Introduction
Colorectal cancer (CRC) is the second leading cause
of cancer-related mortality and was the third most commonly
diagnosed tumor in 2020, according to global cancer statistics
(1,2). The incidence and mortality associated
with colorectal cancer are rapidly increasing, due to environmental
factors, dietary changes, the aging population and germline
genetics (3,4). The incidence of CRC has been
increasing in recent years, and the mortality rates of male and
female patients with CRC in China have increased (5). Therapies for CRC have notably
improved; however, the 5-year survival rate of patients with
metastatic CRC remains extremely low, and metastatic CRC, in most
cases, remains an incurable disease (6). It is thus imperative to exploit new
targets which play crucial roles in carcinogenesis. A more in-depth
understanding the underlying molecular mechanisms may prove to be
helpful for the exploration of novel diagnostic or potential
therapeutic targets for CRC.
Glycosylation, the major post-translational
modification of proteins, regulates a number of biological
processes including cell proliferation, differentiation,
intracellular and intercellular signaling, cell-to-cell
communications and immune recognition (7-9).
Increasing evidence has revealed that aberrant glycosylation plays
crucial roles in tumor proliferation, metastasis, angiogenesis and
immune surveillance, and has been cited as a hallmark of cancer
(10,11). Aberrant glycosylation can stem from
the altered expression of glycosyltransferases, the dysfunction of
glycosyltransferases, donor substrate availability, metabolic
alterations, and modified molecular chaperone activity (12). Fucosylation, performed by
fucosyltransferase, is one of the terminal and important
modifications of glycan (13). The
aberrant expression of fucosyltransferases (FUTs) has been reported
in numerous cancer types and plays vital biological roles in tumor
development (14). The
overexpression of FUT7 has been shown to promote A549 cell
proliferation by activating the EGFR/AKT/mTOR signaling pathway
(15). The high expression of FUT4
has been found to be associated with programmed death-1, immune
response and poor overall survival in operable lung adenocarcinoma
(LUAD) (16). Another study
demonstrated that fucosylation induced by FUT8 was increased in
aggressive prostate cancer cells (17). FUT2 catalyzes fucose in the
α-1,2-linkage at the terminal of glycan; it is also overexpressed
in several cancer types and has been associated with tumor
progression. FUT2 is overexpressed in LUAD and promotes LUAD
metastasis through the epithelial-mesenchymal transition initiated
by TGF-β/Smad signaling (18).
FUT1 and FUT2 play crucial roles in the regulation of the
tumorigenesis, metastasis and cancer stem cell properties of breast
cancer (19). However, the
biological role and molecular mechanisms of action of FUT2 in CRC
remain unknown.
The Wnt signaling pathway plays crucial roles in
cell proliferation, migration and has also been shown to be tightly
associated with cancer. The Wnt/β-catenin pathway, also known as
canonical Wnt signaling, is crucial for intestinal development and
stem cell renewal, and aberrant Wnt/β-catenin signaling is an early
event in CRC development, which has most prominently been described
in CRC (20,21). In recent years, novel insight into
the Wnt/β-catenin pathway was obtained, further clarifying the
regulatory mechanisms of the pathway (21).
In the present study, the effects of FUT2 on CRC
cell proliferation and metastasis were investigated. FUT2 was
overexpressed in CRC tissues and cell lines. The knockdown of FUT2
induced G0/G1 cell cycle arrest and inhibited proliferation,
migration and invasion, while promoting the apoptosis of CRC cells.
Furthermore, FUT2 regulated the proliferation and metastasis of CRC
cells via the Wnt/β-catenin pathway. The findings of the present
study revealed the effects of FUT2 in CRC progression, and may thus
provide a potential treatment strategy for CRC.
Materials and methods
Bioinformatics analysis
Gene Expression Profiling Interactive Analysis
(GEPIA), an online software (http://gepia.cancer-pku.cn) for cancer and normal gene
expression profiling and interactive analysis (22), was used to analyze the gene
expression levels of FUT2 in colon adenocarcinoma (COAD) and rectal
adenocarcinoma (READ). CRC-related data were extracted from The
Cancer Genome Atlas (TCGA) database. In total, 275 CAOD tumor
samples and 349 normal samples, and 92 READ tumor samples and 318
normal samples were selected and analyzed.
CRC specimens
The CRC tissues and their paired adjacent
non-cancerous tissues (28 pairs of tissues; median age of patients,
66.4 years; range, 48-89 years) used in the experiments were
collected from the First Affiliated Hospital of Wenzhou Medical
University between April, 2018 to September, 2018. All cases were
from patients diagnosed with CRC and had not received any treatment
prior to surgery. The present study was approved by the Ethics
Committee of the First Affiliated Hospital of Wenzhou Medical
University (Issuing no. 2019-086). CRC tissue microarrays were
obtained from Alenabio; Taibsbio (http://www.taibsbio.com/), containing 36 pairs of CRC
tissues and adjacent non-tumor tissues, 9 pairs of colorectal
cancer lymph node metastatic tissues and normal lymph node tissues,
and 8 normal colon tissues and 2 normal rectal tissues (DCO1002c).
Clinical staging was based on the International Union for cancer
control/American Joint Committee on Cancer (UICC/AJCC) TNM
classification (7th edition) (23). Written informed consent was
obtained from all participants prior to the commencement of the
study.
Cell lines and cell culture
The human colorectal cancer cell lines, SW-480
(TCHu172), DLD-1 (TCHu134), HCT-116 (TCHu 99) and HCT-8 (TCHu 18),
and the human normal colorectal epithelial cell (FHC; CRL-1831)
were obtained from the Cell Bank of the Chinese Academy of Sciences
(Shanghai, China). The cells were maintained in DMEM (Gibco; Thermo
Fisher Scientific, Inc.) with 10% FBS (fetal bovine serum) (Gibco;
Thermo Fisher Scientific, Inc.). All cells were cultured in 37°C in
a 5% CO2 incubator.
Cell transfection
The knockdown of FUT2 was achieved with short
hairpin RNA (shRNA)-mediated gene silencing using constructed
plasmids containing hairpin FUT2 shRNA sequence (sh-FUT2#1: 5′-GGT
CAG TTA ATT TAG CGG CTC-3′, sh-FUT2#2: 5′-GGT AGG AAT TGT CAC ATA
CCC-3′, sh-NC: 5′-GCT TCG CGC CGT AGT CTT A-3′). For transfection,
the 2.5 µg vectors (pGPU6/GFP/Neo-FUT2 or
pGPU6/GFP/Neo-shNC) (GeneCopoeia, Inc.) were transfected into the
SW-480 and DLD-1 cells using Lipofectamine 3000 reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), with incubation for
15 min at room temperature, according to the manufacturer's
instructions. The cells were cultured in DMEM containing 10% FBS
and 350 ng/ml puromycin (Beijing Solarbio Science & Technology
Co., Ltd.) under 5% CO2 at 37°C for 2 weeks. The
positive clones were verified by using reverse
transcription-quantitative PCR (RT-qPCR) and western blot analysis,
and used in subsequent experiments.
siRNA transfection
For the knockdown of β-catenin, the SW-480 cells
[the cells transfected with shFUT2#1 and the respective control
(NC); 5×105] were cultured in six-well plates to 60%
confluency in DMEM at 37°C and transfected with 100 pmol/µl
siRNA mixed with Lipofectamine 3000 reagent, and incubated for 15
min at room temperature, according to the manufacturer's
instructions (Shanghai GenePharma Co., Ltd.). The siRNA sequences
were as follows: siRNA for β-catenin (si-β-catenin), 5′-GAA TGC CGT
TCG CCT TCA TTA-3′; siRNA for negative control (siNC), 5′-UUC UCC
GAA CGU GUC ACG UTT-3′, (Shanghai GenePharma Co., Ltd.). The cells
were incubated at 37°C for 48 h.
RT-qPCR
Total RNA was extracted from the SW-480 or DLD-1
cells using the RNA prep Pure Cell kit (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol. cDNA
was obtained by reverse transcription using the Prime Script RT
reagent kit (RR047A; Takara Bio, Inc.) The reaction conditions were
as follows: 37°C for 15 min and 85°C for 5 sec. The obtained cDNA
was quantified using the SYBR® Premix Ex Taq™ (Perfect
Real Time) qPCR kit (Takara Bio, Inc.), and detected using an ABI
real-time fluorescent quantitative PCR system (Applied Biosystems;
Thermo Fisher Scientific, Inc.). The PCR was performed as follows:
95°C for 3 min, followed by 40 cycles of denaturation at 95°C for
10 sec and annealing at 60°C for 30 sec. The primers used for
RT-qPCR are listed in Table I. The
2−ΔΔCq method was used to calculate the difference in
gene expression, and GAPDH was used as the internal reference gene
(24).
 | Table IPrimer sequences used for gene
expression analysis. |
Table I
Primer sequences used for gene
expression analysis.
| Gene | Forward
(5′-3′) | Reverse
(5′-3′) |
|---|
| FUT2 |
GTGGTGTTTGCTGGCGATGG |
AAAGATTTTGAGGAAAGGGGAGTCG |
| GAPDH |
GAACATCATCCCTGCCTCTACT |
CCTGCTTCACCACCTTCTTG |
Cell proliferation assay
The Cell Counting Kit-8 (CCK-8) was used to measure
cell viability. In brief, the cells were incubated in 96-well
plates at a density of 5,000 cells/well at 37°C with 5%
CO2. Following cell culture for 24, 48, or 72 h, the
cells were washed with PBS, and 10 µl CCK-8 reagent (Dojindo
Laboratories, Inc.) was added to each well. The samples were
measured using an automatic ELISA plate reader (Varioskan Flash;
Thermo Fisher Scientific, Inc.) at a wavelength of 450 nm.
Colony formation assay
For colony formation assay, the SW-480 and DLD-1
cells were digested, resuspended in complete DMEM and seeded in
six-well plates (500 cells/well) containing 2 ml DMEM. Following
incubation at 37°C for 2 weeks, the cells were fixed with 4%
paraformaldehyde for 20 min, then stained with 0.1% crystal violet
(Beyotime Institute of Biotechnology) at room temperature for 30
min. Images were obtained under a microscope (TS100; Nikon
Corporation), and colonies were counted and analyzed using Image J
software (v1.8.0; National Institutes of Health).
Wound healing assay
SW-480 and DLD-1 cells were seeded in six-well
plates with a density of 1×106 cells/well. Following
cell culture up to 80-90% confluency in 10% serum, the cells were
scratched to form two parallel straining lines, and non-adherent
cells were washed off with PBS. Images of the wounds were
photographed under a microscope (TS100; Nikon Corporation) at 0 and
48 h, respectively, and the wound widths were calculated using
Image J software (v1.8.0; National Institutes of Health). Values
were calculated by applying the following formula: Percentage of
wound closure=1−(widtht/width0).
Transwell migration and invasion
assays
Transwell chambers (24-well; Corning, Inc.) were
used to assess the migratory and invasive ability of the CRC cells
(SW-480 and DLD-1). For the migration assay, cells
(6×104 cells/well) were re-suspended in serum-free DMEM
and seeded into the upper chamber with an 8 µm pore size,
and into the lower chamber, DMEM with 10% FBS was added. Following
cell culture for 24 h at 37°C, the chambers were washed with PBS.
Subsequently, the cells that had migrated to the lower chamber were
fixed with 4% paraformaldehyde at room temperature for 30 min, then
stained with crystal violet (Beyotime Institute of Biotechnology)
at room temperature for 30 min. Finally, images were captured under
a microscope (TS100; Nikon Corporation) and the numbers of cells
were counted.
For the invasion assays, the experimental procedure
applied was similar to that described above for migration, with the
difference that the upper chamber was pre-coated with BD Matrigel
(BD Biosciences) at 37°C for 8 h according to the manufacture's
protocol for the invasion assay, and cells were cultured for 48
h.
Flow cytometry
Flow cytometry was used to assess the cell cycle and
apoptosis. For the cell cycle assay, SW-480 and DLD-1 cells
(1×106 cells/well) were seeded in six-well plates in
DMEM with 10% FBS and cultured for 48 h. Following trypsinization
and washing with PBS, cells were fixed in 70% ethanol at 4°C for 2
h. Subsequently, the samples were resuspended in the staining
buffer with propidium iodide (PI; Nanjing KeyGen Biotech Co., Ltd.)
and RNase A (the ratio of PI to RNase A was 9:1), and incubated in
the dark for 30 min at room temperature. Finally, the samples were
analyzed using a flow cytometer (FACS Arial, BD Biosciences). The
proportion of cells in the S + G2 phase was calculated using FlowJo
v10.8.1 software (BD Biosciences).
For the apoptosis assay, the cells were harvested
and incubated with Annexin V-APC and PI using an Annexin V-APC/PI
Apoptosis Detection kit (Bio-Rad Laboratories, Inc.), according to
the manufacturer's instructions, and analyzed using a flow
cytometer (as described above).
Western blot analysis
The processed cells (SW-480, DLD-1, HCT-116, HCT-8
and FHC) and tissues were lysed in RIPA buffer (Beyotime Institute
of Biotechnology) supplemented with 0.1% protease inhibitors
(MilliporeSigma), and proteins were obtained according to the
manufacturer's protocol. The supernatant was collected by
centrifugation (20,900 × g, 30 min, 4°C). The protein concentration
was measured using a BCA Protein Assay kit (Beyotime Institute of
Biotechnology). For western blot analysis, the extracted protein
(20 µg) was separated by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and
electro-transferred onto polyvinylidene fluoride (PVDF) membranes
(MilliporeSigma). The membranes were blocked with 5% non-fat milk
in Tris buffer saline with 0.1% Tween-20 (TBST) at room temperature
for 2 h, and subsequently incubated with specific primary
antibodies at 4°C overnight. After washing with TBST, the membranes
were incubated with the corresponding secondary antibodies at room
temperature for 1 h, and visualized using an enhanced
chemiluminescence (ECL) reagent (NCM USA). The results were
analyzed using Image J software (v1.8.0; National Institutes of
Health). The primary antibodies used were as follows: FUT2 (cat.
no. sc-100742) was obtained from Santa Cruz Biotechnology, Inc.
Antibodies against C-myc (ab32072), cyclin D1 (ab134175) and
phosphorylated (p-) β-catenin (cat. no. ab47335) were purchased
from Abcam. Primary antibody against glycogen synthase kinase-3β
(GSK3β; cat. no. 12456) was acquired from Cell Signaling
Technology, Inc. Primary antibodies against β-catenin (51067-2-AP),
and Wnt2 (66656-1-Ig) were obtained from Proteintech Group, Inc.
Antibodies against tubulin (db3285), GAPDH (db1209) and β-actin
(db10001) were purchased from Diagbio (http://www.daigebio.com), and used as the loading
controls. HRP-conjugated Affinipure goat anti-rat IgG (A0206) and
HRP-conjugated Affinipure goat anti-rabbit IgG (A0208) were used as
the secondary antibodies and purchased from Beyotime Institute of
Biotechnology.
Immunohistochemistry (IHC)
The paraffin-embedded tissue sections
(5-µm-thick) were purchased from Alenabio; Taibsbio
(http://www.taibsbio.com/). For IHC staining, the
tissue sections were used as previously described (25). Briefly, the tissues were
deparaffinized with xylene, rehydrated with ethanol, followed by
antigen retrieval with sodium citrate solution and endogenous
peroxidase blocking with 3% H2O2 at room
temperature for 30 min. Following blocking with 5% bovine serum
albumin (BSA; Beyotime Institute of Biotechnology), the samples
were incubated with an antibody against FUT2 (ab177239; 1:150;
Abcam) at 4°C overnight, and were subsequently incubated with
biotinylated secondary antibodies (enhancement of enzyme-labeled
goat anti-rabbit IgG; A0279; Beyotime Institute of Biotechnology)
for 1 h at room temperature, treated with 3,3′-diaminobenzidine
(D12384; Merck KGaA), and finally counterstained with hematoxylin
(C0105S; Beyotime Institute of Biotechnology) at room temperature
for 3 min. Images were obtained using a microscope (TS100; Nikon
Corporation).
Immunofluorescence
The immunofluorescence assays were performed as
previously described (26).
Briefly, the processed cells were washed with PBS three times, and
fixed with 4% paraformaldehyde at room temperature for 30 min.
After washing with PBS, the cells were permeabilized with 0.5%
Triton X-100 for 15 min at room temperature, and then blocked with
5% BSA for 2 h at room temperature. Subsequently, the cells were
incubated with an anti-β-catenin antibody (51067-2-AP; Thermo
Fisher Scientific, Inc.) at 4°C overnight, then incubated with the
secondary antibody Cy3-conjugated anti-rabbit antibody (A0516;
Beyotime Institute of Biotechnology) in the dark for 1 h at room
temperature, stained with 0.5 µg/ml DAPI
(4′,6-diamidino-2-phenylindole) (MilliporeSigma) at room
temperature for 10 min to dye the nucleus. The samples were
analyzed using a confocal laser scanning microscope (Nikon
Corporation).
Co-immunoprecipitation
The SW-480 cells (5×106 cells) were lysed
using RIPA buffer as mentioned above. The cell lysate was
centrifuged at 20,900 × g for 20 min at 4°C. The cell lysate was
pre-cleared using sepharose-protein A/G beads (ab193262; Abcam) 4°C
for 2 h, and IgG was used as a control. The supernatant was
collected and incubated with primary antibody (Wnt2, 66656-1-Ig,
1:1,000, Thermo Fisher Scientific, Inc.; FUT2, sc-100742, 1:200,
Santa Cruz Biotechnology) at 4°C overnight, and sepharose-protein
A/G beads were then added and incubated at 4°C for 2 h. The
protein-antibody complexes were collected and resuspended in the
SDS buffer. Finally, the samples were boiled at 100°C for 10 min,
the magnetic beads were removed, and the binding proteins were
obtained for western blot analysis as described above.
Lectin pull-down
The SW-480 cells (5×106 cells) were lysed
using RIPA buffer as mentioned above. Cell lysates ere incubated
with Agarose-bound (Ulex Europaeus Agglutin-1 (UEA-1; AL-1063;
Vector Laboratories, Inc.) and rotated overnight at 4°C. After
washing, the precipitated proteins were further examined using
western blot analysis with Wnt2 antibody. The BCA Protein Assay kit
was used to measure the protein concentration.
Xenograft nude mouse model
BALB/c nude mice (12 females, 4 to 5 weeks old,
weighing 20-22 g) were purchased from the Beijing Vital River
Laboratory Animal Technology Co., Ltd. The mice were housed at
~25°C and 50% humidity with a 12/12-h light/dark cycle, and ad
libitum access to food and water. For the in vivo
subcutaneous tumorigenesis assay, shNC or shFUT2 SW-480 cells (50
µl, 2×107 cells/ml in DMEM) were mixed with 50
µl 50% BD Matrigel and injected into the subcutaneous right
flanks of the BALB/C-nu nude mice (six mice per group). Animal
health and behavior was observed every 2 days throughout the study
period, and the tumors were measured every week. No mice died
during the experiment. Human endpoints were reached when the
maximum tumor volume was >750 mm3. After 30 days, a
total of 12 mice (six per group) were anesthetized using 2%
pentobarbital sodium (60 mg/kg, intraperitoneal injection) followed
by cervical dislocation. The death of all experimental animals was
verified though the cessation of respiration, heartbeat and nerve
reflex. Subsequently, the tumors were collected, weighed and
measured. Tumor volume (mm3) was calculated using the
following formula: Volume=0.5 × length × width2. The
animal experiment was approved by the Laboratory Animal Ethics
Committee of Wenzhou Medical University and Laboratory Animal
Centre of Wenzhou Medical University (reference no.
WYDW2019-0245).
Statistical analysis
Comparisons between two groups were performed using
an unpaired Student's t-test or one-way ANOVA analysis followed by
Tukey's test. Statistical analyses were performed using SPSS
software version 17.0 (SPSS, Inc.). Data are expressed as the mean
± SD of at least three repeated experiments. P<0.05 was
considered to indicate a statistically significant difference.
Results
FUT2 expression is upregulated in
CRC
GEPIA was used in order to analyze the expression
levels of FUT2 in CRC. As demonstrated in Fig. 1A, the expression of FUT2 was
upregulated both in COAD (n=275) and READ (n=92), as compared with
normal colorectal tissue (n=349, n=318, respectively). To further
determine the expression of FUT2 in CRC, the FUT2 protein levels
were detected using western blot analysis in CRC tissues and
matched tumor-adjacent tissues (n=28). The expression of FUT2 was
found to be significantly increased in CRC tissues, compared with
the adjacent non-cancerous tissue (Fig. 1B). The observed double bands for
FUT2 may be the outcome of FUT2 modification or degradation. FUT2
was also evaluated using IHC in 36 pairs of CRC tissues and
adjacent non-cancerous tissues, and nine pairs of CRC lymph node
metastatic tissues and the normal lymph node tissues (Fig. 1C). As shown in Fig. 1D, the expression of FUT2 in CRC
tissues was significantly increased, compared with the adjacent
non-cancerous tissues. The expression of FUT2 in colorectal cancer
lymph node metastatic tissues was also increased, compared with the
normal lymph node tissues. In order to further evaluate the
clinical significance of FUT2, FUT2 expression in the different
stages of CRC was analyzed. Compared to normal colon and rectal
tissues, the expression of FUT2 was increased in stage I, II and
III CRC tissues. However, there was no significant difference among
the stage I, II and III (P>0.05, ANOVA with Tukey's post hoc
test) samples (Fig. 1D),
indicating that the FUT2 level was not associated with the TNM
stage. Furthermore, FUT2 expression was detected in SW-480, DLD-1,
HCT-116, HCT-8 and FHC cells. As demonstrated in Fig. 1E, FUT2 protein expression in CRC
cells (SW-480, DLD-1 and HCT-116) was higher than that in FHC
cells. Overall, these results suggested that the upregulation of
FUT2 may be associated with CRC development and progression.
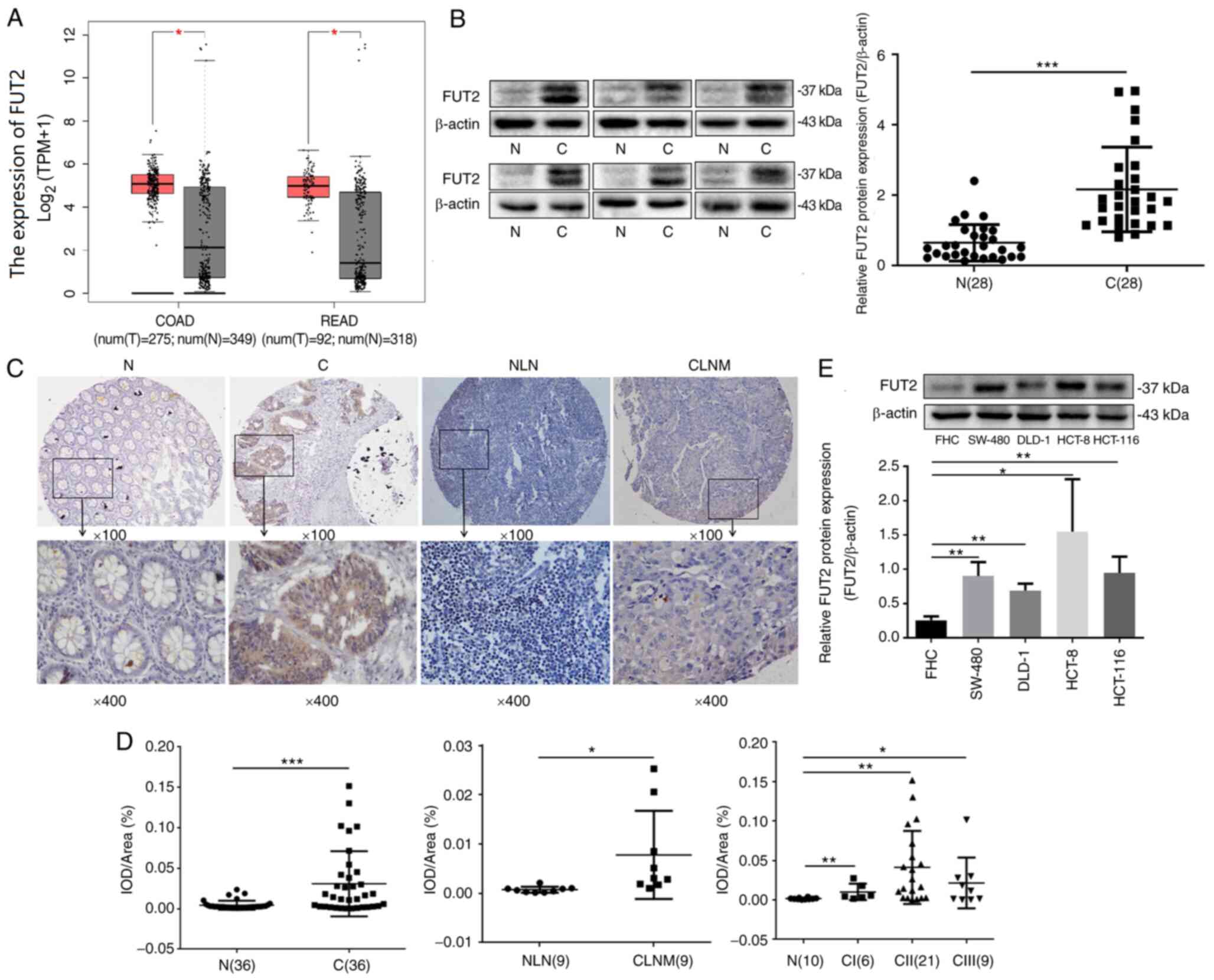 | Figure 1FUT2 is overexpressed in colorectal
cancer tissues and cell lines. (A) Boxplot illustrating the
relative expression of FUT2 in normal, COAD and READ samples. (B)
Evaluation of FUT2 expression in fresh human colorectal cancer
tissues and adjacent normal tissues using western blot analysis. C,
colorectal cancer tissues; N, normal tissues. (C) Representative
images of the expression of FUT2 proteins assessed using
immunohistochemistry. C1, colorectal cancer tissues; N1, adjacent
tissues; CLNM, colorectal cancer lymph node metastatic tissue; NLN,
normal lymph node tissue. Magnification: Upper panels, ×100; lower
panels, ×400. (D) Quantitative analysis of the average MOD of FUT2
staining in normal tissues, colorectal cancer tissues and lymph
node metastatic tissues. N, normal control; CI, cancer stage I;
CII, cancer stage II; CIII, cancer stage III. (E) The expression of
FUT2 in colorectal cancer cell lines and FHC was examined using
western blot analysis. β-actin was used as the loading control.
Data are expressed as the mean ± SD. *P<0.05,
**P<0.01 and ***P<0.001. FUT2,
fucosyltransferase 2; COAD, colon adenocarcinoma; READ, rectal
adenocarcinoma; MOD, mean optical density. |
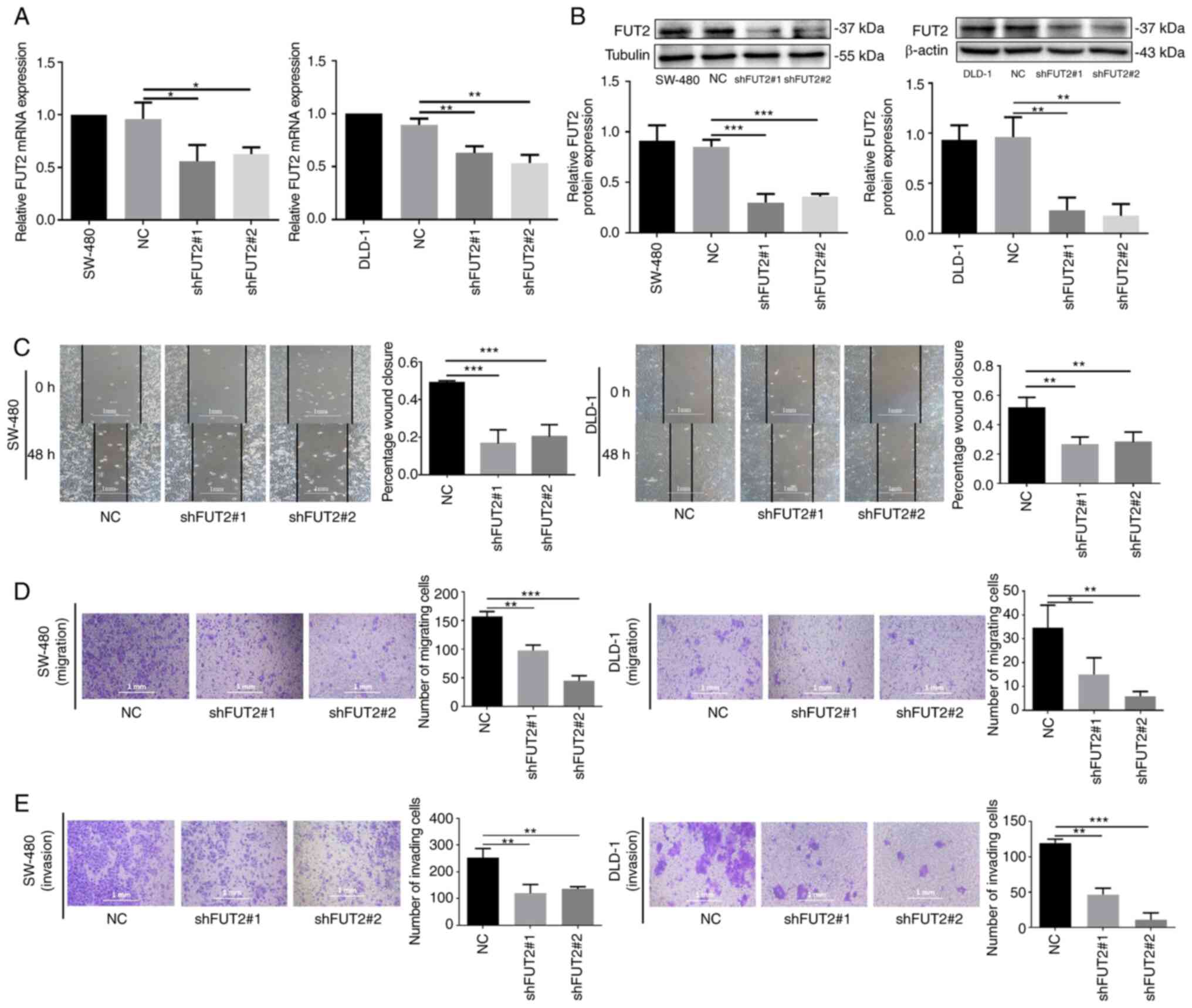
Knockdown of FUT2 suppresses the
migration and invasion of CRC cells
To detect the effects of FUT2 on CRC progression
in vitro, the SW-480 and DLD-1 cell lines were selected for
further experiments. FUT2-knockdown cells (shFUT2; shFUT2#1 and
shFUT2#2) were generated by transfecting the plasmid DNA vector
encoding an shRNA that targets FUT2 for the inhibition of FUT2
expression in CRC cell lines (SW-480 and DLD-1), and their
corresponding scrambled vector (NC) was used as a control. The mRNA
expression levels of FUT2 were markedly decreased in SW-480 cells
in which FUT2 was knocked down (shFUT2#1, shFUT2#2), compared with
the NC cells, and a similar effect was observed in the DLD-1 cells
(Fig. 2A). The results of western
blot analysis revealed that the protein levels of FUT2 were also
significantly decreased in the shFUT2#1 and shFUT2#2 groups, as
compared with the NC group (Fig.
2B).
Wound healing and Transwell assay were then
performed to evaluate the effects of FUT2 on cell migration and
invasion. As depicted in Fig. 2C,
the knockdown of FUT2 significantly inhibited the wound closure,
compared with the control cells (NC), suggesting that FUT2
knockdown inhibited the migration of SW-480 and DLD-1 cells.
Furthermore, Transwell assay revealed that the number of SW-480 and
DLD-1 cells that migrated through the membrane was significantly
decreased in the shFUT2 groups (shFUT#1, shFUT2#2), as compared
with their corresponding NC group (Fig. 2D). Consistent with results of the
wound healing assay, FUT2 knockdown inhibited CRC cell migration.
As depicted in Fig. 2E, Matrigel
invasion assay revealed that the invasive capacity of the SW-480
and DLD-1 cells was also markedly suppressed in the shFUT2 groups
(shFUT#1 and shFUT2#2). These findings suggested that FUT2 promoted
the migration and invasion of CRC cells.
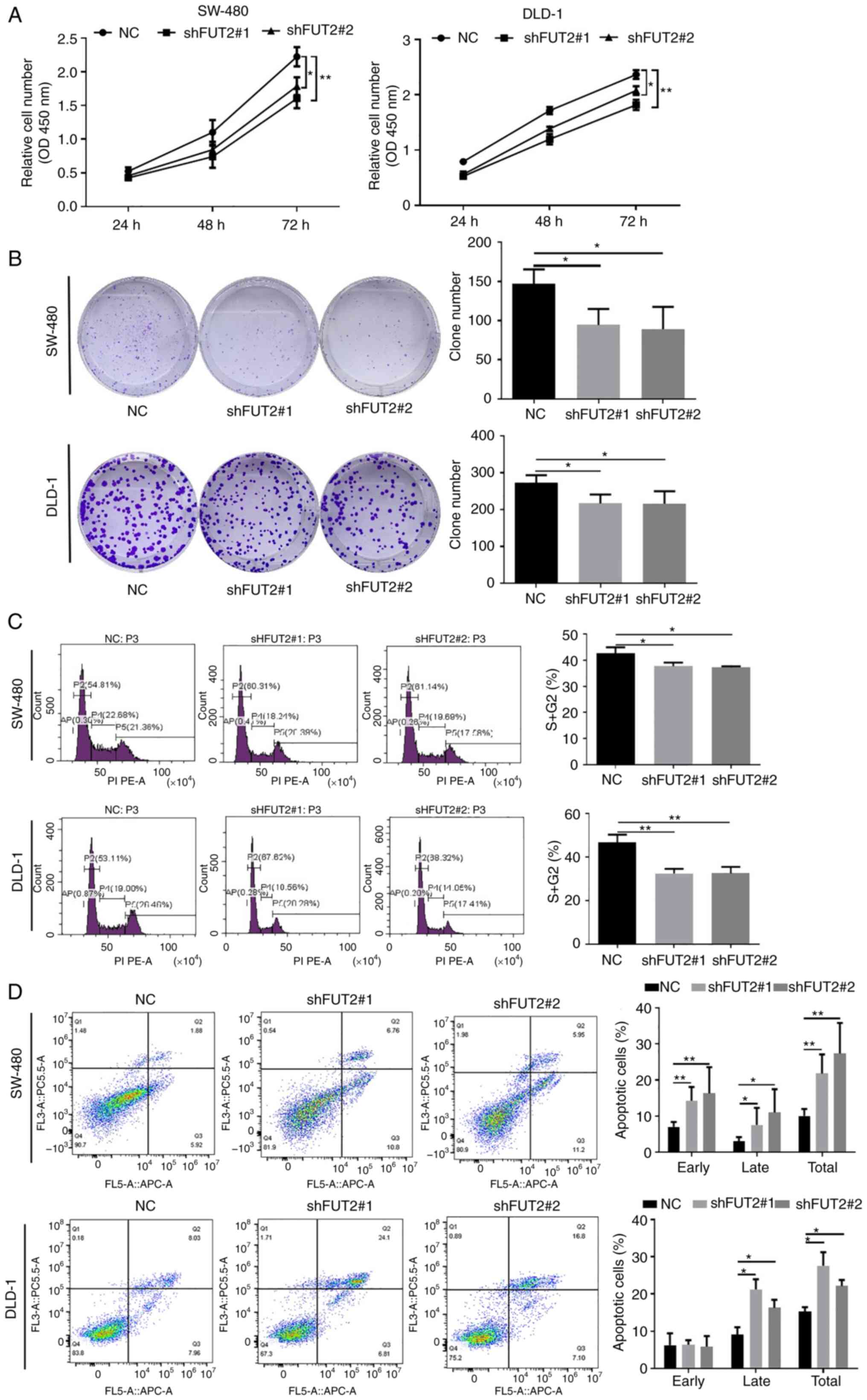
Knockdown of FUT2 inhibits CRC growth in
vitro and in vivo
CCK-8 and plate clone formation assays were then
performed in order to determine the role of FUT2 in the growth of
CRC cells. The knockdown of FUT2 (shFUT2#1 and shFUT2#2)
significantly inhibited the growth of CRC cells (SW-480 and DLD-1),
as shown by CCK-8 assay (Fig. 3A).
Similarly, the number of cell colonies was markedly decreased in
the shFUT2 groups, compared with the NC groups (Fig. 3B). It was thus demonstrated that
the knockdown of FUT2 suppressed the proliferation of CRC
cells.
To further explore the role of FUT2 in CRC, flow
cytometric analysis was used to examine cell cycle progression and
apoptosis. The knockdown of FUT2 led to an increase in the cell
ratio at the G0/G1 phase, with a marked decrease in the number of
cells in the S phase (Fig. 3C),
indicating that the CRC cells subjected to FUT2 knockdown were
arrested at the G0/G1 phase. Furthermore, the results of flow
cytometry demonstrated that the apoptotic indexes were
significantly higher in the shFUT2 groups than the NC groups
(Fig. 3D), indicating that FUT2
knockdown promoted the apoptosis of CRC cells. On the whole, these
results indicated that the knockdown of FUT2 suppressed CRC
progression by inhibiting cell proliferation and promoting
apoptosis.
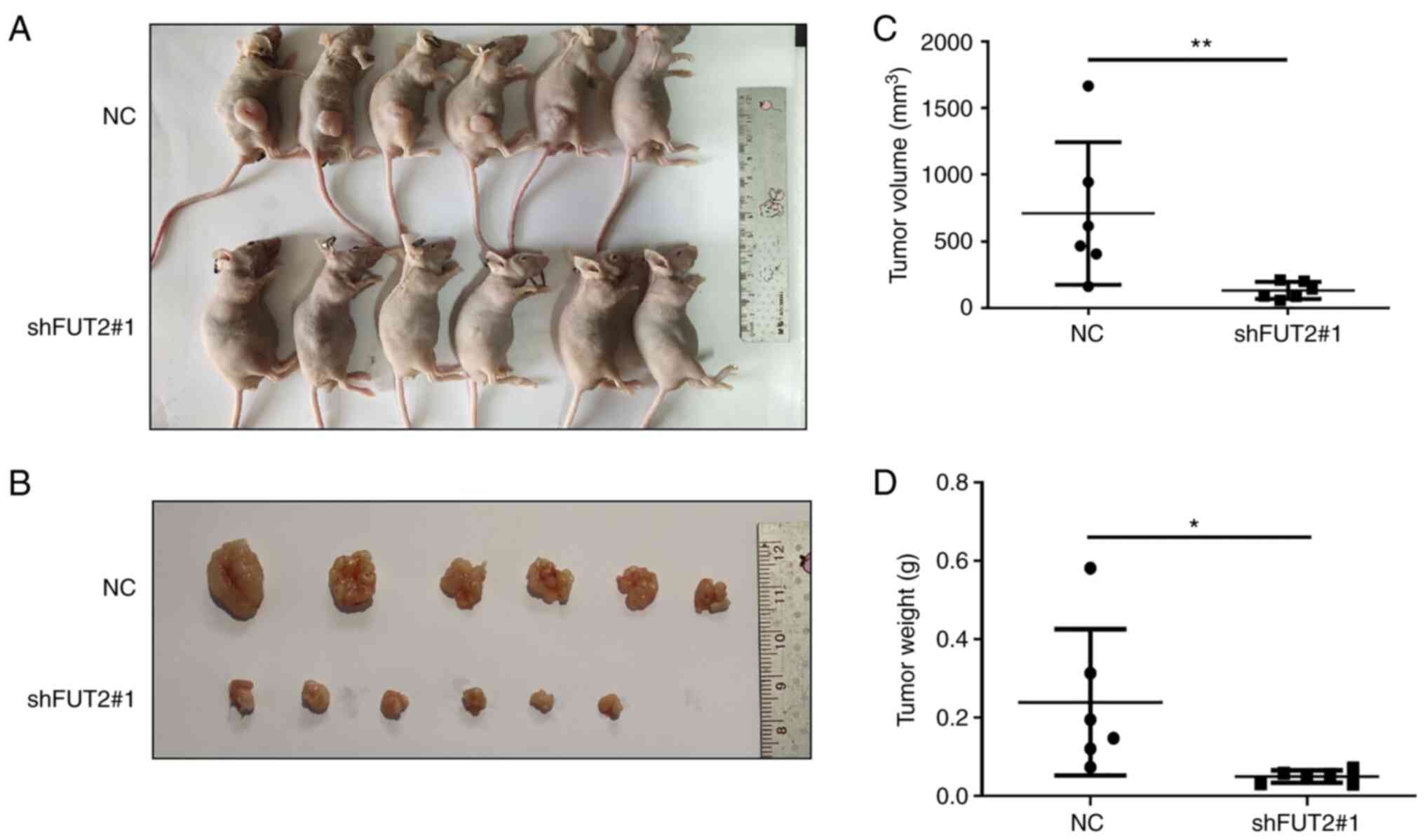
To further assess the effects of FUT2 on CRC in
vivo, a mouse CRC xenograft model was established. As
demonstrated in Fig. 4B, the
harvested tumors in the shFUT2 group were smaller than the tumors
in the NC group. The average lengths of the total number of tumors
in the NC and shFUT2 groups were 12.48±3.39 mm and 9.87±2.53 mm,
respectively. The average widths of the total number of tumors in
the NC and shFUT2 groups were 7.05±1.30 mm and 5.65±0.95 mm,
respectively. The average tumor volume of the shFUT2 group was
710.1 mm3, and the average tumor volume of the NC group
was 131.4 mm3 (Fig.
4C). The average tumor weight of the NC and shFUT2 groups was
0.239 and 0.050 g, respectively (Fig.
4D). FUT2 knockdown significantly inhibited tumor growth in
vivo.
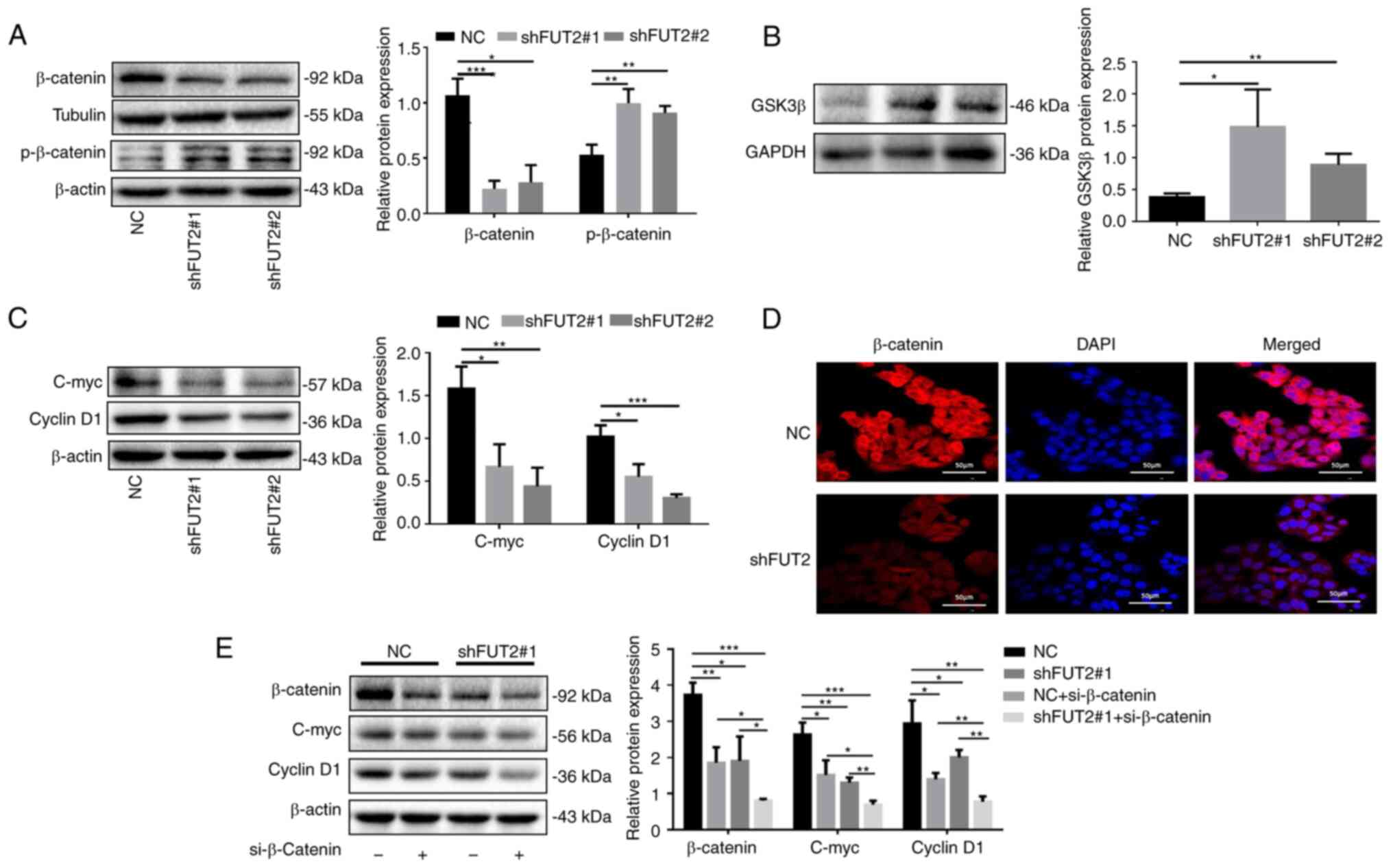
FUT2 promotes CRC progression via
β-catenin signaling
Wnt signaling is tightly associated with cancer and
has most prominently been described for CRC (21). In the present study, to elucidate
the underlying molecular mechanisms of FUT2 in CRC development, the
canonical Wnt/β-catenin signaling was investigated. As depicted in
Fig. 5A, the knockdown of FUT2
markedly decreased the expression of β-catenin and increased the
phosphorylation of β-catenin (Fig.
5A), which is known to be regulated by GSK3β, and subsequent
β-catenin ubiquitination and degradation (27). It was revealed that the expression
of GSK3β was significantly increased in the shFUT2 groups, compared
with the NC group, which suggested that the knockdown of FUT2
promoted GSK3β expression (Fig.
5B). Moreover, the expression levels of cyclin D1 and C-myc
were significantly decreased in the shFUT2 groups (shFUT2#1 and
shFUT2#2), as compared with the NC group (Fig. 5C). To further confirm the effects
of FUT2 on β-catenin signaling, the distribution of β-catenin was
examined using confocal laser microscopy. It was demonstrated that
the level of nuclear β-catenin was markedly reduced following FUT2
knockdown (Fig. 5D). Furthermore,
the knockdown of β-catenin or FUT2 inhibited the expression of
C-myc and cyclin D1, the target genes of β-catenin. The
simultaneous knockdown of β-catenin and FUT2 significantly enhanced
the suppressive effects of β-catenin or FUT2 knockdown on the
expression of C-myc and cyclin D1 in the SW-480 cells (Fig. 5E), suggesting that FUT2 promoted
the expression of C-myc and cyclin D1 via β-catenin signaling.
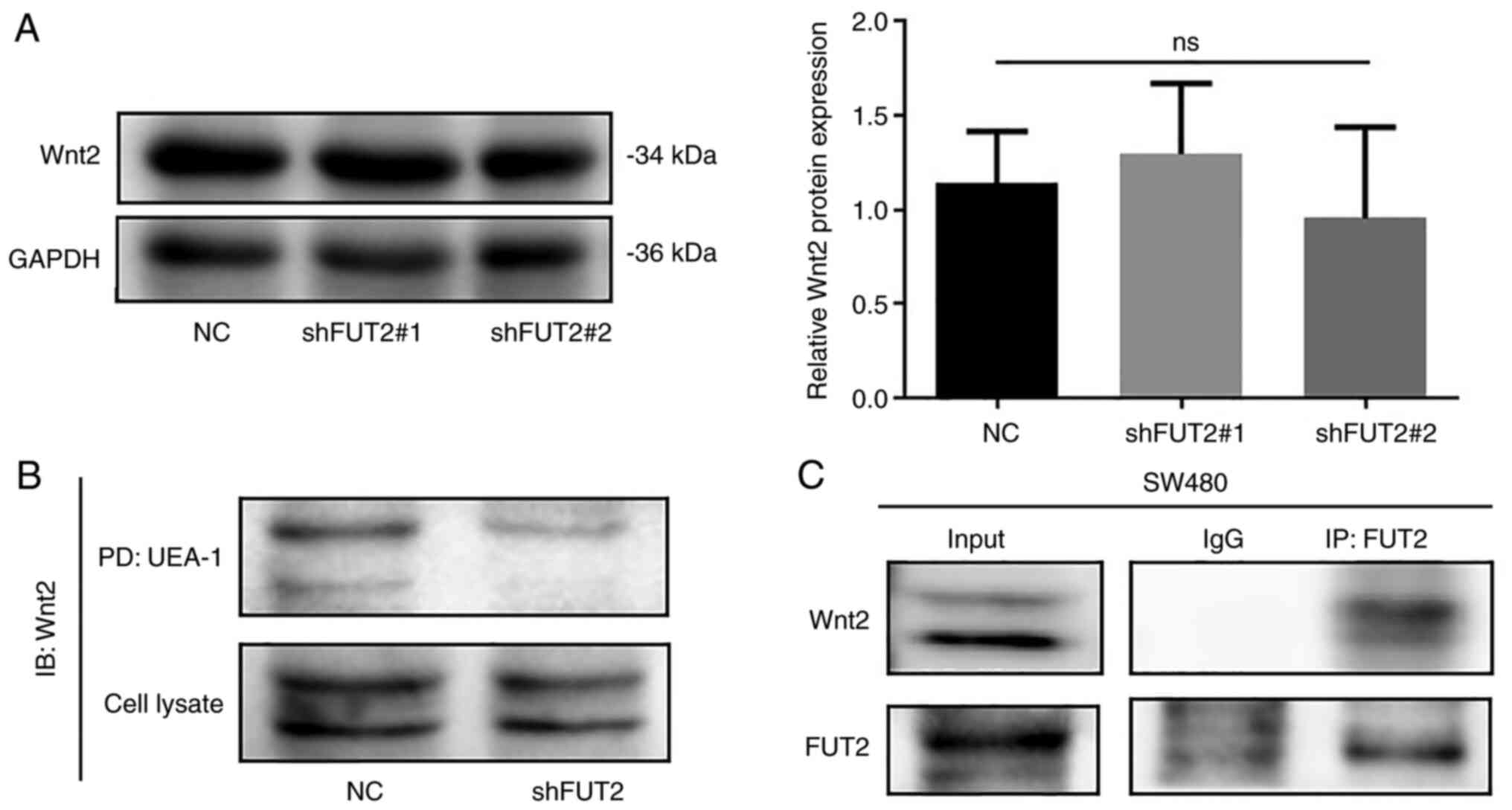
FUT2 regulates the fucosylation of Wnt2
in CRC cells
In the canonical Wnt pathway, the phosphorylation
and degradation of β-catenin are regulated by Wnt (21). In the present study, to further
elucidate the role of FUT2 in the regulation of the Wnt/β-catenin
pathway, lectin pull-down and immunoprecipitation, and western blot
analysis were performed. The results of western blot analysis
revealed that the protein levels of Wnt2 were not affected by FUT2
knockdown (Fig. 6A). UEA-1
pull-down revealed that the knockdown of FUT2 markedly decreased
the fucosylation of Wnt2 (Fig.
6B). Immunoprecipitation and western blot analysis also
demonstrated that FUT2 interacted with Wnt2 (Fig. 6C). These results suggested that the
FUT2-mediated fucosylation of Wnt2 may activate Wnt/β-catenin
signaling.
Discussion
CRC is a disease of the digestive system and is
associated with a high morbidity and mortality, and a poor clinical
outcome. Although the clinical treatment of CRC has improved, the
prognosis of CRC remains poor. Therefore, it is crucial to
elucidate the underlying mechanisms of CRC tumorigenesis and
development, in order to improve diagnosis and the treatment
effects. Cancer progression has been associated with a variation of
protein expression and post-translational modifications (28). Over the past few decades,
accumulating evidence has demonstrated that aberrant glycosylation
plays a vital role in cancer (29). The alteration of glycosylation may
be explained by the dysregulation of glycosyltransferases, which
catalyze glycosylation in order to create glycan structures
(30,31). Recently, it has been revealed that
fucosyl transferases are overexpressed in cancer, including CRC,
and mediate several specific biological functions (32,33).
Herein, based on TCGA data analysis for FUT2 in CRC, FUT2
expression was analyzed using the GEPIA web-portal. The expression
of FUT2 was upregulated in CRC. Furthermore, IHC assay and western
blot analysis revealed that the FUT2 protein levels in CRC tissues
were increased in comparison with those in adjacent non-cancerous
tissues. However, no association with clinical stage was detected.
Furthermore, FUT2 was expressed in CRC cells at significantly
higher levels than in FHC cells. These results thus suggested that
FUT2 may be a positive regulator of the progression of CRC. In the
present study, SW-480 and DLD-1 cells were used to investigate the
functional role and the molecular mechanisms of FUT2 in CRC.
Functionally, the results of the present study
demonstrated that the knockdown of FUT2 attenuated the viability
and growth of CRC cells, and inhibited CRC growth in vivo.
The dysregulation of the cell cycle, as one of the hallmarks of
tumor cells, is associate with limitless tumor cell proliferation
and growth, and the regulation of cell cycle can be used to prevent
cancer development (34). The
present study demonstrated that FUT2 knockdown induced the G0/G1
phase arrest of the SW-480 and DLD-1 cells, which may eventually
decelerate CRC growth. As a type of programmed cell death,
apoptosis is the reverse of cell proliferation and plays a crucial
role in maintaining cell and tissue homeostasis (35). In the present study, the knockdown
of FUT2 significantly increased the apoptosis of CRC cells.
Moreover, FUT2 knockdown inhibited the migratory and invasive
capabilities of the SW-480 and DLD-1 cells. These findings
suggested that FUT2 may be a positive regulator of CRC
development.
The Wnt pathway, a highly conserved and versatile
pathway, is critical for cell differentiation, proliferation,
tissue homeostasis and regeneration, and is involved in almost all
stages of tumor development (36,37).
The Wnt pathway is commonly divided into β-catenin-dependent and
-independent signaling. Known as 'canonical Wnt signaling', the
β-catenin-dependent pathway (Wnt/β-catenin pathway) is considered
as the most crucial and well-characterized pathway, and functions
by regulating the stabilization and nuclear translocation of
β-catenin, thereby regulating cellular processes (38). The stabilization of β-catenin is
considered as one of the 'gatekeepers' of Wnt signaling in
tumorigenesis (39). Herein, the
findings of western blot analysis and confocal laser microscopy
revealed that the knockdown of FUT2 reduced the expression of
β-catenin, cyclin D1 and C-myc in SW-480 cells, indicating that
FUT2 may induce Wnt/β-catenin signaling in CRC cells. The
stabilization of β-catenin is determined by its phosphorylation
status. When Wnt is inactivated, β-catenin is recruited by APC and
delivered to the 'destruction complex', subsequently being degraded
following phosphorylation and ubiquitination (40). The results of the present study
revealed that the knockdown of FUT2 increased the phosphorylation
levels of β-catenin and GSK3β expression, which mediated the
phosphorylation of β-catenin. Furthermore, the simultaneous
downregulation of FUT2 and β-catenin enhanced the suppressive
effect of the sole downregulation of FUT2 on C-myc and cyclin D1
expression. These results indicated that FUT2 promoted CRC cell
growth and metastasis via the Wnt/β-catenin pathway.
Among 19 Wnt ligands in mammals, Wnt2, one of the
WNT gene family members, is overexpressed in human cancers,
including ovarian cancer, lung cancer, pancreatic cancer and CRC
(41,42). Wnt2, as a potential marker in CRC,
plays tumorigenic roles through the induced activation of β-catenin
in CRC (43). In the present
study, FUT2 knockdown did not affect the expression of Wnt2;
however, the fucosylation of Wnt2 was significantly reduced by FUT2
knockdown, and the interaction between FUT2 and Wnt2 was verified
using co-immunoprecipitation/western blot analysis. These results
suggested that the FUT2-mediated fucosylation of Wnt2 activated
Wnt/β-catenin signaling.
In conclusion, the present study demonstrates that
FUT2 is overexpressed in CRC, promoting the tumor growth and
metastasis in CRC, and regulating CRC tumorigenesis via the
Wnt/β-catenin signaling pathway. These findings may indicate the
role of FUT2 as a potential diagnostic or therapeutic target for
CRC.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XC and ZZ participated in the conceptualization of
the study. PL, JL, MD, YL and YZ performed the experiments, data
collection and data interpretation. XC and ZZ prepared and revised
the manuscript. PL and XC confirm the authenticity of all the raw
data. All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the First Affiliated Hospital of Wenzhou Medical
University (Issuing no. 2019-086). Written informed consent was
obtained from all participants prior to the commencement of the
study. The animal experiments were approved (reference no.
WYDW2019-0245) by the Laboratory Animal Ethics Committee of Wenzhou
Medical University and Laboratory Animal Centre of Wenzhou Medical
University (Wenzhou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to acknowledge the Key
Discipline of Zhejiang Province in Medical Technology (First Class,
Category A) for their support (which included technology and
communication platforms, large scale equipment, training).
Funding
The present study was supported by the Wenzhou Science and
Technology Bureau (grant no. Y20210180).
References
|
1
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2022. CA Cancer J Clin. 72:7–33. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Akimoto N, Ugai T, Zhong R, Hamada T,
Fujiyoshi K, Giannakis M, Wu K, Cao Y, Ng K and Ogino S: Rising
incidence of early-onset colorectal cancer-a call to action. Nat
Rev Clin Oncol. 18:230–243. 2021. View Article : Google Scholar
|
|
4
|
Pandurangan AK, Divya T, Kumar K,
Dineshbabu V, Velavan B and Sudhandiran G: Colorectal
carcinogenesis: Insights into the cell death and signal
transduction pathways: A review. World J Gastrointest Oncol.
10:244–259. 2018. View Article : Google Scholar
|
|
5
|
Xia C, Dong X, Li H, Cao M, Sun D, He S,
Yang F, Yan X, Zhang S, Li N and Chen W: Cancer statistics in China
and United States, 2022: Profiles, trends, and determinants. Chin
Med J (Engl). 135:584–590. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huang H, He Y, Li Y, Gu M, Wu M and Ji L:
Eriodictyol suppresses the malignant progression of colorectal
cancer by downregulating tissue specific transplantation antigen
P35B (TSTA3) expression to restrain fucosylation. Bioengineered.
13:5551–5563. 2022. View Article : Google Scholar :
|
|
7
|
Schjoldager KT, Narimatsu Y, Joshi HJ and
Clausen H: Global view of human protein glycosylation pathways and
functions. Nat Rev Mol Cell Biol. 21:729–749. 2020. View Article : Google Scholar
|
|
8
|
Costa AF, Campos D, Reis CA and Gomes C:
Targeting glycosylation: A new road for cancer drug discovery.
Trends Cancer. 6:757–766. 2020. View Article : Google Scholar
|
|
9
|
Rini JM, Moremen KW, Davis BG, Esko JD,
Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, et al:
Glycosyltransferases and glycan-processing enzymes. Essentials of
Glycobiology [Internet]. 4th edition. Cold Spring Harbor Laboratory
Press; Cold Spring Harbor, NY: Chapter 47. 2022
|
|
10
|
Dos Reis JS, da Costa Santos MA, Mendonça
DP, do Nascimento SIM, Barcelos PM, de Lima RG, da Costa KM,
Freire-de-Lima CG, Morrot A, Previato JO, et al: Glycobiology of
cancer: Sugar drives the show. Medicines (Basel). 9:342022.
View Article : Google Scholar
|
|
11
|
Munkley J and Elliott DJ: Hallmarks of
glycosylation in cancer. Oncotarget. 7:35478–35489. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mereiter S, Balmaña M, Campos D, Gomes J
and Reis CA: Glycosylation in the era of cancer-targeted therapy:
Where are we heading? Cancer Cell. 36:6–16. 2019. View Article : Google Scholar
|
|
13
|
Blanas A, Sahasrabudhe NM, Rodríguez E,
van Kooyk Y and van Vliet SJ: Fucosylated antigens in cancer: An
alliance toward tumor progression, metastasis, and resistance to
chemotherapy. Front Oncol. 8:392018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gao Z, Wu Z, Han Y, Zhang X, Hao P, Xu M,
Huang S, Li S, Xia J, Jiang J and Yang S: Aberrant fucosylation of
saliva glycoprotein defining lung adenocarcinomas malignancy. ACS
Omega. 7:17894–17906. 2022. View Article : Google Scholar
|
|
15
|
Liang JX, Gao W and Cai L:
Fucosyltransferase VII promotes proliferation via the EGFR/AKT/mTOR
pathway in A549 cells. Onco Targets Ther. 10:3971–3978. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu C, Li Z, Wang S, Fan Y, Zhang S, Yang
X, Hou K, Tong J, Hu X, Shi X, et al: FUT4 is involved in
PD-1-related immunosuppression and leads to worse survival in
patients with operable lung adenocarcinoma. J Cancer Res Clin
Oncol. 145:65–76. 2019. View Article : Google Scholar
|
|
17
|
Shah P, Wang X, Yang W, Eshghi ST, Sun S,
Hoti N, Chen L, Yang S, Pasay J, Rubin A and Zhang H: Integrated
proteomic and glycoproteomic analyses of prostate cancer cells
reveal glycoprotein alteration in protein abundance and
glycosylation. Mol Cell Proteomics. 14:2753–2763. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Deng G, Chen L, Zhang Y, Fan S, Li W, Lu J
and Chen X: Fucosyltransferase 2 induced epithelial-mesenchymal
transition via TGF-β/Smad signaling pathway in lung
adenocarcinaoma. Exp Cell Res. 370:613–622. 2018. View Article : Google Scholar
|
|
19
|
Lai TY, Chen IJ, Lin RJ, Liao GS, Yeo HL,
Ho CL, Wu JC, Chang NC, Lee AC and Yu AL: Fucosyltransferase 1 and
2 play pivotal roles in breast cancer cells. Cell Death Discov.
5:742019. View Article : Google Scholar :
|
|
20
|
Kramer N, Schmöllerl J, Unger C, Nivarthi
H, Rudisch A, Unterleuthner D, Scherzer M, Riedl A, Artaker M,
Crncec I, et al: Autocrine WNT2 signaling in fibroblasts promotes
colorectal cancer progression. Oncogene. 36:5460–5472. 2017.
View Article : Google Scholar
|
|
21
|
Zhan T, Rindtorff N and Boutros M: Wnt
signaling in cancer. Oncogene. 36:1461–1473. 2017. View Article : Google Scholar
|
|
22
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017. View Article : Google Scholar :
|
|
23
|
Edge SB and Compton CC: The American joint
committee on cancer: The 7th edition of the AJCC cancer staging
manual and the future of TNM. Ann Surg Oncol. 17:1471–1474. 2010.
View Article : Google Scholar
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
25
|
Zhou W, Ma H, Deng G, Tang L, Lu J and
Chen X: Clinical significance and biological function of
fucosyltransferase 2 in lung adenocarcinoma. Oncotarget.
8:97246–97259. 2017. View Article : Google Scholar :
|
|
26
|
Mao Y, Zhang Y, Fan S, Chen L, Tang L,
Chen X and Lyu J: GALNT6 promotes tumorigenicity and metastasis of
breast cancer cell via β-catenin/MUC1-C signaling pathway. Int J
Biol Sci. 15:169–182. 2019. View Article : Google Scholar :
|
|
27
|
Liu J, Xiao Q, Xiao J, Niu C, Li Y, Zhang
X, Zhou Z, Shu G and Yin G: Wnt/β-catenin signaling: Function,
biological mechanisms, and therapeutic opportunities. Signal
Transduct Target Ther. 7:32022. View Article : Google Scholar
|
|
28
|
Deschuyter M, Leger DY, Verboom A,
Chaunavel A, Maftah A and Petit JM: ST3GAL2 knock-down decreases
tumoral character of colorectal cancer cells in vitro and in vivo.
Am J Cancer Res. 12:280–302. 2022.PubMed/NCBI
|
|
29
|
Vajaria BN and Patel PS: Glycosylation: A
hallmark of cancer? Glycoconj J. 34:147–156. 2017. View Article : Google Scholar
|
|
30
|
Tvaroška I: Glycosyltransferases as
targets for therapeutic intervention in cancer and inflammation:
Molecular modeling insights. Chem Pap. 76:1953–1988. 2022.
View Article : Google Scholar
|
|
31
|
Vasconcelos-Dos-Santos A, Oliveira IA,
Lucena MC, Mantuano NR, Whelan SA, Dias WB and Todeschini AR:
Biosynthetic machinery involved in aberrant glycosylation:
Promising targets for developing of drugs against cancer. Front
Oncol. 5:1382015. View Article : Google Scholar :
|
|
32
|
Liang Y, Wang T, Gao R, Jia X, Ji T, Shi
P, Xue J, Yang A, Chen M and Han P: Fucosyltransferase 8 is
overexpressed and influences clinical outcomes in lung
adenocarcinoma patients. Pathol Oncol Res. 28:16101162022.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Blanas A, Zaal A, van der Haaràvila I,
Kempers M, Kruijssen L, de Kok M, Popovic MA, van der Horst JC and
van Vliet SJ: FUT9-driven programming of colon cancer cells towards
a stem cell-like state. Cancers (Basel). 12:25802020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stewart ZA, Westfall MD and Pietenpol JA:
Cell-cycle dysregulation and anticancer therapy. Trends Pharmacol
Sci. 24:139–145. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tang H, Yang P, Yang X, Peng S, Hu X and
Bao G: Growth factor receptor bound protein-7 regulates
proliferation, cell cycle, and mitochondrial apoptosis of thyroid
cancer cells via MAPK/ERK signaling. Mol Cell Biochem. 472:209–218.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hiremath IS, Goel A, Warrier S, Kumar AP,
Sethi G and Garg M: The multidimensional role of the Wnt/β-catenin
signaling pathway in human malignancies. J Cell Physiol.
237:199–238. 2022. View Article : Google Scholar
|
|
37
|
Cebrat M, Strzadala L and Kisielow P: Wnt
inhibitory factor-1: A candidate for a new player in tumorigenesis
of intestinal epithelial cells. Cancer Lett. 206:107–113. 2004.
View Article : Google Scholar
|
|
38
|
Huang HL, Tang GD, Liang ZH, Qin MB, Wang
XM, Chang RJ and Qin HP: Role of Wnt/β-catenin pathway agonist
SKL2001 in Caerulein-induced acute pancreatitis. Can J Physiol
Pharmacol. 97:15–22. 2019. View Article : Google Scholar
|
|
39
|
Shang S, Hua F and Hu ZW: The regulation
of β-catenin activity and function in cancer: Therapeutic
opportunities. Oncotarget. 8:33972–33989. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Stamos JL and Weis WI: The β-catenin
destruction complex. Cold Spring Harb Perspect Biol. 5:a0078982013.
View Article : Google Scholar
|
|
41
|
Jung YS, Jun S, Lee SH, Sharma A and Park
JI: Wnt2 complements Wnt/β-catenin signaling in colorectal cancer.
Oncotarget. 6:37257–37268. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Huang C, Ma R, Xu Y, Li N, Li Z, Yue J, Li
H, Guo Y and Qi D: Wnt2 promotes non-small cell lung cancer
progression by activating WNT/β-catenin pathway. Am J Cancer Res.
5:1032–1046. 2015.
|
|
43
|
Rahiminejad S, Maurya MR, Mukund K and
Subramaniam S: Modular and mechanistic changes across stages of
colorectal cancer. BMC Cancer. 22:4362022. View Article : Google Scholar
|