Pancreatic cancer (PC) is currently the seventh
leading cause of cancer-related mortality, with almost as many
deaths as cases; the highest incidence rates are observed in Europe
and Northern America (1). With the
lowest 5-year survival rate of any cancer type (10%), PC is also
projected to become the second leading cause of cancer-related
mortality in the United States by the year 2030 (2,3). As
pancreatic ductal adenocarcinoma (PDAC) accounts for ~90% of all PC
cases (4), the present review
focuses primarily on this type of PC.
Alternative strategies to combat this lethal disease
are therefore sought after and the targeting of the deregulated
metabolic physiology, which is additionally connected to radio- and
chemo-resistance in PDAC, is one of the promising options (16,17).
The common genomic alterations, such as the activation of the
KRAS oncogene, the deletion of the tumor suppressor gene,
SMAD4, or the transformation of the tumor suppressor, p53,
into a prooncogenic protein significantly stimulate nutrient
acquisition and a variety of metabolic pathways (18). The ability to stratify PDAC tumors
into subgroups with distinct metabolic requirements and
vulnerabilities could offer valuable prognostic information and may
aid to in the selection of specific treatments. For example, some
studies have identified glycogenic and lipogenic subtypes
associated with different survival outcomes and distinct
sensitivity to metabolic inhibitors (19,20).
The ability of PC cells to obtain nutrients from
their surroundings and through autophagic pathways, unique
physiological characteristics of the tumor microenvironment, as
well as the interactions of PC cells with non-cancer cells are also
among the factors that markedly contribute to the extensively
reprogrammed metabolism in PDAC (21). Populations of highly clonogenic,
resistant and metastatic PC stem cells (PCSCs) appear to have
distinct therapeutic vulnerabilities related to specific metabolic
features that could be exploited. Importantly, metabolic plasticity
that allows PCSCs to adapt to environmental changes appears to be
connected to distinct patterns of DNA methylation and so a
disruption of this metabolic-epigenetic crosstalk present within
PDA could represent another therapeutic target (22).
The present review discusses a range of metabolic
and physiological aspects of PC cells, which play a role in the
development and progression of this difficult-to-treat and
aggressive malignancy. The present review also focuses on the
possibility of targeting PDAC-specific metabolic pathways in order
to aid in the development of more effective and personalized
therapies. The metabolic pathways discussed herein as taking place
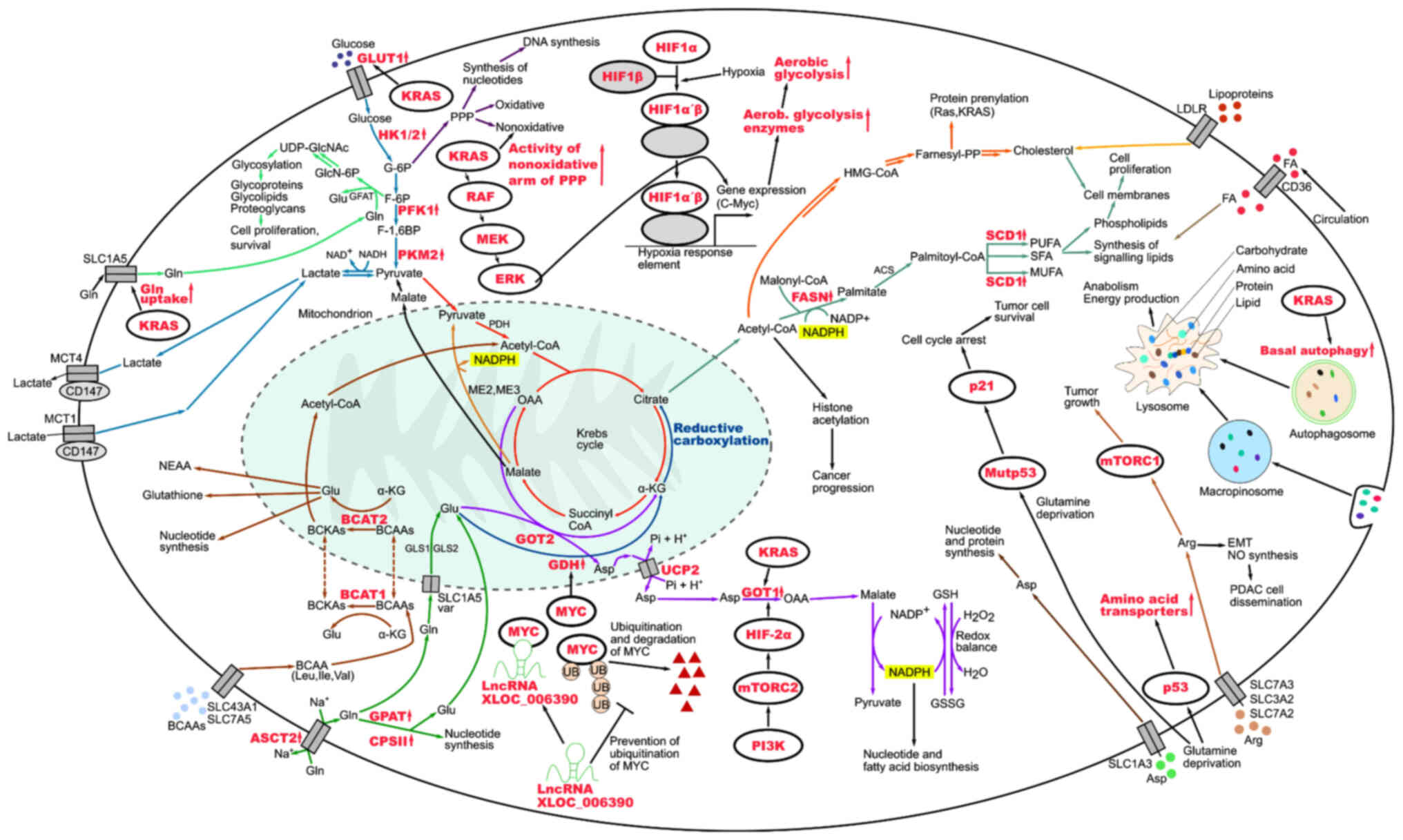
in PC cells are summarized in Fig.
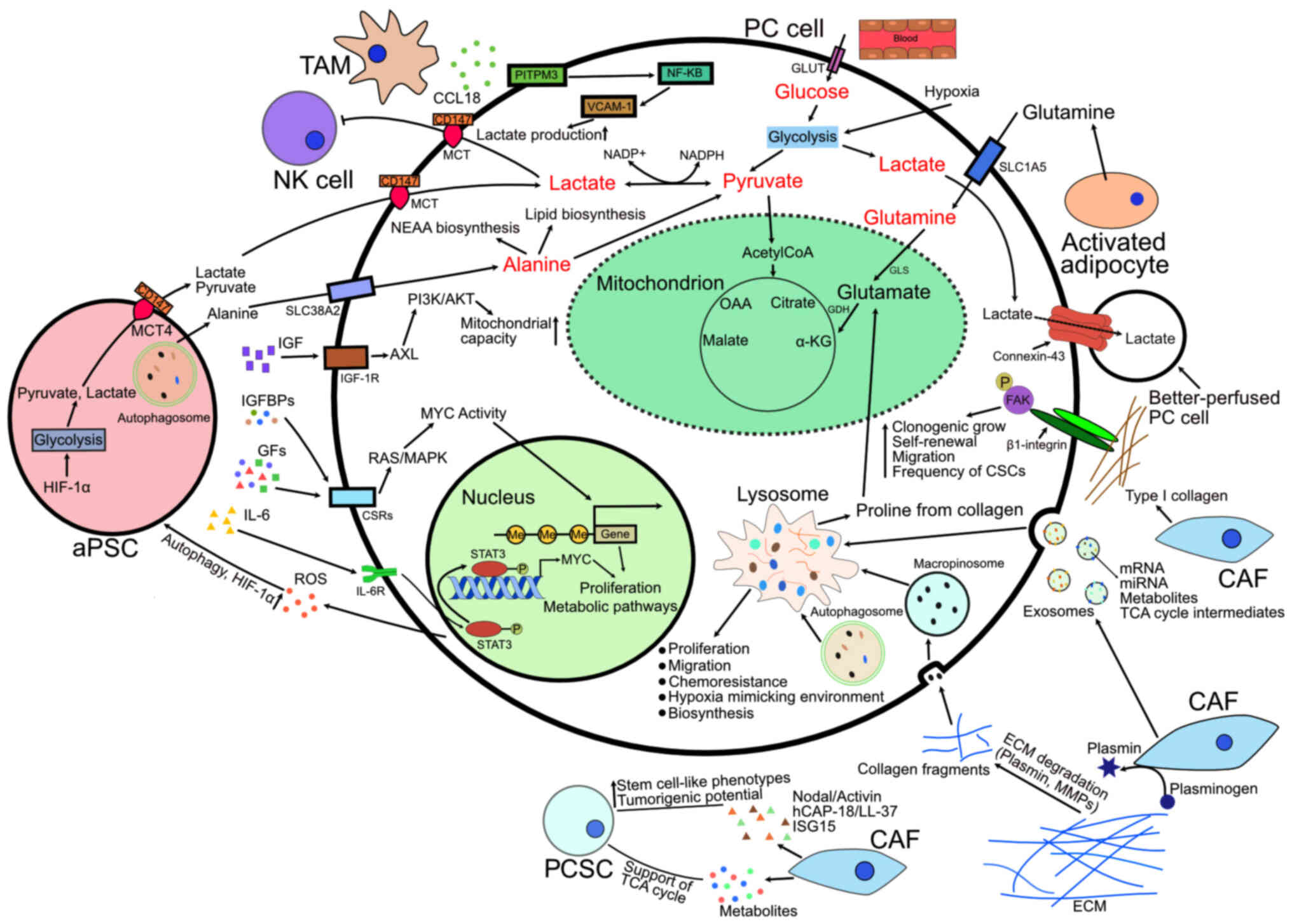
1 and the metabolic crosstalk within the tumor microenvironment
in PC is illustrated in Fig.
2.
The PubMed/MEDLINE, Scopus and Web of Science
databases were searched with search terms including 'cell
metabolism', 'metabolic phenotypes', 'metabolic profiles',
'pancreatic cancer', 'tumor microenvironment', 'stem cells',
'therapeutic/treatment resistance' and 'metabolomics' in various
Boolean combinations as search strings. In order to cover the
selected topic comprehensively, articles were searched without a
limit on their date of publication. Only published, peer-reviewed
articles in the English language were included in the search and
literature analysis. The articles were then manually sorted and
critically evaluated according to relevance. Based on the database
searches, electronic and manual cross-referencing was used to
identify additional relevant sources.
Pancreatic tumors are known to be heterogenic at the
cellular level, with distinct metabolic traits and methods of
nutrient acquisition, which can lead to issues with effective
treatment (23). There is
therefore a need for a better understanding of the biological
characteristics of PC together with an assessment of metabolic
phenotypes of individual tumors in order to lead to the development
of more targeted therapeutic strategies. Patient stratification
strategies are becoming increasingly useful in cancer diagnosis,
prognosis assignment and in creating appropriate treatment
strategies. The comparison of mutational profiles has proven
problematic with questionable associations between mutational
status and disease behavior (24).
Genomic and transcriptomic studies based on
molecular profiling have identified subgroups of PDAC often
overlapping to varying degrees (25-29).
Moffitt et al (26) used
PDAC gene expression microarray data to identify and validate two
tumor subtypes, 'basal-like' and 'classical'. On a molecular level,
the basal-like subtype was similar to basal tumors of the urinary
bladder and breast cancer and had a worse outcome. Notably, using
the algorithmic separation of tumor, stromal and normal gene
expression, they also identified distinct stromal subtypes, which
were defined as 'normal' and 'activated' and were independently
prognostic. These findings demonstrate a particular significance of
the stromal compartment, which has been shown to play crucial roles
in PDAC biology (30-35). It is also becoming clear that
further heterogeneity in expression profiles exists within cells of
a single tumor (36).
PC cells need to be able to proliferate rapidly,
which is complicated by the dense stroma environment, which lacks
adequate vasculature and therefore presents a nutrient- and
oxygen-poor environment. Cells in such conditions undergo extensive
oncogene-directed metabolic reprogramming, which includes a higher
nutrient acquisition, increased glucose utilization through aerobic
glycolysis, an upregulated biosynthesis of lipids and amino acids,
alterations in the redox balance, as well as an activation of
recycling and scavenging pathways (37-41).
In addition to the molecular signatures of PDAC
afore-mentioned, metabolic subtypes with specific metabolic
requirements have also been identified that are of prognostic value
and could aid in the selection of treatments and may thus lead to
more favorable therapeutic outcomes. Based on broad metabolic
profiling, Daemen et al (19) described 'slow proliferating',
'glycolytic' and 'lipogenic' PDAC subtypes, which exhibited
different metabolite levels associated with glycolysis, lipogenesis
and redox pathways. Studying the differences between glycolytic and
lipogenic lines, theys found particularly significant high levels
of phosphoenolpyruvate (PEP) and of the ENO2 mRNA coding for
the glycolytic enzyme neuron-specific enolase (ENO2), which
converts 2-phosphoglycerate to PEP. The two metabolic subtypes also
responded differently to inhibitors of glycolysis and lipid
metabolism. Based on these findings, those authors suggested ENO2
inhibitors as a possible therapeutic option for aggressive, fast
growing glycolytic tumors where ENO2 was found to be
strongly expressed. They also observed a correlation between these
subtypes and those identified by Collisson et al (25), indicating that links indeed exist
between molecular and metabolic subtypes. They used their findings
to create a model, in which epithelial tumors use glucose for the
tricarboxylic acid (TCA) cycle and de novo lipogenesis,
whereas mesenchymal tumors preferentially fuel the TCA cycle by
glutamine with a potentially enhanced vulnerability to ROS-inducing
agents (19).
It should be kept in mind that interactions
involving distinct cell populations within the tumor
microenvironment may also contribute to the complex nature of PDAC
metabolic phenotypes. Single-cell metabolomics can be used to
distinguish different cell types in a heterogeneous cell mixture
and to provide information about metabolic specificities of tumor
cells from clinical cancer tissues (45). Specifically, a high-throughput,
label-free and sensitive dielectric barrier discharge
ionization-mass spectrometry (DBDI-MS) platform has been developed
for the analysis of single-cell metabolites, which identified
deregulated lipid metabolism in PDAC cells and detected a reduction
in lipid content after the inhibition of ATP citrate lyase, a
rate-limiting enzyme for lipid synthesis, which has high mRNA
levels in both patients with PDAC from The Cancer Genome Atlas
dataset, as well as PC cell lines (45). Distinguishing metabolic variations
among the subsets of cell populations on a single-cell level is
also possible via a complex metabolic profiling method single cell
energetic metabolism by profiling translation inhibition (SCENITH)
or by measuring multiple enzymatic activities reflecting different
metabolic pathways at saturating substrate conditions at single
cell resolution (46,47). SCENITH is based around puromycin
labeling and flow cytometry measurements of protein synthesis,
reflective of global cellular metabolic activity. It can be used
directly in heterogeneous human tumor samples to study metabolic
profiles of multiple cell types. The analysis of single cell
enzymatic activities within a native tissue microenvironment relies
on enzyme histochemistry and automated histocytometry to identify
particular cell types and simultaneously characterize intra- and
intercellular metabolic configurations (46,47).
Metabolic phenotypes of cancer cells can be
transient. For example, progressively de-differentiated PC cells
have been shown to undergo a shift from glycolytic to oxidative
metabolism, resulting in a quiescent state. Following the
re-differentiation, these cells regained their proliferative
capacity and glycolytic metabolism, commonly associated with a
greater aggressiveness (48).
Metabolic switches in PDAC cells can occur also as a response to
disruptions caused by treatments (39,49).
It is thus of utmost significance to understand the underlying
mechanisms, as it could help either to prevent, overcome or even
exploit such metabolic plasticity.
The enhanced utilization of glucose by tumor cells
even under normal oxygen levels was observed by Otto Warburg as
early as the 1920s and was later defined as the Warburg effect
(50). Since then, the importance
of the Warburg phenotype in cancer cells has been well-documented.
Its advantages lie in the quick supply of ATP, the support of
increased biosynthetic needs, the modification of chromatin
structure and redox regulation (51,52).
Moreover, the produced and exported lactate contributes to
immunosuppression within the tumor microenvironment (53). To compensate for the relatively low
ATP output compared to mitochondrial oxidative phosphorylation
(OXPHOS), the uptake of glucose can be increased by an
overexpression of the glucose transporter 1 (GLUT1) (54). Additionally, phosphorylation on
Ser226 in GLUT1 was identified as a key event for the enhanced cell
surface localization of GLUT1 and for the regulation of glucose
transport (55). A sustained
elevated glycolytic flux in PDAC is markedly connected with an
increased expression of rate-limiting glycolytic enzymes like
phosphofructokinase 1 and lactate dehydrogenase A (LDHA) (56-58),
and there is also evidence of glycolytic enzymes regulating the
epithelial-mesenchymal transition (EMT) in PC cells, inducing PC
cell invasion and metastasis as well as promoting tumor
angiogenesis (59-63). The Raf/MEK/extracellular
signal-regulated kinase (ERK) signaling pathway activated by
oncogenic KRAS together with hypoxic conditions known to be present
in PDAC increase glucose uptake and the expression of glycolytic
enzymes, which then leads to the establishment of an acidic
microenvironment and the promotion of tumorigenesis (57,64).
The transport of lactate in and out of cells is a
key factor in metabolic adaptations to deal with the unequal access
to vasculature and differing oxygen levels within a tumor. Out of
the group of monocarboxylate transporters (MCTs) belonging to the
SLC16A gene family, which are crucial for controlling
acidity levels within the tumor and the microenvironment, MCT-1 and
MCT-4 were previously shown to be upregulated in PC (65). To correctly function, MCT1 and MCT4
need to be properly inserted into the plasma membrane, which is
facilitated by CD147 glycoprotein (66). MCT-4 is upregulated by hypoxia,
promotes glucose uptake and the production of lactate via
glycolysis. MCT-1 expression on the other hand is dependent on
oxygen and is repressed by hypoxia (67). It facilitates lactate uptake by
cells, which is then used as a substrate in the TCA cycle for ATP
production in OXPHOS (68). Cells
expressing MCT-1 have a well-developed mitochondrial network and
produce large amounts of TCA intermediates and ATP (41). This led to the development of a
model of PDAC where aerobic cells in normoxic regions utilize
OXPHOS fed by lactate produced by hypoxic cells and imported
through MCT-1, leaving glucose to be preferentially used in aerobic
glycolysis by cells growing at low oxygen pressure (65).
A similar collaboration appears to exist between
cancer cells and stromal resident cells. Cancer cells induce
oxidative stress and aerobic glycolysis in cancer-associated
fibroblasts (CAFs) and subsequently use energy-rich metabolites
lactate and pyruvate secreted by CAFs to sustain their own
increased proliferation (69,70).
The Gi-coupled receptor GPR81 was confirmed to be
expressed in several cancer cell lines, including those of PDAC and
to play a critical role in sensing extracellular lactate (71). Its activation leads to an increased
expression of MCTs, CD147 and peroxisome proliferator-activated
receptor-γ co-activator PGC-1α which, apart from its ability to
increase MCT1 expression, engages in a spectrum of biological
processes (72). PGC-1α is
strongly linked to the regulation of glucose and fatty acid
metabolism, fiber type switching in skeletal muscle, adaptive
thermogenesis and heart development as well as the stimulation of
mitochondrial biogenesis (73).
The silencing of GPR81 has been found to negatively affect the
mitochondrial activity of cancer cells grown in cell culture
conditions, with only lactate as an available energy source. Roland
et al (71) also observed
that the loss of GPR81 was associated with a reduced PC tumor
growth and metastasis in vivo. Following these findings
regarding the key role of lactate metabolism in cancer progression,
researchers have begun to explore potential benefits of the
inhibition of MCTs and CD147 in PDAC and beyond (74-78).
Lipids support cancer progression via multiple
mechanisms, including the formation of biomembranes, participation
in cellular signaling and as an energy source (80). Depending on the microenvironmental
conditions, as well as currently active metabolic pathways, PC
cells can direct glucose or glutamine-derived carbons to increase
the synthesis of fatty acids (19). Notably, increased levels of fatty
acid synthase (FASN) involved in de novo lipid synthesis
have been shown to be associated with a worse prognosis of patients
with PC (81). As for the possible
underlying reasons behind this association, FASN was previously
associated with the HER2-PI3K/AKT signaling axis and shown to exert
a prominent effect on the proliferation and migration of cancer
cells (82). Additionally, acidic
extracellular conditions could lie behind the transcriptional
upregulation of FASN by means of epigenetic modifications (83). It is not unreasonable to suggest
that similar processes could be taking place in PC, where the
increased expression of other lipogenic genes has been found
(19). Furthermore, FASN knockdown
has been found to lead to an increased responsiveness of PC cells
to gemcitabine, as well as radiation treatments, thus creating a
link between the overexpression of FASN and treatment-resistant
phenotypes (16). In a more recent
study, a paclitaxel-poly(lactic-co-glycolic acid) nanoparticle
(PPNPs) formulation was able to inhibit de novo lipid
synthesis, alter membrane stability and improve anticancer efficacy
of gemcitabine in PC cells, further emphasizing the importance of
lipid metabolic signaling in PC chemoresistance (84). Aside from FASN, stearoyl-CoA
desaturase1 (SCD1) is another central lipogenic enzyme that
contributes to the progression of cancer. SCD1 is important for
keeping the correct composition of cancer cell membranes, where
monounsaturated phospholipids provide protection from oxidative
stress (85,86). Taken together with the fact that
the inhibition of SCD1 appears to cause obstructions in aberrant
RAS and AKT signaling often involved in the development and
progression of PDAC, it could represent a potential therapeutic
target (87,88).
An increased amount of free, newly synthesized fatty
acids, such as palmitate could be toxic for cancer cells with
apoptosis as the likely outcome. Therefore, a controlled release of
fatty acids from intracellular lipid stores, where they are stored
in the form of triglycerides, by intracellular lipolysis is crucial
for preventing that scenario (89). Apart from de novo synthesis,
PC cells can obtain fatty acids also from the circulation from food
digestion or from fatty acid release from the adipose tissue. This
may present an obstacle, as it could render the aforementioned
inhibition of the endogenous fatty acid synthesis in PDAC
insufficient with regard to antitumor effects, making it hard to
justify as an isolated therapeutic approach. The ability of cancer
cells to use fatty acid uptake in addition to de novo fatty
acid synthesis could help to explain why obesity with
characteristically elevated fatty acid plasma levels and a high-fat
diet are among the potential risk factors for cancer development
and worse clinical outcomes of malignancies (38,90-94).
Cholesterol and its synthetic pathway have numerous
functions in cancer cells, supporting their growth, proliferation
and migration (95). Both the
targeting of exogenous cholesterol uptake in PC cells through the
shRNA silencing of low-density lipoprotein (LDL) receptor (96) and the inhibition of
3-hydroxy-methylglutaryl-CoA reductase (HMG-CoA reductase), a key
enzyme in cholesterol synthesis, by statins has exhibited promising
anticancer effects (97). An
inhibition of HMG-CoA reductase can also disrupt the process of
protein prenylation, an essential process for the activation of
signaling proteins including KRAS, which could be exploited to
further target cancer cells (98).
It is, however, uncertain whether statins may be used as a
monotherapy for cancer, as their therapeutic efficacy in clinical
trials is controversial (99). For
example, pravastatin, a potent HMG-CoA reductase inhibitor, has
been found to significantly prolong the survival of patients with
advanced hepatocellular carcinoma (HCC) in one study (100), while in another clinical study,
the same statin did not improve the survival of patients with
advanced HCC, despite the high doses (40-80 mg/day) administered
(101). Nevertheless, their use,
specifically in combination with other agents, such as FASN
inhibitors, remains of pharmacological interest.
Looking at the metabolic rewiring in PDAC and
specifically, at the alterations of lipid metabolism in the context
of genomic alterations, inactivating mutations of the TP53
gene that often precede the emergence of metastatic tumors stand
out in particular. Recently, mass-spectrometry-based lipidome
profiling in combination with the transcriptomics of in
vitro models and patients with PDAC revealed that the loss of
p53 caused a deregulation of intracellular and secreted
lysophospholipids with the suggestion that this class of lipids may
play a role in p53-mediated non-cell-autonomous molecular
signaling, which causes the remodeling of the cancer
microenvironment and contributes to immune evasion during PDAC
pathogenesis (102).
In addition to rendering the treatment of PDAC more
effective, it is crucial to spot its onset early. Biomarkers with
optimal specificity and sensitivity for an early diagnosis of PC
are therefore much sought after. Within that context, altered serum
levels of several lipid metabolites, such as very-low-density
lipoprotein, LDL, high-density lipoprotein, 3-hydroxybyturate,
lysophosphatidylcholine, phosphatidylethanolamine and bile acids,
such as taurocholic acid were identified (103,104). Recently, Wang et al
(105) combined machine learning
and metabolomics to select lipids, such as diacylglycerol and
lysophosphatidylethanolamine for their detection in PDAC patients'
serum and proposed the potential clinical application of their
liquid chromatography-mass spectrometry-based targeted lipidomics
assay for the effective and accurate auxiliary diagnosis of PDAC.
Analyzing the lipid content in pancreatic tissue could also be used
to distinguish chronic pancreatitis from PC, e.g., through the
employment of NMR spectroscopy (106).
As a consequence of the use of aerobic glycolysis
for ATP production and the conversion of pyruvate to lactate,
cancer cells use other mechanisms to secure the fueling of the
citric acid cycle than the conversion of pyruvate of glycolytic
origin to acetyl-CoA and then to citrate. An increased dependence
on glutaminolysis, which is accompanied by nicotinamide-adenine
dinucleotide phosphate (NADPH) production and changes in the
activity of corresponding enzymes is one particular mechanism
through which cancer cells maintain the proper function of the
Krebs cycle (107). This allows
such cells to reserve glucose-derived carbon for the use in
biosynthetic pathways, such as the pentose phosphate pathway, which
ultimately yields NADPH and substrates for the synthesis of
ribonucleotides (108,109). NADPH, as a glutathione reductase
(GR) co-substrate, plays a role in maintaining the reduced pool of
glutathione important for reactive oxygen species (ROS) scavenging
(110). Stepping aside from the
important role of glutamine metabolism in maintaining the
appropriate levels of ROS in cancer cells, the metabolism of other
amino acids has also been identified as a possible target for the
disruption of the redox homeostasis in PC cells. The inhibition of
the import of oxidized cysteine has been shown to cause a redox
imbalance and subsequent ferroptosis in targeted cells (111). This points at a critical
dependency of PDAC cells on cysteine contribution to the synthesis
of glutathione and coenzyme A to endure elevated lipid ROS
production, typically elicited by oncogenic signaling to stimulate
tumor growth (111).
The mitochondrial production of citrate from
glutamine in a multistep process and its subsequent conversion to
cytosolic acetyl-CoA links glutaminolysis with fatty acid synthesis
to support the growth of cancer cells in vivo (112). Glutamine also serves as a
substrate in the hexosamine phosphate synthesis and is critical for
the replenishment of nucleotides, as well as amino acids pools
(113-115). Bott et al (116) found that in glutamine-deprived
PDAC cells, the glutamate ammonia ligase-mediated de novo
synthesis of glutamine was critical for the transfer of the
terminal amide nitrogen to nucleotides and hexosamines and
subsequently, for their growth. Among the enzymes present in cancer
cells that are responsible for the production of glutamine-derived
α-ketoglutarate are glutamate-dehydrogenase (GDH), alanine
aminotransferase (ALT or GTP), phosphoserine aminotransferase 1 and
aspartate transaminase (AST or GOT) (117). In PC, a so-called non-canonical
glutamine metabolism pathway is prominently used, where glutamine
is first converted to aspartate, which is then converted to
oxaloacetate by GOT1 in the cytoplasm. This NADPH-producing pathway
concludes with the formation of malate and later pyruvate. An
oncogenic form of the KRAS protein has been shown to be associated
with the establishment of this non-canonical pathway of glutamine
metabolism in a study on PDAC (118). This growth advantage-conferring
pathway can also be stimulated by hypoxia, often observed in PDAC
cells: The PI3K/mammalian target of rapamycin complex (mTORC) 2
pathway targets hypoxia-inducible factor 2-α (HIF2-α) with a
consequent transcriptional activation of GOT1 (119). Recently, it has also been
confirmed in PDAC cells that the uncoupling protein 2
(UCP2)-mediated transport of glutamine-derived aspartate from
mitochondria to cytosol is critical for the generation of NADPH in
this pathway. Notably, UCP2 silencing has been shown to markedly
suppress the growth of KRASmut PDAC cells (120).
Multiple research teams have tried to confirm the
critical importance of this non-canonical pathway of glutamine
metabolism in PC. In a previous study, a pan-inhibitor of
transaminases, aminooxyacetate (AOA) negatively affected the growth
of PDAC cell lines; however, non-transformed human pancreatic
ductal cells and human diploid fibroblasts remained largely
unaffected (118). The authors of
that study suggested, however, that the sensitivity of cells to AOA
was dependent on whether or not oncogenic KRAS was present to
support the anabolic metabolism of glutamine. Encouragingly, the
decreased ability of PDAC cells to combat oxidative stress after an
impairment of Gln metabolism could significantly increase the
success rate of the whole approach (118). In another study, the inhibition
of mitochondrial GOT2 led to a marked induction of cyclin-dependent
kinase inhibitor p27-mediated cellular senescence and the growth
suppression of PDAC cells. This effect was not observed in
non-transformed cells (121).
Apart from oncogenic KRAS signaling, other
regulatory mechanisms exist in PDAC that modulate the metabolism of
amino acids and provide for the metabolic needs of cancer cells.
The MYC oncogene positively regulates glutamine uptake and causes a
suppression of microRNAs (miRNAs/miRs) with an inhibitory effect
towards glutaminase (GLS) (122,123). Son et al (118) demonstrated that a chemical
inhibition of GLS had a growth-suppressive effect on both human and
mouse PDAC cells. However, Roux et al (124) observed a decreased activity and
mRNA levels of GLS in the majority of the analyzed PDAC cells.
Those cells had an increased activity, mRNA and protein expression
of glycerol-3-phosphate acyltransferase and carbamoyl phosphate
synthetase II, which allowed them to produce nucleotides and
glutamate, as well as upregulated levels of alanine serine cysteine
transporter 2 responsible for glutamine import. GDH and AST/GOT
were other enzymes with an elevated activity. Another study
demonstrated that the activity of Myc itself could be regulated by
RNA and that expression levels of long non-coding RNA (lncRNA)
XLOC_006390 were specifically associated with changes in glutamate
metabolism, proliferation and migration of PC cells (125). It was found that this specific
lncRNA functioned by binding to c-Myc and preventing ubiquitination
with the final result being the increased stability of the
transcription factor that binds to the promoter of GDH1 and
acts as its transcriptional activator. Of note, He et al
(125) suggested that it was the
XLOC_006390/c-Myc signaling axis resulting in the upregulation of
GDH1 expression that was associated with worse survival times of
patients with PC rather than the expression of other enzymes
involved in the α-ketoglutarate supply.
In conditions with a low glutamine availability, the
transcription factor p53 is a chief participant in pro-survival
signaling and can help cancer cells to overcome such a nutritional
challenge by stimulating the expression of transporters,
facilitating the import of other amino acids. Aspartate and
arginine can be taken up by cells more robustly due to the
involvement of p53, with the latter amino acid capable of
activating the mTORC1, and in turn promoting tumor growth (126,127). In addition to wild-type p53,
mutated p53 can also play a role upon a glutamine withdrawal to
promote tumor cell survival via an induction of p21 resulting in
cell cycle arrest (128). Taken
together, p53 can act in conjunction with mTORC1 either towards
stopping or promoting cell proliferation in a context-dependent
manner, with factors such as the type of stress stimulus and the
mutational status of p53 likely to be involved.
Other tumor suppressors frequently mutated in cancer
cells have been linked to the altered cellular metabolism of
various types of cancer, including PDAC (18). A homozygous deletion of the tumor
suppressor gene locus SMAD4, which occurs in PDAC and can cause the
loss of the malic enzyme 2 (ME2), was previously exploited to
disrupt NADPH production, mitochondrial redox homeostasis and amino
acid metabolism. The simultaneous deletion of both mitochondrial
malic acid enzymes ME2 and ME3 led to a suppressed transcription of
branched-chain aminotransferase ½ and thus impeded the transfer of
amino groups from branched chain amino acids to α-ketoglutarate. As
a consequence, glutamate could not be resynthesized efficiently and
the de novo nucleotide synthesis was also impaired (129). This approach could be potentially
therapeutically beneficial in patients with PDAC with a homozygous
deletion of the SMAD4 locus, who constitute approximately
one-third of PDAC cases and who, at the same time, have ME2
deletions (129).
Aberrant metabolism can lead to epigenetic
alterations associated with an uncontrolled proliferation, as the
activity of chromatin-modulating enzymes involved in the regulation
of DNA transcription is dependent on the presence of cellular
metabolites, such as acetyl-coenzyme A and S-adenosylmethionine
(130-132). Kottakis et al (133) established how a concrete
metabolic state interconnected with epigenetic processes
contributes to the oncogenic transformation in pancreatic cells
with concurrent LKB1 and KRAS mutations. An interrelation was found
between the generation of excess S-adenosylmethionine induced by
the mTOR-dependent channeling of glucose and glutamine-derived
intermediates into the serine-glycine-one carbon pathway, and
transcriptional silencing through DNA methylation (133).
The importance of amino acids is also apparent when
it comes to the transcriptional regulation of the Snail family of
EMT transcription factors (EMT-TFs). These EMT-TFs suppress
E-cadherin, and other epithelial markers and adhesion molecules,
and increase the expression of matrix metalloproteinase (MMPs),
thus stimulating EMT, which often precedes the metastatic spread of
cancer cells (134). Arginine
additionally supports PDAC cell dissemination by serving as the
precursor for the synthesis of nitric oxide (NO), while glutamate
has been shown to stimulate the metastatic spread in PC,
functioning through its neurotransmitter receptors (135,136). Specifically, the depletion of
arginine has been found to impede the migration, adhesion and
invasion of PC cells with a lower expression of EMT-TFs,
extracellular matrix-rebuilding MMPs and an increased expression of
the epithelial marker, E-cadherin (134). The use of arginine deprivation in
the treatment of PC could also achieve more prominent antitumor
effects. It may thus combat the development of therapeutic
resistance with a simultaneous use of cytotoxic agents, such as
gemcitabine or when combined with the inhibition of other metabolic
pathways in PDAC cells, including those belonging to glutamine
metabolism, serine biosynthesis or polyamine biosynthesis (137,138).
Finally, to be able to cope with a situation when
nutritional deficiencies arise, PDAC cells are well-adapted to take
advantage of extracellular nutrient sources available in the
microenvironment. When needed, they can utilize
protein-macropinocytosis to obtain important amino acids by
lysosomal degradation, as well as autophagy to recycle proteins,
macromolecules and whole organelles and to refill essential
nucleotide pools (139-143). Additionally, autophagy in PC
supports immune evasion and is required for proper cystine
transport and cysteine homeostasis by promoting the localization of
the cystine transporter SLC7A11 at the plasma membrane (144,145). Notably, Maertin et al
(140) found that while the basal
autophagic activity of PC cells was relatively high, amino acid
depletion and hypoxia activated autophagy only weakly, if at all.
As regards the precise regulation of the autophagic process, they
confirmed the stimulation of basal autophagy by the oncogenic
activation mutation in KRAS GTPase and by the consequently
stimulated OXPHOS. A limitation of amino acid supply to suppress
OXPHOS was also mentioned as a potential option for the treatment
of PDAC (140).
It is no longer controversial that numerous cancer
cells have fully functional mitochondria performing OXPHOS,
depending on the oxygen availability in the particular tumor site,
the amount of available metabolites capable of fueling the TCA
cycle and the required regulation state. This is contrary to the
previous hypothesis of universally diminished OXPHOS activity with
defective mitochondria whenever glycolysis was found to be
upregulated in tumors (146).
Substrates produced by fatty acid oxidation,
glycolysis and glutaminolysis enter the TCA cycle, which extracts
electrons passed on to NADH and flavoproteins. A constant supply of
electrons is necessary for the creation of a transmembrane proton
gradient and the efficient production of ATP by the electron
transport chain complexes and ATP synthase in the inner
mitochondrial membrane (147).
The transport of these electrons can lead to the generation of ROS
in mitochondrial complexes of the electron transport chain
(148). Oncogene-inducible ROS
can support anchorage-independent cancer cell growth by decreasing
the phosphorylation of ERK1/2 in the ERK MAPK signaling pathway to
levels that are compatible with cellular proliferation and as such,
were described as a factor supporting KRAS-driven tumorigenicity
(149). Attempts to target the
TCA cycle in PDAC to suppress mitochondrial metabolism are
currently ongoing, with a protocol that includes the lipoic acid
analogue, devimistat, which selectively inhibits the TCA cycle by
impairing the activity of pyruvate dehydrogenase and
α-ketoglutarate dehydrogenase, combined with the modified
FOLFIRINOX regimen (150).
Within a cohort of patients with PDAC, tumors with
heterogeneous OXPHOS rates have been discovered and the high OXPHOS
activity with high amounts of complex I mRNAs and proteins could be
used as a helpful biomarker to identify tumors vulnerable to
substances, such as phenformin in combination with traditional
chemotherapy (151). The cause
for optimism with this type of treatments is the fact that they
could be applied independently of the main genetic alterations
forming the causal background of the majority of PDAC cases, such
as KRAS mutations, and that the antitumor effects of phenformin
with gemcitabine were successfully replicated in vivo
(151). It was hypothesized that
after inhibiting OXPHOS at complex I with phenformin, the affected
cells would lose the protection from the stress caused by the
inhibition of DNA synthesis by gemcitabine (151). The clinical confirmation of those
results remains to be performed, at least to the best of our
knowledge. To improve the results of OXPHOS inhibition, combined
treatments using several compounds will likely be required. For
example, Candido et al (152) based their experiments on the
findings of metformin-mediated autophagy induction through the
suppression of mTORC1 activity. Using metformin, they increased the
anti-proliferative effects of the mTOR inhibitor, rapamycin, in
PDAC cell lines (152).
Attention should be paid to the possibility of
potential disease relapses when cancer treatments targeting
mitochondrial metabolism are used. An adaptation to OXPHOS
inhibition by a fraction of tumor cells that can switch to
glycolysis and vice versa was previously described in PCSCs, where
their metabolic phenotype observed at a particular time depended on
MYC/PGC-1α balance (39). These
highly metastatic and treatment-resistant cells capable of tumor
repopulation were previously described as more reliant on the
OXPHOS pathway and thus could represent an interesting cell
subpopulation in PC to be targeted by OXPHOS inhibiting drugs
(17). The inhibition of OXPHOS
could further benefit cancer patients by lowering tumor hypoxia,
which can interfere with the efficacy of treatments. An oxygen
consumption decrease in better accessible normoxic regions could
lead to a higher oxygen tension across the microenvironment of the
whole tumor, leading to an improvement of the anticancer effects in
the initially hypoxic regions (146). Finally, carefully selected
medications aimed at increasing tumor oxygenation could help in the
quest to identify strategies with which to suppress treatment
resistance in a number of cancer cases, as exemplified by an
improved response to radiotherapy following the administration of
metformin in a prostate cancer in a study using xenografts
(153).
The extracellular environment in PC consists of a
desmoplastic tissue composed of collagen fibers, dissolved growth
factors, metabolites and diverse stromal cells, among which CAFs,
stellate cells and activated macrophages are strongly represented
(154). Drug penetration through
this dense, fibrotic tissue barrier can be problematic, which
together with frequently occurring hypovascularity, adds to the
intrinsic chemoresistance of PDAC (154). This desmoplastic environment can
be advantageous for cancer cells, while at the same time forming an
obstacle to their access to growth factors and metabolites and for
the metastatic spread. A proteolytic breakdown of stromal tissue
facilitated by plasminogen activation system, as well as MMPs
expressed by CAFs and tumor cells can help tumor growth and
invasion (155). SerpinB2 was
previously identified as a regulator of stromal remodeling of
collagen in PDAC, with its enzymatic target urokinase plasminogen
activator (uPA) emerging as a novel PDAC prognostic marker
(156). The non-universally
beneficial implications of the fibrotic stroma on PDAC cells have
been reflected in reported findings regarding the role of tumor
microenvironment in PDAC (157-163). Olive et al (160) discovered that the depletion of
tumor-associated stromal tissue by the inhibition of the Hedgehog
cellular signaling pathway enhanced the delivery of chemotherapy in
a mouse model of PC, which led to a transient stabilization of the
disease. By contrast, Croucher et al (155) pointed out the association between
reduced stromal integrity and tumor growth, as well as local
invasion in PC.
Pancreatic stellate cells (PSCs) are primarily
responsible for the maintenance of normal stromal tissue
architecture; however, in the case of a repeated or sustained
pancreatic injury, as well as during PC, they become perpetually
activated. Apart from causing pathological fibrosis, important
supporting roles of activated PSCs towards adjacent cancer cells
have been uncovered (164).
Signals derived from stromal cells in the form of secreted factors
regulate the expression of various genes in PC cells, including
those connected with the cell cycle, DNA replication and metabolic
pathways. Those signals can increase glycolytic metabolites, the
pentose phosphate pathway, nucleic acid synthesis and the TCA
cycle, which translates into the increased viability of PDAC cells
under nutrient-deprived conditions (33). Of note, genes activated by stromal
cues have been found to overlap with genes activated by the
KrasG12D allele, suggesting a cooperation of stromal
components and oncogenes in transcriptionally driven PC progression
(33). Furthermore, oncogenic KRAS
switches on Sonic hedgehog secretion by PDAC cells to activate PSCs
and triggers heterocellular crosstalk, which leads, together with
multiple phosphorylation events via an IGFR1/AXL-AKT axis, to an
increased mitochondrial capacity in tumor cells (35).
In an effort to elucidate the mechanisms through
which the stromal secretome can alter gene expression in PDAC cells
and to identify potential novel therapeutic targets, Sherman et
al (33) uncovered the histone
acetylation at promoter and enhancer regions of functionally
relevant genes as a key mediating event. In their concluding
remarks, they also proposed the hepatocyte growth factor,
insulin-like growth factor binding proteins 2, 3, 7 and
MYC-activating, PSCs-produced Il-6 as possible
microenvironment-derived factors inducing transcriptional and
metabolic changes in the epithelial compartment.
The cooperation between stromal and cancer cells can
take on a different form as PDAC cells can manipulate PSCs to
actively use autophagy and secrete non-essential amino acids
(NEAAs). Those alternative carbon sources are then captured by PDAC
cells and serve to support TCA anaplerosis, lipid and NEAA
biosynthesis when other drivers of these pathways such as glucose
or glutamine become scarce (34).
It has been determined that oxidative stress induced by cancer
cells in the adjacent stromal cells may be behind the autophagy
activation in PSCs with the resultant stromal overproduction of
recycled nutrients benefiting cancer cells by driving mitochondrial
biogenesis and anabolic growth (165). Additionally, the excess stromal
ROS production enhances the antioxidant defense in nearby cancer
cells and contributes to their genomic instability (165). It has also been suggested that
autophagy in CAFs could be induced by ammonia produced by cancer
cells during glutaminolysis and diffused into the microenvironment,
which could lead to a positive feedback, with subsequent high
levels of glutamine secreted into the TME by CAFs increasing the
conversion of glutamine to glutamate and ammonia along with
mitochondrial activity in cancer cells (166,167). A supportive role of the metabolic
scavenging in PDAC is further evidenced by the confirmed existence
of CAF-derived exosomes containing mRNA, miRNA, intact metabolites
and TCA cycle intermediates, which promote PC cell proliferation,
migration and chemoresistance in a mechanism similar to
macropinocytosis under nutrient-depleted conditions (31,168-170). Aside from acting as a direct
source of metabolites, such exosomes could create a
hypoxia-mimicking microenvironment stimulating the reductive
carboxylation of glutamine in cancer cells, the pathway previously
observed in rapidly growing malignant cells and identified to be an
alternative to oxidative metabolism as the major source of citrate
and precursors for macromolecular synthesis (168,171).
Under conditions of nutrient stress, PDAC cells can
also take up collagen fragments from the surrounding stroma either
through macropinocytosis-dependent or independent mechanisms.
Following the breakdown of extracellular matrix proteins,
collagen-derived proline is further metabolized to support the TCA
cycle and to promote PDAC cell survival and proliferation (172). Moreover, following their
activation by PDAC cells, fibroblasts start to produce higher
amount of type I collagen, which enhances integrin-FAK signaling in
PDAC cells, resulting in an increased PDAC clonogenic growth,
self-renewal and the frequency of cancer stem cells (CSCs)
(173).
In addition to stromal fibroblasts, PC cells can
interact with different cell types within the tumor
microenvironment. Adipocytes can acquire a mesenchymal phenotype
characterized by decreased lipid content and energy utilization
after a de-differentiation caused by a co-culture with PC cells.
Adipocytes with this phenotype can increase PC cell aggressiveness
in vitro and can also contribute to matrix remodeling
(174). Meyer et al
(175) established a model of
adipocyte-induced proliferation of PC cells enhanced by
nutrient-poor conditions, demonstrating that adipocytes secreted
glutamine and initiated its transfer to cancer cells after their
own catabolism of the same metabolite was downregulated by PC cells
(175).
Other intercellular interplay found to contribute to
the malignant progression of PDAC involves tumor-associated
macrophages (TAMs). By secreting the cytokine, CCL18, M2 TAMs can
activate the NF-κB signal transduction pathway in PDAC cells and
thus induce their expression of the adhesion molecule, vascular
cell adhesion molecule-1, which subsequently enhances aerobic
glycolysis in PDAC cells. Lactate produced by PC cells reciprocally
facilitates the M2-like polarization of macrophages, thus creating
a positive feedback loop (176).
Due to the ability of lactate to modify the host antitumor immune
response, its increased production contributes to the formation of
an immunosuppressive tumor niche and the improved survival of
metastatic cells, as shown by the improvement of the natural killer
cell cytolytic function towards LDHA-deficient PanO2 cells in mice
(53).
Intercellular interactions in the pancreatic tumor
stroma may also include short-range interactions through gap
junctions or the transfer of mitochondria between PC cells and
other stromal cells, including mesenchymal stem cells (MSCs), as
has been shown for other types of cancer (177). CAFs have been shown to influence
the metabolism and support the malignant progression of non-small
cell lung cancer (NSCLC) cells through connexin-43 gap junctions
(178). Of note, the outward
transfer of mitochondria mediated by cell adhesion and tunneling
nanotubes from Jurkat cells, which had an increased level of ROS
after a cytotoxic treatment, to MSCs was shown to cause
chemoresistance in Jurkat cells (179). Multiple myeloma cells can also
receive mitochondria from their microenvironment, which can lead to
changes in their metabolism, resulting in a more prominent use of
OXPHOS (180). It is possible
that similar intercellular interactions may be present in
pancreatic tumors. In fact, the role of connexin-43 channels in
exporting lactate from glycolytic PDAC cells to sustain their
metabolism and growth when an outward MCT transport is inhibited by
extracellular acidity, as well as to provide substrate for OXPHOS
of cells in better-perfused areas, where there is also a more
favorable transmembrane gradient for MCT-facilitated lactate
off-loading, has already been described (181). It would thus be of interest to
examine whether other such mechanisms play a role in PC tumors.
CSCs are a highly-chemoresistant subpopulation of
cells within tumors capable to self-renew and replenish the cancer
cell population following chemotherapy, which leads to the
recurrence of the disease. The importance of eradicating these
cells to successfully treat cancer has become clear (182-184). A tremendous effort has been place
in identifying potential biomarkers for PCSCs over the years, so
they could be isolated and targeted. For this purpose, multiple
surface markers expressed by these undifferentiated cells were
identified, such as CD44, CD24, EpCAM, CD133, CXCR4, ALDH1 and
hepatocyte growth factor receptor c-MET (185-189). To further expand the options of
the detection and isolation of CSCs in different types of cancer,
Miranda-Lorenzo et al (190) used autofluorescence as an
exclusive marker of CSCs from solid human tumors, including that of
PDAC. Such cells expressed pluripotency-associated genes and
displayed chemoresistance, long-term tumorigenicity and
invasiveness in vivo, traits suggestive of their stem-like
identity. As to what potential advantage could these cells gain
from having this autofluorescent phenotype, e.g., during hostile
conditions after chemotherapy, it was hypothesized that riboflavin
stored in the CSC autofluorescent vesicles could be used for the
synthesis of flavin-dependent coenzymes and flavin nucleotides,
factors important for the setting up of an antioxidant defense
system (191,192). Additionally, certain genes that
are differentially expressed and distinguish stem cells in the
normal pancreas could be potentially used to identify stem-like
cells in various tumors (193).
Advances in the identification and isolation of
PCSCs uncovered their association with PSCs and CAFs. These stromal
cells create a paracrine niche for PCSCs by secreting factors,
including Nodal/Activin, hCAP-18/LL-37 and ISG15, and enhance their
stem cell-like phenotypes along with their tumorigenic potential
(30,194,195).
In keeping with the exceptional ability of PCSCs to
survive stressful conditions, one would predict the occurrence of
specific metabolic adaptations in these cells. Indeed, an increased
utilization of OXPHOS by PCSCs could confer to them some resistance
in situations when glucose or glutamine is limited, as well as
during other situations affecting their specifically adjusted
metabolism, such as the ablation of K-Ras oncogene (17,39).
Importantly, oxygen deprivation typical for the tumor
microenvironment of PDAC does not prevent the ability of PCSCs to
use OXPHOS; instead, hypoxia supports autophagy and favors their
survival and migration (196,197). A higher reliance on OXPHOS could,
however, be an issue for PCSCs as the ROS produced by this pathway
need to be detoxified to maintain their stemness. Jagust et
al (198) demonstrated their
dependency on glutathione metabolism, which was confirmed by the
significantly diminished CSC self-renewal and chemoresistance after
a pharmacological targeting of this antioxidant pathway. Similar to
their non-CSCs counterparts, PCSCs can utilize multiple seemingly
crucial metabolic pathways. The targeting of the non-canonical
glutamine metabolism pathway in PCSCs was shown to negatively
affect their self-renewal, elevate their intracellular levels of
ROS and subsequently increase their radiosensitivity (199). Apart from glutamine, the
inhibition of fatty acid synthesis and mevalonate pathways caused
an anti-proliferative effect in PCSCs that was greater than that in
parental PC cells, suggesting that their utilization of metabolic
substrates could be heterogeneous, often dependent on the
microenvironmental context (200).
Epigenetic alterations connected with distinct
metabolic pathways in PCSCs probably predetermine their metabolic
plasticity, which allows them to respond promptly to different
environmental challenges and enhances their tumorigenicity.
Notably, chromatin modifications linked to distant metastatic
subclones was reversed by targeting a specific metabolic enzyme in
a PDAC study, with those results further supporting
metabolism-epigenome links in cancer (201). Currently, knowledge is expanding
regarding the factors behind the metabolic plasticity of PCSCs,
which needs to be overcome if this highly resistant cellular
subpopulation is to be eliminated in PDAC tumors. The impairment of
mitochondrial ISGylation critical for the recycling of
dysfunctional mitochondria disrupts PaCSC mitochondrial metabolism,
consequently affecting their metabolic plasticity and rendering
them susceptible to a prolonged inhibition with metformin in
vivo (202). Recently, it was
also demonstrated that the unique metabolic signatures of PCSCs
mediate organ-specific metastasis, which further points to the
importance of metabolic programming of this cellular entity
(203).
Therapeutic options for PDAC are still limited
mainly to surgery and cytotoxic chemotherapy. The targeting of the
reprogrammed cancer metabolism provides an innovative therapeutic
strategy to be used also in combination with cytotoxic agents to
achieve greater effects. Such applications already exist.
Devimistat, a drug that inhibits key enzymes for the functioning of
the TCA cycle in tumor cells in combination with the modified
FOLFIRINOX, protocol led to a response rate of 61% in patients with
metastatic PC in an open-label, phase I trial (NCT01835041)
(204) and a phase-III clinical
trial to evaluate efficacy and safety of the same combination of
drugs in patients with metastatic adenocarcinoma of the pancreas is
ongoing (205). The cell
autophagy inhibitor, hydroxychloroquine, in combination with
cytotoxic agents or ERK/MAPK pathway inhibitors is currently being
tested for effectiveness in PC clinical trials (NCT01506973;
NCT04145297; NCT03825289 and NCT04132505).
It is likely that further such therapeutic
strategies can be developed based on a detailed understanding of
the metabolic flexibility and needs of cancer cells together with
the metabolic cooperation and signaling within the tumor
microenvironment.
Not applicable.
Both authors (MZ and JT) conceived the present
review article. MZ collected the literature and drafted the
manuscript. JT made critical revisions to the manuscript. Both
authors have read and approved the final manuscript. Data
authentication is not applicable.
Not applicable.
Not applicable.
Both authors declare that they have no competing
interests.
Not applicable.
No external funding was received.
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2021. CA Cancer J Clin. 71:7–33. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rahib L, Smith BD, Aizenberg R, Rosenzweig
AB, Fleshman JM and Matrisian LM: Projecting cancer incidence and
deaths to 2030: The unexpected burden of thyroid, liver, and
pancreas cancers in the United States. Cancer Res. 74:2913–2921.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sarantis P, Koustas E, Papadimitropoulou
A, Papavassiliou AG and Karamouzis MV: Pancreatic ductal
adenocarcinoma: Treatment hurdles, tumor microenvironment and
immunotherapy. World J Gastrointest Oncol. 12:173–181. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hu JX, Zhao CF, Chen WB, Liu QC, Li QW,
Lin YY and Gao F: Pancreatic cancer: A review of epidemiology,
trend, and risk factors. World J Gastroenterol. 27:4298–4321. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
McGuigan A, Kelly P, Turkington RC, Jones
C, Coleman HG and McCain RS: Pancreatic cancer: A review of
clinical diagnosis, epidemiology, treatment and outcomes. World J
Gastroenterol. 24:4846–4861. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vincent A, Herman J, Schulick R, Hruban RH
and Goggins M: Pancreatic cancer. Lancet. 378:607–620. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Picozzi VJ, Oh SY, Edwards A, Mandelson
MT, Dorer R, Rocha FG, Alseidi A, Biehl T, Traverso LW, Helton WS
and Kozarek RA: Five-year actual overall survival in resected
pancreatic cancer: A contemporary single-institution experience
from a multidisciplinary perspective. Ann Surg Oncol. 24:1722–1730.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Von Hoff DD, Ervin T, Arena FP, Chiorean
EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et
al: Increased survival in pancreatic cancer with nab-paclitaxel
plus gemcitabine. N Engl J Med. 369:1691–1703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Conroy T, Hammel P, Hebbar M, Ben
Abdelghani M, Wei AC, Raoul JL, Choné L, Francois E, Artru P, Biagi
JJ, et al: FOLFIRINOX or gemcitabine as adjuvant therapy for
pancreatic cancer. N Engl J Med. 379:2395–2406. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Upadhrasta S and Zheng L: Strategies in
developing immunotherapy for pancreatic cancer: Recognizing and
correcting multiple immune 'defects' in the tumor microenvironment.
J Clin Med. 8:14722019. View Article : Google Scholar
|
|
12
|
Beatty GL, O'Hara MH, Lacey SF, Torigian
DA, Nazimuddin F, Chen F, Kulikovskaya IM, Soulen MC, McGarvey M,
Nelson AM, et al: Activity of mesothelin-specific chimeric antigen
receptor T cells against pancreatic carcinoma metastases in a phase
1 trial. Gastroenterology. 155:29–32. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
O'Hara M, O'Reilly E, Rosemarie M,
Varadhachary G, Wainberg ZA, Ko A, Fisher GA, Rahma O, Lyman JP,
Cabanski CR, et al: Abstract CT004: A phase Ib study of CD40
agonistic monoclonal antibody APX005M together with gemcitabine
(Gem) and nab-paclitaxel (NP) with or without nivolumab (Nivo) in
untreated metastatic ductal pancreatic adenocarcinoma (PDAC)
patients. Cancer Res. 79(Suppl 13): CT0042019. View Article : Google Scholar
|
|
14
|
Bahary N, Garrido-Laguna I, Cinar P,
O'Rourke MA, Somer BG, Nyak-Kapoor A, Lee JS, Munn D, Paul Kennedy
E, Vahanian NN, et al: Phase 2 trial of the indoleamine
2,3-dioxygenase pathway (IDO) inhibitor indoximod plus
gemcitabine/nab-paclitaxel for the treatment of metastatic pancreas
cancer: Interim analysis. J Clin Oncol. 34(Suppl 15): S30202016.
View Article : Google Scholar
|
|
15
|
Di Federico A, Tateo V, Parisi C, Formica
F, Carloni R, Frega G, Rizzo A, Ricci D, Di Marco M, Palloni A and
Brandi G: Hacking pancreatic cancer: Present and future of
personalized medicine. Pharmaceuticals (Basel). 14:6772021.
View Article : Google Scholar
|
|
16
|
Yang Y, Liu H, Li Z, Zhao Z, Yip-Schneider
M, Fan Q, Schmidt CM, Chiorean EG, Xie J, Cheng L, et al: Role of
fatty acid synthase in gemcitabine and radiation resistance of
pancreatic cancers. Int J Biochem Mol Biol. 2:89–98.
2011.PubMed/NCBI
|
|
17
|
Viale A, Pettazzoni P, Lyssiotis CA, Ying
H, Sánchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V,
et al: Oncogene ablation-resistant pancreatic cancer cells depend
on mitochondrial function. Nature. 514:628–632. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu M, Liu W, Qin Y, Xu X, Yu X, Zhuo Q
and Ji S: Regulation of metabolic reprogramming by tumor suppressor
genes in pancreatic cancer. Exp Hematol Oncol. 9:232020. View Article : Google Scholar
|
|
19
|
Daemen A, Peterson D, Sahu N, McCord R, Du
X, Liu B, Kowanetz K, Hong R, Moffat J, Gao M, et al: Metabolite
profiling stratifies pancreatic ductal adenocarcinomas into
subtypes with distinct sensitivities to metabolic inhibitors. Proc
Natl Acad Sci USA. 112:E4410–E4417. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Karasinska JM, Topham JT, Kalloger SE,
Jang GH, Denroche RE, Culibrk L, Williamson LM, Wong HL, Lee MK,
O'Kane GM, et al: Altered gene expression along the
glycolysis-cholesterol synthesis axis is associated with outcome in
pancreatic cancer. Clin Cancer Res. 26:135–146. 2020. View Article : Google Scholar
|
|
21
|
Halbrook CJ and Lyssiotis CA: Employing
metabolism to improve the diagnosis and treatment of pancreatic
cancer. Cancer Cell. 31:5–19. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Perusina Lanfranca M, Thompson JK, Bednar
F, Halbrook C, Lyssiotis C, Levi B and Frankel TL: Metabolism and
epigenetics of pancreatic cancer stem cells. Semin Cancer Biol.
57:19–26. 2019. View Article : Google Scholar :
|
|
23
|
Perera RM and Bardeesy N: Pancreatic
cancer metabolism: Breaking it down to build it back up. Cancer
Discov. 5:1247–1261. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dal Molin M, Zhang M, De Wilde RF,
Ottenhof NA, Rezaee N, Wolfgang CL, Blackford A, Vogelstein B,
Kinzler KW, Papadopoulos N, et al: Very long-term survival
following resection for pancreatic cancer is not explained by
commonly mutated genes: Results of whole-exome sequencing analysis.
Clin Cancer Res. 21:1944–1950. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Collisson EA, Sadanandam A, Olson P, Gibb
WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L, et al:
Subtypes of pancreatic ductal adenocarcinoma and their differing
responses to therapy. Nat Med. 17:500–503. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Moffitt RA, Marayati R, Flate EL, Volmar
KE, Loeza SG, Hoadley KA, Rashid NU, Williams LA, Eaton SC, Chung
AH, et al: Virtual microdissection identifies distinct tumor- and
stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat
Genet. 47:1168–1178. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao L, Zhao H and Yan H: Gene expression
profiling of 1200 pancreatic ductal adenocarcinoma reveals novel
subtypes. BMC Cancer. 18:6032018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Maurer C, Holmstrom SR, He J, Laise P, Su
T, Ahmed A, Hibshoosh H, Chabot JA, Oberstein PE, Sepulveda AR, et
al: Experimental microdissection enables functional harmonisation
of pancreatic cancer subtypes. Gut. 68:1034–1043. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chan-Seng-Yue M, Kim JC, Wilson GW, Ng K,
Figueroa EF, O'Kane GM, Connor AA, Denroche RE, Grant RC, McLeod J,
et al: Transcription phenotypes of pancreatic cancer are driven by
genomic events during tumor evolution. Nat Genet. 52:231–240. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lonardo E, Frias-Aldeguer J, Hermann PC
and Heeschen C: Pancreatic stellate cells form a niche for cancer
stem cells and promote their self-renewal and invasiveness. Cell
Cycle. 11:1282–1290. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Richards KE, Zeleniak AE, Fishel ML, Wu J,
Littlepage LE and Hill R: Cancer-associated fibroblast exosomes
regulate survival and proliferation of pancreatic cancer cells.
Oncogene. 36:1770–1778. 2017. View Article : Google Scholar :
|
|
32
|
Mejia I, Bodapati S, Chen KT and Díaz B:
Pancreatic adenocarcinoma invasiveness and the tumor
microenvironment: From biology to clinical trials. Biomedicines.
8:4012020. View Article : Google Scholar :
|
|
33
|
Sherman MH, Yu RT, Tseng TW, Sousa CM, Liu
S, Truitt ML, He N, Ding N, Liddle C, Atkins AR, et al: Stromal
cues regulate the pancreatic cancer epigenome and metabolome. Proc
Natl Acad Sci USA. 114:1129–1134. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sousa CM, Biancur DE, Wang X, Halbrook CJ,
Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, et
al: Pancreatic stellate cells support tumour metabolism through
autophagic alanine secretion. Nature. 536:479–483. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tape CJ, Ling S, Dimitriadi M, McMahon KM,
Worboys JD, Leong HS, Norrie IC, Miller CJ, Poulogiannis G,
Lauffenburger DA and Jørgensen C: Oncogenic KRAS regulates tumor
cell signaling via stromal reciprocation. Cell. 165:910–920. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Juiz N, Elkaoutari A, Bigonnet M, Gayet O,
Roques J, Nicolle R, Iovanna J and Dusetti N: Basal-like and
classical cells coexistence in pancreatic cancer revealed by single
cell analysis. bioRxiv. 2020.
|
|
37
|
Suzuki T, Otsuka M, Seimiya T, Iwata T,
Kishikawa T and Koike K: The biological role of metabolic
reprogramming in pancreatic cancer. MedComm (2020). 1:302–310.
2020.
|
|
38
|
Zaidi N, Lupien L, Kuemmerle NB, Kinlaw
WB, Swinnen JV and Smans K: Lipogenesis and lipolysis: The pathways
exploited by the cancer cells to acquire fatty acids. Prog Lipid
Res. 52:585–589. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sancho P, Burgos-Ramos E, Tavera A, Bou
Kheir T, Jagust P, Schoenhals M, Barneda D, Sellers K,
Campos-Olivas R, Graña O, et al: MYC/PGC-1α balance determines the
metabolic phenotype and plasticity of pancreatic cancer stem cells.
Cell Metab. 22:590–605. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kamphorst JJ, Nofal M, Commisso C, Hackett
SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA,
Bar-Sagi D, et al: Human pancreatic cancer tumors are nutrient poor
and tumor cells actively scavenge extracellular protein. Cancer
Res. 75:544–553. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yoshida GJ: Metabolic reprogramming: The
emerging concept and associated therapeutic strategies. J Exp Clin
Cancer Res. 34:1112015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yu M, Zhou Q, Zhou Y, Fu Z, Tan L, Ye X,
Zeng B, Gao W, Zhou J, Liu Y, et al: Metabolic phenotypes in
pancreatic cancer. PLoS One. 10:e01151532015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lomberk G, Blum Y, Nicolle R, Nair A,
Gaonkar KS, Marisa L, Mathison A, Sun Z, Yan H, Elarouci N, et al:
Distinct epigenetic landscapes underlie the pathobiology of
pancreatic cancer subtypes. Nat Commun. 9:19782018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kaoutari AE, Fraunhoffer NA, Hoare O,
Teyssedou C, Soubeyran P, Gayet O, Roques J, Lomberk G, Urrutia R,
Dusetti N and Iovanna J: Metabolomic profiling of pancreatic
adenocarcinoma reveals key features driving clinical outcome and
drug resistance. EBioMedicine. 66:1033322021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu Q, Ge W, Wang T, Lan J,
Martínez-Jarquín S, Wolfrum C, Stoffel M and Zenobi R:
High-throughput single-cell mass spectrometry reveals abnormal
lipid metabolism in pancreatic ductal adenocarcinoma. Angew Chem
Int Ed Engl. 60:24534–24542. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Argüello RJ, Combes AJ, Char R, Gigan JP,
Baaziz AI, Bousiquot E, Camosseto V, Samad B, Tsui J, Yan P, et al:
SCENITH: A flow cytometry-based method to functionally profile
energy metabolism with single-cell resolution. Cell Metab.
32:1063–1075.e7. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Miller A, Nagy C, Knapp B, Laengle J,
Ponweiser E, Groeger M, Starkl P, Bergmann M, Wagner O and Haschemi
A: Exploring metabolic configurations of single cells within
complex tissue microenvironments. Cell Metab. 26:788–800.e6. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ambrosini G, Dalla Pozza E, Fanelli G, Di
Carlo C, Vettori A, Cannino G, Cavallini C, Carmona-Carmona CA,
Brandi J, Rinalducci S, et al: Progressively de-differentiated
pancreatic cancer cells shift from glycolysis to oxidative
metabolism and gain a quiescent stem state. Cells. 9:15722020.
View Article : Google Scholar :
|
|
49
|
Biancur DE, Paulo JA, Małachowska B,
Quiles Del Rey M, Sousa CM, Wang X, Sohn ASW, Chu GC, Gygi SP,
Harper JW, et al: Compensatory metabolic networks in pancreatic
cancers upon perturbation of glutamine metabolism. Nat Commun.
8:159652017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Koppenol WH, Bounds PL and Dang CV: Otto
Warburg's contributions to current concepts of cancer metabolism.
Nat Rev Cancer. 11:325–337. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Liu XS, Little JB and Yuan ZM: Glycolytic
metabolism influences global chromatin structure. Oncotarget.
6:4214–4225. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Liberti MV and Locasale JW: The Warburg
effect: How does it benefit cancer cells? Trends Biochem Sci.
41:211–218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Husain Z, Huang Y, Seth P and Sukhatme VP:
Tumor-derived lactate modifies antitumor immune response: Effect on
myeloid-derived suppressor cells and NK cells. J Immunol.
191:1486–1495. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Du J, Gu J, Deng J, Kong L, Guo Y, Jin C,
Bao Y, Fu D and Li J: The expression and survival significance of
glucose transporter-1 in pancreatic cancer: Meta-analysis,
bioinformatics analysis and retrospective study. Cancer Invest.
39:741–755. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lee EE, Ma J, Sacharidou A, Mi W, Salato
VK, Nguyen N, Jiang Y, Pascual JM, North PE, Shaul PW, et al: A
protein kinase C phosphorylation motif in GLUT1 affects glucose
transport and is mutated in GLUT1 deficiency syndrome. Mol Cell.
58:845–853. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Cheng CS, Tan HY, Wang N, Chen L, Meng Z,
Chen Z and Feng Y: Functional inhibition of lactate dehydrogenase
suppresses pancreatic adenocarcinoma progression. Clin Transl Med.
11:e4672021. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ying H, Kimmelman AC, Lyssiotis CA, Hua S,
Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff
JL, et al: Oncogenic Kras maintains pancreatic tumors through
regulation of anabolic glucose metabolism. Cell. 149:656–670. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Jiang W, Qiao L, Zuo D, Qin D, Xiao J, An
H, Wang Y, Zhang X, Jin Y and Ren L: Aberrant lactate dehydrogenase
A signaling contributes metabolic signatures in pancreatic cancer.
Ann Transl Med. 9:3582021. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yalcin A, Solakoglu TH, Ozcan SC, Guzel S,
Peker S, Celikler S, Balaban BD, Sevinc E, Gurpinar Y and Chesney
JA: 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase-3 is
required for transforming growth factor β1-enhanced invasion of
Panc1 cells in vitro. Biochem Biophys Res Commun. 484:687–693.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ji S, Zhang B, Liu J, Qin Y, Liang C, Shi
S, Jin K, Liang D, Xu W, Xu H, et al: ALDOA functions as an
oncogene in the highly metastatic pancreatic cancer. Cancer Lett.
374:127–135. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Principe M, Borgoni S, Cascione M,
Chattaragada MS, Ferri-Borgogno S, Capello M, Bulfamante S,
Chapelle J, Di Modugno F, Defilippi P, et al: Alpha-enolase (ENO1)
controls alpha v/beta 3 integrin expression and regulates
pancreatic cancer adhesion, invasion, and metastasis. J Hematol
Oncol. 10:162017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Principe M, Ceruti P, Shih NY,
Chattaragada MS, Rolla S, Conti L, Bestagno M, Zentilin L, Yang SH,
Migliorini P, et al: Targeting of surface alpha-enolase inhibits
the invasiveness of pancreatic cancer cells. Oncotarget.
6:11098–11113. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Azoitei N, Becher A, Steinestel K, Rouhi
A, Diepold K, Genze F, Simmet T and Seufferlein T: PKM2 promotes
tumor angiogenesis by regulating HIF-1α through NF-κB activation.
Mol Cancer. 15:32016. View Article : Google Scholar
|
|
64
|
Thews O and Riemann A: Tumor pH and
metastasis: A malignant process beyond hypoxia. Cancer Metastasis
Rev. 38:113–129. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Guillaumond F, Leca J, Olivares O, Lavaut
MN, Vidal N, Berthezène P, Dusetti NJ, Loncle C, Calvo E, Turrini
O, et al: Strengthened glycolysis under hypoxia supports tumor
symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma.
Proc Natl Acad Sci USA. 110:3919–3924. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kirk P, Wilson MC, Heddle C, Brown MH,
Barclay AN and Halestrap AP: CD147 is tightly associated with
lactate transporters MCT1 and MCT4 and facilitates their cell
surface expression. EMBO J. 19:3896–3904. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Semenza GL: Tumor metabolism: Cancer cells
give and take lactate. J Clin Invest. 118:3835–3837.
2008.PubMed/NCBI
|
|
68
|
Sun X, Wang M, Wang M, Yao L, Li X, Dong
H, Li M, Sun T, Liu X, Liu Y and Xu Y: Role of proton-coupled
monocarboxylate transporters in cancer: From metabolic crosstalk to
therapeutic potential. Front cell Dev Biol. 8:6512020. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Pavlides S, Whitaker-Menezes D,
Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro
MC, Wang C, Fortina P, Addya S, et al: The reverse Warburg effect:
Aerobic glycolysis in cancer associated fibroblasts and the tumor
stroma. Cell Cycle. 8:3984–4001. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Benny S, Mishra R, Manojkumar MK and
Aneesh TP: From Warburg effect to reverse Warburg effect; the new
horizons of anti-cancer therapy. Med Hypotheses. 144:1102162020.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Roland CL, Arumugam T, Deng D, Liu SH,
Philip B, Gomez S, Burns WR, Ramachandran V, Wang H,
Cruz-Monserrate Z and Logsdon CD: Cell surface lactate receptor
GPR81 is crucial for cancer cell survival. Cancer Res.
74:5301–5310. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Benton CR, Yoshida Y, Lally J, Han X-X,
Hatta H and Bonen A: PGC-1alpha increases skeletal muscle lactate
uptake by increasing the expression of MCT1 but not MCT2 or MCT4.
Physiol Genomics. 35:45–54. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Cheng CF, Ku HC and Lin H: PGC-1α as a
pivotal factor in lipid and metabolic regulation. Int J Mol Sci.
19:34472018. View Article : Google Scholar
|
|
74
|
Schneiderhan W, Scheler M, Holzmann KH,
Marx M, Gschwend JE, Bucholz M, Gress TM, Seufferlein T, Adler G
and Oswald F: CD147 silencing inhibits lactate transport and
reduces malignant potential of pancreatic cancer cells in in vivo
and in vitro models. Gut. 58:1391–1398. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Lee SH, Hwang HK, Lee WJ and Kang CM: MCT4
as a potential therapeutic target to augment gemcitabine
chemosensitivity in resected pancreatic cancer. Cell Oncol (Dordr).
44:1363–1371. 2021. View Article : Google Scholar
|
|
76
|
Benjamin D, Robay D, Hindupur SK, Pohlmann
J, Colombi M, El-Shemerly MY, Maira SM, Moroni C, Lane HA and Hall
MN: Dual inhibition of the lactate transporters MCT1 and MCT4 Is
synthetic lethal with metformin due to NAD+ depletion in
cancer cells. Cell Rep. 25:3047–3058.e4. 2018. View Article : Google Scholar
|
|
77
|
Sonveaux P, Végran F, Schroeder T, Wergin
MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C,
Jordan BF, et al: Targeting lactate-fueled respiration selectively
kills hypoxic tumor cells in mice. J Clin Invest. 118:3930–3942.
2008.PubMed/NCBI
|
|
78
|
Wu DH, Liang H, Lu SN, Wang H, Su ZL,
Zhang L, Ma JQ, Guo M, Tai S and Yu S: miR-124 suppresses
pancreatic ductal adenocarcinoma growth by regulating
monocarboxylate transporter 1-mediated cancer lactate metabolism.
Cell Physiol Biochem. 50:924–935. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Lau AN, Li Z, Danai LV, Westermark AM,
Darnell AM, Ferreira R, Gocheva V, Sivanand S, Lien EC, Sapp KM, et
al: Dissecting cell-type-specific metabolism in pancreatic ductal
adenocarcinoma. Elife. 9:e567822020. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Koundouros N and Poulogiannis G:
Reprogramming of fatty acid metabolism in cancer. Br J Cancer.
122:4–22. 2020. View Article : Google Scholar :
|
|
81
|
Alo PL, Amini M, Piro F, Pizzuti L,
Sebastiani V, Botti C, Murari R, Zotti G and Di Tondo U:
Immunohistochemical expression and prognostic significance of fatty
acid synthase in pancreatic carcinoma. Anticancer Res.
27:2523–2527. 2007.PubMed/NCBI
|
|
82
|
Li N, Lu H, Chen C, Bu X and Huang P: Loss
of fatty acid synthase inhibits the 'hER2-PI3K/Akt axis' activity
and malignant phenotype of Caco-2 cells. Lipids Health Dis.
12:832013. View Article : Google Scholar
|
|
83
|
Menendez JA, Decker JP and Lupu R: In
support of fatty acid synthase (FAS) as a metabolic oncogene:
Extracellular acidosis acts in an epigenetic fashion activating FAS
gene expression in cancer cells. J Cell Biochem. 94:1–4. 2005.
View Article : Google Scholar
|
|
84
|
Shetty A, Nagesh PKB, Setua S, Hafeez BB,
Jaggi M, Yallapu MM and Chauhan SC: Novel paclitaxel
nanoformulation impairs de novo lipid synthesis in pancreatic
cancer cells and enhances gemcitabine efficacy. ACS Omega.
5:8982–8991. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Tracz-Gaszewska Z and Dobrzyn P:
Stearoyl-CoA desaturase 1 as a therapeutic target for the treatment
of cancer. Cancers (Basel). 11:9482019. View Article : Google Scholar
|
|
86
|
Rysman E, Brusselmans K, Scheys K,
Timmermans L, Derua R, Munck S, Van Veldhoven PP, Waltregny D,
Daniëls VW, Machiels J, et al: De novo lipogenesis protects cancer
cells from free radicals and chemotherapeutics by promoting
membrane lipid saturation. Cancer Res. 70:8117–8126. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Fritz V, Benfodda Z, Rodier G, Henriquet
C, Iborra F, Avancès C, Allory Y, de la Taille A, Culine S, Blancou
H, et al: Abrogation of de novo lipogenesis by stearoyl-CoA
desaturase 1 inhibition interferes with oncogenic signaling and
blocks prostate cancer progression in mice. Mol Cancer Ther.
9:1740–1754. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Eser S, Schnieke A, Schneider G and Saur
D: Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer.
111:817–822. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Menendez JA: Fine-tuning the
lipogenic/lipolytic balance to optimize the metabolic requirements
of cancer cell growth: Molecular mechanisms and therapeutic
perspectives. Biochim Biophys Acta. 1801:381–391. 2010. View Article : Google Scholar
|
|
90
|
Xu M, Jung X, Hines OJ, Eibl G and Chen Y:
Obesity and pancreatic cancer: Overview of epidemiology and
potential prevention by weight loss. Pancreas. 47:158–162. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Li D, Morris JS, Liu J, Hassan MM, Day RS,
Bondy ML and Abbruzzese JL: Body mass index and risk, age of onset,
and survival in patients with pancreatic cancer. JAMA.
301:2553–2562. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Kasenda B, Bass A, Koeberle D, Pestalozzi
B, Borner M, Herrmann R, Jost L, Lohri A and Hess V: Survival in
overweight patients with advanced pancreatic carcinoma: A
multicentre cohort study. BMC Cancer. 14:7282014. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Anderson AS, Renehan AG, Saxton JM, Bell
J, Cade J, Cross AJ, King A, Riboli E, Sniehotta F, Treweek S, et
al: Cancer prevention through weight control-where are we in 2020?
Br J Cancer. 124:1049–1056. 2021. View Article : Google Scholar
|
|
94
|
Jacobs EJ, Newton CC, Patel AV, Stevens
VL, Islami F, Flanders WD and Gapstur SM: The association between
body mass index and pancreatic cancer: Variation by age at body
mass index assessment. Am J Epidemiol. 189:108–115. 2020.
View Article : Google Scholar
|
|
95
|
Chimento A, Casaburi I, Avena P, Trotta F,
De Luca A, Rago V, Pezzi V and Sirianni R: Cholesterol and its
metabolites in tumor growth: Therapeutic potential of statins in
cancer treatment. Front Endocrinol (Lausanne). 9:8072019.
View Article : Google Scholar
|
|
96
|
Guillaumond F, Bidaut G, Ouaissi M,
Servais S, Gouirand V, Olivares O, Lac S, Borge L, Roques J, Gayet
O, et al: Cholesterol uptake disruption, in association with
chemotherapy, is a promising combined metabolic therapy for
pancreatic adenocarcinoma. Proc Natl Acad Sci USA. 112:2473–2478.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Nam GH, Kwon M, Jung H, Ko E, Kim SA, Choi
Y, Song SJ, Kim S, Lee Y, Kim GB, et al: Statin-mediated inhibition
of RAS prenylation activates ER stress to enhance the
immunogenicity of KRAS mutant cancer. J Immunother cancer.
9:e0024742021. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Jiang W, Hu JW, He XR, Jin WL and He XY:
Statins: A repurposed drug to fight cancer. J Exp Clin Cancer Res.
40:2412021. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Osmak M: Statins and cancer: Current and
future prospects. Cancer Lett. 324:1–12. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Kawata S, Yamasaki E, Nagase T, Inui Y,
Ito N, Matsuda Y, Inada M, Tamura S, Noda S, Imai Y and Matsuzawa
Y: Effect of pravastatin on survival in patients with advanced
hepatocellular carcinoma. A randomized controlled trial. Br J
Cancer. 84:886–891. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Lersch C, Schmelz R, Erdmann J, Hollweck
R, Schulte-Frohlinde E, Eckel F, Nader M and Schusdziarra V:
Treatment of HCC with pravastatin, octreotide, or gemcitabine-a
critical evaluation. Hepatogastroenterology. 51:1099–1103.
2004.PubMed/NCBI
|
|
102
|
Butera A, Roy M, Zampieri C, Mammarella E,
Panatta E, Melino G, D'Alessandro A and Amelio I: p53-driven
lipidome influences non-cell-autonomous lysophospholipids in
pancreatic cancer. Biol Direct. 17:62022. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Zhang L, Jin H, Guo X, Yang Z, Zhao L,
Tang S, Mo P, Wu K, Nie Y, Pan Y and Fan D: Distinguishing
pancreatic cancer from chronic pancreatitis and healthy individuals
by (1)H nuclear magnetic resonance-based metabonomic profiles. Clin
Biochem. 45:1064–1069. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Urayama S, Zou W, Brooks K and Tolstikov
V: Comprehensive mass spectrometry based metabolic profiling of
blood plasma reveals potent discriminatory classifiers of
pancreatic cancer. Rapid Commun Mass Spectrom. 24:613–620. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Wang G, Yao H, Gong Y, Lu Z, Pang R, Li Y,
Yung Y, Song H, Liu J, Jin Y, et al: Metabolic detection and
systems analyses of pancreatic ductal adenocarcinoma through
machine learning, lipidomics, and multi-omics. Sci Adv.
7:eabh27242021. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Fang F, He X, Deng H, Chen Q, Lu J, Spraul
M and Yu Y: Discrimination of metabolic profiles of pancreatic
cancer from chronic pancreatitis by high-resolution magic angle
spinning 1H nuclear magnetic resonance and principal components
analysis. Cancer Sci. 98:1678–1682. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Wang Z, Liu F, Fan N, Zhou C, Li D,
Macvicar T, Dong Q, Bruns CJ and Zhao Y: Targeting glutaminolysis:
New perspectives to understand cancer development and novel
strategies for potential target therapies. Front Oncol.
10:5895082020. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
DeBerardinis RJ, Mancuso A, Daikhin E,
Nissim I, Yudkoff M, Wehrli S and Thompson CB: Beyond aerobic
glycolysis: Transformed cells can engage in glutamine metabolism
that exceeds the requirement for protein and nucleotide synthesis.
Proc Natl Acad Sci USA. 104:19345–19350. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Ghanem N, El-Baba C, Araji K, El-Khoury R,
Usta J and Darwiche N: The pentose phosphate pathway in cancer:
Regulation and therapeutic opportunities. Chemotherapy. 66:179–191.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Ju HQ, Lin JF, Tian T, Xie D and Xu RH:
NADPH homeostasis in cancer: Functions, mechanisms and therapeutic
implications. Signal Transduct Target Ther. 5:2312020. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Badgley MA, Kremer DM, Maurer HC,
DelGiorno KE, Lee HJ, Purohit V, Sagalovskiy IR, Ma A, Kapilian J,
Firl CEM, et al: Cysteine depletion induces pancreatic tumor
ferroptosis in mice. Science. 368:85–89. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Hatzivassiliou G, Zhao F, Bauer DE,
Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA and
Thompson CB: ATP citrate lyase inhibition can suppress tumor cell
growth. Cancer Cell. 8:311–321. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Akella NM, Ciraku L and Reginato MJ:
Fueling the fire: Emerging role of the hexosamine biosynthetic
pathway in cancer. BMC Biol. 17:522019. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Tong X, Zhao F and Thompson CB: The
molecular determinants of de novo nucleotide biosynthesis in cancer
cells. Curr Opin Genet Dev. 19:32–37. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Wang Y, Bai C, Ruan Y, Liu M, Chu Q, Qiu
L, Yang C and Li B: Coordinative metabolism of glutamine carbon and
nitrogen in proliferating cancer cells under hypoxia. Nat Commun.
10:2012019. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Bott AJ, Shen J, Tonelli C, Zhan L,
Sivaram N, Jiang YP, Yu X, Bhatt V, Chiles E, Zhong H, et al:
Glutamine anabolism plays a critical role in pancreatic cancer by
coupling carbon and nitrogen metabolism. Cell Rep. 29:1287–1298.e6.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Li T and Le A: Glutamine metabolism in
cancer. The Heterogeneity of Cancer Metabolism. Le A: Springer
International Publishing; Cham: pp. 13–32. 2018, View Article : Google Scholar
|
|
118
|
Son J, Lyssiotis CA, Ying H, Wang X, Hua
S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et
al: Glutamine supports pancreatic cancer growth through a
KRAS-regulated metabolic pathway. Nature. 496:101–105. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Li W, Chen C, Zhao X, Ye H, Zhao Y, Fu Z,
Pan W, Zheng S, Wei L, Nong T, et al: HIF-2α regulates
non-canonical glutamine metabolism via activation of PI3K/mTORC2
pathway in human pancreatic ductal adenocarcinoma. J Cell Mol Med.
21:2896–2908. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Raho S, Capobianco L, Malivindi R, Vozza
A, Piazzolla C, De Leonardis F, Gorgoglione R, Scarcia P, Pezzuto
F, Agrimi G, et al: KRAS-regulated glutamine metabolism requires
UCP2-mediated aspartate transport to support pancreatic cancer
growth. Nat Metab. 2:1373–1381. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Yang S, Hwang S, Kim M, Seo SB, Lee JH and
Jeong SM: Mitochondrial glutamine metabolism via GOT2 supports
pancreatic cancer growth through senescence inhibition. Cell Death
Dis. 9:552018. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Wise DR, Deberardinis RJ, Mancuso A, Sayed
N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon
SB and Thompson CB: Myc regulates a transcriptional program that
stimulates mitochondrial glutaminolysis and leads to glutamine
addiction. Proc Natl Acad Sci USA. 105:18782–18787. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Gao P, Tchernyshyov I, Chang TC, Lee YS,
Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT and
Dang CV: c-Myc suppression of miR-23a/b enhances mitochondrial
glutaminase expression and glutamine metabolism. Nature.
458:762–765. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Roux C, Riganti C, Borgogno SF, Curto R,
Curcio C, Catanzaro V, Digilio G, Padovan S, Puccinelli MP,
Isabello M, et al: Endogenous glutamine decrease is associated with
pancreatic cancer progression. Oncotarget. 8:95361–95376. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
He J, Li F, Zhou Y, Hou X, Liu S, Li X,
Zhang Y, Jing X and Yang L: LncRNA XLOC_006390 promotes pancreatic
carcinogenesis and glutamate metabolism by stabilizing c-Myc.
Cancer Lett. 469:419–428. 2020. View Article : Google Scholar
|
|
126
|
Lowman XH, Hanse EA, Yang Y, Ishak Gabra
MB, Tran TQ, Li H and Kong M: p53 Promotes cancer cell adaptation
to glutamine deprivation by upregulating Slc7a3 to increase
arginine uptake. Cell Rep. 26:3051–3060.e4. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Tajan M, Hock AK, Blagih J, Robertson NA,
Labuschagne CF, Kruiswijk F, Humpton TJ, Adams PD and Vousden KH: A
role for p53 in the adaptation to glutamine starvation through the
expression of SLC1A3. Cell Metab. 28:721–736.e6. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Tran TQ, Lowman XH, Reid MA,
Mendez-Dorantes C, Pan M, Yang Y and Kong M: Tumor-associated
mutant p53 promotes cancer cell survival upon glutamine deprivation
through p21 induction. Oncogene. 36:1991–2001. 2017. View Article : Google Scholar :
|
|
129
|
Dey P, Baddour J, Muller F, Wu CC, Wang H,
Liao WT, Lan Z, Chen A, Gutschner T, Kang Y, et al: Genomic
deletion of malic enzyme 2 confers collateral lethality in
pancreatic cancer. Nature. 542:119–123. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Dai Z, Ramesh V and Locasale JW: The
evolving metabolic landscape of chromatin biology and epigenetics.
Nat Rev Genet. 21:737–753. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Wang G and Han JJ: Connections between
metabolism and epigenetic modifications in cancer. Med Rev.
1:199–221. 2021. View Article : Google Scholar
|
|
132
|
Etchegaray JP and Mostoslavsky R:
Interplay between metabolism and epigenetics: A nuclear adaptation
to environmental changes. Mol Cell. 62:695–711. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Kottakis F, Nicolay BN, Roumane A, Karnik
R, Gu H, Nagle JM, Boukhali M, Hayward MC, Li YY, Chen T, et al:
LKB1 loss links serine metabolism to DNA methylation and
tumorigenesis. Nature. 539:390–395. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Wang H, Li QF, Chow H, Choi S and Leung
YC: Arginine deprivation inhibits pancreatic cancer cell migration,
invasion and EMT via the down regulation of snail, slug, twist, and
MMP1/9. J Physiol Biochem. 76:73–83. 2020. View Article : Google Scholar
|
|
135
|
Wang J, Yang S, He P, Schetter AJ, Gaedcke
J, Ghadimi BM, Ried T, Yfantis HG, Lee DH, Gaida MM, et al:
Endothelial nitric oxide synthase traffic inducer (NOSTRIN) is a
negative regulator of disease aggressiveness in pancreatic cancer.
Clin Cancer Res. 22:5992–6001. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Herner A, Sauliunaite D, Michalski CW,
Erkan M, De Oliveira T, Abiatari I, Kong B, Esposito I, Friess H
and Kleeff J: Glutamate increases pancreatic cancer cell invasion
and migration via AMPA receptor activation and Kras-MAPK signaling.
Int J Cancer. 129:2349–2359. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Gitto SB, Pandey V, Oyer JL, Copik AJ,
Hogan FC, Phanstiel O IV and Altomare DA: Difluoromethylornithine
combined with a polyamine transport inhibitor is effective against
gemcitabine resistant pancreatic cancer. Mol Pharm. 15:369–376.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Kremer JC, Prudner BC, Lange SES, Bean GR,
Schultze MB, Brashears CB, Radyk MD, Redlich N, Tzeng SC, Kami K,
et al: Arginine deprivation inhibits the Warburg effect and
upregulates glutamine anaplerosis and serine biosynthesis in
ASS1-deficient cancers. Cell Rep. 18:991–1004. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Davidson SM, Jonas O, Keibler MA, Hou HW,
Luengo A, Mayers JR, Wyckoff J, Del Rosario AM, Whitman M, Chin CR,
et al: Direct evidence for cancer-cell-autonomous extracellular
protein catabolism in pancreatic tumors. Nat Med. 23:235–241. 2017.
View Article : Google Scholar
|
|
140
|
Maertin S, Elperin JM, Lotshaw E, Sendler
M, Speakman SD, Takakura K, Reicher BM, Mareninova OA, Grippo PJ,
Mayerle J, et al: Roles of autophagy and metabolism in pancreatic
cancer cell adaptation to environmental challenges. Am J Physiol
Gastrointest Liver Physiol. 313:G524–G536. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Saliakoura M, Sebastiano MR, Nikdima I,
Pozzato C and Konstantinidou G: Restriction of extracellular lipids
renders pancreatic cancer dependent on autophagy. J Exp Clin Cancer
Res. 41:162022. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Piffoux M, Eriau E and Cassier PA:
Autophagy as a therapeutic target in pancreatic cancer. Br J
Cancer. 124:333–344. 2021. View Article : Google Scholar :
|
|
143
|
Li J, Chen X, Kang R, Zeh H, Klionsky DJ
and Tang D: Regulation and function of autophagy in pancreatic
cancer. Autophagy. 17:3275–3296. 2021. View Article : Google Scholar :
|
|
144
|
Yamamoto K, Venida A, Yano J, Biancur DE,
Kakiuchi M, Gupta S, Sohn ASW, Mukhopadhyay S, Lin EY, Parker SJ,
et al: Autophagy promotes immune evasion of pancreatic cancer by
degrading MHC-I. Nature. 581:100–105. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Mukhopadhyay S, Biancur DE, Parker SJ,
Yamamoto K, Banh RS, Paulo JA, Mancias JD and Kimmelman AC:
Autophagy is required for proper cysteine homeostasis in pancreatic
cancer through regulation of SLC7A11. Proc Natl Acad Sci USA.
118:e20214751182021. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Ashton TM, Gillies McKenna W,
Kunz-Schughart LA and Higgins GS: Oxidative phosphorylation as an
emerging target in cancer therapy. Clin Cancer Res. 24:2482–2490.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Zong WX, Rabinowitz JD and White E:
Mitochondria and cancer. Mol Cell. 61:667–676. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Lambert A and Brand M: Reactive oxygen
species production by mitochondria. Methods Mol Biol. 554:165–181.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Weinberg F, Hamanaka R, Wheaton WW,
Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger
GR and Chandel NS: Mitochondrial metabolism and ROS generation are
essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA.
107:8788–8793. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Nevala-Plagemann C, Hidalgo M and
Garrido-Laguna I: From state-of-the-art treatments to novel
therapies for advanced-stage pancreatic cancer. Nat Rev Clin Oncol.
17:108–123. 2020. View Article : Google Scholar
|
|
151
|
Masoud R, Reyes-Castellanos G, Lac S,
Garcia J, Dou S, Shintu L, Abdel Hadi N, Gicquel T, El Kaoutari A,
Diémé B, et al: Targeting mitochondrial complex I overcomes
chemoresistance in high OXPHOS pancreatic cancer. Cell Rep Med.
1:1001432020. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Candido S, Abrams SL, Steelman L,
Lertpiriyapong K, Martelli AM, Cocco L, Ratti S, Follo MY, Murata
RM, Rosalen PL, et al: Metformin influences drug sensitivity in
pancreatic cancer cells. Adv Biol Regul. 68:13–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Zannella VE, Dal Pra A, Muaddi H, McKee
TD, Stapleton S, Sykes J, Glicksman R, Chaib S, Zamiara P,
Milosevic M, et al: Reprogramming metabolism with metformin
improves tumor oxygenation and radiotherapy response. Clin Cancer
Res. 19:6741–6750. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Neesse A, Bauer CA, Öhlund D, Lauth M,
Buchholz M, Michl P, Tuveson DA and Gress TM: Stromal biology and
therapy in pancreatic cancer: Ready for clinical translation? Gut.
68:159–171. 2019. View Article : Google Scholar
|
|
155
|
Croucher DR, Saunders DN, Lobov S and
Ranson M: Revisiting the biological roles of PAI2 (SERPINB2) in
cancer. Nat Rev Cancer. 8:535–545. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Harris NLE, Vennin C, Conway JRW, Vine KL,
Pinese M, Cowley MJ, Shearer RF, Lucas MC, Herrmann D, Allam AH, et
al: SerpinB2 regulates stromal remodelling and local invasion in
pancreatic cancer. Oncogene. 36:4288–4298. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Jiang B, Zhou L, Lu J, Wang Y, Liu C, You
L and Guo J: Stroma-targeting therapy in pancreatic cancer: One
coin with two sides? Front Oncol. 10:5763992020. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Rhim AD, Oberstein PE, Thomas DH, Mirek
ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP,
Tattersall IW, et al: Stromal elements act to restrain, rather than
support, pancreatic ductal adenocarcinoma. Cancer Cell. 25:735–747.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
de la Fouchardière C, Gamradt P, Chabaud
S, Raddaz M, Blanc E, Msika O, Treilleux I, Bachy S,
Cattey-Javouhey A, Guibert P, et al: A promising biomarker and
therapeutic target in patients with advanced PDAC: The stromal
protein βig-h3. J Pers Med. 12:6232022. View Article : Google Scholar
|
|
160
|
Olive KP, Jacobetz MA, Davidson CJ,
Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA,
Caldwell ME, Allard D, et al: Inhibition of hedgehog signaling
enhances delivery of chemotherapy in a mouse model of pancreatic
cancer. Science. 324:1457–1461. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Bulle A and Lim KH: Beyond just a tight
fortress: Contribution of stroma to epithelial-mesenchymal
transition in pancreatic cancer. Signal Transduct Target Ther.
5:2492020. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Malchiodi ZX, Cao H, Gay MD, Safronenka A,
Bansal S, Tucker RD, Weinberg BA, Cheema A, Shivapurkar N and Smith
JP: Cholecystokinin receptor antagonist improves efficacy of
chemotherapy in murine models of pancreatic cancer by altering the
tumor microenvironment. Cancers (Basel). 13:49492021. View Article : Google Scholar
|
|
163
|
Lee JJ, Perera RM, Wang H, Wu DC, Liu XS,
Han S, Fitamant J, Jones PD, Ghanta KS, Kawano S, et al: Stromal
response to hedgehog signaling restrains pancreatic cancer
progression. Proc Natl Acad Sci USA. 111:E3091–E3100. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Wu Y, Zhang C, Jiang K, Werner J, Bazhin
AV and D'Haese JG: The role of stellate cells in pancreatic ductal
adenocarcinoma: Targeting perspectives. Front Oncol. 10:6219372021.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Lisanti MP, Martinez-Outschoorn UE,
Chiavarina B, Pavlides S, Whitaker-Menezes D, Tsirigos A,
Witkiewicz A, Lin Z, Balliet R, Howell A and Sotgia F:
Understanding the 'lethal' drivers of tumor-stroma co-evolution:
Emerging role(s) for hypoxia, oxidative stress and
autophagy/mitophagy in the tumor micro-environment. Cancer Biol
Ther. 10:537–542. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Ko YH, Lin Z, Flomenberg N, Pestell RG,
Howell A, Sotgia F, Lisanti MP and Martinez-Outschoorn UE:
Glutamine fuels a vicious cycle of autophagy in the tumor stroma
and oxidative mitochondrial metabolism in epithelial cancer cells:
Implications for preventing chemotherapy resistance. Cancer Biol
Ther. 12:1085–1097. 2011. View Article : Google Scholar
|
|
167
|
Mariño G and Kroemer G: Ammonia: A
diffusible factor released by proliferating cells that induces
autophagy. Sci Signal. 3:pe192010. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Zhao H, Yang L, Baddour J, Achreja A,
Bernard V, Moss T, Marini JC, Tudawe T, Seviour EG, San Lucas FA,
et al: Tumor microenvironment derived exosomes pleiotropically
modulate cancer cell metabolism. Elife. 5:e102502016. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Waldenmaier M, Seibold T, Seufferlein T
and Eiseler T: Pancreatic cancer small extracellular vesicles
(exosomes): A tale of short- and long-distance communication.
Cancers (Basel). 13:48442021. View Article : Google Scholar
|
|
170
|
Takikawa T, Masamune A, Yoshida N, Hamada
S, Kogure T and Shimosegawa T: Exosomes derived from pancreatic
stellate cells: MicroRNA signature and effects on pancreatic cancer
cells. Pancreas. 46:19–27. 2017. View Article : Google Scholar
|
|
171
|
Mullen AR, Wheaton WW, Jin ES, Chen PH,
Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS and
DeBerardinis RJ: Reductive carboxylation supports growth in tumour
cells with defective mitochondria. Nature. 481:385–388. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Olivares O, Mayers JR, Gouirand V,
Torrence ME, Gicquel T, Borge L, Lac S, Roques J, Lavaut MN,
Berthezène P, et al: Collagen-derived proline promotes pancreatic
ductal adenocarcinoma cell survival under nutrient limited
conditions. Nat Commun. 8:160312017. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Begum A, McMillan RH, Chang YT, Penchev
VR, Rajeshkumar NV, Maitra A, Goggins MG, Eshelman JR, Wolfgang CL,
Rasheed ZA and Matsui W: Direct interactions with cancer-associated
fibroblasts lead to enhanced pancreatic cancer stem cell function.
Pancreas. 48:329–334. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Cai Z, Liang Y, Xing C, Wang H, Hu P, Li
J, Huang H, Wang W and Jiang C: Cancer-associated adipocytes
exhibit distinct phenotypes and facilitate tumor progression in
pancreatic cancer. Oncol Rep. 42:2537–2549. 2019.PubMed/NCBI
|
|
175
|
Meyer KA, Neeley CK, Baker NA, Washabaugh
AR, Flesher CG, Nelson BS, Frankel TL, Lumeng CN, Lyssiotis CA,
Wynn ML, et al: Adipocytes promote pancreatic cancer cell
proliferation via glutamine transfer. Biochem Biophys Rep.
7:144–149. 2016.PubMed/NCBI
|
|
176
|
Ye H, Zhou Q, Zheng S, Li G, Lin Q, Wei L,
Fu Z, Zhang B, Liu Y, Li Z and Chen R: Tumor-associated macrophages
promote progression and the Warburg effect via CCL18/NF-kB/VCAM-1
pathway in pancreatic ductal adenocarcinoma. Cell Death Dis.
9:4532018. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Asencio-Barría C, Defamie N, Sáez JC,
Mesnil M and Godoy AS: Direct intercellular communications and
cancer: A snapshot of the biological roles of connexins in prostate
cancer. Cancers (Basel). 11:13702019. View Article : Google Scholar
|
|
178
|
Luo M, Luo Y, Mao N, Huang G, Teng C, Wang
H, Wu J, Liao X and Yang J: Cancer-associated fibroblasts
accelerate malignant progression of non-small cell lung cancer via
connexin 43-formed unidirectional gap junctional intercellular
communication. Cell Physiol Biochem. 51:315–336. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Wang J, Liu X, Qiu Y, Shi Y, Cai J, Wang
B, Wei X, Ke Q, Sui X, Wang Y, et al: Cell adhesion-mediated
mitochondria transfer contributes to mesenchymal stem cell-induced
chemoresistance on T cell acute lymphoblastic leukemia cells. J
Hematol Oncol. 11:112018. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Marlein CR, Piddock RE, Mistry JJ,
Zaitseva L, Hellmich C, Horton RH, Zhou Z, Auger MJ, Bowles KM and
Rushworth SA: CD38-driven mitochondrial trafficking promotes
bioenergetic plasticity in multiple myeloma. Cancer Res.
79:2285–2297. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Dovmark TH, Saccomano M, Hulikova A, Alves
F and Swietach P: Connexin-43 channels are a pathway for
discharging lactate from glycolytic pancreatic ductal
adenocarcinoma cells. Oncogene. 36:4538–4550. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Yu Z, Pestell TG, Lisanti MP and Pestell
RG: Cancer stem cells. Int J Biochem Cell Biol. 44:2144–2151. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Mummery CL, van de Stolpe A, Roelen B and
Clevers H: Chapter 12-Cancer stem cells: Where do they come from
and where are they going? Stem Cells. 3rd. Mummery CL, van de
Stolpe A, Roelen B and Clevers HB: Academic Press; Boston: pp.
299–328. 2021, View Article : Google Scholar
|
|
184
|
Nguyen LV, Vanner R, Dirks P and Eaves CJ:
Cancer stem cells: An evolving concept. Nat Rev Cancer. 12:133–143.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Li C, Heidt DG, Dalerba P, Burant CF,
Zhang L, Adsay V, Wicha M, Clarke MF and Simeone DM: Identification
of pancreatic cancer stem cells. Cancer Res. 67:1030–1038. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Gzil A, Zarębska I, Bursiewicz W, Antosik
P, Grzanka D and Szylberg Ł: Markers of pancreatic cancer stem
cells and their clinical and therapeutic implications. Mol Biol
Rep. 46:6629–6645. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Hermann PC, Huber SL, Herrler T, Aicher A,
Ellwart JW, Guba M, Bruns CJ and Heeschen C: Distinct populations
of cancer stem cells determine tumor growth and metastatic activity
in human pancreatic cancer. Cell Stem Cell. 1:313–323. 2007.
View Article : Google Scholar
|
|
188
|
Rasheed ZA, Yang J, Wang Q, Kowalski J,
Freed I, Murter C, Hong SM, Koorstra JB, Rajeshkumar NV, He X, et
al: Prognostic significance of tumorigenic cells with mesenchymal
features in pancreatic adenocarcinoma. J Natl Cancer Inst.
102:340–351. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Li C, Wu JJ, Hynes M, Dosch J, Sarkar B,
Welling TH, Pasca di Magliano M and Simeone DM: c-Met is a marker
of pancreatic cancer stem cells and therapeutic target.
Gastroenterology. 141:2218–2227.e5. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Miranda-Lorenzo I, Dorado J, Lonardo E,
Alcala S, Serrano AG, Clausell-Tormos J, Cioffi M, Megias D,
Zagorac S, Balic A, et al: Intracellular autofluorescence: A
biomarker for epithelial cancer stem cells. Nat Methods.
11:1161–1169. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Sancho P, Alcala S, Usachov V, Hermann PC
and Sainz B Jr: The ever-changing landscape of pancreatic cancer
stem cells. Pancreatology. 16:489–496. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Depeint F, Bruce WR, Shangari N, Mehta R
and O'Brien PJ: Mitochondrial function and toxicity: Role of the B
vitamin family on mitochondrial energy metabolism. Chem Biol
Interact. 163:94–112. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Rovira M, Scott SG, Liss AS, Jensen J,
Thayer SP and Leach SD: Isolation and characterization of
centroacinar/terminal ductal progenitor cells in adult mouse
pancreas. Proc Natl Acad Sci USA. 107:75–80. 2010. View Article : Google Scholar :
|
|
194
|
Sainz BJ Jr, Alcala S, Garcia E,
Sanchez-Ripoll Y, Azevedo MM, Cioffi M, Tatari M, Miranda-Lorenzo
I, Hidalgo M, Gomez-Lopez G, et al: Microenvironmental
hCAP-18/LL-37 promotes pancreatic ductal adenocarcinoma by
activating its cancer stem cell compartment. Gut. 64:1921–1935.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Sainz B Jr, Martín B, Tatari M, Heeschen C
and Guerra S: ISG15 is a critical microenvironmental factor for
pancreatic cancer stem cells. Cancer Res. 74:7309–7320. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Rausch V, Liu L, Apel A, Rettig T,
Gladkich J, Labsch S, Kallifatidis G, Kaczorowski A, Groth A, Gross
W, et al: Autophagy mediates survival of pancreatic
tumour-initiating cells in a hypoxic microenvironment. J Pathol.
227:325–335. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Zhu H, Wang D, Zhang L, Xie X, Wu Y, Liu
Y, Shao G and Su Z: Upregulation of autophagy by hypoxia-inducible
factor-1α promotes EMT and metastatic ability of CD133+ pancreatic
cancer stem-like cells during intermittent hypoxia. Oncol Rep.
32:935–942. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Jagust P, Alcalá S, Sainz B Jr, Heeschen C
and Sancho P: Glutathione metabolism is essential for self-renewal
and chemoresistance of pancreatic cancer stem cells. World J Stem
Cells. 12:1410–1428. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Li D, Fu Z, Chen R, Zhao X, Zhou Y, Zeng
B, Yu M, Zhou Q, Lin Q, Gao W, et al: Inhibition of glutamine
metabolism counteracts pancreatic cancer stem cell features and
sensitizes cells to radiotherapy. Oncotarget. 6:31151–31163. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Brandi J, Dando I, Pozza ED, Biondani G,
Jenkins R, Elliott V, Park K, Fanelli G, Zolla L, Costello E, et
al: Proteomic analysis of pancreatic cancer stem cells: Functional
role of fatty acid synthesis and mevalonate pathways. J Proteomics.
150:310–322. 2017. View Article : Google Scholar
|
|
201
|
McDonald OG, Li X, Saunders T,
Tryggvadottir R, Mentch SJ, Warmoes MO, Word AE, Carrer A, Salz TH,
Natsume S, et al: Epigenomic reprogramming during pancreatic cancer
progression links anabolic glucose metabolism to distant
metastasis. Nat Genet. 49:367–376. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Alcalá S, Sancho P, Martinelli P, Navarro
D, Pedrero C, Martín-Hijano L, Valle S, Earl J, Rodríguez-Serrano
M, Ruiz-Cañas L, et al: ISG15 and ISGylation is required for
pancreatic cancer stem cell mitophagy and metabolic plasticity. Nat
Commun. 11:26822020. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Nimmakayala RK, Leon F, Rachagani S, Rauth
S, Nallasamy P, Marimuthu S, Shailendra GK, Chhonker YS, Chugh S,
Chirravuri R, et al: Metabolic programming of distinct cancer stem
cells promotes metastasis of pancreatic ductal adenocarcinoma.
Oncogene. 40:215–231. 2021. View Article : Google Scholar
|
|
204
|
Alistar A, Morris BB, Desnoyer R, Klepin
HD, Hosseinzadeh K, Clark C, Cameron A, Leyendecker J, D'Agostino R
Jr, Topaloglu U, et al: Safety and tolerability of the
first-in-class agent CPI-613 in combination with modified
FOLFIRINOX in patients with metastatic pancreatic cancer: A
single-centre, open-label, dose-escalation, phase 1 trial. Lancet
Oncol. 18:770–778. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Philip PA, Buyse ME, Alistar AT, Rocha
Lima CM, Luther S, Pardee TS and Van Cutsem E: A phase III
open-label trial to evaluate efficacy and safety of CPI-613 plus
modified FOLFIRINOX (mFFX) versus FOLFIRINOX (FFX) in patients with
metastatic adenocarcinoma of the pancreas. Future Oncol.
15:3189–3196. 2019. View Article : Google Scholar : PubMed/NCBI
|