Introduction
Neuroblastoma (NB) is a pediatric neuroendocrine
tumor of the sympathetic nervous system. NB arises from
undifferentiated neural crest precursor cells, which results in
tumors emerging mainly in the adrenal medulla or paraspinal
ganglia. It is the most common malignancy diagnosed in the first
year of life and the most common extracranial solid tumor in
childhood (1). NB represents 8-10%
of all pediatric tumors, but results in ~15% of all pediatric
cancer deaths (2,3). Tumors commonly arise in children
<5 years of age with an average age of 18 months at the time of
diagnosis (4). Understanding NB is
a challenge due to the heterogeneity in the course of the disease,
ranging from tumors that show spontaneous regression (even without
treatment) to highly aggressive tumors with a treatment-resistant
phenotype. Patients with high-risk NB (HR-NB) have a very poor
prognosis, with <50% 5-year overall survival rate despite heavy
multimodal treatment, and the survival rate is <10% for relapsed
cases (5). Considering the high
mortality rate associated with HR-NB, increased understanding of
the biology behind this malignancy is important as it may lead to
improvement in patient stratification and ultimately in novel
therapeutic strategies.
Analysis of chromosomal copy number variations
(CNVs) in NB provides relevant information for the diagnosis and
prognosis of the disease. Tumors with numerical aberrations only
(gain/loss of whole chromosomes) and near triploid karyotype are
typically associated with a good prognosis, whereas tumors with
segmental rearrangements and near di- or tetraploid karyotypes
generally have a poor prognosis (6,7).
Recurrent CNVs in NB include loss of chromosome 1p, 3p, 4p and 11q,
gain of 1q, 2p and 17q, and amplification of the MYCN
oncogene; notably, MYCN amplification and 11q-deletion are
associated with more aggressive NB. Small focal alterations have
been shown to target cell cycle genes (e.g., CDKN2A/B, CCND1,
CDK4/6 andMDM2) (8-10) or
genes involved in chromatin remodeling and telomere maintenance
(e.g., ATRX, ARID1A/ARID1B andTERT) (11,12).
Whereas large segmental alterations are common in NB, the somatic
acquisition of small genetic alterations are relatively rare in
sporadic NB, with recurrent alterations mainly found in ALK,
ATRX, PTPN11 andTIAM1 (13-15).
However, as the use of large-scale, high-throughput techniques,
such as next generation sequencing, is expanding, additional
recurrent aberrations that contribute to the development and
progression of NB are expected to be detected (16).
Recent whole genome sequencing (WGS) studies
performed by us and others have detected recurrent structural
variants (SVs) that affect the limbic system-associated membrane
protein (LSAMP) gene located in chromosomal region 3q13.31
(12,17). LSAMP encodes a protein
member of the Ig-like cell adhesion (IgLON) family of proteins,
which also include opioid-binding protein/cell adhesion molecule
like (OPCML), neuronal growth regulator 1 (NEGR1),
neurotrimin (NTM) and IGLON5 (18). All of the IgLON members have been
shown to be expressed during central nervous system development,
and are involved in cell adhesion, neurite outgrowth (19), dendritic arborization, neuronal
development (20) and synapse
formation (21,22). Alterations in IgLONs have been
reported to be associated with mental disorders (23-25)
and also different types of cancer, where they function as tumor
suppressor genes (26-28).
The presence of LSAMP has been observed in neural
crest cells in experiments conducted in chickens (19), indicating that it may be relevant
in the context of NB, as it is considered a malignancy caused by
dysregulation of embryonic neural crest development (29). In addition, LSAMP is reportedly
involved in axon guidance during the limbic system development
(30), neurite outgrowth in dorsal
root ganglia (31) and psychiatric
disorders (24,32). Notably, it has also previously been
reported as a tumor suppressor gene in several types of cancer
(33-44).
Given that LSAMP has been shown to have a
role in neural development and has been implicated as a tumor
suppressor gene in different types of cancer, the present study
hypothesized that LSAMP could have a tumor-suppressing
capacity in NB. This was investigated in the present study by
examining the frequency and extent of recurrent genomic alterations
targeting the LSAMP gene in NB tumors and NB cell lines
through WGS and single nucleotide polymorphism (SNP)-microarrays,
together with in vitro exploration of the functional
implication of LSAMP re-expression or silencing.
Materials and methods
Tumor material and cell lines
All NB samples from Swedish patients (n=35) were
collected after obtaining written informed consent from their
parents or guardians, and were analyzed according to permits
approved by the Karolinska Institutet and the Karolinska University
Hospital ethics committees (Stockholm, Sweden; approval no.
2009/1369-31/1) in agreement with The Declaration of Helsinki.
Sampling was performed during routine clinical procedures, and
treatment was performed according to established national and
international protocols. Demographic and genomic data of patients
are outlined in Table I. The NB
cell lines NB1, NB69, SK-N-AS, SK-N-BE(2), SK-N-F1, SK-N-DZ, SK-N-SH, SH-SY5Y,
KELLY and IMR-32 were obtained from the American Type Culture
Collection. LS and NGP cell lines were kindly provided by Prof.
Manfred Schwab (DKFZ, Heidelberg, Germany), whereas the CLB-GAR,
CLB-BAR, CLB-PE and CLB-GE cell lines were retrieved from the
Centre Léon Bérard (Lyon, France) under a material transfer
agreement. All cell lines were cultured in high-glucose DMEM
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS
(Gibco; Thermo Fisher Scientific, Inc.) at 37°C in an atmosphere
containing 5% CO2, and were routinely verified to be
negative for mycoplasma, bacterial and fungal infection.
 | Table IDemographic and genomic features of
the study cohort. |
Table I
Demographic and genomic features of
the study cohort.
| Characteristic | Whole cohort
(n=35) |
LSAMP-deleted (n=6) | LSAMP not
deleted (n=29) |
|---|
| Sex | | | |
| Female | 17 | 2 | 15 |
| Male | 18 | 4 | 14 |
| Age, months | | | |
| Median age at
diagnosis (range) | 36 (1-265) | 31 (11-63) | 36 (1-265) |
| Median age of time
of sampling (range) | 48 (1-265) | 65 (11-132) | 48 (1-265) |
| Outcome | | | |
| DOD | 11 | 3 | 8 |
| NED | 11 | 1 | 10 |
| AWD | 10 | 2 | 8 |
| NA | 3 | 0 | 3 |
| Genomic
subgroupa | | | |
| MNA | 6 | 1 | 5 |
| MNA +
11q-deleted | 2 | 1 | 1 |
| 11q-deleted | 16 | 2 | 14 |
| 17q-gain | 6 | 1 | 5 |
| Other
structural | 5 | 1 | 4 |
| Genomic
alterations | | | |
| TERT or ATRX | 18 | 3 | 15 |
| ALK-mutation | 4 | 1 | 3 |
| Cell cycle (CCND1,
CDKN2A/B, MDM2 or CDK4) | 6 | 3 | 3 |
| 1p-del | 16 | 1 | 15 |
| 2p-gain | 16 | 5 | 11 |
| Segmental
17q-gain/wcg17 | 28/7 | 6/0 | 22/7 |
| Number of somatic
events | | | |
| Median SV
(range) | 28 (1-89) | 33 (28-89) | 26 (81-85) |
| Median SNV
(range) | 18 (1-81) | 24 (8-37) | 18 (1-81) |
WGS analysis
DNA from primary tumor and corresponding normal
samples from 35 patients were analyzed by WGS, including seven
patients from our recent study where relapsed tumor material was
also subject to sequencing (12).
Tumor DNA was extracted from fresh frozen tissue and constitutional
DNA from the blood using DNeasy Blood and Tissue Kit (Qiagen GmbH)
with sequencing, subsequent bioinformatics handling and variant
filtering performed as described previously (12). Briefly, pair-end sequencing was
conducted on NovaSeq 6000 (Illumina, Inc.) with an average coverage
of at least 60X for tumor material and 30X for constitutional DNA.
The Sentieon TNscope software version 201911 (Sentieon, Inc.,
Mountain View, CA) was used for mapping to hg19, removal of read
duplicates, realignment around InDels and variant calling, whereas
QIAGEN Clinical Insight Interpret software (version 8.1.20210827;
Qiagen GmbH) was used for systematic filtering of called variants.
Copy number profiles were generated using the CANVAS tool (version
1.38.0.1554) (45) and somatic SVs
were called using the Manta tool (version 1.1.1) (46). All called SVs were evaluated by
manual inspection using Integrative Genomics Viewer (version
2.3.81) (47).
SNP-array analysis
CytoScan™ HD-microarrays (cat. no. 901835; Applied
Biosystems; Thermo Fisher Scientific, Inc.) were also used to
analyze copy number changes on the tumor samples from the 35
patients with NB in combination with 16 NB cell lines [NB1, NB69,
CLB-GAR, CLB-BAR, CLB-PE, CLB-GE, SK-N-AS, SK-N-BE(2), SK-N-F1, SK-N-DZ, SK-N-SH, SH-SY5Y,
KELLY, IMR-32, LS and NGP] to corroborate and further explore the
3q13.31 chromosomal region. The arrays were scanned using a
confocal laser scanner, GeneChip Scanner 3000 (Applied Biosystems;
Thermo Fisher Scientific, Inc.) with normalization of the tumor
samples performed in silico with genomic annotation based on
the hg19 human genome. The procedure for handling CytoScan HD
microarrays has been described previously (48).
Sanger sequencing analysis
Primers for LSAMP breakpoint verification
were designed either using Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/)
or placed manually with specificity evaluated through UCSC
In-Silico PCR (https://genome.ucsc.edu/), and were used together with
PLATINUM SuperFI PCR Master Mix (Thermo Fisher Scientific, Inc.)
for PCR amplification, according to the manufacturer's
instructions. Primer sequences are presented in Table SI. Sanger sequencing of amplicons
was performed by Eurofins Genomics following standard conditions.
The sequencing visualization and analysis was performed using
SnapGene viewer software version 5.1 (www.snapgene.com), to verify LSAMP
rearrangements.
Cell transfection, proliferation,
viability and differentiation assays
To evaluate the role of LSAMP in proliferation and
viability, LSAMP was silenced in the LSAMP-expressing
cell lines SH-SY5Y and SK-N-BE(2),
whereas re-/overexpression of LSAMP was conducted in the
KELLY and SK-N-AS cell lines (with homozygous and heterozygous
LSAMP deletion, respectively). Knockdown of LSAMP was
performed using pre-designed short hairpin (sh)RNA lentiviral
particles from Santa Cruz Biotechnology, Inc. LSAMP shRNA
(h) Lentiviral Particles (cat. no. sc-78206-V) were used and
Control shRNA Lentiviral Particles-A (cat. no. sc-108080) were used
as the control, whereas copGFP-Control Lentiviral Particles (cat.
no. sc-108084) was used for optimization and determination of
initial transfection efficiency in parallel experiments.
Transduction using lentiviral particles was performed with
1×105 particles and 1×105 cells,
corresponding to a multiplicity of infection of 1, in 1 ml medium
per well in 12-well plates using Polybrene (cat. no. sc-134220) at
a final concentration of 8 µg/ml. After 48 h transduction,
the medium containing lentiviral particles and Polybrene was
replaced with complete medium lacking Polybrene. Stable cells were
selected by adding puromycin (2 µg/ml) to the medium 72 h
post-transduction, with changes of fresh medium supplemented with
puromycin (2 µg/ml) every 3-4 days during the expansion for
further analysis. To re-/overexpress LSAMP, pCMV6-AC-LSAMP
and pCMV6-AC (control) plasmids (GeneArt; Thermo Fisher Scientific,
Inc.) were used. The GFP-tagged constructs pCMV6-AC-LSAMP-GFP and
pCMV6-AC-GFP were seeded in parallel experiments in order to
determine initial transfection efficiency as well as cellular
protein localization. All constructs were verified by Sanger
sequencing. The transfection was conducted in 10-cm plates
containing 2.5×106 cells/well using 1 µg
respective vector dissolved in Lipofectamine® 3000
(Invitrogen; Thermo Fisher Scientific, Inc.). The medium was
changed 24 h after transfection and 200 µg/ml neomycin was
added 48 h post-transfection for the selection of stable
transfects. The medium was replaced with fresh medium supplemented
with neomycin (200 µg/ml) every 3-4 days and cells were
expanded for further analyses. Real-time measurements of cell
proliferation were performed for 96 h using E-plates in the
xCELLigence RTCA DP Instrument (Agilent Technologies, Inc.)
according to the manufacturer's protocol, which uses impedance for
live monitoring of proliferation. By contrast, cell viability was
monitored after 72 h of seeding using Presto Blue HS (Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol with a
15-min incubation with the Presto Blue HS reagent prior to
absorbance measurement. Each group was assessed four times in
96-well plates and all experiments were repeated three times.
To evaluate the effect of LSAMP silencing on
differentiation capacity, SK-N-BE(2) and SH-SY-5Y cells were seeded at a
density of 750 cells/well in 96-well plates 1 day before replacing
the medium with differentiation medium, which consisted of
high-glucose medium supplemented with 2.5% FBS and 7.5 µM
retinoic acid (RA; MilliporeSigma). High-glucose medium
supplemented with 2.5% FBS and 0.075% EtOH was used as a control.
The medium was then changed 4 days after seeding and neurite
extension was evaluated using a fluorescence microscope after 7
days of RA incubation using a neurite outgrowth staining kit (cat.
no. A15001; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol.
Reverse transcription-quantitative PCR
(RT-qPCR)
RNA was extracted from 14 NB cell lines and
transfected cells (maximum 80% confluence) using RNeasy plus mini
kit (Qiagen GmbH), and was quantified spectrophotometri cally using
a NanoDrop spectrophotometer (NanoDrop; Thermo Fisher Scientific,
Inc.). cDNA synthesis was conducted using the Superscript IV VILO
master mix according to the manufacturer's protocol (Invitrogen;
Thermo Fisher Scientific, Inc.). qPCR was performed using TaqMan™
Fast Advanced Master Mix (Applied Biosystems; Thermo Fisher
Scientific) under standard conditions in a 7500 Real-Time PCR
system (Applied Biosystems; Thermo Fisher Scientific) under the
following conditions: Initial hold at 95°C for 2 min, followed by
40 cycles of denaturation at 95°C for 3 sec and annealing/extension
at 60°C for 30 sec. The following probes were used: LSAMP
pre-designed TaqMan probes covering three different exon boundaries
(Exon 1-2; Hs01082649_m1, exon 2-3; Hs01082650_m1 and exon 4-5;
Hs01082652_m1; Thermo Fisher Scientific, Inc.) and two housekeeping
gene probes, qA-01-0104P5 (ACTB) and qA-01-0105P5
(YWHAC) (both from TATAA Biocenter AB). The housekeeping
genes used, ACTB and YWHAC, were selected and
verified using a human reference gene panel (cat. no. A101) from
TATAA Biocenter AB and analyzed by GeneEx (https://bio.tools/geneex). Gene expression analysis
was performed using the 2−ΔΔCq method (49).
Kaplan-Meier analysis
Kaplan-Meier analysis with calculation of log-rank
test for overall and event-free survival probability in relation to
the expression levels of LSAMP and other members of the
IgLON family (OPCML, NTM, IGLON5 and NEGR1) was
performed using the Kaplan-scan cutoff method in 'R2: Genomic
Analysis and Visualization Platform' (http://r2.amc.nl)
with the publicly available dataset 'Tumor
Neuroblastoma-SEQC-498-rpm-Seqnb1' (n=498). The Kaplan-scan cutoff
method examines every increasing expression value as cutoff for
log-rank test in order to find the optimal segregation point of two
groups based on gene expression. This method then presents the most
statistically significant cutoff with corresponding Bonferroni
corrected P-value together with the initial non-corrected
P-value.
Statistical analysis
The proliferation and viability studies were
performed on at least three repeats per respective cell line, and
included at least four replicates per sample and assay. Cell
proliferation was measured every hour during 96 h. Two-way repeated
measures (RM) ANOVA was used to determine the significance of the
proliferation differences between two groups over time. The
Geisser-Greenhouse ε correction was applied following ANOVA, since
the experiment includes multiple measurements over time, the
sphericity of the sample cannot be assumed. Cell viability was
measured 72 h after cell seeding. Unpaired two-sided t-test
analysis was performed using the normalized mean value of each
assay, to compare the means of two groups, assuming Gaussian
distribution. LSAMP expression levels in the different NB
cell lines were quantified by RT-qPCR in triplicate and qPCR was
repeated three times. Differences in LSAMP expression in
cell lines were analyzed using unpaired two-sided t-test, comparing
the means from two groups with different LSAMP status (wild
type vs. SVs). Comparison of LSAMP expression levels in
human NB tumors with or without MYCN amplification was
performed by one-way ANOVA, whereas the correlation of gene
expression levels of LSAMP, OPCML, NTM, IGLON5 and
NEGR1 was assessed using Pearson correlation coefficient
analysis, both using the 'Tumor Neuroblastoma-SEQC-498-rpm-Seqnb1'
(n=498) dataset (http://r2.amc.nl). All statistical
analyses were performed using GraphPad 9.2.0 (GraphPad Software,
Inc.). P<0.05 was considered to indicate a statistically
significant difference.
Results
Copy number analysis detects recurrent
focal alterations of LSAMP
Through copy number profiling and structural
variation calling from WGS analysis, somatic focal rearrangements
located within chromosomal region 3q13.31 were observed at the
heterozygous level in tumor tissue from six out of the 35
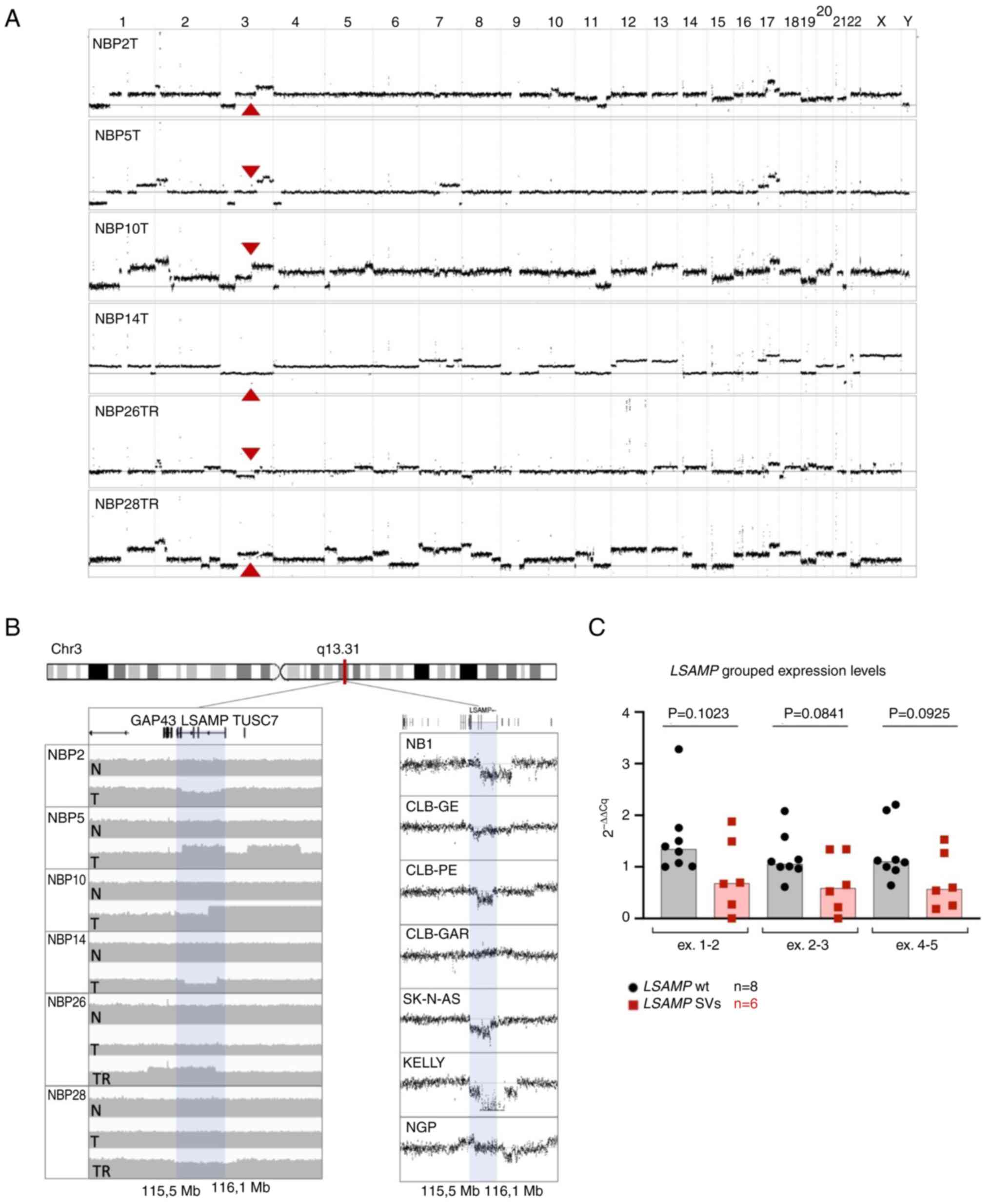
investigated NB tumor samples (Fig. 1A
and B). Analysis of these rearrangements revealed that the
breaks were located within or in the vicinity of the LSAMP
gene (Table II), resulting in
disruption of the gene. SNP-microarray performed on the same tumor
tissue verified these segmental alterations (Fig. S1A and B), whereas the specific
breakpoints in LSAMP were verified by PCR and Sanger
sequencing (Fig. S2).
| Table IIPosition and type of 3q13.31
alteration. |
Table II
Position and type of 3q13.31
alteration.
| Case | 3q13.31 alteration
| Other genomic
features |
|---|
| Type | Position |
|---|
| NBP2 | Deletion |
chr3:115.582.490-116.120.525 | 1p/1q-imbalance,
2p-gain, MNA, 3p-del, 3q-gain, 10q-gain, 11q-del, 17q-gain |
| NBP5 | Deletion |
chr3:116.165.059-116.472.978 | 1p/1q-imbalance,
2p-gain, MNA 3p-del, 3q-gain, 4p-del, 7q-gain, 17q-gain,
TERT SV, CDKN2A-del |
| NBP10 | Translocation |
t(1;3)(114.696.299)(115.952.494) | 1p/1q-imbalance,
2p-gain, 3p-del, 3q-gain, 4p-del, 5p-del, 5q-gain, 11q-del,
17q-gain, 20q-gain, 21q-del, ARID1A SV, TERT SV |
| NBP14 | Deletion |
chr3:115.624.534-116.061.783 | 1q-del,
interstitial 7q-del, 17q-gain, 21q-del, 22q-gain,
ATRX-del |
| NBP26 | Tandem
duplication |
chr3:115.150.006-116.052.713 | 2p-gain, 2q-gain,
interstitial 3q-del, 5q-gain, 6q-gain, 8p-del, 12q-amplification
(CDK4/MDM2), 17q-gain, 18p-del, 19q-gain, interstitial
Xp-gain |
| NBP28 | Deletion |
chr3:115.505.427-116.344.076 | 1p/1q-imbalance,
2p-gain, interstitial 2q-del, 3p-del, 5p-gain, 6q-del, 7p-gain,
8p-gain, 9p-del, 11q-del, 14q-del, 17q-gain, TERT SV |
From the seven tumors included in our previous
study, from which primary and relapsed material was available,
LSAMP rearrangement was present in two. In these two cases,
the rearrangements were present in the relapsed tumors only and not
in the primary tumors (cases NBP26 and NBP28) (10) (Fig.
1B; left panel). WGS did not detect any non-synonymous
mutations in LSAMP among the investigated tumors.
Other large recurrent segmental or genomic
alterations detected in the six NB samples with LSAMP
rearrangement included 1p/1q-imbalance (n=4), 2p-gain (n=5),
3p-deletion (n=4), 4p-deletion (n=2), 17q-gain (n=6), 11q-deletion
(n=3), MYCN-amplification (n=2), and break in proximity of
TERT or in ATRX (n=4) (Fig. 1A; Table II). Major genomic alterations
detected in the full cohort are summarized in Table I. In addition to the primary tumor
material, 16 NB cell lines were analyzed by SNP-microarrays. This
analysis revealed that seven out of the 16 cell lines (NB1, CLB-GE,
CLB-PE, CLB-GAR, SK-N-AS, KELLY and NGP) had segmental alterations
involvingLSAMP, including a homozygous deletion within
LSAMP detected in KELLY (Fig.
1B, right panel; Fig.
S1C).
Segmental alterations are associated with
a decreased gene expression in NB cell lines
In order to analyze whether the expression levels of
LSAMP differed between the cell lines with and without
segmental alterations, TaqMan probes intersecting three different
exon boundaries (exons 1-2, 2-3 and 4-5) were utilized (Fig. S3C). After unpaired two-sided
t-test, no statistically significant differences in LSAMP
expression levels were observed when comparing cell lines with and
without 3q13.31 SVs (Figs. 1C and
S3A); however, a trend of lower
expression was observed in the cell lines with 3q13.31 SVs.
Furthermore, in the case of a homozygous deletion break point
inside of LSAMP, i.e. in the KELLY cell line, no expression
was detected at exons 1-2 and 2-3, and very low expression was
detected at exon 4-5 (Fig.
S3A).
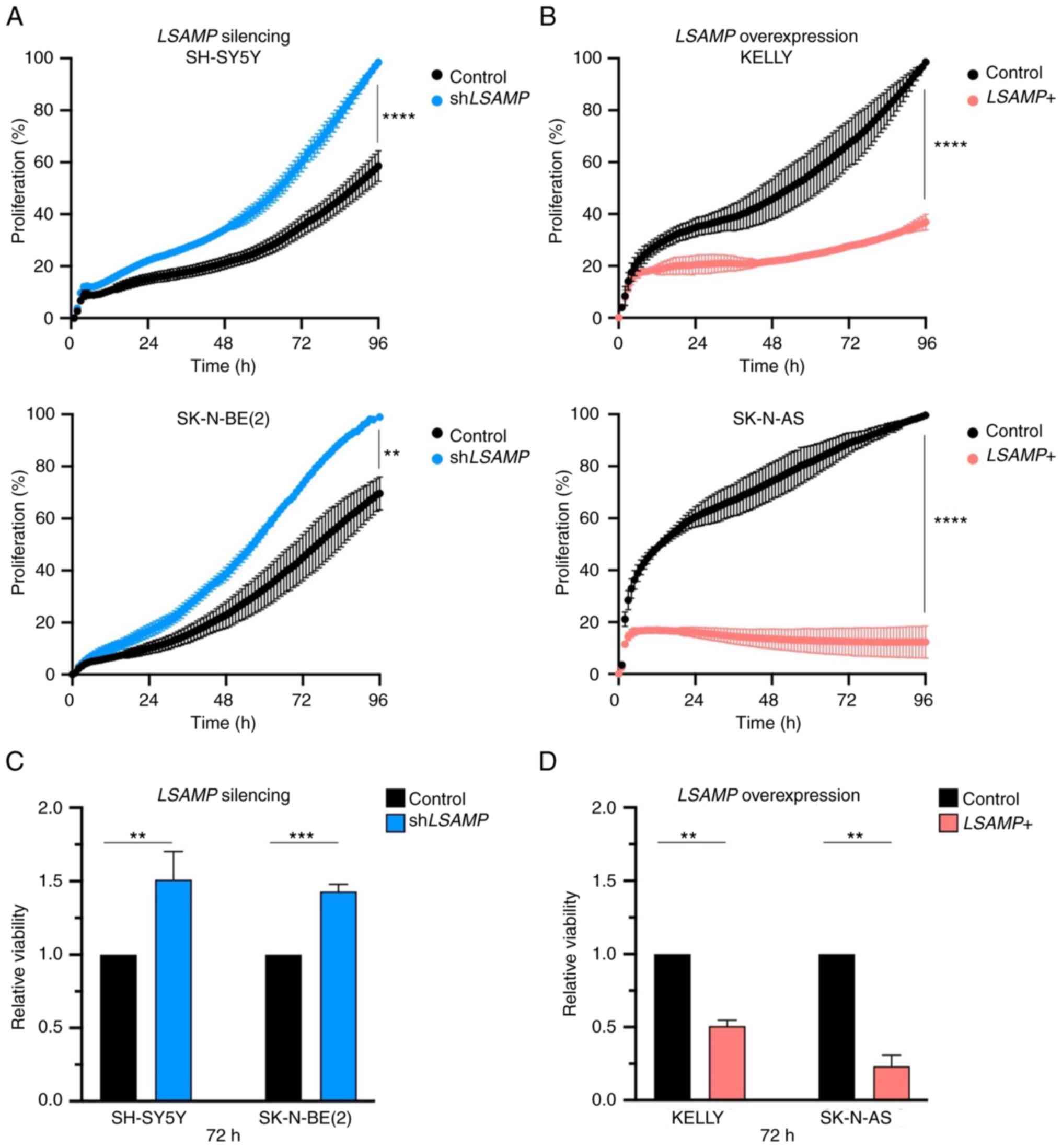
Knockdown of LSAMP restimulates NB cell
proliferation in vitro
To investigate the effect of LSAMP knockdown
on the LSAMP-expressing cell lines SH-SY5Y and
SK-N-BE(2), shRNA lentiviral
particles were used. Initial transduction success for SH-SY5Y and
SK-N-BE(2) cells was ~70% for both
cell lines, as judged by the number of GPF-expressing cells among
control cells transduced with copGFP-Control lentiviral particles
(data not shown). Determination of knockdown efficiency through
qPCR showed a ~50% reduction in LSAMP expression levels in
silenced cells as compared with stable cells transduced with
control shRNA (Fig. S3B, left
panel). After the selection of stable clones, proliferation and
viability analyses were conducted. Knockdown of LSAMP led to
statistically significant increase in proliferation in both SH-SY5Y
and SK-N-BE(2) cell lines after 96
h [two-way RM ANOVA with the Geisser-Greenhouse ε correction;
SH-SY5Y, P<0.0001; SK-N-BE(2),
P=0.045; Fig. 2A]. A significant
difference in viability after 72 h was also seen after LSAMP
knockdown in SH-SY5Y (unpaired t-test; P=0.004) and
SK-N-BE(2) (unpaired t-test;
P=0.0004) cells (Fig. 2C).
Knockdown of LSAMP, with or without the presence of RA, did
not exhibit any major effect on neurite extension in SH-SY5Y and
SK-N-BE(2) cells (Fig. S4).
LSAMP re- and overexpression inhibits NB
proliferation and viability in vitro
In order to investigate how the re-expression and
overexpression of LSAMP can affect cell proliferation and
viability, KELLY cells (in which LSAMP is homozygously
deleted) and SK-N-AS cells (which have a heterozygous LSAMP
deletion) were used, as both display low expression levels of
LSAMP. Initial transfection efficacy for KELLY and SK-N-AS
cells was 70%, as judged by the expression of GFP in cells
transfected with pCMV6-AC-GFP at the same occasion (data not
shown). Evaluation by qPCR showed a significant increase of
LSAMP expression levels in LSAMP-transfected cells as
compared with in pCMV6-AC-tranfected controls (Fig. S3B, right panel). Re- and
overexpression of LSAMP in KELLY and SK-N-AS cells both led
to a significant decrease in cell proliferation after 96 h (two-way
RM ANOVA with Geisser-Greenhouse's ε correction; P<0.0001;
Fig. 2B) as well as reduced
viability after 72 h in KELLY (unpaired t-test; P=0.0022) and
SK-N-AS (unpaired t-test; P=0.0032) cells (Fig. 2D). Regarding KELLY cells, viability
was also significantly reduced after 96 h (P<0.001; Fig. S3D). Moreover, preliminary data of
LSAMP overexpression in SH-SY5Y cells also exhibited a
significant reduction in viability (data not shown).
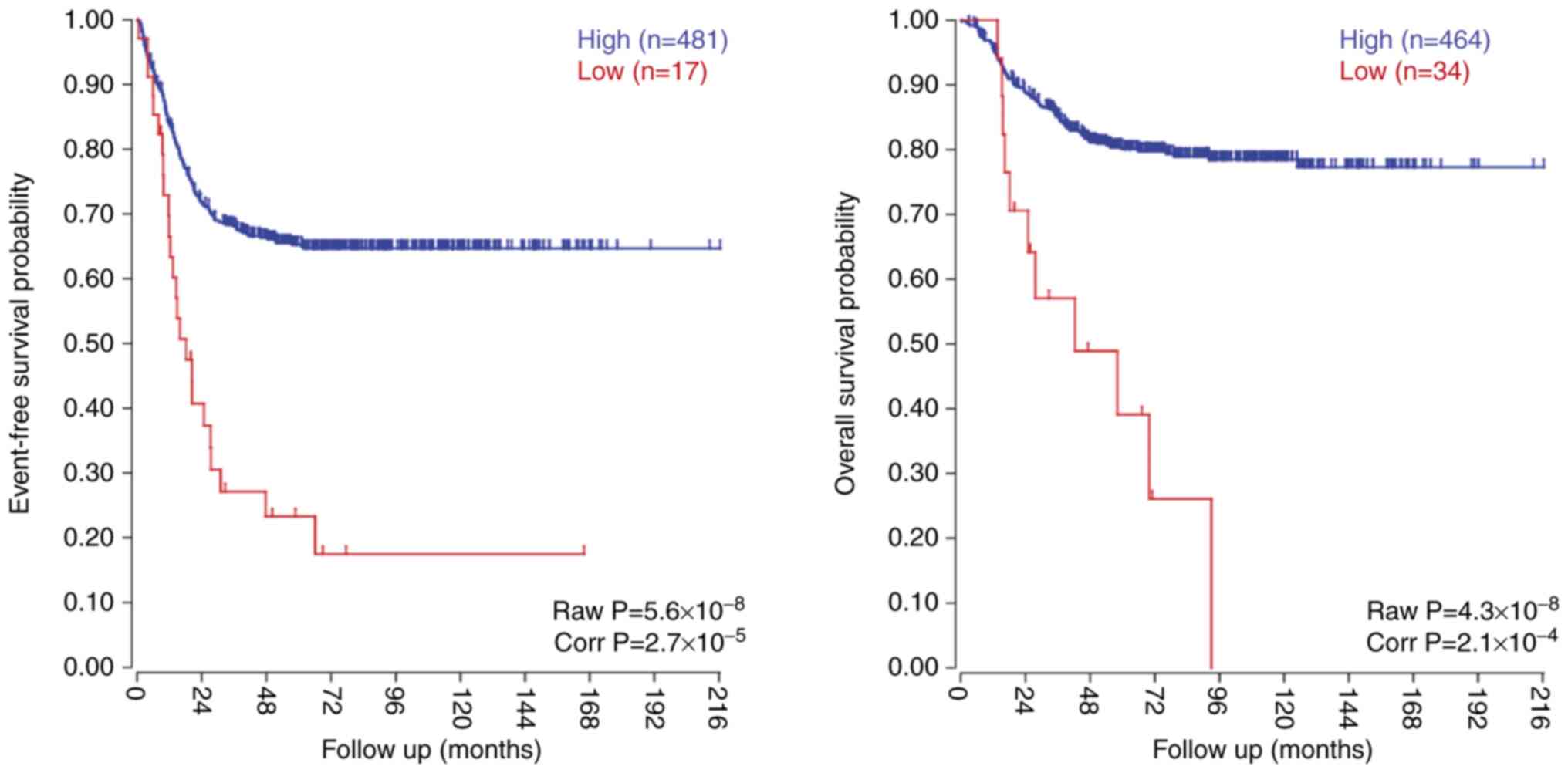
Low expression of LSAMP and other members
of the IgLON family in patients with NB is associated with poor
survival
To investigate the prognostic value of LSAMP
in NB, the expression levels of LSAMP in a publicly
available and validated cohort of 498 NB tumors were studied. In
this dataset, low LSAMP expression was significantly
associated with poor patient overall survival and event-free
survival (Fig. 3). The association
ofLSAMP with poor overall and event-free survival was most
prominent among the non-MYCN-amplified cases and to lesser
extent among the MYCN-amplified cases (Fig. S5A), although no statistically
significant difference in expression was seen when comparing
MYCN-amplified vs. non-MYCN-amplified NB (Fig. S5B and C). Similar results were
seen for other NB cohorts also publicly available from the R2
genomic analysis and visualization platform (data not shown).
Investigation of other members of the IgLON family,
OPCML, NTM, IGLON5 and NEGR1, using the
same NB dataset as aforementioned, revealed that low expression of
these genes was also significantly associated with poor overall and
event-free survival (Fig. S6A).
However, no strong correlation in gene expression was detected
between any of the different IgLON members (Fig. S6B).
Discussion
By WGS and SNP-microarrays, recurrent focal
segmental rearrange ments affecting the LSAMP gene located
at chromosomal region 3q13.31 were de tected in six out of 35 NB
tumor samples (17%) as well as in seven out of 16 NB cell lines
(44%). The LSAMP alterations were co-occurring with several
other genomic alterations, including MYCN-amplification,
1p-deletion, 11q-deletion and 17q-gain, in which the latter was
observed in all cases with LSAMP aberration. The
co-occurrence of 17q-gain indicated that they are a part of a
subgroup associated with a worse prognosis. However, segmental and
numerical gains affecting the long arm of chromosome 17 is the most
common chromosomal aberration in NB present in all genomic
subgroups (7), and thus not unique
for cases with LSAMP alterations. WGS did not detect any
pathogenic or likely pathogenic variants in LSAMP. However,
the genetic etiology with few somatic mutations indicated that NB
is mainly driven by genomic events that cause copy number
alterations, lead to enhancer hijacking or disruption of genes.
Thus, the lack of point mutations in favor of segmental alterations
of LSAMP is not completely surprising.
LSAMP has a selective role in neuronal growth, which
is also interesting from a NB research perspective. In addition,
recurrent alterations in other genes associated with
neuro-development and NB have recently been described by Lopez
et al (17). Notably,
LSAMP has previously been described as a tumor suppressor
gene in osteosarcoma (33-37), ovarian cancer (38), renal cell carcinoma (39), prostatic cancer (40,41),
epithelioid glioblastoma (42),
myeloid leukemia (43) and
recently in lung cancer (44).
Gene expression analysis among NB cell lines indicated that the
average expression levels were lower in LSAMP-rearranged
samples, although no statistically significant difference was
detected between the groups; however, in the cell line KELLY with a
homozygous deletion rearrangement inside LSAMP, no
expression levels were detected at exons 1-2 and 2-4. The low
expression of LSAMP in cell lines lacking the 3q13.31
rearrangement could be due to epigenetic alterations or other
mechanisms, as found in lung cancer and clear cell renal cell
carcinoma (33,38); however, further studies are needed
to clarify this. In order to study the expression and regulation of
LSAMP in NB, knockdown and re-/overexpression experiments
were performed in different NB cell lines. LSAMP knockdown
led to a significant increase in cell proliferation and viability
in the LSAMP wild-type cell lines SH-SY5Y and
SK-N-BE(2). These results are in
concordance with previous publications (33-44),
suggesting that LSAMP could be a putative tumor suppressor
gene also in NB. In addition, re-expression of LSAMP in
KELLY cells (with homozygous LSAMP deletion) and
overexpression of LSAMP in SK-N-AS cells (with heterozygous
LSAMP deletion) inhibited cell proliferation and viability.
The difference in the level of effect on proliferation between
SK-N-AS cells, which had major cell loss, compared with KELLY
cells, which had only a decrease in proliferation, could possibly
be due to the hetero- and homozygous status of LSAMP
deletion in the respective cell line. As evaluation of apoptosis
was not performed, the current study is restricted as we cannot
distinguish between cell death or decreased proliferation as a
cause of decreased cell viability.
The present study also revealed that low expression
of LSAMP was associated with poor overall survival in
patients with NB, which supports a possible role in aggressive
tumor behavior. This association was most evident for the
non-MYCN-amplified tumors. In the present cohort of mainly
high-risk cases, SVs affecting LSAMP were present in
different genomic subgroups (MYCN-amplified,
MYCN-amplified together with 11q-deletion, 11q-deleted,
17q-gain and other structural aberrations). A study of a larger
cohort reported that SVs affecting LSAMP were predominantly
found in non-MYCN-amplified high-risk tumors (17). However, the higher occurrence of
LSAMP SVs in cell lines relative to patient tumor samples
indicated that this alteration might be a later event contributing
to tumor progression rather than initiation. Furthermore, for two
of the tumors, LSAMP rearrangements were detected in the
tumor during relapse, but not at the time of diagnosis. These
findings also supported the hypothesis that LSAMP
alterations are not associated with initial tumor development but
rather contribute to the progression of the disease or the
promotion of cell migration. No major difference in neurite
extension was seen after LSAMP knockdown in NB cell lines.
However, the current study is limited and additional investigations
exploring longer or other differentiation conditions, together with
additional cell lines, is required for a better understanding of
LSAMP in the context of cell differentiation.
LSAMP is a member of the IgLON family, which is a
subgroup of cell adhesion molecules involved in diverse roles in
neuronal development (18), and
which can also function as tumor suppressor genes (26,50-53).
The main members of this family are all located in other
chromosomal regions that are frequently deleted in NB and
associated with poor prognosis; NEGR1 is located in 1p31,
IGLON5 is located in 19q13, and OPCML and NTM
are located in 11q25, a chromosomal region that is commonly in
cluded in 11q-deleted NB and also in a subset of
MYCN-amplified NB (7,16,54,55).
Notably, low expression of these family members was associated with
poor overall and event-free survival, but did not exhibit obvious
correlation with LSAMP expression levels in NB. In addition,
studies in lung cancer have shown that NEGR1 is involved in
the regulation of LSAMP and that LSAMP can be linked
to cancer cell migration through epithelial-mesenchymal transition
(44). A similar finding was
reported in another study in which depletion of NEGR1 led to
increased cell migration and invasion (53). The presence of LSAMP
alterations were detected at the heterozygous level in most cases,
which would imply haploinsufficiency; however, an additional level
of complexity is added from the fact that IgLONs have a conserved
interaction mode with the capability of binding to each other as
homodimers and heterodimers (56).
LSAMP can heterodimerize with either OPCML or NTM, both
11q-localized (57), and if the
dimerization partner of LSAMP is not present, this could also
possibly affect proper function. This suggests that decreased
levels of LSAMP, possibly in combination with decreased
levels of other IgLON members, may facilitate tumorigenesis through
mechanisms affecting proliferation, migration or contact
inhibition. However, the specific role of the different IgLON
family members in NB pathogenesis is so far speculative and
requires further investigation. Furthermore, having in
consideration the highly conserved molecular features of IgLONs, it
would be interesting to study the possible implications of
LSAMP downregulation on MAPK/PI3K signaling, as NEGR1 has
been reported to be related with this relevant pathway in cancer
(18,58).
In conclusion, the present sequencing analyses
detected novel recurrent somatic alterations involving
LSAMP, a cell adhesion molecule that functions in neural
crest development and neurite extension. The in vitro
studies revealed that LSAMP may have an anti-proliferative
function in NB, possibly through the dysfunction of intercellular
adhesion and neural differentiation.
Supplementary Data
Availability of data and materials
The genetic datasets from patients associated with
this manuscript are not publicly available due to protection from
complete disclosure of genome data according to the consent form.
SNP-microarray data generated and/or analyzed during the current
study are available in the Gene Expression Omnibus database under
accession no. GSE209728 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE209728).
Other data-sets used and/or analyzed during the current study are
available from the corresponding author on reasonable request.
Authors' contributions
AMM conceived and designed experiments. AMM, AD, JG
and SF performed the experiments, analyzed and interpreted the
data. PK helped with patient care and obtaining clinical
information. SF designed the study, formulated strategy and
directed the research. AMM and SF confirmed the authenticity of all
the raw data and drafted the manuscript. All authors read and
approved final version of the manuscript.
Ethics approval and consent to
participate
All NB samples from Swedish patients were collected
after obtaining written informed consent from their parents or
guardians, and were analyzed according to permits approved by the
Karolinska Institutet and the Karolinska University Hospital ethics
committees (Stockholm, Sweden; approval no. 2009/1369-31/1) in
agreement with The Declaration of Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This work has been supported by grants from the Assar
Gabrielsson foundation, the Swedish Childhood Cancer Foundation
(grant no. SF: PR2018-099, 15-0061) and the Swedish Research
Council (grant no. SF: 521-2014-3031).
References
|
1
|
Diller GP and Baumgartner H: Sudden
cardiac death during exercise in patients with congenital heart
disease: The exercise paradox and the challenge of appropriate
counselling. Eur Heart J. 37:627–629. 2016. View Article : Google Scholar
|
|
2
|
Stiller CA and Parkin DM: International
variations in the incidence of neuroblastoma. Int J Cancer.
52:538–543. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Park JR, Eggert A and Caron H:
Neuroblastoma: Biology, prognosis, and treatment. Hematol Oncol
Clin North Am. 24:65–86. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
London WB, Castleberry RP, Matthay KK,
Look AT, Seeger RC, Shimada H, Thorner P, Brodeur G, Maris JM,
Reynolds CP and Cohn SL: Evidence for an age cutoff greater than
365 days for neuroblastoma risk group stratification in the
children's oncology group. J Clin Oncol. 23:6459–6465. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maris JM, Hogarty MD, Bagatell R and Cohn
SL: Neuroblastoma. Lancet. 369:2106–2120. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Janoueix-Lerosey I, Schleiermacher G,
Michels E, Mosseri V, Ribeiro A, Lequin D, Vermeulen J, Couturier
J, Peuchmaur M, Valent A, et al: Overall genomic pattern is a
predictor of outcome in neuroblastoma. J Clin Oncol. 27:1026–1033.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Caren H, Kryh H, Nethander M, Sjöberg RM,
Träger C, Nilsson S, Abrahamsson J, Kogner P and Martinsson T:
High-risk neuroblastoma tumors with 11q-deletion display a poor
prognostic, chromosome instability phenotype with later onset. Proc
Natl Acad Sci USA. 107:4323–4328. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guimier A, Ferrand S, Pierron G, Couturier
J, Janoueix-Lerosey I, Combaret V, Mosseri V, Thebaud E, Gambart M,
Plantaz D, et al: Clinical characteristics and outcome of patients
with neuroblastoma presenting genomic amplification of loci other
than MYCN. PLoS One. 9:e1019902014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Martinez-Monleon A, Oberg HK, Gaarder J,
Berbegall AP, Javanmardi N, Djos A, Ussowicz M, Taschner-Mandl S,
Ambros IM, Øra I, et al: Amplification of CDK4 and MDM2: A detailed
study of a high-risk neuroblastoma subgroup. Sci Rep. 12:124202022.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Molenaar JJ, Koster J, Ebus ME, van Sluis
P, Westerhout EM, de Preter K, Gisselsson D, Øra I, Speleman F,
Caron HN and Versteeg R: Copy number defects of G1-cell cycle genes
in neuroblastoma are frequent and correlate with high expression of
E2F target genes and a poor prognosis. Genes Chromosomes Cancer.
51:10–19. 2012. View Article : Google Scholar
|
|
11
|
Ackermann S, Cartolano M, Hero B, Welte A,
Kahlert Y, Roderwieser A, Bartenhagen C, Walter E, Gecht J,
Kerschke L, et al: A mechanistic classification of clinical
phenotypes in neuroblastoma. Science. 362:1165–1170. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fransson S, Martinez-Monleon A, Johansson
M, Sjöberg RM, Björklund C, Ljungman G, Ek T, Kogner P and
Martinsson T: Whole-genome sequencing of recurrent neuroblastoma
reveals somatic mutations that affect key players in cancer
progression and telomere maintenance. Sci Rep. 10:224322020.
View Article : Google Scholar
|
|
13
|
Molenaar JJ, Koster J, Zwijnenburg DA, van
Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J,
Westerman BA, van Arkel J, et al: Sequencing of neuroblastoma
identifies chromothripsis and defects in neuritogenesis genes.
Nature. 483:589–593. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen QR, Bilke S, Wei JS, Whiteford CC,
Cenacchi N, Krasnoselsky AL, Greer BT, Son CG, Westermann F,
Berthold F, et al: cDNA array-CGH profiling identifies genomic
alterations specific to stage and MYCN-amplification in
neuroblastoma. BMC Genomics. 5:702004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vandesompele J, Speleman F, Van Roy N,
Laureys G, Brinskchmidt C, Christiansen H, Lampert F, Lastowska M,
Bown N, Pearson A, et al: Multicentre analysis of patterns of DNA
gains and losses in 204 neuroblastoma tumors: How many genetic
subgroups are there? Med Pediatr Oncol. 36:5–10. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fransson S, Ostensson M, Djos A,
Javanmardi N, Kogner P and Martinsson T: Estimation of copy number
aberrations: Comparison of exome sequencing data with SNP
microarrays identifies homozygous deletions of 19q13.2 and CIC in
neuroblastoma. Int J Oncol. 48:1103–1116. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lopez G, Conkrite KL, Doepner M, Rathi KS,
Modi A, Vaksman Z, Farra LM, Hyson E, Noureddine M, Wei JS, et al:
Somatic structural variation targets neurodevelopmental genes and
identifies SHANK2 as a tumor suppressor in neuroblastoma. Genome
Res. 30:1228–1242. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kubick N, Brosamle D and Mickael ME:
Molecular evolution and functional divergence of the IgLON family.
Evol Bioinform Online. 14:11769343187750812018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kimura Y, Katoh A, Kaneko T, Takahama K
and Tanaka H: Two members of the IgLON family are expressed in a
restricted region of the developing chick brain and neural crest.
Dev Growth Differ. 43:257–263. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pischedda F and Piccoli G: The IgLON
family member Negr1 promotes neuronal arborization acting as
soluble factor via FGFR2. Front Mol Neurosci. 8:892015.
|
|
21
|
Yamada M, Hashimoto T, Hayashi N, Higuchi
M, Murakami A, Nakashima T, Maekawa S and Miyata S: Synaptic
adhesion molecule OBCAM; synaptogenesis and dynamic
internalization. Brain Res. 1165:5–14. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hashimoto T, Maekawa S and Miyata S: IgLON
cell adhesion molecules regulate synaptogenesis in hippocampal
neurons. Cell Biochem Funct. 27:496–498. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Minhas HM, Pescosolido MF, Schwede M,
Piasecka J, Gaitanis J, Tantravahi U and Morrow EM: An unbalanced
translocation involving loss of 10q26.2 and gain of 11q25 in a
pedigree with autism spectrum disorder and cerebellar juvenile
pilocytic astrocytoma. Am J Med Genet A. 161A:787–791. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Karis K, Eskla KL, Kaare M, Täht K, Tuusov
J, Visnapuu T, Innos J, Jayaram M, Timmusk T, Weickert CS, et al:
Altered expression profile of IgLON family of neural cell adhesion
molecules in the dorsolateral prefrontal cortex of schizophrenic
patients. Front Mol Neurosci. 11:82018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Singh K, Jayaram M, Kaare M, Leidmaa E,
Jagomäe T, Heinla I, Hickey MA, Kaasik A, Schäfer MK, Innos J, et
al: Neural cell adhesion molecule Negr1 deficiency in mouse results
in structural brain endophenotypes and behavioral deviations
related to psychiatric disorders. Sci Rep. 9:54572019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ntougkos E, Rush R, Scott D, Frankenberg
T, Gabra H, Smyth JF and Sellar GC: The IgLON family in epithelial
ovarian cancer: Expression profiles and clinicopathologic
correlates. Clin Cancer Res. 11:5764–5768. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xing X, Cai W, Ma S, Wang Y, Shi H, Li M,
Jiao J, Yang Y, Liu L, Zhang X and Chen M: Down-regulated
expression of OPCML predicts an unfavorable prognosis and promotes
disease progression in human gastric cancer. BMC Cancer.
17:2682017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang N, Xu J, Wang Y, Heng X, Yang L and
Xing X: Loss of opioid binding protein/cell adhesion molecule-like
gene expression in gastric cancer. Oncol Lett. 15:9973–9977.
2018.PubMed/NCBI
|
|
29
|
Tomolonis JA, Agarwal S and Shohet JM:
Neuroblastoma pathogenesis: Deregulation of embryonic neural crest
development. Cell Tissue Res. 372:245–262. 2018. View Article : Google Scholar :
|
|
30
|
Zhukareva V, Chernevskaya N, Pimenta A,
Nowycky M and Levitt P: Limbic system-associated membrane protein
(LAMP) induces neurite outgrowth and intracellular Ca2+ increase in
primary fetal neurons. Mol Cell Neurosci. 10:43–55. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sanz RL, Ferraro GB, Girouard MP and
Fournier AE: Ectodomain shedding of limbic system-associated
membrane protein (LSAMP) by ADAM metallopeptidases promotes neurite
outgrowth in DRG neurons. Sci Rep. 7:79612017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Koido K, Traks T, Balotsev R, Eller T,
Must A, Koks S, Maron E, Tõru I, Shlik J, Vasar V and Vasar E:
Associations between LSAMP gene polymorphisms and major depressive
disorder and panic disorder. Transl Psychiatry. 2:e1522012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yen CC, Chen WM, Chen TH, York-Kwan WC,
Chih-Hsueh PC, Chiou HJ, Hung GY, Wu HTH, Wei CJ, Shiau CY, et al:
Identification of chromosomal aberrations associated with disease
progression and a novel 3q13.31 deletion involving LSAMP gene in
osteosarcoma. Int J Oncol. 35:775–788. 2009.PubMed/NCBI
|
|
34
|
Kresse SH, Ohnstad HO, Paulsen EB,
Bjerkehagen B, Szuhai K, Serra M, Schaefer KL, Myklebost O and
Meza-Zepeda LA: LSAMP, a novel candidate tumor suppressor gene in
human osteosarcomas, identified by array comparative genomic
hybridization. Genes Chromosomes Cancer. 48:679–693. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pasic I, Shlien A, Durbin AD, Stavropoulos
DJ, Baskin B, Ray PN, Novokmet A and Malkin D: Recurrent focal
copy-number changes and loss of heterozygosity implicate two
noncoding RNAs and one tumor suppressor gene at chromosome 3q13.31
in osteosarcoma. Cancer Res. 70:160–171. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Baroy T, Kresse SH, Skarn M, Stabell M,
Castro R, Lauvrak S, Llombart-Bosch A, Myklebost O and Meza-Zepeda
LA: Reexpression of LSAMP inhibits tumor growth in a preclinical
osteosarcoma model. Mol Cancer. 13:932014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Smida J, Xu H, Zhang Y, Baumhoer D, Ribi
S, Kovac M, von Luettichau I, Bielack S, O'Leary VB, Leib-Mösch C,
et al: Genome-wide analysis of somatic copy number alterations and
chromosomal breakages in osteosarcoma. Int J Cancer. 141:816–828.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang L, Yuan Y, Lu KH and Zhang L:
Identification of recurrent focal copy number variations and their
putative targeted driver genes in ovarian cancer. BMC
Bioinformatics. 17:2222016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen J, Lui WO, Vos MD, Clark GJ,
Takahashi M, Schoumans J, Khoo SK, Petillo D, Lavery T, Sugimura J,
et al: The t(1;3) breakpoint-spanning genes LSAMP and NORE1 are
involved in clear cell renal cell carcinomas. Cancer Cell.
4:405–413. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Petrovics G, Li H, Stumpel T, Tan SH,
Young D, Katta S, Li Q, Ying K, Klocke B, Ravindranath L, et al: A
novel genomic alteration of LSAMP associates with aggressive
prostate cancer in African American men. EBioMedicine. 2:1957–1964.
2015. View Article : Google Scholar
|
|
41
|
Huang SP, Lin VC, Lee YC, Yu CC, Huang CY,
Chang TY, Lee HZ, Juang SH, Lu TL and Bao BY: Genetic variants in
nuclear factor-kappa B binding sites are associated with clinical
outcomes in prostate cancer patients. Eur J Cancer. 49:3729–3737.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nobusawa S, Hirato J, Kurihara H, Ogawa A,
Okura N, Nagaishi M, Ikota H, Yokoo H and Nakazato Y: Intratumoral
heterogeneity of genomic imbalance in a case of epithelioid
glioblastoma with BRAF V600E mutation. Brain Pathol. 24:239–246.
2014. View Article : Google Scholar
|
|
43
|
Yoon J, Cho EH, Yun JW, Kim HY, Jang JH,
Kim HJ and Kim SH: LSAMP rearrangement in acute myeloid leukemia
with a jumping translocation involving 3q13.31. Ann Lab Med.
41:342–345. 2021. View Article : Google Scholar :
|
|
44
|
Chang CY, Wu KL, Chang YY, Liu YW, Huang
YC, Jian SF, Lin YS, Tsai PH, Hung JY, Tsai YM and Hsu YL: The
downregulation of LSAMP expression promotes lung cancer progression
and is associated with poor survival prognosis. J Pers Med.
11:5782021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Roller E, Ivakhno S, Lee S, Royce T and
Tanner S: Canvas: Versatile and scalable detection of copy number
variants. Bioinformatics. 32:2375–2377. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen X, Schulz-Trieglaff O, Shaw R, Barnes
B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S and Saunders CT:
Manta: Rapid detection of structural variants and indels for
germline and cancer sequencing applications. Bioinformatics.
32:1220–1222. 2016. View Article : Google Scholar
|
|
47
|
Thorvaldsdottir H, Robinson JT and Mesirov
JP: Integrative genomics viewer (IGV): High-performance genomics
data visualization and exploration. Brief Bioinform. 14:178–192.
2013. View Article : Google Scholar :
|
|
48
|
Umapathy G, Guan J, Gustafsson DE,
Javanmardi N, Cervantes-Madrid D, Djos A, Martinsson T, Palmer RH
and Hallberg B: MEK inhibitor trametinib does not prevent the
growth of anaplastic lymphoma kinase (ALK)-addicted neuroblastomas.
Sci Signal. 10:eaam75502017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
50
|
Sellar GC, Watt KP, Rabiasz GJ, Stronach
EA, Li L, Miller EP, Massie CE, Miller J, Contreras-Moreira B,
Scott D, et al: OPCML at 11q25 is epigenetically inactivated and
has tumor-suppressor function in epithelial ovarian cancer. Nat
Genet. 34:337–343. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
51
|
Reed JE, Dunn JR, du Plessis DG, Shaw EJ,
Reeves P, Gee AL, Warnke PC, Sellar GC, Moss DJ and Walker C:
Expression of cellular adhesion molecule 'OPCML' is down-regulated
in gliomas and other brain tumours. Neuropathol Appl Neurobiol.
33:77–85. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Cui Y, Ying Y, van Hasselt A, Ng KM, Yu J,
Zhang Q, Jin J, Liu D, Rhim JS, Rha SY, et al: OPCML is a broad
tumor suppressor for multiple carcinomas and lymphomas with
frequently epigenetic inactivation. PLoS One. 3:e29902008.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kim H, Hwang JS, Lee B, Hong J and Lee S:
Newly identified cancer-associated role of human neuronal growth
regulator 1 (NEGR1). J Cancer. 5:598–608. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Abbasi MR, Rifatbegovic F, Brunner C, Mann
G, Ziegler A, Pötschger U, Crazzolara R, Ussowicz M, Benesch M,
Ebetsberger-Dachs G, et al: Impact of disseminated neuroblastoma
cells on the identification of the relapse-seeding clone. Clin
Cancer Res. 23:4224–4232. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Caron H: Allelic loss of chromosome 1 and
additional chromosome 17 material are both unfavourable prognostic
markers in neuroblastoma. Med Pediatr Oncol. 24:215–221. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Venkannagari H, Kasper JM, Misra A, Rush
SA, Fan S, Lee H, Sun H, Seshadrinathan S, Machius M, Hommel JD and
Rudenko G: Highly conserved molecular features in IgLONs contrast
their distinct structural and biological outcomes. J Mol Biol.
432:5287–5303. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Reed J, McNamee C, Rackstraw S, Jenkins J
and Moss D: Diglons are heterodimeric proteins composed of IgLON
subunits, and Diglon-CO inhibits neurite outgrowth from cerebellar
granule cells. J Cell Sci. 117:3961–3973. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Szczurkowska J, Pischedda F, Pinto B,
Managò F, Haas CA, Summa M, Bertorelli R, Papaleo F, Schäfer MK,
Piccoli G and Cancedda L: NEGR1 and FGFR2 cooperatively regulate
cortical development and core behaviours related to autism
disorders in mice. Brain. 141:2772–2794. 2018.PubMed/NCBI
|