Introduction
Pancreatic cancer is one of the most aggressive
malignancies and highly resistant to currently applied cancer
therapies (1). Even after
curative-intent surgery, cure is exceedingly rare, as demonstrated
by a 5-year overall survival (OS) of less than 20% (2). Effective adjuvant systemic therapies
are necessary to improve survival outcomes.
Previously, the PRODIGE 24/CCTG PA.6 trial reported
an impressive survival benefit with adjuvant modified–FOLFIRINOX
(mFOLFIRINOX) compared with adjuvant gemcitabine (median OS 54 vs.
35 months; P<0.001) (3).
However, due to the high toxicity rate of this treatment regime,
gemcitabine is still being recommended, in particular for patients
with poor or declining performance status.
Resistance to gemcitabine is a major impediment of
successful treatment and is primarily due to molecular mechanisms
limiting the intracellular uptake and metabolic activation of
gemcitabine, and thus, its overall efficacy (4).
Type I IFNs (IFN-α and -β) have been proposed as
potential adjuvant to gemcitabine treatment in order to improve
survival outcomes in patients with pancreatic cancer (5,6).
Type I IFNs are pleiotropic cytokines that were originally
identified as viral replication suppressor. Further
characterization of their biological effect revealed a wide range
of antitumor effect; for instance, direct inhibitory effects on
tumor cells, anti-angiogenesis, enhanced immunogenicity of tumors
as well as other immune stimulatory effects (7,8).
Thereby, type I IFNs induce synergistic effects on pancreatic
cancer cells when co-treated with gemcitabine in vitro
(9,10). Recently, the chemo-sensitizing
effect of IFN-β in pancreatic cancer, formed in immune deficient
mice, was confirmed and upregulation of gemcitabine
transporter-coding genes was identified (9).
Both IFN-α and -β act via the type I IFN receptor
(IFNAR) complex, of which IFNAR-1 and IFNAR-2c are the most
important subunits (11). Binding
to this receptor activates the JAK-STAT signaling pathway, which
subsequently initiates the transcription of numerous
interferon-stimulated genes (ISGs) that are responsible for
mediating the biological activities of type I IFNs.
To accomplish an effect of type I IFNs, the presence
of the IFNAR complex is necessary (11,12). Previously, expression of IFNAR-1
and IFNAR-2c in 91.5 and 68.1% of pancreatic cancer tissues,
respectively, was reported (13).
Although IFN-α and -β bind to the same receptor and
induce signals through similar mechanisms, they have different
binding affinities. Previous studies reported a 50-fold higher
receptor-binding affinity for IFN-β than IFN-α, resulting in more
potent, elicited at much lower concentrations, antitumor effects
(10,14).
Despite strong evidence of the more potent and safer
antitumor effects of IFN-β compared with IFN-α, only a few studies
focused on adjuvant IFN-β therapy in pancreatic cancer. In
addition, the immunomodulatory effects of IFN-α and -β are less
described and primarily investigated in IFN-α. Hence, in the
present study, the potential immunomodulatory effect of IFN-β
towards pancreatic cancer cells was revealed for the first time.
The unique KPC3 cell line was used, derived from the clinically
relevant KPC mouse model, which mimics the immune phenotypic
features, aggressiveness, and gemcitabine-resistant character of
human pancreatic cancer (15,16). Thereby, the present study focused
on expression of gemcitabine transporter- and activating-coding
genes, as potential target to increase gemcitabine efficacies.
Materials and methods
Cells and culture conditions
The mouse KPC3 pancreatic cell line is derived from
a primary pancreatic ductal adenocarcinoma tumor of a female
KrasG12D/+;Trp53R172H/+;Pdx-1-Cre (KPC) mouse and was kindly
provided by Dr van Montfoort (Leiden University Medical Center,
Leiden, The Netherlands) (15).
Origin of cells was confirmed using short tandem repeat profiling
(Powerplex Kit; Promega Corporation). Cells were cultured in
RPMI-1640 medium supplemented with 5%fetal calf serum (FCS),
penicillin (1×105 U/l), and L-glutamine (2 mmol/l). FCS
was purchased from Sigma-Aldrich; Merck KGaA. Media and other
supplements were obtained from Gibco; Thermo Fisher Scientific,
Inc. FCS was purchased from Sigma-Aldrich; Merck KGaA. Cells were
cultured in a 5% CO2 atmosphere at 37°C and routinely
validated as Mycoplasma-free. Culture conditions were as previously
described in detail (17).
After trypsinization, KPC3 cells were plated in
24-well plates at the appropriate density in order to obtain 80%
confluency at the end of the experiment. The next day, incubations
were started in quadruplicate and control cells were
vehicle-treated. Medium and compounds were refreshed after 3 days.
All cell culture experiments were carried out at least twice in
quadruplicate. Mouse recombinant IFN-β-1a (Bio-Connect) and
gemcitabine (Sigma-Aldrich; Merck KGaA) stock dilutions were
diluted in distilled water.
Cell proliferation assay
Effects of IFN-β and/or gemcitabine on cell growth
were assessed by measuring the total DNA amount per well, as a
measure of cell number. After treatment, media were removed and
plates were stored at −20°C until DNA measurement. Measurement of
total DNA was performed with the bisbenzimide fluorescent dye
(Hoechst 33258; Sigma-Aldrich; Merck KGaA) as previously described
in detail (17).
Reverse transcription-quantitative
(RT-q) PCR
RNA isolation, cDNA synthesis and RT-qPCR were
performed as previously described (9), but using different primers (Table SI). Two housekeeping genes were
used to normalize mRNA levels using the Vandesompele method:
hypoxanthine-guanine phosphoribosyl transferase 1 (Hprt1)
and glucuronidase beta (Gusb) (Gibco; ThermoFisher
Scientific, Inc.) (18).
Mice
A total of 28 male C57BL/6 mice (8–10 weeks old)
were purchased from Charles River Laboratories. All mice were
housed in groups of seven and maintained at room temperature on a
daily 12-h light/12-h dark cycle in ventilated cages with
autoclaved bedding. Water and autoclaved laboratory rodent diet
were provided ad libitum. All mouse experiments were
controlled by the animal welfare committee of the Erasmus
University Medical Center (Rotterdam, The Netherlands) and approved
(approval no. AVD101002017867) by the national central committee of
animal experiments, in accordance with the Dutch Act on Animal
Experimentation and European Union (EU) Directive 2010/63/EU.
In vivo experiments
Mice were randomized in four groups (n=7 each) and
subcutaneously injected in the flank with 100,000 low passage
(passage number 3) KPC3 cells in 100 µl PBS/0.1% BSA (Gibco; Thermo
Fisher Scientific, Inc.). Cultured KPC3 cells were harvested at 80%
confluency and only single-cell suspensions of greater than 90%
viability were used for injection. Tumor size and body weight were
measured twice weekly. Tumor volume was calculated as (width^2 ×
length)/2 using a caliper. Treatment was started when tumor volumes
reached ~50 mm3. Mice in the control group and in the
IFN-β monotherapy group received daily an intraperitoneal (i.p.)
injection of 100 µl of distilled water or 10,000 units IFN-β. Mice
randomized to the gemcitabine monotherapy group received two times
a week (day 2 and 5) an i.p. injection of 50 mg/kg gemcitabine.
Mice in the combination group received upon start of the treatment,
daily an injection of 10,000 units IFN-β i.p. and, on day 2 and 5,
an i.p. injection of 50 mg/kg gemcitabine. The effect of
gemcitabine monotherapy in this model has been previously described
(19).
Necropsy procedures
Mice were sacrificed by cervical dislocation under
5% isoflurane anesthesia when tumor volume reached 1,000
mm3 or when the wellbeing of the mice could no longer be
maintained. Tumors were resected during necropsy and tumor volumes
were measured. Tumors were divided into two parts and subsequently
snap-frozen in liquid nitrogen and fixed overnight at 4°C in
freshly prepared 4% formaldehyde solution, and prepared for
paraffin sectioning (5-µm thick).
NanoString analysis
RNA was extracted according to the manufacturer's
instructions using the RNeasy Plus Micro kit (Qiagen). RNA samples
were diluted in RNA free water and stored at −80°C. The 2100
Bioanalyzer (Agilent Technologies, Inc.) was used to measure RNA
Quality Control. Total RNA concentrations were corrected to include
fragments seized between 300 and 4,000 nucleotides. A total of 200
ng RNA was hybridized to the PanCancer IO 360 Panel (NanoString
Technologies, Inc.) at 67°C for 17 h. The advanced analysis module
(version 2.0) of nSolver™ software (version 4.0; NanoString
Technologies, Inc.) was used for data analysis. A total of 8 out of
11 housekeeping genes were selected for normalization with the
geNorm algorithm embedded in the advanced analysis module (Table SII). Expression threshold was
calculated as twice of the average expression of the negative
controls. Gene expression below the threshold in more than 80% of
the samples were excluded from further analysis. Normalized data
were log2 transformed. Differentially expressed genes were
identified with simplified negative binomial models, mixture
negative binomial models, or log-linear models based on the
convergence of each gene. Adjusted P-values were calculated with
the Benjamini Hoghberg method. Genes were considered differentially
expressed when the adjusted P<0.05.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism version 3.0 (GraphPad Software, Inc.). The half maximal
effective concentration (EC50) on cell growth was
calculated using non-linear regression curve fitting program.
Effect of IFN-β was set on 100% and used as control to analyze the
combined effect of IFN-β pre-treatment and gemcitabine in in
vitro experiments. One-way ANOVA followed by Tukey's multiple
comparisons test was used for comparisons between treatment groups.
In all analyses, P<0.05 was considered to indicate a
statistically significant difference. Data are indicated as the
mean ± SEM.
Results
IFN-β sensitivity in KPC3 cells in
vitro
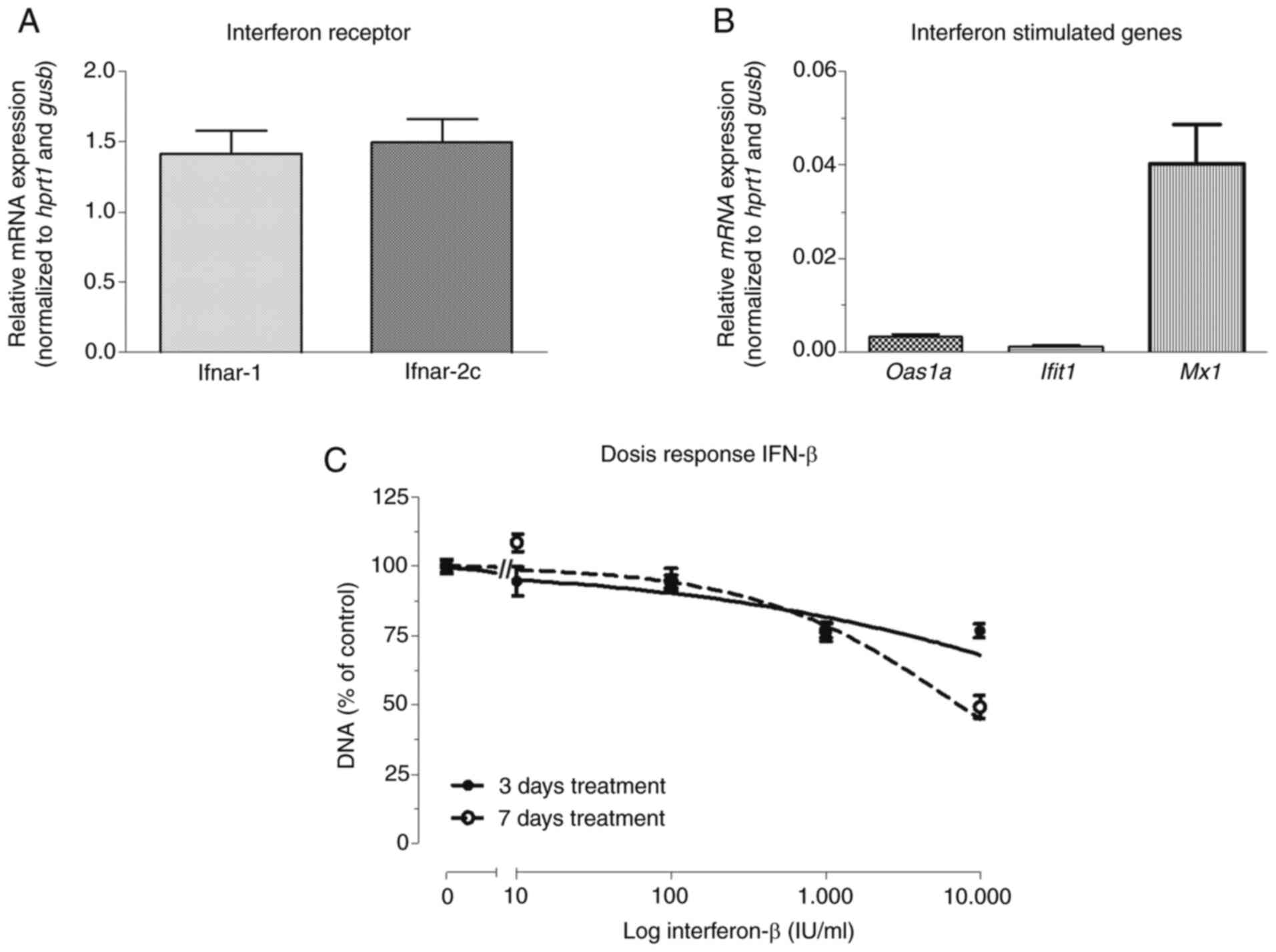
Relative mRNA expression of the IFN
receptor-coding genes, Ifnar-1 and Ifnar-2c, were
comparable in KPC3 cells (1.42±0.16 and 1.50±0.17, respectively)
(Fig. 1A). Baseline expression of
ISGs in untreated KPC3 cells was relatively low (Fig. 1B). The growth-inhibitory effect of
IFN-β was not very potent, as shown by an EC50 >1,000
IU/ml and maximal inhibition of 51% after 7 days (P<0.001 vs.
control) (Fig. 1C).
Effect of IFN-β pre-treatment on the
response to gemcitabine in vitro
Next, it was analyzed whether IFN-β pre-treatment
could sensitize KPC3 cells, reflected by a decrease in the
EC50 value of gemcitabine. Although 72 h after 1,000
IU/ml IFN-β pre-treatment alone had no statistically significant
effect on the cell amount, gemcitabine sensitivity was slightly
increased, as shown by a 1.3-fold decrease in EC50 value
compared with untreated control cells (EC50 1.5 ng/ml
vs. EC50 1.1 ng/ml respectively; P<0.001) (Fig. 2A and C). Expression of
Oas1a and Ifit1 was strongly upregulated following 72
h after 1,000 IU/ml IFN-β treatment (51- and 13-fold increase
respectively; both P<0.001 vs. untreated cells), whereas
expression of Mx1 was not affected by IFN-β treatment
(Fig. 2B).
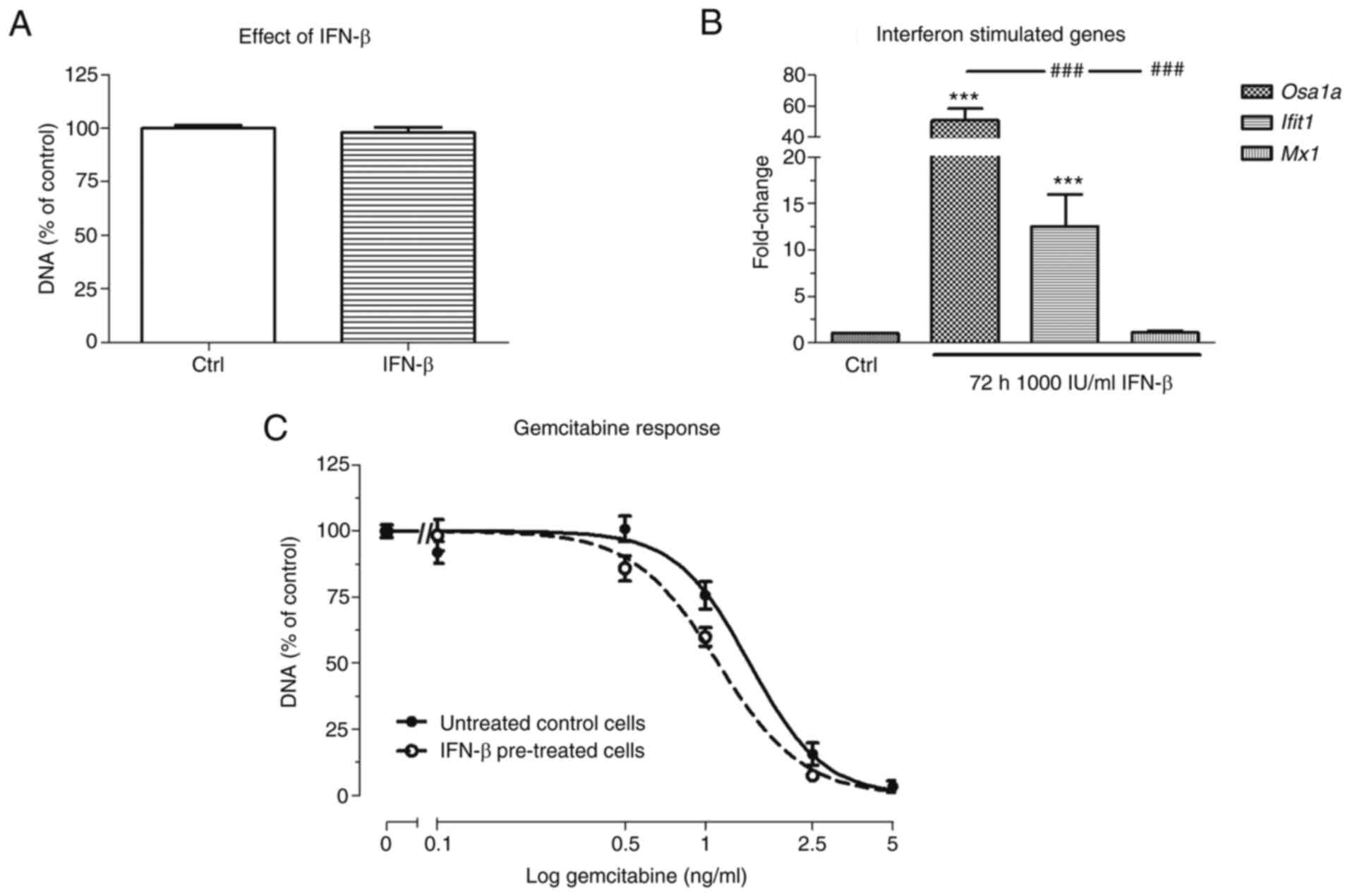 | Figure 2.Effect of IFN-β pre-treatment on
gemcitabine response in KPC3 cells. Cells were pre-treated for 72 h
with 1,000 IU/ml IFN-β, followed by 72 h gemcitabine monotherapy.
(A) Effect of 72 h 1,000 IU/ml IFN-β monotherapy. (B) Fold change
in mRNA expression of Oas1a, Ifit1, and Mx1
between no treatment and after 72 h 1,000 IU/ml IFN-β. (C) Overall
gemcitabine response in untreated control cells (solid line) vs. 72
h IFN-β pre-treated cells (dotted lines). Controls represent
vehicle control for non-IFN-β pre-treated and treatment with IFN-β
for IFN-β pre-treated cells. Values represent the mean ± SEM of at
least two independent experiments in quadruplicate and are shown as
a percentage of control. ***P<0.01 vs. control and
###P<0.01 vs. other gene. IFN-β, interferon-β; Oas1a,
2′-5′ oligoadenylate synthetase 1a; Ifit1, interferon induced
protein with tetratricopeptide repeats 1; Mx1, MX dynamin like
GTPase 1. |
Baseline expression of transporter- and
metabolizing-coding genes in KPC3 cells was low and even
undetectable for Slc28a1 and Slc28a3 (Fig. S1). Expression was not upregulated
after IFN-β treatment (Fig.
S1).
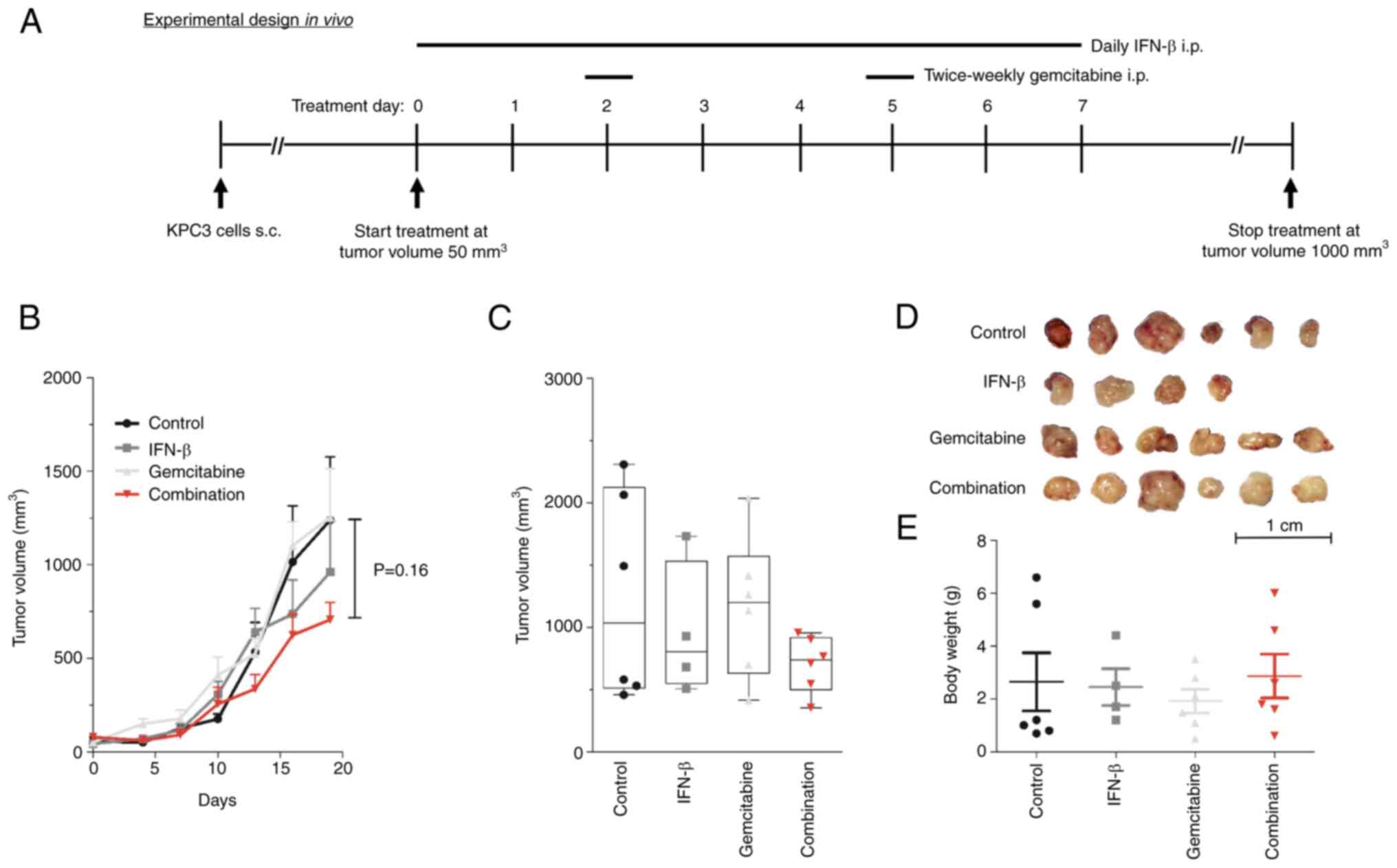
In vivo validation of IFN-β combined
with gemcitabine in immune-competent mice
To study the immunomodulatory antitumor effect of
IFN-β, low-passage KPC3 cells were subcutaneously injected in
C57BL/6 mice. Mice were randomized into four treatment arms: daily
H2O (control), daily 10,000 units IFN-β, twice weekly 50
mg/kg gemcitabine, or the combination of daily 10,000 units IFN-β
plus twice weekly 50 mg/kg gemcitabine (Fig. 3A). All mice were sacrificed at day
21, after which tumors were collected for analysis. None of the
treatment arms resulted in a statistically significant tumor growth
inhibition over time, although lowest tumor volumes were observed
in the combined treatment group, suggesting an additive effect
rather than a synergistic effect (Fig. 3B-D). After 21 days of treatment,
tumor volume was 707±92 mm3 in the combination
treated-mice compared with 1,239±338 mm3 in the vehicle
treated-mice (P=0.16) (Fig. 3C).
Tumor volumes in gemcitabine and IFN-β mono-treated mice were
1,162±232 and 962±271 mm3, respectively (both P>0.05
vs. untreated mice).
No significant weight loss was observed in any of
the treatment groups, indicating that all drugs were well tolerated
(Fig. 3E). Expression of
transporter- and metabolizing-coding genes of gemcitabine was low
in untreated KPC3 tumors and were not affected by any treatment
(data not shown).
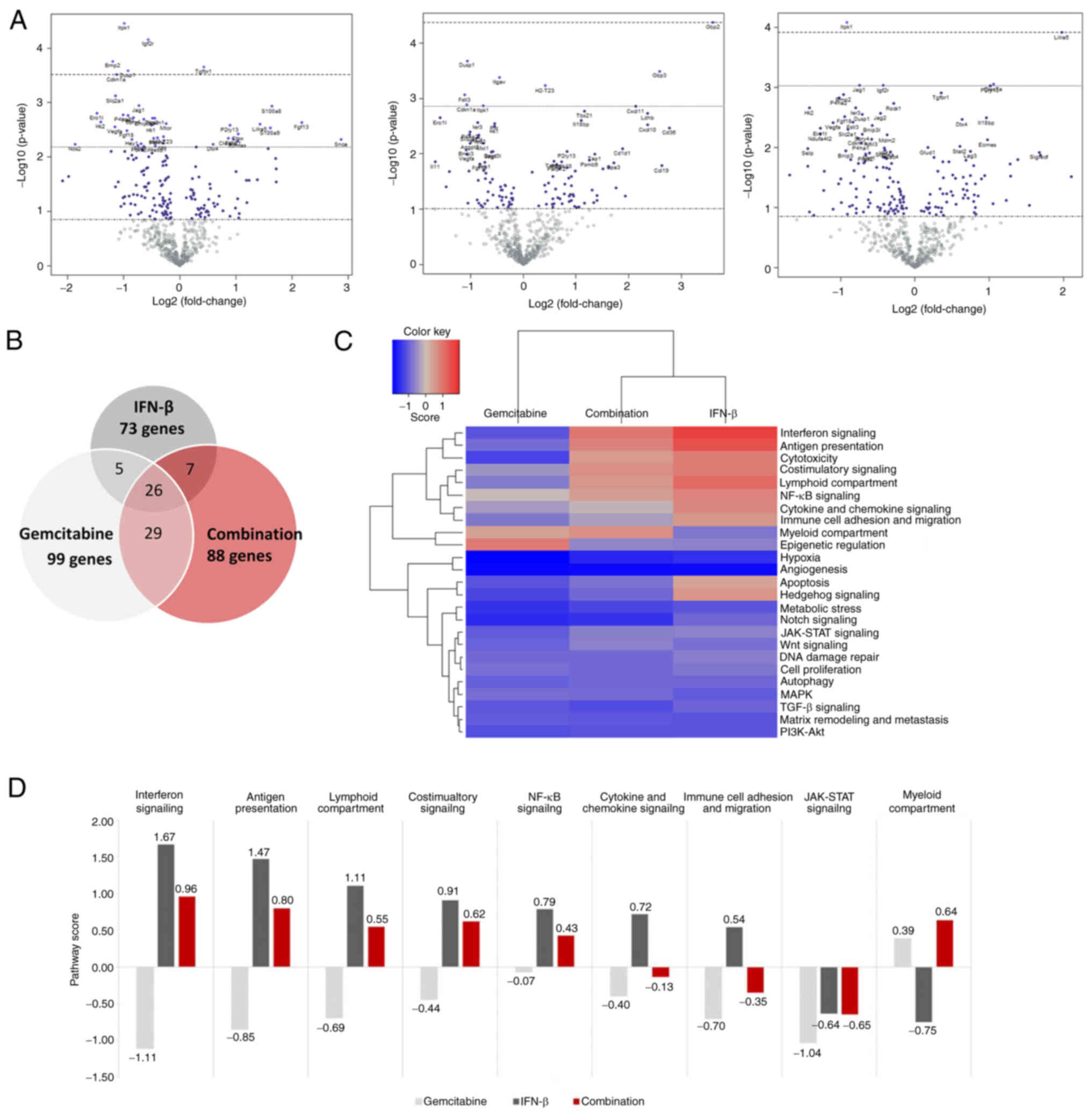
Effect of IFN-β on expression of
immune-related genes
To specifically address the immunomodulatory
capacity of IFN-β, a targeted gene expression array was performed
on tumor samples of treated and untreated mice. Differentially
expressed genes upon treatment compared with untreated mice are
revealed in Fig. 4A. After
adjusting for multiple testing, only a few genes were significantly
altered by the different treatment groups. Specifically,
gemcitabine downregulated expression of Itpk1, Igf2r, Bmp2,
Dusp1, and Cdkn1a, but increased Tgfbr1. IFN-β
only upregulated Gdp2, and in combination-treated tumors,
Itpk1 and Lilra5 were respectively down- and
upregulated (Fig. 4A, Table SIII).
Without adjusting P-values, an extensive number of
genes was found to be differentially expressed (Fig. 4B and Table SIII). In total, 35 genes were
upregulated by IFN-β, while 38 genes were downregulated. In
gemcitabine-treated tumors, 30 and 69 genes were respectively up-
and downregulated. A further 32 genes were upregulated by
combination therapy, whereas 56 genes were downregulated (Fig. 4B). Top ten up- and downregulated
genes are summarized in Table I.
Gemcitabine and combination therapy commonly affected several genes
(Snca, Lilra5, Fgfl3, Siglecf, Prkaa2, Selp, Hk2, Erol1,
Ndufa412 and Vegfa), while IFN-β alone induced a
different gene expression profile (Table I).
IFN-β monotherapy markedly upregulated expression of
several immune-related genes. As a consequence, 7 out of 9
immune-related pathways were upregulated in IFN-β-treated tumors
compared with untreated mice (range pathway score 1.67-0.54)
(Fig. 4C and D). By contrast,
gemcitabine induced a suppressive effect on these pathways (range
pathway score-1.11-0.39), but when co-treated with IFN-β, 6 out of
9 immune-related pathways were increased (range pathway score
0.96-0.43).
Expression of IFN signaling pathway regulators
(Ifi35, Irf1, Irf7 and Stat2) and four ISGs (Gbp2,
Gbp3, H2-d1 and Uba7) were induced by IFN-β alone.
Moreover, expression of several ISG chemokines (Ccl19,
Cxcl10 and Cxcl11) were increased in these tumors
(Table II). In
combination-treated tumors, the regulatory factors Ifi35 and
Trim21 were upregulated as well as two well-known
pro-apoptotic ISGs (Mx1 and Oas1a). By contrast,
gemcitabine downregulated IFN-related genes (H2-K1, H2-T23,
Ifi203, Igf2r and Irf2). Numerous genes involved in
antigen presentation were stimulated by IFN-β; for instance,
Tap1, Tap2, Psmb9 and Psmb10. This effect was less
pronounced in combination-treated tumors and also affected a
different set of genes (Trim21, Ctsw and Lag3).
Remarkably, the myeloid compartment pathway was downregulated by
IFN-β, but stimulated by gemcitabine and combination therapy
through upregulation of Clec7a, Lilra5, and
P2ry13.
Analysis reported no evident differences in
pro-tumor pathways, such as angiogenesis and metastasis, and were
equally inhibited in all treatment groups compared with untreated
tumors (Fig. 4C).
Discussion
Pancreatic cancer is a highly aggressive malignancy
with limited treatment outcomes. Despite promising recent advances
in systemic therapies, for instance mFOLFIRINOX, gemcitabine is
still being recommended for patients with a poor or declining
performance status (3).
Type I IFN-based adjuvant therapies have been widely
studied in pancreatic cancer, primarily due to their potential
synergistic effects when co-treated with gemcitabine (5,6,9,10).
Thereby, type I IFNs induce several direct antitumor effects,
including apoptosis and cell growth arrest as well as critical
immune stimulatory effects on various immune cells (7,8).
To date, studies have primarily focused on the
direct antitumor effects of the less effective IFN-α. The
immunomodulatory effects of IFN-α and -β are less described in
pancreatic cancer and primarily investigated in IFN-α. To the best
of our knowledge, this is the first study evaluating the antitumor
effects of the more promising IFN subtype IFN-β, alone and combined
with gemcitabine, on pancreatic cancer cells in immune competent
mice. The unique KPC3 pancreatic cancer cell line was used,
generated from the clinically relevant and non-immunogenic KPC
mouse model (15).
A trend towards smaller tumor volumes in
combination-treated mice was observed, suggesting an additive
effect rather than synergistic effect. Moreover, tumors displayed a
differential gene expression profile upon treatment. In particular,
IFN-β alone induced expression of numerous immune-related genes,
such as chemokines (Ccl19, Cxcl10 and Cxcl11) and
antigen processors (Tap1 and Tap2). Thereby, expression of
Irf7 was upregulated, which primarily regulates the
immunomodulatory capacities of IFN-β, as well as other IFN
signaling pathway regulators (e.g., Ifi35, Irf1 and
Stat2) (20,21). Notably, Irf1, which
promotes expression of ISGs, and Irf7 are key factors in the
positive feedback regulation of IFN-β production and potentiate ISG
expression as they directly target ISGs, even in the absence of IFN
signaling (22–24). It should be emphasized, however,
that only mRNA expression was evaluated. Further studies
should demonstrate that IFN-β treatment indeed results in effective
antitumor immune responses.
Controversially, gemcitabine induced an immune
suppressive effect, whereas addition of IFN-β to gemcitabine solely
induced a subset of immune-related genes, which was less pronounced
compared with IFN-β mono-treated tumors. Since gemcitabine promotes
the infiltration of particularly M2 macrophages in the tumor
microenvironment, it may have diminished the immune stimulating
effect of IFN-β when administered together (25).
Efficacy of several anticancer therapies, including
chemotherapies, partially depend on an intact type I IFN signaling
for the promotion of both direct (tumor cell inhibition) and
indirect effects (antitumor immune responses) (26). Thus, impaired IFN signaling may as
a consequence contribute to therapy resistance in patients with
cancer. KPC tumors respond poorly to gemcitabine therapy,
particularly orthotopic tumors, which is consistent with clinic
outcomes, as only 5–10% of gemcitabine-treated patients show an
objective radiographic response at the primary tumor site (27). Regarding in vivo research
in KPC mice, the most frequently used gemcitabine concentrations
are 50 and 100 mg/kg (16,28,29).
To avoid any potential toxicity, mice were treated with 50 mg/kg
gemcitabine. As expected, no significant tumor inhibition was
observed with gemcitabine alone. Moreover, type I IFN signaling was
diminished in gemcitabine-treated tumors as several IFN-related
genes were downregulated when compared with untreated tumors (for
instance, H2-K1, H2-T23, Ifi203, Igf2r and Irf2).
Meanwhile, combination therapy increased expression of IFN
regulator factors (for instance, Ifi35 and Trim21) as
well as two well-known pro-apoptotic ISGs (Mx1 and
Oas1a), but was still insufficient to significantly inhibit
tumor growth.
Over the years, numerous studies highlighted the
significant role of immune host-mediated mechanisms in the response
to type I IFNs, even in IFN-resistant tumor cells (30–33). However, most studies used
high-dose intratumoral IFN-β concentrations (33–35). Although intratumoral IFN-β
concentrations were not evaluated in the present study, the
relatively low concentration IFN-β (i.p. administered) as well as
the resistant character of KPC-3 cell to IFN-β therapy may have
resulted in insufficient intratumoral levels to induce significant
tumor growth inhibition. It is plausible that IFN-β will exert
stronger antitumor responses in IFN-β sensitive tumors. In fact, it
has been previously shown that IFN-β combined with gemcitabine
synergistically reduced tumor volumes in immune deficient mice,
bearing an IFN-β sensitive tumor, when compared with untreated mice
(9). Moreover, the relatively
short half-life time of IFN-β may limit sufficient circulating
IFN-β concentrations. Strategies to increase the half-life time of
IFN-β, such as IFN-based conjugates or PEGylated form of IFN-β,
have demonstrated promising results to achieve higher serum
concentration, requiring lower and less frequent doses compared
with the conventional IFNs (36,37).
While recombinant IFN therapies are generally given
as exogenous pharmaceuticals, it is suggested that the autocrine
and paracrine actions of endogenous type I IFNs on tumor growth
control (both the direct and indirect effects) are much stronger.
New IFN-related cancer treatment strategies, such as STING and
RIG-I agonists, have emerged as promising and effective strategies
to produce significant amounts of endogenous type I IFNs and are
currently being examined in (pre-) clinical studies in various
types of cancer, including pancreatic cancer (8). IFN-β gene therapy induced by viral
vectors provides another promising strategy to achieve high
intratumoral IFN-β levels and has demonstrated potent antitumor
efficacy in several pre-clinical cancer models, including
pancreatic cancer, with low toxicities (38–40). Discrepancies between treatment
outcomes largely depend on differences in IFN-β sensitivity,
highlighting the need for accurate biomarkers to predict IFN-β
treatment response. Expression of the active IFN-receptor subunits
(IFNAR-1 and IFNAR-2c) is required to form a high-binding affinity
site and to initiate signal transduction leading to the induction
of ISGs (11,41). However, despite expression of both
receptor subunits, KPC3 cells responded poorly to IFN-β, suggesting
other alterations involving the IFN downstream signaling pathway.
In fact, IFN dysregulation can occur through several mechanisms
such as loss or silencing of key signaling effector proteins and
components (JAKs, STATs and IRFs) or through upregulation of
negative regulators (SOCS1/3) (42,43). A careful evaluation of type I IFN
signaling may provide important insights into sensitivity to IFN-β
therapies and may possibly contribute to a more personalized
medicine approach in the future.
Previously emerged evidence has suggested that type
I IFNs may have dual roles in antitumor immunity, which may be even
harmful and cause further adaptive resistance to therapies,
including chemotherapy, radiotherapy and immune checkpoint blockade
(44–47). Two hypotheses have been proposed
for this controversial effect. First, continuous exposure of type I
IFNs may upregulate PD-L1 expression in tumor cells, which
subsequently promotes immune resistance trough interaction with
PD-1+ immune effector cells (48). Thereby, prolonged type I IFN
stimulation may induce an IFN-related DNA damage resistance
signature (IRDS) that indicates an unfavorable response to
DNA-damaging interventions such as chemotherapy and radiotherapy
(44). Previously, IRDS scoring
strategies have been used to identify patients with breast and lung
cancer and showed higher expression of specific ISGs in poor
responders to chemotherapy (49).
These paradoxical findings are not yet fully understood and may
also differ among types of cancer and depend on the subtype of type
I IFNs (50).
In conclusion, for the first time the
immunomodulatory potential of exogenous IFN-β combined with
gemcitabine was revealed in the immune competent KPC3 mouse
pancreatic model. The interplay between tumor cells and type I IFNs
is complex and still not fully understood. The dynamic role of type
I IFNs should be carefully considered to fully exploit its
therapeutic value as anticancer drug. Further studies should focus
on timing and duration of type I IFN administration as well as
prognostic markers for predicting effective antitumor
responses.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
The authors would like to thank the animal welfare
takers Kim van der Leest and Lisette Dinnessen (Erasmus Medical
Center, Rotterdam, The Netherlands) for their help during the mouse
experiments.
Funding
Funding: No funding was received.
Availability of data and materials
All data are available from the corresponding author
on reasonable request. All raw gene expression array data are
available on https://figshare.com/s/ec1cfa7d1d66f8528ec3.
Authors' contributions
AB, PMVK, CHJVE and LJH conceptualized the study.
AB, PMVK, DAMM and LJH performed formal analysis. CHJVE acquired
funding. AB, PMVK, JD and SVZ conducted investigation. AB, PMVK,
DAMM, CHJVE and LJH provided the methodology. PMVK, CHJVE and LJH
supervised the study. FD validated the data. AB, CHJVE and LJH
performed visualization. AB wrote the original draft. PMVK, DAMM,
FD, CHJVE and LJH wrote, reviewed and edited the manuscript. AB,
PMVK, CHJVE and LJH confirm the authenticity of all the raw data.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was controlled by the animal
welfare committee of the Erasmus University Medical Center
(Rotterdam, The Netherlands) and approved (approval no.
AVD101002017867) by the national central committee of animal
experiments, in accordance with the Dutch Act on Animal
Experimentation and European Union (EU) Directive 2010/63/EU.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rahib L, Smith BD, Aizenberg R, Rosenzweig
AB, Fleshman JM and Matrisian LM: Projecting cancer incidence and
deaths to 2030: The unexpected burden of thyroid, liver, and
pancreas cancers in the United States. Cancer Res. 74:2913–2921.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bengtsson A, Andersson R and Ansari D: The
actual 5-year survivors of pancreatic ductal adenocarcinoma based
on real-world data. Sci Rep. 10:164252020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Conroy T, Hammel P, Hebbar M, Ben
Abdelghani M, Wei AC, Raoul JL, Choné L, Francois E, Artru P, Biagi
JJ, et al: FOLFIRINOX or gemcitabine as adjuvant therapy for
pancreatic cancer. N Engl J Med. 379:2395–2406. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Amrutkar M and Gladhaug IP: Pancreatic
cancer chemoresistance to gemcitabine. Cancers (Basel). 9:1572017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Linehan DC, Tan MC, Strasberg SM, Drebin
JA, Hawkins WG, Picus J, Myerson RJ, Malyapa RS, Hull M, Trinkaus K
and Tan BR Jr: Adjuvant interferon-based chemoradiation followed by
gemcitabine for resected pancreatic adenocarcinoma: A
single-institution phase II study. Ann Surg. 248:145–151. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ohman KA, Liu J, Linehan DC, Tan MC, Tan
BR, Fields RC, Strasberg SM and Hawkins WG: Interferon-based
chemoradiation followed by gemcitabine for resected pancreatic
adenocarcinoma: Long-term follow-up. HPB (Oxford). 19:449–457.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
De Maeyer E and De Maeyer-Guignard J: Type
I interferons. Int Rev Immunol. 17:53–73. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Blaauboer A, Sideras K, van Eijck CHJ and
Hofland LJ: Type I interferons in pancreatic cancer and development
of new therapeutic approaches. Crit Rev Oncol Hematol.
159:1032042021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Blaauboer A, Booy S, van Koetsveld PM,
Karels B, Dogan F, van Zwienen S, van Eijck CHJ and Hofland LJ:
Interferon-beta enhances sensitivity to gemcitabine in pancreatic
cancer. BMC Cancer. 20:9132020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tomimaru Y, Eguchi H, Wada H, Tomokuni A,
Kobayashi S, Marubashi S, Takeda Y, Tanemura M, Umeshita K, Mori M,
et al: Synergistic antitumor effect of interferon-β with
gemcitabine in interferon-α-non-responsive pancreatic cancer cells.
Int J Oncol. 38:1237–1243. 2011.PubMed/NCBI
|
|
11
|
Domanski P and Colamonici OR: The type-I
interferon receptor. The long and short of it. Cytokine Growth
Factor Rev. 7:143–151. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wagner TC, Velichko S, Chesney SK, Biroc
S, Harde D, Vogel D and Croze E: Interferon receptor expression
regulates the antiproliferative effects of interferons on cancer
cells and solid tumors. Int J Cancer. 111:32–42. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Booy S, Hofland LJ, Waaijers AM, Croze E,
van Koetsveld PM, de Vogel L, Biermann K and van Eijck CH: Type I
interferon receptor expression in human pancreatic and
periampullary cancer tissue. Pancreas. 44:99–105. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Booy S, van Eijck CH, Dogan F, van
Koetsveld PM and Hofland LJ: Influence of type-I Interferon
receptor expression level on the response to type-I Interferons in
human pancreatic cancer cells. J Cell Mol Med. 18:492–502. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee JW, Komar CA, Bengsch F, Graham K and
Beatty GL: Genetically engineered mouse models of pancreatic
cancer: The KPC model
[LSL-Kras(G12D/+);LSL-Trp53(R172H/+);Pdx-1-Cre], its variants, and
their application in immuno-oncology drug discovery. Curr Protoc
Pharmacol. 73:14.39.1–14.39.20. 2016.PubMed/NCBI
|
|
16
|
Olive KP, Jacobetz MA, Davidson CJ,
Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA,
Caldwell ME, Allard D, et al: Inhibition of Hedgehog signaling
enhances delivery of chemotherapy in a mouse model of pancreatic
cancer. Science. 324:1457–1461. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Herrera-Martínez AD, Feelders RA, de
Herder WW, Castaño JP, Gálvez Moreno M, Dogan F, van Dungen R, van
Koetsveld P and Hofland LJ: Effects of Ketoconazole on
ACTH-producing and Non-ACTH-producing neuroendocrine tumor cells.
Horm Cancer. 10:107–119. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vandesompele J, De Preter K, Pattyn F,
Poppe B, Van Roy N, De Paepe A and Speleman F: Accurate
normalization of real-time quantitative RT-PCR data by geometric
averaging of multiple internal control genes. Genome Biol.
3:RESEARCH00342002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Blaauboer A, van Koetsveld PM, Mustafa
DAM, Dumas J, Dogan F, van Zwienen S, van Eijck CHJ and Hofland LJ:
The Class I HDAC inhibitor valproic acid strongly potentiates
gemcitabine efficacy in pancreatic cancer by immune system
activation. Biomedicines. 10:5172022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Iwanaszko M and Kimmel M: NF-κB and IRF
pathways: Cross-regulation on target genes promoter level. BMC
Genomics. 16:3072015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Solis M, Goubau D, Romieu-Mourez R, Genin
P, Civas A and Hiscott J: Distinct functions of IRF-3 and IRF-7 in
IFN-alpha gene regulation and control of anti-tumor activity in
primary macrophages. Biochem Pharmacol. 72:1469–1476. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Platanitis E and Decker T: Regulatory
Networks involving STATs, IRFs, and NFκB in inflammation. Front
Immunol. 9:25422018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Marié I, Durbin JE and Levy DE:
Differential viral induction of distinct interferon-alpha genes by
positive feedback through interferon regulatory factor-7. EMBO J.
17:6660–6669. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sato M, Hata N, Asagiri M, Nakaya T,
Taniguchi T and Tanaka N: Positive feedback regulation of type I
IFN genes by the IFN-inducible transcription factor IRF-7. FEBS
Lett. 441:106–110. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Deshmukh SK, Tyagi N, Khan MA, Srivastava
SK, Al-Ghadhban A, Dugger K, Carter JE, Singh S and Singh AP:
Gemcitabine treatment promotes immunosuppressive microenvironment
in pancreatic tumors by supporting the infiltration, growth, and
polarization of macrophages. Sci Rep. 8:120002018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zitvogel L, Galluzzi L, Kepp O, Smyth MJ
and Kroemer G: Type I interferons in anticancer immunity. Nat Rev
Immunol. 15:405–414. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tempero M, Plunkett W, Ruiz Van Haperen V,
Hainsworth J, Hochster H, Lenzi R and Abbruzzese J: Randomized
phase II comparison of dose-intense gemcitabine: Thirty-minute
infusion and fixed dose rate infusion in patients with pancreatic
adenocarcinoma. J Clin Oncol. 21:3402–3408. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Miller BW, Morton JP, Pinese M, Saturno G,
Jamieson NB, McGhee E, Timpson P, Leach J, McGarry L, Shanks E, et
al: Targeting the LOX/hypoxia axis reverses many of the features
that make pancreatic cancer deadly: Inhibition of LOX abrogates
metastasis and enhances drug efficacy. EMBO Mol Med. 7:1063–1076.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cook N, Frese KK, Bapiro TE, Jacobetz MA,
Gopinathan A, Miller JL, Rao SS, Demuth T, Howat WJ, Jodrell DI and
Tuveson DA: Gamma secretase inhibition promotes hypoxic necrosis in
mouse pancreatic ductal adenocarcinoma. J Exp Med. 209:437–444.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Belardelli F and Gresser I: The neglected
role of type I interferon in the T-cell response: Implications for
its clinical use. Immunol Today. 17:369–372. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Belardelli F and Ferrantini M: Cytokines
as a link between innate and adaptive antitumor immunity. Trends
Immunol. 23:201–208. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rizza P, Capone I, Moretti F, Proietti E
and Belardelli F: IFN-α as a vaccine adjuvant: Recent insights into
the mechanisms and perspectives for its clinical use. Expert Rev
Vaccines. 10:487–498. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Spaapen RM, Leung MY, Fuertes MB, Kline
JP, Zhang L, Zheng Y, Fu YX, Luo X, Cohen KS and Gajewski TF:
Therapeutic activity of high-dose intratumoral IFN-β requires
direct effect on the tumor vasculature. J Immunol. 193:4254–4260.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schiavoni G, Sistigu A, Valentini M,
Mattei F, Sestili P, Spadaro F, Sanchez M, Lorenzi S, D'Urso MT,
Belardelli F, et al: Cyclophosphamide synergizes with type I
interferons through systemic dendritic cell reactivation and
induction of immunogenic tumor apoptosis. Cancer Res. 71:768–778.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang X, Zhang X, Fu ML, Weichselbaum RR,
Gajewski TF, Guo Y and Fu YX: Targeting the tumor microenvironment
with interferon-β bridges innate and adaptive immune responses.
Cancer Cell. 25:37–48. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Borden EC: Interferons α and β in cancer:
Therapeutic opportunities from new insights. Nat Rev Drug Discov.
18:219–234. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pepinsky RB, LePage DJ, Gill A,
Chakraborty A, Vaidyanathan S, Green M, Baker DP, Whalley E,
Hochman PS and Martin P: Improved pharmacokinetic properties of a
polyethylene glycol-modified form of interferon-beta-1a with
preserved in vitro bioactivity. J Pharmacol Exp Ther.
297:1059–1066. 2001.PubMed/NCBI
|
|
38
|
Ravet E, Lulka H, Gross F, Casteilla L,
Buscail L and Cordelier P: Using lentiviral vectors for efficient
pancreatic cancer gene therapy. Cancer Gene Ther. 17:315–324. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Endou M, Mizuno M, Nagata T, Tsukada K,
Nakahara N, Tsuno T, Osawa H, Kuno T, Fujita M, Hatano M and
Yoshida J: Growth inhibition of human pancreatic cancer cells by
human interferon-beta gene combined with gemcitabine. Int J Mol
Med. 15:277–283. 2005.PubMed/NCBI
|
|
40
|
Ohashi M, Yoshida K, Kushida M, Miura Y,
Ohnami S, Ikarashi Y, Kitade Y, Yoshida T and Aoki K:
Adenovirus-mediated interferon alpha gene transfer induces regional
direct cytotoxicity and possible systemic immunity against
pancreatic cancer. Br J Cancer. 93:441–449. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Deonarain R, Chan DC, Platanias LC and
Fish EN: Interferon-alpha/beta-receptor interactions: A complex
story unfolding. Curr Pharm Des. 8:2131–2137. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Budhwani M, Mazzieri R and Dolcetti R:
Plasticity of Type I Interferon-mediated responses in cancer
therapy: From Anti-tumor immunity to resistance. Front Oncol.
8:3222018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Patel SJ, Sanjana NE, Kishton RJ,
Eidizadeh A, Vodnala SK, Cam M, Gartner JJ, Jia L, Steinberg SM,
Yamamoto TN, et al: Identification of essential genes for cancer
immunotherapy. Nature. 548:537–542. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Weichselbaum RR, Ishwaran H, Yoon T,
Nuyten DS, Baker SW, Khodarev N, Su AW, Shaikh AY, Roach P, Kreike
B, et al: An interferon-related gene signature for DNA damage
resistance is a predictive marker for chemotherapy and radiation
for breast cancer. Proc Natl Acad Sci USA. 105:18490–18495. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Erdal E, Haider S, Rehwinkel J, Harris AL
and McHugh PJ: A prosurvival DNA damage-induced cytoplasmic
interferon response is mediated by end resection factors and is
limited by Trex1. Genes Dev. 31:353–369. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Post AEM, Smid M, Nagelkerke A, Martens
JWM, Bussink J, Sweep F and Span PN: Interferon-stimulated genes
are involved in Cross-resistance to radiotherapy in
tamoxifen-resistant breast cancer. Clin Cancer Res. 24:3397–3408.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Benci JL, Xu B, Qiu Y, Wu TJ, Dada H,
Twyman-Saint Victor C, Cucolo L, Lee DSM, Pauken KE, Huang AC, et
al: Tumor interferon signaling regulates a multigenic resistance
program to immune checkpoint blockade. Cell. 167:1540–1554.e12.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Terawaki S, Chikuma S, Shibayama S,
Hayashi T, Yoshida T, Okazaki T and Honjo T: IFN-α directly
promotes programmed cell death-1 transcription and limits the
duration of T cell-mediated immunity. J Immunol. 186:2772–2779.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Pitroda SP, Stack ME, Liu GF, Song SS,
Chen L, Liang H, Parekh AD, Huang X, Roach P, Posner MC, et al:
JAK2 inhibitor SAR302503 abrogates PD-L1 expression and targets
therapy-resistant non-small cell lung cancers. Mol Cancer Ther.
17:732–739. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Doherty MR, Cheon H, Junk DJ, Vinayak S,
Varadan V, Telli ML, Ford JM, Stark GR and Jackson MW:
Interferon-beta represses cancer stem cell properties in
triple-negative breast cancer. Proc Natl Acad Sci USA.
114:13792–13797. 2017. View Article : Google Scholar : PubMed/NCBI
|