Cell Biol. 34:3435–3449. 2014.
View Article : Google Scholar
|
|
57
|
Choi YK, Cho SG, Woo SM, Yun YJ, Park S,
Shin YC and Ko SG: Herbal extract SH003 suppresses tumor growth and
metastasis of MDA-MB-231 breast cancer cells by inhibiting
STAT3-IL-6 signaling. Mediators Inflamm. 2014:4921732014.
View Article : Google Scholar
|
|
58
|
Luo RZ, Yuan ZY, Li M, Xi SY, Fu J and He
J: Accumulation of p62 is associated with poor prognosis in
patients with triple-negative breast cancer. Onco Targets Ther.
6:883–888. 2013.
|
|
59
|
Wei Y, Liu D, Jin X, Gao P, Wang Q, Zhang
J and Zhang N: PA-MSHA inhibits the growth of doxorubicin-resistant
MCF-7/ADR human breast cancer cells by downregulating Nrf2/p62.
Cancer Med. 5:3520–3531. 2016. View Article : Google Scholar
|
|
60
|
Fuchinoue F, Hirotani Y, Nakanishi Y,
Yamaguchi H, Nishimaki H, Noda H, Tang XY, Iizuka M, Amano S,
Sugitani M, et al: Overexpression of PGC1α and accumulation of p62
in apocrine carcinoma of the breast. Pathol Int. 65:19–26. 2015.
View Article : Google Scholar
|
|
61
|
Nozaki F, Hirotani Y, Nakanishi Y,
Yamaguchi H, Nishimaki H, Noda H, Tang X, Yamamoto H, Suzuki A,
Seki T and Masuda S: p62 regulates the proliferation of molecular
apocrine breast cancer cells. Acta Histochem Cytochem. 49:125–130.
2016. View Article : Google Scholar
|
|
62
|
Shen P, Chen M, He M, Chen L, Song Y, Xiao
P, Wan X, Dai F, Pan T and Wang Q: Inhibition of ERα/ERK/P62
cascades induces 'autophagic switch' in the estrogen
receptor-positive breast cancer cells exposed to gemcitabine.
Oncotarget. 7:48501–48516. 2016. View Article : Google Scholar
|
|
63
|
Fang J and Starczynowski DT: Genomic
instability establishes dependencies on acquired gene regulatory
networks: A novel role of p62 in myeloid malignancies with del(5q).
Mol Cell Oncol. 2:e10142192015. View Article : Google Scholar
|
|
64
|
Fang J, Barker B, Bolanos L, Liu X, Jerez
A, Makishima H, Christie S, Chen X, Rao DS, Grimes HL, et al:
Myeloid malignancies with chromosome 5q deletions acquire a
dependency on an intrachromosomal NF-κB gene network. Cell Rep.
8:1328–1338. 2014. View Article : Google Scholar
|
|
65
|
Sanz L, Diaz-Meco MT, Nakano H and Moscat
J: The atypical PKC-interacting protein p62 channels NF-kappaB
activation by the IL-1TRAF6 pathway. EMBO J. 19:1576–1586. 2000.
View Article : Google Scholar
|
|
66
|
Teramachi J, Silbermann R, Yang P, Zhao W,
Mohammad KS, Guo J, Anderson JL, Zhou D, Feng R, Myint KZ, et al:
Blocking the ZZ domain of sequestosome1/p62 suppresses myeloma
growth and osteoclast formation in vitro and induces dramatic bone
formation in myeloma-bearing bones in vivo. Leukemia. 30:390–398.
2016. View Article : Google Scholar
|
|
67
|
Sanz L, Sanchez P, Lallena MJ, Diaz-Meco
MT and Moscat J: The interaction of p62 with RIP links the atypical
PKCs to NF-kappaB activation. EMBO J. 18:3044–3053. 1999.
View Article : Google Scholar
|
|
68
|
Rubio N, Verrax J, Dewaele M, Verfaillie
T, Johansen T, Piette J and Agostinis P: p38(MAPK)-regulated
induction of p62 and NBR1 after photodynamic therapy promotes
autophagic clearance of ubiquitin aggregates and reduces reactive
oxygen species levels by supporting Nrf2-antioxidant signaling.
Free Radic Biol Med. 67:292–303. 2014. View Article : Google Scholar
|
|
69
|
Chang KH, Sengupta A, Nayak RC, Duran A,
Lee SJ, Pratt RG, Wellendorf AM, Hill SE, Watkins M, Gonzalez-Nieto
D, et al: p62 is required for stem cell/progenitor retention
through inhibition of IKK/NF-κB/Ccl4 signaling at the bone marrow
macrophage-osteoblast niche. Cell Rep. 9:2084–2097. 2014.
View Article : Google Scholar
|
|
70
|
Milan E, Perini T, Resnati M, Orfanelli U,
Oliva L, Raimondi A, Cascio P, Bachi A, Marcatti M, Ciceri F and
Cenci S: A plastic SQSTM1/p62-dependent autophagic reserve
maintains proteostasis and determines proteasome inhibitor
susceptibility in multiple myeloma cells. Autophagy. 11:1161–1178.
2015. View Article : Google Scholar
|
|
71
|
Riz I, Hawley TS and Hawley RG:
KLF4-SQSTM1/p62-associated prosurvival autophagy contributes to
carfilzomib resistance in multiple myeloma models. Oncotarget.
6:14814–14831. 2015. View Article : Google Scholar
|
|
72
|
Trocoli A, Bensadoun P, Richard E,
Labrunie G, Merhi F, Schläfli AM, Brigger D, Souquere S, Pierron G,
Pasquet JM, et al: p62/SQSTM1 upregulation constitutes a survival
mechanism that occurs during granulocytic differentiation of acute
myeloid leukemia cells. Cell Death Differ. 21:1852–1861. 2014.
View Article : Google Scholar
|
|
73
|
Ségal-Bendirdjian E, Tschan MP, Reiffers J
and Djavaheri-Mergny M: Pro-survival role of p62 during
granulocytic differentiation of acute myeloid leukemia cells. Mol
Cell Oncol. 1:e9700662014. View Article : Google Scholar
|
|
74
|
Goussetis DJ, Gounaris E, Wu EJ, Vakana E,
Sharma B, Bogyo M, Altman JK and Platanias LC: Autophagic
degradation of the BCR-ABL oncoprotein and generation of
antileukemic responses by arsenic trioxide. Blood. 120:3555–3562.
2012. View Article : Google Scholar
|
|
75
|
Zhong Z, Sanchez-Lopez E and Karin M:
Autophagy, inflammation, and immunity: A Troika governing cancer
and its treatment. Cell. 166:288–298. 2016. View Article : Google Scholar
|
|
76
|
Moscat J, Karin M and Diaz-Meco MT: p62 in
cancer: Signaling adaptor beyond autophagy. Cell. 167:606–609.
2016. View Article : Google Scholar
|
|
77
|
Wang X, Du Z, Li L, Shi M and Yu Y: Beclin
1 and p62 expression in non-small cell lung cancer: Relation with
malignant behaviors and clinical outcome. Int J Clin Exp Pathol.
8:10644–10652. 2015.
|
|
78
|
Schläfli AM, Adams O, Galván JA, Gugger M,
Savic S, Bubendorf L, Schmid RA, Becker KF, Tschan MP, Langer R and
Berezowska S: Prognostic value of the autophagy markers LC3 and
p62/SQSTM1 in early-stage non-small cell lung cancer. Oncotarget.
7:39544–39555. 2016. View Article : Google Scholar
|
|
79
|
Huang H, Zhu J, Li Y, Zhang L, Gu J, Xie
Q, Jin H, Che X, Li J, Huang C, et al: Upregulation of SQSTM1/p62
contributes to nickel-induced malignant transformation of human
bronchial epithelial cells. Autophagy. 12:1687–1703. 2016.
View Article : Google Scholar
|
|
80
|
Linares JF, Duran A, Yajima T, Pasparakis
M, Moscat J and Diaz-Meco MT: K63 polyubiquitination and activation
of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol
Cell. 51:283–296. 2013. View Article : Google Scholar
|
|
81
|
Lou JS, Yan L, Bi CW, Chan GK, Wu QY, Liu
YL, Huang Y, Yao P, Du CY, Dong TT and Tsim KW: Yu Ping Feng San
reverses cisplatin-induced multi-drug resistance in lung cancer
cells via regulating drug transporters and p62/TRAF6 signalling.
Sci Rep. 6:319262016. View Article : Google Scholar
|
|
82
|
Lau A, Zheng Y, Tao S, Wang H, Whitman SA,
White E and Zhang DD: Arsenic inhibits autophagic flux, activating
the Nrf2-Keap1 pathway in a p62-dependent manner. Mol Cell Biol.
33:2436–2446. 2013. View Article : Google Scholar
|
|
83
|
Son YO, Pratheeshkumar P, Roy RV, Hitron
JA, Wang L, Zhang Z and Shi X: Nrf2/p62 signaling in apoptosis
resistance and its role in cadmium-induced carcinogenesis. J Biol
Chem. 289:28660–28675. 2014. View Article : Google Scholar
|
|
84
|
Wang Y, Zhang J, Huang ZH, Huang XH, Zheng
WB, Yin XF, Li YL, Li B and He QY: Isodeoxyelephantopin induces
protective autophagy in lung cancer cells via Nrf2-p62-keap1
feedback loop. Cell Death Dis. 8:e28762017. View Article : Google Scholar
|
|
85
|
Xia M, Gonzalez P, Li C, Meng G, Jiang A,
Wang H, Gao Q, Debatin KM, Beltinger C and Wei J: Mitophagy
enhances oncolytic measles virus replication by mitigating
DDX58/RIG-I-like receptor signaling. J Virol. 88:5152–5164. 2014.
View Article : Google Scholar
|
|
86
|
Nihira K, Miki Y, Ono K, Suzuki T and
Sasano H: An inhibition of p62/SQSTM1 caused autophagic cell death
of several human carcinoma cells. Cancer Sci. 105:568–575. 2014.
View Article : Google Scholar
|
|
87
|
Zhang J, Ma K, Qi T, Wei X, Zhang Q, Li G
and Chiu JF: P62 regulates resveratrol-mediated Fas/Cav-1 complex
formation and transition from autophagy to apoptosis. Oncotarget.
6:789–801. 2015. View Article : Google Scholar
|
|
88
|
Xu L, Xu F, Kong Q, Yang T, Tan D, Zhang
X, Li N, Zhao S, Zhao J and Li M: Inhibition of p62/SQSTM1
sensitizes small-cell lung cancer cells to cisplatin-induced
cytotoxicity by targeting NEDD9 expression. Mol Carcinog.
59:967–979. 2020. View Article : Google Scholar
|
|
89
|
Kim MJ, Min Y, Im JS, Son J, Lee JS and
Lee KY: p62 is negatively implicated in the TRAF6-BECN1 signaling
axis for autophagy activation and cancer progression by toll-like
receptor 4 (TLR4). Cells. 9:11422020. View Article : Google Scholar
|
|
90
|
Kim MJ, Min Y, Kwon J, Son J, Im JS, Shin
J and Lee KY: p62 negatively regulates TLR4 signaling via
functional regulation of the TRAF6-ECSIT complex. Immune Netw.
19:e162019. View Article : Google Scholar
|
|
91
|
Li S and Wei Y: Association of HMGB1,
BRCA1 and P62 expression in ovarian cancer and chemotherapy
sensitivity. Oncol Lett. 15:9572–9576. 2018.
|
|
92
|
Iwadate R, Inoue J, Tsuda H, Takano M,
Furuya K, Hirasawa A, Aoki D and Inazawa J: High expression of
SQSTM1/p62 protein is associated with poor prognosis in epithelial
ovarian cancer. Acta Histochem Cytochem. 47:295–301. 2014.
View Article : Google Scholar
|
|
93
|
Ju LL, Zhao CY, Ye KF, Yang H and Zhang J:
Expression and clinical implication of beclin1, HMGB1, p62,
survivin, BRCA1 and ERCC1 in epithelial ovarian tumor tissues. Eur
Rev Med Pharmacol Sci. 20:1993–2003. 2016.
|
|
94
|
Wang J, Garbutt C, Ma H, Gao P, Hornicek
FJ, Kan Q, Shi H and Duan Z: Expression and role of
autophagy-associated p62 (SQSTM1) in multidrug resistant ovarian
cancer. Gynecol Oncol. 150:143–150. 2018. View Article : Google Scholar
|
|
95
|
Bartsch G, Jennewein L, Harter PN,
Antonietti P, Blaheta RA, Kvasnicka HM, Kögel D, Haferkamp A,
Mittelbronn M and Mani J: Autophagy-associated proteins BAG3 and
p62 in testicular cancer. Oncol Rep. 35:1629–1635. 2016. View Article : Google Scholar
|
|
96
|
Iwadate R, Inoue J, Tsuda H, Takano M,
Furuya K, Hirasawa A, Aoki D and Inazawa J: High expression of p62
protein is associated with poor prognosis and aggressive phenotypes
in endometrial cancer. Am J Pathol. 185:2523–2533. 2015. View Article : Google Scholar
|
|
97
|
Darvekar SR, Elvenes J, Brenne HB,
Johansen T and Sjøttem E: SPBP is a sulforaphane induced
transcriptional coactivator of NRF2 regulating expression of the
autophagy receptor p62/SQSTM1. PLoS One. 9:e852622014. View Article : Google Scholar
|
|
98
|
Jung D, Khurana A, Roy D, Kalogera E,
Bakkum-Gamez J, Chien J and Shridhar V: Quinacrine upregulates
p21/p27 independent of p53 through autophagy-mediated
downregulation of p62-Skp2 axis in ovarian cancer. Sci Rep.
8:24872018. View Article : Google Scholar
|
|
99
|
Xia MH, Yan XY, Zhou L, Xu L, Zhang LC, Yi
HW and Su J: p62 suppressed VK3-induced oxidative damage through
Keap1/Nrf2 pathway in human ovarian cancer cells. J Cancer.
11:1299–1307. 2020. View Article : Google Scholar
|
|
100
|
Yan XY, Zhong XR, Yu SH, Zhang LC, Liu YN,
Zhang Y, Sun LK and Su J: p62 aggregates mediated caspase 8
activation is responsible for progression of ovarian cancer. J Cell
Mol Med. 23:4030–4042. 2019. View Article : Google Scholar
|
|
101
|
Chang MA, Morgado M, Warren CR, Hinton CV,
Farach-Carson MC and Delk NA: p62/SQSTM1 is required for cell
survival of apoptosis-resistant bone metastatic prostate cancer
cell lines. Prostate. 74:149–163. 2014. View Article : Google Scholar
|
|
102
|
Falasca L, Torino F, Marconi M, Costantini
M, Pompeo V, Sentinelli S, De Salvo L, Patrizio M, Padula C,
Gallucci M, et al: AMBRA1 and SQSTM1 expression pattern in prostate
cancer. Apoptosis. 20:1577–1586. 2015. View Article : Google Scholar
|
|
103
|
Wang L, Kim D, Wise JT, Shi X, Zhang Z and
DiPaola RS: p62 as a therapeutic target for inhibition of autophagy
in prostate cancer. Prostate. 78:390–400. 2018. View Article : Google Scholar
|
|
104
|
Burdelski C, Reiswich V, Hube-Magg C,
Kluth M, Minner S, Koop C, Graefen M, Heinzer H, Tsourlakis MC,
Wittmer C, et al: Cytoplasmic accumulation of sequestosome 1 (p62)
is a predictor of biochemical recurrence, rapid tumor cell
proliferation, and genomic instability in prostate cancer. Clin
Cancer Res. 21:3471–3479. 2015. View Article : Google Scholar
|
|
105
|
Kim JH, Hong SK, Wu PK, Richards AL,
Jackson WT and Park JI: Raf/MEK/ERK can regulate cellular levels of
LC3B and SQSTM1/p62 at expression levels. Exp Cell Res.
327:340–352. 2014. View Article : Google Scholar
|
|
106
|
Linares JF, Duran A, Reina-Campos M,
Aza-Blanc P, Campos A, Moscat J and Diaz-Meco MT: Amino acid
activation of mTORC1 by a PB1-domain-driven kinase complex cascade.
Cell Rep. 12:1339–1352. 2015. View Article : Google Scholar
|
|
107
|
Jones S, Cunningham DL, Rappoport JZ and
Heath JK: The non-receptor tyrosine kinase Ack1 regulates the fate
of activated EGFR by inducing trafficking to the p62/NBR1
pre-autophagosome. J Cell Sci. 127:994–1006. 2014.
|
|
108
|
Jiang G, Liang X, Huang Y, Lan Z, Zhang Z,
Su Z, Fang Z, Lai Y, Yao W, Liu T, et al: p62 promotes
proliferation, apoptosis-resistance and invasion of prostate cancer
cells through the Keap1/Nrf2/ARE axis. Oncol Rep. 43:1547–1557.
2020.
|
|
109
|
Huang J, Duran A, Reina-Campos M, Valencia
T, Castilla EA, Müller TD, Tschöp MH, Moscat J and Diaz-Meco MT:
Adipocyte p62/SQSTM1 suppresses tumorigenesis through opposite
regulations of metabolism in adipose tissue and tumor. Cancer Cell.
33:770–784.e6. 2018. View Article : Google Scholar
|
|
110
|
Valencia T, Kim JY, Abu-Baker S,
Moscat-Pardos J, Ahn CS, Reina-Campos M, Duran A, Castilla EA,
Metallo CM, Diaz-Meco MT and Moscat J: Metabolic reprogramming of
stromal fibroblasts through p62-mTORC1 signaling promotes
inflammation and tumorigenesis. Cancer Cell. 26:121–135. 2014.
View Article : Google Scholar
|
|
111
|
Chang MA, Patel V, Gwede M, Morgado M,
Tomasevich K, Fong EL, Farach-Carson MC and Delk NA: IL-1β induces
p62/SQSTM1 and represses androgen receptor expression in prostate
cancer cells. J Cell Biochem. 115:2188–2197. 2014. View Article : Google Scholar
|
|
112
|
Chen K, Zeng J, Xiao H, Huang C, Hu J, Yao
W, Yu G, Xiao W, Xu H and Ye Z: Regulation of glucose metabolism by
p62/SQSTM1 through HIF1α. J Cell Sci. 129:817–830. 2016.
|
|
113
|
Liu XD, Yao J, Tripathi DN, Ding Z, Xu Y,
Sun M, Zhang J, Bai S, German P, Hoang A, et al: Autophagy mediates
HIF2α degradation and suppresses renal tumorigenesis. Oncogene.
34:2450–2460. 2015. View Article : Google Scholar
|
|
114
|
Li T, Jiang D and Wu K: p62 promotes
bladder cancer cell growth by activating KEAP1/NRF2-dependent
antioxidative response. Cancer Sci. 111:1156–1164. 2020. View Article : Google Scholar
|
|
115
|
Liu JL, Chen FF, Lung J, Lo CH, Lee FH, Lu
YC and Hung CH: Prognostic significance of p62/SQSTM1 subcellular
localization and LC3B in oral squamous cell carcinoma. Br J Cancer.
111:944–954. 2014. View Article : Google Scholar
|
|
116
|
Kuo WL, Sharifi MN, Lingen MW, Ahmed O,
Liu J, Nagilla M, Macleod KF and Cohen EE: p62/SQSTM1 accumulation
in squamous cell carcinoma of head and neck predicts sensitivity to
phosphatidylinositol 3-kinase pathway inhibitors. PLoS One.
9:e901712014. View Article : Google Scholar
|
|
117
|
Liang L, Luo H, He Q, You Y, Fan Y and
Liang J: Investigation of cancer-associated fibroblasts and p62
expression in oral cancer before and after chemotherapy. J
Craniomaxillofac Surg. 46:605–610. 2018. View Article : Google Scholar
|
|
118
|
Yoshihara N, Takagi A, Ueno T and Ikeda S:
Inverse correlation between microtubule-associated protein
1A/1B-light chain 3 and p62/sequestosome-1 expression in the
progression of cutaneous squamous cell carcinoma. J Dermatol.
41:311–315. 2014. View Article : Google Scholar
|
|
119
|
Shah P, Trinh E, Qiang L, Xie L, Hu WY,
Prins GS, Pi J and He YY: Arsenic induces p62 expression to form a
positive feedback loop with Nrf2 in human epidermal keratinocytes:
Implications for preventing arsenic-induced skin cancer. Molecules.
22:1942017. View Article : Google Scholar
|
|
120
|
Colunga A, Bollino D, Schech A and
Aurelian L: Calpain-dependent clearance of the autophagy protein
p62/SQSTM1 is a contributor to ΔPK oncolytic activity in melanoma.
Gene Ther. 21:371–378. 2014. View Article : Google Scholar
|
|
121
|
Yeh LY, Liu CJ, Wong YK, Chang C, Lin SC
and Chang KW: miR-372 inhibits p62 in head and neck squamous cell
carcinoma in vitro and in vivo. Oncotarget. 6:6062–6075. 2015.
View Article : Google Scholar
|
|
122
|
Yamanaka T, Tosaki A, Kurosawa M,
Matsumoto G, Koike M, Uchiyama Y, Maity SN, Shimogori T, Hattori N
and Nukina N: NF-Y inactivation causes atypical neurodegeneration
characterized by ubiquitin and p62 accumulation and endoplasmic
reticulum disorganization. Nat Commun. 5:33542014. View Article : Google Scholar
|
|
123
|
Wang C, Chen S, Yeo S, Karsli-Uzunbas G,
White E, Mizushima N, Virgin HW and Guan JL: Correction: Elevated
p62/SQSTM1 determines the fate of autophagy-deficient neural stem
cells by increasing superoxide. J Cell Biol. 212:8792016.
View Article : Google Scholar
|
|
124
|
Wang C, Chen S, Yeo S, Karsli-Uzunbas G,
White E, Mizushima N, Virgin HW and Guan JL: Elevated p62/SQSTM1
determines the fate of autophagy-deficient neural stem cells by
increasing superoxide. J Cell Biol. 212:545–560. 2016. View Article : Google Scholar
|
|
125
|
Ivankovic D, Chau KY, Schapira AH and Gegg
ME: Mitochondrial and lysosomal biogenesis are activated following
PINK1/parkin-mediated mitophagy. J Neurochem. 136:388–402. 2016.
View Article : Google Scholar
|
|
126
|
Su J, Liu F, Xia M, Xu Y, Li X, Kang J, Li
Y and Sun L: p62 participates in the inhibition of NF-κB signaling
and apoptosis induced by sulfasalazine in human glioma U251 cells.
Oncol Rep. 34:235–243. 2015. View Article : Google Scholar
|
|
127
|
Zeng RX, Zhang YB, Fan Y and Wu GL:
p62/SQSTM1 is involved in caspase-8 associated cell death induced
by proteasome inhibitor MG132 in U87MG cells. Cell Biol Int.
38:1221–1226. 2014. View Article : Google Scholar
|
|
128
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013. View Article : Google Scholar
|
|
129
|
Qiang L, Zhao B, Ming M, Wang N, He TC,
Hwang S, Thorburn A and He YY: Regulation of cell proliferation and
migration by p62 through stabilization of Twist1. Proc Natl Acad
Sci USA. 111:9241–9246. 2014. View Article : Google Scholar
|
|
130
|
Qiang L and He YY: Autophagy deficiency
stabilizes TWIST1 to promote epithelial-mesenchymal transition.
Autophagy. 10:1864–1865. 2014. View Article : Google Scholar
|
|
131
|
Bertrand M, Petit V, Jain A, Amsellem R,
Johansen T, Larue L, Codogno P and Beau I: SQSTM1/p62 regulates the
expression of junctional proteins through epithelial-mesenchymal
transition factors. Cell Cycle. 14:364–374. 2015. View Article : Google Scholar
|
|
132
|
Jiang X, Huang Y, Liang X, Jiang F, He Y,
Li T, Xu G, Zhao H, Yang W, Jiang G, et al: Metastatic prostate
cancer-associated P62 inhibits autophagy flux and promotes
epithelial to mesenchymal transition by sustaining the level of
HDAC6. Prostate. 78:426–434. 2018. View Article : Google Scholar
|
|
133
|
Grassi G, Di Caprio G, Santangelo L, Fimia
GM, Cozzolino AM, Komatsu M, Ippolito G, Tripodi M and Alonzi T:
Autophagy regulates hepatocyte identity and
epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions
promoting snail degradation. Cell Death Dis. 6:e18802015.
View Article : Google Scholar
|
|
134
|
Geng Y, Ju Y, Ren F, Qiu Y, Tomita Y,
Tomoeda M, Kishida M, Wang Y, Jin L, Su F, et al: Insulin receptor
substrate 1/2 (IRS1/2) regulates Wnt/β-catenin signaling through
blocking autophagic degradation of dishevelled2. J Biol Chem.
289:11230–11241. 2014. View Article : Google Scholar
|
|
135
|
Ma JB, Hu SL, Zang RK, Su Y, Liang YC and
Wang Y: MicroRNA-487a promotes proliferation of esophageal cancer
cells by inhibiting p62 expression. Eur Rev Med Pharmacol Sci.
23:1502–1512. 2019.
|
|
136
|
Zhong JH, Xiang X, Wang YY, Liu X, Qi LN,
Luo CP, Wei WE, You XM, Ma L, Xiang BD and Li LQ: The lncRNA SNHG16
affects prognosis in hepatocellular carcinoma by regulating p62
expression. J Cell Physiol. 235:1090–1102. 2020. View Article : Google Scholar
|
|
137
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar
|
|
138
|
Adamik J, Silbermann R, Marino S, Sun Q,
Anderson J L, Zhou D, Xie XQ, Roodman GD and Galson DL: XRK3F2
inhibition of p62-ZZ domain signaling rescues myeloma-induced
GFI1-driven epigenetic repression of the Runx2 gene in
pre-osteoblasts to overcome differentiation suppression. Front
Endocrinol (Lausanne). 9. pp. 3442018, View Article : Google Scholar
|
|
139
|
Li Y, Li Y, Yin J, Wang C, Yang M, Gu J,
He M, Xu H, Fu W, Zhang W, et al: A mitophagy inhibitor targeting
p62 attenuates the leukemia-initiation potential of acute myeloid
leukemia cells. Cancer Lett. 510:24–36. 2021. View Article : Google Scholar
|
|
140
|
Andersen AN, Landsverk OJ, Simonsen A,
Bogen B, Corthay A and Øynebråten I: Coupling of HIV-1 antigen to
the selective autophagy receptor SQSTM1/p62 promotes
T-cell-mediated immunity. Front Iunol. 7:1672016.
|
|
141
|
Venanzi F, Shifrin V, Sherman M, Gabai V,
Kiselev O, Komissarov A, Grudinin M, Shartukova M,
Romanovskaya-Romanko EA, Kudryavets Y, et al: Broad-spectrum
anti-tumor and anti-metastatic DNA vaccine based on p62-encoding
vector. Oncotarget. 4:1829–1835. 2013. View Article : Google Scholar
|
|
142
|
Gabai V, Venanzi FM, Bagashova E, Rud O,
Mariotti F, Vullo C, Catone G, Sherman MY, Concetti A, Chursov A,
et al: Pilot study of p62 DNA vaccine in dogs with mammary tumors.
Oncotarget. 5:12803–12810. 2014. View Article : Google Scholar
|
|
143
|
Ponomarenko DM, Klimova ID, Chapygina YA,
Dvornichenko VV, Zhukova NV, Orlova RV, Manikhas GM, Zyryanov AV,
Burkhanova LA, Badrtdinova II, et al: Safety and efficacy of p62
DNA vaccine ELENAGEN in a first-in-human trial in patients with
advanced solid tumors. Oncotarget. 8:53730–53739. 2017. View Article : Google Scholar
|