Introduction
Cancer is one of the leading causes of mortality
worldwide, as it accounted for almost 10 million deaths in 2020, or
nearly one in six deaths, with the most common cancers being
breast, lung, colon and rectum and prostate cancers (1). Primary liver cancer is also a
challenging global health concern, as more than one million
individuals are estimated to be affected annually by the year 2025.
The most common type of liver cancer is hepatocellular carcinoma
(HCC), the incidence of which has been increasing worldwide, mostly
due to chronic viral hepatitis B infection (2). Recently, non-alcohol-related
steatohepatitis has also rapidly emerged as another etiological
concern (3,4).
In common clinical practice, HCC is diagnosed using
non-invasive criteria, including a serum alpha-fetoprotein (AFP) or
ultrasound test, and the treatment applied may vary, depending on
the overall tumor burden and underlying liver disease severity
(2). However, novel evidence
points towards the importance of histology and of the
characterization of the molecules that drive pathogenesis, to
identify druggable targets (5,6).
Moreover, immunotherapies that instigate the host immunity to
induce a systemic response against tumors currently offer much
clinical promise (7). Therefore,
the clinical classification of HCC using appropriate biomarkers
accompanied by treatment is important for the improvement of of the
prognosis of patients with HCC.
The majority of malignant tumors can be recognized
by the host immune-surveillance defensive system, namely, natural
killer (NK) and T-cells; however, cancer cells evolve to acquire
genetic instabilities and other associated 'hallmarks' that can
enable persistent growth and immune evasion from NK or T-cells, in
spite of their presence near tumors (8). Unlike T-cells, relatively few B-cells
are found in tumor infiltrates and have been rarely studied.
However, their existence and functionality have been recently
suggested as important prognostic factors for the response to
immunotherapy (9). Furthermore,
the plasma cells present in tumor infiltrates produce large amounts
of cytokines and tumor-associated (TA) antibodies, even with
reduced cell counts, and these soluble factors can serve as
significant biomarkers (9).
TA antibodies have been examined in patients with
cancer for several decades. The levels of serum autoantibodies
against TA self-antigens have been proposed as early markers of
cancer (10-12). Moreover, serum autoantibodies can
serve as potent prognostic markers at later stages of disease
(13), which may be related to the
prognostic significance of tumor-infiltrating B-cells (14,15).
Therefore, screening significant autoantibody biomarkers related to
cancer and identifying appropriate combined therapies may be
necessary for precision cancer medicine in the future.
In the present study, a bromodomain-containing
protein 2 (BRD2) autoantibody was reported as a novel TA
autoantibody biomarker for HCC. B-cell hybridoma pool obtained from
the HBx-transgenic HCC tumor-bearing mouse model (16-18)
was screened using human liver cancer cells, and one TA
autoantibody, XC246, was obtained. Using the purified XC246
autoantibody, its target antigen was identified as BRD2, a
transcriptional regulator of diverse genes, and the neo-antigenic
properties of BRD2 in liver cancer cells were analyzed. The present
study also screened the mimicries of the epitope on the autoantigen
BRD2 from a random cyclic peptide library and used it to develop a
serum autoantibody enzyme-linked immunosorbent assay (ELISA).
Finally, the significance of the BRD2 autoantibody as a cancer
diagnostic marker was evaluated using cyclic peptide ELISA and
compared with other serum biomarkers.
Materials and methods
HCC-associated autoantibody
A monoclonal TA auto-antibody, XC246, was prepared
from a B-cell hybridoma generated from the splenocytes of
HBx-transgenic mice bearing HCC tumors (18). The isotype of the XC246
autoanti-body was determined as IgM using an isotyping kit (Thermo
Fisher Scientific, Inc.). The XC246 autoantibody was purified from
hybridoma-cell culture media using protein L agarose (Thermo Fisher
Scientific, Inc.) and applied in further studies after analysis,
using SDS-PAGE and western blotting.
Cell lines
The following human cancer cell lines were obtained
from the American Type Culture Collection (ATCC) or the Korean Cell
Line Bank (KCLB): The liver cancer cells, HepG2 (ATCC HB-8065),
Hep3B (ATCC HB-8064), PLC/PRF/5 (ATCC CRL-8024), Huh7 (KCLB 60104)
and SK-HEP1 (ATCC HTB-52); the gastric cancer cells, SNU638 (KCLB
00638); the lung cancer cells, A549 (ATCC CRM-CCL-185); the
colorectal cancer cells, HT-29 (ATCC HTB-38); the prostate cancer
cells, LNCaP/LN3 (KCLB 80018); the cervical cancer cells, HeLa
(ATCC CRM-CCL-2); and the breast cancer cells, SK-BR-3 (ATCC
HTB-30). The cells were cultured in Dulbecco's modified Eagle's
medium (DMEM) or RPMI-1640 (Invitrogen; Thermo Fisher Scientific,
Inc.) supplemented with 10% inactivated fetal bovine serum (FBS;
Sigma-Aldrich). Conditioned media containing exosomes were prepared
from 70 to 80% confluent HepG2 cells, which were cultured in
complete DMEM for 48 h with 10% exosome-free FBS (System
Biosciences, LLC). The cell lines (HepG2, HT-29 and SK-HEP1) were
authenticated using short tandem repeat (STR) analysis by the
Korean Cell Line Bank (KCLB), and the STR profile results are
presented in Data S1.
The expression of recombinant human BRD2
in E. coli
For the expression of full-length and a series of
truncated BRD2 proteins (hBRD2: NM_005104.4) with N-terminal
3xFLAG-tag [(DYKDDDDK)x3] were cloned into the pE28a(+) vector
(Novagen, Sigma-Aldrich) using NdeI and XhoI.
Expression vectors were transformed into E. coli strain
SHuffle® T7 (New England Biolabs, cat. no. C3029J). The
E. coli SHuffle® T7 transformants were grown at
30°C for 16 h in 2xYT broth (16 g Bacto Tryptone, 10 g Bacto Yeast
Extract, 5 g sodium chloride in 1 l distilled water, pH 7.0)
containing kanamycin (50 µg/ml). The culture was diluted
100-fold into fresh 2xYT medium and grown in a shaking incubator at
30°C. When the OD600 of the cultures reached 2.0,
isopropyl-β-D-thiogalactopyranoside (IPTG; Sigma-Aldrich cat. no.
16758) was added to a final concentration of 1 mM and incubation
was continued at 25°C for 22 h. Cells were harvested by
centrifugation at 3,000 x g for 30 min at 4°C. The cell pellets
were solubilized in 5X SDS-PAGE sample buffer and used for further
analysis.
SDS-PAGE and western blot analysis
The purified antibody or epitope-conjugated bovine
serum albumin (BSA) was analyzed by using 10% SDS-PAGE and
Coomassie brilliant blue staining. Western blot analysis of cell or
tissue lysates was performed as previously described (18). Total cell or tissue lysates were
prepared in radioimmunoprecipitation assay (RIPA) buffer containing
a protease inhibitor cocktail (Sigma-Aldrich). The liver tissues
from the H-ras mice were already available as part of a previous
study (18). For subcellular
fractionation, cells were lysed using NE-PER™ Nuclear and
Cytoplasmic Extraction Reagents kit (Thermo Fisher Scientific, Inc.
cat. no 78833). Exosomes were collected from the conditioned media
using an exosome isolation kit ExoQuick-TC (System Biosciences,
LLC, cat. no. EXOTC50A-1) or by ultra-centrifugation at 100,000 x g
for 1 h at 4°C, and lysed in RIPA buffer (17). The protein concentration was
determined using Bradford reagent (Bio-Rad Laboratories, Inc.).
Equal amounts of protein (10 µg per lane) were analyzed
using western blotting with the indicated primary antibodies.
Briefly, proteins were separated by 12% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and
transferred onto PVDF membranes (MERCK, cat. no. IPVH00010). After
blocking with 5% BSA in Tris-buffered saline with 0.1% Tween-20
(TBST) at 25°C for 60 min, the membranes were probed with primary
antibodies at 25°C for 2 h. After washing with TBST, the membranes
were probed with HRP-conjugated secondary antibodies. The membranes
were then washed with TBST and antigens were detected with enhanced
chemiluminescence reagent (MERCK, cat. no. GERPN2106). β-actin or
GAPDH was probed as loading controls. The primary antibodies used
in this study were as follows: BRD2 (Novus Biologicals, cat. no.
NBP1-84310, NBP1-30475; 1:1,000 dilution), AICAR
transformylase/inosine monophosphate cyclohydrolase (ATIC; Thermo
Fisher Scientific, cat. no. MA1-086; 1:500 dilution), programmed
cell death 6-interacting protein (ALIX; exosomal marker; Merck
Millipore, cat. no. ABC1435; 1:500 dilution), calnexin (endoplasmic
reticulum marker; Santa Cruz Biotechnology, cat. no. sc-46669;
1:1,000 dilution), GAPDH (Santa Cruz Biotechnology, cat. no.
sc-47724; 1:5,000 dilution), and β-actin (Santa Cruz Biotechnology,
cat. no. sc-8432; 1:5,000 dilution). Anti-mouse IgG-horseradish
peroxidase (HRP; Cell Signaling Technology, cat. no. #7076S;
1:2,500 dilution) or anti-rabbit IgG-HRP (Cell Signaling
Technology, Inc. cat. no. 7074S; 1:2,500 dilution) were used as
secondary antibodies. Band intensities were quantified using ImageJ
v1.52a (National Institutes of Health), and the relative intensity
compared with that of β-actin or GAPDH was calculated. For the
immunoprecipitation analysis of the XC246 antigen, 600 µg of
HepG2 cell lysate was incubated with XC246 antibody-conjugated
agarose beads (bead volume, 20 µl) for 16 h at 4°C; after a
brief washing the beads in RIPA buffer by centrifugation at 1,000 x
g for 30 sec at 4°C, the agarose beads were analyzed by western
blotting. For the proteomics analysis of the XC246 antigen, 500
µl of antibody-conjugated beads was used for
immunoprecipitation with 9.69 mg of SNU638 cell lysate. As a
control, blank agarose beads without the immobilized antibody were
used. XC246 antibody-conjugated agarose beads were prepared using a
co-immunoprecipitation kit (Thermo Fisher Scientific, cat. no.
26149). For competitive western blot analysis, 5 µg of the
XC246 primary antibody in 10 ml of 5% skim milk in TBS was
pre-incubated with epitope-peptide-conjugated BSA (2 µg) at
25°C for 90 min and then used as the primary antibody for probing
the blot.
Flow cytometric analysis
Cells were fixed and permeabilized with BD
Cytofix/Cytoperm solution (BD Biosciences) at 4°C for 30 min. The
cells were then incubated with the XC246 primary antibody solution
and secondary reagent goat anti-mouse IgG F(ab')2-PE (Thermo Fisher
Scientific, cat no. A10543; 1:400 dilution) at 4°C for 40 min. The
stained cells were analyzed on a FACSCalibur (BD Biosciences)
instrument, and the obtained data were analyzed using the CellQuest
software v6.0 (BD Biosciences). When determining whether the
autoantibody-mimotope display phages could compete with the target
cellular antigen for antibody binding, the XC246 primary antibody
was pre-incubated with each mimotope-display phage at 25°C for 60
min and then used as primary antibody solution.
Immunofluorescence
Cells were plated on glass coverslips and, after an
additional 48 h, were used for immunofluorescence detection, as
described previously (17). Cells
were fixed and permeabilized with BD Cytofix/Cytoperm solution (BD
Biosciences), washed with BD Cytoperm/wash solution (BD
Biosciences), and incubated with the primary antibody in
wash-solution (5 µg/ml) at 4°C for 16 h. After washing with
wash-solution, the cells were treated with goat anti-mouse
IgGAM-rhodamine (Abcam, cat. no. ab6004) or anti-rabbit IgG-FITC
(Abcam, cat. no. ab6798) in wash-solution (1:1,000 dilution) at
25°C for 1 h, followed by mounting on a DAPI-containing mounting
medium (Vector Laboratories, cat. no. H-1200). Confocal microscopic
analysis was performed using a Zeiss LSM 510 Meta microscope (Zeiss
AG). The staining with an anti-FBXO2 antibody (mouse IgM; Santa
Cruz Biotechnology, cat. no. sc-393873) was performed under the
conditions described above to confirm the accessibility of the IgM
antibody to the nucleus.
Immunohistochemistry (IHC)
A paraffin-embedded human HCC BioMax Array (cat. no.
LV1201b) was purchased from US BioMax Inc. Biopsy features included
age, sex, organ or anatomic site involved, grading and pathological
diagnosis (H&E-stained sections). The HCC tissue array was
comprised of HCC or HCC-related liver disease tissues and normal
liver tissues as follows: i) 10 cases of benign tumor (cavernous
hemangioma); ii) 14 cases of normal liver tissues; iii) 25 cases of
hepatocellular carcinoma (stage I, n=1; stage II, n=16; stage III,
n=5; stage IV, n=3); iv) 29 cases of cirrhosis; v) 22 cases of
chronic hepatitis; and vi) 13 cases of fatty degeneration. US
Biomax states that 'All tissue is collected under the highest
ethical standards with the donor being informed completely and with
their consent. We make sure we follow standard medical care and
protect the donors' privacy. All human tissues are collected under
HIPPA approved protocols. All animal tissues are collected under
IACUC protocol. All samples have been tested negative for HIV and
hepatitis B or their counterparts in animals, and approved for
commercial product development'. The Public Institutional Review
Board of the Ministry of Health and Welfare reviewed the contents
of the present study and confirmed that research using
commercialized human tissue falls under the exemption categories
specified by Bioethics and Safety Act in Korea (IRB, approval no.:
P01-202008-31-009; Republic of Korea). The immunohistochemical
staining for XC246 antigen was performed using XC246 monoclonal
autoantibody (2 µg/ml) as previously described (16). The IHC staining of all tissues was
performed under identical conditions, as tissues with all
pathological characteristics were present on one slide. The
endogenous peroxidase activity was blocked by treatment with 4%
H2O2/methanol for 10 min, and this was
followed by remaining staining protocol. The photomicrographs were
acquired at x200 or x400 magnification using a multi-view
fluorescence microscope (BX51; Olympus Corporation). The DAB
intensity of each staining using DAB kit (GBI Labs, cat. no. D22-6)
was quantified by ImageJ v1.52i software (National Institutes of
Health) and plotted.
Mass spectrometry (MS) analysis of the
XC246 antigen
For the enrichment of the antigen against the XC246
autoantibody, a SNU638 cell lysate prepared with RIPA cell lysis
buffer was immunoprecipitated using XC246 antibody-conjugated beads
as described above. Subsequently, the beads were treated with 0.1 M
glycine buffer (pH 2.5) to elute the target antigen, and the eluate
was neutralized with 1.0 M Tris buffer (pH 8.0). The eluates were
then separated using 10% SDS-PAGE, followed by western blotting or
Coomassie brilliant blue staining at 25°C for 2 h. The
Coomassie-stained band corresponding to the XC246 antigen band was
confirmed using western blotting. The XC246 antigen band was then
excised and in-gel digested with trypsin (Promega Corporation).
Protein identification was performed via ESI-TRAP mass spectrometry
(LTQ Orbitrap XL Hybrid Ion Trap Mass Spectrometer; Thermo Fisher
Scientific, Inc.) and Mascot database search at the Korea Basic
Science Institute (Ochang, Korea).
Knockdown of candidate antigens and
reverse transcription polymerase chain reaction (RT-PCR)
To verify whether the candidate proteins identified
by mass spectrometric analysis corresponded to the XC246 antigen,
the HepG2 cells were transfected with siRNAs, targeting the
candidate genes (Bioneer Corporation and Thermo Fisher Scientific
Inc.) using Lipofectamine RNAimax reagent (Thermo Fisher Scientific
Inc.), followed by western blot analysis using the XC246 anti-body.
The sequences of the siRNAs were as follows: siRNA for the
elongation factor 2 (si-EEF2) si-sense, 5′-GAG AUG UAU GUG GCC AAG
Utt-3′ and antisense, 5′-ACU UGG CCA CAU ACA UCU Ctt-3′; siRNA for
the isoform 3 of unconventional myosin 1c (si-MYO1C) sense, 5′-GAC
CAA GAC AGC CCU CAG Utt-3′ and antisense, 5′-ACU GAG GGC UGU CUU
GGU Ctt-3′; si-BRD2 sense, 5′-CUG GGA GUC UUG AGC CUA Att-3′ and
antisense, 5′-UUA GGC UCA AGA CUC CCA Gga-3′; si-control sense, CCU
ACG CCA CCA AUU UCG U(dTdT) and antisense, ACG AAA UUG GUG GCG UAG
G(dTdT). The siRNA-treated cells were analyzed 72 h after
transfection. RT-PCR was performed using primer pairs purchased
from Bioneer Corporation, as follows: EEF2 sense, 5′-GTG GAG AAC
GTG AAC GTC ATC-3′ and antisense, 5′-GAA GGT GCG TGG CAG CTT CT-3′;
MYO1C sense, 5′-CGC TAC CGG GCG TCG GCC-3′ and antisense, 5′-CGT
GCG CAG TGC TCG GT AC-3′; BRD2 sense, 5′-AGA GTC CTC CAG TGA GGA
AAG-3′ and antisense, 5′-CCA CTG CCA CTT GCT TTC TTG-3′. GAPDH
sense, 5′-CCA ATA TGA TTC CAC CCA TGG C-3′ and antisense, 5′-GCT
GAT GAT CTT GAG GCT GTT G-3′. Briefly, total RNA was extracted from
the siRNA transfectants using the RNeasy Plus mini kit (Qiagen,
Germany, cat. no. 74134) following the manufacturer′s protocol.
Reverse transcription (RT) was performed using the GoScript™
Reverse Transcription Mix kit (Promega Corporation, cat. no.
A2791). PCR was carried out using 2X Taq PCR Pre-Mix (BIOFACT, cat.
no. ST302-10h) using a Verti 96 well Thermal Cycler (Invitrogen;
Thermo Fisher Scientific, Inc.) which was performed by an initial
denaturing step at 95°C for 5 min followed by 27 cycles of
denaturation at 95°C for 10 sec, 55-62°C for 30 sec for annealing
and elongation at 72°C for 30 sec. At the end of additional
elongation at 72°C for 5 min, PCR products were analyzed by 1%
agarose gel electrophoresis. The relative levels of each PCR
product were quantified using ImageJ v1.53k software and normalized
to that of GAPDH.
Biopanning of the XC246-specific cyclic
peptide mimotopes
For the selection of the mimotope specific to XC246
auto-antibody, the phage-display random cyclic peptide library
Ph.D.™-CX7C (New England Biolabs, Inc.) was used, as
previously described (16-18). Panning was repeated four times, and
the mimotope sequences were determined by sequencing the selected
phages according to the manufacturer's instructions. DNA sequencing
was performed by Bioneer Corporation based on Sanger method using
-96gIII sequencing primer (5′-CCC TCA TAG TTA GCG TAA CG-3′).
ELISA
ELISA was performed as previously described, with
some modifications (17). Briefly,
ELISA plates (Nunc Maxisorp; Thermo Fisher Scientific, Inc.) were
coated with the indicated amount of antigen [cyclic peptide
epitope-display M13 phages or epitope-miniPEG2-conjugated BSA] in
PBS (pH 7.4) overnight at 4°C and blocked with Protein-Free
Blocking Buffer (PFBB; Thermo Fisher Scientific, Inc.). The
CX7C peptide mimotopes were synthesized and cyclized via
terminal cysteine residues. The cyclized peptides were then coupled
to miniPEG2 spacer via the free amine residue at the amino terminus
of the peptide. The PEG-conjugated cyclic peptides were synthesized
and purified by Peptron. Cyclic peptide-miniPEG2-conjugated BSA was
prepared using the EDC coupling method (Thermo Fisher Scientific,
Inc.). The miniPEG2-conjugated BSA without the peptide epitope was
used as the control antigen. The primary anti-body or secondary
reagent solution was prepared in PFBB. Anti-mouse IgGAM-HRP (Thermo
Fisher Scientific, Inc.) was used as the secondary reagent. To
detect the human autoantibody in patients' sera, ELISA plates were
coated with XC246p9-miniPEG-conjugated BSA at 500 ng/well. After
blocking with a buffer containing 1% polyvinyl alcohol (molecular
weight, 14-15k; MilliporeSigma) and 1% Ficoll P400 (MilliporeSigma)
in TBS, the plates were treated with human sera (1:50 dilution in
blocking buffer) at 37°C for 90 min and detected using
HRP-conjugated anti-human IgGAM antibody (Thermo Fisher Scientific,
Inc.; 1:2,000 dilution). Serum AFP was quantified using a human
alpha-Fetoprotein Quantikine ELISA kit (R&D Systems, cat. no.
DAFP00). The present study using human HCC and normal serum samples
was conducted after receiving an exemption from the Public
Institutional Review Board of the Ministry of Health and Welfare
(IRB No.: P01-202008-31-009; Republic of Korea). Human serum
samples were obtained from the National Biobank of Korea, which is
supported by the Ministry of Health and Welfare. Serum samples were
kept at −80°C until further use.
Statistical analysis
All data are presented as the mean ± standard
deviation (SD) and were analyzed using a two-tailed unpaired
Student's t-test or one-way ANOVA followed by the Bonferroni
post-hoc test. The results of ELISA were evaluated using receiver
operating characteristics (ROC) analysis, leading to estimates of
the area under the ROC curve (AUC), with 95% confidence intervals
(CIs). The correlation between the biomarkers was assessed using
Pearson's correlation analysis. Statistical analyses were performed
using the Prism 7 software (GraphPad Software, Inc.). P<0.05 was
considered to indicate a statistically significant difference.
Results
The TA autoantibody XC246 identified in
the HBx-tg HCC mouse model displays an increase in target antigen
expression in human HCC tissues
HBx-transgenic mice, which exhibit spontaneous
generation of liver cancer at 10-13 months after birth, have been
proven to be suitable for the study of human HCC (19-21).
Previously, three HCC-associated autoantibodies were identified by
the authors using B-cell hybridoma cells constructed using
tumor-bearing HBx-transgenic mice (16-18).
In the present study, a monoclonal TA autoantibody, which was
termed XC246, was identified, and its antigenic characteristics
were analyzed. Firstly, the presence of an antigen reactive to the
XC246 autoantibody in human cancer cells was examined using flow
cytometry. For the staining of intracellular antigens, cells were
fixed and mildly permeabilized with a paraformaldehyde/saponin
solution, to maintain the conformational epitopes on the target
antigens. The XC246 TA autoantibody in hybridoma-cultured media
reacted to human liver cancer cells, including HepG2 and Huh7 cells
(Fig. 1A). The colorectal cancer
HT-29 cells also exhibited reactivity to the XC246 autoantibody.
These results indicated that TA autoantigen against XC246 TA
autoantibody generated in a mouse HCC tumor model may also be
expressed in human tumor cell lines. The isotype of the XC246
antibody was determined using an isotyping kit as being IgM with a
kappa light chain (data not shown). For the identification and
characterization of the target antigen of the XC246 TA
autoantibody, the antibody was purified from hybridoma-cultured
media using protein L agarose. SDS-PAGE analysis of the purified
antibody showed the presence of IgM heavy and light chains
(Fig. 1B).
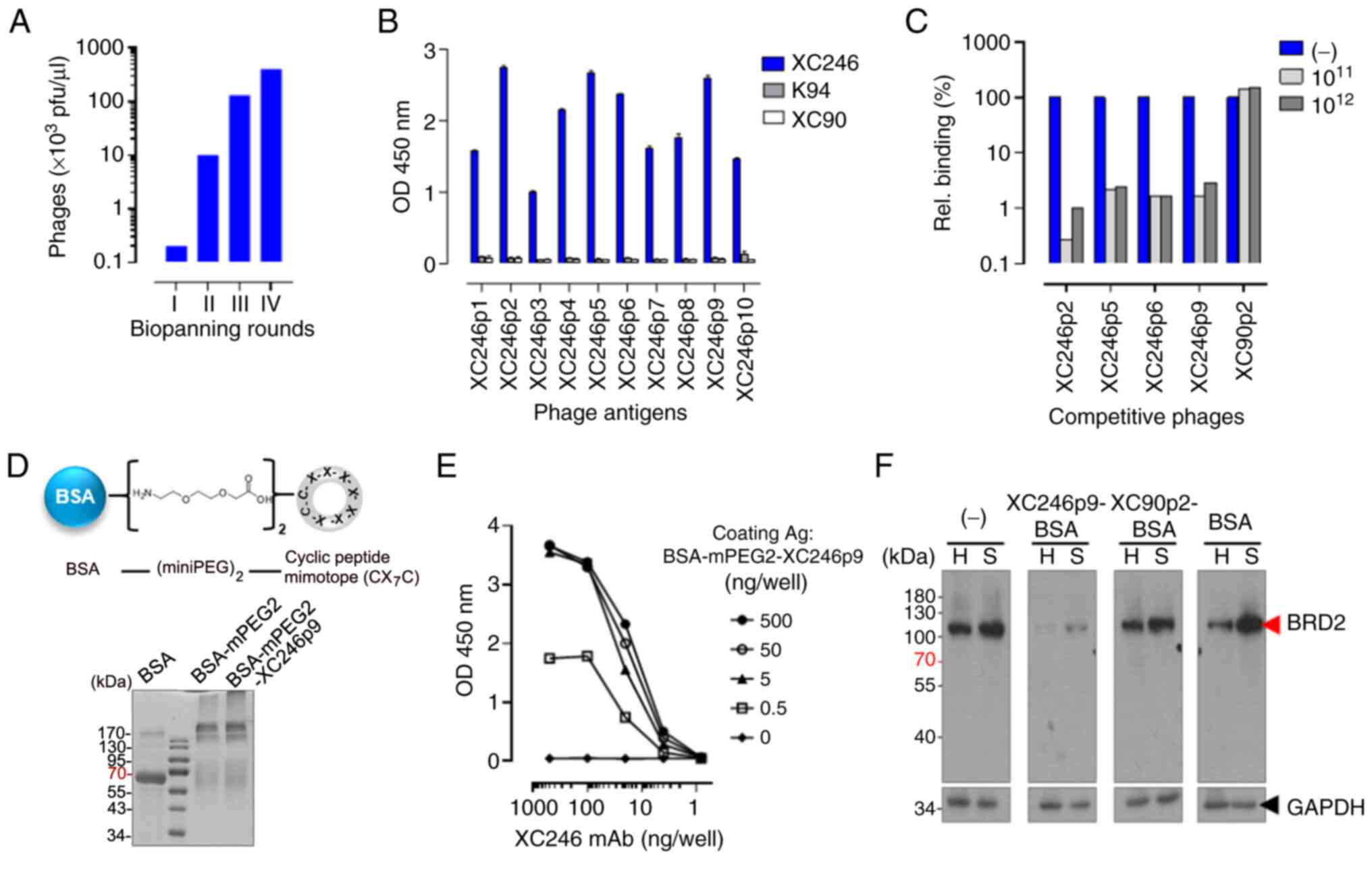
 | Figure 1The tumor-associated monoclonal
autoantibody, XC246, was identified in the HBx-Tg HCC mouse model.
(A) Flow cytometric analysis of intracellular stained tumor cell
lines with the XC246 autoantibody. Fixed and permeabilized cells
were treated with XC246 hybridoma-cultured media, followed by
staining with PE-labeled anti-mouse IgG. β-Actin staining was also
performed, and the relative ratio of XC246 to β-actin staining was
plotted. (B) SDS-PAGE analysis of purified XC246 monoclonal
autoantibody. Purified XC246 antibody (5 µg) was treated
with reducing SDS-PAGE sample buffer and separated on 10% SDS-PAGE.
The Coomassie blue-stained gel revealed the immunoglobulin µ
heavy chain with a molecular weight of 72 kDa and the light chain
with a molecular weight of 25 kDa. BSA (5 µg) was loaded as
a control. (C) Western blot analysis of XC246 antigen expression in
various human tumor cell lines (cell lysates; 10 µg per
lane). GAPDH served as an internal control. The red arrow indicates
the XC246 antigen. (D) Immunofluorescence staining of the XC246
antigen in human cancer cells (human liver cancer cell lines HepG2
and Huh7; gastric cancer cell line SNU638). Fixed and permeabilized
cells were treated with purified XC246 antibody, followed by
staining with Rhodamine-labeled anti-mouse IgG antibody. (E)
Expression of XC246 Ag in liver tissues of H-ras12V-tg mice. The
liver tissue lysates (50 µg) of wild-type mice (n=3) or
tumor-bearing H-ras12V-tg mice (n=6) were separated on 10%
SDS-PAGE, and western blots were probed with the XC246
autoantibody. Band intensities were quantified using ImageJ
software, and the values were normalized to those of β-actin. (F)
IHC of a human liver tissue microarray. A tissue microarray
containing benign tumor (liver hemangioma; n=10), normal (n=14),
HCC (n=25), cirrhosis (n=29), chronic hepatitis (n=22), or fatty
liver (n=13) tissues was stained with the XC246 antibody (0.5
µg/ml). Representative images of IHC staining are presented.
H&E stains are included in Fig. S1. Scale bar, 50 µm.
The DAB intensities of IHC images were quantified using ImageJ
software, and the quantified values were plotted. Statistical
significance was determined using (E) a two-tailed Student's t-test
or (F) one-way ANOVA followed by the Bonferroni post-hoc test.
*P<0.05, ***P<0.001 and
****P<0.0001. ns, not significant (P>0.05); HCC,
hepatocellular carcinoma; BSA, bovine serum albumin; IHC,
immunohistochemistry. |
The target antigen of XC246 antibody was analyzed by
western blot analysis of human tumor cell lines. As depicted in
Fig. 1C, the XC246 antibody
reacted to a specific antigen (termed XC246 Ag) with a molecular
weight of about 110 kDa. XC246 Ag was ubiquitously and highly
expressed in various human tumor cells, including liver cancer
(HepG2), gastric cancer (SNU638) and colorectal cancer (HT-29);
however, its expression was unusually low in Hep3B cells. XC246 Ag
was noted to be mainly localized to the cytoplasm, as demonstrated
by tumor cell immunofluorescence staining (Fig. 1D). Of note, XC246 antibody detected
a target antigen of ~110 kDa in liver tumor tissues of H-ras12V
transgenic mice (Fig. 1E). The
same antigen was also detected in non-transgenic mice; however, its
expression was lower than that in tumor tissues (P≤0.05; Fig. 1E), which implies that the
upregulation of the XC246 antigen is associated with tumorigenesis.
XC246 antigen expression was also higher in human HCC tissues than
in normal liver tissues, as shown by immunohistochemical staining
(P≤0.001; Figs. 1F and S1). Of note, its expression was
significantly increased in patients with chronic hepatic liver
diseases (P≤0.0001), including cirrhosis and chronic hepatitis.
Overall, these results suggested that the
HCC-associated autoantibody XC246 may react with a specific antigen
expressed in the cytoplasm of human or mouse tumor cells with a
molecular weight of ~110 kDa and that its expression was increased
in HCC and chronic liver diseases.
The target antigen of the TA autoantibody
XC246 is BRD2, a transcriptional regulator secreted from tumor
cells as an exosomal component
The target antigen of the XC246 autoantibody was
identified using MS analysis. The lysate of SNU638 cells was
immunoprecipitated using XC246 anti-body-conjugated agarose beads,
and its eluate was separated on preparative 10% SDS-PAGE. As a
control, the immunoprecipitates using agarose beads without
antibodies were used. A tenth of the immunoprecipitates was
analyzed using western blot analysis, revealing that the XC246
antigen was enriched in the immunoprecipitated fraction (Fig. 2A). The protein band on the
preparative SDS-PAGE that corresponded to the immuno-stained
antigen was then excised, in-gel digested and analyzed via ESI-TRAP
MS (Fig. 2A). The isoelectric
point (pI) of the XC246 antigen was also examined by
two-dimensional gel electrophoresis of a total HepG2 cell lysate,
followed by western blot analysos using the XC246 antibody; the pI
of the 110-kDa antigen was estimated to be ~9 (Fig. S2). The identified proteins in MS
analysis that exhibited a high %mol/score are listed in Table I.
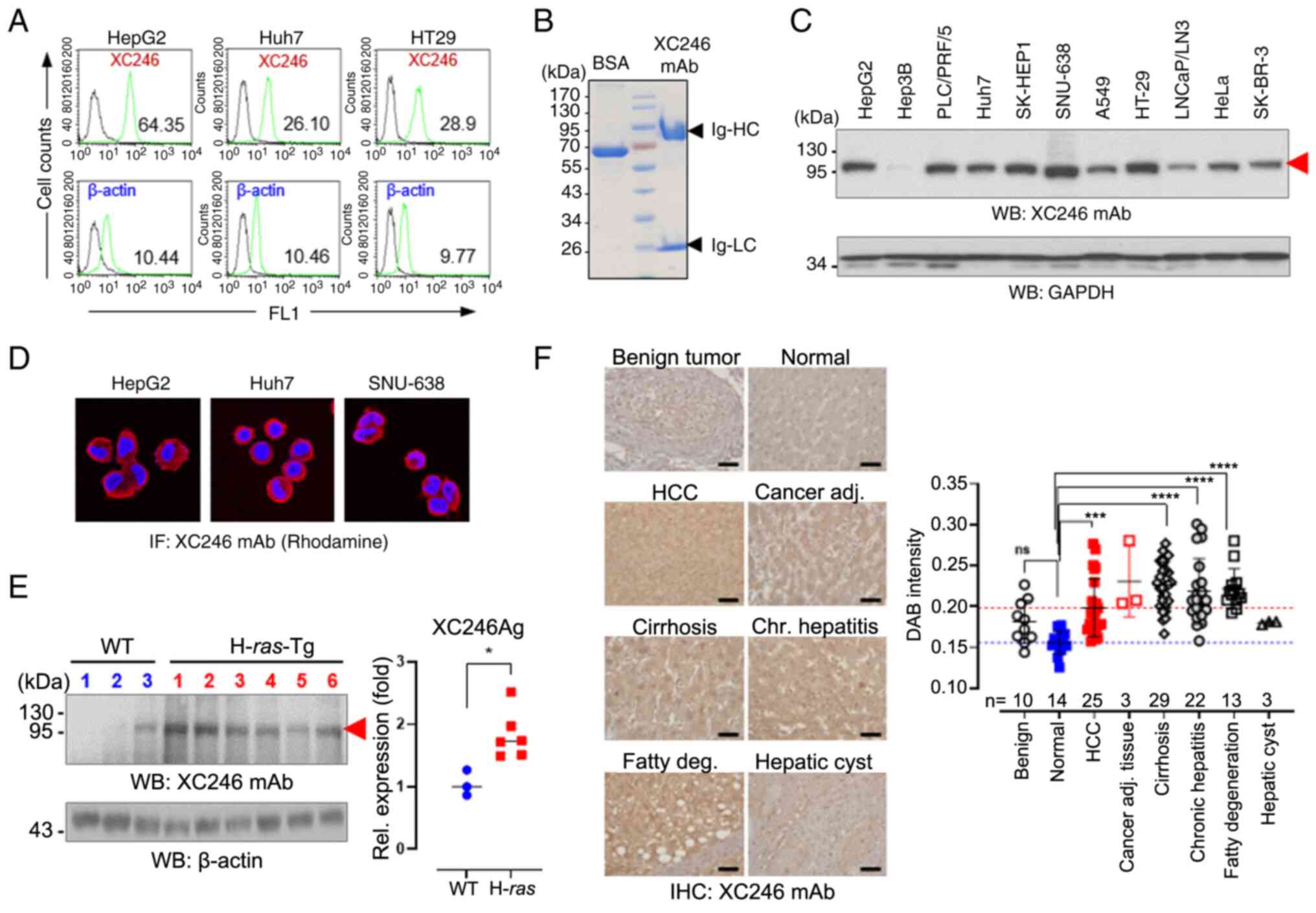
 | Figure 2The target antigen of the XC246
autoantibody was identified as BRD2. (A) Preparative 10% SDS-PAGE
was performed to isolate the XC246 antigen, and in-gel digestion
was carried out for mass spectrometric-based protein
identification. A preparative SDS-PAGE gel for western blotting was
divided into two sections and blotted separately. The western
blotting result is a combined image of two blots, with a dotted
line representing the edges of two images. The protein band
containing the XC246 antigen confirmed by western blotting was
excised (indicated by the red arrow) and in-gel digested with
trypsin. The proteins identified by mass spectrometric analysis are
listed in Table I. (B) Validation
of the XC246 antigen as BRD2 by an RNA interference assay. HepG2
cells were transfected with siRNAs for candidate genes (EEF2, MYO1C
and BRD2), and their cell lysates were examined by western blotting
with the XC246 antibody. The knockdown of target genes was
confirmed using reverse transcription polymerase chain reaction or
western blotting. GAPDH was used as an internal control. (C)
Immunoprecipitation analysis for the verification of the XC246
antigen as BRD2. The HepG2 cell lysate was immunoprecipitated with
XC246 antibody-conjugated agarose beads and analyzed by western
blotting with an anti-BRD2 or the XC246 antibody.
Immunoprecipitates obtained using agarose beads without antibody
conjugation were used as the control. Red arrows indicate the XC246
antigen or BRD2. (D) Immunofluorescence staining of the XC246
antigen in HepG2 cells. Fixed and permeabilized cells were treated
with purified XC246 antibody or an anti-BRD2 antibody, followed by
staining with FITC- or RDM-labeled anti-mouse IgG. To visualize the
nuclei, cells were stained with DAPI. To verify the nuclear
permeability of stained cells, an IgM-type mouse antibody (FBXO2
antibody) was also employed. (E) Western blot analysis of the
intracellular distribution of the XC246 antigen or BRD2. Total cell
lysates, subcellular fractions (cytosolic or nuclear fractions),
and exosome lysates were prepared as described in the 'Materials
and methods' and analyzed using western blotting. The blots were
probed with the XC246 autoantibody, anti-BRD2 antibody, or
anti-ATIC antibody. Each target antigen is indicated by colored
arrows (red: XC246 and exosome XC246 antigen; blue: BRD2; green:
ATIC). BRD2, bromodomain-containing protein 2; RDM, rhodamine;
ATIC, AICAR transformylase/inosine monophosphate
cyclohydrolase. |
 | Table IMass spectrometric analysis of XC246
antigen. |
Table I
Mass spectrometric analysis of XC246
antigen.
| Accession
number | Identified
proteins | Molecular mass
(Da) | pI | % mola/scoreb
|
|---|
| Band-1 | Band-2 |
|---|
| P13639 | EEF2 | 96,246 | 6.41 | 0.9864/555 | 4.002/1645 |
| P49588 | AARS | 107,484 | 5.34 | 0.9059/771 | 2.906/1883 |
| A0A140T9E9 | BRD2 | 88,664 | 9.13 | - | 2.7652/1357 |
| Q14697-2 | GANAB | 109,825 | 5.82 | - | 1.6993/934 |
| P53618 | COPB1 | 108,214 | 5.72 | 0.9461/603 | 1.6692/779 |
| K7EJE8 | LONP1 | 93,637 | 6.08 | - | 1.6692/848 |
| P22314 | UBA1 | 118,858 | 5.49 | 0.2416/274 | 1.3876/1532 |
| P34932 | HSPA4 | 95,127 | 5.11 | 0.4328/397 | 1.3273/789 |
| Q7KZF4 | SND1 | 102,618 | 6.74 | 0.3624/328 | 1.3172/1019 |
| E9PLK3 | NPEPPS | 103,720 | 5.41 | 0.8455/450 | 1.2971/734 |
| P55060 | CSE1L | 111,145 | 5.51 | 0.4731/313 | 1.2267/709 |
| O00159-3 | MYO1C | 120,352 | 9.5 | 0.3624/307 | 1.1966/1007 |
| Q9Y678 | COPG1 | 98,967 | 5.32 | 0.4127/185 | 1.1262/683 |
| P33991 | MCM4 | 97,068 | 6.28 | 0.6543/273 | 1.0357/558 |
| P19367 | HK1 | 103,561 | 6.36 | 0.0906/65 | 1.0055/579 |
Based on the protein data acquired, including
molecular weight and pI, three candidate proteins (EEF2, BRD2 and
MYO1C) were selected for further validation. The downregulation of
the XC246 antigen following the knockdown of each candidate gene
was examined. The knockdown of BRD2 in HepG2 cells clearly reduced
the expression of the XC246 antigen, as demonstrated using western
blot analysis with the XC246 antibody. By contrast, the knockdown
of the remaining candidates (EEF2 or MYO1C) had no or only a
minimal effect on XC246 antigen levels (Fig. 2B). BRD2 was also confirmed as the
XC246 antigen by analysis of the immunoprecipitates obtained with
the XC246 antibody (Fig. 2C). The
commercial anti-BRD2 antibody detected the target antigen with a
molecular weight of ~110 kDa in cell lysate input or flow-through
fractions. It also detected the immunoprecipitates obtained with
the XC246 antibody, the molecular weight of which was identical to
that of the BRD2 protein. Collectively, these results confirmed
that the XC246 antigen, which induced the expression of the TA
autoantibody in the HCC mouse model, was BRD2.
The BRD2 gene encodes a transcriptional regulator
that belongs to the bromodomain and extra-terminal domain (BET)
family of proteins. BRD2 associates with transcription complexes
and acetylated chromatin during mitosis; it then selectively binds
to the acetylated lysine 12 residue of histone H4 via its two
bromodomains (22). Decreased BRD2
expression has been associated with longevity-promoting processes
(23,24), whereas increased BRD2 expression
can promote cancer in murine hematopoietic cells and B lymphocytes
(25). The Cancer Genome Atlas
(TCGA) has demonstrated that BRD2 expression is elevated across 32
distinct tumor types and established BRD2 as a promising drug
target for human cancers (26).
The downregulation of BRD2 in HeLa cells leads to a 60% increase in
tumor-suppressing p53 levels (27), which also supports the notion that
increased BRD2 promotes cancer growth. Hence, the overexpression of
BRD2 in HCC can be oncogenic, whereas the inhibition of the
activity of BRD2 limits cancer progression. More importantly,
Kaplan-Meier analysis of overall survival in patients with HCC by
their BRD2 protein expression levels revealed BRD2 as a poor
prognostic marker for liver cancer (Fig. S3). The mouse BRD2 protein
(NP_001191902) presents with 96% amino acid sequence identity (97%
similarity) to human BRD2 (NP_005095), which is composed of 801
amino acids. The difference between the mouse and human BRD2
protein sequences was primarily found between the 590 and 630th
amino acids of human BRD2, a linker region located between the
nuclear localization signal and the extra-terminal (ET) domain. The
sequences of other domains were well conserved (Fig. S4), on which the target sequence of
the XC246 antibody may be located.
The protein function of BRD2 mentioned above raised
a question about its cellular localization. BRD2 is a
transcriptional regulator that functions mainly in the nucleus.
However, the XC246 antigen was immunostained with the XC246
antibody mainly in the cytoplasmic region, as demonstrated in
Fig. 1D. To examine the cellular
localization of the XC246 antigen or BRD2, intracellular staining
with a commercial anti-BRD2 antibody or the XC246 autoantibody was
performed. The accessibility of XC246 antibody, a mouse pentameric
IgM, within the nucleus was confirmed by control staining with an
anti-FBXO2 antibody, the isotype of which is IgM. As depicted in
Fig. 2D, the commercial anti-BRD2
anti-body stained BRD2 mainly within the nucleus. This antibody
also stained the cytoplasmic region, although at reduced levels. By
contrast, XC246 autoantibody stained the cytoplasmic region mainly
(Figs. 1D and 2D). However, the nuclear staining of
XC246 is more evident in Fig. 2D
compared with Fig. 1D, which may
be caused by different staining conditions or image capture
settings. The reactivity of XC246 autoantibody to the intracellular
fractions was examined again using western blot analysis (Figs. 2E and S5). The XC246 autoantibody stained the
110-kDa antigen with a similar ratio in cytoplasmic or nuclear
fractions of HepG2 cells separated on the blot; however, the
commercial anti-BRD2 antibody stained cytoplasmic BRD2 at half the
strength observed for nuclear BRD2 in the same blot. These results
imply that these two antibodies against BRD2 have different
epitopes on the BRD2 protein. In addition, the XC246 autoantibody
is more reactive to cytoplasmic BRD2, which may possess specific
post-translational modifications related to its cytoplasmic
localization.
Another important aspect of TA autoantigens is their
exposure to immune cells. The increased intracellular antigen must
be exposed to the immune cells to induce a specific immune
response, ultimately leading to the production of TA
autoantibodies. The release of cellular components after cell death
or necrosis accompanying tumorigenesis has been assumed to be a
mechanism to expose intracellular proteins because most of the TA
autoantigens reported to date are intracellular proteins, including
p53 (28,29). However, a previous study on TA
exosomes revealed that the intracellular components included in
exosomes can be exposed to immune cells without cell death
(30). In addition, the
tumor-derived exosomes can stimulate or suppress immune responses
(30,31). As described above, the XC246
antigen appears to be a post-translationally modified BRD2 mainly
localized in the cytoplasm. The exosomes secreted by HepG2 cells
were examined using western blot analysis to confirm the secretion
of BRD2 via exosomes. ATIC (16),
another TA autoantigen previously studied, was detected in the
exosomes, as shown in Fig. 2E,
which confirms that the TA exosome is a supplier of TA antigens.
However, the BRD2 protein of 110 kDa was not detected in the
exosomal fraction of HepG2 cells using XC246 or a commercial
anti-BRD2 antibody. Instead of the full-sized BRD2 form, the XC246
antibody detected a protein band of ~65 kDa (Fig. 2E), whereas the commercial anti-BRD2
antibody did not detect it. The immunogen of the commercial
anti-BRD2 antibody has been reported as the 179-259 amino acids of
BRD2. Epitope mapping analysis using a series of truncated
recombinant BRD2 proteins revealed that the commercial anti-BRD2
antibody recognized the region between residues 179 and 205 of
BRD2. It also revealed that the XC246 autoantibody recognized the
region between residues 206 and 229 of BRD2 (Fig. S6). Based on these results, it was
concluded that the exosomal BRD2, a truncated BRD2 with a molecular
weight of about 65 kDa and containing 206-229 amino acid residues
of BRD2, is a post-translationally modified form of BRD2 that
confers a neo-epitope for generating autoantibodies.
Epitope mimicry against the XC246
antibody screened from a phage-display random cyclic heptapeptide
library can be used as antigenic determinants to detect TA
autoantibodies instead of the cellular BRD2 antigen
While epitope mapping of XC246 antibody was
performed by using recombinant BRD2 proteins and western blot
analysis, these recombinant proteins are not appropriate for the
detection of serum autoantibody using ELISA, due to their
relatively low affinity to the anti-body, as demonstrated in a
previous study by the authors (16). In addition, some post-translational
modifications that confer neo-epitopes for generating
autoantibodies were anticipated. Therefore, high-affinity mimotopes
were screened, which are the mimics of the conformational antigenic
determinants, from a random cyclic peptide CX7C library,
as described in previous studies (16-18,32,33).
The biopanning of the M13 phage library containing
1011 cyclic peptides was repeated four rounds with the
XC246 auto-antibody, consequently enriching the cyclic peptide
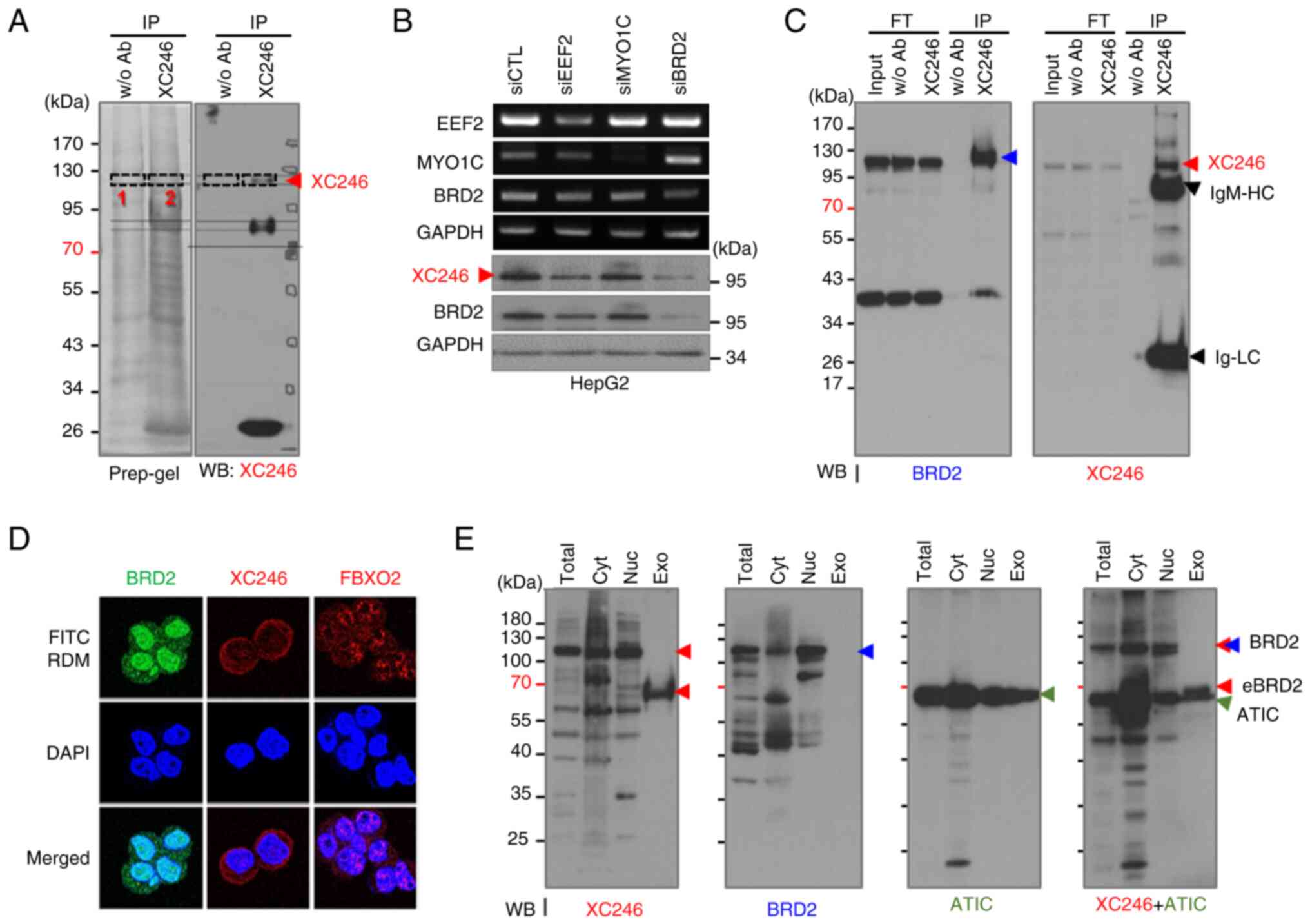
display M13 phages specific to the XC246 antibody (Fig. 3A). The cyclic peptide sequences
displayed on selected phages were determined by sequencing 20
selected M13 phage clones, and 10 different epitopes were confirmed
(Table II). These phages
displayed high reactivity only to the XC246 autoantibody in ELISA,
not to other TA autoantibodies, i.e., K94 (32) or XC90 (17) (Fig.
3B). Among the selected peptide epitopes, the XC246p2, XC246p5,
XC246p6, and XC246p9 sequences showed high reactivity to the XC246
autoantibody (Fig. 3B, Table II). The XC246 antibody-specific
epitopes had the consensus sequence of
C-XSXXLPX-C (Table II). XC246-specific phages
competitively blocked XC246 antibody binding to HepG2 cells, as
demonstrated by FACS analysis (Fig.
3C), indicating that the selected cyclic peptide with high
affinity to the XC246 autoantibody can mimic the specific epitope
on the cellular antigen BRD2. However, linearization of the cyclic
peptide by treatment with a reducing agent resulted in the loss of
those antigenic properties (Fig.
S7), indicating that the conformational properties of the
mimotope are critical for antibody binding.
| Table IIEpitope peptide sequences (C-X7-C*)
of selected phages against XC246 autoantibody. |
Table II
Epitope peptide sequences (C-X7-C*)
of selected phages against XC246 autoantibody.
| Phage antigen | Phage epitope
sequence (-CX7C*-) |
|---|
| XC246p1 | C-SSMFLPS-C |
| XC246p2 | C-SSQWLPF-C |
| XC246p3 | C-TSALFPW-C |
| XC246p4 | C-VSASFPF-C |
| XC246p5 | C-NQVAYPW-C |
| XC246p6 | C-FSALYPW-C |
| XC246p7 | C-CRLRWPH-C |
| XC246p8 | C-TSSFFPH-C |
| XC246p9 | C-TSVFLPH-C |
| XC246p10 | C-STAMALV-C |
To construct an ELISA for the detection of serum
BRD2 autoantibodies, a sufficient supply of target antigen is
needed. Epitope-display M13 phages can be used as coating antigens,
as described in previous studies (32,33);
however, phage amplification and purification are cumbersome
processes. Therefore, synthetic cyclic peptide-conjugated BSA was
prepared and used as a coating antigen to display the
auto-antigen-mimic epitope peptides. For the synthesis of epitope
peptide, the XC246p9 sequence was selected (Table II), which does not contain less
stable amino acids than others, including Q, M, W, N and D, which
are prone to side reactions. Peptide epitope was cyclized by
disulfide bonding between the two cysteine residues located at the
N-terminus and C-terminus of the 9-mer peptide. Two miniPEG were
conjugated to the N-terminus of the peptide as a spacer, and
miniPEG2-linked cyclic peptides were conjugated to BSA via the EDC
reaction (Fig. 3D).
MiniPEG2-conjugated BSA without a peptide epitope was also prepared
and used as a control antigen (Fig.
S8). BSA-miniPEG2-XC246p9 demonstrated high affinity to the
XC246 autoantibody, as shown using ELISA (Figs. 3E). BSA-miniPEG2-XC246p9 also
blocked the binding of the XC246 autoantibody to the endogenous
antigen in tumor cells (HepG2 or SNU638), as revealed by
competitive western blot analysis (Fig. 3F).
Collectively, the TA autoantibody XC246, which was
generated in the HCC model mouse via an autoimmune response to the
TA antigen BRD2, reacted with the XC246p9 cyclic peptide epitope
with high affinity, and XC246p9 peptide-conjugated BSA was prepared
for the detection of the BRD2 autoantibody in human serum.
Human serum ELISA using the
BSA-conjugated BRD2-mimic epitope can differentiate patients with
HCC from control subjects
Human serum ELISA for the BRD2 autoantibody
biomarker was performed using XC246p9 peptide-conjugated BSA under
conditions optimized for each step as follows:
BSA-mini-PEG2-XC246p9 was employed as a coating antigen in ELISA
using a Maxisorp plate. BSA-mini-PEG2 was also used as a negative
control antigen. For blocking non-specific reactions of human sera
to the antigen-coated plates, the plates were treated with a
blocking buffer containing PVP, Ficoll and PFBB, as described in
the 'Materials and methods'. Human serum samples were
heat-inactivated at 50°C for 5 min and diluted 50-fold in blocking
buffer. The sera of 118 patients with HCC and 91 healthy controls
were analyzed for the BRD2 autoantibody biomarker. The patients'
sera with cirrhosis (n=32) and benign cancer (n=3) were also
analyzed. The level of BRD2 autoantibody biomarker was determined
as the difference in OD between the ELISA for BSA-mini-PEG2-XC246p9
and that for BSA-mini-PEG2. As demonstrated in Fig. 4A, the BRD2 autoantibody biomarker
was significantly elevated in the sera of patients with HCC
compared with healthy subjects. The sensitivity of ELISA was 64.41%
and the specificity 82.42% for the cut-off value of 0.1514, with an
AUC value of 0.7761 [95% CI: 0.7136-0.8386, P<0.0001] (Fig. 4B).
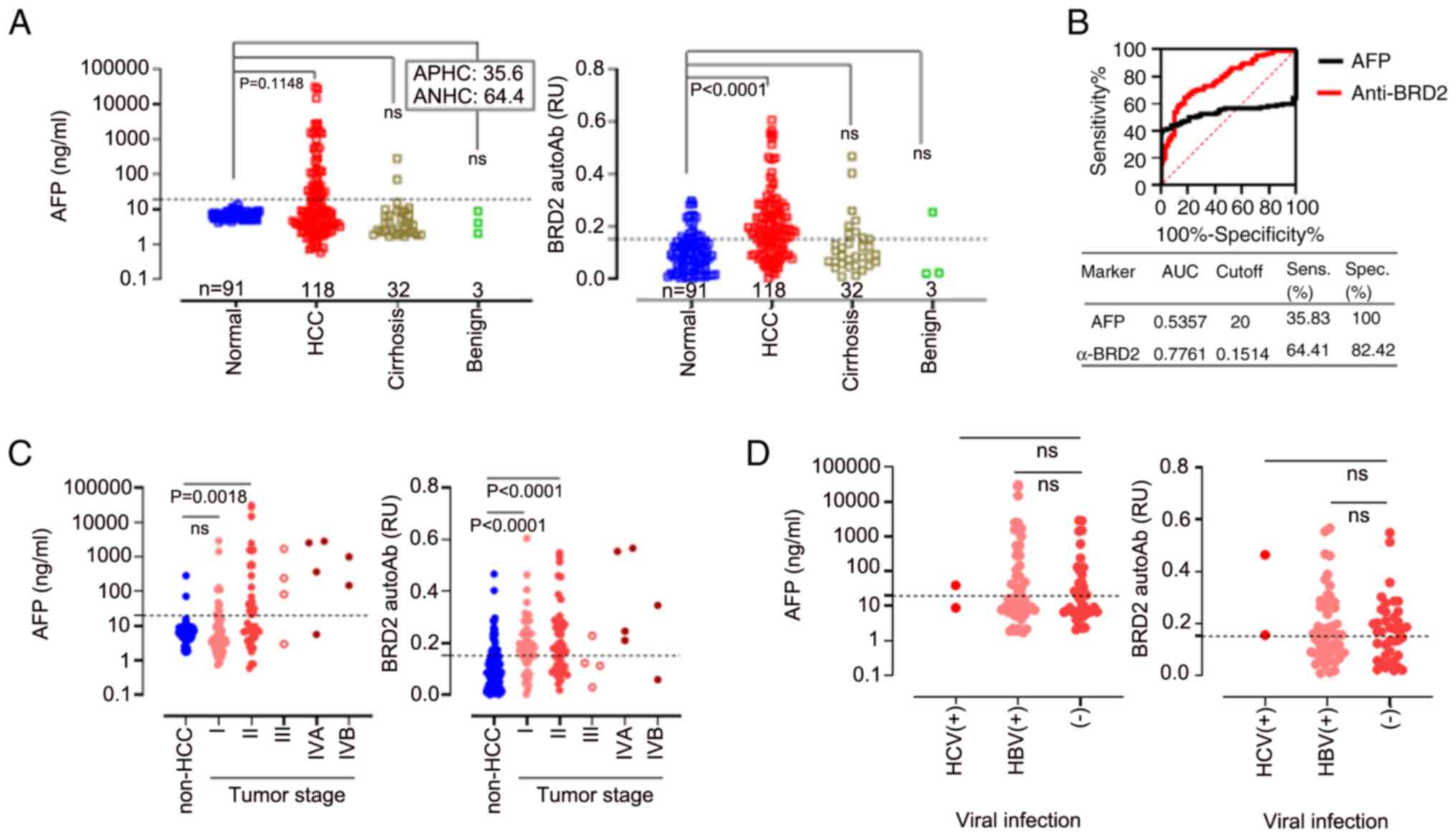 | Figure 4Human serum BRD2 autoantibody ELISA
using BSA-miniPEG2-XC246p9 differentiated patients with HCC from
non-HCC subjects. (A) AFP test and BRD2 autoantibody ELISA using
BSA-miniPEG2-XC246p9 in sera of patients with HCC, as well as
non-tumor subjects. The sample distribution was as follows: Control
(n=91), HCC (n=118), cirrhosis (n=32) and benign liver cancer
(n=3). Serum AFP levels were measured using a commercial
quantification kit. The CV of AFP was 20 ng/ml, and the proportion
of AFP-positive or -negative HCC (APHC or ANHC) is indicated within
the box. The specific binding of the serum autoantibody to the
XC246p9 epitope (anti-BRD2 response) was described as the
difference in OD between the ELISA with BSA-miniPEG2-XC246p9 and
that with BSA-miniPEG2. (B) The ROC curve analysis revealed the
diagnostic sensitivity and specificity of each biomarker. All
experiments were performed in duplicate and repeated at least three
times. (C) Serum AFP and BRD2 autoantibody response related to
tumor stage. The non-HCC group included control, cirrhosis, and
benign liver cancer samples. (D) Serum AFP and BRD2 autoantibody
response related to viral infection. The clinicopathological
features of the participants are described in detail in Table III. ns, not significant
(P>0.05); BRD2, bromodomain-containing protein 2; BSA, bovine
serum albumin; HCC, hepatocellular cancer; CV, cut-off value; APHC,
AFP-positive HCC; ANHC, AFP-negative HCC; AFP, serum
alpha-fetoprotein. |
AFP levels, a most widely used HCC biomarker, were
also analyzed in the same HCC cohort. All healthy controls had a
serum AFP levels below the cut-off value of 20 ng/ml (Fig. 4A). However, not all patients with
HCC had a serum AFP level above the cut-off value. AFP levels
>20 ng/ml were detected only in 35.6% (42/118) of the patients
with HCC and in 6.25% (2/32) of those with cirrhosis. A ROC curve
analysis for the serum AFP test yielded an AUC value of 0.5357 [95%
CI, 0.4514-0.6200; P=0.3746]. The sensitivity was 35.83%, and the
specificity was 100% for the cut-off value of 20 ng/ml AFP
(Fig. 4B).
The detection patterns of these markers were
dependent on the tumor stage. The AFP levels progressively
increased in patients with HCC as the tumor stage advanced
(Fig. 4C). However, BRD2
autoantibody levels were significantly high even in tumor stage I
(P≤0.0001), maintaining those levels throughout all tumor stages
(Fig. 4C). The presence or absence
of viral infection in patients with HCC was not associated with
these two markers in this cohort (Fig.
4D).
The simultaneous detection of AFP and
BRD2 autoantibody in patient sera improves the accuracy of the HCC
diagnosis
AFP is a typical protein biomarker of HCC that is
secreted into the blood via a signal peptide from liver tissue. By
contrast, TA autoantibody biomarkers appeared to be induced by TA
anti-gens, that are released as components of the tumor exosome.
The release mechanism of the tumor exosome is different from that
of secretory proteins, which may reflect other aspects of tumor
characteristics. The correlation between the anti-BRD2 response and
AFP was analyzed using Pearson's correlation analysis, expecting
that AFP and TA autoantibody biomarkers would represent different
characteristics of tumors. The correlation between the BRD2
autoantibody and AFP in sera was very low (Fig. 5A left panel; Pearson's coefficient,
r=0.04240; P=0.5097), suggesting that the appearance of these two
serum markers is regulated by unrelated mechanisms. However, the
BRD2 autoantibody responses in patients with HCC were highly
correlated with another HCC autoantibody biomarker, the ATIC
autoantibody (Fig. 5A right panel;
Pearson's coefficient, r=0.7749; P<0.0001). The ATIC
autoantibody (16) was detected in
the same cohort, using the BSA-miniPEG2-XC154p1 antigen which was
prepared according to the procedures used for the generation of the
BSA-conjugated XC246p9 antigen. This biomarker was observed to be
significantly elevated in the serum of patients with HCC as
compared with that of healthy subjects (Fig. S9A), with an AUC value of 0.8262
[Fig. S9B; 95% CI, 0.7700-0.8824,
P<0.0001]. The sensitivity of the ELISA for ATIC autoantibody
was 64.41%, and the specificity was 78.02% for the cut-off value of
0.6125 (Fig. S9B). The
correlation between the ATIC autoantibody and the BRD2 autoantibody
detection in sera was strong (Pearson's coefficient, r=0.7749;
P<0.0001), although their responses were not identical (Fig. 5A). Pearson's correlation analysis
also was performed on the individual cohorts. As shown in Fig. S10, the AFP and anti-BRD2
autoantibody biomarker pairs exhibited correlation coefficients of
-0.025, 0.105, 0.217 and -0.226 in the HCC, normal, cirrhosis and
benign cohorts, respectively, indicating that there was no
significant correlation between the two biomarkers in all cohorts.
By contrast, the autoantibody biomarker pair (anti-BRD2 and
anti-ATIC) demonstrated positive correlation coefficients (0.795,
0.602, 0.706, and 0.460) in all cohorts, confirming the correlation
between autoantibody biomarkers.
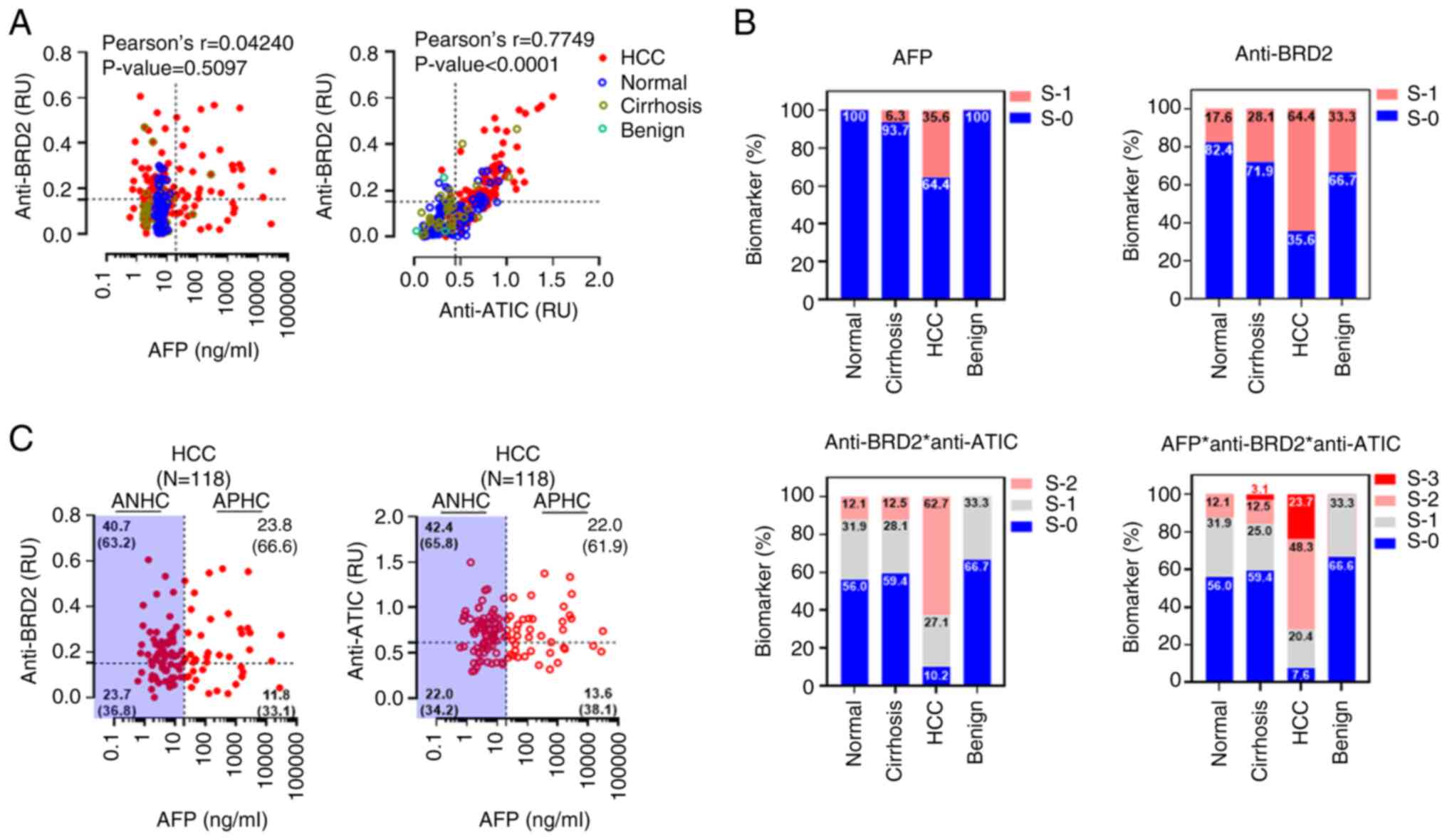 | Figure 5Combined analysis of serum
autoantibody biomarkers with AFP enhanced the diagnostic accuracy
of HCC. (A) Pearson's analysis of the correlations between the BRD2
autoantibody biomarker and the AFP or ATIC autoantibodies. The
dotted lines represent the cutoff value of each biomarker
diagnosis. The results of Pearson's analysis in individual cohorts
are depicted in Fig S10. (B) Combined analysis of HCC biomarkers,
AFP, BRD2 autoantibody, or ATIC autoantibody. The diagnostic values
of each biomarker shown in panel A (AFP, anti-BRD2, and anti-ATIC)
were simplified as either S-1 or S-0 according to whether their
detection values surpassed or fail the cut-off value. Subsequently,
the diagnostic values of each biomarker or their combination were
analyzed. For the combined analysis of these markers, the unified
diagnostic indexes of a serum sample were simply added and
designated triple-negative samples as S-0, single-positive samples
as S-1, double-positive samples as S-2, and triple-positive samples
as S-3. The numbers on the plots represent the percentage of
corresponding subjects. (C) Scattered plot analysis of HCC
biomarker responses depending on AFP and autoantibody biomarker.
The numbers in each quadrant represent the percentage of each case
among patients with HCC (n=118). The numbers in the parentheses are
the proportion of autoantibody biomarker-positive or -negative
samples among ANHC or APHC cases. HCC, hepatocellular cancer; BRD2,
bromodomain-containing protein 2; AFP, serum alpha-fetoprotein;
ATIC, AICAR transformylase/inosine monophosphate cyclohydrolase;
S-1, responsive; S-0, non-responsive; S-2, double positive; S-3,
triple positive; ANHC, AFP-negative HCC; APHC, AFP-positive
HCC. |
The simultaneous detection of cancer biomarkers
representing the different properties of cancer would enhance
diagnostic accuracy. To examine the effects of the combined
analysis of BRD2 or ATIC autoantibody biomarker and AFP, the
response of each biomarker was simplified to positive or negative
and scored as 1 or 0, depending on whether the detection value was
higher or lower than the cutoff value. The combined analysis of
these biomarkers was performed by the simple addition of each
score, which resulted in values of 0, 1, 2, or 3 (Fig. 5B). The proportion of AFP-positive
patients with HCC in the present cohort was 35.6%, and the
percentage of BRD2 autoantibody-positive patients with HCC was
64.4%. The simultaneous detection of the two autoantibody
biomarkers (anti-BRD2 and anti-ATIC) increased the proportion of
biomarker-positive HCC patients (score 1 and 2) to 89.8%. In
addition, the combined detection of AFP and two TA autoantibody
biomarkers (score 1, 2 and 3) could be detected in up to 92.4% of
the patients with HCC. However, a notable proportion of
autoantibody-biomarker-positive cases (score 1 and 2) was also
observed among the normal healthy participant cohort (44.0%).
The diagnosis of HCC is straightforward when
significantly increased serum AFP levels and definitive imaging
features are present. However, AFP-negative hepatic cancer (ANHC)
is not as easily diagnosed, as the majority of ANHCs are early and
small HCCs, often without typical imaging characteristics (34). The diagnosis of ANHC is crucial in
clinical practice, because ANHC accounts for nearly half of HCC
cases and has a better prognosis compared to AFP-positive HCC
(APHC). The BRD2 autoantibody biomarker was analyzed in patients
with ANHC or APHC from the present HCC cohort (Fig. 5C). In the APHC group, 66.6% of the
patients with HCC exhibited a positive BRD2 autoantibody biomarker
response. The BRD2 autoantibody biomarker was also detected as
positive in 63.2% of ANHC cases. The ATIC autoantibody response in
APHC or ANHC cases was similar to that of the BRD2 autoantibody.
However, ~20% of all patients with HCC were determined to be
double-negative for these biomarkers (Fig. 5C).
Discussion
Patients with HCC are known to be frequently
asymptomatic, and the appearance of symptoms can signal the
development of severe disease. Therefore, the early diagnosis of
HCC followed by effective treatment is currently critical for
improving the prognosis and reducing the associated economic burden
(34). AFP is by far the most
widely used serum HCC biomarker. However, its sensitivity for HCC
diagnosis is only 41-65% with a specificity of 80-94% (34).
The present study demonstrated that the anti-BRD2
response can be used to diagnose liver cancer that is not be
screened by AFP (Fig. 5C). In the
cohort used in the present study, AFP was detected only in 35.6% of
the patients with HCC, whereas the BRD2 autoantibody was present in
64.4% of the patients with HCC. Among the AFP-positive patients
with HCC, 66.6% were positive for the BRD2 auto-antibody and 63.2%
of the AFP-negative patients with HCC were also positive for the
BRD2 autoantibody biomarker. Non-responders to AFP, as well as the
anti-BRD2 antibody biomarker comprised the remaining 23.7% of the
patients with HCC. However, the fraction of non-responders to the
HCC biomarkers was decreased to 7.6% by additional detection with
the ATIC autoantibody (Fig. 5B).
TA autoantibodies are biologically amplified signals, corresponding
to TA antigens and hence may be measurable early on, designating
them as promising early biomarkers. However, the antibody response
to each antigen depends on the individual's immune system, and the
amount of antibody will not be exactly proportional to the amount
of antigen. Therefore, in cancer diagnosis using auto-antibody
biomarkers, it is necessary to concurrently measure various
autoantibodies in order to increase accuracy (11,12).
The majority of autoantibody cancer biomarker studies intend to
propose a multiple diagnostic autoantibody panel (11,12).
From the results of the present study, it can also be confirmed
that the diagnostic efficiency is improved by simultaneously
measuring two autoantibodies and AFP and the potential of multiplex
detection of autoantibody biomarkers with AFP as a liver cancer
diagnosis.
Additional verification is required of whether TA
auto-antibodies are detected even in in individuals classified as
normal without HCC. In the present study, the corresponding
individuals appeared to be exposed to abnormal antigens released
from tissues, which induce antigen-specific auto-antibodies,
although their association with HCC is not evidenced. Autoantibody
detection in normal subjects can be an important signal predicting
inflammations or liver diseases. Follow-up of the serum donor may
be required to confirm the disease-related characteristics of the
serum donor.
TA autoantigens, which confer neo-epitopes to the
immune system, can be oncogenic drivers or support tumorigenesis.
The overexpression of the TA antigen ATIC (16) can function as an oncogenic gene
that promotes survival, proliferation, and migration by targeting
AMPK-mTOR-S6 K1 signaling (35).
The genetic deletion of fatty acid synthase (FASN), which is
another TA antigen (33),
suppresses the hepatocarcinogenesis driven by AKT and AKT/c-Met
proto-oncogenes in mice (36),
which implicates the crucial role of FASN during tumorigenesis. The
abundance of BRD2, as well as its high genetic alternation rate
(~19%) in patients with HCC have been shown to be associated with a
worse overall survival (37).
However, the oncogenic properties of upregulated BRD2 in liver
cancer have not been directly confirmed, which can be examined in
further studies.
Although the characteristics of the neo-epitope that
induce BRD2 autoantibody, including genetic alteration,
post-translational modification, abundance, or localization, were
not defined in detail in the present study, evidence was provided,
suggesting the presence of a neo-epitope on oncogenic BRD2. The
BRD2 autoantibody obtained from the HCC mouse model, XC246,
exhibited an unexpected pattern of intracellular staining for BRD2
in tumor cells. In contrast to the commercial anti-BRD2 antibody,
which stains BRD2 mainly in the nucleus, the BRD2 autoantibody,
XC246, stained the cytoplasmic BRD2. Furthermore, the XC246
antibody detected the exosomal BRD2, which was not detected using
the commercial antibody. It is anticipated that these
characteristics of BRD2 are related to the neo-epitope, which
generates the autoantibody.
To develop a detection method for the serum BRD2
auto-antibody, the structural features of the neo-epitope must be
reflected in the capture antigen used in ELISA. Therefore, the
epitope mimicries from a conformational peptide library were
screened, using the BRD2 autoantibody, and then applying it as an
antigen in serum TA autoantibody ELISA, instead of the recombinant
BRD2 protein. It has been previously demonstrated that cyclic
peptide epitopes fused to streptavidin are sufficient to capture
serum autoantibodies (16-18). In the present study, a synthetic
cyclic peptide epitope-conjugated antigen was prepared, in order to
simplify the preparation process of the coating antigen. In
addition, BSA was used as a carrier protein to reduce non-specific
binding in the human serum ELISA. Synthetic cyclic peptide epitopes
fully mimicked the cellular antigenic property, as revealed by
ELISA and competitive western blotting and were proven to be
appropriate for serum ELISA. The ATIC autoantibody was also
detected using peptide epitope-conjugated BSA instead of the
streptavidin conjugate used in a previous study (16). These results demonstrated that
BSA-conjugated mimotope peptides are sufficient for detecting
specific autoantibodies, offering the possibility of its
application to the detection of other autoantigenic epitope mimics
(16-18,32,33).
In conclusion, the present study suggested that the
BRD2 autoantibody can be used as an HCC-associated biomarker and
the feasibility of serum BRD2 autoantibody ELISA using a specific
conformational epitope against the BRD2 autoantibody. Serum BRD2
autoantibody ELISA was also proposed as an accompanying test to
serum AFP detection for the enhancement of HCC diagnostic
efficiency. Further studies on the serum BRD2 autoantibody using
large and well-defined cohorts are necessary to corroborate its
usefulness for cancer diagnosis.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Author's contributions
CKH, WHL and EWC designed the experiments. CKH, WHL,
IP and YSC performed the experiments and collected the data. CKH,
WHL, YSC, KJL and EWC analyzed and interpreted the data. CKH, WHL
and EWC drafted the manuscript. CKH and EWC revised the manuscript
critically. CKH, WHL and YSC confirm the authenticity of all the
raw data. All the authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The Public Institutional Review Board of the
Ministry of Health and Welfare reviewed the contents of the present
study and confirmed that research using commercialized human tissue
falls under the exemption categories specified by Bioethics and
Safety Act in Korea (IRB No. P01-202008-31-009; Republic of Korea).
The study followed the ethical guidance of Ministry of Health and
Welfare in Republic of Korea.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the Korea Research Institute
of Bioscience and Biotechnology Research Initiative Program (grant
no. KGM9942213) and the National Research Foundation of Korea grant
(grant no. NRF-2020R1A2C2014433) funded by the Ministry of Science
and ICT of Korea.
References
|
1
|
World Health Organization (WHO): Cancer.
WHO; Geneva: 2022, https://www.who.int/news-room/fact-sheets/detail/cancer.
Accessed February 3, 2022.
|
|
2
|
Philips CA, Rajesh S, Nair DC, Ahamed R,
Abduljaleel JK and Augustine P: Hepatocellular carcinoma in 2021:
An exhaustive update. Cureus. 13:e192742021.PubMed/NCBI
|
|
3
|
Wong RJ, Cheung R and Ahmed A:
Nonalcoholic steatohepatitis is the most rapidly growing indication
for liver transplantation in patients with hepatocellular carcinoma
in the U.S. Hepatology. 59:2188–2195. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Anstee QM, Reeves HL, Kotsiliti E, Govaere
O and Heikenwalder M: From NASH to HCC: Current concepts and future
challenges. Nat Rev Gastroenterol Hepatol. 16:411–428. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Forner A, Reig M and Bruix J:
Hepatocellular carcinoma. Lancet. 391:1301–1314. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hartke J, Johnson M and Ghabril M: The
diagnosis and treatment of hepatocellular carcinoma. Semin Diagn
Pathol. 34:153–159. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Marshall HT and Djamgoz MBA:
Immuno-oncology: Emerging targets and combination therapies. Front
Oncol. 8:3152018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vinay DS, Ryan EP, Pawelec G, Talib WH,
Stagg J, Elkord E, Lichtor T, Decker WK, Whelan RL, Kumara HMCS, et
al: Immune evasion in cancer: Mechanistic basis and therapeutic
strategies. Semin Cancer Biol. 35(Suppl): S185–S198. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sharonov GV, Serebrovskaya EO, Yuzhakova
DV, Britanova OV and Chudakov DM: B cells, plasma cells and
antibody repertoires in the tumour microenvironment. Nat Rev
Immunol. 20:294–307. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Macdonald IK, Parsy-Kowalska CB and
Chapman CJ: Autoantibodies: Opportunities for early cancer
detection. Trends Cancer. 3:198–213. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kobayashi M, Katayama H, Fahrmann JF and
Hanash SM: Development of autoantibody signatures for common
cancers. Semin Immunol. 47:1013882020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
de Jonge H, Iamele L, Maggi M, Pessino G
and Scotti C: Anti-cancer auto-antibodies: Roles, applications and
open issues. Cancers (Basel). 13:8132021. View Article : Google Scholar :
|
|
13
|
Sexauer D, Gray E and Zaenker P:
Tumour-associated autoantibodies as prognostic cancer biomarkers-a
review. Autoimmun Rev. 21:1030412022. View Article : Google Scholar
|
|
14
|
Wouters MCA and Nelson BH: Prognostic
significance of tumor-infiltrating B cells and plasma cells in
human cancer. Clin Cancer Res. 24:6125–6135. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zheng M, Li YM, Liu ZY, Zhang X, Zhou Y,
Jiang JL, Zhu P, Yang XM, Tang J and Chen ZN: Prognostic landscape
of tumor-infiltrating T and B cells in human cancer. Front Immunol.
12:7313292022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Heo CK, Hwang HM, Lim WH, Lee HJ, Yoo JS,
Lim KJ and Cho EW: Cyclic peptide mimotopes for the detection of
serum Anti-ATIC autoantibody biomarker in hepato-cellular
carcinoma. Int J Mol Sci. 21:97182020. View Article : Google Scholar
|
|
17
|
Heo CK, Hwang HM, Lee HJ, Kwak SS, Yoo JS,
Yu DY, Lim KJ, Lee S and Cho EW: Serum anti-EIF3A autoantibody as a
potential diagnostic marker for hepatocellular carcinoma. Sci Rep.
9:110592019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hwang HM, Heo CK, Lee HJ, Kwak SS, Lim WH,
Yoo JS, Yu DY, Lim KJ, Kim JY and Cho EW: Identification of
anti-SF3B1 auto-antibody as a diagnostic marker in patients with
hepatocellular carcinoma. J Transl Med. 16:1772018. View Article : Google Scholar
|
|
19
|
Zhang XD, Wang Y and Ye LH: Hepatitis B
virus X protein accelerates the development of hepatoma. Cancer
Biol Med. 11:182–190. 2014.PubMed/NCBI
|
|
20
|
Hsieh A, Kim HS, Lim SO, Yu DY and Jung G:
Hepatitis B viral X protein interacts with tumor suppressor
adenomatous polyposis coli to activate Wnt/β-catenin signaling.
Cancer Lett. 300:162–172. 2011. View Article : Google Scholar
|
|
21
|
Yu DY, Moon HB, Son JK, Jeong S, Yu SL,
Yoon H, Han YM, Lee CS, Park JS, Lee CH, et al: Incidence of
hepatocellular carcinoma in transgenic mice expressing the
hepatitis B virus X-protein. J Hepatol. 31:123–132. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
National Library of Medicine (NIH): BRD2
bromodomain containing 2. NIH; Bethesda, MD: 2022, https://www.ncbi.nlm.nih.gov/gtr/genes/6046/. Updated
October 9, 2022.
|
|
23
|
Loganathan SN, Tang N, Fleming JT, Ma Y,
Guo Y, Borinstein SC, Chiang C and Wang J: BET bromodomain
inhibitors suppress EWS-FLI1-dependent transcription and the IGF1
autocrine mechanism in Ewing sarcoma. Oncotarget. 7:43504–43517.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sun LY, Spong A, Swindell WR, Fang Y, Hill
C, Huber JA, Boehm JD, Westbrook R, Salvatori R and Bartke A:
Growth hormone-releasing hormone disruption extends lifespan and
regulates response to caloric restriction in mice. Elife.
2:e010982013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Belkina AC, Blanton WP, Nikolajczyk BS and
Denis GV: The double bromodomain protein Brd2 promotes B cell
expansion and mitogenesis. J Leukoc Biol. 95:451–460. 2014.
View Article : Google Scholar :
|
|
26
|
Pathak S, Stewart WCL, Burd CE, Hester ME
and Greenberg DA: Brd2 haploinsufficiency extends lifespan and
healthspan in C57B6/J mice. PLoS One. 15:e02349102020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hnilicova J, Hozeifi S, Stejskalova E,
Duskova E, Poser I, Humpolickova J, Hof M and Staněk D: The
C-terminal domain of Brd2 is important for chromatin interaction
and regulation of transcription and alternative splicing. Mol Biol
Cell. 24:3557–3568. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Suppiah A and Greenman J: Clinical utility
of anti-p53 auto-antibody: Systematic review and focus on
colorectal cancer. World J Gastroenterol. 19:4651–4670. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zaenker P, Gray ES and Ziman MR:
Autoantibody production in cancer-the humoral immune response
toward autologous antigens in cancer patients. Autoimmun Rev.
15:477–483. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kato T, Fahrmann JF, Hanash SM and
Vykoukal J: Extracellular vesicles mediate B cell immune response
and are a potential target for cancer therapy. Cells. 9:15182020.
View Article : Google Scholar :
|
|
31
|
Hao Q, Wu Y, Wu Y, Wang P and Vadgama JV:
Tumor-derived exosomes in tumor-induced immune suppression. Int J
Mol Sci. 23:14612022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Heo CK, Hwang HM, Ruem A, Yu DY, Lee JY,
Yoo JS, Kim IG, Yoo HS, Oh S, Ko JH and Cho EW: Identification of a
mimotope for circulating anti-cytokeratin 8/18 antibody and its
usage for the diagnosis of breast cancer. Int J Oncol. 42:65–74.
2013. View Article : Google Scholar :
|
|
33
|
Heo CK, Woo MK, Yu DY, Lee JY, Yoo JS, Yoo
HS, Ko JH, Kim JM, Choi JY, Kim IG, et al: Identification of
autoantibody against fatty acid synthase in hepatocellular
carcinoma mouse model and its application to diagnosis of HCC. Int
J Oncol. 36:1453–1459. 2010.PubMed/NCBI
|
|
34
|
Wang T and Zhang KH: New blood biomarkers
for the diagnosis of AFP-negative hepatocellular carcinoma. Front
Oncol. 10:13162020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li M, Jin C, Xu M, Zhou L, Li D and Yin Y:
Bifunctional enzyme ATIC promotes propagation of hepatocellular
carcinoma by regulating AMPK-mTOR-S6 K1 signaling. Cell Commun
Signal. 15:522017. View Article : Google Scholar
|
|
36
|
Che L, Pilo MG, Cigliano A, Latte G,
Simile MM, Ribback S, Dombrowski F, Evert M, Chen X and Calvisi DF:
Oncogene dependent requirement of fatty acid synthase in
hepatocellular carcinoma. Cell Cycle. 16:499–507. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen YR, Ouyang SS, Chen YL, Li P, Xu HW
and Zhu SL: BRD4/8/9 are prognostic biomarkers and associated with
immune infiltrates in hepatocellular carcinoma. Aging (Albany NY).
12:17541–17567. 2020. View Article : Google Scholar
|
|
38
|
Kudo M, Kitano M, Sakurai T and Nishida N:
general rules for the clinical and pathological study of primary
liver cancer, nationwide follow-up survey and clinical practice
guidelines: The outstanding achievements of the liver cancer study
group of Japan. Dig Dis. 33:765–770. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Korean Liver Cancer Study Group (KLCSG);
National Cancer Center, Korea (NCC): 2014.Korean liver cancer study
group-national cancer center Korea practice guideline for the
management of hepatocellular carcinoma. Korean J Radiol.
16:465–522. 2015.
|