Introduction
Breast cancer is a highly prevalent malignancy among
women and remains fatal in spite of the decrease in mortality rate
in recent years (1). Clinically,
three types of biomarkers are used to classify breast cancer:
Estrogen receptor α (ER), progesterone receptor (PR) and epidermal
growth factor receptor 2 (HER-2) (2,3).
Among them, Luminal A subtype highly expresses ER and PR, lacks
HER-2 expression and has the characteristics of low proliferation
rate and good prognosis (4).
Luminal B subtype is similar to Luminal A subtype, but lacks PR
expression. It demonstrates higher proliferation rate, worse
prognostic effect and tolerance to hormone therapy (5). The HER-2 subtype is characterized by
HER-2 overexpression, which can be further divided into HER-2
positive and HER-2 negative according to the expression of ER. It
is more resistant to chemotherapeutics and the prognosis is worse
than that of the Luminal subtypes. Triple negative breast cancer
(TNBC) is characterized by absence of HER-2, PR and ER expression.
It has strong chemotherapy resistance and metastasis. The prognosis
of patients with triple-negative breast cancer is extremely poor
and relapse common (6,7). Distant organ metastasis, especially
to the brain, bones, lungs and liver, is a characteristic feature
of breast cancer (8,9). Currently, metastatic breast cancer
is treated by surgery, radiotherapy and adjuvant chemotherapy.
Surgical resection has inherent disadvantages and is associated
with high recurrence rates (10,11), whereas radiotherapy and
chemotherapy have a number of adverse effects, such as cardiac
damage caused by combined chemoradiotherapy (12,13). Therefore, it is critical to
identify novel therapeutic targets for metastatic triple negative
breast cancer (TNBC).
The ribosomal regulatory protein RRS1 was discovered
in Saccharomyces cerevisiae by Tsuno et al in 1999
(14). The human RRS1 gene is
located on chromosome 8q13.1 and contains only one exon (15). RRS1 regulates ribosome
biosynthesis by recruiting 5S ribonucleoprotein (RNP) to form the
pre-60S ribosomal subunit with ribosomal production factor 2 (Rpf2)
and promoting the maturation of 25S rRNA (16-18). RRS1 also mediates the export of
pre-60S ribosomal subunit from the nucleolus to the cytoplasm.
Thus, depletion of RRS1 results in the accumulation of the pre-60
subunit in the nucleoplasm, eventually stalling ribosome
biosynthesis (18). Studies show
that RRS1 also regulates chromosome rearrangement during mitosis
(19) and telomere aggregation
(20) and serves an important
role in delayed cell aging (21)
and in the development of Huntington's disease (22). In addition, RRS1 is abnormally
expressed in various cancers (23-30). Through binding of RPL11, which
inhibits the interaction between RPL11 and murine doubleminute 2
(MDM2), it promotes the proliferation of breast cancer cells and
reduces p53 levels (26).
Knocking down RRS1 in breast cancer cells significantly reduced
their proliferation rates by inducing cell cycle arrest (26).
RPL11 is a component of 5S ribonucleoprotein
particles (5S RNP) and is translocated to the nucleus along with
RPL5 by the nuclear import protein Syo1, where they bind to 5S rRNA
(31). RPL11 regulates MDM2
(32), which prevents p53
accumulation during cellular stress by inducing protein
ubiquitination and degradation (33-37). Furthermore, RPL11 suppresses c-Myc
activity through a negative feedback pathway (38), which inhibits the transcription of
downstream genes regulating cell growth, proliferation, metabolism,
apoptosis, differentiation and ribosomal biosynthesis (39), whereas c-Myc can transcriptionally
activate RPL11 (40). In
addition, miR-150 and miR-383 suppresses 5S rRNA in esophageal
squamous cell carcinoma cells, which strengthening RPL11-c-Myc
interaction and reducing c-Myc-induced proliferation (41). C-myc regulates the epithelial
mesenchymal transition (EMT) of various tumor cells (42-47), which allows the cells to detach
from the primary tumors, extravasate and metastasize to distant
organs (48).
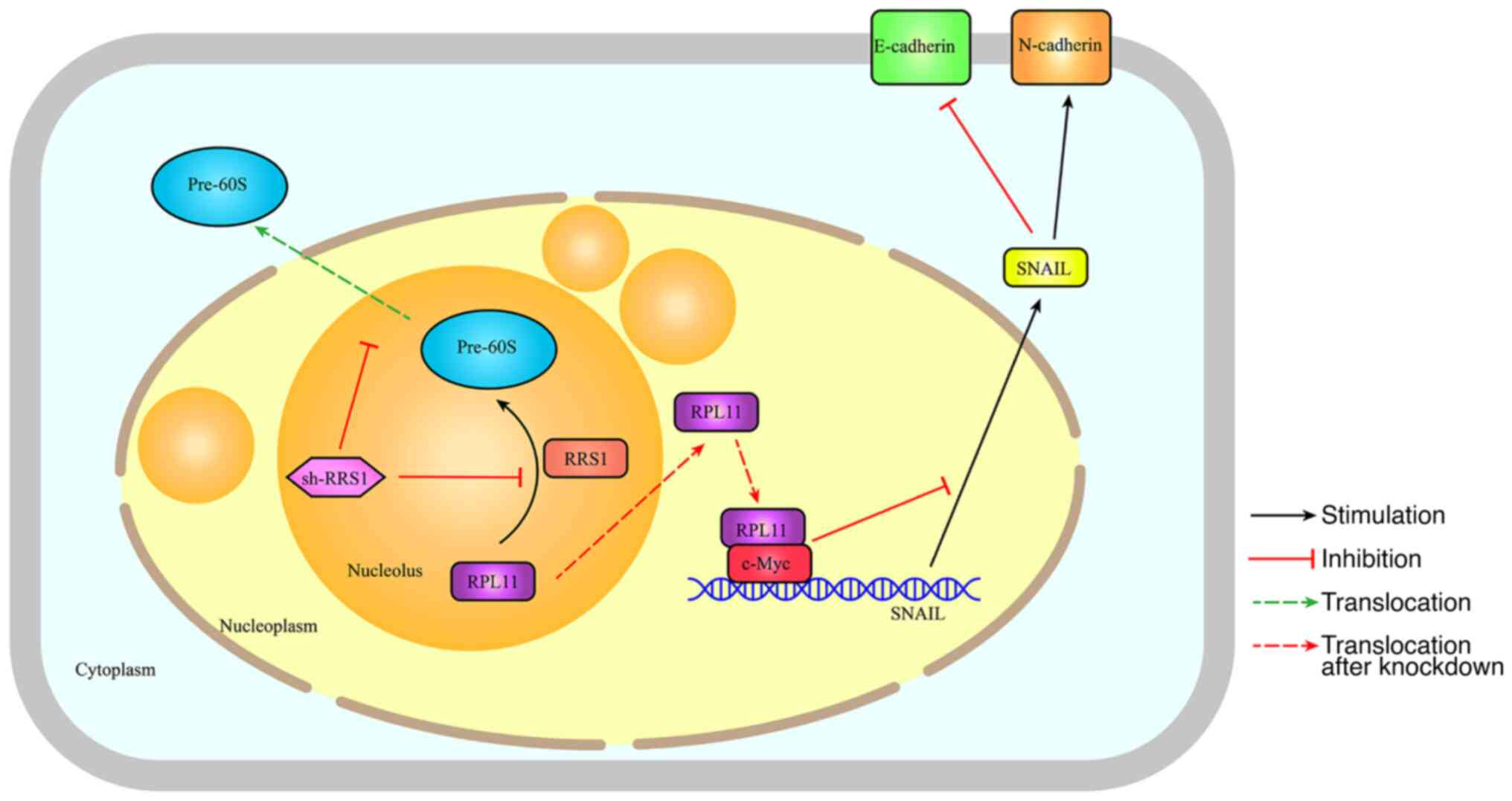
The present study found that RRS1 regulated the
invasion and metastasis of breast cancer cells through the
RPL11-c-Myc-SNAIL axis. RRS1 knockdown disrupted ribosomal
biosynthesis and prevented the export of RPL11 from the nucleus.
The accumulation of RPL11 in the nucleoplasm inhibited
c-Myc-dependent transcription of EMT-related genes. The findings
revealed a new role of RRS1 in regulating breast cancer cell
invasion and metastasis and demonstrate its potential as a
therapeutic target.
Materials and methods
Cell line authentication
The breast cancer cell lines BT549, MDA-MB-231,
MDA-MB-468, HMEC and MCF-7 was purchased from Procell Life Science
& Technology Co., Ltd. in March, 2021. The cell line was
verified in March, 2021 and the follow-up experiments were
performed in the same month. Briefly, DNA was extracted from
1×106 cells using Chelex100 and 20 STR sites and sex
loci were identified using the 21 CELL ID System (Procell Life
Science & Technology Co., Ltd.), including D19S433, D5S818,
D21S11, D18S51, D6S1043, D3S1358, D13S317, D7S820, D16S539, CSF1PO,
PentaD, vWA, D8S1179, TPOX, PentaE, TH01, D12S391, D2S1338, D1656
and Amelogenin1. The PCR products were analyzed with the ABI3130x1
genetic analyzer and Gene Mapper IDX software (Applied Biosystems;
Thermo Fisher Scientific, Inc.) and compared with the ATCC, DSMZ,
JCRB and Cellosaurus databases. BT549, MDA-MB-231, MDA-MB-468 are
Triple negative breast cancer breast cancer. MCF-7 is Luminal A
breast cancer. 293T cells were from the collection of our research
group.
Cell culture and transfection
All the cell lines were cultured in Dulbecco's
modified Eagle's medium (DMEM) supplemented with 10% FBS (Shanghai
ExCell Biology, Inc.) and 1% penicillin and streptomycin solution
at 37°C. On reaching 20-30% confluency, the cells were transduced
with the GV493-GFP lentivirus RNAi expression system (Shanghai
GeneChem Co., Ltd.) expressing RPL11-shRNA or scrambled shRNA with
Hitrans A&P (Shanghai GeneChem Co., Ltd.). The multiplicity of
infection (MOI) was 10. The virus was transfected with serum-free
medium for 10 h at 37°C. The medium was changed and replaced with
complete medium and subsequent experiments were carried out after
culturing for 48 h at 37°C. The siRNAs and scrambled sequence for
siRNAs were synthesized by Sangon Biotech Co., Ltd. The target
sequences for RPL11 were as follows: Forward 5′-GGU GCU GGA GUA UGA
GUU ATT-3′ and reverse 5′-UAA CUC AUA CUC CCG CAC CTT-3′. The cell
was transfected with siRNA using RNAFit (Hanbio Biotechnology Co.,
Ltd.) at 37°C according to the manufacturer's protocol.
Western blotting
Total protein was extracted from cells using RIPA
buffer (Shandong Sparkjade Scientific Instruments Co., Ltd.) and
quantified using a BCA kit (Beijing Solarbio Science &
Technology Co., Ltd.). Then 30 µg of protein per sample was
resolved by 10% SDS-PAGE and blotted onto PVDF membranes. After
blocking with 5% BSA (Beijing Solarbio Science & Technology
Co., Ltd., A8020) for 2 h at room temperature, the membranes were
incubated overnight with primary antibodies against RRS1 (1:1,000;
Abcam; cat. no. ab188161), c-Myc (1:1,000; Abcam; cat. no.
ab32072), NPM1 (1:1,000; Proteintech; cat. no. 60096-1-Ig), Lamin
B1 (1:1,000; Proteintech; cat. no. 12987-1-AP), N-cadherin
(1:1,000; Proteintech; cat. no. 22018-1-AP), E-cadherin (1:1,000;
Proteintech; cat. no. 20874-1-AP), c-Myc (1:1,000; Proteintech;
cat. no. 67447-1-Ig), RPL11 (1:1,000; Proteintech; cat. no.
16277-1-AP), Vimentin (1:1,000; Proteintech; cat. no. 10366-1-AP),
RPL23 (1:1,000; ABclona; cat. no. A4292) and Snail (1:1,000;
ABclona; cat. no. A11794) at 4°C, followed by the secondary
antibodies at room temperature (1:3,000, Bioss; cat. nos.
bs-40296G-HRP and bs-40295G-HRP) for 1 h. The positive bands were
visualized using enhanced chemiluminescence (ECL) kit and the bands
were measured using ImageJ (v1.53, National Institutes of
Health).
Cell proliferation assay
BT549 cells were seeded into 96-well plates at the
density of 2,000 cells/well. After 1, 2, 3, 4, 5 days of incubation
at 37°C, 10 µl of CCK8 solution (Beijing Solarbio Science
& Technology Co., Ltd.) solution was added to each well and the
cells were incubated for 2 h at 37°C. The absorbance at 450 nm was
measured using a microplate reader.
Reverse transcription-quantitative (RT-q)
PCR analysis
Total RNA was extracted by adding 1 ml of
TRIzol® Reagent (Thermo Fisher Scientific, Inc.) per
2×106 cells and using M-MLV Reverse Transcriptase
(HiScript® III All-in-one RT SuperMix kit, Vazyme
Biotech Co., Ltd.) reverse transcription 1 µg. qPCR was
performed using the ChamQ Universal SYBR qPCR Master Mix (Vazyme
Biotech Co., Ltd.) on ABI QuantStudio 3 (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The PCR amplification conditions were:
Pre-denaturing at 95°C for 30 sec, followed by 45 cycles of
denaturing at 95°C for 30 sec, annealing at 60°C for 30 sec and the
melting curve was the system default. The relative gene expression
levels were normalized to GAPDH. The primer sequences were as
follows: RRS1 Forward: 5′-CCC TAC CGG ACA CCA GAG TAA-3′, Reverse:
5′-CCG AAA AGG GGT TGA AAC TTC C-3′; GAPDH Forward: 5′-AGA AGG CTG
GGG CTC ATT TG-3′, Reverse: 5′-AGG GGC CAT CCA CAG TCT TC-3′. The
above experiments followed the manufacturer′s protocol. The
relative expression levels were calculated using the
2−∆∆Cq method (49).
Transwell and invasion assay
Transwell units were used to measure the migration
and invasion capacity of BT549 cells in vitro. BT549 cells
were seeded into the upper compartment of Transwell inserts
(Corning, Inc.) at the density of 10,000 cells/well in serum-free
media. For the invasion assay, the inserts were coated with
Matrigel (35 µl per Transwell unit; diluted 1:8 with
serum-free DMEM) at 4°C and incubated at 37°C for 2 h to gel the
Matrigel. The lower compartments were filled with complete media.
After 48 h of culture, the cells remaining on the upper surface of
the membranes were removed with a cotton swab and those that
migrated/invaded through the membranes were fixed with 4%
paraformaldehyde for 10 min at room temperature and then stained
with 0.1% hexamethylpararosaniline for 10 min at room temperature.
Then three fields of view were randomly selected under an inverted
microscope, images captured at ×100 and ×200 magnification, with
counting and data analyzed from the ×100 image. Migration assay is
used to detect the ability of cells to metastasize and invasion
assay is used to detect the ability of cells to lyse the cell
matrix.
Scratch test
The cells were seeded into a 6-well plate at the
density of 2×105 cells/well and cultured until 90%
confluent. Three lines were scratched on the monolayers with a 200
µl sterile pipette tip and were cultivated in serum-free
medium. The scratched area was measured using ImageJ (v1.53,
National Institutes of Health) at 0, 12, 24 and 48 h and the
migration rate was calculated as (scratch area at 0 h-scratch area
at 12, 24, 48 h)/scratch area at 0 h ×100%.
Co-immunoprecipitation (Co-IP)
Co-IP was performed with anti-c-Myc (1:1,000;
Proteintech; cat. no. 67447-1-Ig) and anti-RPL11 (1:1,000;
Proteintech; cat. no. 16277-1-AP) antibodies using the Classic
Magnetic Protrin A/G IP/Co-IP kit (Epizyme, Inc.) according to the
manufacturer's protocol. The kit contained Protein A/G magnetic
beads, lysis/wash buffer, SDS-PAGE protein loading buffer (5X),
elution buffer, neutralization buffer. Lysis/wash buffer and PMSF
(1:100, Solarbio, P0100) were added at a ratio of 30 µl per
1.0×105 cells, mixed well and incubated on ice for 30
min (mixing several times during this period); collected by
centrifugation (4°C; 12,000 × g; 10 min) with supernatant placed on
ice for later use. Then 500 µl of the prepared sample was
added to a 1.5 ml EP tube, followed by 4 µg antibody and
incubated on a flip mixer (4°C overnight) to form antigen-antibody
complexes. Magnetic bead suspension (25 µl) was placed into
a 1.5 ml EP tube and 500 µl of lysis/wash buffer added, the
magnetic beads were resuspended by gently pipetting and then let
stand on a magnetic stand for 1 min. When the magnetic beads were
adsorbed to the sidewall of the EP tube, the supernatant was
aspirated and this step repeated twice. The antigen-antibody
complex was added to the pretreated magnetic beads and incubated on
an inversion mixer (4°C overnight). Then it was stood on the
magnetic stand for 1 min, until the magnetic beads were adsorbed on
the side wall of the EP tube. The supernatant was aspirated and
discarded and what remained in the centrifuge tube was the
antigen-antibody-magnetic bead complex. Lysis/rinse buffer (500
µl) was added to the antigen-antibody-magnetic bead complex,
the magnetic beads resuspended by gently pipetting and agitation
and then allowed to stand on the magnetic stand for 1 min until the
magnetic beads were adsorbed to the side-wall of the centrifuge
tube. The supernatant was aspirated and discarded and this step
repeated twice. An appropriate amount of 5XSDS-PAGE loading buffer
was added to the antigen-antibody-magnetic bead complex, mixed well
and heated at 100°C for 10 min. Following cooling, the EP tube was
placed on a magnetic stand for 1 min. After the magnetic beads were
adsorbed on the side wall of the EP tube, the supernatant was
collected and detected by SDS-PAGE. The immuno-precipitates were
detected by western blotting as described above.
Luciferase activity assay
The dual-luciferase reporter gene detected the
relationship between the transcription factor c-Myc and the Snail
promoter and the 3′untranslated regions (UTRs) 2,000 bp upstream of
the Snail transcription start site. The RPL11 and c-Myc
overexpression plasmids and the luciferase SNAIL-pro reporter
plasmid and Renilla luciferase plasmid were designed and
constructed by Sangon Biotech Co., Ltd. LipoFiter 3.0 (Hanbio
Biotechnology Co., Ltd.) was used for transfection according to the
manufacturer's protocol. The BT549 cells were co-transfected with
these constructs and lysed 48 h after transfection. Luciferase
activity was measured using a luciferase detection kits (TransGen
Biotech Co., Ltd.; cat. no. FR201-01) on a SpectraMax i3x
Microplate Reader (Molecular Devices, LLC). The method of
normalization was firefly luciferase activity comparison with
Renilla luciferase activity.
Immunofluorescence
BT549 cells were seeded onto a round coverslip
(Biosharp; cat. no. BS-14-RC) at the density of 2,000 cells/well.
After 24 h, the cells were fixed with 4% paraformaldehyde for 10
min, permeabilized for with 2% Triton X-100 10 min, blocked in 10%
goat serum (Wuhan Boster Biological Technology, Ltd.; cat. no.
AR0009) for 30 min (the above steps were carried out at room
temperature) and then incubated overnight with anti-RRS1 (1:200,
Abcam; cat. no. ab188161), anti-RPL11 (1:200, Proteintech; cat. no.
16277-1-AP), anti-c-Myc (1:50, Thermo, MA5-12080), anti-NPM (1:200,
Proteintech; cat. no. 60096-1-Ig), anti-Lamin B1 (1:200,
Proteintech 12987-1-AP) and anti-Fibrillarin/U3 RNP (1:200,
ABclona; cat. no. A0850) at 4°C. Subsequently, the cells were
incubated with the appropriate fluorescent secondary antibody
(1:200, ABclona; cat. no. AS039 and AS011) for 1 h at room
temperature. Nuclei were stained using DAPI (Beijing Solarbio
Science & Technology Co., Ltd.; cat. no. C0065) for 10 min at
room temperature according to the manufacturer's protocol. The
stained cells were viewed with a laser-scanning confocal microscope
(Leica Stellaris 5; Leica Microsystems GmbH), 3 fields of view were
randomly selected, observed and images captured at ×200 and ×630
magnification.
Cellular fractionation
BT549 cells were harvested and 6×106
cells were resuspended in 500 µl hypotonic buffer. After 10
min incubation on ice, they were centrifuged at 500 × g for 10 min
at 4°C, and the supernatant containing the cytoplasmic fraction
aspirated. The precipitate was resuspended in 300 µl S1
buffer, layered on 300 µl S2 buffer and centrifuged at 4°C,
1,500 × g for 5 min. The precipitated nuclear fraction was
resuspended in 300 µl S2 buffer, sonicated for 30 sec with
60 sec intervals for 5 cycles at 4°C, 25 kHz, 60 W, layered on 300
µl S3 buffer and centrifuged at 4°C, 3,000 × g for 10 min.
The nucleoplasmic fraction in the supernatant was separated and the
precipitated nucleolar fraction was resuspended in RIPA buffer.
Protease inhibitor cocktail was added throughout.
Reagent composition: Hypotonic buffer: 10 mM HEPES
(pH 7.9), 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT S1 buffer:
0.25 M sucrose, 10 mM MgCl2 S2 buffer: 0.35 M sucrose,
0.5 mM MgCl2 S3 buffer: 0.88 M sucrose, 0.05 mM
MgCl2
Statistical analyses
The data was analyzed using the SPSS v13.0 program
(SPSS, Inc.). All data was expressed as mean ± standard deviation
(SD) and statistical significance was determined through one-way
ANOVA analysis with Bonferroni post hoc test. P<0.05 was
considered to indicate a statistically significant difference. Each
experiment was repeated three times.
Results
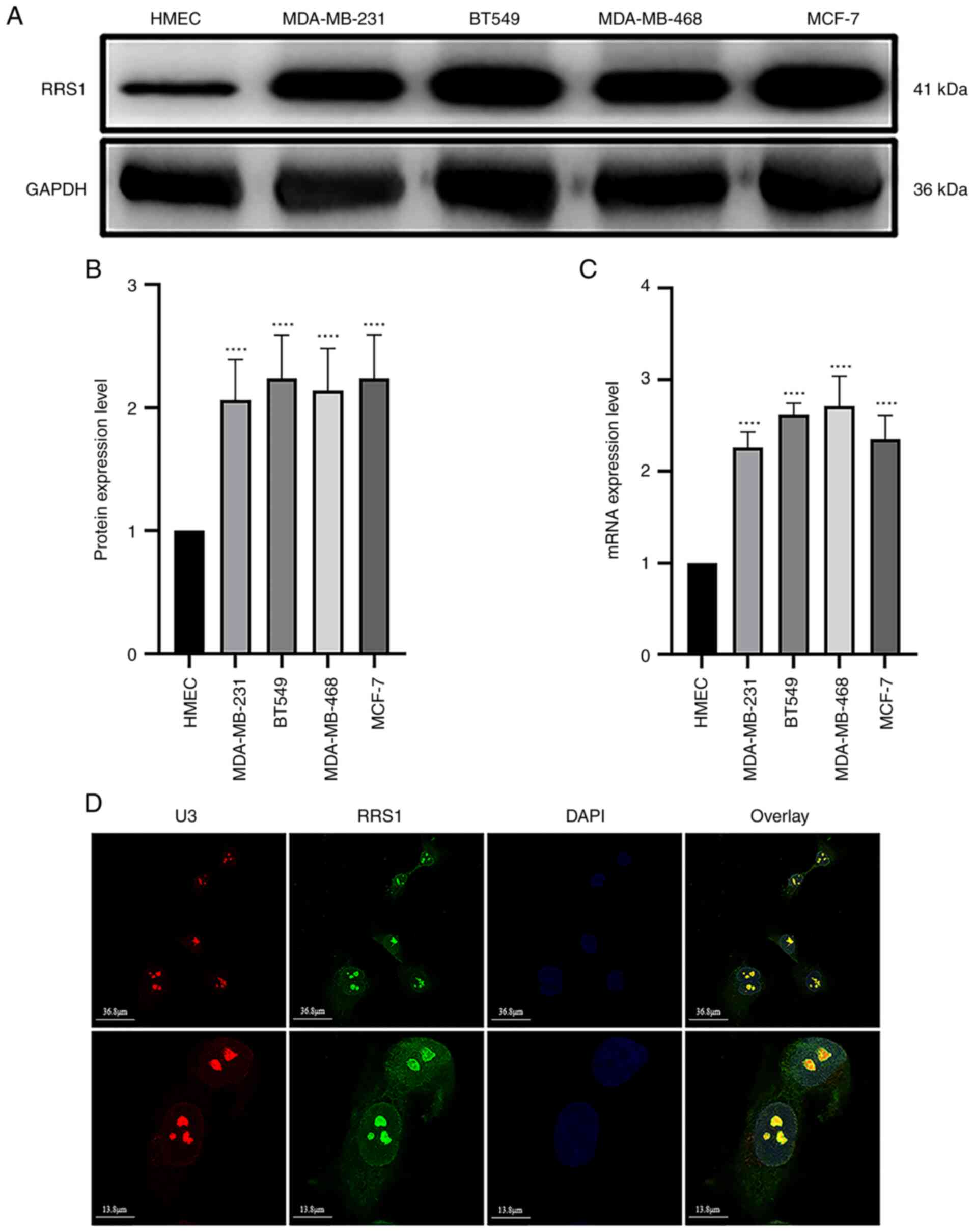
RRS1 is upregulated in breast cancer cell
lines
Tumor cells have a significantly higher rate of
ribosome biosynthesis compared to normal cells in order to sustain
rapid proliferation, which coincides with increased levels of RRS1.
Consistent with this, we found that the expression of RRS1 mRNA and
protein were significantly higher in four breast cancer cell lines
(MDA-MB-231, BT549, MDA-MB-468 and MCF-7) compared to normal
mammary epithelial cells (HMEC; Fig.
1A) and the highest levels were detected in the BT549 cells
(Fig. 1B and C). Fibrillarin/U3
RNP as a nucleolar marker can accurately locate the nucleolus,
immunofluorescence assay using in BT549 cells indicated that RRS1
was mainly located in the nucleus and nucleolus, followed by the
cytoplasm (Fig. 1D). The
sub-cellular localization of RRS1 was consistent with its role in
ribosome biosynthesis.
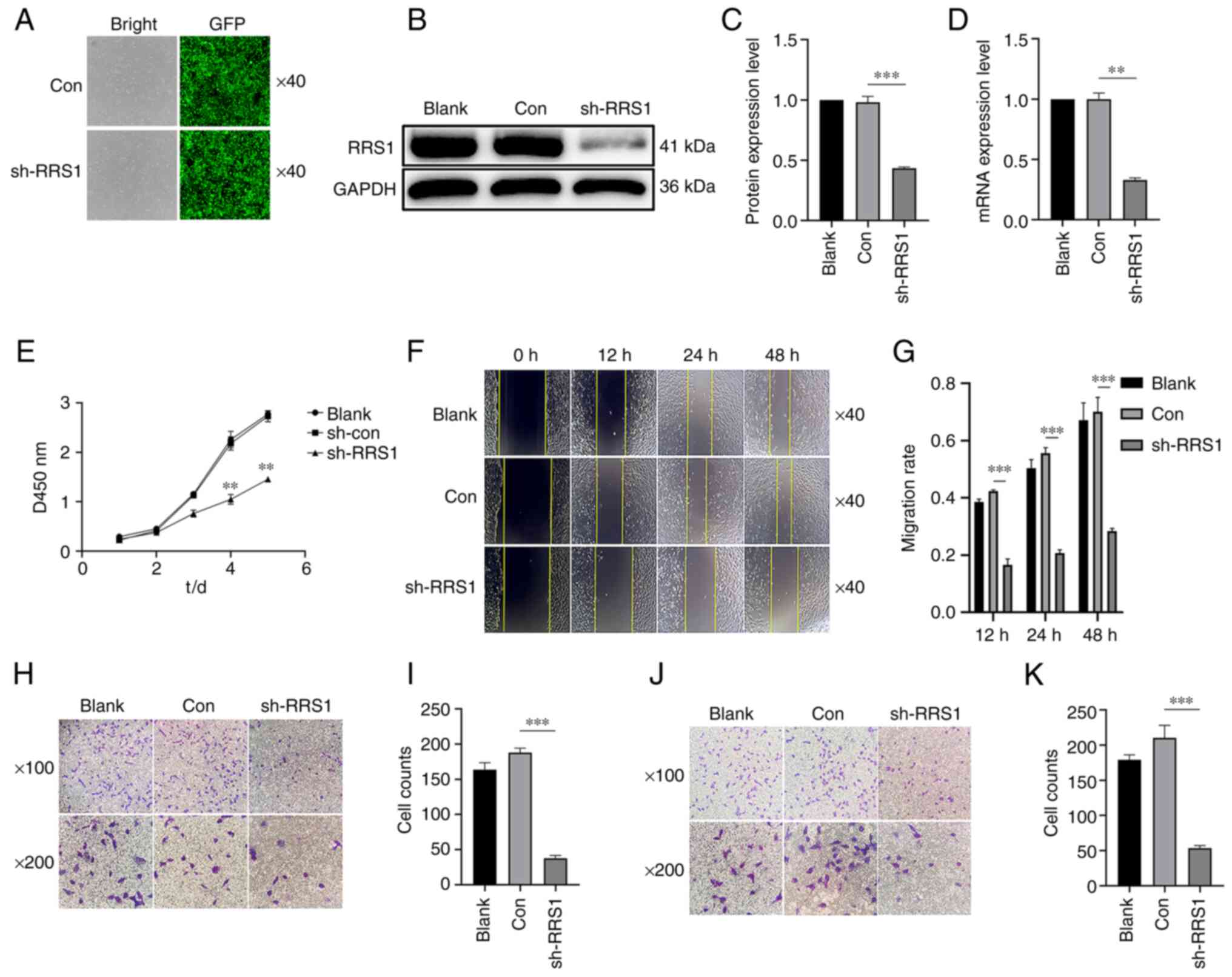
RRS1 knockdown inhibited the
proliferation, invasion and metastasis of BT549 cells
To determine the biological relevance of RRS1 in
breast cancer progression, the present study knocked down RRS1 in
BT-549 cells using an sh-RNA construct (Fig. 2A) and the knockdown efficiency was
validated by the reduced protein and mRNA levels of RRS1 (Fig. 2B-D). RRS1 knockdown significantly
decreased cell proliferation rates (Fig. 2E), as well as the migration and
invasion abilities in vitro (Fig. 2F-K). Similarly, in the
triple-negative breast cancer cell line MDA-MB-231, knocking down
RRS1 also inhibited its invasion and metastasis ability (Fig. S1). Taken together, RRS1 is a key
factor promoting the malignant phenotype of breast cancer
cells.
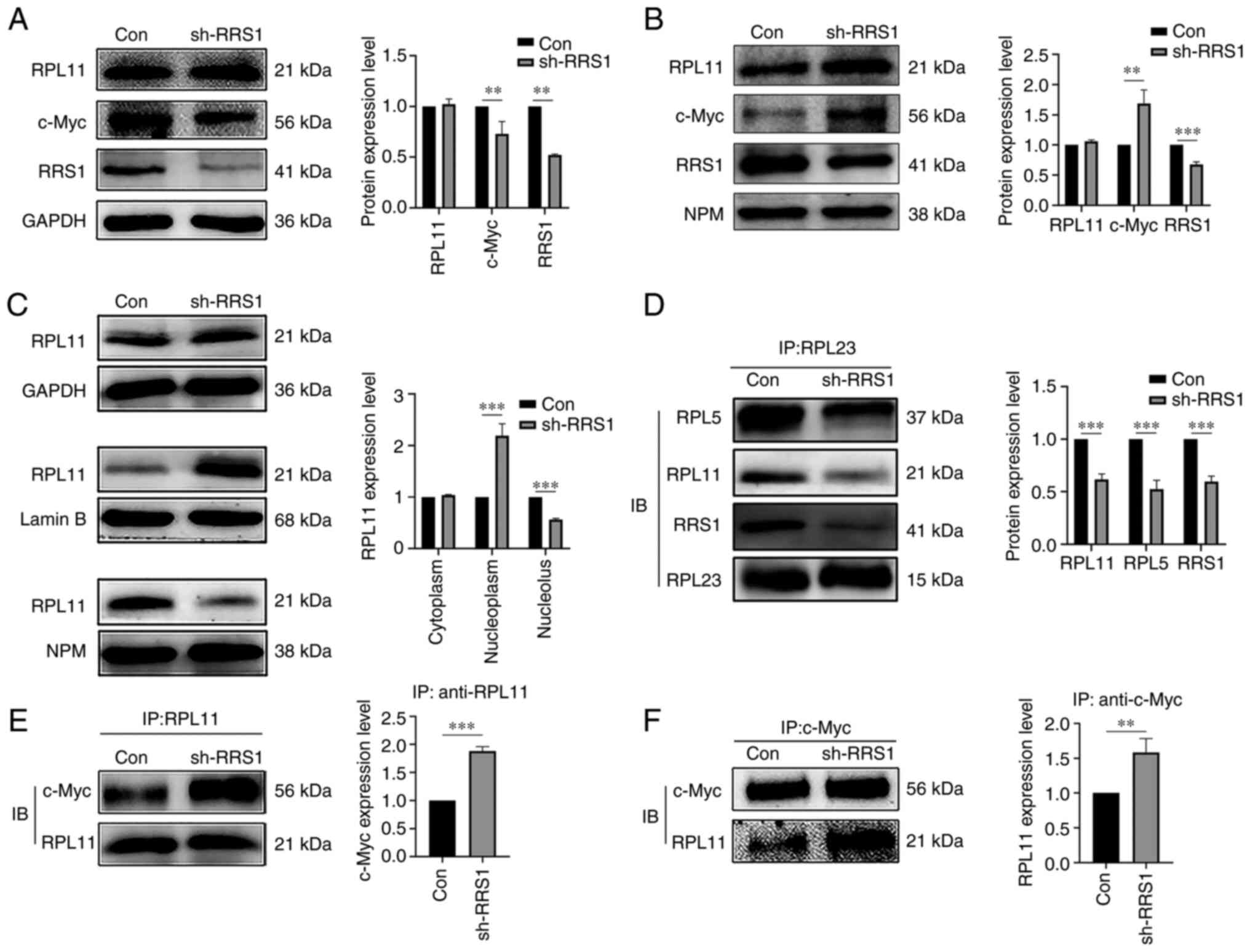
RRS1 knockdown disrupted the ribosome
assembly by altering the cellular localization of RPL11
RRS1 recruits RPL11 to the nucleolus during the
synthesis of large ribosomal subunits, which are then exported to
the cytoplasm for further maturation. Cytoplasmic and nuclear
proteins were analyzed to determine whether RRS1 affected the
cellular localization of RPL11 and it was found that knocking down
RRS1 did not alter the levels of RPL11 in the cytoplasmic or
nuclear fractions (Fig. 3A and
B). By contrast, RRS1 knockdown significantly decreased RPL11
levels in the nucleolus and increased that it in nucleoplasm
(Fig. 3C). Rea1 disengages the
Rsa4 and Rpf2/RRS1 complex from Pre-60S, which converts 5S RNP to
the correct configuration and exports it out of the nucleus.
Deletion of RRS1 can inhibit the interaction between Rea1 and Rsa4
(50). Consistent with this, the
present study found that the knockdown of RRS1 significantly
decreased the interaction of RPL23 (a marker protein of Pre-60S)
with RPL11 and RPL5 (Fig. 3D).
Taken together, RRS1 disrupted ribosomal assembly by preventing the
incorporation of RPL11 into the Pre-60S subunit, which in turn led
to its dissociation into the nucleoplasm. The dissociation of RPL11
from the ribosomal complex allows it to bind to c-Myc and inhibit
its transactivation (41). There
is also evidence that RRS1 knockout inhibits the nuclear export of
RPL11 and increases its accumulation in the nucleus (30). Consistent with these previous
observations, the present study found that RRS1 knockdown
significantly enhanced the interaction between RPL11 and c-Myc
(Fig. 3E and F). Furthermore, the
absence of RRS1 also increased the c-Myc accumulation in the
nucleus, with a concomitant decrease in its cytoplasmic levels
(Fig. 3A and B). These findings
suggested that knocking down RRS1 enhances the interaction between
RPL11 and c-Myc.
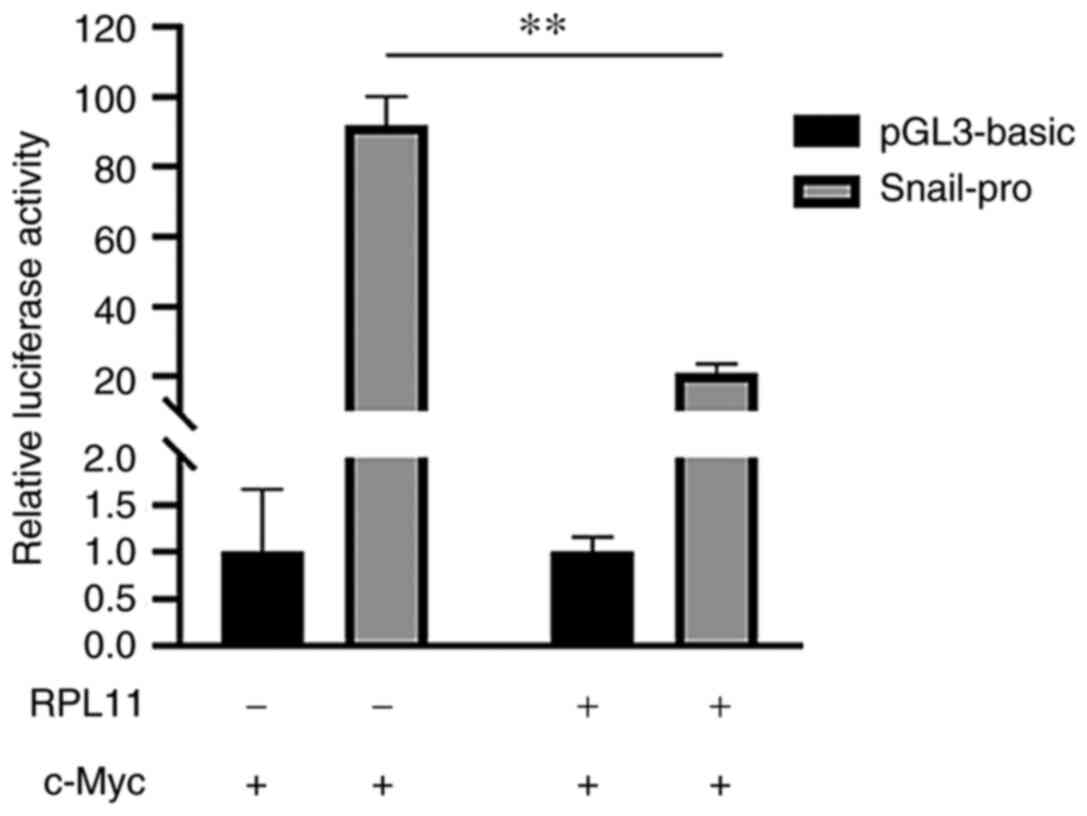
RPL11 inhibits c-Myc-driven SNAIL
transcription
A previous study showed that c-Myc could promote the
EMT, invasion and metastasis of tumor cells by activating the
mesenchymal transcription factor SNAIL (51). As RPL11 inhibits c-Myc
transactivation (38), the
present study next examined whether RPL11 also affected
c-Myc-dependent SNAIL transcription using the luciferase reporter
assay. As shown in Fig. 4, while
overexpression of c-Myc increased luciferase activity in 293T
cells, the expression of RPL11 inhibited the function of c-Myc and
reduced the increase in luciferase activity (Fig. 4). This indicated that RPL11
reduces SNAIL transcription by inhibiting c-Myc activity and may
therefore block the EMT process.
RPL11 inhibits EMT during RRS1
knockdown
EMT is a dynamic process in which serves an
important role in tumor invasion and metastasis. Consistent with
our hypothesis, RRS1 knockdown increased the expression of the
epithelial marker E-cadherin (Fig.
5) and Cytokeratin (Fig. S2)
and reduced that of the mesenchymal markers such as SNAIL,
N-cadherin and vimentin in BT549 cell (Fig. 5). Simultaneous knockdown of RRS1
and RPL11 restored the levels of the mesenchymal markers and
downregulated E-cadherin (Fig.
5). Similar results were obtained in the MDA-MB-231 cell line
(Fig. S3). Taken together, RRS1
may promotes EMT of breast cancer cells through the RPL11-c-Myc
axis, which in turn promotes tumor invasion and metastasis.
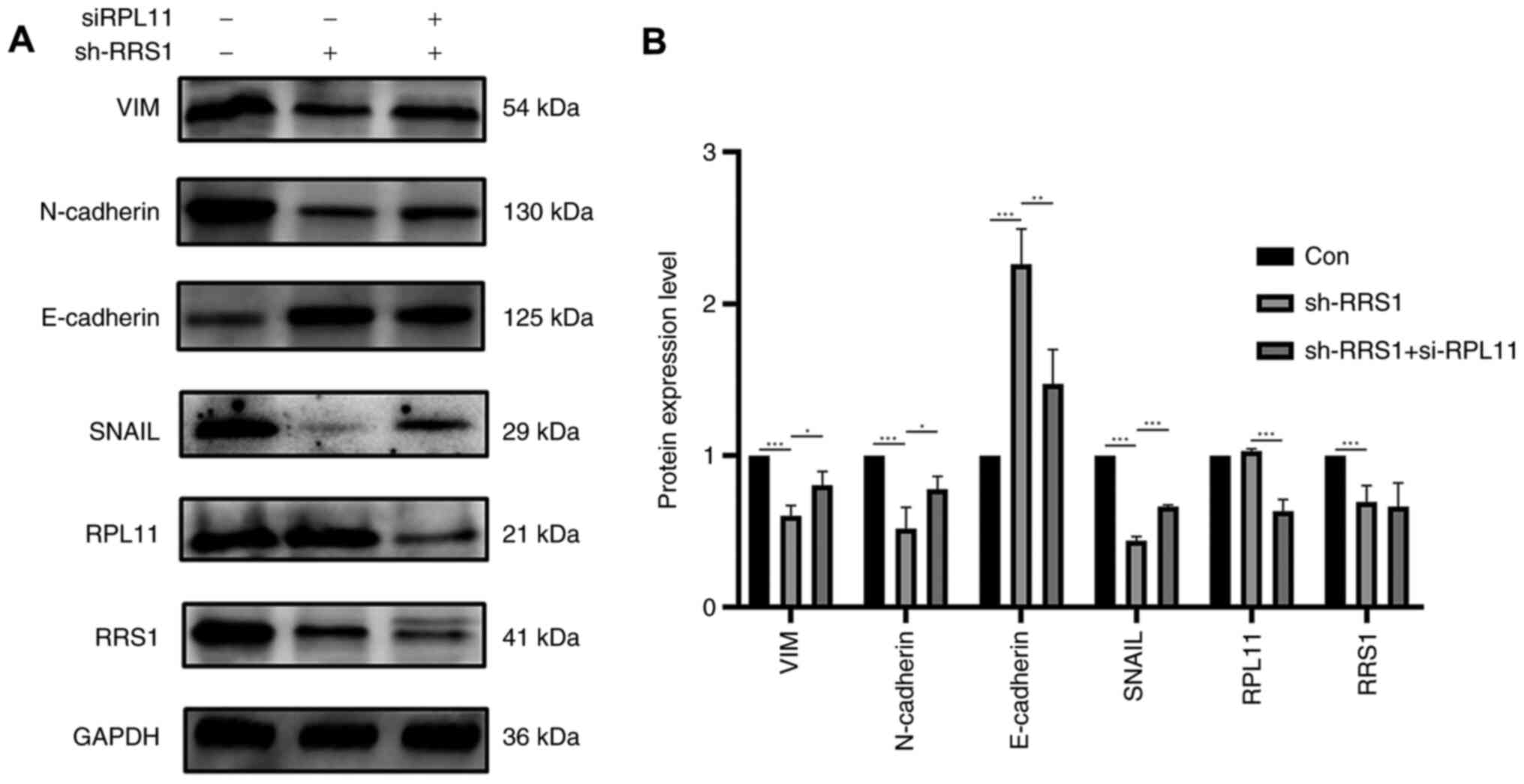 | Figure 5RPL11 reversed EMT which inhibited by
RRS1 reduction. After knockdown of RRS1, cells were lysed to
extract total protein and western blotting assays using indicated
antibodies. (A) Depletion of RRS1 inhibited SNAIL and EMT
processes. (B) RRS1 knockdown and used siRNA to interfere with
RPL11 expression, reversing the inhibition of RRS1 depletion on
SNAIL and EMT processes, *P<0.05,
**P<0.01, ***P<0.001. RPL11, ribosome
protein L11; EMT, epithelial mesenchymal transition; RRS1,
regulator of ribosome synthesis 1; VIM, vimentin; Con, control; si,
short interfering; sh, short hairpin. |
Discussion
Breast cancer has a high mortality rate due to its
metastatic nature (52). The
clinical outcomes of surgery, radiotherapy, chemotherapy, endocrine
therapy and targeted therapy are sub-optimal and the survival rate
of breast cancer patients remains poor (53). The aim of the present study was to
determine the role and underlying mechanism of RRS1 in the invasion
and metastasis of breast cancer cells.
Increased ribosome biosynthesis is a prerequisite
for sustaining the high proliferation rates of tumor cells
(54,55). RRS1 is a regulator of ribosome
biosynthesis and shows high expression levels in multiple tumors
(14,24-28,30). Likewise, the present study also
detected significantly higher levels of RRS1 in the breast cancer
cell lines compared to normal breast epithelial cells. Knocking
down RRS1 inhibited the invasion, migration and proliferation of
BT549 cells, indicating that RRS1 functions as an oncogene in
breast cancer. From a previous study, miRNA-148a inhibits
proliferation, migration and invasion by downregulating the
expression of RRS1 and promotes the apoptosis of cervical cancer
cells (23). RRS1 forms a complex
with Rpf2, which recruits 5S RNP onto the Pre-60S subunit. RPL5
also interacts with Rsa4, which integrates the 5S RNP onto Pre-60S
(56). Subsequently, Rea1, an
AAA+ (ATPases associated with various cellular activities) family
member, interacts with the ubiquitin-like (UBL) domain of Rsa4 and
releases Rsa4 in an ATP-dependent manner, which is followed by the
release of the Rpf2/RRS1 complex (50,57,58). This rotates the conformation of 5S
RNP, which fixes it on Pre-60S and is necessary for Pre-60S export.
RRS1 ensures correct binding of the Rsa4 UBL domain with Rea1
(50). In the absence of RRS1
therefore, Pre-60S maturation is blocked and it is released from
the nucleolus to the nucleoplasm and cannot be exported to the
cytoplasm (18,50). The present study found that while
RRS1 knockdown did not affect the total content of RPL11, it
shifted the localization of RPL11 from the nucleolus to the
nucleoplasm. Consistent with this, RPL23 also accumulated in the
nucleus after RRS1 knockdown. In addition, deletion of RRS1 is
known to inhibit the incorporation of RPL11 and RPL5 into Pre-60S,
thereby affecting Pre-60S assembly (18). Taken together, RRS1 depletion
prevents the incorporation of 5S RNP into Pre-60S, which hinders
the nuclear export of pre ribosomes and inhibits ribosome
biosynthesis.
C-Myc is a pro-survival protein (59), which is often overexpressed in
tumor cells (60,61). RPL11 can regulate the expression
of c-Myc at both mRNA and protein levels (38,62). Dai et al (38) found that RPL11 could bind to the
Myc box II (MB II) of c-Myc at c-Myc target gene promoter and
inhibit the histone H4 acetylation at c-Myc target nucleolin gene
promoter and recruitment of c-Myc coactivator TRRAP, finally
reducing the transcription of the c-Myc downstream target gene. In
addition, RPL11 can bind to 3′-UTR at c-Myc mRNA, recruit
microRNA-induced silencing complex (miRISC) assembled by mir-24 and
combine with Ago2 promote the interaction of miRISC and 3′-UTR,
leading to c-Myc mRNA degradation (63). RPL11 and RPL5 can also suppress
c-Myc expression in a synergistic manner (62). The present study found that RRS1
knockdown increased the interaction between RPL11 and c-Myc and
inhibited the transactivation of SNAIL by c-Myc. Similarly,
knocking down RRS1 reduced the levels of SNAIL, N-cadherin and VIM
proteins and increased that of E-cadherin. Knockdown of RPL11
reversed the inhibitory effect of RRS1 deletion. Thus, RRS1
regulated the function of c-Myc and EMT of breast cancer cells
through RPL11.
To summarize, the present study showed that RRS1 may
promote the EMT and metastasis of breast cancer cells by regulating
ribosome assembly and biosynthesis. Depletion of RRS1 prevented
nuclear export of the Pre-60s subunit and increased the
accumulation of RPL11 in the nucleoplasm. This enhanced the
interaction between RPL11 and c-Myc and decreased the transcription
of the c-Myc target SNAIL, eventually inhibiting EMT (Fig. 6). Therefore, the RPL11-c-Myc-SNAIL
axis may as a potential therapeutic target in breast cancer.
However, it remains to expound the effect of RRS1 on the
RPL11-c-Myc-SNAIL axis and its role in breast cancer.
Supplementary Data
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
LH provided the study conception and design. RW
analyzed the data, edited the figures and wrote the manuscript. CP
and JS contributed to data curation. RW, YH, QW, LD, YC, JZ, LZ, LW
performed the experiments. LH reviewed the manuscript. LH and RW
confirm the authenticity of all the raw data. All authors
contributed to the article and all authors reviewed and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 81472542) and Chinese Medicine
Projects in Shandong Province (grant no. 2021Z036).
References
|
1
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2021. CA Cancer J Clin. 71:7–33. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Roulot A, Héquet D, Guinebretière JM,
Vincent-Salomon A, Lerebours F, Dubot C and Rouzier R: Tumoral
heterogeneity of breast cancer. Ann Biol Clin (Paris). 74:653–660.
2016.
|
|
3
|
Yeo SK and Guan JL: Breast cancer:
Multiple Subtypes within a Tumor? Trends Cancer. 3:753–760. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gao JJ and Swain SM: Luminal A breast
cancer and molecular assays: A review. Oncologist. 23:556–565.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ades F, Zardavas D, Bozovic-Spasojevic I,
Pugliano L, Fumagalli D, de Azambuja E, Viale G, Sotiriou C and
Piccart M: Luminal B breast cancer: Molecular characterization,
clinical management, and future perspectives. J Clin Oncol.
32:2794–2803. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Prat A, Pineda E, Adamo B, Galván P,
Fernández A, Gaba L, Díez M, Viladot M, Arance A and Muñoz M:
Clinical implications of the intrinsic molecular subtypes of breast
cancer. Breast. 24(Suppl 2): S26–S35. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yin L, Duan JJ, Bian XW and Yu SC:
Triple-negative breast cancer molecular subtyping and treatment
progress. Breast Cancer Res. 22:612020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hao S, Ha L, Cheng G, Wan Y, Xia Y,
Sosnoski DM, Mastro AM and Zheng SY: A Spontaneous 3D
Bone-On-a-Chip for bone metastasis study of breast cancer cells.
Small. 14:e17027872018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Watase C, Shiino S, Shimoi T, Noguchi E,
Kaneda T, Yamamoto Y, Yonemori K, Takayama S and Suto A: Breast
cancer brain metastasis-overview of disease state, treatment
options and future perspectives. Cancers (Basel). 13:10782021.
View Article : Google Scholar
|
|
10
|
Maughan KL, Lutterbie MA and Ham PS:
Treatment of breast cancer. Am Fam Physician. 81:1339–1346.
2010.PubMed/NCBI
|
|
11
|
Elsayed M, Alhussini M, Basha A and Awad
AT: Analysis of loco-regional and distant recurrences in breast
cancer after conservative surgery. World J Surg Oncol. 14:1442016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Salata C, deAlmeida CE, Ferreira-Machado
SC, Barroso RC, Nogueira LP, Mantuano A, Pickler A, Mota CL and de
Andrade CBV: Preliminary pre-clinical studies on the side effects
of breast cancer treatment. Int J Radiat Biol. 97:877–887. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Taylor CW and Kirby AM: Cardiac
Side-effects from breast cancer radiotherapy. Clin Oncol (R Coll
Radiol). 27:621–629. 2015. View Article : Google Scholar
|
|
14
|
Tsuno A, Miyoshi K, Tsujii R, Miyakawa T
and Mizuta K: RRS1, a conserved essential gene, encodes a novel
regulatory protein required for ribosome biogenesis in
Saccharomyces cerevisiae. Mol Cell Biol. 20:2066–2074. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hua Y, Song J, Peng C, Wang R, Ma Z, Zhang
J, Zhang Z, Li N and Hou L: Advances in the relationship between
regulator of ribosome Synthesis 1 (RRS1) and diseases. Front Cell
Dev Biol. 9:6209252021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Granneman S and Baserga SJ: Ribosome
biogenesis: Of knobs and RNA processing. Exp Cell Res. 296:43–50.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Baßler J and Hurt E: Eukaryotic ribosome
assembly. Annu Rev Biochem. 88:281–306. 2019. View Article : Google Scholar
|
|
18
|
Zhang J, Harnpicharnchai P, Jakovljevic J,
Tang L, Guo Y, Oeffinger M, Rout MP, Hiley SL, Hughes T and
Woolford JL Jr: Assembly factors Rpf2 and Rrs1 recruit 5S rRNA and
ribosomal proteins rpL5 and rpL11 into nascent ribosomes. Genes
Dev. 21:2580–2592. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gambe AE, Matsunaga S, Takata H,
Ono-Maniwa R, Baba A, Uchiyama S and Fukui K: A nucleolar protein
RRS1 contributes to chromosome congression. FEBS Lett.
583:1951–1956. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Horigome C, Okada T, Shimazu K, Gasser SM
and Mizuta K: Ribosome biogenesis factors bind a nuclear envelope
SUN domain protein to cluster yeast telomeres. EMBO J.
30:3799–3811. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Miyoshi K, Tsujii R, Yoshida H, Maki Y,
Wada A, Matsui Y, Toh-E A and Mizuta K: Normal assembly of 60S
ribosomal subunits is required for the signaling in response to a
secretory defect in Saccharomyces cerevisiae. J Biol Chem.
277:18334–18339. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Carnemolla A, Fossale E, Agostoni E,
Michelazzi S, Calligaris R, De Maso L, Del Sal G, MacDonald ME and
Persichetti F: Rrs1 is involved in endoplasmic reticulum stress
response in Huntington disease. J Biol Chem. 284:18167–18173. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Y, Sun B, Zhao L, Liu Z, Xu Z, Tian
Y and Hao C: Up-regulation of miRNA-148a inhibits proliferation,
invasion, and migration while promoting apoptosis of cervical
cancer cells by down-regulating RRS1. Biosci Rep.
39:BSR201818152019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang J, Li Z, Zuo C, Xie Q, Li H, Jia J,
Zhen Z, Qi R, Li Z, Liu D and Sun B: Knockdown of RRS1 by
lentiviral-mediated RNAi promotes apoptosis and suppresses
proliferation of human hepatocellular carcinoma cells. Oncol Rep.
38:2166–2172. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hua YN, Song JL, Ma ZL, Wu L, Zhang Z,
Zhang L, Li N, Cong SB and Hou L: Effect of RRS1 gene knockdown on
BT549 cell line proliferation and apoptosis in breast cancer.
Neoplasma. 66:28–32. 2019. View Article : Google Scholar
|
|
26
|
Song J, Ma Z, Hua Y, Xu J, Li N, Ju C and
Hou L: Functional role of RRS1 in breast cancer cell proliferation.
J Cell Mol Med. 22:6304–6313. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen F, Jin Y, Feng L, Zhang J, Tai J, Shi
J, Yu Y, Lu J, Wang S, Li X, et al: RRS1 gene expression involved
in the progression of papillary thyroid carcinoma. Cancer Cell Int.
18:202018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
XL Wu, Yang ZW, He L, Dong PD, Hou MX,
Meng XK, Zhao HP, Wang ZY, Wang F, Baoluri, et al: RRS1 silencing
suppresses colorectal cancer cell proliferation and tumorigenesis
by inhibiting G2/M progression and angiogenesis. Oncotarget.
8:82968–82980. 2017. View Article : Google Scholar
|
|
29
|
Ma Y, Yan F, Wei W, Deng J, Li L, Liu L
and Sun J: MicroRNA-598 inhibits the growth and maintenance of
gastric cancer stem-like cells by down-regulating RRS1. Cell Cycle.
18:2757–2769. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cao P, Yang A, Li P, Xia X, Han Y, Zhou G,
Wang R, Yang F, Li Y, Zhang Y, et al: Genomic gain of RRS1 promotes
hepatocellular carcinoma through reducing the RPL11-MDM2-p53
signaling. Sci Adv. 7:eabf43042021. View Article : Google Scholar :
|
|
31
|
Calviño FR, Kharde S, Ori A, Hendricks A,
Wild K, Kressler D, Bange G, Hurt E, Beck M and Sinning I:
Symportin 1 chaperones 5S RNP assembly during ribosome biogenesis
by occupying an essential rRNA-binding site. Nat Commun.
6:65102015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang Y, Wolf GW, Bhat K, Jin A, Allio T,
Burkhart WA and Xiong Y: Ribosomal protein L11 negatively regulates
oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress
checkpoint pathway. Mol Cell Biol. 23:8902–8912. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Haupt Y, Maya R, Kazaz A and Oren M: Mdm2
promotes the rapid degradation of p53. Nature. 387:296–299. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Momand J, Zambetti GP, Olson DC, George D
and Levine AJ: The mdm-2 oncogene product forms a complex with the
p53 protein and inhibits p53-mediated transactivation. Cell.
69:1237–1245. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bursać S, Brdovčak MC, Pfannkuchen M,
Orsolić I, Golomb L, Zhu Y, Katz C, Daftuar L, Grabušić K, Vukelić
I, et al: Mutual protection of ribosomal proteins L5 and L11 from
degradation is essential for p53 activation upon ribosomal
biogenesis stress. Proc Natl Acad Sci USA. 109:20467–20472. 2012.
View Article : Google Scholar
|
|
36
|
Horn HF and Vousden KH: Cooperation
between the ribosomal proteins L5 and L11 in the p53 pathway.
Oncogene. 27:5774–5784. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang HT, Chen TY, Weng CW, Yang CH and
Tang MS: Acrolein preferentially damages nucleolus eliciting
ribosomal stress and apoptosis in human cancer cells. Oncotarget.
7:80450–80464. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dai MS, Arnold H, Sun XX, Sears R and Lu
H: Inhibition of c-Myc activity by ribosomal protein L11. EMBO J.
26:3332–3345. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
van Riggelen J, Yetil A and Felsher DW:
MYC as a regulator of ribosome biogenesis and protein synthesis.
Nat Rev Cancer. 10:301–309. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dai MS, Sears R and Lu H: Feedback
regulation of c-Myc by ribosomal protein L11. Cell Cycle.
6:2735–2741. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang X, Ren Y, Wang Z, Xiong X, Han S, Pan
W, Chen H, Zhou L, Zhou C, Yuan Q and Yang M: Down-regulation of 5S
rRNA by miR-150 and miR-383 enhances c-Myc-rpL11 interaction and
inhibits proliferation of esophageal squamous carcinoma cells. FEBS
Lett. 589:3989–3997. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gao X, Liu X, Lu Y, Wang Y, Cao W, Liu X,
Hu H and Wang H: PIM1 is responsible for IL-6-induced breast cancer
cell EMT and stemness via c-myc activation. Breast Cancer.
26:663–671. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yang J, Wu SP, Wang WJ, Jin ZR, Miao XB,
Wu Y, Gou DM, Liu QZ and Yao KT: A novel miR-200c/c-myc negative
regulatory feedback loop is essential to the EMT process, CSC
biology and drug sensitivity in nasopharyngeal cancer. Exp Cell
Res. 391:1118172020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tao L, Shu-Ling W, Jing-Bo H, Ying Z, Rong
H, Xiang-Qun L, Wen-Jie C and Lin-Fu Z: MiR-451a attenuates
doxorubicin resistance in lung cancer via suppressing
epithelialmesenchymal transition (EMT) through targeting c-Myc.
Biomed Pharmacother. 125:1099622020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu N, Wang Z, Liu D and Xie P:
HOXC13-AS-miR-122-5p-S ATB1-C-Myc feedback loop promotes migration,
invasion and EMT process in glioma. Onco Targets Ther.
12:7165–7173. 2019. View Article : Google Scholar :
|
|
46
|
Lin X, Sun R, Zhao X, Zhu D, Zhao X, Gu Q,
Dong X, Zhang D, Zhang Y, Li Y and Sun B: C-myc overexpression
drives melanoma metastasis by promoting vasculogenic mimicry via
c-myc/snail/Bax signaling. J Mol Med (Berl). 95:53–67. 2017.
View Article : Google Scholar
|
|
47
|
Wang K, Zheng J, Yu J, Wu Y, Guo J, Xu Z
and Sun X: Knockdown of MMP-1 inhibits the progression of
colorectal cancer by suppressing the PI3K/Akt/c-myc signaling
pathway and EMT. Oncol Rep. 43:1103–1112. 2020.PubMed/NCBI
|
|
48
|
Nieto MA, Huang RY, Jackson RA and Thiery
JP: EMT: 2016. Cell. 166:21–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
50
|
Madru C, Lebaron S, Blaud M, Delbos L,
Pipoli J, Pasmant E, Réty S and Leulliot N: Chaperoning 5S RNA
assembly. Genes Dev. 29:1432–1446. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Smith AP, Verrecchia A, Fagà G, Doni M,
Perna D, Martinato F, Guccione E and Amati B: A positive role for
Myc in TGFbeta-induced Snail transcription and
epithelial-to-mesenchymal transition. Oncogene. 28:422–430. 2009.
View Article : Google Scholar
|
|
52
|
Azamjah N, Soltan-Zadeh Y and Zayeri F:
Global trend of breast cancer mortality rate: A 25-year study.
Asian Pac J Cancer Prev. 20:2015–2020. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Henson KE, McGale P, Darby SC, Parkin M,
Wang Y and Taylor CW: Cardiac mortality after radiotherapy,
chemotherapy and endocrine therapy for breast cancer: Cohort study
of 2 million women from 57 cancer registries in 22 countries. Int J
Cancer. 147:1437–1449. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Penzo M, Montanaro L, Treré D and
Derenzini M: The ribosome biogenesis-cancer connection. Cells.
8:552019. View Article : Google Scholar
|
|
55
|
Nait Slimane S, Marcel V, Fenouil T, Catez
F, Saurin JC, Bouvet P, Diaz JJ and Mertani HC: Ribosome biogenesis
alterations in colorectal cancer. Cells. 9:23612020. View Article : Google Scholar :
|
|
56
|
Baßler J, Paternoga H, Holdermann I, Thoms
M, Granneman S, Barrio-Garcia C, Nyarko A, Lee W, Stier G, Clark
SA, et al: A network of assembly factors is involved in remodeling
rRNA elements during preribosome maturation. J Cell Biol.
207:481–498. 2014. View Article : Google Scholar
|
|
57
|
Ulbrich C, Diepholz M, Bassler J, Kressler
D, Pertschy B, Galani K, Böttcher B and Hurt E: Mechanochemical
removal of ribosome biogenesis factors from nascent 60S ribosomal
subunits. Cell. 138:911–922. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Micic J, Li Y, Wu S, Wilson D, Tutuncuoglu
B, Gao N and Woolford JL Jr: Coupling of 5S RNP rotation with
maturation of functional centers during large ribosomal subunit
assembly. Nat Commun. 11:37512020. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Davis AC, Wims M, Spotts GD, Hann SR and
Bradley A: A null c-myc mutation causes lethality before 10.5 days
of gestation in homozygotes and reduced fertility in heterozygous
female mice. Genes Dev. 7:671–682. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Destefanis F, Manara V and Bellosta P: Myc
as a regulator of ribosome biogenesis and cell competition: A link
to cancer. Int J Mol Sci. 21:40372020. View Article : Google Scholar :
|
|
61
|
Wu H, Yang TY, Li Y, Ye WL, Liu F, He XS,
Wang JR, Gan WJ, Li XM, Zhang S, et al: Tumor necrosis factor
receptor-associated Factor 6 promotes hepatocarcinogenesis by
interacting with histone deacetylase 3 to enhance c-Myc Gene
expression and protein stability. Hepatology. 71:148–163. 2020.
View Article : Google Scholar
|
|
62
|
Liao JM, Zhou X, Gatignol A and Lu H:
Ribosomal proteins L5 and L11 co-operatively inactivate c-Myc via
RNA-induced silencing complex. Oncogene. 33:4916–4923. 2014.
View Article : Google Scholar
|
|
63
|
Challagundla KB, Sun XX, Zhang X, DeVine
T, Zhang Q, Sears RC and Dai MS: Ribosomal protein L11 recruits
miR-24/miRISC to repress c-Myc expression in response to ribosomal
stress. Mol Cell Biol. 31:4007–4021. 2011. View Article : Google Scholar : PubMed/NCBI
|