1. Introduction
Glioma is the most prevalent type of primary brain
tumor, accounting for ~75% of malignant central nervous system
(CNS) tumors in adults (1). The
treatments recommended at present for this aggressive neoplasm
include maximal safe surgical resection followed by radiotherapy
paired with temozolomide chemotherapy. The addition of tumor
treatments from other fields to conventional therapies has also led
to improvements in the prognosis of patients (2). However, the intricate biological
properties of glioma have restricted the effectiveness of the
multiple therapeutic modalities and patients continue to exhibit
eventual tumor relapse, with the disease undergoing a dismal
course. The median survival rate of glioblastoma multiforme (GBM),
the highest grade of glioma (World Health Organization grade IV),
which constitutes >50% of the entity, remains at ~16 months,
with near-universal lethality (3,4).
Despite the huge efforts that have been made in
investigating the intrinsic nature of aggressive tumor behaviors,
the processes of rapid proliferation, infiltrative growth and
resistance to therapeutics of glioma remain poorly understood.
However, emerging evidence has revealed that the tumor
microenvironment (TME), where gliomas interact with non-glioma
brain cells, provides a substantial basis for glioma progression
(5). The complex network consists
of multiple non-glioma cell types, including glial cells, immune
cells, vascular cells and neurons. These distinct sets of cells may
be subverted by gliomas through various mechanisms to form a
microenvironment that is conducive to tumor development (6). Preliminary studies have pointed to
the possibility that interfering with oncogenic interactions
between glioma and non-malignant cells may suppress disease
progression (7-9).
Given that glioma infiltrates extensively within the
brain and spinal cord and rarely metastasizes outside the CNS,
neurons as a crucial component of the glioma milieu potentially
confer important microenvironmental dependencies to the
pathogenesis of the tumor (10).
The crosstalk between glioma and peritumor neurons may be mediated
in different ways, which involve electrochemical synapses, secreted
factors, tumor microtubes (TMs) and extracellular vesicles
(11). The bidirectional
interactions provide plentiful scope for malignant cells to
develop; henceforth, the tumor-infiltrated brain becomes
physiologically disorganized, a process that eventually facilitates
glioma growth.
The present review provides an overview of various
aspects of the communication between glioma and neurons, and
readers will develop further insight into the regulatory mechanisms
of glioma-neuronal interactions. Furthermore, potential targets
involved in the network are highlighted and novel concepts and
technologies are described that may be integrated for the design of
novel therapeutic strategies.
2. Neuronal regulation in glioma
progression
Neuron-glioma synapses (NGSs), providing a
substantial basis for communication between the two entities, are
mostly found in the glioma infiltration zone (12). The synaptic contacts may be
categorized into three morphological types that have diverse
functional properties: i) A single contact on a glioma cell; ii) a
multi-synaptic contact to both the neuron and the glioma cell; and
iii) a peri-synaptic contact between two neurons accompanied by a
glioma cell contact to the synaptic cleft (12). These electrochemical synapses may
be regulated by neurotransmitters (Fig. 1), ion channels, TMs and gap
junctions, which exert a marked influence on glioma (13).
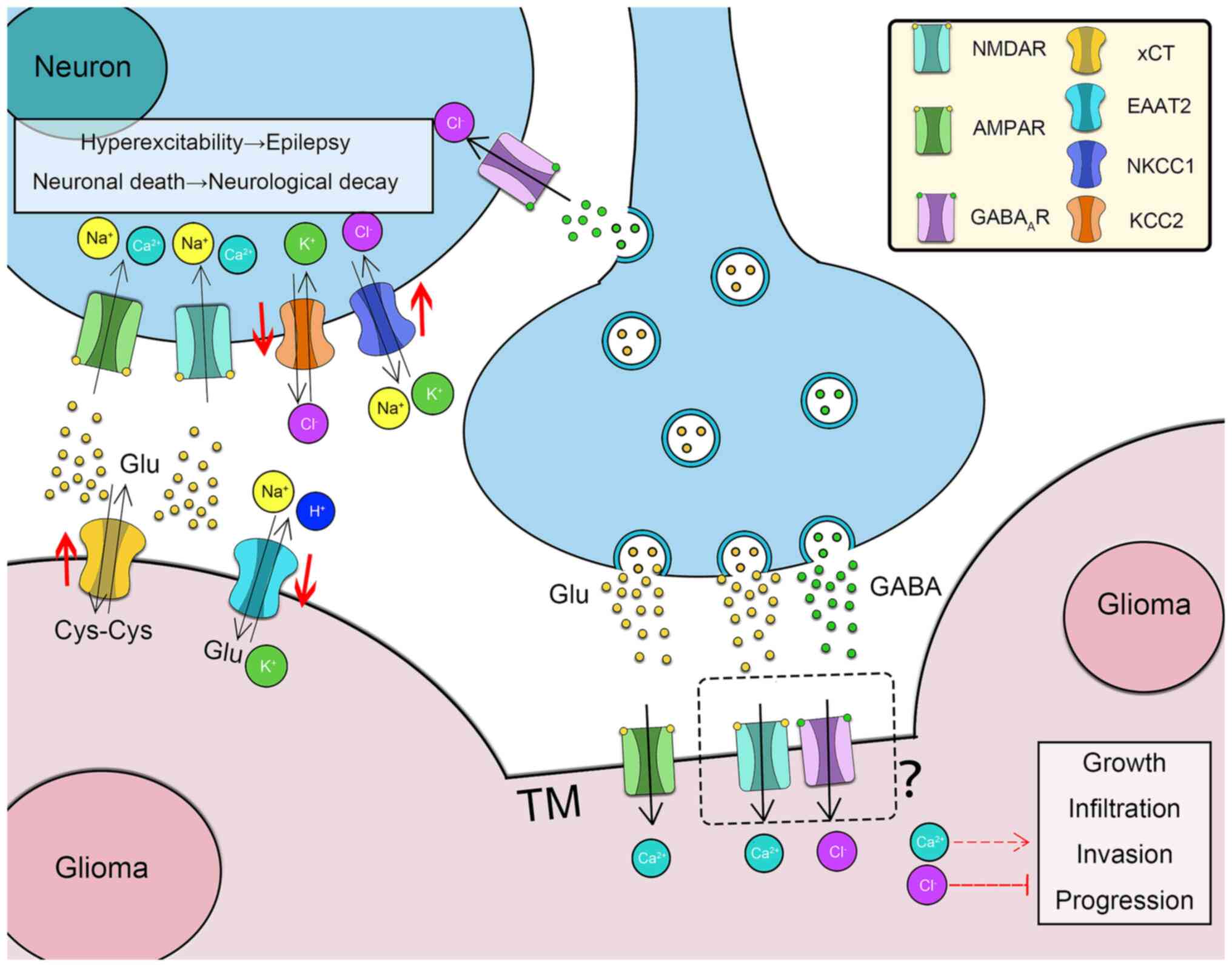 | Figure 1Roles of neurotransmitters in
neuron-glioma interactions. The enrichment of Glu in the glioma
microenvironment is regulated via xCT overexpression and EAAT2
inhibition. Glu activates adjacent neurons by binding to AMPARs and
NMDARs. The high concentration of Glu leads to hyperexcitability
and cell death of adjacent neurons, resulting in neurological decay
and tumor-associated epilepsy. The expression of NKCC1 and KCC2 in
para-tumoral neurons is downregulated and upregulated,
respectively. The intracellular concentration of Cl− in
neurons is consequently high. GABA may depolarize the para-tumoral
neurons and cause epilepsy. Neurons form synapses with the TMs of
glioblastoma multiforme. Upon binding to AMPARs/NMDARs, Glu
promotes glioma progression via influx of Ca2+, whereas
GABA inhibits glioma development via influx of Cl-. xCT,
cystine/glutamate antiporter; EAAT2, excitatory amino acid
transporter 2; Glu, glutamate; Cys, cystine; AMPAR,
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor;
NMDAR, N-methyl-D-aspartate receptor; GABAAR, GABAA receptor;
NKCC1, Na+-K+-Cl- cotransporter 1; KCC2,
K+-Cl- cotransporter 2; GABA, γ-aminobutyric acid; TM,
tumor microtube. |
Neurotransmitters and receptors
γ-aminobutyric acid (GABA) is an inhibitory
neurotransmitter in the adult brain that induces GABAergic
signaling mediated via Cl− influx through activating
GABA receptors (GABARs), predominantly GABAARs (14). A previous study reported on the
aberrant expression of chloride extruder
K+-Cl− cotransporter 2 (KCC2) and
Na+-K+-Cl− cotransporter 1
(NKCC1), which was discovered in the peritumoral area following
GABAergic signaling (15). The
dysregulation of KCC2 and NKCC1 in the adjacent neurons led to an
increasing intracellular Cl− concentration, which
resulted in Cl− efflux. However, it was indicated that a
high concentration of glutamate in the TME was able to counteract
the inhibitory role of GABA via downregulating KCC2 levels
(16). Another study reported that
glioma cells expressed GABAARs when co-cultured with
neurons in vitro, and activation of GABAARs
resulted in inhibition of glioma-cell proliferation (11). Activation of GABAAR with
the exogenous agonist muscimol, however, failed to elicit further
inhibition of glioma-cell proliferation. That study attributed the
phenomenon to the fact that GABA seemingly acts on glioma stem
cells (GSCs) (17). The
overexpression of diazepam-binding inhibitor (DBI) in glioma was
reported to inhibit GABAARs, thereby promoting
gliomagenesis. Despite a dearth of GABAARs,
overexpressed DBI facilitated GBM growth through a lipid metabolism
pathway mediated by GABA (18).
Therefore, employing agonists or antagonists to disrupt the
Cl− current in glioma cells may offer a strategy to
inhibit the progress of tumor development.
Glutamate is the predominant excitatory
neurotransmitter, which is synthesized and secreted by neurons
(19). The association between
glutamate receptors and glutamate-induced Ca2+ signaling
was demonstrated through the upregulation of the Ca2+
permeable-α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid
receptor (AMPAR) in glioma, which increased the rate of
Ca2+ influx. The elevated Ca2+ concentration
serves to activate the pro-oncogenic Akt and ERK/MAPK signaling
pathways (20). It was reported
that glutamatergic synapses were formed between neurons and glioma
cells (termed neurogliomal synapses), and AMPARs are expressed on
post-synapses. AMPAR-mediated neuronal activity has been indicated
to induce tumor invasion and growth (12). AMPARs comprise four different types
of subunits: GluA1, GluA2, GluA3 and GluA4 (21). Blockade of GluA1 and GluA2
overexpression reduced the proliferation rate of glioma cells by
inducing apoptosis and decreasing cell invasiveness and migration
(22). A study has demonstrated
the role of auxiliary subunits (of the transmembrane AMPAR
regulatory protein, cystine-knot AMPAR modulating protein and
cornichon homolog families) of AMPARs in promoting gliomagenesis
(23).
Activation of N-methyl-D-aspartate receptors
(NMDARs) in GBM by extracellular glutamate may potentially boost
tumor expansion in vivo (24). However, the expression of NMDARs on
neurogliomal synapses and the further impacts on glioma development
remain elusive. Previously published studies, however, have
confirmed the inhibitory effects on glioma growth via NMDARs
(25,26). Metabotropic glutamate receptors
(mGluRs) consist of three groups of G-proteins: Groups I (mGluR1
and -5), II (mGluR2 and -3) and III (mGluR4 and -6-8). The group
III receptor antagonists were observed to exert their anti-tumor
effects in various types of tumor, including hepatoma, melanoma and
non-small-cell lung cancer (26).
Riluzole has been reported to promote apoptosis in glioma cells via
antagonizing mGlu3, which results in the suppression of ERK
activation (27). At present,
evidence is lacking; however, in terms of whether mGluRs are
expressed on neurogliomal synapses, although mGluRs are closely
associated with glioma progression, may suggest the involvement of
other neuron-glioma interactions.
Serotonin and dopamine are predominant
neurotransmitters in the CNS. Of note, a previous study suggested a
correlation between depression and gliomagenesis, probably due to
the shared molecular pathways and gene networks (28). Dopamine receptor D4 is a receptor
that is involved in autophagy and apoptosis, which interferes with
glioma-cell proliferation (29).
Serotonin exerts an impact on the glutamatergic system through
influencing AMPARs and NMDARs (30). Selective serotonin reuptake
inhibitor (SSRI) has been indicated to impair glioma cell growth
via inducing autophagy and apoptosis (31), although such effects were also
observed in patients with a history of long-term drug therapy using
tricyclic antidepressants (32).
In those patients, the morbidity rate of glioma was reduced. As
serotonin receptors are also expressed in glioma cells, it is
tempting to hypothesize that SSRI may also exert an influence
through impacting neuron-glioma synapses (33).
Ion channels
As mentioned above, ion currents are involved in
neuron-glioma crosstalk. Ion channels exist in the CNS, primarily
Na+, K+, Ca2+ and Cl−
channels, which regulate cell migration, invasion, proliferation
and apoptosis (34). Specifically,
the K+ channel members include voltage-gated
K+ channels (VGKC or Kv), calcium-activated
K+ channels (KCa) and ATP-sensitive K+
channels (KATP). Voltage-gated calcium channels (VGCCs) are the
most important members of the Ca2+ channel group,
consisting of the L-, T-, N- and P/Q-types.
The migration of neural progenitor cells and stem
cells is influenced by the electric current generated by the local
field potential (35). It was
recently indicated that the electric current directs the migration
and invasion of glioma cells (36). The electrotaxis processes of GBM
cell lines are regulated by voltage-gated channels. For instance,
in T98G cells, electrotaxis was demonstrated to be mediated via
R-type VGCCs, whereas in U251 cells, it was mediated via P/Q-type
VGCCs. By contrast, in both T98G and U251 cells, electrotaxis was
regulated via A-type VGKCs and acid-sensing ion channels (ASICs)
(37). Inhibition of ASICs by
benzamil significantly impaired the directedness of the
electrotaxis of U251 and T98G cells (37). Of note, P/Q- and N-type VGCC
inhibitors derived from spider venom were indicated to markedly
inhibit tumor growth in vivo (38). Collectively, these data suggest
that electric fields exert important effects on glioma-cell
invasion and migration. Therefore, targeting ion channels may be a
potential therapeutic strategy for glioma.
Ion channels are employed by cells to regulate their
volumes for the purpose of cell migration. Glioma cells accumulate
Cl− intracellularly via NKCC1, whereas Cl−
channel protein 3 (CLC3) serves to regulate the Cl−
efflux (39). To balance the
Cl− efflux, glioma cells express KCa1.1 and KCa3.1
channels, which regulate K+ influx via Ca2+
activation (39). Chlorotoxin, a
small neurotoxin of 36 amino acids, causes the internalization of
CLC family members, thereby impeding glioma-cell invasion (40). A previous study suggested that
specific inhibition of KCa3.1 by TRAM-34 leads to a reduction in
the migration and infiltration rates of U87, GL261 and U251 GBM
cells (41). Furthermore, blockade
of KCa1.1 inhibited the radiation- and hypoxia-induced migration of
GBM cells (42).
Apart from cell migration and invasion, ion channels
are also involved in regulating the cell cycle, proliferation and
apoptosis (39). A previous study
revealed that glioma-cell proliferation was inhibited following the
blockade of Kv channels by 4-aminopyridine (43). Inhibition of Kv1.1 by KAaH2, a
homologous Kv1 blocker from scorpion venom, impaired U87-cell
proliferation (44).
Ca2+-activated K+ channels (BK channels) have
also been indicated to be involved in regulating proliferation
(45). However, the association
between neuronal activity and ion channels in terms of how they
influence glioma proliferation remains inadequately understood.
Hypothetically speaking, neuronal hyperexcitability may promote
glioma proliferation, since the activation of ion channels by
electric signals is essential for downstream pathway signaling.
Considering all of this evidence, ion channels have been indicated
to act as an important bridge between neuronal activity and glioma
progression, although their role still requires to be confirmed and
fully elucidated in further studies.
TMs and gap junctions
TMs are a type of tumor protrusion consisting of
F-actin and microtubules formed by gliomas, which permit
cell-to-cell material transportation (46). Targeted patch-clamp recordings have
suggested that a spontaneous excitatory post-synaptic potential
arises in glioma cells cocultured with neurons, which induces
further Ca2+ influx (12). Importantly, TM connectivity is
responsible for the distribution of Ca2+ ions, which
serve as crucial messengers of glioma activity throughout the
glioma syncytium (12).
Furthermore, NGSs significantly promote glioma invasion through
Ca2+ signaling, and potentiate glioma proliferation
through AMPAR activation (12). In
a Drosophila glioma model, Frizzled 1 receptors were
indicated to be highly expressed within TMs, where glioma cells
were able to vampirize Wingless-related integration site (WNT)
ligands from neurons (47) (the
process 'vampirization' is defined in that article). The depletion
of WNT from neurons led to glioma proliferation and expansion
through the JNK/matrix metalloproteinase (MMP) pathway, conversely
resulting in a reduction of neuronal synapse activity (47). TM-associated gap junctions have
also been indicated to amplify the extracellular K+
current and to promote tumor proliferation (48), suggesting that the TM network
potentiates the pro-tumoral effects of NGSs.
In cell-cell communication, gap junctions, which
consist of connexin (Cx) proteins, form conductive pores in the
plasma membranes between adjacent cells that allow the
transportation of cellular material (49). Cx43-based gap junctions and
neuronal growth-associated protein-43 (GAP-43) have been indicated
to be essential for TM formation (50). Depletion of GAP-43 and Cx43 led to
both an impairment of the ability of TMs to form connections and
tumor-cell volume reduction in vivo (51). In a subsequent study, suppressing
Cx43 with a peptide, TAT-Cx43266-283, inhibited glioma-cell
invasiveness, reduced the stemness properties of GSCs and prolonged
the survival rate of mice bearing GSC-derived gliomas (52). IMM, a small molecule that is able
to induce F-actin polymerization, was demonstrated to hinder TM
formation and to prevent glioma invasiveness (53). It is noteworthy that the role of
Cx43, a tumor suppressor, appears to be somewhat paradoxical in
terms of the overall picture (54). According to a previously published
meta-analysis, Cx43 expression was reported to improve the overall
survival rate in patients with glioma (55). Hence, the function of Cx43 in
glioma should be interpreted with caution in further studies. TM
formation was also indicated to be dependent on the EGFR/PI3K
signaling pathway, which induces actin cytoskeleton remodeling and
initiates TM expansion. The microtubule-targeted agent BAL101553
has entered into clinical trials (Table I). Therefore, targeting
TM-associated gap junctions or other pathways may prove to be
useful in terms of treating glioma.
 | Table IPivotal clinical trials concerning
the treatment of gliomas with neural influence. |
Table I
Pivotal clinical trials concerning
the treatment of gliomas with neural influence.
| ClinicalTrials gov
Identifier | Country | Conditions | Phase | Estimated
enrollment |
Allocation/masking | Intervention
model | Intervention | Status/results |
|---|
| NCT00064363 | US | Brain and central
nervous system tumors | II | 30 | Open label | - | Drug:
Talampanel | Talampanel was
well-tolerated as single agent. The PFS6 was 4.6 and 0% for the
initial 22 GBM patients and 8 AG patients, respectively The median
PFS was 5.9 weeks for GBM and 8.9 weeks for AG patients. The median
OS was 13 weeks for GBM patients and 14 months for AG patients |
| NCT00267592 | US | GBM | II | 72 | N/A/Open label | Single group
assignment | Drug: Talampanel
Radiation: RT 5 days a week+ Drug: TMZ 75 mg Drug: Adjuvant TMZ 200
mg | Talampanel was
well-tolerated in combination with RT plus TMZ The mOS was 18.3
months |
| NCT03295396 | US | Glioma | II | 95 | Non-
randomized/open label | Single group
assignment | Drug: ONC201 | ONC201 was safe The
best response to RANO-HGG or RANO-LGG was 30% Duration of response
to RANO-HGG was median 52.7 weeks |
| NCT00040573 | US | Glioma brain
neoplasm | I | 18 | Non-
randomized | Single group
assignment | Drug:
131I-TM-601 | A single dose of 10
mCi I131-TM-601 was well tolerated for 0.25 to 1.0 mg
TM-601. Median survival time was 25.7 weeks for patients in panel 1
(0.25-mg dose), 77.6 weeks in panel 2 (0.50-mg dose), 23.6 weeks in
panel 3 (1.00-mg dose) and 27.0 weeks in all three dosing
groups |
| NCT01753713 | US | Adult giant cell
glioblastoma, adult glioblastoma, adult gliosarcoma, recurrent
adult brain tumor | II | 33 | Non-
randomized/Open label | Parallel
assignment | Drug: Dovitinib,
Other: Laboratory biomarker analysis | PFS6 in Arm 1 was
12±6%; Time to progression in Arm 2 was 0.7-1.8 months |
| NCT04295759 | US | GBM anaplastic
astrocytoma, anaplastic oligodendroglioma, DIPG, high-grade
astrocytoma, NOS, CNS primary tumor, NOS (malignant glioma) | I | 28 | N/A/Open label | Single group
assignment | Drug: INCB7839 | Recruiting |
| NCT03250299 | US | Gliobla- stoma,
MGMT- unmethylated glioblastoma | I | 30 | Non-
randomized/Open label | Sequential
assignment | Drug: Microtubule-
targeted agent BAL101553; Radiation: Radiation therapy; Other:
Laboratory biomarker analysis; Other: Pharmacological study | Recruiting |
| NCT02880371 | US | Advanced solid
tumors | II | 19 | Non-
randomized/Open label | Single group
assignment | drug: ARRY-382;
Drug: Pembrolizumab ARRY-382 plus | The recommended
phase 2 dose of ARRY-382 in combination with Pembrolizumab was 300
mg QD pembrolizumab were safe. Stable disease was the best response
observed for patients in the PD-1/PD- L1 IR, prOVCA and PDA
cohorts: 8 (42.1%), 4 (36.4%), and 5 (18.5%) patients,
respectively. Median PFS (95% CI) was 1.4, 1.6 and 2.1 months in
the PDA, PD1/PD-L1 IR and prOVCA cohorts, respectively |
| NCT01349036 | US | Recurrent
glioblastoma | II | 38 | Non-
randomized/Open label | Single group
assignment | Drug: PLX3397 | PLX3397 was
well-tolerated. No significant improvement in PFS was observed |
Neuronal secretion
The activity of cortical projection neurons promotes
glioma growth and progression through neuronal secretion.
Neurotrophins (NTs), neuroligins (NLGNs), neurotransmitters and
mitogens have all been demonstrated to have participatory roles in
neuron-glioma communication (56).
In addition to direct (or synaptic) neuronal secretion, glioma
cells have also been indicated to be regulated by indirect (or
non-synaptic) paracrine and autocrine signaling pathways (57).
NTs
NTs are growth factors that are expressed in the
nervous system and have an impact on neural growth, survival and
biological function. The four types of NTs comprise nerve growth
factor (NGF), brain-derived neurotrophic factor (BDNF),
neurotrophin 3 (NT3) and neurotrophin 4/5 (NT4/5). NGF has a
preferential affinity for tyrosine kinase receptor (Trk)A, whereas
BDNF and NT4 have a preference towards TrkB and NT3 has an affinity
for TrkC (58).
In the CNS, NGF is secreted predominantly in the
cortex, hippocampus and pituitary gland (59). The specific NGF receptors TrkA and
p75NTR have been indicated to be expressed in gliomas
(60). The effects of NGF on
glioma cells, however, appear to be somewhat paradoxical. Ectopic
treatment of the pediatric low-grade glioma cell (PLGG) line Res259
with NGF led to inhibition of cell growth, whereas NGF treatment
promoted the growth of another PLGG cell line, Res186 (61). A different study suggested that NGF
stimulated U87-cell proliferation through the NOTCH1 receptor
signaling pathway (62). The
underlying impact of NGF on glioma development, however, requires
to be further clarified.
Barreda Tomás et al (63) observed that both progenitor BDNF
(pro-BDNF) and BDNF were expressed in glutamatergic and GABAergic
neurons of the mouse cortex. Another study demonstrated through an
immunocytochemical analysis that BDNF and its receptor, TrkB, are
extrasynaptic, whereas BDNF is preferentially located at the
glutamatergic synapse (64). It
was suggested that BDNF may exert its effects on glioma through
NGSs and the paracrine signaling pathway. Targeting BDNF by
specific microRNAs inhibited glioma invasion, migration and
proliferation (65), indicating
the promoting role of BDNF in glioma malignancy. BDNF also induced
the synthesis of GluA1 subunits and the synaptic incorporation of
CP-AMPARs in primary hippocampal neurons (66). As mentioned above, a high
concentration of glutamate in the glioma microenvironment confers
neuron hyperexcitability, leading to epilepsy (67). Although AMPARs have been indicated
to participate in glutamate-induced epilepsy, BDNF may promote
glioma-associated epilepsy due to its capability of regulating the
synthesis and incorporation of AMPARs. In addition, glutamate was
indicated to stimulate the production of BDNF in neurons (68), which may, in turn, facilitate
glioma progression. Of note, pro-BDNF has been observed to exert an
inhibitory effect on glioma. A previous study reported that the
ratio of pro-BDNF to BDNF was decreased in high-grade glioma,
whereas the expression of pro-BDNF was increased. Pro-BDNF was also
found to inhibit the growth and invasion of glioma cells via
p75NTR (69).
NT-3/TrkC signaling has also been indicated to be
necessary for the induction of glioma-cell death through the
inhibition of autophagy under hypoxic conditions (70), suggesting a life-supporting role of
NT-3/TrkC signaling in GBM in the state of hypoxia. The effect of
NT-4/5 on glioma cells, however, remains elusive, and this requires
further investigation.
NLGN3
NLGN3 exerts an important role in synaptic function
and maturation by binding presynaptic neurexin (71). Venkatesh et al (71) suggested that spontaneous neuronal
activity in the cortex induces NLGN3 secretion and promotes
glioma-cell proliferation (71).
Potentiated neuronal activity also led to an increase in NLGN3
cleavage mediated by MMPs, which may contribute towards mGluR
activation (72). In the clinic,
NLGN3 expression was observed to be positively correlated with
oscillatory brain activity and this was negatively associated with
progression-free survival of patients with glioma (73). NLGN3 not only caused an increase in
its own expression, but it also led to an upregulation of the
sheddase A disintegrin and metalloproteinase domain-containing
protein 10 (ADAM10) in glioma cells (74). Abundant evidence has indicated the
pivotal role of NLGN3 in supporting glioma malignancy. In an
NLGN3-deficient brain model, human glioma xenografts were indicated
to both survive longer and not exhibit any clear signs of
glioma-cell infiltration (75),
indicating that glioma malignancy may depend on the presence of
NLGN3.
NLGN3 activates several oncogenic signaling
pathways, including the PI3K/mTOR pathway, and induces
transcriptional changes, including the upregulation of numerous
synapse-associated genes (76).
NLGN3 has also been indicated to induce the expression of Tweety
homologue-1, which has a role in the construction of the glioma
microtube network in high-grade glioma (77). In addition, protein kinase C
(PKC)-induced NLGN3 cleavage is dependent on MMPs, particularly
MMP3 and MMP9. MMP3/9 blockade led to inhibition of PKC-induced
NLGN3 (78). Collectively, these
results suggested that MMP3/9 inhibition may be effective in terms
of glioma suppression. Patients with GBM were also indicated to
harbor high levels of NLGN3 in the deep regions of the brain, which
may partly explain the high recurrence rate of GBM (73).
NLGN3 is cleaved from both cortical neurons and
oligodendrocyte precursor cells via ADAM10, the release of which
from neurons is dependent on neuronal activity (75). ADAM10 inhibitors have been reported
to prevent the release of NLGN3 and to block glioma growth in
vivo (75). These data
indicate that targeting NLGN3 for gliomas may be a putative
therapeutic strategy. Generally speaking, preventing the release of
NLGN3 into the glioma microenvironment through the use of ADAM10
inhibitors, blocking certain targets in oncogenic signaling
pathways and silencing NLGN3-associated genes are three
transformative methods for the treatment of glioma (Fig. 2).
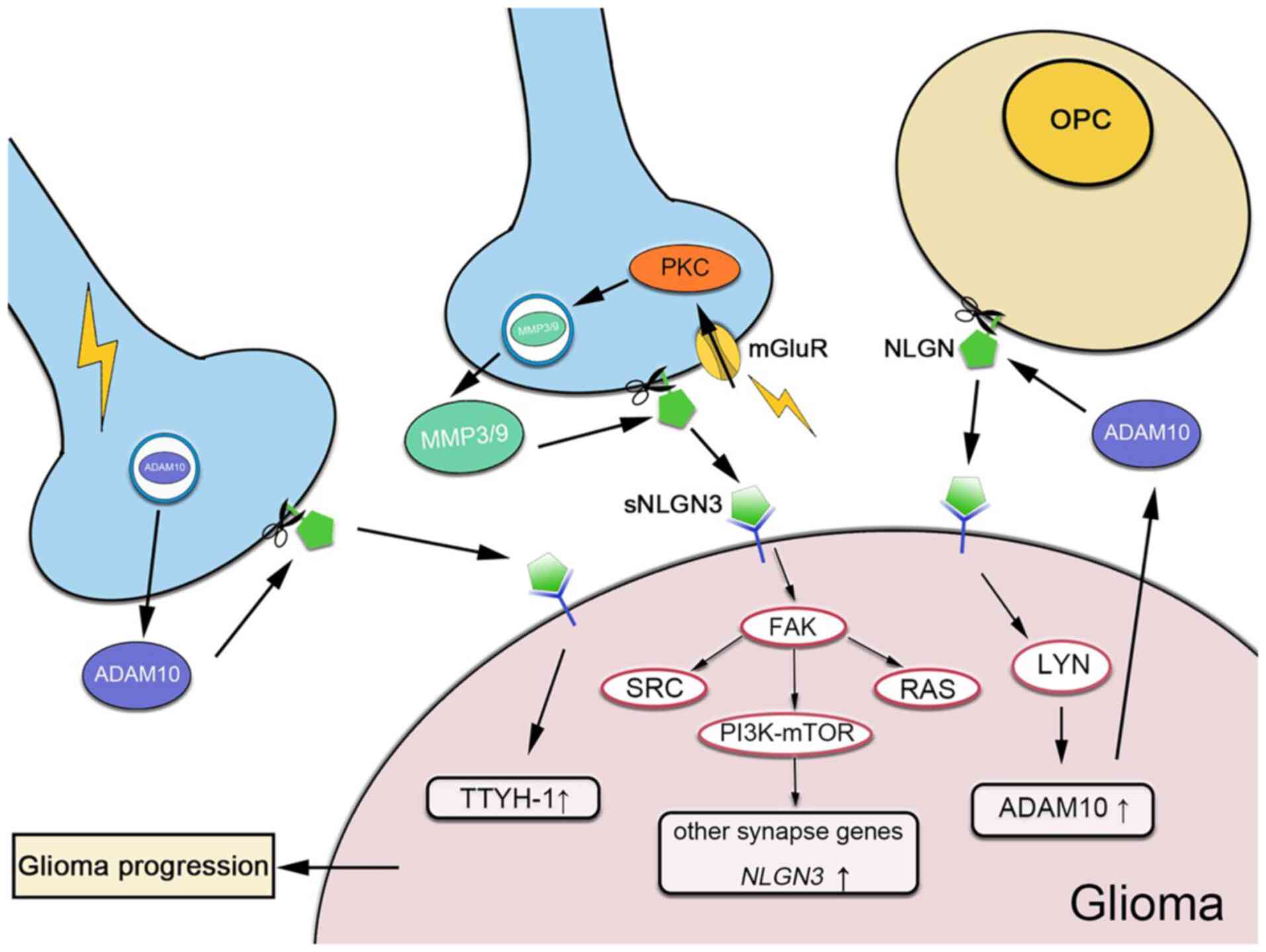 | Figure 2Mechanism of NLGN3 in neuron-glioma
interactions. Neurons and OPCs release sNLGN3 through ADAM10 and/or
MMP cleavage. In neurons, the release of NLGN3 depends on neuronal
activity. The expression of ADAM10 is mediated by neuronal
activity. mGluR activation induces the expression of MMP3/9 through
PKC. sNLGN3 is able to activate NLGN3 and other synapse genes
through the PI3K-mTOR pathway, whereas ADAM10 expression is
upregulated through the Lyn kinase signaling pathway, which
increases TTYH-1 expression. NLGN3, neuroligin-3; OPCs,
oligodendrocyte precursor cells; sNLGN3, soluble NLGN3; ADAM10, A
disintegrin and metalloproteinase domain 10; mGluRs, metabotropic
glutamate receptors; MMP3/9, matrix metal-loproteinase 3/9; PKC,
protein kinase C; TTYH-1, Tweety homologue-1; Proto-oncogene
tyrosine-protein kinase SRC, SRC; Focal adhesion kinase, FAK. |
Extracellular vesicles (EVs) in
neuron-glioma interactions
EVs have an indispensable role in cell-cell
interaction in the brain microenvironment. A previous study
observed how the development of rat cortical neurons led to the
secretion of exosomes containing GluA2/3 subunits of AMPARs
(79). Similar patterns of exosome
secretion were also observed in the differentiated neurons
(80). A previous study indicated
that exosomes derived from cortical neurons only bind to neurons
and not to glial cells (81),
suggesting that neuron-derived EVs (NEVs) mediate neuron-to-glioma
communication indirectly. This indicated that the secretion of
exosomes is activity-dependent and is specifically mediated by
glutamatergic activity involving AMPAR and NMDAR (79,80).
The glioma microenvironment renders neurons hyperexcitable due to
the high glutamate concentration (67). The hyperexcitable neurons are able
to promote the secretion of NEVs, which is chiefly regulated by
glutamatergic activity. In a previously published study on human
neural cultures where MECP2 was knocked down (termed MECP2LOF
neural cultures), NEVs were observed to increase neuron
proliferation and cell numbers (82). These results suggested that NEVs
contain neuroprotective proteins that fulfill an important role in
regulating neural circuits and neurogenesis. The impact of NEVs on
gliomagenesis, however, remains elusive. It is possible that
neurogenesis may increase the formation of NGSs and potentiate
neuronal activity to promote glioma growth.
In addition to NEVs, glioma also influences neuronal
functions via glioma-derived EVs (GEVs). A previous study suggested
that GEVs enhanced the frequency of neuronal spontaneous synaptic
responses (83). This indicated
that GEV-induced neuron hyperexcitability may promote NLGN3 levels,
leading to glioma progression.
3. Retroaction of glioma cells on
neurons
As mentioned above, neuron-glioma interaction is a
bidirectional process. Neuronal activity facilitates glioma
formation through the regulation of precursors, electrochemical
signaling pathways and neuronal secretion. Reciprocally, neuronal
activity is activated by gliomas in various ways, i.e., through the
release of neurotransmitters, promotion of synaptogenesis and
remodeling of neurons in the microenvironment, as well as other
possible mechanisms (77).
Clinical symptoms of glioma-associated neuronal
excitability include cortical hyperexcitability and seizure
activity (84). The
cystine/glutamate antiporter is the key protein involved in
glutamate secretion and this is significantly upregulated in GBM
cells. At the same time, excitatory amino acid transporter 2, which
is responsible for the re-uptake of glutamate, is downregulated in
gliomas (67). The high
concentration of glutamate within the peritumoral microenvironment
leads to hyperexcitability in adjacent neurons and
glioma-associated epilepsy (67).
A previous study also reported that the firing of peritumoral
GABAergic interneurons initiated interictal-like activity, which is
a characteristic of pre-operative seizures in patients with glioma.
This may result from a low expression of KCC2 and a high expression
of NKCC1 (85). Another study
demonstrated that KCC2 expression was downregulated by glioma via
increasing the intracellular Zn2+ concentration in
neurons, eliciting GABA-dependent depolarization of co-cultured
neurons (86). These data implied
that glioma may contribute towards epilepsy by influencing both
glutamatergic and GABAergic signaling in neurons.
Subsequently, a study conducted using a xenograft
model demonstrated a positive correlation between synaptogenic
properties and glioma progression (33). A high concentration of glutamate
contributes towards neuronal death, which frees up space in which
tumors may grow (67).
Furthermore, glioma cells were indicated to disrupt normal
neuron-glial communication, leading to neuronal degeneration and
neurological decay. This pathophysiological process has been
indicated to be more rapid and aggressive in GBM (87).
Gliomas are able to influence the surrounding
neurons by expressing and releasing trophic factors. The expression
of NGF was positively correlated with the glioma grade and
negatively correlated with the median survival time (88). A different study indicated that the
exogenous application of NGF promoted glutamatergic synapse
activity via binding to Trk receptors. This promotion of
glutamatergic synapse activity was regulated by potentiating the
pre-synaptic release of glutamate (89). These data suggested that NGF
released by glioma cells may enhance the glutamatergic NGSs and
promote glioma growth. BDNF was deemed to be an important trophic
factor involved in synaptogenesis, synaptic plasticity and
neuroprotection (90). It has been
reported that depression is a concomitant syndrome of glioma
(28) and is associated with
decreased levels of BDNF (91). It
was hypothesized that glioma may downregulate BDNF, thereby
supporting its own survival and influencing neuronal function
(28). In a Drosophila
model, GBM cells were indicated to produce ImpL2, an antagonist of
the insulin signaling pathway, leading to mitochondrial damage and
the loss of synapses in adjacent neurons. The progression of GBM
has also been observed to be associated with attenuation of the
insulin signaling pathway in neurons (37).
Glioma retroaction on neurons accounts for several
neurological disorders, including epilepsy, depression and
neurodegenerative diseases. The present review has provided several
suggestions of potential therapeutic targets that may be used to
prevent neuronal aberrations and in anti-glioma strategies.
4. Perspectives and future directions
Immunoregulation
In the glioma microenvironment, microglia and
tumor-associated macrophages (TAMs) are the dominant immune cells
assisting neuronal regulation in glioma cells.
Microglia/microphages exert their immune-modulating effects through
mediating the neurotransmitter functioning processes, as well as
via releasing regulatory EVs. Novel immunotherapies may be devised
based on neuron-microglia/macrophage-glioma interactions.
Microglia regulate synapse formation by forming
direct contacts with glioma cells and secreting growth factors,
such as BDNF and interleukin-10 (IL-10) (92). On the other hand, GSCs were
observed to regulate the secretion of IL-10 from microglia,
suggesting the possible presence of a bidirectional communicative
process. Through this bilateral talk, the secretion of IL-10 by
microglia was altered, which may interfere with normal synapse
formation (93). It was reported
that activated microglia were able to increase glutamatergic
synapses on neurons with perineuronal 'nets' (94), possibly via microglial chemokine
C-X-X-X-C motif ligand 1 (CX3CL1) signaling (95), promoting glioma growth and
invasion. It was indicated that depletion of GluA2 on the
microglial membrane resulted in an increase in the expression of
tumor necrosis factor-α (TNF-α), which, in turn, may enhance AMPAR
levels on the neuronal membrane (96). An increased expression of AMPARs on
neurons promoted neuron hyperexcitability and death in an
environment containing a high concentration of glutamate. As the
overexpression of GluA2 inhibits GBM proliferation (97), this treatment was able to both
ameliorate the TME and protect the neurons simultaneously. In
addition, IL-β and TNF-α secreted by microglia were indicated to
decrease the levels of mGlu5 in astrocytes, which impaired
glutamate uptake and led to a further increase in the
extra-cellular glutamate concentration (98). GABA was also indicated to exert
anti-tumor effects via GABA-GABAAR in the
neuron-microglia-tumor axis (11).
Microglia-released IL-1β and TNF-α inhibited GABAergic synaptic
activities in neurons, which supported glioma growth (99). Taken together, TAMs act as a bridge
in neuron-glioma interactions via pro-inflammatory cytokines acting
as mediators. Targeting microglia/macrophages therefore offers a
promising strategy for the treatment of glioma.
Colony-stimulating factor 1 (CSF-1) is produced by
glioma cells and is highly expressed in TAMs (100). CSF-1 inhibition was observed to
significantly reduce both the number of TAMs and glioma progression
in vivo (101), whereas
administration of PLX3397, a CSF-1 inhibitor, had no efficacy in
recurrent GBM according to a phase II clinical trial (101).
EVs fulfill an important role as secondary
messengers within microglia-glioma communication networks. GEVs
under hypoxic conditions skew macrophages towards the M2-type,
which was subsequently indicated to support the proliferation and
migration of U87 cells in vitro and in vivo (102). Microglia have a critical role in
the 'tripartite synapse', which is formed by an astrocyte and two
neurons. The normal functions of the astrocyte-neuron crosstalk are
supported by microglia and neurons in turn, which regulates the
activation and motility of the microglia (95). Neurons maintain the homeostatic
phenotype of microglia via CX3CL1-CX3CR signaling (95,103), which may have an important role
in glioma progression by regulating the function of microglia.
Reciprocally, microglia-derived EVs (MEVs) also mediate neuronal
growth and activity by increasing the miniature excitatory
postsynaptic current via the membrane components of MEVs. In
addition, MEVs have been demonstrated to exert roles in
neuritogenesis and neuroprotection, which may potentiate the
neuron-glioma circuit. In addition, inhibition of neutral
sphingomyelinase 2 successfully impeded the release of EVs from the
brain and aggravated the symptoms in a Parkinson's disease mouse
model, suggesting that the crosstalk between neurons, microglia and
glioma cells based on EVs may be a possible option for therapeutic
intervention (104).
It is theoretically feasible that viro-immunotherapy
may be utilized to reverse the immunosuppressive environment by
introducing oncolytic viruses (OVs). OVs exert antitumor effects
through polarizing TAMs towards the M1 phenotype, which has the
effect of upregulating the pro-inflammatory response (105). A pro-inflammatory TME leads to
inhibition of glioma growth. It is also possible that engineered
MEVs are used to manipulate the neuron-glioma circuit for better
tumor suppression and neuroprotection.
Neural stem cell (NSC)-based delivery
systems
NSCs have the property of tropism towards glioma and
these are dependent on interactions between growth factors and
receptors (106). This property
of NSCs paves the way towards a novel approach of developing
NSC-based drug delivery systems. On this basis, murine NSCs
transfected with adenoviral vector containing TNF-related
apoptosis-inducing ligand (TRAIL) genes were able to successfully
migrate towards glioma cells and express TRAIL in vivo,
thereby significantly increasing the rate of tumor apoptosis
(107). Furthermore,
IL-23-expressing NSCs derived from bone marrow stem cells have been
utilized to treat glioma in an animal model, where the survival
time of mice was significantly prolonged; these effects were
attributed to the potentiated activity of CD8+ T cells, CD4+ T
cells and natural killer cells (108). Recently, researchers have loaded
NSCs with oncolytic viruses to obtain more effective therapeutic
effects on gliomas. Batalla-Covello et al (109) observed that the virus-loaded NSCs
successfully migrated towards glioma and exerted anti-glioma
effects in vivo. These findings demonstrated the feasibility
and efficacy of using an NSC-based delivery system. It is
noteworthy that researchers have observed a close contact (possibly
physical) between NSCs and glioma cells through the use of
immunohistochemistry and fluorescence immunohistochemistry
(107-111). More importantly, Benmelouka et
al (108) observed that the
transplanted NSCs were capable of differentiating into neurons,
astrocytes and oligodendrocytes in vivo. Therefore, NSCs may
incorporate into the neuron-glioma circuit or neuron-glial network
and form NGSs between neurons and gliomas. To use NSCs as a vehicle
to deliver drugs, including small-molecule inhibitors, nucleotides
such as miRNAs and oligonucleotides to target receptors and
channels with both precision and efficiency may prove to be
beneficial in the treatment of glioma, and this approach warrants
further investigation.
Optogenetic modulation
The application of emergent optogenetic tools with
their great precision and high safety provides the possibility of
treating gliomas by neuronal manipulation. Optogenetic tools are
able to modulate the activation of receptors on the neuronal
membrane, which affects the tumor behavior via neuron-glioma
activities (15). In a recent
exploratory study, Trks were engineered with a photosensory core
module of DrBphP, named Dr-Trk opto-kinase, which was successfully
suppressed by far-red (FR) light and reactivated by near-infrared
light (112). It is notable that
the optical modulation may be limited to a single molecule of
Dr-Trk, whereas, by contrast, even selective chemical receptor
tyrosine kinase inhibitors usually suppress several targets
simultaneously. Furthermore, the inhibitory effects of FR light
were similar to pharmacological inhibition (112). In addition to the effects of
modulation on membrane receptors, optogenetic technology may also
be used to regulate neuronal activity. It was demonstrated that
optical stimulation on GABAergic interneurons resulted in impaired
glioma proliferation. The sensory stimulation promoted glioma cell
growth, whereas this effect was limited to glioma cells in the
visual cortex (15). Based on the
insight gained on neuron-glioma interaction, optogenetic technology
harbors great therapeutic potential for the future due to its
non-invasiveness and accuracy.
Co-ulture of neuron organoid and
glioma
The neuron organoids, mostly derived from embryonic
stem cells or induced pluripotent cells, recapitulate multiple
structures and functions similar to those found in the human brain
(113). The co-culture system
provided a suitable microenvironment for glioma development and
interplay with other TME determinants, including astrocytes,
oligodendrocytes, microglia and neurons (114). Brain organoids and GSCs
constitute an ad hoc three-dimensional system, which may be
used to further verify the functions of above-mentioned neuronal
interacting factors, including neurotransmitters and transsynaptic
proteins (including NLGN3 and its modifying enzyme, ADAM10). The
super-resolution imaging technique revealed that GSCs develop an
enhanced tropism for mature neurons by forming direct hemi-synapses
containing pre-synaptic vesicles, for which a complete explanation
of the internal structure awaits further investigation (115). Of note, the co-culture system
provides an 'off-the-wall' methodology for advanced studies on
glioma invasion and screening anti-invasive compounds. Single-cell
transcriptomics combined with neural organoids have provided a
novel perspective in terms of studying glioma heterogeneity and
invasion, as well as the transcriptomic characterization of
surrounding organoid cells upon glioma interaction. Therefore,
specific transcriptional changes underlying therapeutic targets
were denoted to enable effective drugs to be screened in a
patient-specific manner (116).
An up-to-date study has accumulated evidence of cognitive and
electrophysiological neural responses in patients with glioma to
analyze the impacts of functional integration on clinical outcomes.
The results obtained suggested that gliomas with increased
functional connectivity are correlated with higher aggressiveness
through remodeling functional neural circuits in certain cortex
areas (117). It is theoretically
feasible that the co-culture of neuron organoids and glioma, as a
more characteristic and accessible tool, may be applied to study
the integration of glioma with neural networks and this technology
may give rise to a therapeutic strategy that affords cognitive
outcomes and survival.
Availability of data and materials
Publicly available datasets were analyzed in this
study. The summary table of clinical trials included data from
https://clin-icaltrials.gov/.
Authors' contributions
TH, HS, JC and HW conceptualized this study. TH,
HS, MZ, CC, QH, YS, SW and XZ performed the literature search and
drafted the manuscript. TH and HS completed the visualization. All
authors participated in reviewing the paper and HW and QH mainly
performed revision of the manuscript. JC and HW conducted project
administration and funding acquisition. All authors have read and
agreed to the published version of the manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Dr Chengxu Jiang from the Neurosurgery Department,
MD Anderson Cancer Center (Houston, USA) provided instructions on
academic writing and helped polish the language.
Funding
This study was funded by the National Natural Science Foundation
of China (grant no. 81902538), the Shanghai Sailing Program (grant
no. 19YF1448200), the Natural Science Foundation of Hubei Province,
China (grant no. 2021CFB168) and Yuying Project of General Hospital
Of Central Theater Command (grant no. ZZYCZ202103).
References
|
1
|
Lapointe S, Perry A and Butowski NA:
Primary brain tumours in adults. Lancet. 392:432–446. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rominiyi O, Vanderlinden A, Clenton SJ,
Bridgewater C, Al-Tamimi Y and Collis SJ: Tumour treating fields
therapy for glioblastoma: Current advances and future directions.
Br J Cancer. 124:697–709. 2021. View Article : Google Scholar :
|
|
3
|
Tan AC, Ashley DM, López GY, Malinzak M,
Friedman HS and Khasraw M: Management of glioblastoma: State of the
art and future directions. CA Cancer J Clin. 70:299–312. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Louis DN, Perry A, Wesseling P, Brat DJ,
Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM,
Reifenberger G, et al: The 2021 WHO classification of tumors of the
central nervous system: A summary. Neuro Oncol. 23:1231–1251. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Klemm F, Maas RR, Bowman RL, Kornete M,
Soukup K, Nassiri S, Brouland JP, Iacobuzio-Donahue CA, Brennan C,
Tabar V, et al: Interrogation of the microenvironmental landscape
in brain tumors reveals disease-specific alterations of immune
cells. Cell. 181:1643–1660.e17. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Broekman ML, Maas SLN, Abels ER, Mempel
TR, Krichevsky AM and Breakefield XO: Multidimensional
communication in the microenvirons of glioblastoma. Nat Rev Neurol.
14:482–495. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mohme M and Neidert MC: Tumor-specific T
cell activation in malignant brain tumors. Front Immunol.
11:2052020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wei J, Chen P, Gupta P, Ott M, Zamler D,
Kassab C, Bhat KP, Curran MA, de Groot JF and Heimberger AB: Immune
biology of glioma-associated macrophages and microglia: Functional
and therapeutic implications. Neuro Oncol. 22:180–194. 2020.
|
|
9
|
Venkatesh HS: The neural regulation of
cancer. Science. 366:9652019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
De Luca C, Virtuoso A, Papa M, Certo F,
Barbagallo GMV and Altieri R: Regional development of glioblastoma:
The anatomical conundrum of cancer biology and its surgical
implication. Cells. 11:13492022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Parmigiani E, Scalera M, Mori E, Tantillo
E and Vannini E: Old stars and new players in the brain tumor
microenvironment. Front Cell Neurosci. 15:7099172021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Venkataramani V, Tanev DI, Strahle C,
Studier-Fischer A, Fankhauser L, Kessler T, Körber C, Kardorff M,
Ratliff M, Xie R, et al: Glutamatergic synaptic input to glioma
cells drives brain tumour progression. Nature. 573:532–538. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lim-Fat MJ and Wen PY: Glioma progression
through synaptic activity. Nat Rev Neurol. 16:6–7. 2020. View Article : Google Scholar
|
|
14
|
Kirmse K and Zhang C: Principles of
GABAergic signaling in developing cortical network dynamics. Cell
Rep. 38:1105682022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tantillo E, Vannini E, Cerri C, Spalletti
C, Colistra A, Mazzanti CM, Costa M and Caleo M: Differential roles
of pyramidal and fast-spiking, GABAergic neurons in the control of
glioma cell proliferation. Neurobiol Dis. 141:1049422020.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Melgarejo da Rosa M: Communication of
glioma cells with neuronal plasticity: What is the underlying
mechanism? Neurochem Int. 141:1048792020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Radin DP and Tsirka SE: Interactions
between tumor cells, neurons, and microglia in the glioma
microenvironment. Int J Mol Sci. 21:84762020. View Article : Google Scholar :
|
|
18
|
Jung E, Alfonso J, Osswald M, Monyer H,
Wick W and Winkler F: Emerging intersections between neuroscience
and glioma biology. Nat Neurosci. 22:1951–1960. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hasselmo ME, Alexander AS, Hoyland A,
Robinson JC, Bezaire MJ, Chapman GW, Saudargiene A, Carstensen LC
and Dannenberg H: The unexplored territory of neural models:
Potential guides for exploring the function of metabotropic
neuromodulation. Neuroscience. 456:143–158. 2021. View Article : Google Scholar
|
|
20
|
Pei Z, Lee KC, Khan A, Erisnor G and Wang
HY: Pathway analysis of glutamate-mediated, calcium-related
signaling in glioma progression. Biochem Pharmacol. 176:1138142020.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Charsouei S, Jabalameli MR and
Karimi-Moghadam A: Molecular insights into the role of AMPA
receptors in the synaptic plasticity, pathogenesis and treatment of
epilepsy: Therapeutic potentials of perampanel and antisense
oligonucleotide (ASO) technology. Acta Neurol Belg. 120:531–544.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cull-Candy SG and Farrant M:
Ca2+ -permeable AMPA receptors and their auxiliary
subunits in synaptic plasticity and disease. J Physiol.
599:2655–2671. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dohrke JN, Watson JF, Birchall K and
Greger IH: Characterizing the binding and function of TARP
γ8-selective AMPA receptor modulators. J Biol Chem.
295:14565–14577. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Müller-Längle A, Lutz H, Hehlgans S, Rödel
F, Rau K and Laube B: NMDA receptor-mediated signaling pathways
enhance radiation resistance, survival and migration in
glioblastoma cells-A potential target for adjuvant radiotherapy.
Cancers (Basel). 11:5032019. View Article : Google Scholar
|
|
25
|
Nepali K, Hsu TI, Hsieh CM, Lo WL, Lai MJ,
Hsu KC, Lin TE, Chuang JY and Liou JP: Pragmatic recruitment of
memantine as the capping group for the design of HDAC inhibitors: A
preliminary attempt to unravel the enigma of glioblastoma. Eur J
Med Chem. 217:1133382021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wan Z, Sun R, Liu YW, Li S, Sun J, Li J,
Zhu J, Moharil P, Zhang B, Ren P, et al: Targeting metabotropic
glutamate receptor 4 for cancer immunotherapy. Sci Adv.
7:eabj42262021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Blyufer A, Lhamo S, Tam C, Tariq I,
Thavornwatanayong T and Mahajan SS: Riluzole: A neuroprotective
drug with potential as a novel anti-cancer agent (review). Int J
Oncol. 59:952021. View Article : Google Scholar :
|
|
28
|
Mugge L, Mansour TR, Crippen M, Alam Y and
Schroeder J: Depression and glioblastoma, complicated concomitant
diseases: A systemic review of published literature. Neurosurg Rev.
43:497–511. 2020. View Article : Google Scholar
|
|
29
|
Dolma S, Selvadurai HJ, Lan X, Lee L,
Kushida M, Voisin V, Whetstone H, So M, Aviv T, Park N, et al:
Inhibition of dopamine receptor D4 impedes autophagic flux,
proliferation, and survival of glioblastoma stem cells. Cancer
Cell. 29:859–873. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Caffino L, Mottarlini F, Targa G, Verheij
MMM, Fumagalli F and Homberg JR: Responsivity of serotonin
transporter knockout rats to short and long access to cocaine:
Modulation of the glutamate signalling in the nucleus accumbens
shell. Br J Pharmacol. 179:3727–3739. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bi J, Khan A, Tang J, Armando AM, Wu S,
Zhang W, Gimple RC, Reed A, Jing H, Koga T, et al: Targeting
glioblastoma signaling and metabolism with a re-purposed
brain-penetrant drug. Cell Rep. 37:1099572021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tao F, Zhu J, Duan L, Wu J, Zhang J, Yao
K, Bo J and Zu H: Anti-inflammatory effects of doxepin
hydrochloride against LPS-induced C6-glioma cell inflammatory
reaction by PI3K-mediated Akt signaling. J Biochem Mol Toxicol.
34:e224242020. View Article : Google Scholar
|
|
33
|
Otto-Meyer S, DeFaccio R, Dussold C,
Ladomersky E, Zhai L, Lauing KL, Bollu LR, Amidei C, Lukas RV,
Scholtens DM and Wainwright DA: A retrospective survival analysis
of glioblastoma patients treated with selective serotonin reuptake
inhibitors. Brain Behav Immun Health. 2:1000252020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Takayasu T, Kurisu K, Esquenazi Y and
Ballester LY: Ion channels and their role in the pathophysiology of
gliomas. Mol Cancer Ther. 19:1959–1969. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
He L, Sun Z, Li J, Zhu R, Niu B, Tam KL,
Xiao Q, Li J, Wang W, Tsui CY, et al: Electrical stimulation at
nanoscale topography boosts neural stem cell neurogenesis through
the enhancement of autophagy signaling. Biomaterials.
268:1205852021. View Article : Google Scholar
|
|
36
|
Arvind R, Chandana SR, Borad MJ,
Pennington D, Mody K and Babiker H: Tumor-treating fields: A fourth
modality in cancer treatment, new practice updates. Crit Rev Oncol
Hematol. 168:1035352021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tsai HF, IJspeert C and Shen AQ:
Voltage-gated ion channels mediate the electrotaxis of glioblastoma
cells in a hybrid PMMA/PDMS microdevice. APL Bioeng. 4:0361022020.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nicoletti NF, Erig TC, Zanin RF, Roxo MR,
Ferreira NP, Gomez MV, Morrone FB and Campos MM: Pre-clinical
evaluation of voltage-gated calcium channel blockers derived from
the spider P. nigriventer in glioma progression. Toxicon.
129:58–67. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Catacuzzeno L, Sforna L, Esposito V,
Limatola C and Franciolini F: Ion channels in glioma malignancy.
Rev Physiol Biochem Pharmacol. 181:223–267. 2021. View Article : Google Scholar
|
|
40
|
Vannini E, Mori E, Tantillo E, Schmidt G,
Caleo M and Costa M: CTX-CNF1 recombinant protein selectively
targets glioma cells in vivo. Toxins (Basel). 13:1942021.
View Article : Google Scholar
|
|
41
|
Catacuzzeno L and Franciolini F: Role of
KCa3.1 channels in modulating Ca2+ oscillations during
glioblastoma cell migration and invasion. Int J Mol Sci.
19:29702018. View Article : Google Scholar
|
|
42
|
Rosa P, Catacuzzeno L, Sforna L, Mangino
G, Carlomagno S, Mincione G, Petrozza V, Ragona G, Franciolini F
and Calogero A: BK channels blockage inhibits hypoxia-induced
migration and chemoresistance to cisplatin in human glioblastoma
cells. J Cell Physiol. 233:6866–6877. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ru Q, Li WL, Xiong Q, Chen L, Tian X and
Li CY: Voltage-gated potassium channel blocker 4-aminopyridine
induces glioma cell apoptosis by reducing expression of
microRNA-10b-5p. Mol Biol Cell. 29:1125–1136. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Aissaoui D, Mlayah-Bellalouna S, Jebali J,
Abdelkafi-Koubaa Z, Souid S, Moslah W, Othman H, Luis J, ElAyeb M,
Marrakchi N, et al: Functional role of Kv1.1 and Kv1.3 channels in
the neoplastic progression steps of three cancer cell lines,
elucidated by scorpion peptides. Int J Biol Macromol.
111:1146–1155. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hu L, Li LL, Lin ZG, Jiang ZC, Li HX, Zhao
SG and Yang KB: Blockage of potassium channel inhibits
proliferation of glioma cells via increasing reactive oxygen
species. Oncol Res. 22:57–65. 2014. View Article : Google Scholar
|
|
46
|
Roehlecke C and Schmidt MHH: Tunneling
nanotubes and tumor microtubes in cancer. Cancers (Basel).
12:8572020. View Article : Google Scholar
|
|
47
|
Portela M, Venkataramani V, Fahey-Lozano
N, Seco E, Losada-Perez M, Winkler F and Casas-Tintó S:
Glioblastoma cells vampirize WNT from neurons and trigger a JNK/MMP
signaling loop that enhances glioblastoma progression and
neurodegeneration. PLoS Biol. 17:e30005452019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Venkatesh HS, Morishita W, Geraghty AC,
Silverbush D, Gillespie SM, Arzt M, Tam LT, Espenel C, Ponnuswami
A, Ni L, et al: Electrical and synaptic integration of glioma into
neural circuits. Nature. 573:539–545. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Epifantseva I and Shaw RM: Intracellular
trafficking pathways of Cx43 gap junction channels. Biochim Biophys
Acta Biomembr. 1860:40–47. 2018. View Article : Google Scholar
|
|
50
|
Pinto G, Brou C and Zurzolo C: Tunneling
nanotubes: The fuel of tumor progression? Trends Cancer. 6:874–888.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Osswald M, Jung E, Sahm F, Solecki G,
Venkataramani V, Blaes J, Weil S, Horstmann H, Wiestler B, Syed M,
et al: Brain tumour cells interconnect to a functional and
resistant network. Nature. 528:93–98. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Jaraíz-Rodríguez M, Talaverón R,
García-Vicente L, Pelaz SG, Domínguez-Prieto M,
Aacute;lvarez-Vázquez A, Flores-Hernández R, Sin WC, Bechberger J,
Medina JM, et al: Connexin43 peptide, TAT-Cx43266-283, selectively
targets glioma cells, impairs malignant growth, and enhances
survival in mouse models in vivo. Neuro Oncol. 22:493–504. 2020.
View Article : Google Scholar
|
|
53
|
Pettee KM, Becker KN, Alberts AS, Reinard
KA, Schroeder JL and Eisenmann KM: Targeting the mDia
formin-assembled cytoskeleton is an effective anti-invasion
strategy in adult high-grade glioma patient-derived neurospheres.
Cancers (Basel). 11:3922019. View Article : Google Scholar
|
|
54
|
Sinyuk M, Mulkearns-Hubert EE, Reizes O
and Lathia J: Cancer connectors: connexins, gap junctions, and
communication. Front Oncol. 8:6462018. View Article : Google Scholar
|
|
55
|
Zhang C, Liu CF, Chen AB, Yao Z, Li WG, Xu
SJ and Ma XY: Prognostic and clinic pathological value of Cx43
expression in glioma: A meta-analysis. Front Oncol. 9:12092019.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Filbin MG and Segal RA: How neuronal
activity regulates glioma cell proliferation. Neuro Oncol.
17:1543–1544. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yao M, Ventura PB, Jiang Y, Rodriguez FJ,
Wang L, Perry JSA, Yang Y, Wahl K, Crittenden RB, Bennett ML, et
al: Astrocytic trans-differentiation completes a multicellular
paracrine feedback loop required for medulloblastoma tumor growth.
Cell. 180:502–520.e19. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Griffin N, Faulkner S, Jobling P and
Hondermarck H: Targeting neurotrophin signaling in cancer: The
renaissance. Pharmacol Res. 135:12–17. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Rocco ML, Soligo M, Manni L and Aloe L:
Nerve growth factor: Early studies and recent clinical trials. Curr
Neuropharmacol. 16:1455–1465. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Franzese O, Di Francesco AM, Meco D,
Graziani G, Cusano G, Levati L, Riccardi R and Ruggiero A: hTERT
transduction extends the lifespan of primary pediatric low-grade
glioma cells while preserving the biological response to NGF.
Pathol Oncol Res. 27:6123752021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Meco D, Di Francesco AM, Melotti L,
Ruggiero A and Riccardi R: Ectopic nerve growth factor prevents
proliferation in glioma cells by senescence induction. J Cell
Physiol. 234:6820–6830. 2019. View Article : Google Scholar
|
|
62
|
Park JC, Chang IB, Ahn JH, Kim JH, Song
JH, Moon SM and Park YH: Nerve growth factor stimulates
glioblastoma proliferation through Notch1 receptor signaling. J
Korean Neurosurg Soc. 61:441–449. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Barreda Tomás FJ, Turko P, Heilmann H,
Trimbuch T, Yanagawa Y, Vida I and Münster-Wandowski A: BDNF
expression in cortical GABAergic interneurons. Int J Mol Sci.
21:15672020. View Article : Google Scholar
|
|
64
|
Li YF: A hypothesis of monoamine
(5-HT)-glutamate/GABA long neural circuit: Aiming for fast-onset
antidepressant discovery. Pharmacol Ther. 208:1074942020.
View Article : Google Scholar
|
|
65
|
Liu S, Jiang T, Zhong Y and Yu Y: miR-210
inhibits cell migration and invasion by targeting the brain-derived
neurotrophic factor in glioblastoma. J Cell Biochem. Feb
11–2019.Epub ahead of print.
|
|
66
|
Wang J, Gao F, Cui S, Yang S, Gao F, Wang
X and Zhu G: Utility of 7,8-dihydroxyflavone in preventing
astrocytic and synaptic deficits in the hippocampus elicited by
PTSD. Pharmacol Res. 176:1060792022. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lange F, Hörnschemeyer J and Kirschstein
T: Glutamatergic mechanisms in glioblastoma and tumor-associated
epilepsy. Cells. 10:12262021. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Keifer J: Regulation of AMPAR trafficking
in synaptic plasticity by BDNF and the impact of neurodegenerative
disease. J Neurosci Res. 100:979–991. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Gorick CM, Saucerman JJ and Price RJ:
Computational model of brain endothelial cell signaling pathways
predicts therapeutic targets for cerebral pathologies. J Mol Cell
Cardiol. 164:17–28. 2022. View Article : Google Scholar
|
|
70
|
Jawhari S, Bessette B, Hombourger S,
Durand K, Lacroix A, Labrousse F, Jauberteau MO, Ratinaud MH and
Verdier M: Autophagy and TrkC/NT-3 signaling joined forces boost
the hypoxic glioblastoma cell survival. Carcinogenesis. 38:592–603.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Venkatesh HS, Johung TB, Caretti V, Noll
A, Tang Y, Nagaraja S, Gibson EM, Mount CW, Polepalli J, Mitra SS,
et al: Neuronal activity promotes glioma growth through
neuroligin-3 secretion. Cell. 161:803–816. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Bemben MA, Nguyen TA, Li Y, Wang T, Nicoll
RA and Roche KW: Isoform-specific cleavage of neuroligin-3 reduces
synapse strength. Mol Psychiatry. 24:145–160. 2019. View Article : Google Scholar
|
|
73
|
Liu R, Qin XP, Zhuang Y, Zhang Y, Liao HB,
Tang JC, Pan MX, Zeng FF, Lei Y, Lei RX, et al: Glioblastoma
recurrence correlates with NLGN3 levels. Cancer Med. May
18–2018.Epub ahead of print.
|
|
74
|
Dang NN, Li XB, Zhang M, Han C, Fan XY and
Huang SH: NLGN3 upregulates expression of ADAM10 to promote the
cleavage of NLGN3 via activating the LYN pathway in human gliomas.
Front Cell Dev Biol. 9:6627632021. View Article : Google Scholar :
|
|
75
|
Venkatesh HS, Tam LT, Woo PJ, Lennon J,
Nagaraja S, Gillespie SM, Ni J, Duveau DY, Morris PJ, Zhao JJ, et
al: Targeting neuronal activity-regulated neuroligin-3 dependency
in high-grade glioma. Nature. 549:533–537. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Li Z, Gao W, Fei Y, Gao P, Xie Q, Xie J
and Xu Z: NLGN3 promotes neuroblastoma cell proliferation and
growth through activating PI3K/AKT pathway. Eur J Pharmacol.
857:1724232019. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Fumagalli A, Heuninck J, Pizzoccaro A,
Moutin E, Koenen J, Séveno M, Durroux T, Junier MP, Schlecht-Louf
G, Bachelerie F, et al: The atypical chemokine receptor 3 interacts
with Connexin 43 inhibiting astrocytic gap junctional intercellular
communication. Nat Commun. 11:48552020. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Derks J, Wesseling P, Carbo EWS,
Hillebrand A, van Dellen E, de Witt Hamer PC, Klein M, Schenk GJ,
Geurts JJG, Reijneveld JC and Douw L: Oscillatory brain activity
associates with neuroligin-3 expression and predicts progression
free survival in patients with diffuse glioma. J Neurooncol.
140:403–412. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Gu J, Jin T, Li Z, Chen H, Xia H, Xu X and
Gui Y: Exosomes expressing neuronal autoantigens induced immune
response in antibody-positive autoimmune encephalitis. Mol Immunol.
131:164–170. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Kumari M and Anji A: Small but
mighty-exosomes, novel inter-cellular messengers in
neurodegeneration. Biology (Basel). 11:4132022.
|
|
81
|
Chivet M, Javalet C, Laulagnier K, Blot B,
Hemming FJ and Sadoul R: Exosomes secreted by cortical neurons upon
glutamatergic synapse activation specifically interact with
neurons. J Extracell Vesicles. 3:247222014. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Sharma P, Mesci P, Carromeu C, McClatchy
DR, Schiapparelli L, Yates JR III, Muotri AR and Cline HT: Exosomes
regulate neurogenesis and circuit assembly. Proc Natl Acad Sci USA.
116:16086–16094. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Gao X, Zhang Z, Mashimo T, Shen B, Nyagilo
J, Wang H, Wang Y, Liu Z, Mulgaonkar A, Hu XL, et al: Gliomas
interact with non-glioma brain cells via extracellular vesicles.
Cell Rep. 30:2489–2500.e5. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Yu K, Lin CJ, Hatcher A, Lozzi B, Kong K,
Huang-Hobbs E, Cheng YT, Beechar VB, Zhu W, Zhang Y, et al: PIK3CA
variants selectively initiate brain hyperactivity during
gliomagenesis. Nature. 578:166–171. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Wang J, Liu W, Xu W, Yang B, Cui M, Li Z,
Zhang H, Jin C, Xue H and Zhang J: Comprehensive analysis of the
oncogenic, genomic alteration, and immunological landscape of
cation-chloride cotransporters in pan-cancer. Front Oncol.
12:8196882022. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Al Awabdh S, Donneger F, Goutierre M,
Séveno M, Vigy O, Weinzettl P, Russeau M, Moutkine I, Lévi S, Marin
P and Poncer JC: Gephyrin interacts with the K-Cl cotransporter
KCC2 to regulate its surface expression and function in cortical
neurons. J Neurosci. 42:166–182. 2022. View Article : Google Scholar :
|
|
87
|
Moreira Franco YE, Alves MJ, Uno M,
Moretti IF, Trombetta-Lima M, de Siqueira Santos S, Dos Santos AF,
Arini GS, Baptista MS, Lerario AM, et al: Glutaminolysis dynamics
during astrocytoma progression correlates with tumor
aggressiveness. Cancer Metab. 9:182021. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Lisi L, Ciotti GMP, Chiavari M, Martire M
and Navarra P: The effects of CHF6467, a new mutated form of NGF,
on cell models of human glioblastoma. A comparison with wild-type
NGF. Growth Factors. 40:37–45. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Rosso P, Fico E, Mesentier-Louro LA,
Triaca V, Lambiase A, Rama P and Tirassa P: NGF eye administration
recovers the TrkB and glutamate/GABA marker deficit in the adult
visual cortex following optic nerve crush. Int J Mol Sci.
22:100142021. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Colucci-D'Amato L, Speranza L and
Volpicelli F: Neurotrophic factor BDNF, physiological functions and
therapeutic potential in depression, neurodegeneration and brain
cancer. Int J Mol Sci. 21:77772020. View Article : Google Scholar :
|
|
91
|
Carniel BP and da Rocha NS: Brain-derived
neurotrophic factor (BDNF) and inflammatory markers: Perspectives
for the management of depression. Prog Neuropsychopharmacol Biol
Psychiatry. 108:1101512021. View Article : Google Scholar
|
|
92
|
Caponegro MD, Oh K, Madeira MM, Radin D,
Sterge N, Tayyab M, Moffitt RA and Tsirka SE: A distinct microglial
subset at the tumor-stroma interface of glioma. Glia. 69:1767–1781.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Wei J, Song R, Sabbagh A, Marisetty A,
Shukla N, Fang D, Najem H, Ott M, Long J, Zhai L, et al:
Cell-directed aptamer therapeutic targeting for cancers including
those within the central nervous system. Oncoimmunology.
11:20628272022. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Wegrzyn D, Freund N, Faissner A and Juckel
G: Poly I:C activated microglia disrupt perineuronal nets and
modulate synaptic balance in primary hippocampal neurons in vitro.
Front Synaptic Neurosci. 13:6375492021. View Article : Google Scholar :
|
|
95
|
Szepesi Z, Manouchehrian O, Bachiller S
and Deierborg T: Bidirectional microglia-neuron communication in
health and disease. Front Cell Neurosci. 12:3232018. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Moraes CA, Zaverucha-do-Valle C, Fleurance
R, Sharshar T, Bozza FA and d'Avila JC: Neuroinflammation in
sepsis: Molecular pathways of microglia activation. Pharmaceuticals
(Basel). 14:4162021. View Article : Google Scholar
|
|
97
|
Raffaele S, Lombardi M, Verderio C and
Fumagalli M: TNF production and release from microglia via
extracellular vesicles: Impact on brain functions. Cells.
9:21452020. View Article : Google Scholar :
|
|
98
|
Milanese M, Bonifacino T, Torazza C,
Provenzano F, Kumar M, Ravera S, Zerbo AR, Frumento G, Balbi M,
Nguyen TPN, et al: Blocking glutamate mGlu5 receptors
with the negative allosteric modulator CTEP improves disease course
in SOD1G93A mouse model of amyotrophic lateral
sclerosis. Br J Pharmacol. 178:3747–3764. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Boakye PA, Tang SJ and Smith PA: Mediators
of neuropathic pain; focus on spinal microglia, CSF-1, BDNF, CCL21,
TNF-α, Wnt ligands, and interleukin 1β. Front Pain Res (Lausanne).
2:6981572021. View Article : Google Scholar
|
|
100
|
Anagnostakis F and Piperi C: Targeting
options of tumor-associated macrophages (TAM) activity in gliomas.
Curr Neuropharmacol. Jan 20–2022.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Barca C, Foray C, Hermann S, Herrlinger U,
Remory I, Laoui D, Schäfers M, Grauer OM, Zinnhardt B and Jacobs
AH: The colony stimulating factor-1 receptor (CSF-1R)-mediated
regulation of microglia/macrophages as a target for neurological
disorders (glioma, stroke). Front Immunol. 12:7873072021.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Qian M, Wang S, Guo X, Wang J, Zhang Z,
Qiu W, Gao X, Chen Z, Xu J, Zhao R, et al: Hypoxic glioma-derived
exosomes deliver microRNA-1246 to induce M2 macrophage polarization
by targeting TERF2IP via the STAT3 and NF-κB pathways. Oncogene.
39:428–442. 2020. View Article : Google Scholar
|
|
103
|
Counil H and Krantic S: Synaptic activity
and (neuro)inflammation in Alzheimer's disease: Could exosomes be
an additional link? J Alzheimers Dis. 74:1029–1043. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Zhu C, Bilousova T, Focht S, Jun M, Elias
CJ, Melnik M, Chandra S, Campagna J, Cohn W, Hatami A, et al:
Pharmacological inhibition of nSMase2 reduces brain exosome release
and α-synuclein pathology in a Parkinson's disease model. Mol
Brain. 14:702021. View Article : Google Scholar
|
|
105
|
Blitz SE, Kappel AD, Gessler FA, Klinger
NV, Arnaout O, Lu Y, Peruzzi PP, Smith TR, Chiocca EA, Friedman GK
and Bernstock JD: Tumor-associated macrophages/microglia in
glioblastoma oncolytic virotherapy: A double-edged sword. Int J Mol
Sci. 23:18082022. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Adamus T, Hung CY, Yu C, Kang E, Hammad M,
Flores L, Nechaev S, Zhang Q, Gonzaga JM, Muthaiyah K, et al:
Glioma-targeted delivery of exosome-encapsulated antisense
oligonucleotides using neural stem cells. Mol Ther Nucleic Acids.
27:611–620. 2021. View Article : Google Scholar
|
|
107
|
Ehtesham M, Kabos P, Gutierrez MA, Chung
NH, Griffith TS, Black KL and Yu JS: Induction of glioblastoma
apoptosis using neural stem cell-mediated delivery of tumor
necrosis factor-related apoptosis-inducing ligand. Cancer Res.
62:7170–7174. 2002.PubMed/NCBI
|
|
108
|
Benmelouka AY, Munir M, Sayed A, Attia MS,
Ali MM, Negida A, Alghamdi BS, Kamal MA, Barreto GE, Ashraf GM, et
al: Neural stem cell-based therapies and glioblastoma management:
Current evidence and clinical challenges. Int J Mol Sci.
22:22582021. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Batalla-Covello J, Ngai HW, Flores L,
McDonald M, Hyde C, Gonzaga J, Hammad M, Gutova M, Portnow J,
Synold T, et al: Multiple treatment cycles of neural stem cell
delivered oncolytic adenovirus for the treatment of glioblastoma.
Cancers (Basel). 13:63202021. View Article : Google Scholar
|
|
110
|
van Solinge TS, Nieland L, Chiocca EA and
Broekman MLD: Advances in local therapy for glioblastoma-taking the
fight to the tumour. Nat Rev Neurol. 18:221–236. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Luzzi S, Giotta Lucifero A, Brambilla I,
Trabatti C, Mosconi M, Savasta S and Foiadelli T: The impact of
stem cells in neuro-oncology: Applications, evidence, limitations
and challenges. Acta Biomed. 91(Suppl 7): S51–E60. 2020.
|
|
112
|
Leopold AV, Chernov KG, Shemetov AA and
Verkhusha VV: Neurotrophin receptor tyrosine kinases regulated with
near-infrared light. Nat Commun. 10:11292019. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Hwang JW, Loisel-Duwattez J, Desterke C,
Latsis T, Pagliaro S, Griscelli F, Bennaceur-Griscelli A and Turhan
AG: A novel neuronal organoid model mimicking glioblastoma (GBM)
features from induced pluripotent stem cells (iPSC). Biochim
Biophys Acta Gen Subj. 1864:1295402020. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Mariappan A, Goranci-Buzhala G,
Ricci-Vitiani L, Pallini R and Gopalakrishnan J: Trends and
challenges in modeling glioma using 3D human brain organoids. Cell
Death Differ. 28:15–23. 2021. View Article : Google Scholar
|
|
115
|
Goranci-Buzhala G, Mariappan A, Gabriel E,
Ramani A, Ricci-Vitiani L, Buccarelli M, D'Alessandris QG, Pallini
R and Gopalakrishnan J: Rapid and efficient invasion assay of
glioblastoma in human brain organoids. Cell Rep. 31:1077382020.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Krieger TG, Tirier SM, Park J, Jechow K,
Eisemann T, Peterziel H, Angel P, Eils R and Conrad C: Modeling
glioblastoma invasion using human brain organoids and single-cell
transcriptomics. Neuro Oncol. 22:1138–1149. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Krishna S, Choudhury A, Seo K, Ni L,
Kakaizada F, Lee A, Aabedi A, Cao C, Sudharshan R, Egladyous A, et
al: Glioblastoma remodeling of neural circuits in the human brain
decreases survival. bioRxiv. Feb 19–2021.
|














