Introduction
Glioblastoma (GBM), the most common type of human
primary brain tumor, accounts for 81% of malignant tumors in the
central nervous system (1,2).
GBM has been classified by World Health Organization as a grade IV
glioma and it has a high recurrence rate (3,4).
In a Swiss population-based study, the survival rate of patients
with newly diagnosed GBM was ~18% at 1 year and only 3% at 2 years
(5,6). Despite the availability of state of
the art multimodality treatments, the median survival of GBM
patients is 12-15 months (7,8).
The current treatments for GBM include neurosurgical resection,
radiotherapy and pharmacotherapy (9,10).
However, these therapies fail tosuccessfully treat GBM due to
various reasons (11). One of the
main reasons is the high tendency of the tumor to invade the
surrounding healthy brain tissues (12). Another reason is the multi-drug
resistance encountered during GBM chemotherapy (13). Therefore, there is an urgent need
for novel and/or improved drugs or treatments to improve the
survival of patients with GBM.
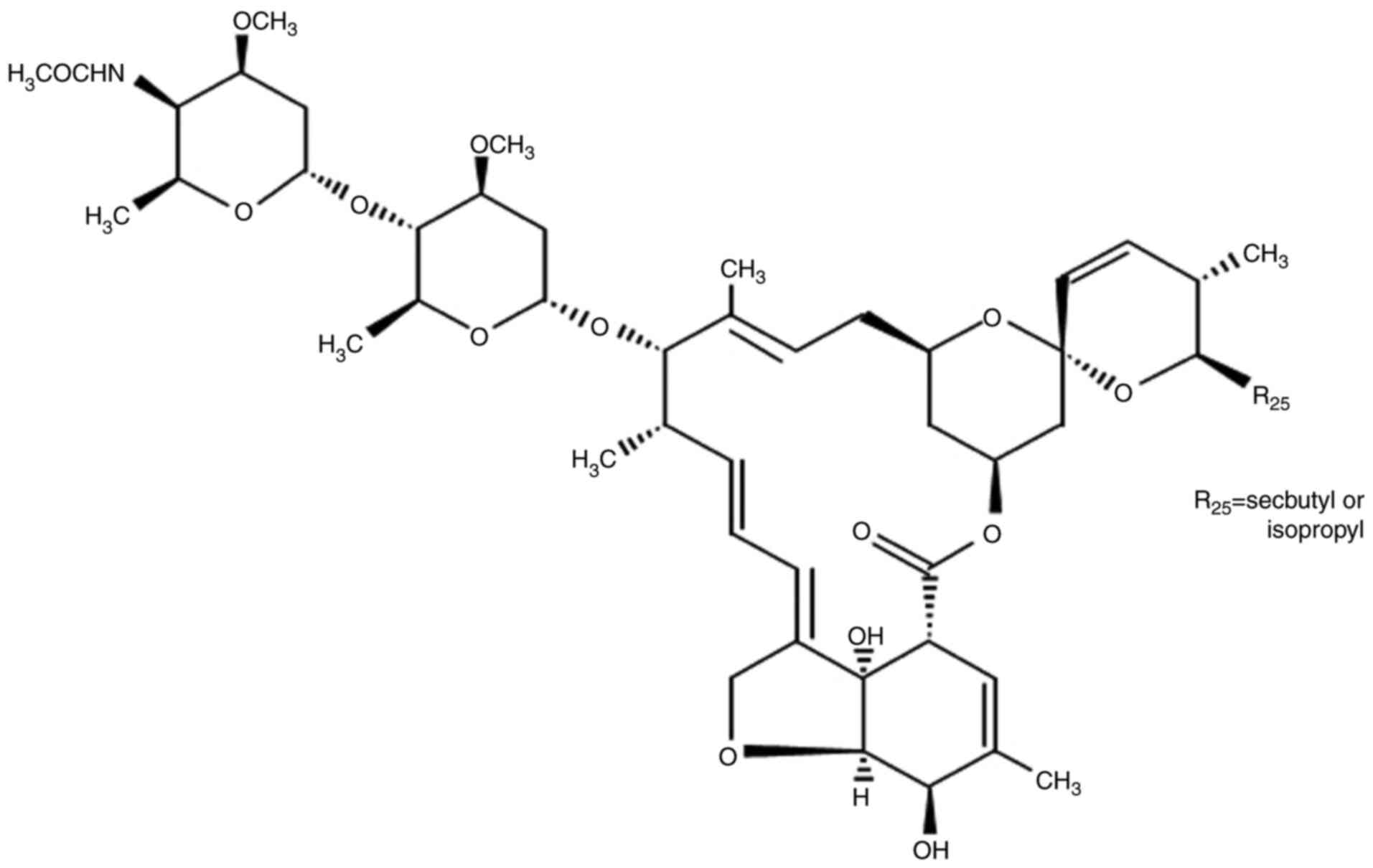
Doramectin (DRM) is a genetically modified
avermectin (AVM), which is produced by actinomycetes (fungi) and
has potent anthelmintic and insecticidal activities (14,15). DRM is one of the most widely used
macrocyclic lactone endo/ectoparasiticides worldwide (16). It has been demonstrated that DRM
could reverse the multidrug resistance of cancer cells (17). In addition, it has been reported
that macrocyclic lactones are well-tolerated agents, which are used
to treat a large number of animals for parasitic infections
(18,19). Macrocyclic lactone
endo/ectoparasiticides have also been demonstrated to suppress
certain activities in human cancer cells (20). For instance, certain studies,
including our previous study, have revealed that ivermectin (IVM)
and AVM induce apoptosis and autophagy in glioma, HeLa and Ehrlich
carcinoma cells (21-23). DRM is a third-generation
derivative of the AVM class of macrolides. Unlike AVM, it has a
cyclohexyl group in the C25 position of the AVM ring (Fig. 1). In addition, DRM is absorbed
more quickly, and has a longer lasting effect and plasma half-life
in animals compared with IVM and AVM (24). Therefore, it was hypothesized that
DRM may have similar effects on cancer cells and may be a superior
anticancer drug compared with existing anticancer drugs.
Programmed cell death has attracted considerable
attention from researchers as a tumor suppression mechanism
(25,26). This biological process emerged
during anticancer therapies, such as radiation, chemotherapy and
certain targeted therapies (27).
There are two main morphologically distinctive forms of programmed
cell death. Apoptosis and autophagy (28,29). Autophagy is a highly conserved
mechanism of eukaryotic cells that serves essential roles in
development, tissue homeostasis and diseases (30). A number of studies have suggested
that enforced overactivation of autophagy will lead to cell death
in certain contexts. For instance, excessive endoplasmic
reticulum-specific autophagy mediated by the autophagy receptor
FAM134B results in cell death in HeLa cells (31) and SH003 activates autophagic cell
death by activating ATF4 and inhibiting G9a under hypoxia in
gastric cancer cells (32).
It is well known that autophagy is characterized by
the formation of double-membrane vesicles, called autophagosomes
(33). In addition, a large
number of studies have demonstrated that autophagy can inhibit
glioma growth (34,35). However, the effects and mechanisms
of DRM in inducing autophagy in GBM cells remain unclear.
The aim of the present study was to explore the
roles of DRM in inducing autophagy in GBM cell lines cultured in
vitro and in vivo, and to analyze the potential
mechanisms of the observed effects using proteomics analysis. In
addition, the present study attempted to elucidate the association
between DRM-induced autophagy and apoptosis in GBM cells in
vitro and in vivo.
Materials and methods
Cell lines, reagents and antibodies
U87 human GBM cell lines and C6 rat GBM cells were
obtained from China Infrastructure of Cell Line Resources,
Institute of Basic Medical Sciences, Chinese Academy of Medical
Sciences (Beijing, China) and have been authenticated by short
tandem repeat (STR) profiling. In addition, STR profiling
identified that the original U87 cell line was established in 1968
at the University of Uppsala (RRID: CVCL-GP63). These cells were
cultured in DMEM (cat. no. 12100-046; Gibco; Thermo Fisher
Scientific, Inc.) supplemented 10% new-fetal bovine serum (cat. no.
23022-8615; Zhejiang Tianhang Biotechnology Co., Ltd.) and 100 U/ml
penicillin/streptomycin (cat. no. C0222; Beyotime Institute of
Biotechnology). The cells were placed in an incubator at 37°C with
5% CO2. DRM was purchased from Sigma-Aldrich; Merck
KGaA. DMSO (cat. no. D2650) and chloroquine (CQ; cat. no. C6628)
were purchased from Merck KGaA. The antibodies used were as
follows: Autophagy-related 5 (Atg5; dilution, 1:1,000; cat. no.
12994; Cell Signaling Technology, Inc.), LC3 (detects both LC3I and
LC3II; dilution, 1:1,000; cat. no. 4599; Cell Signaling Technology,
Inc.), p62 (dilution, 1:1,000; cat. no. 23214; Cell Signaling
Technology, Inc.), Ki-67 (dilution, 1:1,000; cat. no. 9027; Cell
Signaling Technology, Inc.) and β-actin (dilution, 1:1,000; cat.
no. 4970; Cell Signaling Technology, Inc.). The Goat Anti-Rabbit
IgG secondary antibody was obtained from OriGene Technologies, Inc.
(dilution, 1: 2,000; cat. no. TA130015). All antibodies were
dissolved antibody dilution buffer (cat. no. A1800; Beijing
Solarbio Science & Technology Co., Ltd.). GFP-LC3 plasmid and
GFP plasmid were obtained from BioVector NTCC, Inc.
Transmission electron microscopy
(TEM)
The cells were incubated with or without DRM for 48
h. The culture medium was discarded, and the cells were washed
three times with PBS. They were then fixed overnight with 2.5%
glutaraldehyde at 4°C and subsequently fixed with 1% osmium
tetroxide for 1-2 h at 4°C. Next, the cells were dehydrated using a
graded series of ethanol solutions (30, 50, 70, 90 and 100%) for 10
min at a time, and embedded in Epoxy Embedding medium (Merck KGaA)
and polymerized at 37°C for 24h. Finally, it was dyed with 3%
uranium acetate and lead citrate for 30 min at room temperature.
The ultrathin sections (0.1 µm) were observed using a
transmission electron microscope (H7650; Hitachi, Ltd.). Treatment
without DRM was used as a control.
Plasmid transfection
The cells were cultured in 6-well plates at a
density of 2×105 cells/well for 24 h. The cells were
transfected with green fluorescent protein 3 µg GFP-LC3 (2.5
µg/µl) plasmid and 3 µg empty plasmid (4.8
µg/µl) containing no LC3 protein using
Lipofectamine® 2000 (cat. no. 11668019; Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. Subsequently, transfected cells were incubated in
serum-free medium at 37°C for 4 h. After 4 h, the transfected cells
were incubated with fresh medium or fresh medium containing DRM for
48 h. After 48 h, the DRM-treated transfected cells were then
visualized under an inverted fluorescence microscope (Olympus
Corporation). Furthermore, at 48 h after transfection, transfected
cells without DRM treatment were harvested using RIPA lysis buffers
(cat. no. P0013B; Beyotime Institute of Biotechnology) supplemented
with protease inhibitors (cat. no. ST506; Beyotime Institute of
Biotechnology). Next, total proteins were used for western blot
analysis. DRM-treated transfected cells were not harvested.
Treatment without DRM or empty plasmid were used as a control.
Western blot analysis
U87 and C6 cells were incubated with various
concentrations of DRM (0, 5, 10 and 15 µM) at 37°C for 48 h.
Tumor tissue was ground to powder in a mortar containing liquid
nitrogen. The DRM-treated cells and tumor tissue powder were
harvested in RIPA lysis buffers (cat. no. P0013B; Beyotime
Institute of Biotechnology) supplemented with protease inhibitors
(cat. no. ST506; Beyotime Institute of Biotechnology). Next, the
concentration of total proteins was determined using a BCA assay
kit (cat. no. P0012; Beyotime Institute of Biotechnology). Total
proteins (30 µg protein/lane) were separated by 12% sodium
dodecyl sulphate-polyacrylamide gel electrophoresis and transferred
to polyvinylidene fluoride membranes (Millipore Sigma; Merck KGaA).
The membranes were then blocked with 5% fat-free milk in
TBS-0.1%Tween 20 buffer for 2 h at 37°C. Next, the blots were
incubated with primary antibodies overnight at 4°C. Subsequently,
the protein bands were probed with HRP-conjugated goat anti-rabbit
IgG secondary antibody (dilution, 1:2,000; cat. no. TA130015) for
1.5 h at room temperature. Then, each band was visualized using
BeyoECL Moon (cat. no. P0018 FM; Beyotime Institute of
Biotechnology). Finally, the protein bands were analyzed using
Image Lab™ Software (version 5.2.1; Bio-Rad Laboratories, Inc.) on
a ChemiDoc XRS+(Bio-Rad Laboratories, Inc.).
Analysis of autophagy with CQ
The autophagy inhibitor CQ was also used to block
autophagy at a final concentration of 15 µM. Briefly, U87
and C6 cells (1×105 cells/well) in a 6-well plate were
pretreated with CQ (15 µM) at 37°C for 2 h, and then cells
were treated with DRM (15 µM) at 37°C for 48 h. Cells were
collected for MTT, colony formation, western blotting, DAPI
staining and Annexin V-FITC/PI staining analysis.
MTT assay
U87 and C6 cells were seeded into 96-well plates at
a density of 3×103 cells/well and incubated overnight at
37°C for 24 h. DMEM supplemented with DRM alone or together with
the autophagy inhibitor CQ was added to the 96-well plates for 48
h. MTT (1 mg/ml dissolved in PBS; 100 µl/well; cat. no.
M2128; Merck KGaA) was then added to each well and cells were
incubated for an additional 4 h at 37°C. MTT was carefully removed
and 150 µl DMSO was added. Absorbance was measured on a
microplate reader (BioTek Instruments, Inc.) at a wave length of
450 nm. The cell viability inhibition rate (IR) was calculated as
the ratio between the OD of the CQ, CQ+DRM and DRM group and the OD
of the control group. Cells in the control group were not treated
with DRM and CQ.
Colony formation assay
U87 and C6 cells were seeded into 6-well plates at a
density of 3×102 cells/well. Following overnight
incubation at 37°C, U87 and C6 cells were treated with DRM (15
µM) alone or together with the autophagy inhibitor CQ (15
µM) at 37°C for 48 h. The culture medium was discarded, and
the cells were washed with PBS three times. The cells were then
incubated with fresh medium at 37°C. After 2 weeks, the colonies
were fixed with 4% formaldehyde for 30 min at room temperature and
stained with 0.4% crystal violet for 10 min at room temperature.
Subsequently, the counts of cell colonies were manually scored. The
number of colonies was counted and quantified. Colony formation
quantification was performed using ImageJ software (version 2.0;
National Institutes of Health). A colony was defined as >50
cells. Cells in the control group were not treated with DRM and
CQ.
DAPI staining
The cells were seeded into 6-well plates at a
density of 3×102 cells/well and treated with 15
µM DRM alone or together with an autophagy inhibitor (CQ, 15
µM) for 48 h at 37°C. The cells were harvested, washed twice
with PBS, fixed with 4% formaldehyde for 10 min at room temperature
and stained with DAPI (cat. no. D8471; Merck KGaA) staining
solution according to the manufacturer's instructions for 10 min at
37°C. The images were immediately captured using a fluorescence
microscope (Olympus Corporation). Images were analyzed using Image
Pro Plus v. 5.1 software (Media Cybernetics, Inc.). Cells in the
control group were not treated with DRM and CQ.
Annexin V-FITC/PI staining for cell
apoptosis
An Annexin V-FITC/PI apoptosis detection kit (cat.
no. C1062S; Beyotime Institute of Biotechnology) was used to
distinguish apoptotic cells from normal cells. U87 and C6 cells
were incubated with DRM alone or together with the autophagy
inhibitor CQ for 48 h. Cells were harvested and stained with the
FITC-labeled Annexin V and PI for 15 min in the dark. Flow
cytometry was conducted immediately to detect apoptotic cell
populations. A BD Biosciences FACS Calibur flow cytometer (BD
Biosciences) was immediately used to detect apoptotic cell
populationsand data were analyzed using Flow Jo 7.6.2 (Tree Star,
Inc.).
RNA isolation
The total RNA of each sample was extracted using a
High Pure RNA Isolation Kit (cat. no. 1828665; Roche Diagnostics)
according to the manufacturer's instructions. The total RNA of
triplicate samples treated with or without DRM (15 µM) for
48 h at 37°C was then sent to Wuhan Boyue Zhihe Biotechnology Co.,
Ltd. RNA samples were then digested with RNase free DNase I (cat.
no. 89836; Invitrogen; Thermo Fisher Scientific, Inc.) to eliminate
residual genomic DNA, and the digestion products were purified
using magnetic beads (Axygen; Corning, Inc.). Then, the quality and
integrity of the total RNA were assessed using an Agilent 2100
Bioanalyzer (Agilent Technologies, Inc.) and 1.2% agarose gel
electrophoresis. The RNA concentration was measured using a Nano
Drop 2000 instrument (Nano Drop Technologies; Thermo Fisher
Scientific, Inc.). High-quality RNA samples with OD260/280 ratios
ranging between 1.8 and 2.2 and OD260/230 ≥1.8-2.2, RNA integrity
number (RIN) ≥7 and total RNA concentration ≥50 ng/µl were
used for library preparation. After the RNA sample passed the test,
the eukaryotic mRNA (in the case of prokaryote, the mRNA was
enriched by removing rRNA through the kit) was enriched using
magnetic beads with oligo (DT). RNA sequencing libraries were
generated using the KAPA stranded RNA-Seq Kit with RiboErase (HMR;
cat. no. KK8483; Kapa Biosystems; Roche Diagnostics) with
multiplexing primers according to the manufacturer's protocol.
Subsequently, a fragment buffer was added to break the mRNA into
short segments. mRNA was used as a template to synthesize a
single-stranded cDNA with six base random primers. Then, buffer,
dNTPs, DNA polymer I and RNase H were added to synthesize
double-stranded cDNA. Then, the double-stranded cDNA was purified
with AMPure XP beads. The purified double-stranded cDNA was first
repaired with A-tail and connected with the sequencing connector;
then, the fragment size was selected using AMPure XP beads.
Subsequently, the second strand of U-containing cDNA was degraded
with user enzyme so that the final sequencing information came from
the first strand of cDNA, thus preserving the strand orientation of
mRNA. Finally, the PCR amplification was carried out, and the PCR
products were purified with AMPure XP beads to obtain the
chain-specific cDNA library. After the construction of the library,
qubit 3.0 fluorometer (cat. no. Q33216; Thermo Fisher Scientific,
Inc.) was used for preliminary quantification, and the library was
diluted to 1 ng/µl. Then, Qsep100 was used to detect the
insert size of the library. After the insert size met the
expectation, qPCR was used to accurately quantify the effective
concentration of the library (the effective concentration of the
library was >2 nM), so as to ensure the quality of the library.
After the library passed the inspection, different libraries were
pooled according to the requirements of effective concentration and
target off line data volume, following which HiSeq sequencing was
carried out. Paired end sequencing was performed using an Illumina
Hiseq X Ten with a read length of 150 bp. A total of 1 µg
total RNA was used for each Illumina library preparation. FastQC
v0.11.9 software (Babraham Bioinformatics; https://www.bioinformatics.babraham.ac.uk/projects/fastqc/)
was used for quality analysis of the sequencing data in this
project. The number of reads per gene was calculated using
featureCounts V1.6.0 (featureCounts is a tool in the Subread v2.0.1
package; http://subread.sourceforge.net).
Data processing and functional
annotation
Gene expression was standardized by fragments per
kilobase per million mapped fragments using featureCounts v1.5.0.
(36). The following analyses
were conducted on differentially expressed genes (DEGs): i) A Venn
diagram was drawn to identify common and unique DEGs between the
groups using Venny 2.1.1 (https://bioinfogp.cnb.csic.es/tools/venny/) and a
Volcano plot generated by ggplot2 v.3.0.0 (37) in R Package 3.5.3 (https://neuroconductor.org/neurocLite.R)
was used to analyze the screening of differentially expressed genes
between samples; ii) Genesis 1.8.1 (38) was used for hierarchical
clustering, it was used to compare the expression of DEGs in DRM
vs. control groups using cluster analysis software; and iii) Gene
Ontology (GO; http://www.geneontology.org) and Kyoto Encyclopedia of
Genes and Genomes (KEGG; http://www.genome.jp/), pathway annotation and
enrichment analyses were carried out using the cluster profiler
package 3.10.1 (https://bioconductor.org/packages/clusterProfiler/) in
R Package 3.5.3 (https://neuro-conductor.org/neurocLite.R) (39). Pathview in KEGG was used to
analyze data (https://www.kegg.jp/pathway/map04140 and https://www.kegg.jp/kegg/mapper/color.html).
Animal models
All animal experiments were carried out at Harbin
Weike Biotechnology Co., Ltd. The housing conditions were as
follows: 24°C, 50-60% humidity and 12:12 h light-dark cycle.
Animals were given free access to commercial rat pellet diet and
tap water. A total of 36 female BALB/c mice were obtained from
Beijing Vital River Laboratory Animal Technology Co., Ltd. Each
BALB/c mice weighed about 17±1.58 g and was 6 weeks old. After 1
week of feeding in a sterile environment, 100 µl PBS
containing 2.0×106 C6 cells were injected subcutaneously
into the flanks of nude mice. After 10 days, tumor-bearing mice
were allocated to four groups and treated with either saline (100
µl; control group), CQ (20 mg/kg/day CQ in 100 µl; CQ
group), DRM (14 mg/kg/day DRM in 100 µl; DRM group) or
DRM+CQ (20 mg/kg/day CQ combined with 14 mg/kg/day DRM in 100
µl; DRM+CQ group) for 24 days. The four treatments were
injected intraperitoneally into mice daily. Health and behavior of
all nude mice were monitored on a daily basis. The length (L) and
width (W) of tumors were measured and calculated every 3 days
(V=1/2 × L × W2). After 24 days, the tumor volume of
mice in the control group made it difficult to live, including
difficulty drinking, eating and moving, which brought discomfort to
the life of mice. At the same time, the tumor volume had reached
the purpose of the present study. First, all mice were humanely
euthanized by intraperitoneal injection of Nembutal (150 mg/kg).
Then, death was confirmed using cervical dislocation as a secondary
method of euthanasia. All mice showed no vital signs and tumors
were extracted for immunostaining and weighing. The maximum tumor
volume in the present study was 760 mm3. The experiment
progressed 42 days from the injection of mice to euthanasia of
mice. There were no mice that died accidentally during the
experiment, and the mice showed healthy vital signs.
Immunohistochemical staining
All specimens were fixed in 10% neutral-buffered
formalin for 48 h at room temperature and embedded in paraffin.
Next, 5-µm-thick sections were dewaxed and rehydrated. All
specimens were dewaxed in xylene and rehydrated in a graded series
of ethanol solution at room temperature for 5 min at a time.
Sections were treated with 0.01 mol/l boiling sodium citrate buffer
for 10 min. Subsequently, sections were treated with 3%
H2O2 at room temperature for 30 min. Next,
they were incubated in QuickBlock™ Blocking Buffer (cat. no. P0260;
Beyotime Institute of Biotechnology) at 37°C for 20 min. Then, they
were incubated in LC3 (dilution, 1:6,000; cat. no. 4599; Cell
Signaling Technology, Inc.), p62 (dilution, 1:250; cat. no. 23214;
Cell Signaling Technology, Inc.) and Ki-67 (dilution, 1:800; cat.
no. 9027; Cell Signaling Technology, Inc.) primary antibodies for
90 min at 37°C. With an HRP-conjugated goat anti-rabbit IgG
(dilution, 1:1,000; cat. no. 40295G; BIOSS) at 37°C for 90 min. The
sections were incubated with a DAB Horseradish Peroxidase Color
Development Kit (cat. no. P0202; Beyotime Institute of
Biotechnology) at room temperature for 30 min, and then washed with
distilled water two times. Finally, the sections were counter
stained with hematoxylin for 10 min at room temperature and the
slides were observed under a light microscope (D5100; Nikon
Corporation) and analyzed using ImageJ software (version 2.0;
National Institutes of Health). The number of positive cell nuclei
in 30 random fields from randomly chosen tumor sections for each
animal was counted at a magnification of ×400.
TUNEL assay
Cell apoptosis was assessed in vivo using a
TUNEL assay. An In Situ Cell Death Detection kit (cat. no.
11684817910; Roche Diagnostics GmbH) was used according to the
manufacturer's instructions. All specimens were fixed in 10%
neutral-buffered formalin for 48 h at room temperature and embedded
in paraffin. Sections were first dewaxed in xylene and rehydrated
in ethanol solution, after incubation with proteinase K working
solution (20 µM) for 30 min at room temperature. The tumor
sections were washed with PBS twice. The slides were exposed to
TUNEL reaction mixture prepared freshly for 1 h at 37°C in the
dark. Then, the slide were rinsed three times with PBS. All slides
were analyzed in a drop of PBS under a fluorescence microscope
(Olympus Corporation) and analyzed using ImageJ software (version
2.0; National Institutes of Health). The number of positive cell
nuclei in 30 random fields from randomly selected tumor sections
for each animal was counted at a magnification of ×400.
Statistical analysis
For quantification, Ki-67, Atg5, p62 and LC3
staining intensity was measured from the number of positive cell
nuclei in 25% fields using ImageJ software (version 2.0; National
Institutes of Health). All areas were chosen randomly from all
sections. The intensity of bands in western blotting was also
measured by ImageJ software (National Institutes of Health). All
data are presented as the mean ± SD of at least three independent
experiments. The acquired experimental data were analyzed using
SPSS 17.0 software (SPSS, Inc.) and GraphPad Prism 5.0 (GraphPad
Software, Inc). The differences between two groups were analyzed
using an unpaired Student's t-test. Student's t-test was used when
two groups were compared and one-way ANOVA was used when several
groups were compared. The Tukey method was used as the post hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
DRM increases autophagy in GBM cells
Various methods were used to determine the effect of
DRM on the levels of autophagy in GBM cells. TEM results revealed
accumulation of autolysosomes and autophagosomes in the DRM group,
but not in the control group (Fig.
2A). Furthermore, LC3 puncta were observed using the GFP-LC3
transient transfection assay. Microscopy images indicated that DRM
increased the distribution and number of LC3 puncta (Fig. 2B). Additionally, LC3I/LC3II
protein expression after transfection was detected by western
blotting. The results demonstrated that LC3I/LC3II protein
expression increased after transfection without DRM treatment
(Fig. 2C). LC3 is a marker of
autophagy. During the initiation of autophagy, LC3I enzymatically
decomposes a small polypeptide segment and transforms it into LC3II
(40). The ratio of LC3II/LC3I
can be used to estimate the level of autophagy (41). At the same time, p62 and Atg5
proteins are important indicators of regulation of LC3I and LC3II
protein conversion (42,43). Western blot analysis demonstrated
that DRM induced the accumulation of LC3I/LC3II and Atg5 in U87 and
C6 cells when the DRM concentration was 15 µM. In addition,
DRM (15 µM) induced degradation of p62 in U87 and C6 cells
(Fig. 2D-G). In combination,
these data suggested that DRM could promote autophagy initiation in
GBM cells.
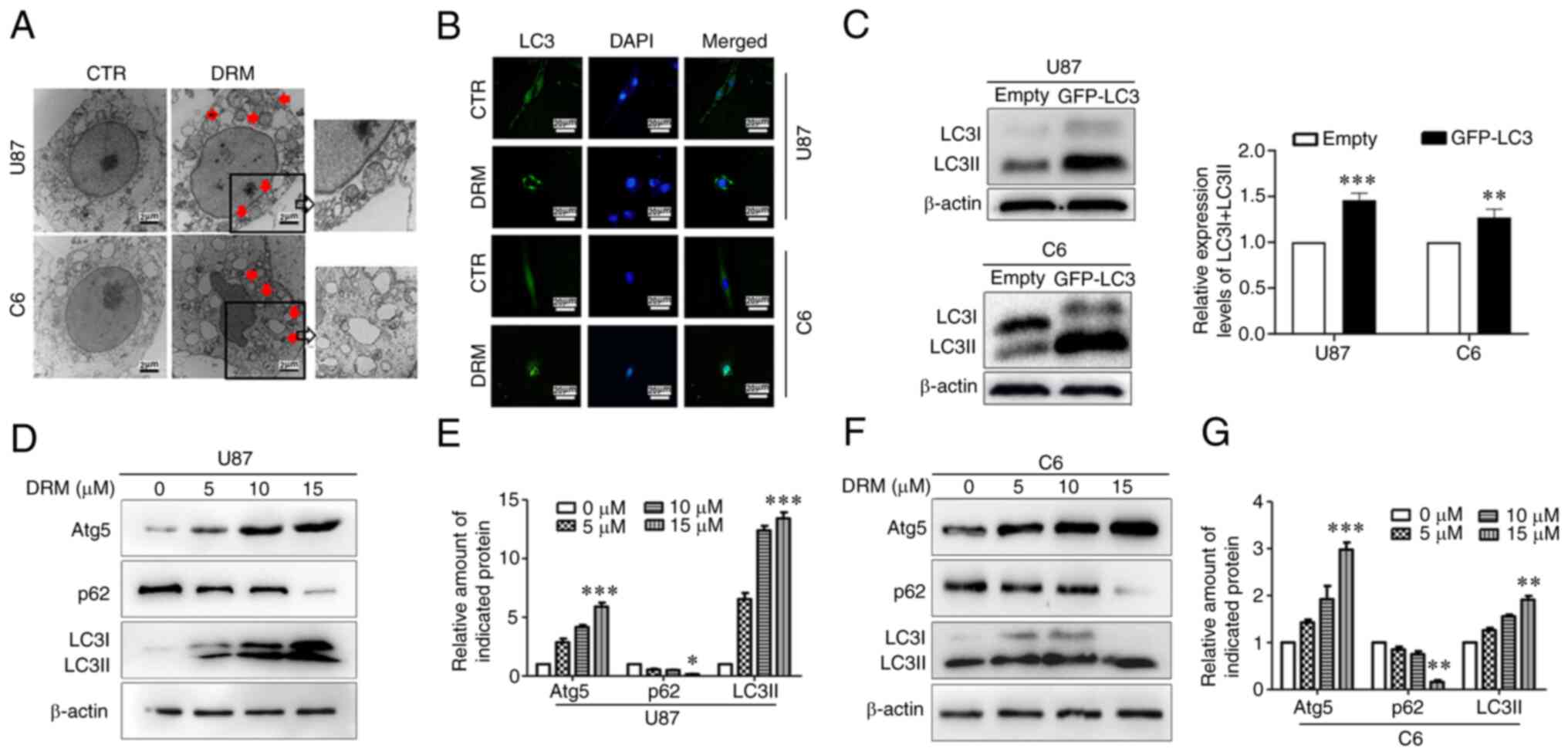 | Figure 2DRM-induced autophagy in U87 and C6
cells. (A) Transmission electron microscopy revealed autophagosome
accumulation in U87 and C6 cells treated with 0 or 15 µM DRM
for 48 h. Red arrows indicate autophagosomes. Scale bar, 2
µm. (B) Fluorescence microscopy using GFP-LC3 as a measure
of the autophagic response in U87 and C6 cells treated with 0 or 15
µM DRM for 48 h. Scale bar, 20 µm. (C) U87 and C6
cells were transfected with the GFP-LC3 plasmid and empty plasmid,
and LC3I/LC3II and β-actin protein expression was examined by
western blot, and semi-quantitative analysis of the protein
expression levels of LC3I/LC3II and β-actin indifferent groups was
performed. (D) U87 cells were treated with different concentrations
of DRM (0, 5, 10 and 15 µM) for 48 h. Western blot analysis
was performed to detect protein expression levels of Atg5, p62,
LC3I/LC3II and β-actin. (E) Graphical representation of
semi-quantitative analysis of autophagic proteins in U87 cells. (F)
Protein expression level of Atg5, p62, LC3I/LC3II and β-actin in C6
cells treated with different concentrations of DRM (0, 5, 10 and 15
µM) for 48 h, as determined by western blot. (G) Graphical
representation of semi-quantitative analysis of autophagic proteins
in C6 cells. The results are presented as the mean ± SD, n≥3.
*P<0.05, **P<0.01 and
***P<0.001 vs. empty or 0 µM. Atg5,
autophagy-related 5; CTR, control; DRM, doramectin; GFP, green
fluorescent protein. |
DRM-induced autophagy decreases
proliferation in GBM cells
The effect of DRM-induced autophagy on GBM cells was
further explored. Western blot analysis was conducted to detect the
protein expression levels of p62 and LC3I/LC3II in the presence of
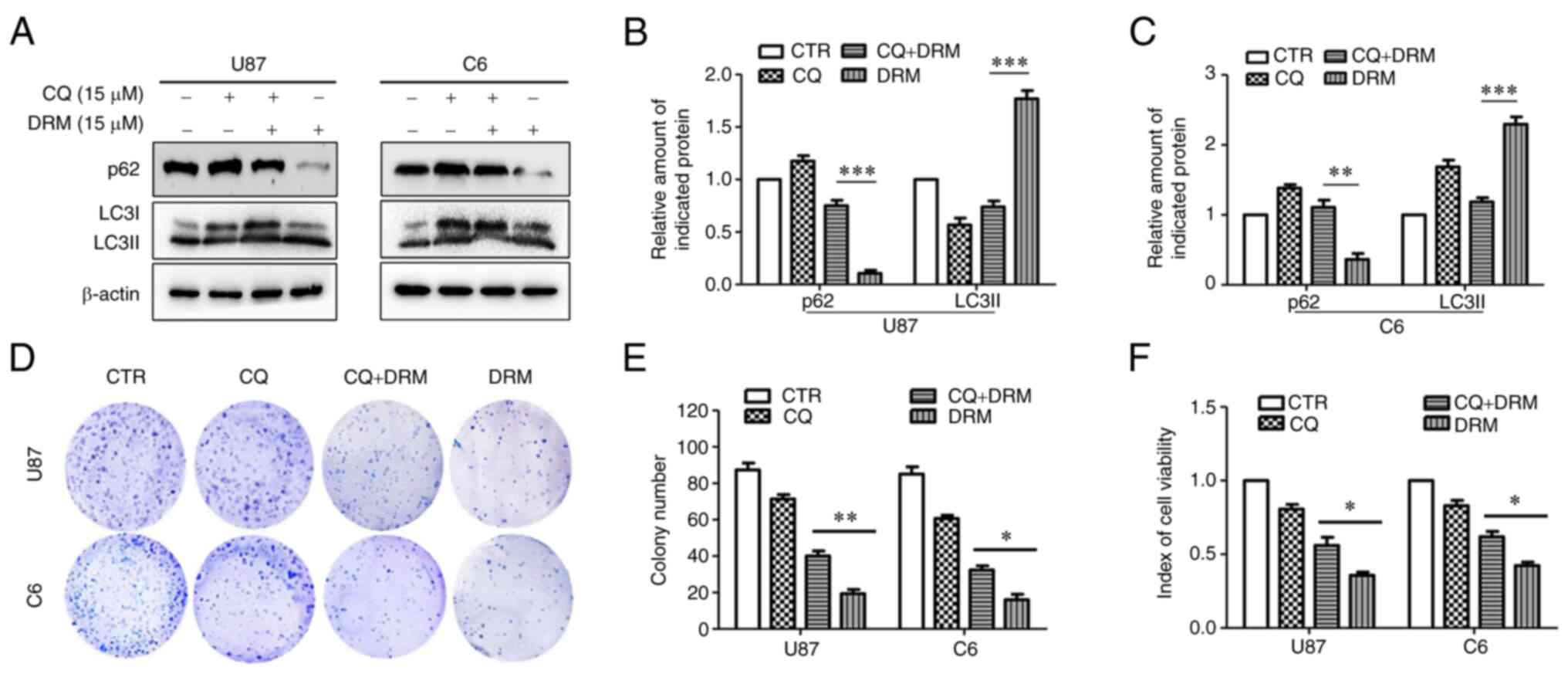
the autophagy inhibitor CQ. As expected, the autophagy inhibitor CQ
partially inhibited DRM-induced cell autophagy (Fig. 3A-C). In the DRM+CQ group compared
with the DRM group, the LC3I/LC3II protein expression was
decreased. Subsequently, MTT and colony formation assays were used
to assess the viability and colony formation ratio of GBM cells.
According to MTT assay results, DRM significantly reduced cell
viability compared with DRM combined with CQ in U87 and C6 cells
(Fig. 3F). As shown in Fig. 3D and E, treatment with DRM
combined with CQ was associated with a significant increase in the
colony formation ability of U87 and C6 cells compared with
treatment with DRM only. In conclusion, these results indicated
that DRM-induced autophagy could reduce the proliferation of GBM
cells.
Inhibition of autophagy decreases
DRM-induced apoptosis
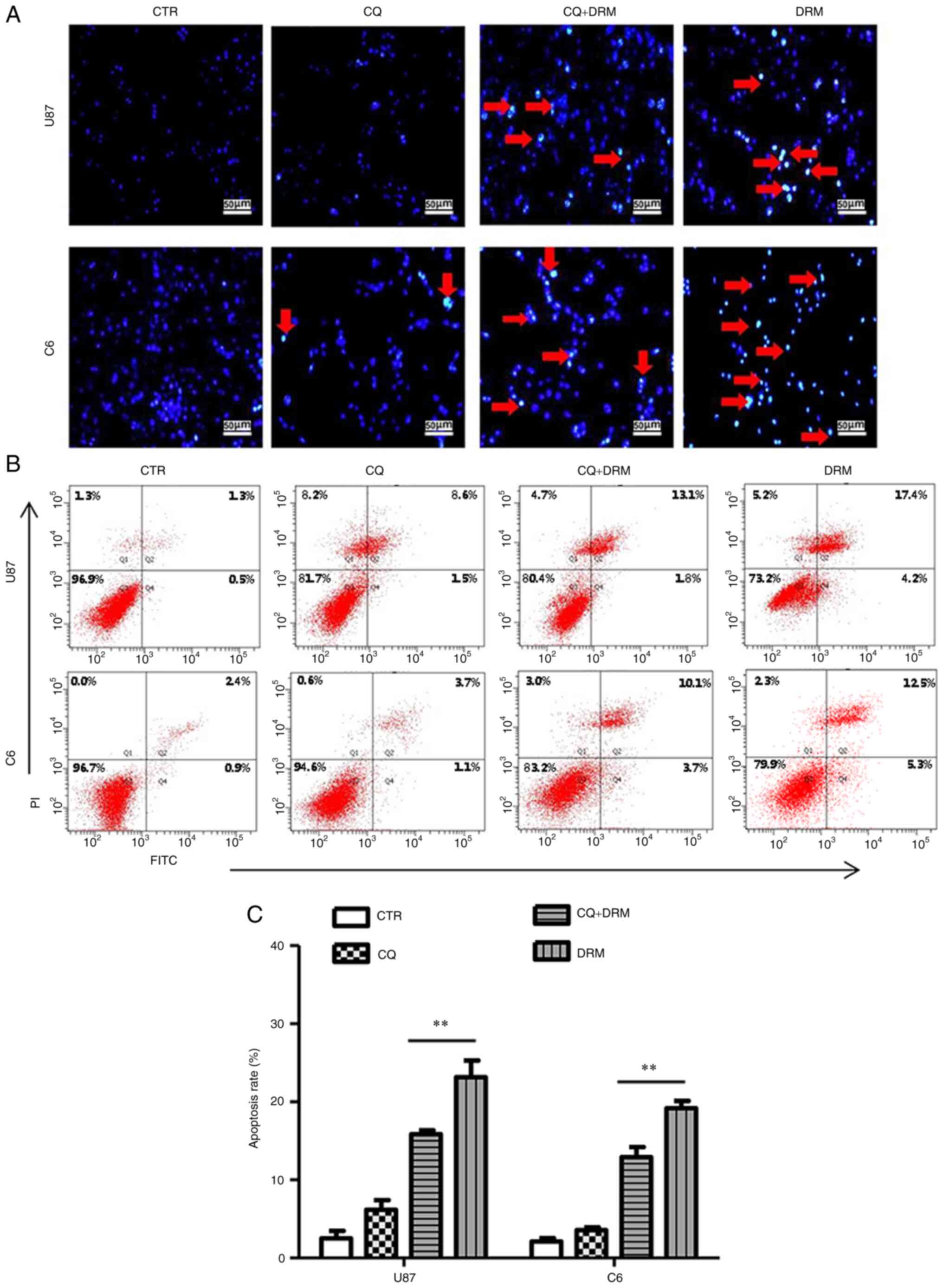
The effect of DRM-induced autophagy on GBM cell
apoptosis was further investigated. First, DAPI staining was
performed to explore the role of autophagy in DRM-induced DNA
double-strand breaks. In Fig. 4A,
the fragmented DNA was increased in the DRM only group compared
with the DRM+CQ group. Subsequently, apoptosis was analyzed by flow
cytometry. Apoptosis was increased in the DRM group compared with
the DRM+CQ, CQ and control groups (Fig. 4B and C). These findings suggested
that DRM-induced autophagy could promote apoptosis in GBM
cells.
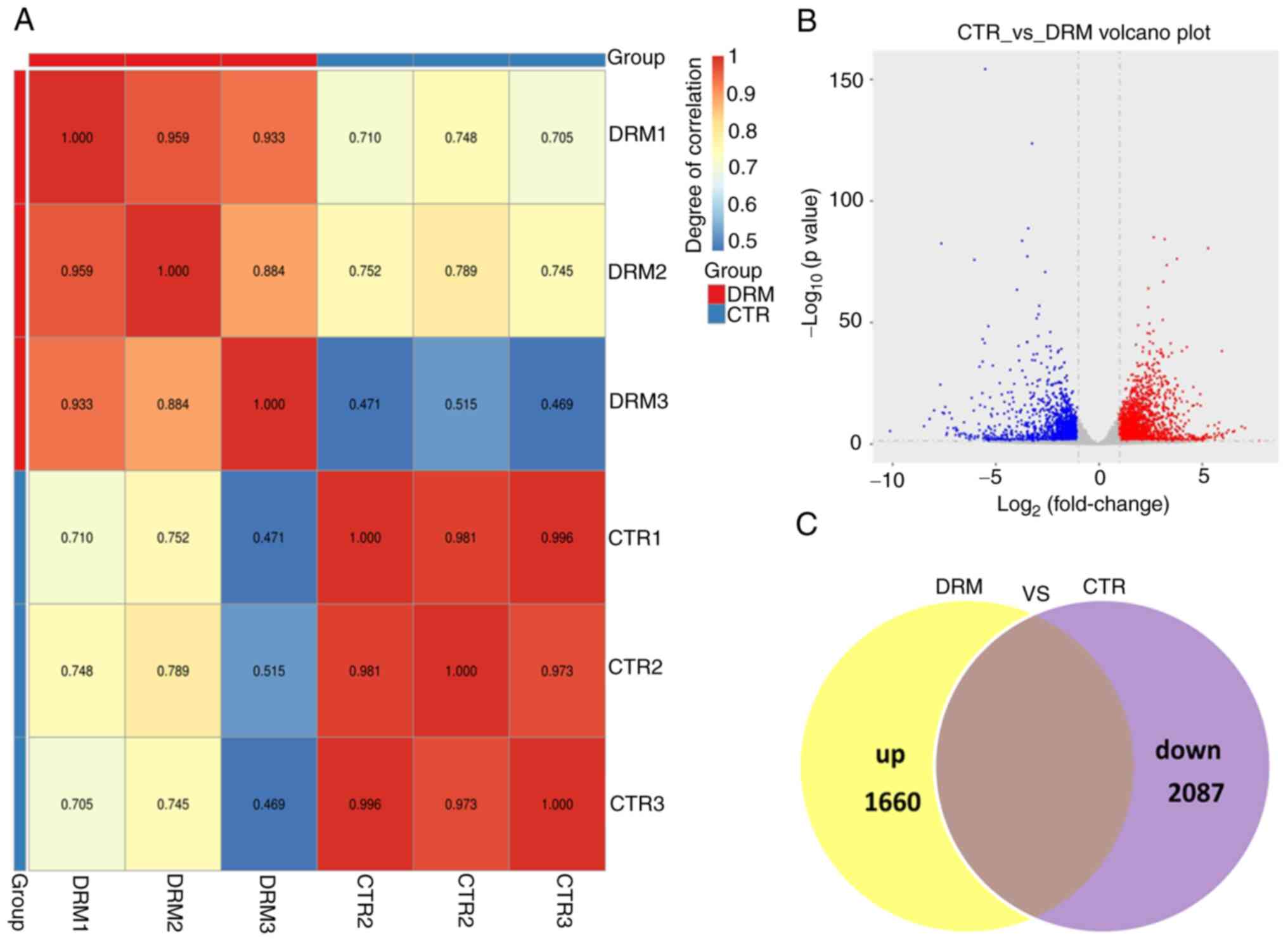
Analysis of DEGs in DRM-treated C6
cells
Comparison of DRM-treated C6 cells with untreated C6
cells revealed a higher degree of differential expression of genes.
First, to ensure the reliability of the sequencing results, the
inter-sample correlation was analyzed (Fig. 5A). As shown by the Venn diagram
and Volcano plot in Fig. 5B and
C, the transcript expression profiles identified 3,747 DEGs,
including 1,660 upregulated and 2,087 downregulated genes.
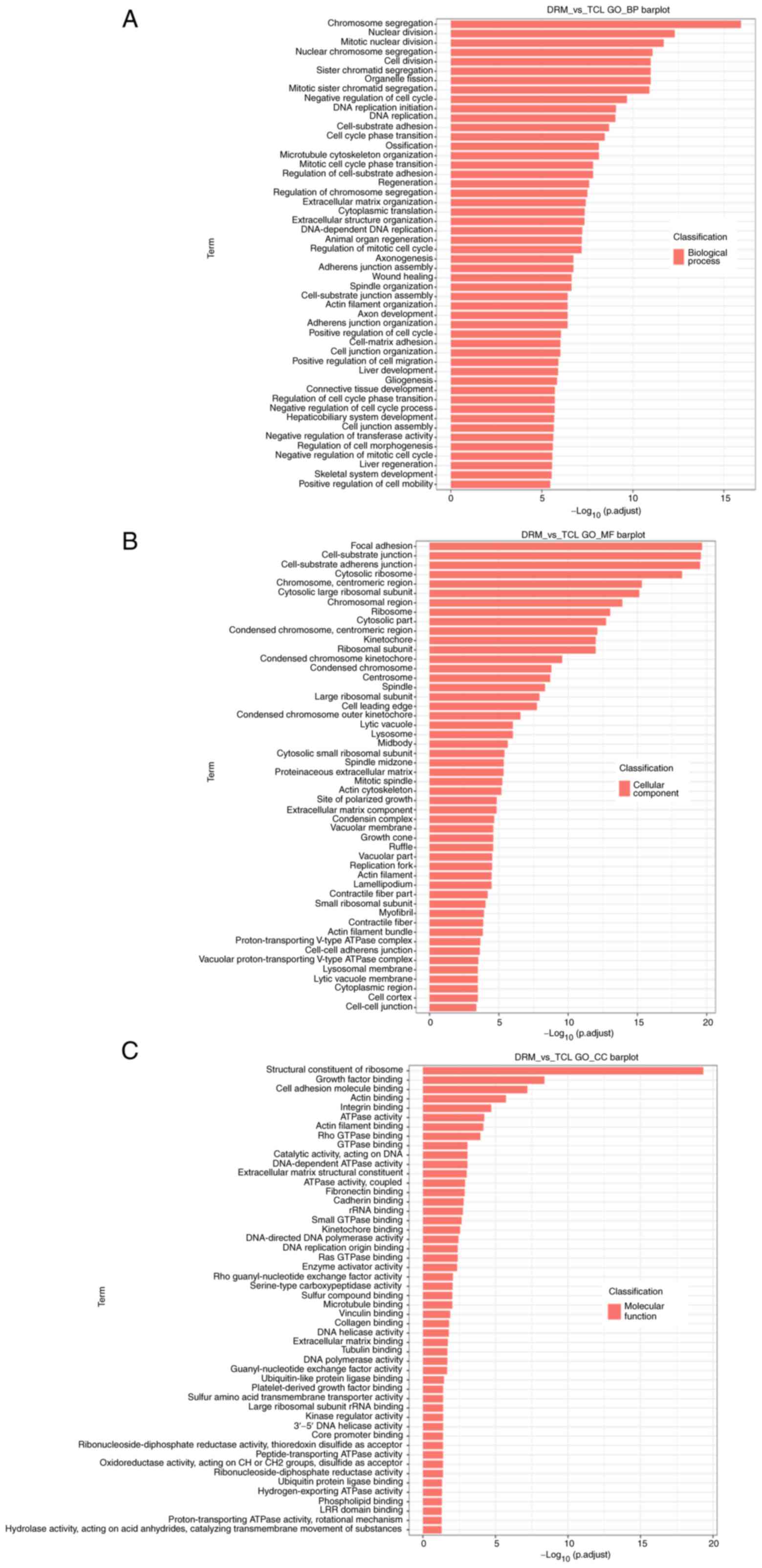
GO enrichment analysis of DEGs
To further explore the potential functions of DEGs,
the DEGs in the treatment groups were examined by GO functional
enrichment analysis in relation to the control group. Each group of
DEGs covered three aspects of biology. The top 50 functionally
enriched classes in each group are shown in Fig. 6. With regards to biological
processes, the DEGs were mainly enriched in 'chromosome
segregation', 'nuclear division', 'mitotic nuclear division',
'nuclear chromosome segregation' and 'cell division' (Fig. 6A). With regards to cellular
components, the DEGs were mainly enriched in 'focal adhesion',
'cell-substrate junction', 'cell-substrate adherens junction',
'cytosolic ribosome' and 'chromosome, centromeric region' (Fig. 6B). With regards to molecular
functions, the DEGs were mainly enriched in 'structural constituent
of ribosome', 'growth factor binding', 'cell adhesion molecule
binding', 'actin binding' and 'integrin binding' (Fig. 6C).
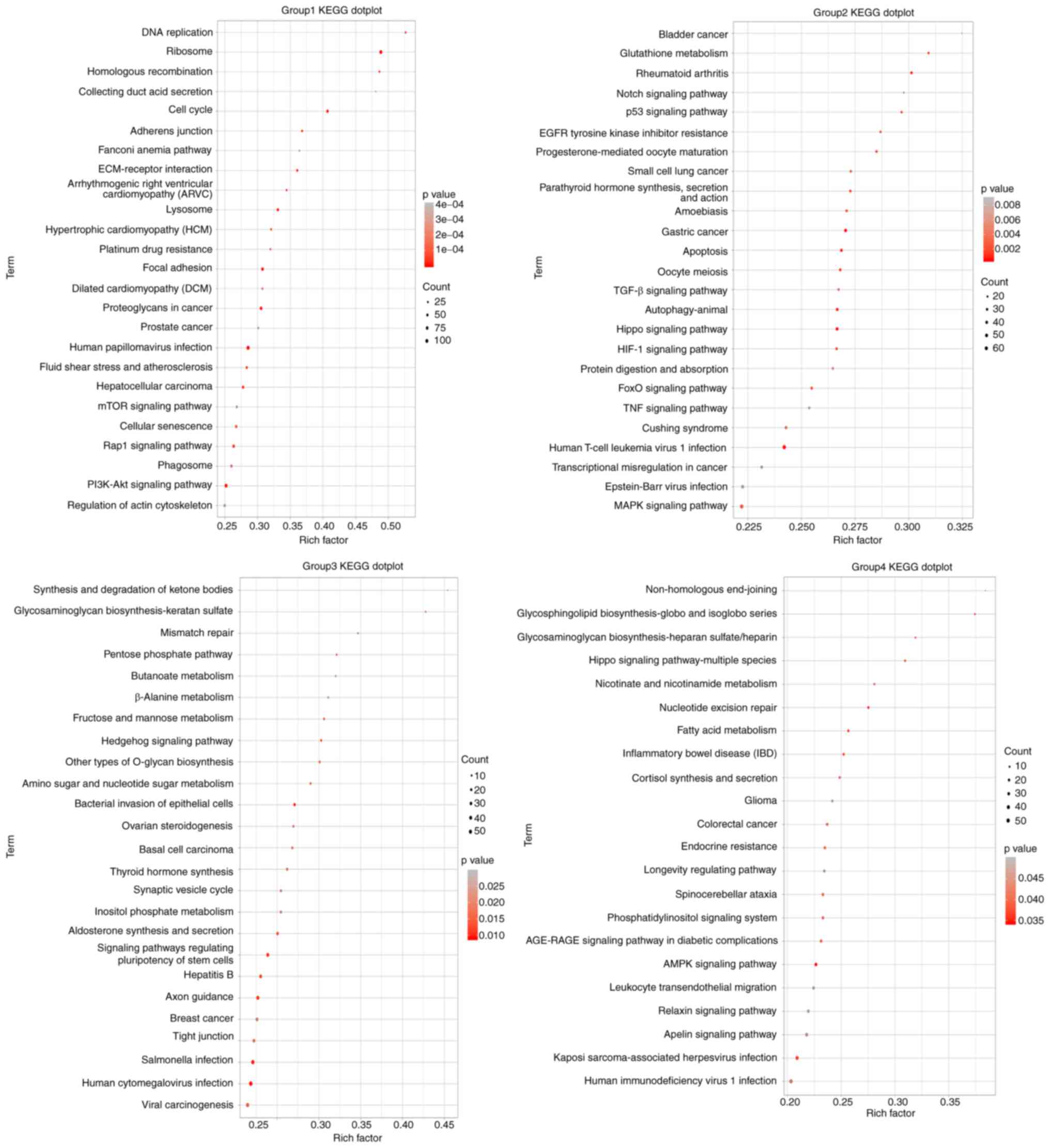
KEGG signaling pathway enrichment
analysis of DEGs
In order to further explore the effect of DRM on GBM
signaling pathways, KEGG functional enrichment analysis of DEGs was
also performed. The results in Fig.
7 revealed that the autophagy signaling pathway was not the
only pathway that was enriched. Multiple signaling pathways
associated with autophagy and apoptosis, such as 'DNA replication'
and the 'p53 signaling pathway', 'mTOR signaling pathway',
'PI3K/Akt signaling pathway' and 'MAPK signaling pathway', were
significantly enriched in C6 DRM-treated cells. A number of
autophagy-related pathways were changed, indicating that the
autophagy pathway was altered in the DRM group compared with the
control group and that the DEGs in these pathway categories may be
closely associated with DRM-induced autophagy. The top 100
signaling pathways that were altered after DRM treatment of C6
cells were randomly shown.
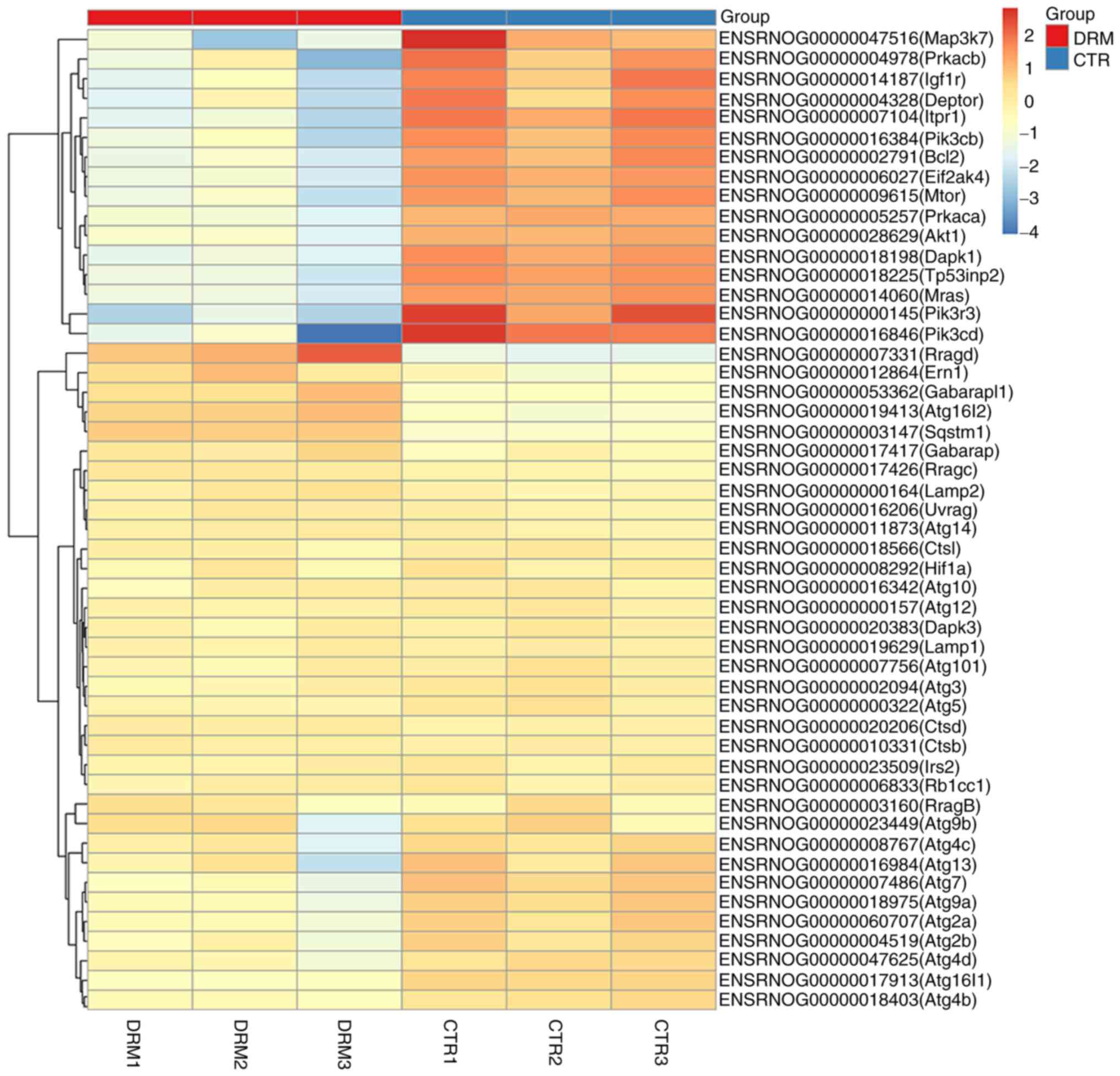
Analysis of DEGs involved in the
autophagy signaling pathway
DEGs involved in autophagy are shown in the heat map
in Fig. 8. Following treatment of
C6 cells with DRM for 48 h, 51 DEGs were shown. In Fig. 9, a number of the genes that
influence and control autophagy were shown, and these were also
shown in the heat map, for example, a series of autophagy-related
genes (Atgs), mTOR, AKT and pI3K, were identified.
DRM-induced autophagy suppresses C6 cell
xenograft growth in vivo
The present study further determined the effect of
DRM-induced autophagy on the regulation of GBM xenograft growth
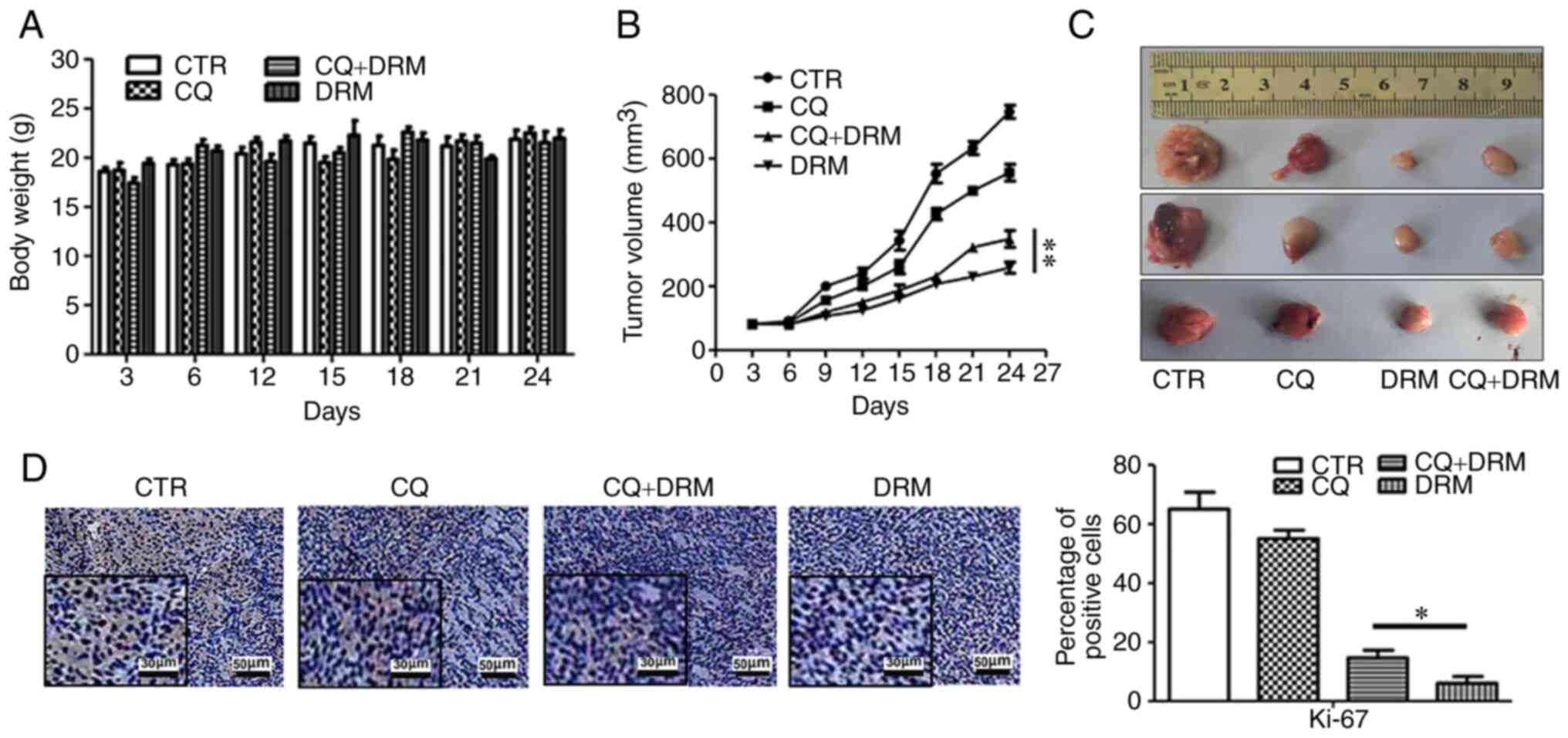
in vivo. No obvious differences in the weight of the mice
were observed between the DRM, CQ and CQ+DRM groups and the control
group (Fig. 10A). When comparing
the two DRM intervention groups, significant suppression of tumor
growth was observed in the CQ-untreated group compared with the
DRM+CQ-treated group (Fig. 10B and
C). To further verify that DRM-induced autophagy can inhibit
GBM cell proliferation, immunohistochemical analysis of tumor
sections was performed. As shown in Fig. 10D, compared with the CQ+DRM
group, the expression levels of Ki-67, a marker of cell
proliferation, were decreased in the DRM group. These results
indicated that DRM-induced autophagy was involved in the reduction
of tumor growth in a mouse xenograft model of GBM.
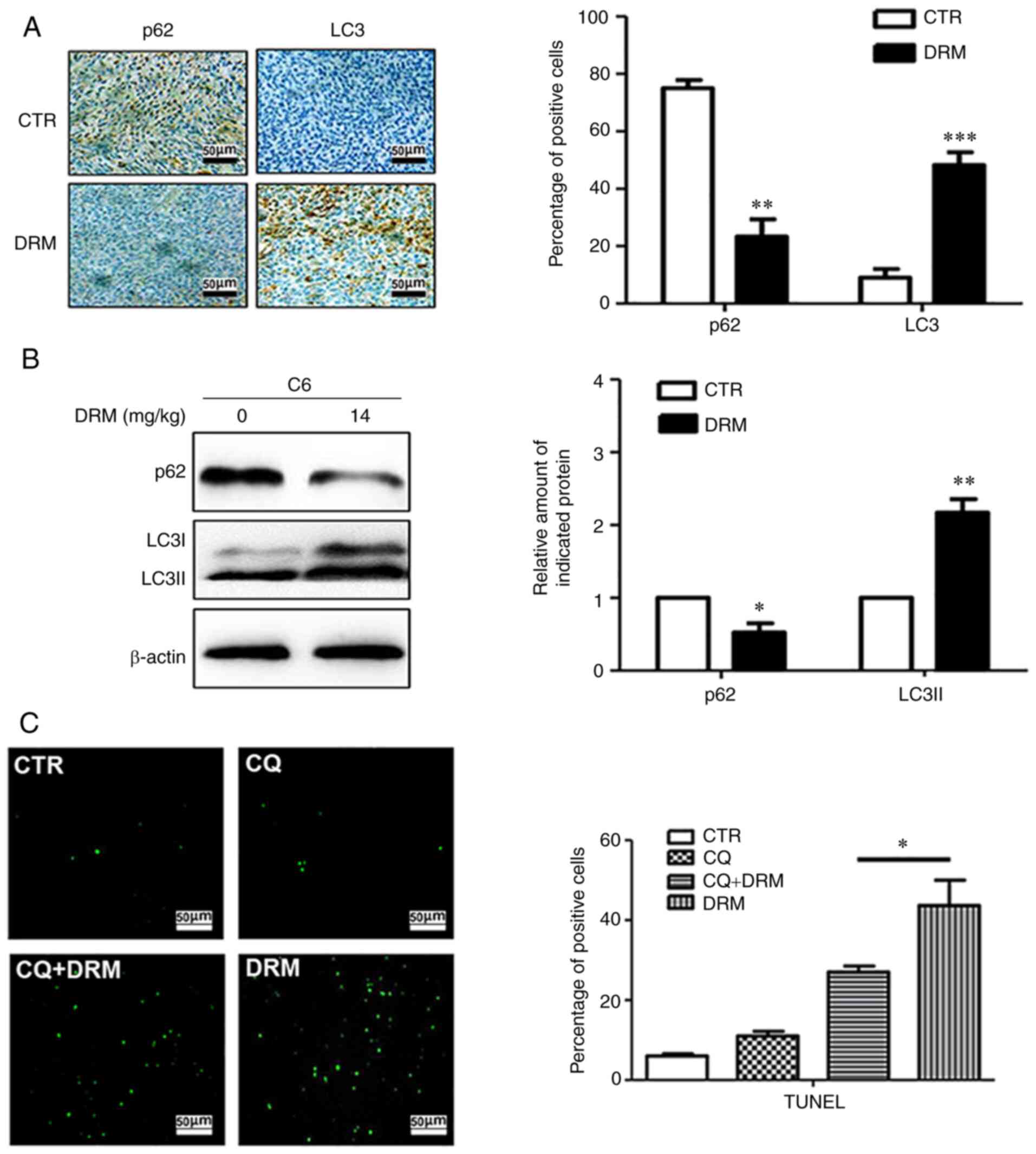
DRM-induced autophagy promotes apoptosis
in vivo
To further detect the effect of DRM on autophagy
in vivo, immunohistochemical staining and western blot
analysis were performed. Immunohistochemical staining indicated
that fewer p62-positive and more LC3-positive cells were observed
in the tumors from DRM-treated mice compared with those from
control mice (Fig. 11A).
Similarly, western blot analysis revealed that the LC3I/LC3II
protein was significantly elevated and P62 protein was
significantly reduced in DRM-treated mice compared with the control
mice (Fig. 11B). These results
demonstrated that treatment with DRM triggered autophagy in GBM
cells in vivo. The association between DRM-induced autophagy
and apoptosis was also explored in the nude mouse xenograft model.
According to the TUNEL assay, an increased number of apoptotic
cells were identified in the DRM-treated group compared with the
CQ+DRM group (Fig. 11C),
indicating that DRM-induced autophagy could enhance tumor
apoptosis.
Discussion
The antitumor activity of macrocyclic lactones has
been a research hotspot in recent years (44). Numerous studies have demonstrated
that macrocyclic lactones inhibit the proliferation of cancer cells
(45-47). In our previous study, it was
demonstrated that IVM and AVM of the AVM family not only inhibited
the proliferation of glioma cells via evoking apoptosis but also
induced autophagy in glioma cells (21,48). Furthermore, IVM has been used to
treat filariasis and kill ixodesscapularis ticks feeding on humans
(49). In the present study, DRM,
a more effective and less toxic drug from the AVM family, was used
to treat GBM. Different from studies on GBM and other macrocyclic
drugs, the present study adopted other research methods to explore
the effects of DRM on GBM. For instance, transcriptome analysis,
which can explain the molecular mechanism of DRM-induced apoptosis
and autophagy in GBM cells, was conducted. Whether DRM can induce
autophagy of GBM cells remains to be studied further. The present
study revealed that DRM induced autophagy in vitro and in
vivo. In addition, DRM-induced autophagy served an important
role in suppressing GBM cell proliferation. It was further
demonstrated that DRM modulated a number of pathways and genes
involved in autophagy, thereby affecting the initiation of
autophagy.
In the present study, TEM and a GFP-LC3 transient
transfection assay demonstrated that DRM not only induced the
formation of autophagosomes but also induced the formation of a
higher number of GFP-LC3 puncta on U87 and C6 cells. In addition,
LC3 exists in both its soluble (LC3I) and autophagosome-related
(LC3II) forms, and LC3I can transform into LC3II (40). The LC3II/LC3I ratio is used to
assess the levels of autophagy (41,42). During the process of autophagy,
the p62 protein is involved in the conversion of LC3I to LC3II
(50). As expected, DRM-induced
autophagy was identified by western blot analysis, as revealed by
the increase in LC3I/LC3II and the decrease in p62 protein
expression. This result demonstrated that DRM can induce autophagy
in GBM cells in vivo.
The functional relationship between autophagy and
cell survival is complex, with recent evidence suggesting that
autophagy is a double-edged sword in cell death (51). On the one hand, it is considered
an essential mechanism of protection and survival. On the other
hand, it is also regarded as a type of programmed cell death
(52,53). However, the specific mechanism of
the dual role of autophagy needs to be explored further. A study
has reported that it is cell line-dependent (54). Some studies have suggested that
different drugs had opposite effects in the same cell lines. For
example, IVM-induced autophagy increased cell proliferation in
glioma (48). However,
5-methoxypsoralen induced glioma cell death by inhibiting autophagy
(55). Therefore, DRM, a
potential anticancer agent, is required to further explore the role
of autophagy in cell death. In the present study, the autophagy
inhibitor CQ was used to clarify the specific mechanisms involved
in the effects of DRM-induced autophagy on GBM cells. When CQ
enters the lysosome, it becomes protonated because of the low pH
within the lysosome, and accumulation of the protonated form of
chloroquine with in the lysosome leads to less acidic conditions
and thereby decreased lysosomal function (56). First, autophagic flux assays
demonstrated that CQ markedly increased LC3II expression in
CQ-pretreated cells. In addition, MTT and colony formation assays
were conducted to determine the effect of DRM-induced autophagy on
inhibition of U87 and C6 cell viability and colony formation ratio.
These results demonstrated that DRM-induced autophagy might be one
of the reasons for the inhibition of proliferation in GBM cell
lines.
It is well-known that the functional relationship
between apoptosis and autophagy is complex (57). They suppress or promote one
another or unilaterally promote or inhibit one another. Autophagy
and apoptosis have been demonstrated to be interconnected by
crosstalk between several molecular nodes (58). Research has demonstrated that Atg5
can be cleaved by calpain, which interacts with Bcl-XL and promotes
cytochrome c release with caspase activation and apoptosis
(59). The association between
DRM-induced autophagy and apoptosis in GBM cells is not well
understood, but the present study revealed that DRM induced GBM
cell apoptosis via the mitochondria-dependent pathway, which is
highly regulated by Bcl-2 family members. In addition, the present
study demonstrated that DRM increased Atg5 protein expression,
suggesting that DRM-induced autophagy can affect apoptotic changes.
In order to clarify the association between autophagy and
apoptosis, the autophagy inhibitor CQ was used to understand the
role of DRM-induced autophagy in the apoptosis of U87 and C6 cells.
Flow cytometry demonstrated that DRM-induced autophagy could
increase the apoptotic rate in U87 and C6 cells. DAPI staining
confirmed this conclusion. Therefore, it was demonstrated that
DRM-induced autophagy promoted the increase in apoptosis in GBM
cells.
The molecular mechanisms of the initiation of
autophagy are not yet fully understood. However, a number of
studies have demonstrated that numerous signaling pathways may be
involved in the initiation of autophagy (60-62). Therefore, the regulation of the
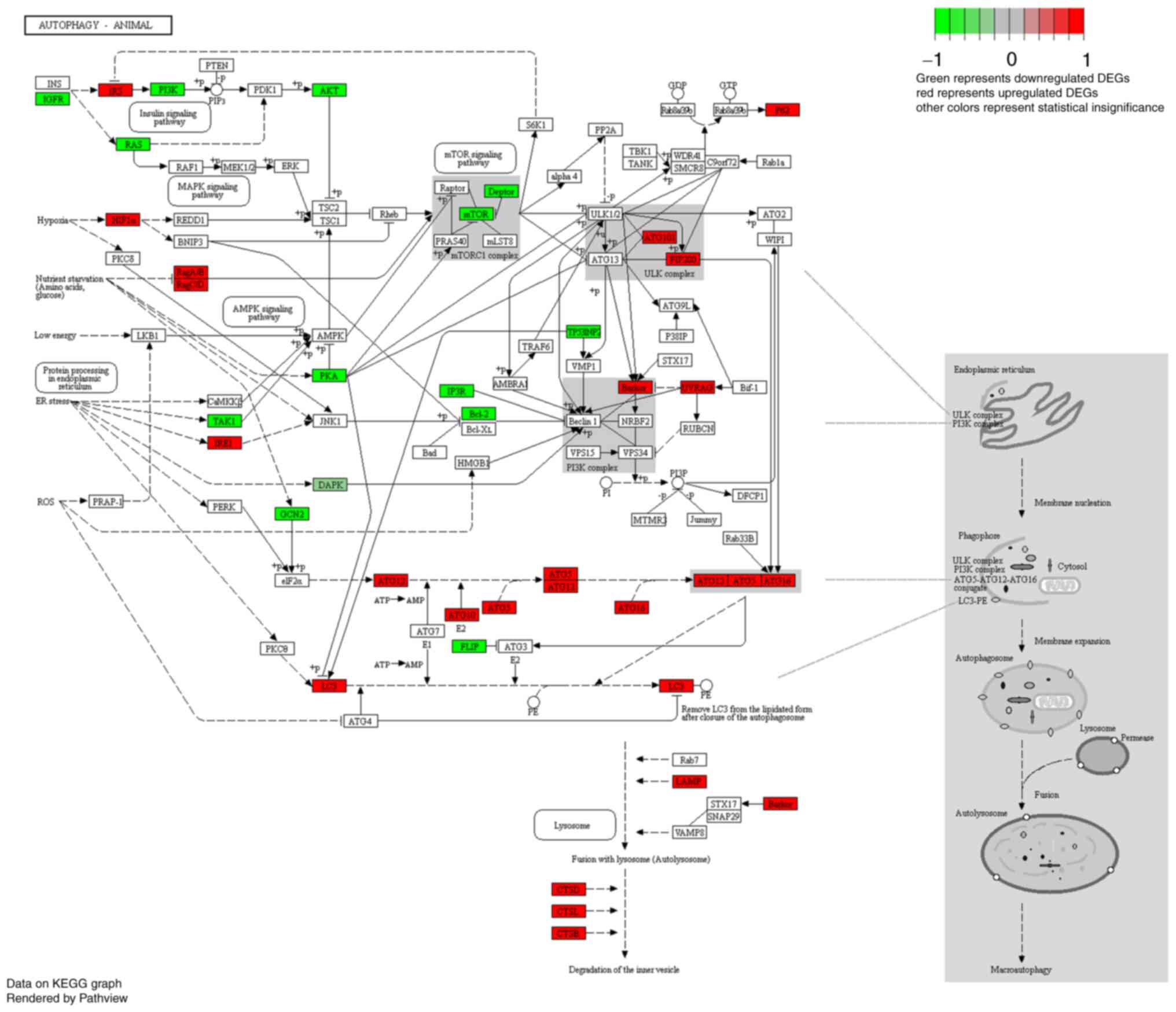
autophagy pathway was investigated using the KEGG database. mTOR
was identified as a protein that serves a key regulatory role in
the formation and maturation of autophagosomes, and the PI3K/AKT
and AMP-activated protein kinase (AMPK) signaling pathways are also
involved in the initiation of autophagy. In brief, a large number
of genes are involved in regulating autophagy in cells. A
transcriptome analysis was conducted on C6 cells to provide a
genome-wide view of biological responses to DRM exposure. At the
molecular level, transcriptome analysis revealed that a large
number of significant DEGs were enriched in autophagy-related
pathways. Autophagosome formation is dependent on the covalent
attachment of a series of Atg proteins during protein
ubiquitination (43,63). It was demonstrated that DRM
induced autophagy in GBM cells at the molecular level. KEGG
analysis results demonstrated that a number of autophagy pathways
were altered in the DRM group compared with the control group,
including the autophagy-animal, PI3K/AKT, lysosome, phagosome,
mTOR, MAPK, AMPK and DNA replication signaling pathways.
Previously, the PI3K/AKT/mTOR pathway, as a critical regulator of
autophagy, has been reported to be involved in the initiation and
promotion of a series of pathological disorders in tumors (64). The PI3K/AKT/mTOR pathway was
identified as the most enriched pathway in KEGG analysis, and the
levels of PI3K, AKT and mTOR proteins were markedly reduced. This
finding suggested that DRM-induced autophagy was mainly caused by
the attenuation of PI3K/AKT/mTOR phosphorylation in GBM cells.
Transcriptome analysis provided further evidence of DRM-induced
autophagy. Our follow-up study will be based on the DEGs identified
in the present study, which will serve as a foundation for
exploring the other effects of DRM on GBM further.
To acquire more reliable evidence to support and
verify the in vitro experimental findings, a xenograft nude
mouse model was used to clarify the underlying molecular mechanisms
of DRM autophagy in GBM cells in vivo. In the previous
study, a subcutaneous injection of IVM was administered to a
xenograft nude mouse model at a dose of 14 mg/kg, and the toxicity
was found to be acceptable and the dose safe (19,45). Therefore, the same drug dosage was
used in the in vivo experiment. A number of clinical trials
have used CQ alone or in combination with other chemotherapies for
the treatment of cancer (65,66). In the present study, the effect of
DRM+CQ was tested in a xenograft nude mouse model. The in
vivo experiments revealed that the tumor volume was
significantly smaller in the DRM group than in the DRM+CQ group.
Immunohistochemical analysis of Ki-67 activity also confirmed this
conclusion. In addition, a TUNEL assay revealed that DRM-induced
autophagy increased cell apoptosis in vivo. It was further
demonstrated that DRM could induce autophagy in vivo. These
findings were consistent with those of previous in vitro
studies. Additionally, our research team has also examined the
inhibitory effect of DRM on other tumors. For example, research has
demonstrated that the inhibitory effect on breast cancer cells was
not obvious (17). Esophageal
cancer is inhibited by DRM (Li et al, unpublished data).
Although DRM was found to have an effect on GBM cell proliferation
and apoptosis in our previous experimental exploration (Chen et
al, unpublished data), the lack of specific experimental data
in this regard is a limitation of the present study. In future
studies, the effects of DRM on GBM cells will be explored further
to provide more data support for DRM as a novel drug for the
treatment of cancer.
In conclusion, it was first demonstrated that DRM
induced autophagy in U87 and C6 GBM cells in vitro and in
vivo. In addition, the present study demonstrated that DRM
altered a number of pathways involved in autophagy in C6 cells, and
induced GBM cell autophagy mainly by blocking PI3K, AKT and mTOR at
the molecular level. In addition, autophagy could inhibit GBM cell
proliferation and apoptosis in vitro and in vivo. In
combination, these findings provided a theoretical basis for the
clinical application of DRM in the treatment of GBM.
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the Sequence Read Archive repository
(https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA726037).
Other data generated or analyzed during this study are included in
this published article.
Authors' contributions
CC conceived and designed the project, interpreted
the results and wrote the manuscript. AG, HL, RQ and XL conceived
and designed the project and assisted in writing the manuscript.
CC, LW, SD and ZC designed and performed the experiments. XM, ZL,
QW and JM performed the statistical analysis, created the figures,
and wrote and edited the manuscript. CC, AG and HL confirmed the
authenticity of the raw data. All authors have read and approved
the final manuscript.
Ethics approval and consent to
participate
All procedures involving animals were performed in
accordance with the ethical standards of the Harbin Weike
Biotechnology research committee (Harbin Weike Biotechnology Co.,
Ltd., Harbin, China). All animal experiments conformed to the
European Parliament Directive (2010/63/EU). The protocol was
approved by the Management and Welfare of Experimental Animal
Ethics Committee of Harbin Vic Biological Technology Development
Co., Ltd. (approval no. JL-SD-0370; Harbin, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Professor Saadia
Khilji (Department of Cellular and Molecular Medicine, Faculty of
Medicine, University of Ottawa, Ottawa, Ontario, Canada) for
assisting in the preparation of the manuscript.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 82071368), the Academic Backbone
Foundation of Northeast Agricultural University (grant no. 19XG20)
and the Outstanding Youth Funding of Harbin Medical University
(grant no. HYD2020JQ0016).
References
|
1
|
Hanif F, Muzaffar K, Perveen K, Malhi SM
and Simjee ShU: Glioblastoma multiforme: A review of its
epidemiology and pathogenesis through clinical presentation and
treatment. Asian Pac J Cancer Prev. 18:3–9. 2017.PubMed/NCBI
|
|
2
|
Strom QT, Gittleman H, Xu J, Kromer C,
Wolinsky Y, Kruchko C and Barnholtz-Sloan JS: CBTRUS statistical
report: Primary brain and other central nervous system tumors
diagnosed in the United States in 2009-2013. Neuro Oncol. 18(Suppl
5): v1–v75. 2016. View Article : Google Scholar
|
|
3
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Parsons DW, Jones S, Zhang X, Lin JC,
Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et
al: An integrated genomic analysis of human glioblastoma
multiforme. Science. 321:1807–1812. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ohgaki H, Dessen P, Jourde B, Horstmann S,
Nishikawa T, Di Patre PL, Burkhard C, Schüler D, Probst-Hensch NM,
Maiorka PC, et al: Genetic pathways to glioblastoma: A
population-based study. Cancer Res. 64:6892–6899. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tykocki T and Eltayeb M: Ten-year survival
in glioblastoma. A systematic review J Clin Neurosci. 54:7–13.
2018. View Article : Google Scholar
|
|
7
|
Sathornsumetee S and Rich JN: Designer
therapies for glioblastoma multiforme. Ann N Y Acad Sci.
1142:108–132. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Witthayanuwat S, Pesee M, Supaadirek C,
Supakalin N, Thamronganantasakul K and Krusun S: Survival analysis
of glioblastoma multiforme. Asian Pac J Cancer Prev. 19:2613–2617.
2018.PubMed/NCBI
|
|
9
|
Preusser M, de Ribaupierre S, Wöhrer A,
Erridge SC, Hegi M, Weller M and Stupp R: Current concepts and
management of glioblastoma. Ann Neurol. 70:9–21. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Anjum K, Shagufta BI, Abbas SQ, Patel S,
Khan I, Shah SA, Akhter N and Hassan SS: Current status and future
therapeutic perspectives of glioblastoma multiforme (GBM) therapy:
A review. Biomed Pharmacother. 92:681–689. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stupp R, Hegi ME, Gilbert MR and
Chakravarti A: Chemoradiotherapy in malignant glioma: Standard of
care and future directions. J ClinOncol. 25:4127–4136. 2007.
View Article : Google Scholar
|
|
12
|
Carson KA, Grossman SA, Fisher JD and Shaw
EG: Prognostic factors for survival in adult patients with
recurrent glioma enrolled onto the new approaches to brain tumor
therapy CNS consortium phase I and II clinical trials. J Clin
Oncol. 25:2601–2606. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Abe T, Mori T, Wakabayashi Y, Nakagawa M,
Cole SP, Koike K, Kuwano M and Hori S: Expression of multidrug
resistance protein gene in patients with glioma after chemotherapy.
J Neurooncol. 40:11–18. 1998. View Article : Google Scholar
|
|
14
|
Wang JB, Pan HX and Tang GL: Production of
doramectin by rational engineering of the avermectin biosynthetic
pathway. Bioorg Med Chem Lett. 21:3320–3323. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Murayama N, Shibata K and Nagata M:
Efficacy of weekly oral doramectin treatment in canine demodicosis.
Vet Rec. 167:63–64. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Taylor LF and Hodge A: Impact of a single
treatment of injectable doramectin on weight gain post weaning in
beef heifers and steers in central Queensland, Australia. Aust Vet
J. 97:185–190. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gao A, Wang X, Xiang W, Liang H, Gao J and
Yan Y: Reversal of P-glycoprotein-mediated multidrug resistance in
vitro by doramectin and nemadectin. J Pharm Pharmacol. 61:393–399.
2010. View Article : Google Scholar
|
|
18
|
Kramer L, Crosara S, Gnudi G, Genchi M,
Mangia C, Viglietti A and Quintavalla C: Wolbachia, doxycycline and
macrocyclic lactones: New prospects in the treatment of canine
heartworm disease. Vet Parasitol. 254:95–97. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ballweber LR and Baeten LA: Use of
macrocyclic lactones in cattle in the USA. Curr Pharm Biotechnol.
13:1061–1169. 2012. View Article : Google Scholar
|
|
20
|
Melotti A, Mas C, Kuciak M, Lorente-Trigos
A, Borges I and Ruiz i Altaba A: The river blindness drug
ivermectin and related macrocyclic lactones inhibit WNT-TCF pathway
responses in human cancer. EMBO Mol Med. 6:1263–1278. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Song D, Liang H, Qu B, Li YJ, Liu JJ,
Zhang YN, Li L, Hu L, Zhang XT, Zhang X and Gao A: Ivermectin
inhibits the growth of glioma cells by inducing cell cycle arrest
and apoptosis in vitro and in vivo. J Cell Biochem. 120:622–633.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang Y, Luo M, Xu W, Yang M, Wang B, Gao
J, Li Y and Tao L: Avermectin confers its cytotoxic effects by
inducing DNA damage and mitochondria-associated apoptosis. J Agric
Food Chemm. 64:6895–6902. 2016. View Article : Google Scholar
|
|
23
|
Drinyaev VA, Mosin VA, Kruglyak EB, Novik
TS, Sterlina TS, Ermakova NV, Kublik LN, Levitman MKh,
Shaposhnikova VV and Korystov YN: Antitumor effect of avermectins.
Eur J Pharmacol. 501:19–23. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Williams JC, Loyacano AF, DeRosa A, Gurie
J, Clymer BC and Guerino F: Comparison of persistent anthelmintic
efficacy of topical formulations of doramectin, ivermectin,
eprinomectin and moxidectin against naturally acquired nematode
infections of beef calves. Vet Parasitol. 85:277–288. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rozali EN, Hato SV, Robinson BW, Lake RA
and Lesterhuis WJ: Programmed death ligand 2 in cancer-induced
immune suppression. Clin Dev Immunol. 2012:6563402012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hu J, Liu X, Hughes D, Esteva FJ, Liu B,
Chandra J and Li S: Herceptin conjugates linked by EDC boost direct
tumor cell death via programmed tumor cell necrosis. PLoS One.
6:e232702011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nordmann N, Hubbard M, Nordmann T,
Sperduto PW, Clark HB and Hunt MA: Effect of gamma knife
radiosurgery and programmed cell death 1 receptor antagonists on
metastatic melanoma. Cureus. 9:e19432017.
|
|
28
|
Speidel D: Transcription-independent p53
apoptosis: An alternative route to death. Trends Cell Biol.
20:14–24. 2010. View Article : Google Scholar
|
|
29
|
Klionsky DJ, Abeliovich H, Agostinis P,
Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA,
Ballabio A, et al: Guidelines for the use and interpretation of
assays for monitoring autophagy in higher eukaryotes. Autophagy.
4:151–175. 2010. View Article : Google Scholar
|
|
30
|
Kim KH and Lee MS: Autophagy-a key player
in cellular and body metabolism. Nat Rev Endocrinol. 10:322–337.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liao Y, Duan B, Zhang Y, Zhang X and Xia
B: Excessive ER-phagy mediated by the autophagy receptor FAM134B
results in ER stress, the unfolded protein response, and cell death
in HeLa cells. J Biol Chem. 294:20009–20023. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim TW, Cheon C and Ko SG: SH003 activates
autophagic cell death by activating ATF4 and inhibiting G9a under
hypoxia in gastric cancer cells. Cell Death Dis. 11:7172020.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Klionsky DJ, Cuervo AM and Seglen PO:
Methods for monitoring autophagy from yeast to human. Autophagy.
3:181–206. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang J, Qi Q, Zhou W, Feng Z, Huang B,
Chen A, Zhang D, Li W, Zhang Q, Jiang Z, et al: Inhibition of
glioma growth by flavokawain B is mediated through endoplasmic
reticulum stress induced autophagy. Autophagy. 14:2007–2022. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Howarth A, Madureira PA, Lockwood G,
Storer LC, Grundy R, Rahman R, Pilkington GJ and Hill R: Modulating
autophagy as a therapeutic strategy for the treatment of paediatric
high-grade glioma. Brain Pathol. 29:707–725. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liao Y, Smyth GK and Shi W: featureCounts:
An efficient general purpose program for assigning sequence reads
to genomic features. Bioinformatics. 30:923–930. 2014. View Article : Google Scholar
|
|
37
|
Maag JL: gganatogram: An R package for
modular visualisation of anatograms and tissues based on ggplot2.
F1000Res. 7:15762018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sturn A, Quackenbush J and Trajanoski Z:
Genesis: Cluster analysis of microarray data. Bioinformatics.
18:207–208. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yu G, Wang LG, Han Y and He QY:
ClusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tanida I, Ueno T and Kominami E: LC3 and
autophagy. Methods Mol Biol. 445:77–88. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Onorati AV, Dyczynski M, Ojha R and
Amaravadi RK: Targeting autophagy in cancer. Cancer. 124:3307–3318.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tanida I, Ueno T and Kominami E: LC3
conjugation system in mammalian autophagy. Int J Biochem Cell Biol.
36:2503–2518. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mizushima N, Yoshimori T and Ohsumi Y: The
role of Atg proteins in autophagosome formation. Annu Rev Cell Dev
Biol. 27:107–132. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gallardo F, Mariamé B, Gence R and
Tilkin-Mariamé AF: Macrocyclic lactones inhibit nasopharyngeal
carcinoma cells proliferation through PAK1 inhibition and reduce in
vivo tumor growth. Drug Des Devel Ther. 12:2805–2814. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Markowska A, Kaysiewicz J, Markowska J and
Huczyński A: Doxycycline, salinomycin, monensin and ivermectin
repositioned as cancer drugs. Bioorg Med Chem Lett. 29:1549–1554.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Juarez M, Schcolnik-Cabrera A and
Duenas-Gonzalez A: The multitargeted drug ivermectin: From an
antiparasitic agent to a repositioned cancer drug. Am J Cancer Res.
8:317–331. 2018.PubMed/NCBI
|
|
47
|
Zhang X, Zhang G, Zhai W, Zhao Z, Wang S
and Yi J: Inhibition of TMEM16A Ca2+-activated Cl-channels by
avermectins is essential for their anticancer effects. Pharmacol
Res. 156:1047632020. View Article : Google Scholar
|
|
48
|
Liu J, Liang H, Chen C, Wang X, Qu F, Wang
H, Yang K, Wang Q, Zhao N, Meng J and Gao A: Ivermectin induces
autophagy-mediated cell death through the AKT/mTOR signaling
pathway in glioma cells. Biosci Rep. 39:BSR201924892019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sheele JM, Ford LR, Tse A, Chidester B,
Byers PA and Sonenshine DE: The use of ivermectin to kill
ixodesscapularis ticks feeding on humans. Wilderness Environ Med.
25:29–34. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Xin P, Xu W, Zhu X, Li C, Zheng Y, Zheng
T, Cheng W and Peng Q: Protective autophagy or autophagic death:
Effects of BEZ235 on chronic myelogenous leukemia. Cancer Manag
Res. 11:7933–7951. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Pyo JO, Yoo SM, Ahn HH, Nah J, Hong SH,
Kam TI, Jung S and Jung YK: Overexpression of Atg5 in mice
activates autophagy and extends lifespan. Nat Commun. 4:23002013.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Levy JM, Towers CG and Thorburn A:
Targeting autophagy in cancer. Nat Rev Cancer. 17:528–542. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Baehrecke EH: Autophagy: Dual roles in
life and death? Nature Rev Mol Cell Biol. 6:505–510. 2005.
View Article : Google Scholar
|
|
55
|
Guo H, He Y, Bu C and Peng Z: Antitumor
and apoptotic effects of 5-methoxypsoralen in U87MG human glioma
cells and its effect on cell cycle, autophagy and PI3K/Akt
signaling pathway. Arch Med Sci. 15:1530–1538. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Mauthe M, Orhon I, Rocchi C, Zhou XD, Luhr
M, Hijlkema KJ, Coppes RP, Engedal N, Mari M and Reggiori F:
Chloroquine inhibits autophagic flux by decreasing
autophagosome-lysosome fusion. Autophagy. 14:1435–1455. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lalaoui N, Lindqvist LM, Sandow JJ and
Ekert PG: The molecular relationships between apoptosis, autophagy
and necroptosis. Semin Cell Dev Biol. 39:63–69. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
McCoy F, Hurwitz J, McTavish N, Paul I,
Barnes C, O'Hagan B, Odrzywol K, Murray J, Longley D, McKerr G and
Fennell DA: Obatoclax induces Atg7-dependent autophagy independent
of beclin-1 and BAX/BAK. Cell Death Dis. 1:e1082010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lin JZ, Wang WW, Hu TT, Zhu GY, Li LN,
Zhang CY, Xu Z, Yu HB, Wu HF and Zhu JG: FOXM1 contributes to
docetaxel resistance in castration-resistant prostate cancer by
inducing AMPK/mTOR-mediated autophagy. Cancer Lett. 469:481–489.
2020. View Article : Google Scholar
|
|
61
|
Liu JZ, Hu YL, Feng Y, Guo YB, Liu YF,
Yang JL, Mao QS and Xue WJ: Rafoxanide promotes apoptosis and
autophagy of gastric cancer cells by suppressing PI3K/Akt/mTOR
pathway. Exp Cell Res. 385:1116912019. View Article : Google Scholar
|
|
62
|
Ding R, Wang X, Chen W, Li Z, Wei AL, Wang
QB, Nie AH and Wang LL: WX20120108, a novel IAP antagonist, induces
tumor cell autophagy via activating ROS-FOXO pathway. Acta
Pharmacol Sin. 40:1466–1479. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Tsuboyama K, Koyama-Honda I, Sakamaki Y,
Koike M, Morishita H and Mizushima N: The ATG conjugation systems
are important for degradation of the inner autophagosomal membrane.
Science. 354:1036–1041. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Xu Z, Han X, Ou D, Liu T, Li Z, Jiang G,
Liu J and Zhang J: Targeting PI3K/AKT/mTOR-mediated autophagy for
tumor therapy. Appl Microbiol Biotechnol. 104:575–587. 2020.
View Article : Google Scholar
|
|
65
|
Höglund R, Moussavi Y, Ruengweerayut R,
Cheomung A, Äbelö A and Na-Bangchang K: Population pharmacokinetics
of a three-day chloroquine treatment in patients with Plasmodium
vivax infection on the Thai-Myanmar border. Malar J. 15:1292016.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Maycotte P, Aryal S, Cummings CT, Thorburn
J, Morgan MJ and Thorburn A: Chloroquine sensitizes breast cancer
cells to chemotherapy independent of autophagy. Autophagy.
8:200–212. 2012. View Article : Google Scholar : PubMed/NCBI
|