1. Introduction
Endometrial cancer (EC) is the most prevalent
gynecologic cancer in developed countries and its incidence rate
has rapidly increased in recent years (1). The most common histological subtype
is endometrioid adenocarcinoma originating from the endometrial
glands. The mainstay of treatment includes total hysterectomy and
bilateral salpingo-oophorectomy, followed by adjuvant treatment
according to the final histology and stage (2). Although the prognosis of patients
with an early diagnosis of EC is favorable, there are fewer choices
and shorter median overall survival (OS) for patients with
recurrent or metastatic diseases (3).
N6-methyladenosine (m6A) is the most abundant RNA
modification in mammalian mRNA and has a crucial role in the
occurrence and development of various diseases, particularly
malignant tumors (4,5). As a hot topic in epigenetics, m6A
modification is essential for regulating various biological
processes, such as splicing, translation and stability of mRNA
through related regulators, thus affecting the proliferation,
invasion, metastasis and self-renewal of tumor cells (6,7). In
recent years, the function of m6A in the pathogenesis and
progression of diseases has attracted considerable attention,
particularly in the prevention and treatment of malignant tumors.
It is anticipated that m6A and the associated regulators will
emerge as new therapeutic targets and prognostic indicators
(8).
Although the exploration of RNA-based therapy is in
its infancy, it has already gained widespread acceptance, as
methylated RNA molecules have important roles in regulating almost
all aspects of cellular biology and may be specifically identified
(9), indicating that therapies
based on RNA modifications may be a valuable method in the field of
cancer treatment. Recent studies reported that the expression of
multiple m6A regulators is up- or downregulated in EC and that m6A
methylation and related regulators are critical to the pathogenesis
and progression of EC. This review discusses the relationship
between m6A methylation modification, related regulators and EC in
the hope that the related findings will provide novel avenues and
approaches for the prevention, early diagnosis and treatment of
EC.
2. EC
ECs are a group of epithelial malignancies that
occur in the endometrium, with adenocarcinoma originating from the
endometrial glands being the most common. It is a major malignancy
of the female reproductive tract and the most prevalent
gynecological cancer in developed countries. In recent years, its
incidence has increased worldwide (2,10,11),
threatening the health and well-being of women (12).
EC is usually divided into two histological types,
with a significant prognostic difference between them. Type I is
estrogen-dependent and may occur under continuous estrogen
stimulation in the absence of progesterone antagonism, resulting in
abnormal proliferation of endometrial epithelial cells (13). Type I EC is frequently associated
with obesity, hypertension, diabetes, anovulatory uterine bleeding
and long-term use of single estrogen or tamoxifen. Type I EC is
histologically classified as well-differentiated to moderately
differentiated tumors, with at least 90% of cases expressing
moderate to high levels of the estrogen receptor. Type I tumors are
characterized by phosphatase and tensin homolog deletion and
mutations in phosphatidylinositol-4,5-bisphosphate 3-kinase
catalytic subunit α, KRAS and β-catenin, as well as microsatellite
instability (MSI) (14,15). By contrast, type II EC is not
related to hyperestrogenemia or endometrial hyperplasia, frequently
occurs in nonobese women and is unrelated to metabolic or endocrine
disorders. Histologically, type II tumors are poorly differentiated
and are most commonly of plasmacytotic, clear cell or
carcinosarcoma subtypes. They are clinically aggressive and are
related to a more advanced stage at initial presentation and a
higher risk of recurrence (16).
Type II tumors are not estrogen-related and are usually
distinguished by a genetic alteration in p53, HER2/neu, p16 and
E-cadherin (14,15).
Recent studies have indicated that despite the
dualistic typing of EC, there are a considerable number of cases
that exhibit an intersection of molecular characteristics, and not
all cases are completely consistent with the pathological
characteristics. Therefore, through genome sequencing analysis, EC
may instead be divided into four subtypes according to the
molecular characteristics: DNA polymerase epsilon (POLE)
ultramutated, MSI, copy-number high (CN high) and CN low (17,18).
Molecular typing has a high predictive value for the prognosis of
EC (19). It has been indicated
that 60% of POLE ultramutated EC cases are high-grade endometrioid
lesions with favorable prognostic outcomes, while the prognosis of
CN-high is the most unfavorable (20-22).
With the continuous growth of the population, the
incidence of EC has increased rapidly in the past decade due to the
high prevalence of obesity and metabolic syndrome (23,24).
Obesity is more closely associated with the progression of EC than
any other type of female-specific cancer. The rate of obesity in
the population is increasing and more than half of all EC cases are
currently attributable to obesity, which is regarded as an
independent risk factor for EC (13). This relationship may be largely
explained by the prevalence of a high level of estrogen in obese
women. Obesity is also related to a high level of insulin. Higher
levels of insulin and estrogen are associated with the risk of EC
(25,26). However, the pathogenesis of EC
remains to be fully elucidated. Therefore, further research on the
mechanisms underlying the development of EC remains crucial for the
development of scientifically effective preventative and
therapeutic measures.
3. m6A
Relatively recently, m6A has become a topic of
intense interest in the field of epigenetics. It is of great
significance in the development and progression of a diverse range
of diseases, particularly malignant tumors (4,5). m6A
refers to a specific methylation modification formed by the
catalysis of the sixth nitrogen atom (N) on adenine (A) by
methyltransferase. The m6A modification sites are primarily
distributed close to the termination codon and the 3′-untranslated
region (3′UTR) (27). They are
widely found in mRNAs and non-coding RNAs (ncRNAs) (28), having important roles in
influencing the biological behavior of RNAs. m6A exists in a
variety of RNAs but is most abundant in eukaryotic mRNAs (29,30),
adding additional complexity to the RNA world. This modification is
dynamic and reversible, and it is catalyzed by three main types of
regulator: It may be installed by methyltransferases
[methyltransferase-like 3 (METTL3), METTL14, Wilms tumor
1-associated protein (WTAP), vir-like m6A
methyltransferase-associated protein (VIRMA, also called KIAA1429),
RNA binding motif protein 15/15B (RBM15/15B) and zinc finger CCCH
domain-containing protein 13 (ZC3H13)], erased by demethylases [fat
mass and obesity-associated protein (FTO), AlkB homolog 5 (ALKBH5)]
and interacts with RNA-binding proteins such as the YT521-B
homology (YTH) domain family, heterogeneous nuclear
ribonucleoprotein (HNRNP) protein family and insulin-like growth
factor 2 mRNA binding proteins (IGF2BP) family to exert their
biological effects. By influencing several phases of an mRNA′s
life, m6A alterations and associated regulators have critical roles
in terms of gene expression (31-33).
The regular functions of these regulators are significant and
abnormal expression is clearly associated with human cancer
(7).
m6A writers
METTL3, METTL14 and WTAP form the m6A
methyltransferase core complex, and are also referred to as
'writers'. METTL3 is the catalytic core enzyme of this complex
(34). METTL14 is the structural
support partner of METTL3 and has a structural role in stabilizing
METTL3 and recognizing target RNAs. Together, they form a stable
heterodimeric core complex that allows m6A to be deposited on
mammalian nuclear RNAs (35,36).
WTAP has a crucial role in the localization of METTL3/14 at the
nuclear speckles and may interact with this complex to influence
this methylation (34,35,37).
In addition to the core components, there are several additional
regulatory factors involved in the m6A methylation process. For
instance, it was discovered that the elements linked to WTAP in
mammalian cells include VIRMA (KIAA1429) and HAKAI. VIRMA mediates
preferential mRNA methylation in the 3'UTR and close to the
termination codon. VIRMA enlists the methyltransferase core complex
as its catalytic core member to control region-selective
methylation (38). RBM15/15B binds
to U-rich sequences preferentially to attract the m6A complex and
may encourage the methylation of particular RNAs (39). ZC3H13 is a CCCH zinc finger protein
that suppresses the growth of tumors by influencing the Ras-ERK
signaling pathway (40). WTAP,
VIRMA and HAKAI are anchored in the nucleus by ZC3H13, which
regulates m6A methylation and mESC self-renewal (41). The functions of m6A writers are
summarized in Table I.
 | Table IFunctions of m6A 'writers'. |
Table I
Functions of m6A 'writers'.
| Regulator | Effect on m6A
modification | (Refs.) |
|---|
| METTL3 | The catalytic core
of methyltransferase | (34) |
| METTL14 | Forms a heterodimer
with METTL3 and catalyze m6A modification | (35,36) |
| WTAP | Recruits METTL3 and
METTL14 into the nuclear speckles | (37) |
| KIAA1429
(VIRMA) | Interacts with WTAP
and attaches m6A to the 3' UTR | (38) |
| RBM15/15B | Recruits the
methyltransferase complex | (39) |
| ZC3H13 | Promotes the WTAP
localization and m6A deposition | (41) |
m6A erasers
To date, FTO and ALKBH5 are the only two m6A
demethylases that have been reported (42). These 'erasers' are members of the
AlkB dioxygenases family and require oxygen, ferrous ion and
α-ketoglutarate to function. FTO, the first m6A demethylase, was
identified in 2011 as being effective in removing m6A modifications
from RNA, indicating that m6A RNA methylation is reversible
(43). FTO is localized in the
cytoplasm and nucleus with different substrate preferences at these
two sites (44). FTO knockdown may
increase the level of m6A in mRNA, whereas FTO overexpression may
decrease the level of m6A in mRNA. Recent research has revealed
that FTO preferentially mediates pre-mRNA alternative splicing and
3'UTR processing (45).
Furthermore, FTO is closely related to weight growth and fat in
humans (46).
ALKBH5, the second eraser, was subsequently
discovered in 2013. ALKBH5 is localized to nuclear speckles and
contributes to the assembly of mRNA processing factors that
regulate gene expression by affecting RNA metabolism, pre-mRNA
processing, mRNA decay and translation (47,48).
ALKBH5 deficiency increases m6A levels, whereas ALKBH5
overexpression decreases m6A levels in mRNA. Aberrant expression of
either FTO or ALKBH5 affects m6A levels, which then influence
certain biological processes in tumor cells through a complex
series of mechanisms. The functions of m6A erasers are listed in
Table II.
| Table IIFunctions of m6A 'erasers'. |
Table II
Functions of m6A 'erasers'.
| Regulators | Effect on m6A
modification | (Refs.) |
|---|
| FTO | Removes m6A
modification, regulates pre-mRNA alternative splicing and 3' UTR
processing | (43,45) |
| ALKBH5 | Removes m6A
modification, regulates RNA metabolism, pre-mRNA processing, mRNA
decay and translation | (48) |
m6A readers
The m6A recognition protein regulates the relevant
biological behaviors of mRNA and performs corresponding functions
by reading m6A methylation. The YTH family may directly identify
m6A methylation and its binding leads to changes in the translation
and stability of m6A-modified RNAs. Members of the YTH family
include YTHDC1-2 and YTHDF1-3 (49).
YTHDF2 may accelerate the degradation of m6A
methylated transcripts by directly recruiting the CCR4-NOT
deadenylase complex (50,51). In addition, YTHDF2 may prevent FTO
from demethylating the 5'UTR, stabilizing the methylation levels in
cells (52). YTHDF1 may facilitate
the translation efficiency of m6A-modified transcripts, attaches to
the m6A sites near the termination codon and improves the
translation of target RNAs by cooperating with the translation
initiation mechanism (53,54). YTHDF3 cooperates with YTHDF1 to
promote RNA translation, while accelerating mRNA degradation by
cooperating with YTHDF2, suggesting a cooperative relationship
between YTHDF proteins (55,56).
YTHDC1 is widely distributed in the nucleus with multiple
regulatory functions. YTHDC1 recruits a variety of splicing factors
to promote exon inclusion. YTHDC1 may accelerate the nuclear export
of m6A-modified mRNAs (57,58).
In addition, YTHDC1 may silence the X chromosome (39), and encourage the degradation of
certain transcripts (59). YTHDC2
increases the translation efficiency and decreases the abundance of
mRNAs by identifying methylated mRNAs (60).
In addition to the YTH structural domain family, the
HNRNP family and IGF2BPs serve as m6A readers and may recognize m6A
modifications (49). HNRNPA2B1
promotes the processing of primary microRNAs (miRNAs) in an
m6A-dependent manner (61).
Furthermore, HNRNPC and HNRNPG influence mRNA abundance and
splicing by dealing with transcripts with the m6A modification. The
RNA secondary structure is impacted by m6A, which makes it easier
for transcripts to bind to HNRNPC and HNRNPG and modulate mRNA
abundance and splicing (62,63).
IGF2BPs are conserved m6A-binding proteins that enhance the
stability of their target mRNAs and improve translation efficiency
in an m6A-dependent manner, impacting gene regulation and cancer
biology (64). The functions of
m6A readers are summarized in Table
III.
| Table IIIFunctions of m6A 'readers'. |
Table III
Functions of m6A 'readers'.
| Regulators | Effect on m6A
modification | (Refs.) |
|---|
| YTH domain
family | | |
| YTHDF1 | Facilitates the
translation of m6A-modified RNA | (53) |
| YTHDF2 | Accelerates the
degradation of m6A-modified RNA | (50) |
| YTHDF3 | Facilitates the
translation and degradation of m6A-modified RNA | (55,56) |
| YTHDC1 | Regulates the
splicing and nuclear export of m6A-modified RNA | (57,58) |
| YTHDC2 | Increases the
translation efficiency of m6A-modified RNA | (60) |
| HNRNP family | | |
| HNRNPA2B1 | Promotes the
processing of primary miRNA | (61) |
| HNRNPC and
HNRNPG | Regulate the
abundance and splicing of m6A-modified RNA | (62,63) |
| IGF2BP1-3 | Promotes the
stability of m6A-modified RNA | (64) |
4. M6A and EC
According to reports, m6A methylation may mediate
post-transcriptional gene expression in EC by regulating functional
gene components, particularly gene promoters and the 3'UTR. Further
research revealed that highly methylated genes were associated with
insulin resistance (IR), while genes with low levels of methylation
were notably enriched in extracellular matrix (ECM) tissues and
adhesive plaques (65). Therefore,
m6A methylation regulates EC progression by focusing on IR and
ECM-related genes.
A growing number of studies suggested that m6A
regulatory factors are associated with tumors, which may function
as oncogenes or tumor suppressor genes to participate in the
proliferation, invasion and metastasis of tumor cells (31,66).
It was determined that the expression of multiple m6A regulators
was up- or downregulated in EC, and that m6A methylation and the
expression of the related regulators are critical to the
pathogenesis and progression of EC.
In addition, multiple signaling pathways are
actively functioning in EC cells, including the MAPK signaling
pathway (67,68) and the PI3K/AKT/mTOR signaling
pathway (69,70). Research has demonstrated that
altering the degree of m6A modification may influence the activity
of these signaling pathways and the downstream targets, thereby
stimulating the proliferation and invasion of EC cells.
These signaling pathways are involved in numerous
physiological and pathological activities. Future studies should
thus focus on controlling the activity of these signaling pathways
by m6A methylation, as aberrant activation of these pathways may be
a significant oncogenic driver of human malignancies. m6A
methylation is involved in various biological processes in EC by
affecting RNA metabolism and participating in the regulation of RNA
expression, translation and decay (71).
The following is a summary of the primary
contributions of certain significant m6A regulators in the
occurrence and progression of EC (Fig.
1).
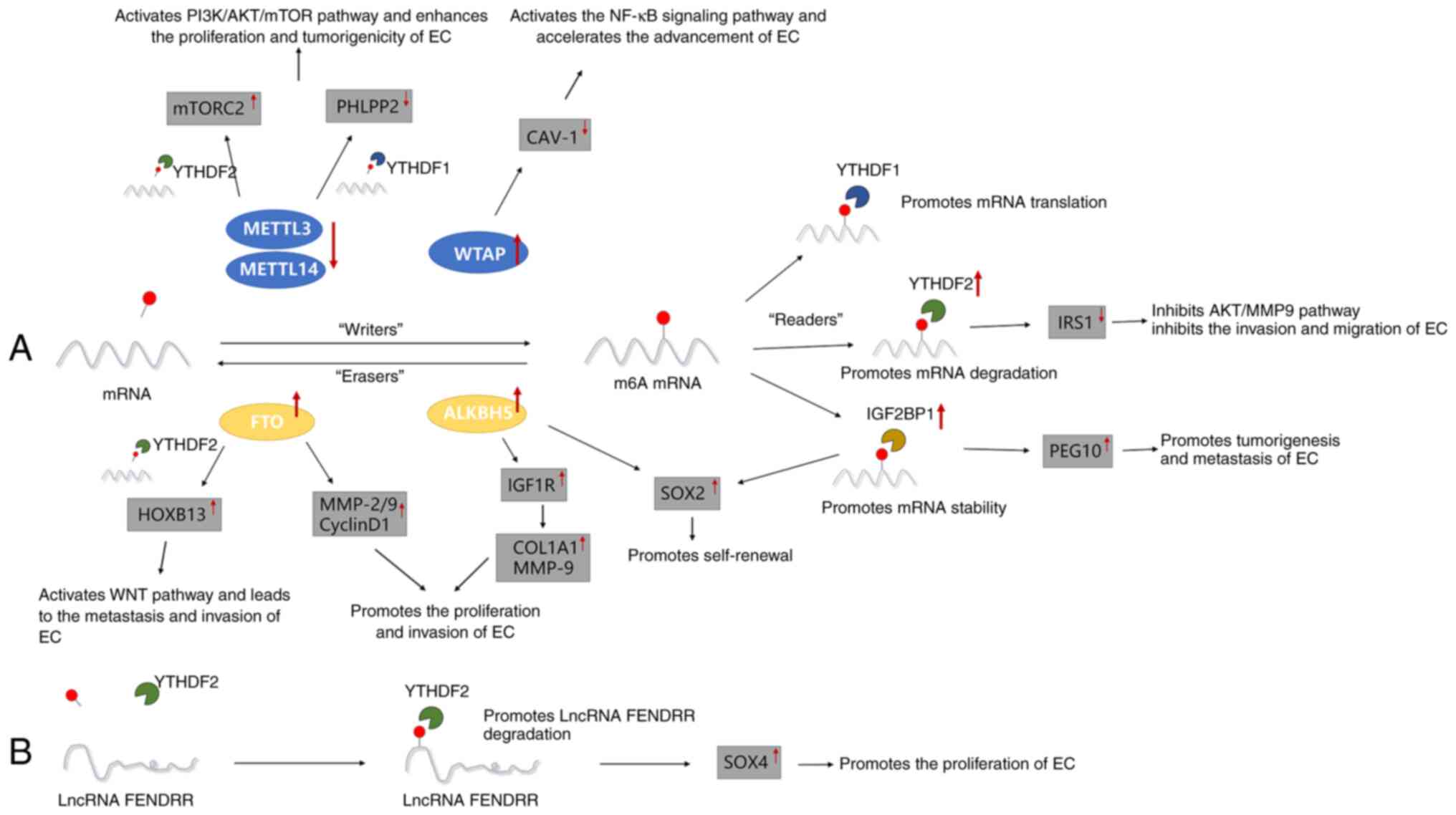 | Figure 1Major roles of different m6A
regulators in the occurrence and development of EC. (A) m6A
methylation regulators METTL3/14, WTAP, FTO, ALKBH5, YTHDF1/2 and
IGF2BP1 promote or inhibit the proliferation, invasion, metastasis
and self-renewal of EC by regulating the m6A-related pathway. (B)
m6A methylation regulator YTHDF2 promotes the proliferation of EC
by downregulating the expression of lncRNA-FENDRR. EC, endometrial
cancer; METTL3/14, methyltransferase-like 3/14; mTORC2, mammalian
target of rapamycin complex 2; PHLPP2, pleckstrin homology domain
and leucine-rich repeat protein phosphatase 2; WTAP, Wilms' tumor
1-associated protein; CAV1, caveolin-1; FTO, fat mass and
obesity-associated protein; ALKBH5, AlkB homolog 5; HOXB13,
homeobox B13; MMP-2/9, matrix metalloproteinases-2/9; IGF1R,
insulin-like growth factor 1 receptor; COL1A1, collagen type I
alpha 1; SOX2, sex-determining region Y-box 2; YTHDF1/2, YT521-B
homology domain family 1/2; IRS1, insulin receptor substrate 1;
IGF2BP1, IGF2 mRNA-binding protein 1; PEG10, paternally expressed
gene 10; lncRNA, long non-coding RNA; SOX4, SRY-related HMG box
transcription factor 4; m6A, N6-methyladenosine. |
METTL3 in EC
The multiple functions, mechanisms and abnormal
regulation of METTL3 are associated with a wide range of human
tumors. METTL3 is related to a variety of processes in neoplasm
progression, including proliferation, aggressiveness, metastasis
and drug resistance (72). These
outcomes are mediated by stem cell self-renewal, miRNA processing
via DGCR8, EMT, apoptosis, the PI3K/AKT pathway and recruitment of
eIF3h. Most potential mechanisms involve multiple m6A-dependent
signaling pathways, including the PI3K/AKT pathway. METTL3 knockout
resulted in a decrease in m6A and thus facilitated the
proliferation and invasive ability of tumor cells by activating the
PI3K-AKT signaling pathway. METTL3 has different roles in different
types of cancers (73). In most
cancer tissues, METTL3 is found to be upregulated and carcinogenic,
such as lung cancer (74),
leukemia (75,76), gastric cancer (77,78)
and ovarian cancer (79). By
contrast, METTL3 is downregulated in renal cell carcinoma where it
acts as a tumor suppressor (80).
Even in the same type of cancer, certain studies
have indicated mutually contradictory outcomes. For instance,
METTL3 is highly upregulated and associated with unfavorable
prognosis in bladder cancer (81,82).
On the contrary, other studies revealed that the absence of METTL3
notably stimulated the growth of bladder cancer cells and acted as
a tumor suppressor gene in the disease (83). Likewise, Cai et al (84) found that METTL3, as an oncogene,
was upregulated in breast cancer. Instead, Wu et al
(85) reported that METTL3, as a
tumor suppressor, was downregulated in breast cancer.
According to Liu et al (86), loss of function mutations in
METTL14 or decreased METTL3 expression may underlie the fact that
m6A methylation levels in ~70% of EC were much lower than those in
normal endometrial tissues.
The levels of m6A modifications were reduced due to
the decreased expression of METTL3 and METTL14, which increased the
proliferation and tumorigenicity of EC. According to further
research, a reduction in the levels of m6A methylation may boost
cell growth by regulating vital enzymes in the AKT signaling
pathway. Dysfunctional AKT signaling may lead to cancer, IR, type-2
diabetes, autoimmune diseases and cardiovascular disease, as well
as inflammatory and neurological disorders (87,88).
The genomic investigation demonstrated that the PI3K/AKT/mTOR
signaling pathway, essential for cell metabolism, growth and
survival (89), is typically
upregulated in EC (69).
In terms of the mechanism, the major AKT Ser473
kinase is the mTORC2. AKT lacking Ser473 phosphorylation is active
but the activity is markedly reduced, and phosphorylation of Ser473
stabilizes both Thr308 phosphorylation and the activation state of
AKT. The decrease in m6A methylation levels may prevent the decay
of the mRNA encoding the mTORC2 complex, which is promoted by
YTHDF2, increasing the expression of mTORC2. mTORC2 is a positive
regulator of AKT activation and promotes AKT activation by
phosphorylating the serine residue at position 473 (69,87,89).
On the contrary, m6A methylation reduction reduces PHLPP2
translation promoted by YTHDF1. PHLPP2 is a negative regulatory
factor in AKT activation and inhibits the AKT signaling pathway
through dephosphorylation of a serine residue at position 473.
Since the loss of PHLPP activity leads to the hyperphosphorylation
of AKT, it stands to reason that the expression of PHLPP1/2 is
reduced or lost in several types of cancer (87).
Therefore, the decrease of m6A mRNA methylation will
affect a variety of AKT pathway components and promote the
proliferation and tumorigenicity of tumor cells by activating the
AKT signaling pathway. The possible mechanism is presented in
Fig. 2. On the contrary, the
enhanced proliferation brought about by a reduction in m6A
methylation was reversed by inhibiting AKT activation.
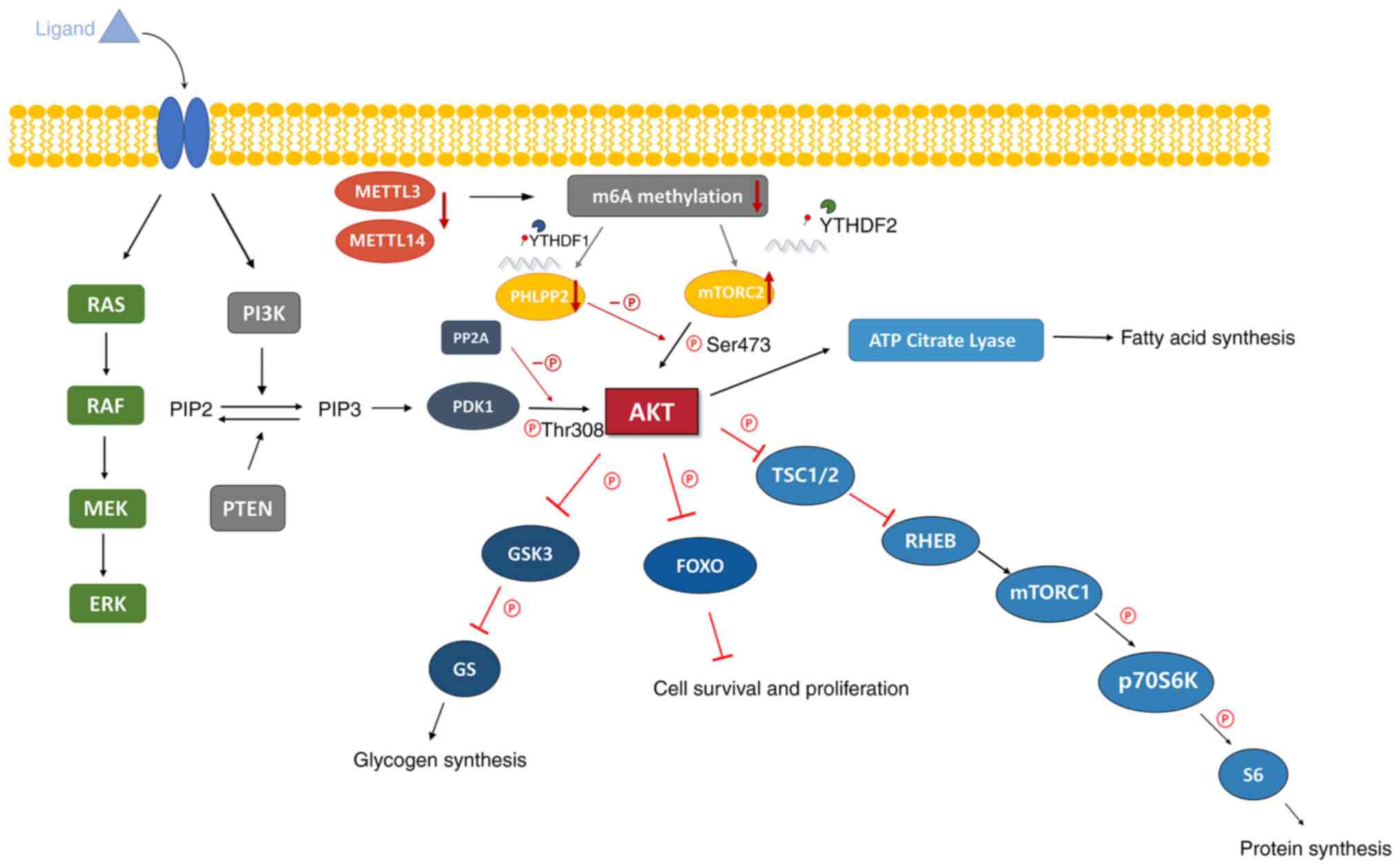 | Figure 2The levels of m6A modifications were
reduced due to the decreased expression of METTL3 and METTL14,
which influenced the expression of PHLPP2 and mTORC2 through
YTHDF1/2, activating AKT signaling pathway to promote the
proliferation and tumorigenicity of tumor cells. PI3K/AKT signaling
pathway. AKT is a serine and threonine kinase, which leads to the
phosphorylation of serine and threonine residues on target
proteins. The growth factor binds to its ligands and activates RTK
or GPCR on the cell membrane, so as to activate PI3K. The activated
PI3K phosphorylates PIP2 and converts it into PIP3, which increases
the level of PIP3, activates PDK1 and activates AKT. i) AKT
phosphorylates GSK3, which is an inhibitor of glycogen synthesis,
and inhibits its activity, resulting in the activation of GS and
increasing glycogen synthesis. ii) AKT inhibits FOXO, which
inhibits cell survival and proliferation, thereby increasing cell
survival and proliferation. iii) AKT phosphorylates TSC1/2 and
inhibits its negative regulation of RHEB, which is an activator of
mTORC1, thereby activating mTORC1, resulting in the activation of
P70S6K and S6 and promoting protein synthesis. iv) AKT activates
ATP citrate lyase and promotes fatty acid synthesis. RTK, receptor
tyrosine kinase; GPCR, G protein coupled receptors; PI3K,
phosphoinositide 3-kinase; PIP2, phosphatidylinositol
4,5-biphosphate; PIP3, phosphatidylinositol (3-5)-triphosphate; PDK1,
3-phosphoinositide-dependent kinase 1; GSK3, glycogen synthase
kinase 3; FOXO, forkhead box O; TSC1/2, Tuberous Sclerosis Complex
1/2; RHEB, Ras homologue enriched in brain; mTORC1/2, mammalian
target of rapamycin complex 1/2; P70S6K, protein 70 S6 kinase;
METTL3/14, methyltransferase-like 3/14; YTHDF1/2, YT521-B homology
domain family 1/2; PHLPP2, pleckstrin homology domain and
leucine-rich repeat protein phosphatase 2; PP2A, protein
phosphatase 2 A. |
In conclusion, the reduction in m6A methylation may
be an oncogenic mechanism in EC and the PI3K/AKT/mTOR pathway may
serve as a therapeutic target, suggesting that altering the
activity of AKT through an m6A-dependent approach may provide novel
approaches for the treatment of malignancies (86).
However, according to Ralser et al (90), the expression levels of METTL3 were
increased, which had prognostic significance and was connected to
the short OS of patients with EC. They speculated that increased
METTL3 expression may result in lower sensitivity to platinum-based
chemotherapy, which is the first-line chemotherapeutic regimen for
advanced EC.
As the main catalytic enzyme in m6A methylation,
METTL3 functions via a complex mechanism and involves a variety of
signaling pathways. The exact mechanisms in cancer remain to be
completely elucidated and require further research.
WTAP in EC
WTAP is a nuclear protein that functions as a
mammalian splicing factor. An increasing body of knowledge has
indicated that WTAP, as an oncogene, is closely related to
different malignancies. For instance, WTAP is substantially
expressed in high-grade serous ovarian cancer (HGSOC), where it is
significantly correlated with lymph node metastasis and a poor
prognostic outcome. It is a prognostic marker of HGSOC and
regulates the progression of ovarian cancer cells (91). In diffuse large B-cell lymphoma
tissues, WTAP is continuously upregulated, promoting cell growth
and counteracting apoptosis, and WTAP was able to form a complex
with BCL6 via Hsp90 (92). WTAP
physically binds to the 3'UTR of CDK2 transcripts and increases the
stability of those transcripts and is significantly overexpressed
and serves as an oncogene in renal cell cancer (93). WTAP is overexpressed in
hepatocellular carcinoma and is closely associated with poor
prognosis (94). In gastric
cancer, WTAP enhances the stability of HK2 mRNA and promotes the
Warburg effect and tumor cell proliferation (95).
However, it remains to be fully elucidated how WTAP
may affect EC. Li et al (96) studied the expression levels of WTAP
in cancer tissues and para-cancerous tissues from a patient with
EC. They observed that WTAP was markedly overexpressed in cancer
tissues and associated with poor prognosis. In vivo and
in vitro, cell proliferation, invasion and migration were
enhanced, and EC apoptosis was reduced, indicating a higher degree
of malignancy and unfavorable survival outcomes. Cell invasion and
migration may be significantly decreased by WTAP knockdown. When
WTAP was knocked out, the expression of CAV-1, a possible WTAP
target, was increased and the enrichment of m6A and METTL3 in its
3'UTR was lowered. In addition, CAV1 prevented activation of the
NF-κB signaling pathway. In conclusion, WTAP may methylate the
3'UTR of CAV-1 and inhibit its expression in order to activate the
NF-κB signaling pathway, thus accelerating the advancement of
EC.
FTO in EC
Previous reports have indicated that FTO is
abundantly expressed in various human malignancies (97) and increases cancer cell metabolism,
thus leading to tumorigenesis and chemotherapeutic resistance
(98). The elevated expression of
FTO in bladder tumor tissue and its association with a poorer
prognosis highlights the potential of FTO in the diagnosis and/or
prognosis of this disease (99).
FTO is involved in maintaining self-renewal and immune evasion of
cancer stem cells. FTO is overexpressed in leukemia and FTO
inhibition renders leukemia cells susceptible to T-cell
cytotoxicity and thus overcome immune escape (100), indicating that FTO is a
prospective therapeutic target.
There has been a constant rise in both the incidence
and mortality of EC. This trend is largely the result of the
worldwide obesity epidemic. Obesity is more strongly associated
with the progression of EC than any other group of female cancers
(13). Despite the fact that the
connection between obesity and EC has been confirmed by numerous
studies, the molecular mechanisms underlying this association have
not been fully elucidated.
FTO is overexpressed in EC and serves as an
indicator of poor prognosis (101). Recently, it was reported that FTO
removes m6A modification from HOXB13 mRNA, prevents YTHDF2-mediated
HOXB13 from being degraded, enhances its expression, and
accelerates the metastasis and invasion of EC (102), which are significant biological
processes that contribute to poor prognosis (103). Mechanistically, HOXB13 is a
homeobox transcription factor whose expression is increased in EC,
leading to an increase in invasive capacity. It is possible that
HOXB13 contributes to the promotion of tumor metastasis during the
course of tumor development. FTO may catalyze the demethylation of
the HOXB13 mRNA 3' UTR region, which inhibits the recognition of
m6A modifications by YTHDF2, preventing the degradation of HOXB13
mRNA, increasing its expression and thereby activating the WNT
signaling pathway, and thus promoting EC invasion and
metastasis.
In addition, growing evidence suggests a connection
between long-term estrogen exposure and the development of type I
EC. Estrogen is still regarded as a significant factor in abnormal
hyperplasia and tumorigenesis, as in the absence of progesterone
antagonism, constant estrogen stimulation results in abnormal
hyperplasia of endometrial epithelial cells (104).
Zhang et al (101) discovered that estradiol may
induce FTO expression and upregulate MMP-2, MMP-9 and cyclin D1
expression by binding to estrogen receptors (ER) and activating the
PI3K/AKT and MAPK signaling pathways, promoting the proliferation
and invasion of EC cells.
Zhu et al (105) further determined that estrogen
may promote FTO protein nuclear localization and promote
proliferation through the mTOR signaling pathway in EC cells. The
possible mechanism is presented in Fig. 3.
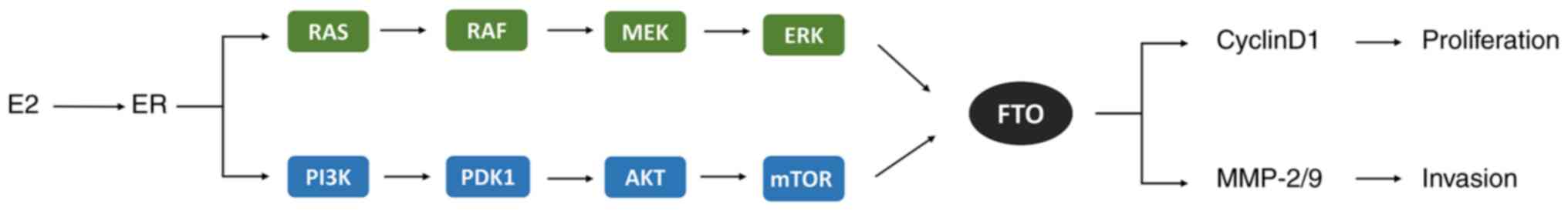 | Figure 3E2 induces FTO overexpression by
binding to the ER and activating the PI3K/AKT and MAPK signaling
pathways to reduce m6A methylation and upregulate MMP-2, MMP-9 and
cyclin D1 molecules, promoting the proliferation and invasion in
endometrial cancer cells. E2, estradiol; ER, estrogen receptor;
MAPK, mitogen-activated protein kinase; RAS, a small GTP-binding
protein; RAF, MAP kinase kinase kinase (MAPKKK); MEK, MAP kinase
kinase (MAPKK); ERK, extracellular signal-regulated kinase (MAPK);
PI3K, phosphoinositide 3-kinase; PDK1, 3-phosphoinositide-dependent
kinase 1; mTOR, mammalian target of rapamycin complex; FTO, fat
mass and obesity-associated protein; MMP-2/9, matrix
metalloproteinases-2/9. |
ALKBH5 in EC
ALKBH5, another RNA demethylase, is involved in the
pathological processes of several types of cancer. In glioblastoma
stem-like cells (GSCs), ALKBH5 expression is upregulated, which in
turn increases FOXM1 expression, maintaining the tumorigenicity of
GSCs and being indicative of poor prognosis (106). Similarly, under intermittent
hypoxia, ALKBH5 downregulated m6A modification, increased FOXM1
expression and promoted the proliferation and invasion of lung
adenocarcinoma cells (107).
ALKBH5 promotes tumorigenesis and self-renewal of tumor stem cells
in acute myeloid leukemia (AML) and its increased expression is
also associated with poor prognostic outcomes in AML (108). In addition, ALKBH5 has been
reported as a target of HIF-1α (109). Hypoxia induces ALKBH5 expression
in breast cancer cells. HIF-dependent ALKBH5 stabilizes NANOG mRNA
by removing m6A methylation and induces a breast cancer stem cell
phenotype (110). By contrast,
ALKBH5 levels were low in most pancreatic cancer (PC) samples.
Deletion of ALKBH5 is a feature of the occurrence and adverse
clinicopathological findings of patients with PC. ALKBH5 activates
PER1 through m6A demethylation in a YTHDF2-dependent manner,
thereby inhibiting tumor proliferation and invasion and preventing
PC progression (111). In
conclusion, ALKBH5 exerts differential effects in different tumor
types through complex mechanisms. ALKBH5 is significantly
upregulated in EC. Pu et al (112) found that downregulation ALKBH5
inhibited the growth and invasive ability of EC cells and IGF1R
expression regulation is a crucial intermediate mechanism of these
ALKBH5-mediated alterations in endometrial cell invasion.
Mechanistically, ALKBH5 primarily regulates the demethylation of
IGF1R, thereby stabilizing IGF1R mRNA and promoting its
translation, and activating the IGF1R signaling pathway, which in
turn stimulated the production of COL1A1 and MMP9 and promoted the
proliferation and invasion of EC, indicating that it is a potential
target for the treatment of EC.
Furthermore, alterations to IGF1 expression and
signaling are crucial for regulating normal uterine physiology
(112). Elevated levels of IGF1
and hyperinsulinemia are involved in the pathogenesis of EC.
Endometrial hyperplasia is associated with increased insulin and
IGF1 receptor expression, which increases the susceptibility of
these cells to insulin and IGF1 and promotes the hyperactivity of
MAPK and PI3K/AKT/mTOR signaling frequently observed in EC
(13).
Chen et al (113) indicated that ALKBH5 promoted SOX2
transcription through HIF-dependent m6A demethylation, maintaining
the EC stem cell (ECSC) status and tumor characteristics. ECSCs are
stem cell-like cells with the ability to differentiate and
self-renew, which are essential for the progression of EC. Under
hypoxic conditions, the levels of ALKBH5 are significantly
increased in ECSCs, which decreases m6A levels and promotes SOX2
expression. SOX2 is a core stem cell transcription factor and
mediates the early steps of tumorigenesis. In summary, these
studies suggest that the HIF-ALKBH5-SOX2 axis mediated by m6A
methylation has a crucial function in the process of ECSC expansion
under hypoxic conditions. ALKBH5, as a promising therapeutic
target, highlights novel avenues for the clinical treatment of
malignant tumors.
YTHDF2 in EC
YTHDF2 is an m6A reader protein that accelerates the
degradation of m6A-modified transcripts (50). Several tumors have been indicated
to abnormally express YTHDF2, including ovarian cancer (114,115), cervical cancer (116), gastric carcinoma (117) and hepatocellular carcinoma
(118).
YTHDF2 was significantly upregulated in EC.
According to Hong et al (119), YTHDF2 knockdown markedly
accelerated endometrial cell proliferation, migration and invasion.
Conversely, overexpression of YTHDF2 exerted a significant
inhibitory role, indicating that YTHDF2 may inhibit the activity of
EC cells. Furthermore, they confirmed that knockdown of YTHDF2
increased MMP9 expression. In subsequent studies, based on m6A-seq
data, it was indicated that insulin receptor substrate 1 (IRS1) had
m6A methylation modifications in EC. By immunoprecipitation, the
presence of m6A sites on the IRS1 transcript was confirmed and it
was indicated that YTHDF2 inhibited the activity of EC cells likely
through inhibition of MMP9 expression in an IRS1-dependent
manner.
IRSs, including IRS1 and IRS2, have a central role
in the insulin signaling cascade by linking insulin and IGF1R to
PI3K/AKT activation (120).
According to reports, IRS1 is critical for cancer cell hyperplasia
and mediates drug resistance, while IRS2 primarily affects the
motility and metastasis of cancer cells (121). IRS1 and IRS2 phosphorylation
attracts downstream factors to activate the MAPK and PI3K cascades
(122).
Mechanistically, YTHDF2 promotes the degradation of
IRS1 mRNA, thereby inhibiting its expression, leading to the
inhibition of the AKT/MMP9 signaling pathway, ultimately inhibiting
the activity of EC cells (119).
When YTHDF2 was knocked down, IRS1 expression was increased and the
AKT signaling pathway was activated. These results indicate that
YTHDF2 may be a tumor suppressor.
Conversely, Shen et al (123) found that the degradation of long
ncRNA (lncRNA) FENDRR mediated by YTHDF2 stimulated cell
proliferation in EC. NcRNAs are associated with m6A modification,
thereby affecting gene expression and the development of cancer
(124). LncRNA FENDRR is a
well-known tumor suppressor gene in several malignancies, including
breast cancer (125), colon
cancer (126) and prostate cancer
(127). Dysregulation of lncRNA
FENDDR is closely associated with the development of several types
of cancer, including EC. Recently, lncRNA FENDRR was identified as
an aberrantly expressed molecule in ERα-related EC (68), although the mechanisms of action
remain elusive.
According to Shen et al (123), EC tissues had higher levels of
m6A methylation of the lncRNA FENDRR, whilst exhibiting lower
levels of lncRNA FENDRR expression. In vitro and in
vivo experiments indicated that YTHDF2 recognized the
m6A-modified lncRNA FENDRR and promoted its degradation.
Overexpression of lncRNA FENDRR inhibited the proliferation and
promoted the apoptosis of the EC cells by decreasing SOX4 protein
levels.
In conclusion, the higher levels of m6A modification
of lncRNA FENDRR in EC tissues promote the degradation of lncRNA
FENDRR, reducing its expression by recruiting YTHDF2. Subsequently,
SOX4 protein accumulates, which promotes the proliferation of EC
cells. These findings suggest that YTHDF2 may be an oncogene.
In addition, atypical endometrial
hyperplasia/endometrioid intraepithelial neoplasia (EAH/EIN) has a
higher risk of developing into endometrioid adenocarcinoma (EMAC)
than endometrial hyperplasia without atypia (EHWA) and is
considered a precancerous lesion of EMAC.
According to Bian et al (128), YTHDF2 was weakly expressed in the
normal endometrium and EHWA, whilst being upregulated in EAH/EIN
and EMAC, indicating that YTHDF2 may be a valuable marker for
differentiating between EAH/EIN and EHWA, which makes earlier
clinical detection and intervention of these precancerous lesions
possible.
IGF2BP1 in EC
IGF2BPs, including IGF2BP1/2/3, are a distinct group
of m6A readers that are able to identify m6A modifications
(64). IGF2BPs influence gene
regulation and cancer biology by enhancing the stability and
storage of their target mRNAs. IGF2BP1 is a conserved oncogenic
driver with significantly upregulated expression in various types
of cancer (129,130).
It was discovered that IGF2BP1 has a critical part
in the regulation of EC through the recognition of m6A-modified
PEG10 mRNA. IGF2BP1 mRNA expression was significantly higher in EC
tissues than in normal tissues and it was associated with
unfavorable prognosis, tumor grade and stage (131). IGF2BP1 regulates the tumor cell
cycle and tumor growth and is able to stimulate or inhibit cell
proliferation when it is upregulated or knocked down,
respectively.
Zhang et al (132) indicated that IGF2BP1 is able to
recognize and interact with PEG10 mRNA and promote PEG10 mRNA
stability and expression. The stability of PEG10 mRNA and protein
expression were both markedly decreased when IGF2BP1 was silenced.
They further indicated that PABPC1 functions in this mechanism in a
synergistic manner. PABPC1 is a typical poly-A binding protein that
is able to bind to and stabilize mRNA, preventing the mRNA tail
from being cut off by nucleases (133). PEG10 is suspected to be an
oncogene that has a role in tumor cell proliferation, apoptosis and
metastasis (134). Previous
research suggested that PEG10 is overexpressed in several diseases,
including liver cancer, pancreatic cancer and bladder cancer
(135-137).
According to research by Zhang et al
(132), PEG10 was upregulated in
EC and was directly associated with the survival rate.
Mechanistically, IGF2BP1 recognizes the m6A modification of the
3'UTR of PEG10 mRNA and recruits PABPC1 to synergistically
stabilize PEG10 mRNA to promote its protein expression and
proliferation of EC cells. Furthermore, a significant portion of
the PEG10 protein binds to the p16 and p18 promoter regions,
limiting RNA and protein expression and increasing cell cycle
progression.
According to Xue et al (67), MEK1 citrullination caused by PADI2
activates ERK1/2 and helps IGF2BP1 to stabilize SOX2 mRNA in EC. In
fact, several different malignancies have been reported to exhibit
upregulation of PADIs compared with the respective healthy tissues
(138). PADI2 converts arginine
to citrulline; its expression is positively associated with the
development of EC and it is required for proliferation, migration
and invasion.
Mechanistically, PADI2 interacts with and catalyzes
MEK1 citrullination, which activates ERK1/2, thereby increasing the
expression of IGF2BP1. IGF2BP1 enhances the stability of SOX2 mRNA.
There is an enrichment of three nonredundant m6A sites (GGACH)
around the 3'UTR of the SOX2 gene and m6A modification on these
three sites stabilizes SOX2 mRNA and increases its protein
expression. IGF2BP1 binds to these three m6A sites to maintain the
stability of the transcripts and prevent SOX2 mRNA degradation.
Aberrant expression of IGF2BP1 mediated by PADI2/MEK1/ERK signaling
leads to the accumulation of SOX2, supporting the malignant state
of EC. Thus, patients with EC may benefit from a therapeutic
strategy that targets the PADI2/MEK1/ERK/IGF2BP1 axis (67).
Other m6A regulators in EC
The prognosis of EC is tightly associated with
changes in the m6A regulator, whose alterations may serve as a
valid and trustworthy marker for EC prognosis (139,140). According to research by Ma et
al (131), the expression of
ZC3H13, YTHDC1 and METTL14 in EC tissues was significantly
decreased, which were thus considered potential diagnostic and
prognostic biomarkers for EC. The expression of ZC3H13, YTHDC1 and
METTL14 in EC tissues is positively correlated with PD-L1
expression; PD-L1 interacts with PD-1 to suppress anti-tumor
immunity, while blocking PD-L1/PD-1 interaction significantly
enhances the anti-tumor immune response (141), indicating that blocking these
proteins may enhance the effects of immunotherapy.
According to Zhai et al (142), RBM15/15B, YTHDF1 and IGF2BP1/2
are upregulated in endometrial adenocarcinoma. By contrast, FTO,
KIAA1429, METTL14, ZC3H13 and YTHDC1 expression was downregulated.
In their study, decreased FTO expression in tumors conflicted with
previous findings; they speculated that this discrepancy may be
related to the pathological type of EC used in their study. RBM15,
FTO and YTHDF1 were identified as prognostic biomarkers in EC that
may be involved in cell cycle regulation, affect RNA processing and
translation, and contribute to tumor-associated processes and
prognosis of endometrial adenocarcinoma. However, the exact
mechanisms of these regulators have remained to be fully elucidated
and further study is required.
5. Conclusions and future perspectives
At present, the primary treatment of EC comprises
surgery, radiation and chemotherapy. These conventional treatment
methods may result in trauma for patients and the adverse reactions
of radiotherapy and chemotherapy are well documented, which
seriously affect the quality of a patient's life.
As a hot topic in epigenetics, m6A modifications
have attracted considerable attention, providing us with novel
ideas and methods for the treatment of EC. A total of three types
of regulators carry out the dynamic reversible m6A modification
process: Methyltransferases (referred to as writers), demethylases
(referred to as erasers) and m6A binding proteins (referred to as
readers). Through these regulators, splicing, translation,
stability and the decay of mRNAs are regulated. Oncogenes and tumor
suppressor genes are regulated by m6A modifications, which have an
impact on the occurrence and development of malignancies. At the
same time, m6A modification may affect its role in cancer by
regulating the levels of m6A modifications and the expression of
regulators. Therefore, m6A regulators are anticipated to become
potential targets for cancer therapy. Recent research has indicated
that the expression of multiple m6A regulators is either up- or
downregulated in EC, and m6A methylation, as well as related
regulators, are critical in the development and progression of
EC.
Despite the fact that m6A modifications were
initially identified in the 1970s (143), its function was not investigated
in detail until ~2012. The development of high-throughput
sequencing technologies has made it easier than ever to study RNA
modifications (144) and has
provided a necessary basis for elucidating the unique molecular
characteristics of transcriptomes on m6A (145). Future studies using m6A-seq and
MeRIP will help to deeper analyze m6A regulators in EC. By
determining the genetic risk prognosis model to forecast the rate
of survival of patients with EC, certain m6A regulatory factors may
be employed as promising markers to predict clinical outcomes of
cancer patients and offer a theoretical foundation or target for
the treatment of EC.
The present review summarized the primary functions
of the significant m6A regulators in EC, with the aim of providing
novel methods and approaches for the diagnosis or prognosis of EC.
To date, certain small chemical molecule inhibitors that target m6A
regulators have demonstrated considerable promise for preventing
the growth of cancer (7). For
instance, R-2-hydroxyglutarate (R-2HG) reduces FTO activity and
raises m6A levels in R-2HG-sensitive leukemia cells, further
reducing the stability of MYC/CEBPA transcripts and finally
inhibiting the activity of leukemia cells, promoting cell cycle
arrest and apoptosis, and exerting anti-leukemic effects (146). Carbonic anhydrase IV interacts
with WTAP and promotes WTAP protein degradation, facilitates the
transcriptional activity of WT1, an inhibitor of the WNT pathway,
and inhibits the occurrence and development of colon cancer
(147). In general, m6A
regulator-specific inhibitors offer a potentially valuable
alternative approach for cancer treatment.
The research on m6A in EC is still in its early
stages and there are numerous up- and downstream m6A regulators
whose precise mechanisms remain elusive and require to be
investigated. In addition, the changes in the levels of certain m6A
regulators were demonstrated to be closely related to the prognosis
of EC and different types of EC with different molecular
characteristics also have different prognoses. We speculate whether
there are any differences in m6A RNA methylation levels, the
expression of regulatory factors, and related mechanisms in
different molecular types of EC. At present, the research on RNA
m6A methylation in different molecular types of EC is still
insufficient. It is hypothesized that m6A regulators will serve as
valuable targets for cancer therapy and offer novel approaches for
treating malignancies. However, the clinical application of
m6A-based cancer treatment requires a considerable amount of
research before these treatments may be used in the clinic.
Availability of data and materials
Not applicable.
Authors' contributions
JT and JG designed the study. SS selected/searched
the literature and drafted the manuscript. NL and QC revised the
manuscript. All authors have read and approved the final
manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This work was supported by a special research fund for
gynecological oncology 'Le Foundation' (grant no.
KH-2021-LZJJ-001).
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Crosbie EJ, Kitson SJ, McAlpine JN,
Mukhopadhyay A, Powell ME and Singh N: Endometrial cancer. Lancet.
399:1412–1428. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
MacKay HJ, Freixinos VR and Fleming GF:
Therapeutic targets and opportunities in endometrial cancer: Update
on endocrine therapy and nonimmunotherapy targeted options. Am Soc
Clin Oncol Educ Book. 40:1–11. 2020.PubMed/NCBI
|
|
4
|
Yang J, Chen J, Fei X, Wang X and Wang K:
N6-methyladenine RNA modification and cancer. Oncol Lett.
20:1504–1512. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhou Z, Lv J, Yu H, Han J, Yang X, Feng D,
Wu Q, Yuan B, Lu Q and Yang H: Mechanism of RNA modification
N6-methyladenosine in human cancer. Mol Cancer. 19:1042020.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang G, Sun Z and Zhang N: Reshaping the
role of m6A modification in cancer transcriptome: A review. Cancer
Cell Int. 20:3532020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Han X, Wang M, Zhao YL, Yang Y and Yang
YG: RNA methylations in human cancers. Semin Cancer Biol.
75:97–115. 2021. View Article : Google Scholar
|
|
8
|
Shen S, Zhang R, Jiang Y, Li Y, Lin L, Liu
Z, Zhao Y, Shen H, Hu Z, Wei Y and Chen F: Comprehensive analyses
of m6A regulators and interactive coding and non-coding RNAs across
32 cancer types. Mol Cancer. 20:672021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu AM, Jian C, Yu AH and Tu MJ: RNA
therapy: Are we using the right molecules? Pharmacol Ther.
196:91–104. 2019. View Article : Google Scholar :
|
|
10
|
Arend RC, Jones BA, Martinez A and
Goodfellow P: Endometrial cancer: Molecular markers and management
of advanced stage disease. Gynecol Oncol. 150:569–580. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gu B, Shang X, Yan M, Li X, Wang W, Wang Q
and Zhang C: Variations in incidence and mortality rates of
endometrial cancer at the global, regional, and national levels,
1990-2019. Gynecol Oncol. 161:573–580. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ryan NAJ, Glaire MA, Blake D,
Cabrera-Dandy M, Evans DG and Crosbie EJ: The proportion of
endometrial cancers associated with Lynch syndrome: A systematic
review of the literature and meta-analysis. Genet Med.
21:2167–2180. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Onstad MA, Schmandt RE and Lu KH:
Addressing the role of obesity in endometrial cancer risk,
prevention, and treatment. J Clin Oncol. 34:4225–4230. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hecht JL and Mutter GL: Molecular and
pathologic aspects of endometrial carcinogenesis. J Clin Oncol.
24:4783–4791. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bansal N, Yendluri V and Wenham RM: The
molecular biology of endometrial cancers and the implications for
pathogenesis, classification, and targeted therapies. Cancer
Control. 16:8–13. 2009. View Article : Google Scholar
|
|
16
|
Brooks RA, Fleming GF, Lastra RR, Lee NK,
Moroney JW, Son CH, Tatebe K and Veneris JL: Current
recommendations and recent progress in endometrial cancer. CA
Cancer J Clin. 69:258–279. 2019.PubMed/NCBI
|
|
17
|
León-Castillo A, Gilvazquez E, Nout R,
Smit VT, McAlpine JN, McConechy M, Kommoss S, Brucker SY, Carlson
JW, Epstein E, et al: Clinicopathological and molecular
characterisation of 'multiple-classifier' endometrial carcinomas. J
Pathol. 250:312–322. 2020. View Article : Google Scholar
|
|
18
|
Bell DW and Ellenson LH: Molecular
genetics of endometrial carcinoma. Annu Rev Pathol. 14:339–367.
2019. View Article : Google Scholar
|
|
19
|
Wang M and Hui P: A timely update of
immunohistochemistry and molecular classification in the diagnosis
and risk assessment of endometrial carcinomas. Arch Pathol Lab Med.
145:1367–1378. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cancer Genome Atlas Research Network;
Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H,
Robertson AG, Pashtan I, Shen R, et al: Integrated genomic
characterization of endometrial carcinoma. Nature. 497:67–73. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Murali R, Soslow RA and Weigelt B:
Classification of endometrial carcinoma: more than two types.
Lancet Oncol. 15:e268–e278. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Winterhoff B, Thomaier L, Mullany S and
Powell MA: Molecular characterization of endometrial cancer and
therapeutic implications. Curr Opin Obstet Gynecol. 32:76–83. 2020.
View Article : Google Scholar
|
|
23
|
Ferlay J, Colombet M, Soerjomataram I,
Dyba T, Randi G, Bettio M, Gavin A, Visser O and Bray F: Cancer
incidence and mortality patterns in Europe Estimates for 40
countries and 25 major cancers in 2018. Eur J Cancer. 103:356–387.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
McAlpine JN, Temkin SM and Mackay HJ:
Endometrial cancer: Not your grandmother's cancer. Cancer.
122:2787–2798. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hazelwood E, Sanderson E, Tan VY, Ruth KS,
Frayling TM, Dimou N, Gunter MJ, Dossus L, Newton C, Ryan N, et al:
Identifying molecular mediators of the relationship between body
mass index and endometrial cancer risk: A mendelian randomization
analysis. BMC Med. 20:1252022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Merritt MA, Strickler HD, Hutson AD,
Einstein MH, Rohan TE, Xue X, Sherma ME, Brinton LA, Yu H, Miller
DS, et al: Sex hormones, insulin, and insulin-like growth factors
in recurrence of high-stage endometrial cancer. Cancer Epidemiol
Biomarkers Prev. 30:719–726. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Meyer KD, Saletore Y, Zumbo P, Elemento O,
Mason CE and Jaffrey SR: Comprehensive analysis of mRNA methylation
reveals enrichment in 3' UTRs and near stop codons. Cell.
149:1635–1646. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang H, Weng H and Chen J: m6A
modification in coding and non-coding RNAs: Roles and therapeutic
implications in cancer. Cancer Cell. 37:270–288. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Huang H, Weng H and Chen J: The biogenesis
and precise control of RNA m6A methylation. Trends
Genet. 36:44–52. 2020. View Article : Google Scholar
|
|
30
|
Wang T, Kong S, Tao M and Ju S: The
potential role of RNA N6-methyladenosine in cancer progression. Mol
Cancer. 19:882020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
He L, Li H, Wu A, Peng Y, Shu G and Yin G:
Functions of N6-methyladenosine and its role in cancer. Mol Cancer.
18:1762019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hu Y, Wang S, Liu J, Huang Y, Gong C, Liu
J, Xiao Y and Yang S: New sights in cancer: Component and function
of N6-methyladenosine modification. Biomed Pharmacother.
122:1096942020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
He PC and He C: m6 A RNA methylation: From
mechanisms to therapeutic potential. EMBO J. 40:e1059772021.
View Article : Google Scholar
|
|
34
|
Lin S, Choe J, Du P, Triboulet R and
Gregory RI: The m(6)A methyltransferase METTL3 promotes translation
in human cancer cells. Mol Cell. 62:335–345. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang
L, Jia G, Yu M, Lu Z, Deng X, et al: A METTL3-METTL14 complex
mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem
Biol. 10:93–95. 2014. View Article : Google Scholar :
|
|
36
|
Huang J, Dong X, Gong Z, Qin LY, Yang S,
Zhu YL, Wang X, Zhang D, Zou T, Yin P and Tang C: Solution
structure of the RNA recognition domain of METTL3-METTL14
N6-methyladenosine methyltransferase. Protein Cell.
10:272–284. 2019. View Article : Google Scholar
|
|
37
|
Ping XL, Sun BF, Wang L, Xiao W, Yang X,
Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, et al: Mammalian WTAP is
a regulatory subunit of the RNA N6-methyladenosine
methyltransferase. Cell Res. 24:177–189. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang
Z, Cheng T, Gao M, Shu X, Ma H, et al: VIRMA mediates preferential
m6A mRNA methylation in 3'UTR and near stop codon and
associates with alternative polyadenylation. Cell Discov. 4:102018.
View Article : Google Scholar
|
|
39
|
Patil DP, Chen CK, Pickering BF, Chow A,
Jackson C, Guttman M and Jaffrey SR: m(6)A RNA methylation promotes
XIST-mediated transcriptional repression. Nature. 537:369–373.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhu D, Zhou J, Zhao J, Jiang G, Zhang X,
Zhang Y and Dong M: ZC3H13 suppresses colorectal cancer
proliferation and invasion via inactivating Ras-ERK signaling. J
Cell Physiol. 234:8899–8907. 2019. View Article : Google Scholar
|
|
41
|
Wen J, Lv R, Ma H, Shen H, He C, Wang J,
Jiao F, Liu H, Yang P, Tan L, et al: Zc3h13 regulates nuclear RNA
m6A methylation and mouse embryonic stem cell
self-renewal. Mol Cell. 69:1028–1038.e6. 2018. View Article : Google Scholar
|
|
42
|
Shen D, Wang B, Gao Y, Zhao L, Bi Y, Zhang
J, Wang N, Kang H, Pang J, Liu Y, et al: Detailed resume of RNA
m6A demethylases. Acta Pharm Sin B. 12:2193–2205. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang
Y, Yi C, Lindahl T, Pan T, Yang YG and He C: N6-methyladenosine in
nuclear RNA is a major substrate of the obesity-associated FTO. Nat
Chem Biol. 7:885–887. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wei J, Liu F, Lu Z, Fei Q, Ai Y, He PC,
Shi H, Cui X, Su R, Klungland A, et al: Differential
m6A, m6Am m, and m1A
demethylation mediated by FTO in the cell nucleus and cytoplasm.
Mol Cell. 71:973–985.e5. 2018. View Article : Google Scholar
|
|
45
|
Bartosovic M, Molares HC, Gregorova P,
Hrossova D, Kudla G and Vanacova S: N6-methyladenosine demethylase
FTO targets pre-mRNAs and regulates alternative splicing and 3'-end
processing. Nucleic Acids Res. 45:11356–11370. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gao S, Li X, Zhang M, Zhang N, Wang R and
Chang J: Structural characteristics of small-molecule inhibitors
targeting FTO demethylase. Future Med Chem. 13:1475–1489. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zheng G, Dahl JA, Niu Y, Fedorcsak P,
Huang CM, Li CJ, Vågbø CB, Shi Y, Wang WL, Song SH, et al: ALKBH5
is a mammalian RNA demethylase that impacts RNA metabolism and
mouse fertility. Mol Cell. 49:18–29. 2013. View Article : Google Scholar :
|
|
48
|
Qu J, Yan H, Hou Y, Cao W, Liu Y, Zhang E,
He J and Cai Z: RNA demethylase ALKBH5 in cancer: From mechanisms
to therapeutic potential. J Hematol Oncol. 15:82022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhao Y, Shi Y, Shen H and Xie W:
m6A-binding proteins: The emerging crucial performers in
epigenetics. J Hematol Oncol. 13:352020. View Article : Google Scholar
|
|
50
|
Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M,
Ma J and Wu L: YTHDF2 destabilizes m(6)A-containing RNA through
direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun.
7:126262016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Liu J, Gao M, Xu S, Chen Y, Wu K, Liu H,
Wang J, Yang X, Wang J, Liu W, et al: YTHDF2/3 are required for
somatic reprogramming through different RNA deadenylation pathways.
Cell Rep. 32:1081202020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR
and Qian SB: Dynamic m(6)A mRNA methylation directs translational
control of heat shock response. Nature. 526:591–594. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang X, Zhao BS, Roundtree IA, Lu Z, Han
D, Ma H, Weng X, Chen K, Shi H and He C: N(6)-methyladenosine
modulates messenger RNA translation efficiency. Cell.
161:1388–1399. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chen Z, Zhong X, Xia M and Zhong J: The
roles and mechanisms of the m6A reader protein YTHDF1 in tumor
biology and human diseases. Mol Ther Nucleic Acids. 26:1270–1279.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu
PJ, Liu C and He C: YTHDF3 facilitates translation and decay of
N6-methyladenosine-modified RNA. Cell Res. 27:315–328.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Li A, Chen YS, Ping XL, Yang X, Xiao W,
Yang Y, Sun HY, Zhu Q, Baidya P, Wang X, et al: Cytoplasmic
m6A reader YTHDF3 promotes mRNA translation. Cell Res.
27:444–447. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Li S, Qi Y, Yu J, Hao Y, He B, Zhang M,
Dai Z, Jiang T, Li S, Huang F, et al: Nuclear Aurora kinase A
switches m6A reader YTHDC1 to enhance an oncogenic RNA
splicing of tumor suppressor RBM4. Signal Transduct Target Ther.
7:972022. View Article : Google Scholar
|
|
58
|
Kim GW, Imam H and Siddiqui A: The RNA
binding proteins YTHDC1 and FMRP regulate the nuclear export of
N6-methyladenosine-modified hepatitis B virus
transcripts and affect the viral life cycle. J Virol.
95:e00097212021. View Article : Google Scholar
|
|
59
|
Shima H, Matsumoto M, Ishigami Y, Ebina M,
Muto A, Sato Y, Kumagai S, Ochiai K, Suzuki T and Igarashi K:
S-adenosylmethionine synthesis is regulated by selective
N6-adenosine methylation and mRNA degradation involving
METTL16 and YTHDC1. Cell Rep. 21:3354–3363. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y,
Qi M, Lu Z, Shi H, Wang J, et al: Ythdc2 is an
N6-methyladenosine binding protein that regulates
mammalian spermatogenesis. Cell Res. 27:1115–1127. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Alarcón CR, Goodarzi H, Lee H, Liu X,
Tavazoie S and Tavazoie SF: HNRNPA2B1 is a mediator of
m(6)A-dependent nuclear RNA processing events. Cell. 162:1299–1308.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Liu N, Dai Q, Zheng G, He C, Parisien M
and Pan T: N(6)-methyladenosine-dependent RNA structural switches
regulate RNA-protein interactions. Nature. 518:560–564. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Liu N, Zhou KI, Parisien M, Dai Q,
Diatchenko L and Pan T: N6-methyladenosine alters RNA structure to
regulate binding of a low-complexity protein. Nucleic Acids Res.
45:6051–6063. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Huang H, Weng H, Sun W, Qin X, Shi H, Wu
H, Zhao BS, Mesquita A, Liu C, Yuan CL, et al: Recognition of RNA
N6-methyladenosine by IGF2BP proteins enhances mRNA
stability and translation. Nat Cell Biol. 20:285–295. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Song K, Xu H and Wang C: The role of
N6-methyladenosine methylation in the progression of endometrial
cancer. Cancer Biother Radiopharm. Oct 14–2020.Epub ahead of print.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Lan Q, Liu PY, Haase J, Bell JL,
Hüttelmaier S and Liu T: The critical role of RNA m6A
methylation in cancer. Cancer Res. 79:1285–1292. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Xue T, Liu X, Zhang M, E Q, Liu S, Zou M,
Li Y, Ma Z, Han Y, Thompson P and Zhang X: PADI2-catalyzed MEK1
citrullination activates ERK1/2 and promotes IGF2BP1-mediated SOX2
mRNA stability in endometrial cancer. Adv Sci (Weinh).
8:20028312021. View Article : Google Scholar
|
|
68
|
Liu A, Zhang D, Yang X and Song Y:
Estrogen receptor alpha activates MAPK signaling pathway to promote
the development of endometrial cancer. J Cell Biochem.
120:17593–17601. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Barra F, Evangelisti G, Ferro Desideri L,
Di Domenico S, Ferraioli D, Vellone VG, De Cian F and Ferrero S:
Investigational PI3K/AKT/mTOR inhibitors in development for
endometrial cancer. Expert Opin Investig Drugs. 28:131–142. 2019.
View Article : Google Scholar
|
|
70
|
Wang Y, Yin L and Sun X: CircRNA
hsa_circ_0002577 accelerates endometrial cancer progression through
activating IGF1R/PI3K/Akt pathway. J Exp Clin Cancer Res.
39:1692020. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Zhao BS, Roundtree IA and He C:
Post-transcriptional gene regulation by mRNA modifications. Nat Rev
Mol Cell Biol. 18:31–42. 2017. View Article : Google Scholar
|
|
72
|
Hu C, Liu J, Li Y, Jiang W, Ji D, Liu W
and Ma T: Multifaceted roles of the N6-methyladenosine
RNA methyltransferase METTL3 in cancer and immune microenvironment.
Biomolecules. 12:10422022. View Article : Google Scholar
|
|
73
|
Zheng W, Dong X, Zhao Y, Wang S, Jiang H,
Zhang M, Zheng X and Gu M: Multiple functions and mechanisms
underlying the role of METTL3 in human cancers. Front Oncol.
9:14032019. View Article : Google Scholar
|
|
74
|
Wei W, Huo B and Shi X: miR-600 inhibits
lung cancer via downregulating the expression of METTL3. Cancer
Manag Res. 11:1177–1187. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Vu LP, Pickering BF, Cheng Y, Zaccara S,
Nguyen D, Minuesa G, Chou T, Chow A, Saletore Y, MacKay M, et al:
The N6-methyladenosine (m6A)-forming enzyme
METTL3 controls myeloid differentiation of normal hematopoietic and
leukemia cells. Nat Med. 23:1369–1376. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Barbieri I, Tzelepis K, Pandolfini L, Shi
J, Millán-Zambrano G, Robson SC, Aspris D, Migliori V, Bannister
AJ, Han N, et al: Promoter-bound METTL3 maintains myeloid leukaemia
by m6A-dependent translation control. Nature.
552:126–131. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Liu T, Yang S, Sui J, Xu SY, Cheng YP,
Shen B, Zhang Y, Zhang XM, Yin LH, Pu YP and Liang GY: Dysregulated
N6-methyladenosine methylation writer METTL3 contributes to the
proliferation and migration of gastric cancer. J Cell Physiol.
235:548–562. 2020. View Article : Google Scholar
|
|
78
|
Yue B, Song C, Yang L, Cui R, Cheng X,
Zhang Z and Zhao G: METTL3-mediated N6-methyladenosine modification
is critical for epithelial-mesenchymal transition and metastasis of
gastric cancer. Mol Cancer. 18:1422019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Hua W, Zhao Y, Jin X, Yu D, He J, Xie D
and Duan P: METTL3 promotes ovarian carcinoma growth and invasion
through the regulation of AXL translation and epithelial to
mesenchymal transition. Gynecol Oncol. 151:356–365. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Li X, Tang J, Huang W, Wang F, Li P, Qin
C, Qin Z, Zou Q, Wei J, Hua L, et al: The M6A methyltransferase
METTL3: Acting as a tumor suppressor in renal cell carcinoma.
Oncotarget. 8:96103–96116. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Cheng M, Sheng L, Gao Q, Xiong Q, Zhang H,
Wu M, Liang Y, Zhu F, Zhang Y, Zhang X, et al: The m6A
methyltransferase METTL3 promotes bladder cancer progression via
AFF4/NF-κB/MYC signaling network. Oncogene. 38:3667–3680. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Han J, Wang JZ, Yang X, Yu H, Zhou R, Lu
HC, Yuan WB, Lu JC, Zhou ZJ, Lu Q, et al: METTL3 promote tumor
proliferation of bladder cancer by accelerating pri-miR221/222
maturation in m6A-dependent manner. Mol Cancer. 18:1102019.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zhao S, Liu J, Nanga P, Liu Y, Cicek AE,
Knoblauch N, He C, Stephens M and He X: Detailed modeling of
positive selection improves detection of cancer driver genes. Nat
Commun. 10:33992019. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Cai X, Wang X, Cao C, Gao Y, Zhang S, Yang
Z, Liu Y, Zhang X, Zhang W and Ye L: HBXIP-elevated
methyltransferase METTL3 promotes the progression of breast cancer
via inhibiting tumor suppressor let-7g. Cancer Lett. 415:–19. 2018.
View Article : Google Scholar
|
|
85
|
Wu L, Wu D, Ning J, Liu W and Zhang D:
Changes of N6-methyladenosine modulators promote breast cancer
progression. BMC Cancer. 19:3262019. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Liu J, Eckert MA, Harada BT, Liu SM, Lu Z,
Yu K, Tienda SM, Chryplewicz A, Zhu AC, Yang Y, et al:
m6A mRNA methylation regulates AKT activity to promote
the proliferation and tumorigenicity of endometrial cancer. Nat
Cell Biol. 20:1074–1083. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Manning BD and Toker A: AKT/PKB signaling:
Navigating the network. Cell. 169:381–405. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Xue C, Li G, Lu J and Li L: Crosstalk
between circRNAs and the PI3K/AKT signaling pathway in cancer
progression. Signal Transduct Target Ther. 6:4002021. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Engelman JA, Luo J and Cantley LC: The
evolution of phosphatidylinositol 3-kinases as regulators of growth
and metabolism. Nat Rev Genet. 7:606–619. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Ralser DJ, Condic M, Klümper N, Ellinger
J, Staerk C, Egger EK, Kristiansen G, Mustea A and Thiesler T:
Comprehensive immunohistochemical analysis of N6-methyladenosine
(m6A) writers, erasers, and readers in endometrial cancer. J Cancer
Res Clin Oncol. Jun 22–2022.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Yu HL, Ma XD, Tong JF, Li JQ, Guan XJ and
Yang JH: WTAP is a prognostic marker of high-grade serous ovarian
cancer and regulates the progression of ovarian cancer cells. Onco
Targets Ther. 12:6191–6201. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Kuai Y, Gong X, Ding L, Li F, Lei L, Gong
Y, Liu Q, Tan H, Zhang X, Liu D, et al: Wilms' tumor 1-associating
protein plays an aggressive role in diffuse large B-cell lymphoma
and forms a complex with BCL6 via Hsp90. Cell Commun Signal.
16:502018. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Tang J, Wang F, Cheng G, Si S, Sun X, Han
J, Yu H, Zhang W, Lv Q, Wei JF and Yang H: Wilms' tumor
1-associating protein promotes renal cell carcinoma proliferation
by regulating CDK2 mRNA stability. J Exp Clin Cancer Res.
37:402018. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Chen Y, Peng C, Chen J, Chen D, Yang B, He
B, Hu W, Zhang Y, Liu H, Dai L, et al: WTAP facilitates progression
of hepatocellular carcinoma via m6A-HuR-dependent epigenetic
silencing of ETS1. Mol Cancer. 18:1272019. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Yu H, Zhao K, Zeng H, Li Z, Chen K, Zhang
Z, Li E and Wu Z: N6-methyladenosine (m6A)
methyltransferase WTAP accelerates the Warburg effect of gastric
cancer through regulating HK2 stability. Biomed Pharmacother.
133:1110752021. View Article : Google Scholar
|
|
96
|
Li Q, Wang C, Dong W, Su Y and Ma Z: WTAP
facilitates progression of endometrial cancer via CAV-1/NF-κB axis.
Cell Biol Int. 45:1269–1277. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Azzam SK, Alsafar H and Sajini AA: FTO m6A
demethylase in obesity and cancer: Implications and underlying
molecular mechanisms. Int J Mol Sci. 23:38002022. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Deng X, Su R, Stanford S and Chen J:
Critical enzymatic functions of FTO in obesity and cancer. Front
Endocrinol (Lausanne). 9:3962018. View Article : Google Scholar
|
|
99
|
Tao L, Mu X, Chen H, Jin D, Zhang R, Zhao
Y, Fan J, Cao M and Zhou Z: FTO modifies the m6A level of MALAT and
promotes bladder cancer progression. Clin Transl Med. 11:e3102021.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Su R, Dong L, Li Y, Gao M, Han L,
Wunderlich M, Deng X, Li H, Huang Y, Gao L, et al: Targeting FTO
suppresses cancer stem cell maintenance and immune evasion. Cancer
Cell. 38:79–96.e11. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Zhang Z, Zhou D, Lai Y, Liu Y, Tao X, Wang
Q, Zhao G, Gu H, Liao H, Zhu Y, et al: Estrogen induces endometrial
cancer cell proliferation and invasion by regulating the fat mass
and obesity-associated gene via PI3K/AKT and MAPK signaling
pathways. Cancer Lett. 319:89–97. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Zhang L, Wan Y, Zhang Z, Jiang Y, Lang J,
Cheng W and Zhu L: FTO demethylates m6A modifications in HOXB13
mRNA and promotes endometrial cancer metastasis by activating the
WNT signalling pathway. RNA Biol. 18:1265–1278. 2021. View Article : Google Scholar :
|
|
103
|
Lewczuk Ł, Pryczynicz A and
Guzińska-Ustymowicz K: Cell adhesion molecules in endometrial
cancer-a systematic review. Adv Med Sci. 64:423–429. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Delaunay S and Frye M: RNA modifications
regulating cell fate in cancer. Nat Cell Biol. 21:552–559. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Zhu Y, Shen J, Gao L and Feng Y: Estrogen
promotes fat mass and obesity-associated protein nuclear
localization and enhances endometrial cancer cell proliferation via
the mTOR signaling pathway. Oncol Rep. 35:2391–2397. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Zhang S, Zhao BS, Zhou A, Lin K, Zheng S,
Lu Z, Chen Y, Sulman EP, Xie K, Bögler O, et al: m6A
demethylase ALKBH5 maintains tumorigenicity of glioblastoma
stem-like cells by sustaining FOXM1 expression and cell
proliferation program. Cancer Cell. 31:591–606.e6. 2017. View Article : Google Scholar
|
|
107
|
Chao Y, Shang J and Ji W:
ALKBH5-m6A-FOXM1 signaling axis promotes proliferation
and invasion of lung adenocarcinoma cells under intermittent
hypoxia. Biochem Biophys Res Commun. 521:499–506. 2020. View Article : Google Scholar
|
|
108
|
Shen C, Sheng Y, Zhu AC, Robinson S, Jiang
X, Dong L, Chen H, Su R, Yin Z, Li W, et al: RNA demethylase ALKBH5
selectively promotes tumorigenesis and cancer stem cell
self-renewal in acute myeloid leukemia. Cell Stem Cell.
27:64–80.e9. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Thalhammer A, Bencokova Z, Poole R,
Loenarz C, Adam J, O'Flaherty L, Schödel J, Mole D, Giaslakiotis K,
Schofield CJ, et al: Human AlkB homologue 5 is a nuclear
2-oxoglutarate dependent oxygenase and a direct target of
hypoxia-inducible factor 1α (HIF-1α). PLoS One. 6:e162102011.
View Article : Google Scholar
|
|
110
|
Zhang C, Samanta D, Lu H, Bullen JW, Zhang
H, Chen I, He X and Semenza GL: Hypoxia induces the breast cancer
stem cell phenotype by HIF-dependent and ALKBH5-mediated
m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci USA.
113:E2047–E2056. 2016. View Article : Google Scholar
|
|
111
|
Guo X, Li K, Jiang W, Hu Y, Xiao W, Huang
Y, Feng Y, Pan Q and Wan R: RNA demethylase ALKBH5 prevents
pancreatic cancer progression by posttranscriptional activation of
PER1 in an m6A-YTHDF2-dependent manner. Mol Cancer. 19:912020.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Pu X and Gu Z and Gu Z: ALKBH5 regulates
IGF1R expression to promote the proliferation and tumorigenicity of
endometrial cancer. J Cancer. 11:5612–5622. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Chen G, Liu B, Yin S, Li S, Guo Y, Wang M,
Wang K and Wan X: Hypoxia induces an endometrial cancer stem-like
cell phenotype via HIF-dependent demethylation of SOX2 mRNA.
Oncogenesis. 9:812020. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Li J, Wu L, Pei M and Zhang Y: YTHDF2, a
protein repressed by miR-145, regulates proliferation, apoptosis,
and migration in ovarian cancer cells. J Ovarian Res. 13:1112020.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Xu F, Li J, Ni M, Cheng J, Zhao H, Wang S,
Zhou X and Wu X: FBW7 suppresses ovarian cancer development by
targeting the N6-methyladenosine binding protein YTHDF2.
Mol Cancer. 20:452021. View Article : Google Scholar
|
|
116
|
Li Z, Luo Q, Wang H, Liu Y, Feng X, Li Z
and Yi P: Knockdown of YTH N6-methyladenosine RNA
binding protein 2 (YTHDF2) inhibits cell proliferation and promotes
apoptosis in cervical cancer cells. Xi Bao Yu Fen Zi Mian Yi Xue Za
Zhi. 36:255–263. 2020.In Chinese. PubMed/NCBI
|
|
117
|
Shen X, Zhao K, Xu L, Cheng G, Zhu J, Gan
L, Wu Y and Zhuang Z: YTHDF2 inhibits gastric cancer cell growth by
regulating FOXC2 signaling pathway. Front Genet. 11:5920422021.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Zhong L, Liao D, Zhang M, Zeng C, Li X,
Zhang R, Ma H and Kang T: YTHDF2 suppresses cell proliferation and
growth via destabilizing the EGFR mRNA in hepatocellular carcinoma.
Cancer Lett. 442:252–261. 2019. View Article : Google Scholar
|
|
119
|
Hong L, Pu X, Gan H, Weng L and Zheng Q:
YTHDF2 inhibit the tumorigenicity of endometrial cancer via
downregulating the expression of IRS1 methylated with
m6A. J Cancer. 12:3809–3818. 2021. View Article : Google Scholar :
|
|
120
|
Reuveni H, Flashner-Abramson E, Steiner L,
Makedonski K, Song R, Shir A, Herlyn M, Bar-Eli M and Levitzki A:
Therapeutic destruction of insulin receptor substrates for cancer
treatment. Cancer Res. 73:4383–4394. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Ganeff C, Chatel G, Munaut C, Frankenne F,
Foidart JM and Winkler R: The IGF system in in-vitro human
decidualization. Mol Hum Reprod. 15:27–38. 2009. View Article : Google Scholar
|
|
122
|
Hopkins BD, Goncalves MD and Cantley LC:
Insulin-PI3K signalling: An evolutionarily insulated metabolic
driver of cancer. Nat Rev Endocrinol. 16:276–283. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Shen J, Feng XP, Hu RB, Wang H, Wang YL,
Qian JH and Zhou YX: N-methyladenosine reader YTHDF2-mediated long
noncoding RNA FENDRR degradation promotes cell proliferation in
endometrioid endometrial carcinoma. Lab Invest. 101:775–784. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Luo L, Zhen Y, Peng D, Wei C, Zhang X, Liu
X, Han L and Zhang Z: The role of N6-methyladenosine-modified
non-coding RNAs in the pathological process of human cancer. Cell
Death Discov. 8:3252022. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Li Y, Zhang W, Liu P, Xu Y, Tang L, Chen W
and Guan X: Long non-coding RNA FENDRR inhibits cell proliferation
and is associated with good prognosis in breast cancer. Onco
Targets Ther. 11:1403–1412. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Liu J and Du W: LncRNA FENDRR attenuates
colon cancer progression by repression of SOX4 protein. Onco
Targets Ther. 12:4287–4295. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Zhang YQ, Chen X, Fu CL, Zhang W, Zhang
DL, Pang C, Liu M and Wang JY: FENDRR reduces tumor invasiveness in
prostate cancer PC-3 cells by targeting CSNK1E. Eur Rev Med
Pharmacol Sci. 23:7327–7337. 2019.PubMed/NCBI
|
|
128
|
Bian PP, Liu SY, Luo QP and Xiong ZT:
YTHDF2 is a novel diagnostic marker of endometrial adenocarcinoma
and endometrial atypical hyperplasia/intraepithelial neoplasia.
Pathol Res Pract. 234:1539192022. View Article : Google Scholar
|
|
129
|
Müller S, Bley N, Glaß M, Busch B,
Rousseau V, Misiak D, Fuchs T, Lederer M and Hüttelmaier S: IGF2BP1
enhances an aggressive tumor cell phenotype by impairing
miRNA-directed downregulation of oncogenic factors. Nucleic Acids
Res. 46:6285–6303. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Sun CY, Cao D, Du BB, Chen CW and Liu D:
The role of Insulin-like growth factor 2 mRNA-binding proteins
(IGF2BPs) as m6A readers in cancer. Int J Biol Sci.
18:2744–2758. 2022. View Article : Google Scholar :
|
|
131
|
Ma J, Yang D and Ma XX: Immune
infiltration-related N6-methyladenosine RNA methylation regulators
influence the malignancy and prognosis of endometrial cancer. Aging
(Albany NY). 13:16287–16315. 2021. View Article : Google Scholar
|
|
132
|
Zhang L, Wan Y, Zhang Z, Jiang Y, Gu Z, Ma
X, Nie S, Yang J, Lang J, Cheng W and Zhu L: IGF2BP1 overexpression
stabilizes PEG10 mRNA in an m6A-dependent manner and promotes
endometrial cancer progression. Theranostics. 11:1100–1114. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Nicholson AL and Pasquinelli AE: Tales of
detailed poly(A) tails. Trends Cell Biol. 29:191–200. 2019.
View Article : Google Scholar
|
|
134
|
Xie T, Pan S, Zheng H, Luo Z, Tembo KM,
Jamal M, Yu Z, Yu Y, Xia J, Yin Q, et al: PEG10 as an oncogene:
Expression regulatory mechanisms and role in tumor progression.
Cancer Cell Int. 18:1122018. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Peng YP, Zhu Y, Yin LD, Zhang JJ, Wei JS,
Liu X, Liu XC, Gao WT, Jiang KR and Miao Y: PEG10 overexpression
induced by E2F-1 promotes cell proliferation, migration, and
invasion in pancreatic cancer. J Exp Clin Cancer Res. 36:302017.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Li Y, Guo D, Lu G, Mohiuddin Chowdhury
ATM, Zhang D, Ren M, Chen Y, Wang R and He S: LncRNA SNAI3-AS1
promotes PEG10-mediated proliferation and metastasis via decoying
of miR-27a-3p and miR-34a-5p in hepatocellular carcinoma. Cell
Death Dis. 11:6852020. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Kawai Y, Imada K, Akamatsu S, Zhang F,
Seiler R, Hayashi T, Leong J, Beraldi E, Saxena N, Kretschmer A, et
al: Paternally expressed gene 10 (PEG10) promotes growth, invasion,
and survival of bladder cancer. Mol Cancer Ther. 19:2210–2220.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Chang X, Han J, Pang L, Zhao Y, Yang Y and
Shen Z: Increased PADI4 expression in blood and tissues of patients
with malignant tumors. BMC Cancer. 9:402009. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Pang X, Zhang X, Huang Y and Qian S:
Development and validation of m6A regulators' prognostic
significance for endometrial cancer. Medicine (Baltimore).
100:e265512021. View Article : Google Scholar
|
|
140
|
Wang Y, Ren F, Song Z, Wang X and Ma X:
Multiomics profile and prognostic gene signature of m6A regulators
in uterine corpus endometrial carcinoma. J Cancer. 11:6390–6401.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Han Y, Liu D and Li L: PD-1/PD-L1 pathway:
Current researches in cancer. Am J Cancer Res. 10:727–742.
2020.PubMed/NCBI
|
|
142
|
Zhai J, Li S, Li Y and Du Y: Data mining
analysis of the prognostic impact of N6-methyladenosine
regulators in patients with endometrial adenocarcinoma. J Cancer.
12:4729–4738. 2021. View Article : Google Scholar :
|
|
143
|
Huisman B, Manske G, Carney S and Kalantry
S: Functional dissection of the m6A RNA modification. Trends
Biochem Sci. 42:85–86. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Yang Y, Hsu PJ, Chen YS and Yang YG:
Dynamic transcriptomic m6A decoration: Writers, erasers,
readers and functions in RNA metabolism. Cell Res. 28:616–624.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Xu Z, Peng B, Cai Y, Wu G, Huang J, Gao M,
Guo G, Zeng S, Gong Z and Yan Y: N6-methyladenosine RNA
modification in cancer therapeutic resistance: Current status and
perspectives. Biochem Pharmacol. 182:1142582020. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Su R, Dong L, Li C, Nachtergaele S,
Wunderlich M, Qing Y, Deng X, Wang Y, Weng X, Hu C, et al: R-2HG
exhibits anti-tumor activity by targeting
FTO/m6A/MYC/CEBPA signaling. Cell. 172:90–105.e23. 2018.
View Article : Google Scholar
|
|
147
|
Zhang J, Tsoi H, Li X, Wang H, Gao J, Wang
K, Go MY, Ng SC, Chan FK, Sung JJ and Yu J: Carbonic anhydrase IV
inhibits colon cancer development by inhibiting the Wnt signalling
pathway through targeting the WTAP-WT1-TBL1 axis. Gut.
65:1482–1493. 2016. View Article : Google Scholar
|














