1. Introduction
N6-methyladenosine (m6A), discovered in the 1970s,
is a type of methylation modification that occurs on the sixth
nitrogen atom of adenosine, which is the major form of mammalian
mRNA methylation (1). In mammalian
cells, each mRNA molecule contains three m6A residues on average
and m6A is mostly enriched around the stop codon, near the long
internal exon, and in the 3′-untranslated region (3′-UTR), which is
involved in almost all steps of RNA metabolism (2-4).
The increased proliferative capability of tumor
cells and their enhanced ability of invasive migration may lead to
an increase in tumor malignancy, which affects the prognosis of
patients. Tumor metabolic reprogramming, as one of the important
tumor markers, provides energy supply for tumor growth and fulfills
an important role in tumor progression (5). Ferroptosis, an intracellular
iron-dependent form of regulated cell death that is distinct from
apoptosis, necrosis and autophagy, also affects tumor progression.
Recent studies have revealed that m6A-associated factors are able
to alter the proliferative, invasive and migratory capabilities of
tumor cells, regulate tumor metabolism and participate in the
process of ferroptosis in tumors, thereby affecting tumor
progression (6-9). Furthermore, m6A-associated factors
have been shown to alter the tumor immune microenvironment,
providing novel insight into, and strategies for, the immunotherapy
of tumors, and inhibitors of m6A-associated proteins are also
anticipated to potentially serve as novel and promising anti-cancer
agents in the future treatment of cancer.
Given the important role of m6A in cancer research,
collating and summarizing new research findings on the role of m6A
in cancer, and presenting these latest findings and developmental
trends, are essential for researchers to gain a deeper
understanding of the latest developments on m6A in cancer research.
However, the present crop of reviews that have been published on
the role of m6A in cancer may broadly be divided into two major
categories: One that summarizes the mechanism of action of m6A in
only one type of cancer, and the other that, although summarizing
the mechanism of action of m6A in multiple types of cancer, either
focuses on the effects of m6A on tumor progression or on its
effects on tumor metabolism.
The present review first provided a comprehensive
summary of the mechanisms associated with m6A modification in
various types of cancer (i.e., tumor progression and tumor
metabolism), highlighting the importance of m6A in regulating tumor
metabolism and influencing tumor progression. Furthermore, the
association between m6A and ferroptosis, another current hot topic
in this area of research, was explored. Finally, the important
roles of m6A in immunotherapy and clinical targeted therapy were
emphasized, with the aim of showing how targeting m6A is expected
to bring new hope for cancer treatment.
2. m6A modifications
m6A modifications have been indicated to have
participatory roles in tumor metabolism, tumor immune regulation
and ferroptosis, and the entire process comprises a series of
proteins that are classified into three categories, namely
methyltransferases, demethylases and m6A recognition binding
proteins, also known as writers, erasers and readers, respectively
(10) (Fig. 1).
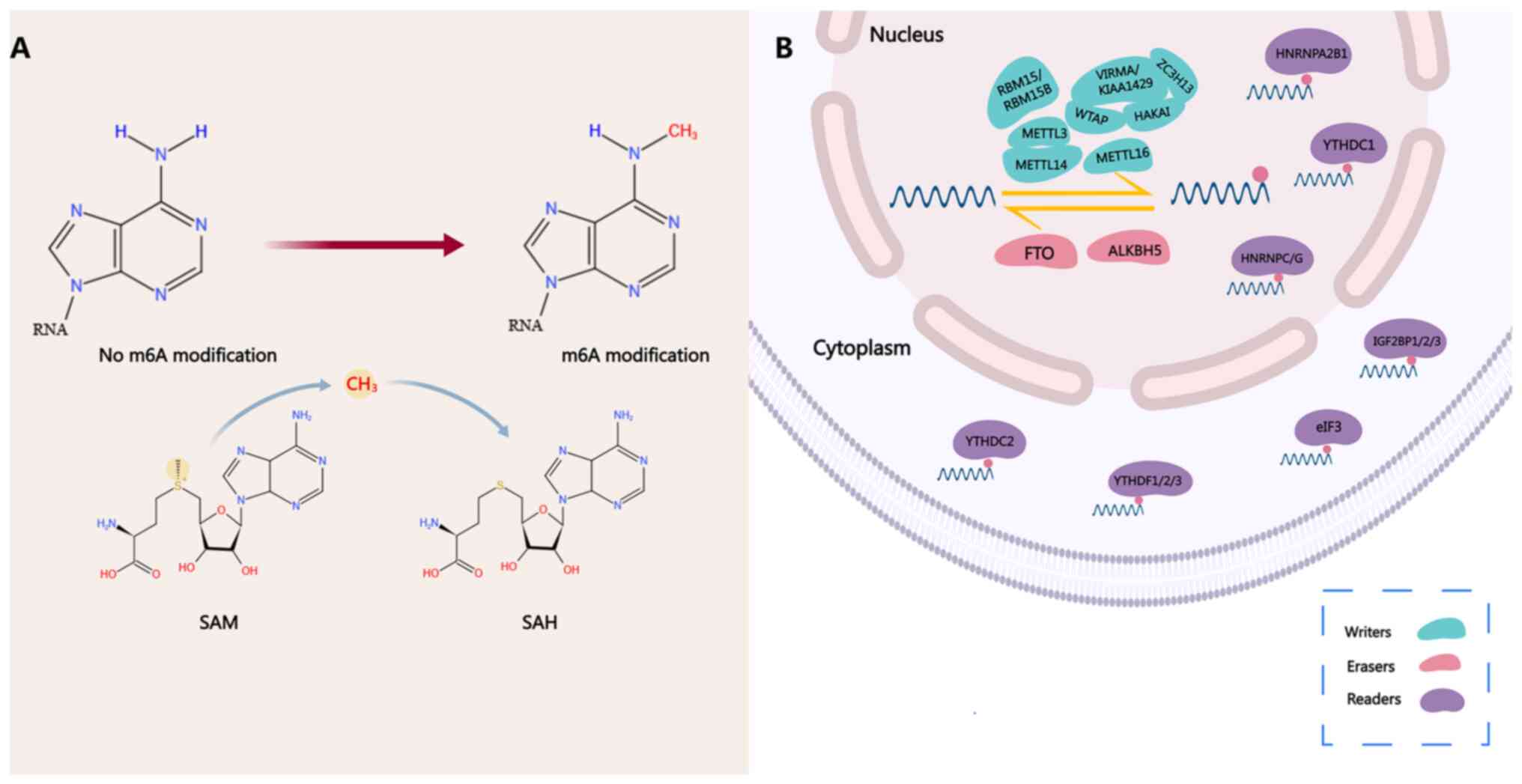 | Figure 1Process of m6A modification. (A) SAM
is a methyl donor for almost all eukaryotic cell methylation
reactions and it maintains RNA methylation through the activity of
RNA methyltransferase. Methyltransferases catalyze the transfer of
a methyl group from SAM to the N-6 position of adenosine. At the
same time, after demethylation, SAM is converted into SAH. (B) m6A
modification is a dynamic and reversible process. The modification
of m6A is catalyzed by a methyltransferase complex consisting of
METTL3 and METTL14 and its cofactors WTAP, RBM15/RBM15B,
VIRMA/KIAA1429, ZC3H13, HAKAI and METTL16 ('writers'), wherein
METTL3 and METTL4 constitute the catalytic subunit m6A-METTL
complex, and WTAP, VIRMA/KIAA1429, ZC3H13 and HAKAI constitute the
regulatory subunit m6A-METTL-associated complex. The m6A
modification can be removed by the demethylating enzymes FTO and
ALKBH5 ('erasers'). m6A functions by recruiting m6A binding
proteins ('readers') such as YTHDF1/2/3, YTHDC1/2, IGF2BP1/2/3,
HNRNPA2B1 and HNRNPC/G, which participate in mRNA transcription,
translation, splicing and degradation processes, thereby regulating
their stability. YTHDC1 is associated with RNA splicing and RNA
nuclear output in the nucleus; HNRNPA2B1 and HNRNPC/G are
associated with RNA splicing and miRNA processing in the nucleus;
IGF2BP1/2/3 is associated with RNA stability; YTHDC2, YTHDF1/3 and
eIF3 are associated with RNA translation in the cytoplasm; and
YTHDF2/3 is associated with RNA attenuation in the cytoplasm. SAM,
S-adenosylmethionine; SAH, S-adenosylhomocysteine; m6A,
N6-methyladenosine; METTL3/4, methyltransferase-like 3/4; WTAP,
Wilms tumor 1-associated protein; FTO, fat mass and
obesity-associated protein; ALKBH5, α-ketoglutarate-dependent
dioxygenase homolog 5; IGF2BP1/2/3, IGF2 binding protein 1/2/3;
YTHDF1/2/3, YTH domain family protein 1/2/3; HNRNPA2B1,
heterogeneous nuclear ribonucleoprotein A2B1; eIF3, eukaryotic
initiation factor 3; RBM15/RBM15B, RNA-binding motif protein
15/RNA-binding motif protein 15B; VIRMA, Vir like M6A
methyltransferase associated; HAKAI, E3 ubiquitin-protein ligase
Hakai; ZC3H13, zinc finger CCCH domain-containing protein 13. |
m6A writers/methyltransferases
The installation of m6A on mRNA or non-coding RNA
may be catalyzed by a methyltransferase complex composed of several
proteins, and these enzymes that are able to catalyze the transfer
of a methyl group from S-adenosylmethionine (SAM) to the N-6
position of adenosine are called methyltransferases, also known as
'writers' (11). In 1994, Bokar
et al (12) purified
methyltransferase into a protein complex for the first time,
thereby identifying it as a multi-component complex. To date, the
following methyltransferases have been identified:
Methyltransferase-like 3 (METTL3), METTL14, RNA-binding motif
protein 15 (RBM15)/RBM15B, Wilms tumor 1-associated protein (WTAP),
Vir like M6A methyltransferase associated (VIRMA)/KIAA1429, the E3
ubiquitin-protein ligase Hakai (HAKAI), zinc finger CCCH
domain-containing protein 13 (ZC3H13) and METTL16.
METTL3 promotes the translation of human oncogenes
and drives the progression of many types of tumors (including lung
cancer, gastric cancer and pancreatic cancer) (13-15).
METTL14 can not only catalyze the methylation of m6A RNA, but it
also forms a stable METTL3-METTL14 heterodimer complex with METTL3
and participates in the methylation modification of mammalian m6A
through mediating the deposition of m6A on mammalian cell nuclear
RNA (16). The METTL3-METTL14
heterodimer complex fulfills a crucial role in the m6A methylation
modification process. The METTL3-METTL14 heterodimer is able to
interact with WTAP without methylation activity, thereby affecting
the deposition of m6A (16). It
may also be directed towards specific targets and modify m6A with
the assistance of RBM15/RBM15B without catalytic function (17), and can also be recruited by
VIRMA/KIAA1429 to guide the process of methylation modification
(18).
In addition, WTAP, as the regulatory subunit of m6A
methyltransferase, has been shown to be involved in regulating the
recruitment of m6A methyltransferase complex to specific mRNA
targets, thereby affecting the activity of methyltransferase in
mammals (19).
The methyltransferase HAKAI is also an E3 ubiquitin
ligase that promotes the process of epithelial-mesenchymal
transition (EMT) in tumors through mediating downregulation of the
expression levels of E-cadherin, which is essential to maintain the
stability of m6A mRNA (20). The
zinc finger protein ZC3H13 is also a key methyltransferase, which
assists in the anchorage of WTAP, Virilizer (KIAA1429 human
homologous protein) and HAKAI in the nucleus to promote the process
of m6A methylation; thereby, ZC3H13 has an important role in
regulating RNA m6A methylation in the nucleus (21). METTL16 is a conserved U6 small
nuclear RNA (U6 snRNA) methyltransferase that adds m6A tags on to
U6 snRNA and SAM synthase precursor mRNA, and is able to regulate
methyl donor SAM homeostasis through activating methionine
adenosyltransferase 2A splicing and regulating SAM synthase
(22,23).
m6A erasers/demethylases
m6A demethylase is an enzyme that is able to
directly remove the m6A modification from mRNA, hence the name
'eraser'. In 2011, Jia et al (24) discovered the first demethylase,
which also functions as an obesity genetic factor, namely fat mass
and obesity-associated protein (FTO), which was shown to have
oxidative demethylation activity through a series of in
vitro experiments, effectively reversing the m6A modification
on mRNA via oxidation and demethylation (24). Although FTO was initially
considered to be associated with weight gain and obesity, more
recent studies have focused on its role in tumor formation and
progression.
Soon after, in 2013 Zheng et al (25) identified a second demethylase,
α-ketoglutarate-dependent dioxygenase homolog 5 (ALKBH5), which was
shown to affect mRNA export and RNA metabolism through the
oxidative demethylation of abundant m6A residues on mRNA. FTO has
been shown to be localized in the nucleus and cytoplasm, whereas
ALKBH5 is localized in the nucleus and co-localizes with nuclear
spots; therefore, both FTO and ALKBH5 are able to exert an effect
on the splicing of mRNA (25,26).
The last m6A demethylase to be discussed herein is ALKBH3, although
its substrates are predominantly transfer RNAs (27). Taken together, the various reports
published on demethylases to date have suggested that their
presence dictates that m6A RNA methylation modification is a
reversible process that can be dynamically regulated.
m6A readers/m6A-binding proteins
m6A-binding proteins, also known as 'readers', are a
class of proteins that are able to recognize and bind to
m6A-modified RNA, and yield specific phenotypic results through
regulating a variety of processes. Dominissini et al
(3) demonstrated that proteins
containing YTH domains were m6A-binding proteins, including YTH
domain family protein 1/2/3 (YTHDF1/2/3) and YTH domain 1/2
(YTHDC1/2).
In addition, insulin-like growth factor 2
mRNA-binding proteins (IGF2BPs, including the family members
IGF2BP1/2/3) also belong to the m6A-binding protein family, which
have been indicated to support the malignant state of cancer cells
by promoting the stability of mRNA and/or increasing its storage
(28).
Eukaryotic initiation factor 3 (eIF3), which
fulfills a core role in mammalian cell translation initiation, has
also been identified as an m6A-binding protein. Heterogeneous
nuclear ribonucleoprotein A2B1 (HNRNPA2B1) binds directly to m6A
sites in the transcriptome, and considering that it regulates the
variable splicing of exons in a set of transcripts, it is therefore
identified as an m6A-tagged nuclear reader and effector (29).
3. Targeting m6A in tumor growth and
metastasis
Numerous studies have confirmed that METTL3 is able
to promote the proliferation of tumor cells, increase the incidence
of tumor metastasis and have a significant positive role in tumor
progression. METTL3 may participate in all stages of the RNA life
cycle via regulating m6A modifications in mRNA; it has been shown
to promote the growth and invasion of bladder cancer cells through
the ALF transcription elongation factor 4/NF-κB/MYC signaling
pathway in an m6A-dependent manner, and it has also been shown to
promote the progression of bladder cancer by regulating non-coding
RNA, i.e., it promotes the maturation of pri-microRNA (miR)221/222,
thereby enhancing the proliferation of bladder cancer cells
(30,31). In addition, the 'writer'
methyltransferase METTL3 is able to combine with the 'reader'
IGF2BP2 to maintain the expression of sex-determining region Y
(SRY)-box 2 (SOX2) in colorectal cancer cells in an m6A-dependent
manner, thereby exerting a pro-cancer effect through their working
together (32). METTL3 also exerts
an active role in gastric cancer. METTL3 stabilizes and enhances
the mRNA expression of zinc finger MYM-type containing 1 (ZMYM1)
through an m6A-HuR-dependent pathway, which leads to the binding of
ZMYM1 to the C-terminal binding protein/lysine-specific demethylase
1/corepressor of RE1 silencing transcription factor complex,
thereby targeting and repressing E-cadherin transcription, leading
to an increase in EMT and promoting the metastasis of gastric
cancer cells (33).
METTL14 is an N6-adenosine methyltransferase that is
able to affect tumor growth through regulating the function of RNA.
In certain cases, the 'writer' METTL14 has been demonstrated to
promote tumor progression in an m6A-dependent manner. In pancreatic
cancer, overexpression of METTL14 can directly act on the
downstream target p53 apoptosis effector related to PMP22 (PERP; a
p53 target gene) through m6A modification, leading to a decrease in
PERP levels and a marked enhancement of the growth and metastasis
of pancreatic cancer cells (34).
However, in most cases, METTL14 is involved in tumor biological
processes as a tumor suppressor. It was shown that knockdown of
METTL14 in colorectal cancer leads to reduced m6A-methylation
levels of X inactivation-specific transcript (XIST), a newly
identified long non-coding RNA (lncRNA), and enhanced XIST
expression, resulting in the enhanced proliferation and invasion of
colorectal cancer cells (35).
Furthermore, METTL14 was also found to inhibit the metastasis of
colorectal cancer through mediating the m6A modification of
SRY-related high-mobility-group box 4 (SOX4) mRNA (36). In addition, METTL14-mediated
degradation of both XIST and SOX4 mRNA was shown to be dependent on
the YTHDF2-dependent pathway of the m6A reader protein (35,36).
METTL14 may also inhibit the growth and metastasis of gastric
cancer via stabilizing the expression of phosphatase and tensin
homologue mRNA or inactivating the PI3K/AKT/mTOR pathway and the
EMT (37,38).
It should be noted that ZC3H13 and METTL14 are
similar: Both proteins may function as tumor suppressor genes to
mediate the tumor immune response and fulfill a role in promoting
immunosuppression in breast cancer cells (39). WTAP, an oncogenic protein, has been
shown to promote the progression of hepatocellular carcinoma
through mediating m6A modification to silence ETS proto-oncogene 1
(40).
KIAA1429, a scaffold protein that functions as a
catalytic core component of the bridging m6A methyltransferase
complex, also promotes the progression of hepatocellular carcinoma
by inducing m6A methylation at the 3′-UTR of the pre-mRNA of the
oncogene GATA binding protein 3 (GATA3), leading to the degradation
of GATA3 pre-mRNA and thereby promoting tumor invasion and
metastasis (18). In addition, the
expression levels of KIAA1429 and its m6A were found to be
abnormally high in lung adenocarcinoma, thereby regulating the
expression of mucin 3A in an m6A-modified manner and promoting the
proliferation, invasion and migration of lung adenocarcinoma cells
(41).
METTL16 has also been shown to serve a
non-methylation-dependent role in promoting the formation of the
80S translation initiation complex through the interaction of its
methyltransferase structural domain with eIF3a/b and ribosomal RNA,
thereby promoting the growth, migration and invasion of
hepatocellular carcinoma cells (42).
As the first identified demethylase, FTO was shown
to act as a positive regulator of the progression and metastasis of
a variety of different types of tumor. In HER2-positive breast
cancer cells, FTO was found to accelerate tumor migration and
invasion through the FTO/miR-181b-3p/ADP ribosylation factor like
GTPase 5B signaling pathway (43).
As an oncogene, FTO has been shown to be associated with the
prognosis of bladder cancer and it regulates the methylation of
metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) to
promote tumor cell growth; subsequently, MALAT1 interacts with
miR-384 to further regulate MAL2 expression, thereby promoting the
progression of bladder cancer (44). During metastasis, the expression
level of FTO is abnormally increased in endometrial cancer tissues,
thereby promoting the expression of homeobox (HOX)B13, which
activates the WNT signaling pathway, further enhancing endometrial
cancer metastasis by removing the methylation modification of the
3′-UTR region of HOXB13 mRNA and eliminating the recognition of
this region by YTHDF2 (45).
ALKBH5, another m6A demethylase, has been shown to
promote the progression of multiple types of solid tumors and to
promote the self-renewal of cancer stem cells (46). In epithelial ovarian cancer, ALKBH5
has been shown to mediate the m6A demethylation of Janus kinase 2
(JAK2) mRNA, leading to the inhibition of YTHDF2-mediated mRNA
degradation and the maintenance of JAK2 mRNA expression;
furthermore, ALKBH5 joins with its upstream transcription factor,
HOXA10, in a regulatory loop to maintain ALKBH5 and HOXA10
overexpression in tumors, thereby activating the JAK2/STAT3
signaling pathway to promote tumor growth (47). In addition, ALKBH5 has been shown
to enhance both the stability of Bcl-2 mRNA and the interaction
between Bcl-2 and Beclin1, a key protein in autophagy, through
catalyzing m6A demethylation, which subsequently promoted the
proliferation and migration of ovarian cancer cells (48). When breast cancer cells are exposed
to hypoxic conditions, ALKBH5 expression has been shown to be
promoted in a hypoxia-inducible factor-dependent manner.
Overexpressed ALKBH5 may enhance the stability of NANOG mRNA,
thereby increasing its expression level and inducing an increase in
the breast cancer stem cell phenotype via mediating m6A
demethylation, which leads to the promotion of breast cancer
progression (49). Overexpression
of ALKBH5 in gastric cancer has been shown to induce the
demethylation of nuclear paraspeckle assembly transcript 1, leading
to an upregulation of its expression level, which induces
upregulation of the expression of EZH2, ultimately leading to the
enhancement of invasion and metastasis of gastric cancer cells
(50). However, it is notable that
numerous studies have demonstrated that ALKBH5 also has a role in
inhibiting tumor progression. In pancreatic cancer cells,
overexpression of ALKBH5 has been shown to interfere with Wnt
signaling through inhibiting the m6A modification of Wnt inhibitory
factor 1, which leads to an attenuation of the ability of
pancreatic cancer cells to proliferate and migrate, and also
enhances their sensitivity to chemotherapeutic agents (51). A subsequent study revealed that
inhibition of ALKBH5 expression in pancreatic cancer leads to
downregulation of the mRNA level of the tumor suppressor period
circadian regulator 1 in an m6A-YTHDF2-dependent manner, a process
that promotes the progression of pancreatic cancer (52). Therefore, several studies have
demonstrated how ALKBH5 fulfills a dual role in regulating tumor
progression; that is, it may exert different roles in different
types of tumor by regulating various biological processes.
m6A readers likewise influence tumor progression.
The YTH structural domain family, including YTHDF1/2/3, is able to
preferentially recognize m6A-modified mRNAs, thereby exerting an
important role in tumor progression.
YTHDF1 has been shown to promote the growth and
metastasis of ovarian cancer cells and to fulfill a key oncogenic
role in ovarian cancer, in which the underlying mechanism may be
that YTHDF1 regulates the protein expression of its downstream
target, eIF3c, and its translation in an m6A-dependent manner,
which, in turn, affects the progression of ovarian cancer (53).
In addition, it was found that YTHDF2 exerts a key
role in the m6A-dependent regulatory mechanism of glioblastoma, and
it may be involved in the malignant progression of glioblastoma
through multiple pathways. On the one hand, YTHDF2 may promote
glioblastoma growth via stabilizing MYC and VEGFA transcription and
targeting IGF2BP3 through m6A RNA modification (54); on the other hand, the protein
expression level of YTHDF2 is regulated by the EGFR/SRC/ERK
signaling pathway in glioblastoma, and its regulation of liver X
receiver α and HIV type I enhancer binding protein 2 occurs in an
m6A-dependent manner, ultimately promoting the proliferation and
invasion of tumor cells (55).
Overexpression of YTHDF2 promotes the growth and metastasis of
prostate cancer cells, probably since YTHDF2 both promotes the mRNA
degradation of the tumor suppressors phospholysine phosphohistidine
inorganic pyrophosphate phosphatase and NK3 homeobox 1, and
regulates AKT phosphorylation to promote prostate cancer
progression (56). The expression
level of YTHDF2 has also been shown to be associated with the
prognosis of patients with hepatocellular carcinoma, and it both
promotes the hepatocellular carcinoma stem cell phenotype and
enhances tumor metastasis through increasing the level of m6A in
the 5′-UTR of OCT4 mRNA (57).
However, it is noteworthy that YTHDF2 is also able to act as a
tumor suppressor in certain cases. It was found that overexpressed
YTHDF2 was able to inhibit the growth and proliferation of
hepatocellular carcinoma cells and induce apoptosis via promoting
the degradation of EGFR mRNA, thereby hindering the progression of
stem cell carcinoma (58).
Accordingly, YTHDF2 fulfills different roles in different
tumors.
The expression level of YTHDF3 has been shown to be
associated with the development of brain metastases from breast
cancer. It was found that overexpression of YTHDF3 both led to an
increase in the binding of eIF3a to ST6GALNAC5, GJA1 and EGFR, and
promoted the translation of these transcripts, thereby exerting a
key role in the process of breast cancer brain metastasis (59). In colorectal cancer, growth
arrest-specific 5 (GAS5) has been shown to inhibit the growth of
colorectal cancer cells by phosphorylating Yes-associated protein
(YAP) to induce its ubiquitination and degradation, whereas YTHDF3
is able to reverse the tumor suppression effects via binding to
m6A-modified GAS5 and degrading GAS5, thereby leading to the
progression of colorectal cancer (60).
The role of 'reader' YTHDC1 on RNA splicing is of
great value in tumor progression (61). In lung cancer, nuclear Aurora
kinase A was found to induce YTHDC1 to splice the tumor suppressor
RNA-binding protein 4 in an abnormal manner, which provides a novel
target for the target-directed treatment of lung cancer (62).
In general, YTHDC2 acts as a tumor suppressor gene
with low expression in tumor tissues, including non-small cell lung
cancer and head and neck squamous cell carcinoma (63,64).
In lung cancer tissue, YTHDC2 has a role in maintaining the
stability of tumor suppressor cyclindromatosis through m6A
modification, such that the downregulation of YTHDC2 promotes the
proliferation and invasion of lung cancer cells (65).
IGF2BPs are upregulated in most tumors and are
associated with tumor growth (66). IGF2BP1 has been shown to enhance
the expression of serum response factor through binding to a
conserved 3′-UTR and acting in an m6A-dependent manner, thereby
enhancing the invasive phenotype of tumors (67). In addition, IGF2BP1 has been shown
to exert a positive role in the progression of endometrial cancer.
IGF2BP1 and the mRNA stabilizer polyadenylate-binding protein 1 are
able to jointly maintain the stability of paternally expressed 10
mRNA and promote its expression, thereby accelerating the
proliferation of endometrial cancer cells (68). Therefore, IGF2BP1 may be a
potential target for the treatment of endometrial cancer.
Considering IGF2BP2, this protein is typically
involved in regulating the stability of mRNA, exerting a positive
role in tumor cell proliferation, invasion and metastasis. IGF2BP2
acts as an m6A reader that is able to identify the m6A methylation
of the lncRNA differentiation antagonizing non-protein coding RNA,
thereby delaying its mRNA degradation, increasing its stability and
promoting its expression, which ultimately achieves the goal of
promoting the stem cell characteristics of pancreatic cancer and
accelerating the growth of pancreatic cancer (69). In colorectal cancer, IGF2BP2 also
acts as a tumor promoter, recognizing YAP mRNA modifications and
upregulating the expression of ErbB2 by increasing its stability,
thereby promoting the proliferation, migration and invasion of
colorectal cancer cells and inhibiting their apoptosis (70).
The expression level of IGF2BP2 was found to be
associated with whether or not head and neck squamous cell
carcinoma has lymph node metastasis. The mRNA of Slug, an
EMT-associated protein, was found to be recognized and bound by
IGF2BP2, an interaction that regulates Slug expression and
maintains its mRNA stability, thereby promoting the lymphatic
metastasis of head and neck squamous cell carcinoma cells (71).
The 'reader' IGF2BP3 also has an m6A reading role in
colorectal cancer. It was demonstrated to promote tumor cell
proliferation and tumor angiogenesis via reading the m6A
modifications of cyclin D1 and VEGF (72). A high expression level of IGF2BP3
in bladder cancer is frequently indicative of a poor prognosis and
IGF2BP3 is able to enhance bladder cancer cell proliferation,
promote cell cycle progression and inhibit apoptosis through
activating the JAK/STAT pathway (73).
Of note, abnormal expression of circular RNAs
(circRNAs) may affect m6A modifications (74). By interacting with IGF2BP1, the
tumor suppressor circPTPRA was indicated to not only downregulate
the expression of MYC and fascin actin-bundling protein 1, but also
to impede the recognition of its downstream m6A-modified RNA by
IGF2BP1, ultimately inhibiting the proliferation, migration and
invasion of breast cancer cells (75). Similarly, overexpression of
circNDUFB2 was observed to reduce the stability of IGF2BPs via the
ubiquitin-proteasome pathway, leading to a significant inhibition
of growth and metastasis in non-small cell lung cancer (76).
In general, m6A-associated enzymes have been shown
to have a wide range of clinical significance in tumor growth and
metastasis. Current research on the mechanism of m6A in tumor
progression provides novel ideas and targets for the treatment of
tumors. m6A-associated enzymes themselves are potential targets for
a variety of tumor treatments, which should help patients to
achieve a better prognosis (Table
I).
 | Table IRole of m6A enzyme in tumor
proliferation, invasion and migration. |
Table I
Role of m6A enzyme in tumor
proliferation, invasion and migration.
| m6A | Cancer type | Role | Regulated
target | (Refs.) |
|---|
| METTL3 | Bladder cancer | Oncogene | AFF4/NF-κB/MY | (30) |
| Bladder cancer | Oncogene | pri-miR221/222 | (31) |
| Colorectal
cancer | Oncogene | SOX2 | (32) |
| Gastric cancer | Oncogene | ZMYM1 | (33) |
| METTL14 | Pancreatic
cancer | Oncogene | PERP | (34) |
| Colorectal
cancer | Tumor
suppressor | XIST | (35) |
| Colorectal
cancer | Tumor
suppressor | SOX4 | (36) |
| Gastric cancer | Tumor
suppressor | PI3K/AKT/mTOR | (37) |
| Stomach
adenocarcinoma | Tumor
suppressor | PTEN | (38) |
| WTAP | Hepatocellular
carcinoma | Oncogene | ETS1 | (40) |
| KIAA1429 | Hepatocellular
carcinoma | Oncogene | GATA3 | (18) |
| Lung
adenocarcinoma | Oncogene | MUC3A | (41) |
| METTL16 | Hepatocellular
carcinoma | Oncogene | eIF3a/b | (42) |
| FTO | Breast cancer | Oncogene | miR-181b-3p | (43) |
| Bladder cancer | Oncogene | MALAT1 | (44) |
| Endometrial
cancer | Oncogene | HOXB13 | (45) |
| ALKBH5 | Epithelial ovarian
cancer | Oncogene | HOXA10, JAK2 | (47) |
| Epithelial ovarian
cancer | Oncogene | BCL-2 | (48) |
| Breast cancer | Oncogene | NANOG | (49) |
| Gastric cancer | Oncogene | NEAT1 | (50) |
| Pancreatic
cancer | Tumor
suppressor | WIF-1 | (51) |
| Pancreatic
cancer | Tumor
suppressor | PER1 | (52) |
| YTHDF1 | Ovarian cancer | Oncogene | EIF3C | (53) |
| YTHDF2 | Glioblastoma | Oncogene | MYC, IGF2BP3 | (54) |
| Glioblastoma | Oncogene | LXRA, HIVEP2 | (55) |
| Prostate
cancer | Oncogene | LHPP, NKX3-1 | (56) |
| Hepatocellular
carcinoma | Oncogene | OCT4 | (57) |
| Hepatocellular
carcinoma | Tumor
suppressor | EGFR | (58) |
| YTHDF3 | Breast cancer | Oncogene | ST6GALNAC5, GJA1,
EGFR | (59) |
| Colorectal
cancer | Oncogene | GAS5 | (60) |
| YTHDC1 | Lung cancer | Oncogene | RBM4 | (62) |
| YTHDC2 | Lung cancer | Tumor
suppressor | CYLD | (65) |
| IGF2BP1 | Endometrial
cancer | Oncogene | PEG10 | (68) |
| IGF2BP2 | Pancreatic
cancer | Oncogene | DANCR | (69) |
| Colorectal
cancer | Oncogene | YAP | (70) |
| Head and neck
squamous cell carcinoma | Oncogene | Slug | (71) |
| IGF2BP3 | Colon cancer | Oncogene | CCND1, VEGF | (72) |
| Bladder cancer | Oncogene | JAK/STAT | (73) |
4. Targeting m6A in tumor metabolism
When tumor cells face challenging conditions such as
nutrient deficiency and hypoxia, they frequently carry out energy
metabolism reprogramming to meet their own energy needs (77). The change of the energy metabolism
mode is critically important for tumor occurrence, tumor cell
growth and proliferation, and thus, metabolic reprogramming is
regarded as a novel feature of tumors (78).
m6A and glucose metabolism
The Warburg effect, also known as aerobic
glycolysis, as the name suggests, indicates that tumor cells
preferentially undergo glycolysis even in the presence of oxygen,
rather than providing energy for cell growth through the oxidative
phosphorylation pathway. This metabolic approach gives tumor cells
the ability to obtain energy through anaerobic glycolysis,
providing the prerequisite energy for tumor cell proliferation and
metastasis (79). Recent studies
have confirmed that m6A-associated proteins are involved in the
regulation of glycolysis in tumor cells (Fig. 2).
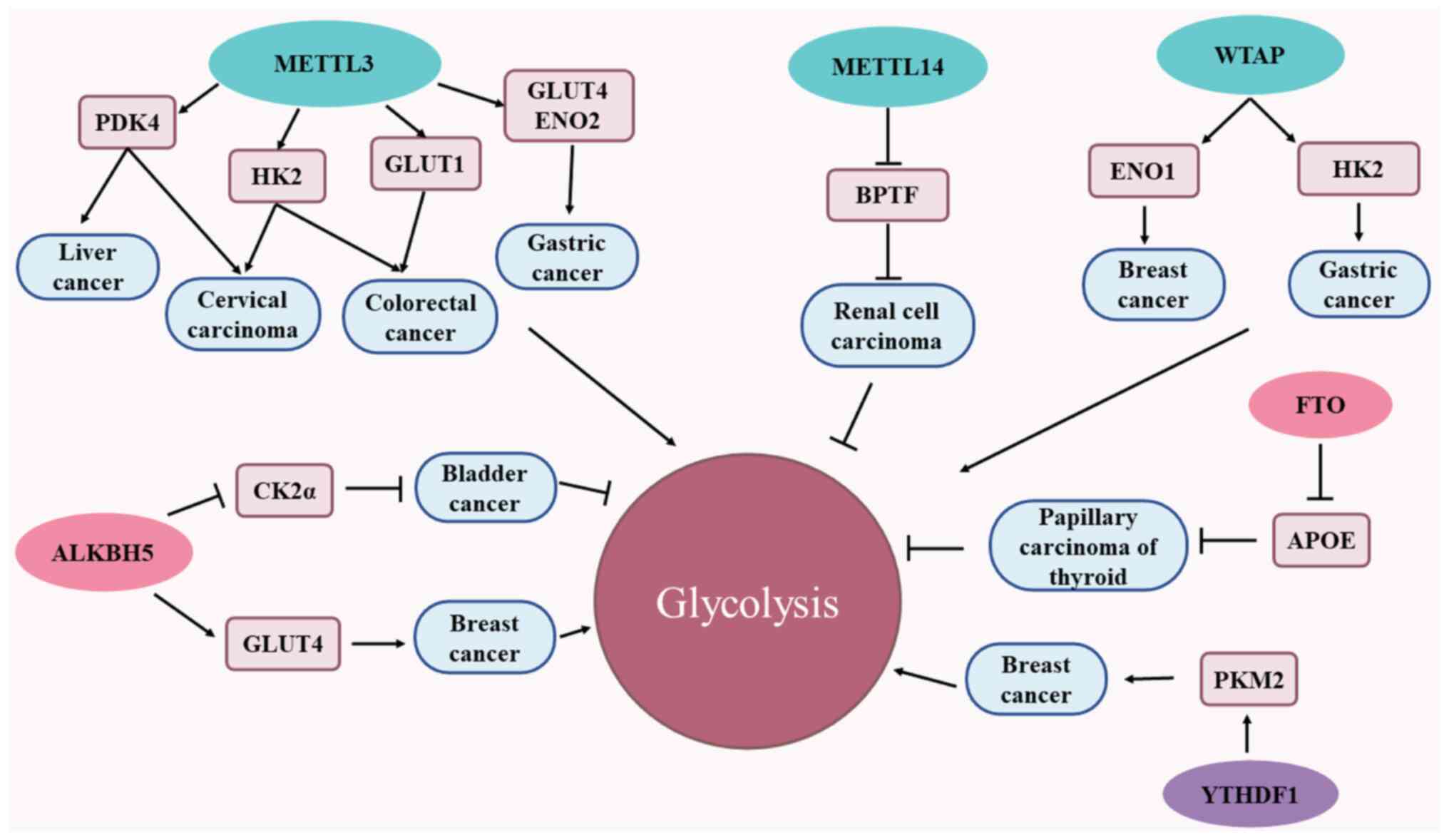 | Figure 2m6A methylases that participate in
cancer glycolytic metabolic pathways. Cancer cells rely on
glycolytic pathways to meet their energy needs for growth and
proliferation, and m6A-associated enzymes participate in the cancer
glycolytic pathway. The m6A methyltransferase METTL3 is able to
promote glycolysis and tumor progression through m6A
modification-mediated downstream targets (PDK4, GLUT1, HK2, GLUT1,
GLUT4 and ENO2). METTL14 inhibits glycolysis and tumor progression
by accelerating the degradation of BPTF mRNA. WTAP is able to
mediate the glycolytic process of tumors and promote tumor
progression through either upregulating ENO1 expression or
enhancing the stability of HK2 mRNA in an m6A-dependent manner.
ALKBH5 has a dual effect on the glycolytic pathway, on one hand
attenuating CK2α, and on the other hand increasing the stability of
GLUT4 mRNA, thereby promoting the glycolysis of breast cancer
cells. FTO inhibits glycolysis and thyroid cancer growth by
reducing the stability of apoE mRNA in an m6A-dependent manner.
Finally, YTHDF1 enhances glycolysis by improving the protein
translation of PKM2, thereby promoting the progression of breast
cancer. apoE, apolipoprotein E; METTL3/4, methyltransferase-like
3/4; WTAP, Wilms tumor 1-associated protein; FTO, fat mass and
obesity-associated protein; ALKBH5, α-ketoglutarate-dependent
dioxygenase homolog 5; PDK4, pyruvate dehydrogenase kinase 4;
GLUT1/4, glucose transporter 1/4; YTHDF1/2/3, YTH domain family
protein 1/2/3; HK2, hexokinase 2; ENO2, enolase 2; BPTF,
bromodomain PHD finger transcription factor; CK2α, casein kinase
2α; m6A, N6-methyladenosine; PKM2, pyruvate kinase M2; ENO2,
enolase 2. |
METTL3, as a tumor driver, promotes both the
proliferative and invasive abilities of tumor cells, as well as
cellular glycolysis, driving tumor progression. METTL3 is able to
positively regulate the glycolysis of tumor cells through promoting
the expression of pyruvate dehydrogenase kinase 4 (PDK4), and the
stability of PDK4 mRNA is maintained by IGF2BP3, which exerts a
positive role in promoting the growth of cervical cancer cells and
liver cancer cells (80). METTL3,
an important m6A-regulated enzyme in colorectal cancer cells, is
also closely associated with glycolysis in colorectal cancer. It is
able to mediate the downstream target glucose transporter protein 1
(GLUT1) through m6A modification, thereby regulating mechanistic
target of rapamycin complex 1 (mTORC1) signaling, promoting glucose
metabolism and increasing glucose uptake and lactate production
(81). In addition, METTL3 has
been shown to regulate the expression of hexokinase 2 (HK2) and
GLUT1, and maintain both the level and stability of HK2 and GLUT1
mRNA, depending on its m6A methyltransferase activity and m6A
reader IGF2BP2/3, thereby regulating glucose metabolism in
colorectal cancer and promoting tumor progression (7). The expression level of METTL3 in
gastric cancer cells may also be increased through inducing the
activation of H3K27 acetylation. An increase in the expression
level of METTL3 not only promotes the proliferation, metastasis and
tumor vascular growth of gastric cancer cells, but also enhances
the expression of glucose transporter 4 (GLUT4) and enolase 2
(ENO2) through the m6A modification of hepatoma-derived growth
factor (HDGF) mRNA, thereby enhancing the glycolytic rate of
gastric cancer cells and promoting tumor growth and liver
metastasis (14). In cervical
cancer, METTL3 also has a role in promoting glycolysis. The
underlying mechanism may involve METTL3 regulating its downstream
target HK2 and enhancement of the stability of HK2 in an
m6A-dependent manner mediated by YTHDF1 (82).
The expression of METTL14 has been shown to be
down-regulated in renal cell carcinoma, particularly during the
late metastatic stages. Since METTL14 is able to accelerate the
degradation of bromodomain PHD finger transcription factor (BPTF)
mRNA, a low expression level of METTL14 may activate ENO2 and SRC
proto-oncogene nonreceptor tyrosine kinase (SRC) through an
upregulation of the expression of BPTF, thereby promoting the
occurrence of glycolysis and metastasis of renal cell carcinoma,
and accelerating the progression of renal cell carcinoma (83).
WTAP fulfills the role of 'writer' in m6A
modification, and is also able to influence the Warburg effect of
cells and tumor progression. In breast cancer, C5aR1-positive
neutrophils have been shown to induce WTAP phosphorylation at
serine-341 through over-activating the ERK1/2 signal pathway to
stabilize the expression of WTAP protein, thereby upregulating ENO1
expression and enhancing the glycolytic activity of breast cancer
in a WTAP m6A-dependent manner (84). WATP has also been shown to improve
the stability of HK2 mRNA via its interaction with the 3′-UTR m6A
site, thereby mediating the glycolytic process and promoting the
progression of gastric cancer (85). Therefore, high expression levels of
WATP are associated with poor prognosis of patients with gastric
cancer.
The regulation of FTO on tumor progression is not a
case of simple inhibition or promotion. The expression of FTO has
been shown to be abnormally increased in certain tumors (such as
bladder cancer and endometrial cancer) to promote tumor
progression, whereas overexpression of FTO in other tumors has the
effect of inhibiting glycolysis, thereby inhibiting tumor cell
proliferation and hindering tumor progression. A low expression
level of FTO in papillary thyroid carcinoma leads to improvement in
the stability and expression of apolipoprotein E (APOE) by
enhancing the binding rate of IGF2BP2 to APOE mRNA, thereby
promoting both the uptake of glucose by tumor cells and tumor
growth (86). Taken together, the
effects of FTO on tumor action may depend on the different
downstream target molecules and the specific tissue
environment.
Low expression levels of ALKBH5 have been found to
be associated with poor prognosis in bladder cancer, as this
reduces the glucose utilization of, and lactate production by,
tumor cells via attenuating the mRNA stability of casein kinase 2α
and inhibiting glycolysis in an m6A-dependent manner, ultimately
inhibiting not only tumor progression, but also enhancing the
sensitivity of bladder cancer cells to chemotherapeutic agents such
as cisplatin (87). By contrast,
the upregulation of ALKBH5 expression not only increases resistance
to HER2-targeted therapy in patients with HER2-positive breast
cancer, but it also promotes the m6A demethylation of GLUT4 mRNA
under the influence of YTHDF2 to increase GLUT4 mRNA stability,
thereby promoting glycolysis in breast cancer cells (88).
In addition to methylases and demethylases that
participate in tumor cell glycolysis, m6A-binding proteins also
have an important role in the glycolytic process. YTHDF1 is a key
driving factor for the development of breast cancer, able to
enhance the proliferation and invasion of breast cancer cells and
inhibit apoptosis. In addition, YTHDF1 can enhance glycolysis
through increasing the protein translation of PKM2, thereby
promoting the progression of breast cancer (89).
Therefore, abnormal expression of m6A-associated
proteins in tumor cells is able to affect glycolysis, leading to
changes in glucose uptake and lactic acid production, affecting ATP
production, and ultimately influencing the progression of
tumors.
m6A and lipid metabolism
For tumor cells, fatty acids are also an important
source of energy and lipid metabolism also has an important role in
tumor progression (90). For
mammals, there are two main sources of lipids: One through food
intake (exogenous), and the other by means of de novo lipid
synthesis (endogenous), where de novo lipid synthesis is
also an important marker of tumor metabolism. Fatty acids are
important products of lipid metabolism that are essential for tumor
cell proliferation, and thus, reducing fatty acid levels by
disrupting their production and increasing their degradation may
provide an effective approach for tumor treatment (91). The lipid metabolism has been shown
to improve the energy source for the growth and proliferation of
tumor cells, improve the invasive and migratory capabilities of
tumors and also fulfill an important role in tumor progression
(92,93). Overall, it is indicated that the
m6A modification also exerts an important role in the lipid
metabolism of tumors.
The methylase METTL3 has been observed to promote
the upregulation of the expression of LINC00958, an
adipogenesis-associated lncRNA, through mediated m6A modification,
which subsequently led to increased proliferation, invasion and
migration of hepatocellular carcinoma cells, and increased
adipogenesis via the miR-3619-5p/HDGF pathway, driving the
progression of hepatocellular carcinoma (94).
The demethylase FTO has also been indicated to be
involved in regulating lipid metabolism in the liver, which is
dependent upon m6A demethylation to promote the generation of large
amounts of fat in liver cells, consequently leading to large
amounts of lipid deposition (95).
In hepatocellular carcinoma cells, deletion of FTO leads to
decreased stability and downregulation of the expression level of
fatty acid synthase mRNA, a lipid metabolism gene that is
influenced by m6A modification, which further downregulates the
protein expression levels of acetyl coenzyme A carboxylase and
ATP-citrate lyase, ultimately inhibiting ab initio lipid
synthesis and promoting tumor cell apoptosis, impeding the
progression of hepatocellular carcinoma (96). In addition, FTO has also been shown
to be involved in the regulation of lipid metabolism in esophageal
cancer, and FTO may increase the formation of lipid droplets in
esophageal cancer by promoting the translation efficiency of
HSD17B11 through YTHDF1, thereby promoting the proliferation,
migration and stemness of esophageal cancer cells and enhancing
their tumorigenicity (97).
As the current understanding of the mechanisms
associated with the role of m6A-associated enzymes in tumor lipid
metabolism deepens, novel therapeutic ideas will be pursued for
tumor treatment, and ultimately, new therapeutic strategies will be
adopted to intervene with the lipid uptake of tumor cells, or to
develop novel anti-cancer drugs by discovering new targets for
intervention, thereby improving the already existing therapeutic
interventions for tumors.
m6A and glutamine metabolism
In addition to the Warburg effect and fatty acid
metabolism, another common alteration in tumor metabolism is
increased glutamine metabolism. Glutamine metabolism exerts a
positive regulatory effect on the tricarboxylic acid cycle and
nucleotide and fatty acid biosynthesis in tumor cells (98). Glutamine, the most abundant amino
acid in human blood, in its high-level state, provides a ready
source of carbon and nitrogen for the growth of tumor cells
(99). For tumor cells to
continuously proliferate, this process is dependent on glutamine to
meet the metabolic demands of the tumor cells; therefore, targeting
glutamine catabolism has been a key research direction in tumor
therapy. Recent studies have also identified the mechanism
underlying the role of m6A-associated enzymes in glutamine
metabolism.
When glutamine catabolism is inhibited, tumor cells
are able to survive through activating autophagy, a process in
which m6A serves an important role in the regulation of activating
transcription factor 4 (ATF4). FTO was found to be upregulated upon
inhibition of glutamine catabolism, which served to maintain the
stability, prolong the half-life and upregulate the expression of
ATF4 mRNA, subsequently promoting the transcription of
DNA-damage-inducible transcript 4 and inducing inactivation of the
mTOR signaling pathway, thereby promoting cellular autophagy and
maintaining tumor survival and growth (100).
Deficiency of the tumor suppressor von Hippel-Lindau
(VHL) is a hallmark feature of renal clear cell carcinoma, and
glutamine has been shown to support the growth and survival of
VHL-deficient renal clear cell carcinoma cells (101); however, FTO is highly expressed
in VHL-deficient renal clear cell carcinoma cells and regulates the
expression of the glutamine transporter protein SLC1A5 in an
m6A-dependent manner, suggesting that FTO fulfills an important
role in the metabolic reprogramming (glutamine metabolism) of
VHL-deficient renal clear cell carcinoma through mediating SLC1A5
(102).
m6A readers are also involved in the regulation of
tumor glutamine metabolism. YTHDF1, a potential oncogene, is highly
expressed in the colorectum, subsequently leading to a decrease in
tumor sensitivity to cisplatin, the underlying mechanism of which
may be that YTHDF1 is able to bind directly to the 3′-UTR of
glutaminase-1 (GLS1) mRNA, thereby promoting protein translation
(103). Therefore, activation of
the GLS1-glutamine metabolic axis mediated by YTHDF1 is a key step
in YTHDF1-mediated cisplatin resistance in colorectal cancer,
suggesting that inhibition of glutamine metabolism, in combination
with cisplatin therapy, exerts a synergistic effect in the
treatment of tumors. YTHDC2 was also found to exhibit antitumor
activity in lung adenocarcinoma via promoting the degradation and
decay of solute carrier family 1 member 5 (SLC7A1) mRNA in an
m6A-dependent manner through the YTH structural domain and by
inhibiting cystine uptake, resulting in impaired oxidation in lung
adenocarcinoma cells and ultimately inhibiting tumor progression
(104).
Since the growth and proliferation of tumor cells
depend on glutamine metabolism, targeting glutamine metabolism may
become a new strategy for tumor therapies.
5. Targeting m6A in ferroptosis
Ferroptosis, a recently discovered form of
programmed cell death, is mainly characterized by iron-dependent
reactive oxygen species production and the accumulation of lipid
peroxidation products. Investigating ferroptosis, as a key tumor
suppressor mechanism, therefore provides a new research direction
for tumor treatment. A large number of studies have now revealed
the regulatory mechanism of m6A in ferroptosis (Fig. 3).
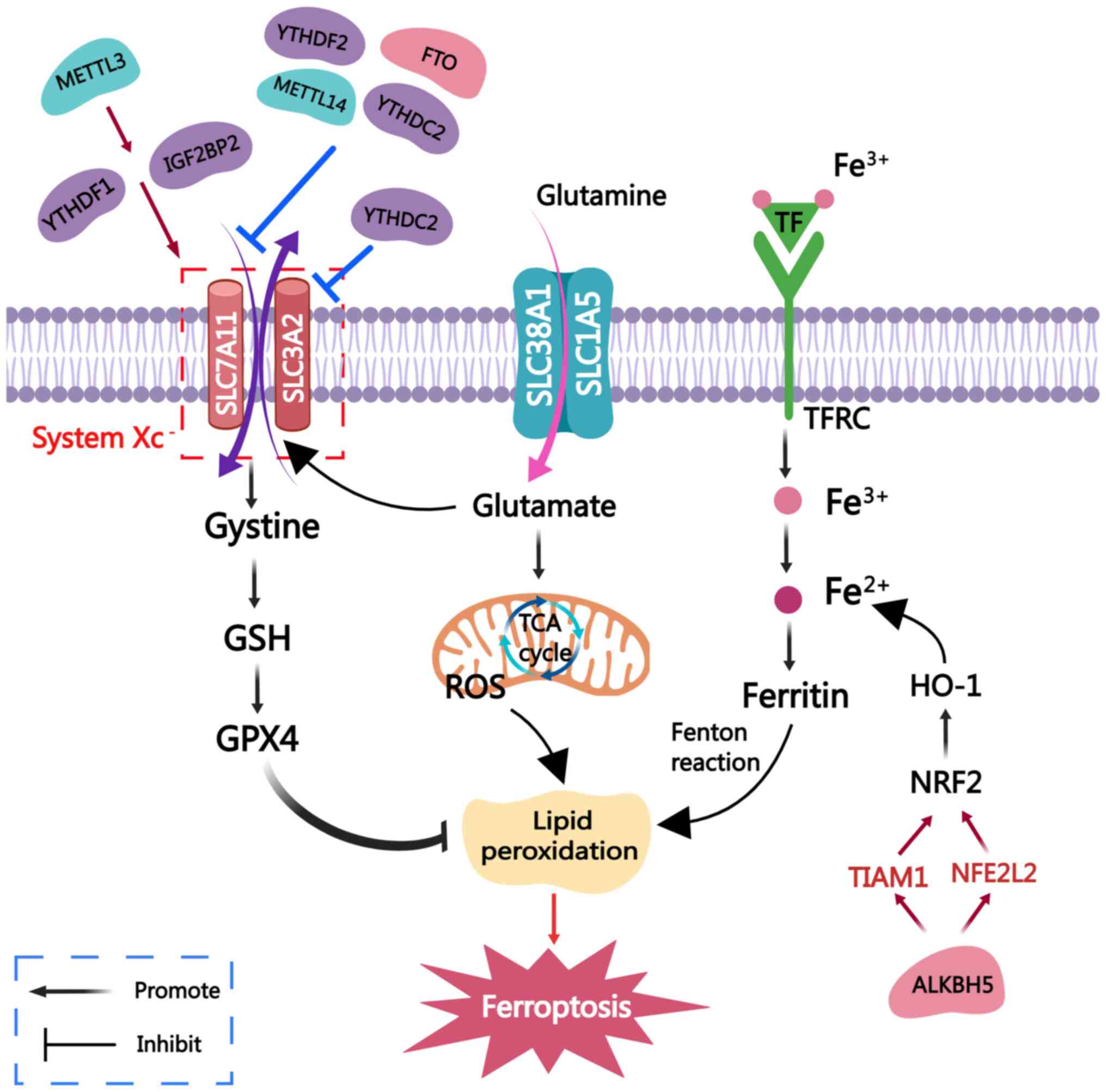 | Figure 3m6A methylases that participate in
ferroptosis. Ferroptosis is a form of cell death characterized by
an accumulation of Fe2+ ions and lipid peroxide
overload. The mechanism of ferroptosis is as follows: i) Iron ions
are inputted into the cells, converted into divalent iron and
accumulate in large amounts, promoting the occurrence of lipid
peroxidation through the Fenton reaction; ii) the systemXc-system
(SLC3A2+SLC7A11) is presented: The SystemXc−-GSH-GPX4
axis, wherein GPX4 inhibits lipid peroxidation; iii)
SLC38A1+SLC1A5: Glutamine enters the cell through glutamine
transporters to generate glutamate, which enters the mitochondria
and participates in the TCA cycle to generate mitochondrial ROS,
thereby promoting ferroptosis. m6A-associated enzymes can
participate in the ferroptotic pathway of tumors, thereby
regulating tumor progression. METTL3 inhibits ferroptosis by
enhancing the stability of SLC7A11 mRNA through YTHDF1 and IGF2BP2,
respectively. METTL14, FTO, YTHDF2 and YTHDC2 inhibit SLC7A11,
thereby inducing ferroptosis. YTHDC2 can also increase the
sensitivity of tumor cells to ferroptosis by inhibiting SLC3A2.
Finally, ALKBH5 induces ferroptosis by mediating the
m6A-TIAM1-Nrf2/HO-1 signaling pathway, thereby hindering tumor
progression. SLC3A2, solute carrier family 3 member 2; METTL3/4,
methyltransferase-like 3/4; FTO, fat mass and obesity-associated
protein; ALKBH5, α-ketoglutarate-dependent dioxygenase homolog 5;
IGF2BP1/2/3, IGF2 binding protein 1/2/3; YTHDF1/2/3, YTH domain
family protein 1/2/3; GSH, glutathione; GPX4, GSH peroxidase 4;
ROS, reactive oxygen species; m6A, N6-methyladenosine; TIAM1, TIAM
Rac1 associated GEF 1; Nrf2, nuclear factor erythroid 2-related
factor 2; HO-1, heme oxygenase 1; NFE2L2, NFE2 like bZIP
transcription factor 2; TCA, tricarboxylic acid; TF, transferrin;
TFRC, TF receptor. |
System Xc-is a glutamate-cystine reverse transport
system that includes two subunits, namely SLC7A11 and SLC3A2, and
inhibition of system Xc-has been shown to induce ferroptosis
(105,106). SLC7A11 is overexpressed in a
variety of tumor types, and may promote tumor growth through
inhibiting ferroptosis (107).
SLC3A2, a chaperone protein of SLC7A11, may also support the
function of SLC7A11, leading to the prevention of cellular lipid
peroxidation (108).
METTL3 was found to stabilize SLC7A11 mRNA, which
subsequently promotes its translation by recruiting the
YTHDF1-mediated m6A modification, thereby promoting lung
adenocarcinoma cell proliferation and inhibiting cellular
ferroptosis (109). In
hepatoblastoma, SLC7A11 mRNA is also regulated by METTL3 through
the modification of m6A. Furthermore, METTL3/IGF2BP2 have not only
been shown to enhance the stability of SLC7A11 mRNA through m6A
modification, but they also upregulate its expression via
inhibiting the process of deadenylation, thereby enhancing the
ferroptosis resistance of the tumor cells (110).
In contrast to the tumor-promoting effect of METTL3,
METTL14 frequently has a tumor-inhibiting role via inducing
ferroptosis. Under normoxic conditions, METTL14 targets the
methylation of m6A at the 3′-UTR of SLC7A11 mRNA to promote the
degradation of SLC7A11 mRNA. At the same time, YTHDF2 can also
recognize SLC7A11 mRNA and promote its degradation, thereby
inducing the occurrence of ferroptosis of tumor cells and
inhibiting the progression of hepatocellular carcinoma (111).
The expression level of FTO has also been shown to
be downregulated in papillary thyroid carcinoma, and this was
negatively correlated with SLC7A11; therefore, FTO functions as a
tumor suppressor gene. Overexpressed FTO was shown to inhibit
SLC7A11 expression via mediating m6A methylation, thereby promoting
the ferroptosis of thyroid papillary carcinoma cells and inhibiting
tumor cell growth (112).
Similarly, the demethylase ALKBH5, which is
downregulated in thyroid cancer, induces ferroptosis through
mediating the m6A-TIAM Rac1 associated GEF 1-nuclear factor
erythroid 2-related factor 2 (Nrf2)/heme oxygenase-1 signaling
pathway, thereby hindering the progression of thyroid cancer
(113). ALKBH5 also acts as an
oncogenic factor in head and neck squamous cell carcinoma,
mediating ferroptosis in tumor cells. Deletion of ALKBH5 not only
significantly reduces the sensitivity of tumor cells to ferroptosis
activators, but also induces the binding of NFE2 like bZIP
transcription factor 2/NRF2 transcripts to IGF2BP2, thereby
delaying its mRNA degradation and enhancing protein expression,
ultimately achieving the goal of inhibiting ferroptosis in tumor
cells (114).
YTHDF2 is also involved in the regulation of tumor
ferroptosis as an m6A reader. YTHDF2 can form a cystathionine
β-synthase (CBS) mRNA stabilizing lncRNA (CBSLR)/YTHDF2/CBS complex
with the lncRNA CBSLR under hypoxic conditions, thereby
destabilizing CBS mRNA in an m6A-regulated manner and preventing
gastric cancer cells from entering into ferroptosis in
oxygen-deficient states, which provides a target for the treatment
of refractory hypoxic tumors (115).
In addition, the RNA-binding protein YTHDC2 also
acts as a ferroptosis inducer in lung adenocarcinoma, where it is
able to both directly inhibit SLC7A11 in an m6A-dependent manner
and indirectly inhibit SLC3A2 through destabilizing HOXA13 mRNA,
leading to an increase in the sensitivity of lung adenocarcinoma
cells to ferroptosis (116).
Therefore, the m6A modification exerts a key role in
mediating tumor ferroptosis, and in particular, the regulatory role
mediated by m6A modification on SLC7A11 is of great research
interest. Therefore, there is a need to further focus on mRNA m6A
methylation and its functional role in the regulation of
ferroptosis in numerous types of tumor, and to develop different
therapeutic strategies for the different regulatory mechanisms of
m6A-associated enzymes in tumor ferroptosis, which may also bring
new insight into the treatment of tumors.
6. Targeting m6A in tumor immunotherapy
In recent years, the treatment of solid tumors is no
longer merely limited to radiotherapy and chemotherapy, but
immunotherapy has become one of the most common treatment methods,
among which the programmed cell death protein 1 (PD-1)/programmed
cell death ligand 1 (PD-L1) axis is considered as the main target
of immunotherapy (117). PD-1 is
mainly expressed on activated T cells and it binds to PD-L1 in
tumor cells and in the tumor microenvironment to mediate
immunosuppression (118).
Targeting PD-1/PD-L1 inhibition is a strategy for restoring the
immune response of T cells to tumor cells, and this has provided a
key breakthrough for certain types of tumor therapies (119). Furthermore, studies have
identified that m6A methylation regulators may be key enzymes that
regulate the expression of PD-1/PD-L1 and mediate immune
infiltration (Fig. 4).
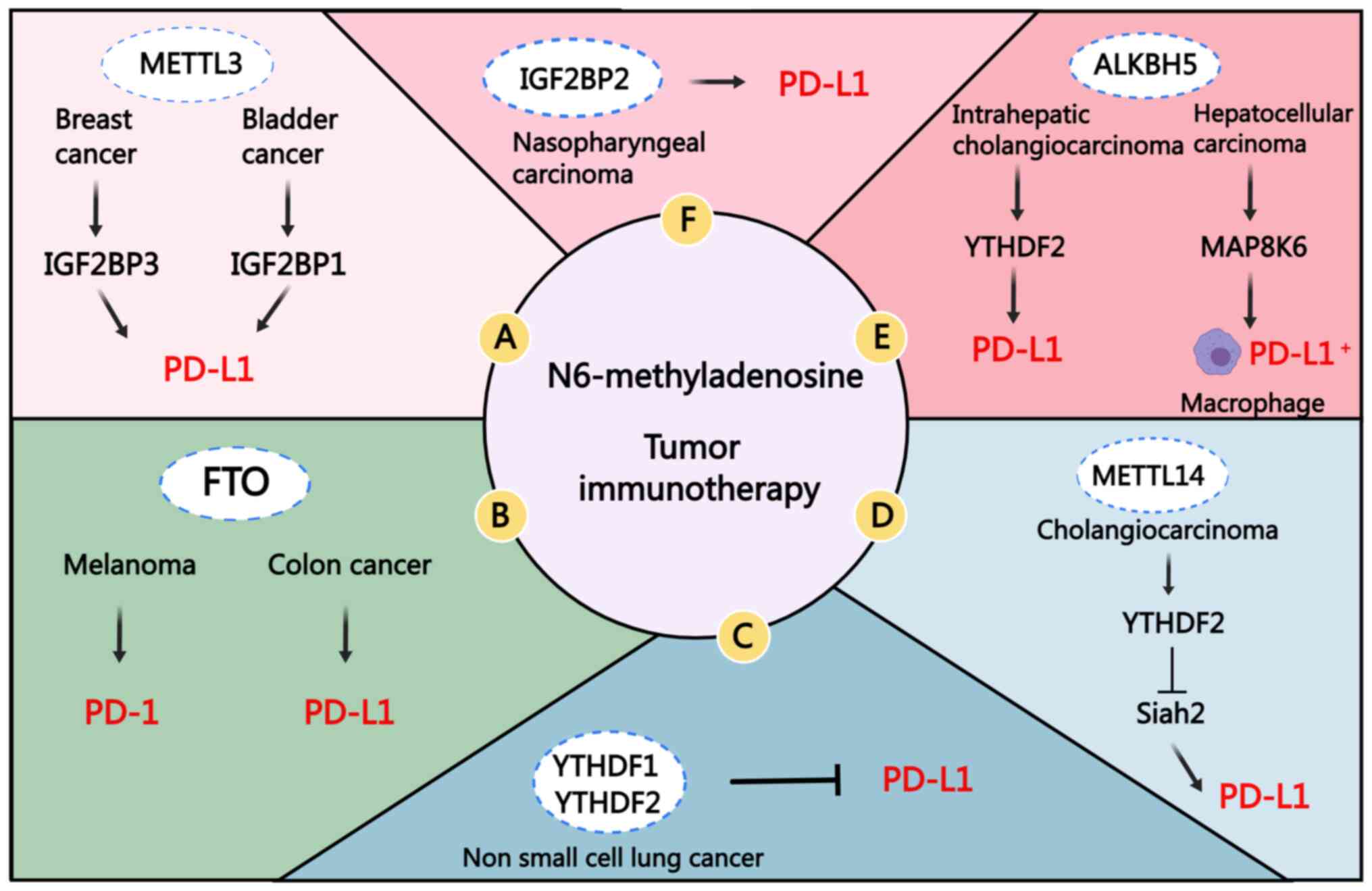 | Figure 4Role of m6A methylases in tumor
immunotherapy. (A) METTL3 can inhibit anti-tumor T-cell activation
through both IGF2BP3- and IGF2BP1-mediated PD-L1 mRNA stability and
upregulation of its expression. (B) FTO can promote the expression
of PD-1 and PD-L1 in an m6A-dependent manner. (C) The expression
levels of YTHDF1 and YTHDF2 in non-small cell lung cancer are
inversely proportional to PD-L1. (D) METTL14 mainly promotes the
degradation of its downstream target Siah2 mRNA in a
YTHDF2-dependent manner, thereby mediating immune escape. (E) The
overexpression of ALKBH5 in intrahepatic cholangiocarcinoma
maintains the stability of PD-L1 mRNA through YTHDF2. In
hepatocellular carcinoma, ALKBH5 regulates MAP8K6 expression in an
m6A-dependent manner and promotes the recruitment of PD-L1+
tumor-related macrophages. (F) IGF2BP2 can positively regulate the
expression of PD-L1, and inhibition of the expression of IGF2BP2
can achieve the effect of inhibiting PD-L1. METTL3/4,
methyltransferase-like 3/4; FTO, fat mass and obesity-associated
protein; ALKBH5, α-ketoglutarate-dependent dioxygenase homolog 5;
IGF2BP1/2/3, IGF2 binding protein 1/2/3; YTHDF1/2/3, YTH domain
family protein 1/2/3; PD-1, programmed cell death protein 1; PD-L1,
programmed cell death ligand 1; m6A, N6-methyladenosine. |
Macrophages are found in almost all tissues and were
one of the first 'immune' cells to appear in evolution (120). Macrophages are also prevalent in
the solid tumor micro-environment, where immune evasion of the
tumor cells occurs (121). It was
found that deletion of METTL3 in bone marrow cells promotes tumor
growth in vivo, even enhancing tumor invasion and
metastasis, leading to the increased infiltration of
tumor-associated macrophages and regulatory T cells in tumors, and
an attenuation of PD-1 blockade therapy (122). In addition, it has been found
that deletion of METTL3 in mouse T cells may affect the homeostasis
and differentiation of naive T cells (123). Therefore, m6A modification is
helpful for maintaining the homeostasis and function of immune
cells, and this may open up a new avenue for tumor immunotherapy.
PD-L1, as a downstream target of METTL3, is directly regulated by
METTL3 in terms of the m6A modification of its mRNA. In addition,
METTL3 has also been shown to upregulate PD-L1 expression and
maintain its mRNA stability in an m6A-IGF2BP3-dependent manner,
thereby inhibiting the activation of anti-tumor T cells and
promoting immune escape of tumors (124). Therefore, studying the
METTL3/IGF2BP3 axis is of great significance for tumor
immunotherapy. METTL3 has also been shown to regulate immune escape
in bladder cancer in an m6A-dependent manner according to the
following mechanism: Activation of the JNK signaling pathway
upregulates the expression of METTL3, thereby promoting tumor
immune escape through regulating PD-L1 resistance to
CD8+ T-cell cytotoxicity and, in the process, the reader
IGF2BP1 mediates the mRNA stability of PD-L1 and its expression
level (125).
METTL14 is also able to mediate tumor immune escape
according to a mechanism that depends on m6A modifications. In
cholangiocarcinoma, METTL14 directly regulates its downstream
target seven in absentia homolog 2 (Siah2) through promoting Siah2
mRNA degradation mediated via YTHDF2, leading to a decrease in
Siah2 protein expression levels and stable PD-L1 protein
expression, which thereby inhibits T-cell expansion-mediated immune
escape (126).
FTO, as a cancer-promoting factor of melanoma, has
been shown to promote the proliferation, invasion and migration of
melanoma cells. The loss of FTO was found to lead to an increase in
the m6A enrichment of PD-1 and a decrease in its stability, and
YTHDF2 was subsequently shown to mediate its mRNA decay (127). In addition, FTO was also shown to
regulate PD-L1 mRNA and its expression through m6A modification in
colon cancer (128). Hence,
inhibition of the FTO pathway combined with PD-1 blockade
immunotherapy may provide a new research direction for the
treatment of tumors.
In the immune microenvironment of intrahepatic
cholangiocarcinoma, overexpression of ALKBH5 was found, on the one
hand, to regulate the level of m6A modification in the 3′-UTR of
PD-L1 mRNA and to maintain the stability of PD-L1 mRNA by YTHDF2;
on the other hand, it may also upregulate the expression of PD-L1
in monocytes/macrophages and reduce the infiltration of bone
marrow-derived suppressor-like cells, thereby providing a possible
new approach for tumor immunotherapy (129). In hepatocellular carcinoma,
ALKBH5 was shown to regulate MAP8K6 expression in an m6A-dependent
manner, promoting the recruitment of PD-L1+
tumor-associated macrophages (130). Therefore, ALKBH5 overexpressed in
hepatocellular carcinoma likewise has a role in regulating the
tumor immune microenvironment.
YTHDF1, as a reader of m6A-modified mRNA, is
associated with immune cell infiltration in the tumor
microenvironment and serves an important role in immune regulation;
it can also regulate the expression level of immune checkpoint
genes through different signal transduction pathways, thereby
allowing the identification of ideal targets for immunotherapy and
providing a theoretical basis for combined molecular targeting
therapies (131). The deletion of
YTHDF1 in dendritic cells was shown to lead to inhibition of the
expression of lysosomal proteases, thereby attenuating antigen
degradation and achieving improved cross-presentation and
cross-activation of CD8+ T cells (132). The expression levels of YTHDF1
and YTHDF2 in non-small cell lung cancer were found to be
proportional to the number of tumor-infiltrating lymphocytes, and
inversely proportional to PD-L1 expression, indicating that high
expression of YTHDF1 and YTHDF2 in non-small cell lung cancer is a
predictor of improved prognosis for patients with tumors (133).
In hypopharyngeal carcinoma, inhibition of IGF2BP2
expression led to inhibition of PD-1/PD-L1, whereas inhibiting
PD-L1 also leads to downregulation of IGF2BP2 expression levels,
thereby inhibiting tumor growth (134). Although the mechanism of action
has yet to be fully elucidated, targeting IGF2BP2 or using PD-L1
inhibitors may provide an effective means for the treatment of
hypopharyngeal cancer.
In general, it has been shown that the association
of m6A-related enzymes with the tumor immune system is crucial for
the treatment of clinical tumors. However, at the present time,
there remains a lack of systematic and comprehensive studies on the
roles of m6A-associated enzymes in the tumor immune
microenvironment, and their impact on human tumor immunotherapy.
More detailed experimental studies are required to explore the
potential mechanisms of m6A-associated enzymes in human tumor
immunotherapy, and to provide a solid theoretical basis and novel
insight for immunotherapy of tumors.
7. Potential clinical applications for
targeting m6A-associated proteins
In recent years, the findings of studies associated
with the mechanism of action of m6A-related proteins in tumor
progression have led to the realization that m6A-associated
proteins may be used as cancer therapeutic targets and this insight
has motivated an increasing number of researchers to investigate
their clinical applications (Fig.
5).
 | Figure 5Inhibitors targeting m6A-associated
proteins for cancer therapy. (A) As a potent and selective METTL3
inhibitor, STM2457 is able to impair the progression of AML and ICC
by targeting METTL3. (B) BTYNB can effectively inhibit the
proliferation of OvCa and melanoma cells expressing IGF2BP1. (C)
FB23, FB23-2, CS1, CS2 and R-2HG, as FTO inhibitors, can exert
anti-leukemia activity by inhibiting FTO activity. (D)
5-Hydroxy-1-[3-(trifluorome thyl) phenyl]-1H-pyrazole-3-carboxylic
acid (20m) is a highly selective inhibitor of ALKBH5. The
inhibitors Ena15 and Ena21 can inhibit the proliferation of
GBM-derived cell lines by inhibiting the activity of ALKBH5.
Finally, the inhibitors
2-[(1-hydroxy-2-oxo-2-phenylethyl)sulfonyl]acid and
4-{[(furan-2-yl) methyl]amino}-1,2-diazinane-3,6-dione can inhibit
the proliferation of leukemia cells by inhibiting ALKBH5. (E) The
small-molecule compound CWI1-2 can inhibit the carcinogenic effect
of IGF2BP2 and inhibit the progression of acute myeloid leukemia.
FTO, fat mass and obesity-associated protein; ALKBH5,
α-ketoglutarate-dependent dioxygenase homolog 5; IGF2BP1/2/3, IGF2
binding protein 1/2/3; METTL3/4, methyltransferase-like 3/4; OvCa,
ovarian cancer; GBM, glioblastoma multiforme; AML, acute myeloid
leukemia; ICC, intrahepatic cholangiocarcinoma; STM2457, a specific
small molecule inhibitor of METTL3; FB23, FTO demethylase inhibitor
FB23; FB23-2, FTO demethylase inhibitor FB23-2; CS1, FTO
demethylase inhibitor CS1; CS2, FTO demethylase inhibitor CS2;
R-2HG, R-enantiomer of 2-hydroxyglutarate; Ena15 and Ena21, ALKBH5
inhibitor; BTYNB, 2-{[(5-bromo-2-thienyl)methylene]amino}
benzamide; CWI1-2, small molecule inhibitors of IGF2BP2. |
STM2457, a potent and selective inhibitor of METTL3,
was shown to hinder the development of acute myeloid leukemia
through catalytically targeting the inhibition of METTL3 to achieve
certain therapeutic effects (135). In addition, for intra-hepatic
cholangiocarcinoma with high expression of METTL3, treatment of
intrahepatic cholangiocarcinoma cells with STM2457 led to
inhibition of the proliferation and enhanced rates of apoptosis
(136). Therefore, STM2457, as a
METTL3 inhibitor with anti-cancer cell-proliferation potential, may
be used to treat cancers with high METTL3 expression, bringing new
hope for the treatment of such tumors.
Given the pro-cancer role mediated by FTO in tumors,
the development of targeted FTO inhibitors also has broad potential
for cancer therapy. FB23 and FB23-2 are small-molecule FTO
inhibitors and meclofenamic acid derivatives have been developed
that exert anti-proliferative effects and are effective therapeutic
strategies for the treatment of acute myeloid leukemia (137). A previously published study
demonstrated how computer screening and the experimental validation
of compounds in the National Cancer Institute Developmental
Therapeutics Program library led to the identification of two small
molecules with FTO-inhibiting activity, namely CS1 and CS2, which
also exerted anti-leukemic effects (138). In addition, R-2-hydroxyglutarate
was also found to inhibit the proliferation of leukemic cells,
exerting anti-leukemic effects via inhibiting FTO activity
(139). Therefore, the research
goal is to confirm the potential therapeutic effects of these FTO
inhibitors in other types of cancer with high FTO expression.
What is known about the mechanism underlying the
action of the demethylase ALKBH5 in cancer indicates that ALKBH5 is
expected to become an important target for cancer treatment, and
thus, it is imperative to develop an effective and selective ALKBH5
inhibitor. It was found that
5-hydroxy-1-[3-(trifluoromethyl)phenyl]-1H-pyrazole-3-car-boxylic
acid exhibited high selectivity for ALKBH5 and its administration
led to inhibition of m6A demethylation (140), although further experiments are
required to confirm its role in cancer therapy. Through
high-throughput screening, two novel inhibitors of ALKBH5, Ena15
and Ena21, were identified to inhibit cell proliferation of
glioblastoma multiforme-derived cell lines via inhibiting ALKBH5D
activity, thereby exerting anti-tumor effects and impeding tumor
progression (141). Furthermore,
2-[(1-hydroxy-2-oxo-2-phenylethyl)sulfanyl]acetic acid and
4-{[(furan-2-yl)methyl]amino}-1,2-diazinane-3,6-dione are ALKBH5
inhibitors that were also identified through high-throughput
screening, which exert inhibitory effects on the proliferation of
leukemia cells at low micromolar concentrations (142), once again providing a new
approach for the development of anti-cancer therapies.
IGF2BP1 frequently acts as a cancer-promoting
factor, which exerts an active role in the progression of cancer.
It has been reported that the IGF2BP1 inhibitor BTYNB not only
inhibits the proliferation of melanoma and ovarian cancer cells by
breaking the bond between IGF2BP1 and MYC mRNA (143), but also inhibits the
proliferation of tumor cells and the growth of solid tumors by
reducing the binding of IGF2BP1 to E2F1 mRNA (144). It has been observed that BTYNB,
as a special type of IGF2BP1 inhibitor, is able to inhibit tumor
cell proliferation through blocking the IGF2BP1-RNA association,
which brings new hope for the treatment of IGF2BP1-driven
tumors.
A recent study screened CWI1-2, a small-molecule
compound that is able to inhibit the pro-cancer effects of IGF2BP2,
and demonstrated through a series of in vivo and ex
vivo experiments that it exhibited appreciable anti-leukemic
effects, indicating the great potential of CWI1-2 as an
anti-leukemia drug (145).
In general, the targeted clinical application of
m6A-associated proteins remains in the initial stages of research.
Therefore, there is an urgent need to perform additional studies to
increase the current understanding of the functions and mechanisms
of m6A-associated proteins in cancer regulation, so as to provide a
solid theoretical basis for the development of drugs targeting
m6A-associated proteins. Furthermore, the role of targeting
m6A-associated proteins may be further explored in terms of
synergistic cancer therapies, combining them with traditional drugs
for cancer treatment. In conclusion, the development and screening
of efficient and selective inhibitors of m6A-associated proteins
for use in anti-cancer therapy through extensive basic and clinical
experiments should bring new promise and hope for the future of
cancer treatment.
8. Conclusions and future prospects
In recent years, research on the m6A methylation of
RNA has helped improve our understanding of the potential
underlying mechanisms and its functional role in tumor progression
and tumor immunotherapy. The present review has summarized the
basic functional characteristics of m6A-associated proteins,
outlining the effects of m6A-associated proteins on the processes
of tumor cell proliferation, migration and invasion, the regulation
of tumor metabolism, the potential mechanisms involved in
ferroptosis and their impact on tumor immunotherapy, through which
targeting m6A has been highlighted as a promising new avenue for
future cancer treatment.
Of note, the abnormal m6A methylation levels
exhibited by different tumor tissues have provided insight into the
dual role of m6A methylation: i) The same m6A-associated protein
may exert different roles in different tumor tissues; ii) different
m6A-associated proteins fulfill different roles in the same tumor
tissue; iii) certain genes exert a pro-cancer role following m6A
methylation, while others exert a pro-cancer role after
demethylation; iv) the same m6A-associated protein may fulfill
different roles in regulating different downstream targets in the
same tumor tissue. Therefore, a deep understanding of the complex
regulatory mechanisms of m6A methylation should provide a solid
theoretical basis for predicting patient prognosis, investigating
targeted therapies for m6A-associated proteins and for improving
anti-PD-1/PD-L1 responsiveness.
Although a large number of studies on m6A
methylation modifications have been published to date, the current
knowledge in this area remains incomplete. More in-depth and
comprehensive studies on m6A-associated proteins are required to
reveal their underlying biological mechanisms in tumor development
and to establish how they regulate the tumor immune
microenvironment. In addition, a deeper understanding of the dual
roles served by m6A in cancer progression (i.e., cancer promotion
and cancer inhibition) is required, and our hope is that more
efficient and selective drugs targeting m6A-associated proteins
will be developed and screened for cancer treatment in the near
future in order to improve patient prognosis.
Availability of data and materials
Not applicable.
Authors' contributions
XP, YTW, QJ and SQF conceived the study and wrote
the manuscript. HZ, LMC, HWT and MTW collated the data. XW and MX
(corresponding author) revised and edited the manuscript. All
authors have read and approved the final manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This work was supported by the National Natural Science
Foundation of China (grant no. 82072754), the Science and
Technology project of Jiangsu Provincial Health Commission (grant
no. M2020011), the Key R&D Programs of Jiangsu Province (grant
no. BE2018689), the Key R&D Programs of Zhenjiang City (grant
no. SH2018033), the National Natural Science Foundation of China
(grant no. 32170910), the Natural Science Foundation of Jiangsu
Province (grant no. BK20211124) and the Zhenjiang Key Research and
Development Program (grant no. SH2021037).
Abbreviations:
|
m6A
|
N6-methyladenosine
|
|
UTR
|
untranslated region
|
|
MTC
|
methyltransferase complex
|
|
SAM
|
S-adenosylmethionine
|
|
METTL3
|
methyltransferase-like 3
|
|
RBM15
|
RNA-binding motif protein 15
|
|
WTAP
|
Wilms tumor 1-associated protein
WTAP
|
|
ZC3H13
|
zinc finger CCCH domain-containing
protein 13
|
|
U6 snRNA
|
U6 small nuclear RNA
|
|
FTO
|
fat mass and obesity-associated
protein
|
|
ALKBH5
|
α-ketoglutarate-dependent dioxygenase
homolog 5
|
|
IGF2BPs
|
insulin-like growth factor 2
mRNA-binding proteins
|
|
eIF3
|
eukaryotic initiation factor 3
|
|
HNRNPA2B1
|
heterogeneous nuclear
ribonucleoprotein A2B1
|
|
XIST
|
X inactivation-specific
transcript
|
|
80S TIC
|
80S translation initiation
complex
|
|
MALAT1
|
metastasis-associated lung
adenocarcinoma transcript 1
|
|
HOXA10
|
homeobox A10
|
|
BCSC
|
breast cancer stem cell
|
|
WIF-1
|
Wnt inhibitory factor 1
|
|
PER1
|
period circadian regulator 1
|
|
LXRA
|
liver X receiver α
|
|
HIVEP2
|
human immunodeficiency virus type I
enhancer binding protein 2
|
|
CYLD
|
cyclindromatosis
|
|
YAP
|
Yes-associated protein
|
|
EMT
|
epithelial mesenchymal transition
|
|
PDK4
|
pyruvate dehydrogenase kinase 4
|
|
GLUT1
|
glucose transporter protein 1
|
|
HK2
|
hexokinase 2
|
|
GLUT4
|
glucose transporter 4
|
|
ENO2
|
enolase 2
|
|
BPTF
|
bromodomain PHD finger transcription
factor
|
|
SRC
|
SRC proto-oncogene nonreceptor
tyrosine kinase
|
|
APOE
|
apolipoprotein E
|
|
CK2
|
casein kinase 2
|
|
ATF4
|
activating transcription factor 4
|
|
GLS1
|
glutaminase-1
|
|
PD-1
|
programmed cell death protein 1
|
|
PD-L1
|
programmed cell death ligand 1
|
|
Siah2
|
seven in absentia homolog 2
|
|
ICP
|
immune checkpoint
|
|
AML
|
acute myeloid leukemia
|
|
GBM
|
glioblastoma multiforme
|
|
OvCa
|
ovarian cancer
|
References
|
1
|
Desrosiers R, Friderici K and Rottman F:
Identification of methylated nucleosides in messenger RNA from
Novikoff hepatoma cells. Proc Natl Acad Sci USA. 71:3971–3975.
1974.
|
|
2
|
Perry RP, Kelley DE, Friderici K and
Rottman F: The methylated constituents of L cell messenger RNA:
Evidence for an unusual cluster at the 5′ terminus. Cell.
4:387–394. 1975.
|
|
3
|
Dominissini D, Moshitch-Moshkovitz S,
Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K,
Jacob-Hirsch J, Amariglio N, Kupiec M, et al: Topology of the human
and mouse m6A RNA methylomes revealed by m6A-seq. Nature.
485:201–206. 2012.
|
|
4
|
Meyer KD, Saletore Y, Zumbo P, Elemento O,
Mason CE and Jaffrey SR: Comprehensive analysis of mRNA methylation
reveals enrichment in 3′ UTRs and near stop codons. Cell.
149:1635–1646. 2012.
|
|
5
|
Pavlova NN and Thompson CB: The emerging
hallmarks of cancer metabolism. Cell Metab. 23:27–47. 2016.
|
|
6
|
Zhang B, Wu Q, Li B, Wang D, Wang L and
Zhou YL: m6A regulator-mediated methylation modification
patterns and tumor microenvironment infiltration characterization
in gastric cancer. Mol Cancer. 19:532020.
|
|
7
|
Shen C, Xuan B, Yan T, Ma Y, Xu P, Tian X,
Zhang X, Cao Y, Ma D, Zhu X, et al: m6A-dependent
glycolysis enhances colorectal cancer progression. Mol Cancer.
19:722020.
|
|
8
|
Liu Y, Liang G, Xu H, Dong W, Dong Z, Qiu
Z, Zhang Z, Li F, Huang Y, Li Y, et al: Tumors exploit FTO-mediated
regulation of glycolytic metabolism to evade immune surveillance.
Cell Metab. 33:1221–1233.e11. 2021.
|
|
9
|
Shen M, Li Y, Wang Y, Shao J, Zhang F, Yin
G, Chen A, Zhang Z and Zheng S: N6-methyladenosine
modification regulates ferroptosis through autophagy signaling
pathway in hepatic stellate cells. Redox Biol. 47:1021512021.
|
|
10
|
Uddin MB, Wang Z and Yang C: The
m6A RNA methylation regulates oncogenic signaling
pathways driving cell malignant transformation and carcinogenesis.
Mol Cancer. 20:612021.
|
|
11
|
Li Q, Ni Y, Zhang L, Jiang R, Xu J, Yang
H, Hu Y, Qiu J, Pu L, Tang J and Wang X: HIF-1α-induced expression
of m6A reader YTHDF1 drives hypoxia-induced autophagy and
malignancy of hepatocellular carcinoma by promoting ATG2A and ATG14
translation. Signal Transduct Target Ther. 6:762021.
|
|
12
|
Bokar JA, Rath-Shambaugh ME, Ludwiczak R,
Narayan P and Rottman F: Characterization and partial purification
of mRNA N6-adenosine methyltransferase from HeLa cell nuclei.
Internal mRNA methylation requires a multisubunit complex. J Biol
Chem. 269:17697–11704. 1994.
|
|
13
|
Lin S, Choe J, Du P, Triboulet R and
Gregory RI: The m(6)A methyltransferase METTL3 promotes translation
in human cancer cells. Mol Cell. 62:335–345. 2016.
|
|
14
|
Wang Q, Chen C, Ding Q, Zhao Y, Wang Z,
Chen J, Jiang Z, Zhang Y, Xu G, Zhang J, et al: METTL3-mediated
m6A modification of HDGF mRNA promotes gastric cancer
progression and has prognostic significance. Gut. 69:1193–1205.
2020.
|
|
15
|
Xia T, Wu X, Cao M, Zhang P, Shi G, Zhang
J, Lu Z, Wu P, Cai B, Miao Y and Jiang K: The RNA m6A
methyltransferase METTL3 promotes pancreatic cancer cell
proliferation and invasion. Pathol Res Pract. 215:1526662019.
|
|
16
|
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang
L, Jia G, Yu M, Lu Z, Deng X, et al: A METTL3-METTL14 complex
mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem
Biol. 10:93–95. 2014.
|
|
17
|
Coker H, Wei G, Moindrot B, Mohammed S,
Nesterova T and Brockdorff N: The role of the Xist 5′ m6A region
and RBM15 in X chromosome inactivation. Wellcome Open Res.
5:312020.
|
|
18
|
Lan T, Li H, Zhang D, Xu L, Liu H, Hao X,
Yan X, Liao H, Chen X, Xie K, et al: KIAA1429 contributes to liver
cancer progression through N6-methyladenosine-dependent
post-transcriptional modification of GATA3. Mol Cancer.
18:1862019.
|
|
19
|
Ping XL, Sun BF, Wang L, Xiao W, Yang X,
Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, et al: Mammalian WTAP is
a regulatory subunit of the RNA N6-methyladenosine
methyltransferase. Cell Res. 24:177–189. 2014.
|
|
20
|
Bawankar P, Lence T, Paolantoni C,
Haussmann IU, Kazlauskiene M, Jacob D, Heidelberger JB, Richter FM,
Nallasivan MP, Morin V, et al: Hakai is required for stabilization
of core components of the m6A mRNA methylation
machinery. Nat Commun. 12:37782021.
|
|
21
|
Wen J, Lv R, Ma H, Shen H, He C, Wang J,
Jiao F, Liu H, Yang P, Tan L, et al: Zc3h13 regulates nuclear RNA
m6A methylation and mouse embryonic stem cell
self-renewal. Mol Cell. 69:1028–1038.e6. 2018.
|
|
22
|
Pendleton KE, Chen B, Liu K, Hunter OV,
Xie Y, Tu BP and Conrad NK: The U6 snRNA m6A
methyltransferase METTL16 regulates SAM synthetase intron
retention. Cell. 169:824–835.e14. 2017.
|
|
23
|
Ruszkowska A: METTL16,
methyltransferase-like protein 16: Current insights into structure
and function. Int J Mol Sci. 22:21762021.
|
|
24
|
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang
Y, Yi C, Lindahl T, Pan T, Yang YG and He C: N6-methyladenosine in
nuclear RNA is a major substrate of the obesity-associated FTO. Nat
Chem Biol. 7:885–887. 2011.
|
|
25
|
Zheng G, Dahl JA, Niu Y, Fedorcsak P,
Huang CM, Li CJ, Vågbø CB, Shi Y, Wang WL, Song SH, et al: ALKBH5
is a mammalian RNA demethylase that impacts RNA metabolism and
mouse fertility. Mol Cell. 49:18–29. 2013.
|
|
26
|
Zhao X, Yang Y, Sun BF, Shi Y, Yang X,
Xiao W, Hao YJ, Ping XL, Chen YS, Wang WJ, et al: FTO-dependent
demethylation of N6-methyladenosine regulates mRNA splicing and is
required for adipogenesis. Cell Res. 24:1403–1419. 2014.
|
|
27
|
Chen Z, Qi M, Shen B, Luo G, Wu Y, Li J,
Lu Z, Zheng Z, Dai Q and Wang H: Transfer RNA demethylase ALKBH3
promotes cancer progression via induction of tRNA-derived small
RNAs. Nucleic Acids Res. 47:2533–2545. 2019.
|
|
28
|
Huang H, Weng H, Sun W, Qin X, Shi H, Wu
H, Zhao BS, Mesquita A, Liu C, Yuan CL, et al: Recognition of RNA
N6-methyladenosine by IGF2BP proteins enhances mRNA
stability and translation. Nat Cell Biol. 20:285–295. 2018.
|
|
29
|
Alarcón CR, Goodarzi H, Lee H, Liu X,
Tavazoie S and Tavazoie SF: HNRNPA2B1 is a mediator of
m(6)A-dependent nuclear RNA processing events. Cell. 162:1299–1308.
2015.
|
|
30
|
Cheng M, Sheng L, Gao Q, Xiong Q, Zhang H,
Wu M, Liang Y, Zhu F, Zhang Y, Zhang X, et al: The m6A
methyltransferase METTL3 promotes bladder cancer progression via
AFF4/NF-κB/MYC signaling network. Oncogene. 38:3667–3680. 2019.
|
|
31
|
Han J, Wang JZ, Yang X, Yu H, Zhou R, Lu
HC, Yuan WB, Lu JC, Zhou ZJ, Lu Q, et al: METTL3 promote tumor
proliferation of bladder cancer by accelerating pri-miR221/222
maturation in m6A-dependent manner. Mol Cancer. 18:1102019.
|
|
32
|
Li T, Hu PS, Zuo Z, Lin JF, Li X, Wu QN,
Chen ZH, Zeng ZL, Wang F, Zheng J, et al: METTL3 facilitates tumor
progression via an m6A-IGF2BP2-dependent mechanism in
colorectal carcinoma. Mol Cancer. 18:1122019.
|
|
33
|
Yue B, Song C, Yang L, Cui R, Cheng X,
Zhang Z and Zhao G: METTL3-mediated N6-methyladenosine modification
is critical for epithelial-mesenchymal transition and metastasis of
gastric cancer. Mol Cancer. 18:1422019.
|
|
34
|
Wang M, Liu J, Zhao Y, He R, Xu X, Guo X,
Li X, Xu S, Miao J, Guo J, et al: Upregulation of METTL14 mediates
the elevation of PERP mRNA N6 adenosine methylation
promoting the growth and metastasis of pancreatic cancer. Mol
Cancer. 19:1302020.
|
|
35
|
Yang X, Zhang S, He C, Xue P, Zhang L, He
Z, Zang L, Feng B, Sun J and Zheng M: METTL14 suppresses
proliferation and metastasis of colorectal cancer by
down-regulating oncogenic long non-coding RNA XIST. Mol Cancer.
19:462020.
|
|
36
|
Chen X, Xu M, Xu X, Zeng K, Liu X, Pan B,
Li C, Sun L, Qin J, Xu T, et al: METTL14-mediated
N6-methyladenosine modification of SOX4 mRNA inhibits tumor
metastasis in colorectal cancer. Mol Cancer. 19:1062020.
|
|
37
|
Liu X, Xiao M, Zhang L, Li L, Zhu G, Shen
E, Lv M, Lu X and Sun Z: The m6A methyltransferase METTL14 inhibits
the proliferation, migration, and invasion of gastric cancer by
regulating the PI3K/AKT/mTOR signaling pathway. J Clin Lab Anal.
35:e236552021.
|
|
38
|
Yao Q, He L, Gao X, Tang N, Lin L, Yu X
and Wang D: The m6A methyltransferase METTL14-mediated
N6-methyladenosine modification of PTEN mRNA inhibits tumor growth
and metastasis in stomach adenocarcinoma. Front Oncol.
11:6997492021.
|
|
39
|
Gong PJ, Shao YC, Yang Y, Song WJ, He X,
Zeng YF, Huang SR, Wei L and Zhang JW: Analysis of
N6-methyladenosine methyltransferase reveals METTL14 and ZC3H13 as
tumor suppressor genes in breast cancer. Front Oncol.
10:5789632020.
|
|
40
|
Chen Y, Peng C, Chen J, Chen D, Yang B, He
B, Hu W, Zhang Y, Liu H, Dai L, et al: WTAP facilitates progression
of hepatocellular carcinoma via m6A-HuR-dependent epigenetic
silencing of ETS1. Mol Cancer. 18:1272019.
|
|
41
|
Zhao W and Xie Y: KIAA1429 promotes the
progression of lung adenocarcinoma by regulating the m6A level of
MUC3A. Pathol Res Pract. 217:1532842021.
|
|
42
|
Su R, Dong L, Li Y, Gao M, He PC, Liu W,
Wei J, Zhao Z, Gao L, Han L, et al: METTL16 exerts an
m6A-independent function to facilitate translation and
tumorigenesis. Nat Cell Biol. 24:205–216. 2022.
|
|
43
|
Xu Y, Ye S, Zhang N, Zheng S, Liu H, Zhou
K, Wang L, Cao Y, Sun P and Wang T: The FTO/miR-181b-3p/ARL5B
signaling pathway regulates cell migration and invasion in breast
cancer. Cancer Commun (Lond). 40:484–500. 2020.
|
|
44
|
Tao L, Mu X, Chen H, Jin D, Zhang R, Zhao
Y, Fan J, Cao M and Zhou Z: FTO modifies the m6A level of MALAT and
promotes bladder cancer progression. Clin Transl Med.
11:e3102021.
|
|
45
|
Zhang L, Wan Y, Zhang Z, Jiang Y, Lang J,
Cheng W and Zhu L: FTO demethylates m6A modifications in HOXB13
mRNA and promotes endometrial cancer metastasis by activating the
WNT signalling pathway. RNA Biol. 18:1265–1278. 2021.
|
|
46
|
Shen C, Sheng Y, Zhu AC, Robinson S, Jiang
X, Dong L, Chen H, Su R, Yin Z, Li W, et al: RNA demethylase ALKBH5
selectively promotes tumorigenesis and cancer stem cell
self-renewal in acute myeloid leukemia. Cell Stem Cell.
27:64–80.e9. 2020.
|
|
47
|
Nie S, Zhang L, Liu J, Wan Y, Jiang Y,
Yang J, Sun R, Ma X, Sun G, Meng H, et al: ALKBH5-HOXA10
loop-mediated JAK2 m6A demethylation and cisplatin resistance in
epithelial ovarian cancer. J Exp Clin Cancer Res. 40:2842021.
|
|
48
|
Zhu H, Gan X, Jiang X, Diao S, Wu H and Hu
J: ALKBH5 inhibited autophagy of epithelial ovarian cancer through
miR-7 and BCL-2. J Exp Clin Cancer Res. 38:1632019.
|
|
49
|
Zhang C, Samanta D, Lu H, Bullen JW, Zhang
H, Chen I, He X and Semenza GL: Hypoxia induces the breast cancer
stem cell phenotype by HIF-dependent and ALKBH5-mediated
m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci USA.
113:E2047–E2056. 2016.
|
|
50
|
Zhang J, Guo S, Piao HY, Wang Y, Wu Y,
Meng XY, Yang D, Zheng ZC and Zhao Y: ALKBH5 promotes invasion and
metastasis of gastric cancer by decreasing methylation of the
lncRNA NEAT1. J Physiol Biochem. 75:379–389. 2019.
|
|
51
|
Tang B, Yang Y, Kang M, Wang Y, Wang Y, Bi
Y, He S and Shimamoto F: m6A demethylase ALKBH5 inhibits
pancreatic cancer tumorigenesis by decreasing WIF-1 RNA methylation
and mediating Wnt signaling. Mol Cancer. 19:32020.
|
|
52
|
Guo X, Li K, Jiang W, Hu Y, Xiao W, Huang
Y, Feng Y, Pan Q and Wan R: RNA demethylase ALKBH5 prevents
pancreatic cancer progression by posttranscriptional activation of
PER1 in an m6A-YTHDF2-dependent manner. Mol Cancer. 19:912020.
|
|
53
|
Liu T, Wei Q, Jin J, Luo Q, Liu Y, Yang Y,
Cheng C, Li L, Pi J, Si Y, et al: The m6A reader YTHDF1 promotes
ovarian cancer progression via augmenting EIF3C translation.
Nucleic Acids Res. 48:3816–3831. 2020.
|
|
54
|
Dixit D, Prager BC, Gimple RC, Poh HX,
Wang Y, Wu Q, Qiu Z, Kidwell RL, Kim LJY, Xie Q, et al: The RNA m6A
reader YTHDF2 maintains oncogene expression and is a targetable
dependency in glioblastoma stem cells. Cancer Discov. 11:480–499.
2021.
|
|
55
|
Fang R, Chen X, Zhang S, Shi H, Ye Y, Shi
H, Zou Z, Li P, Guo Q, Ma L, et al: EGFR/SRC/ERK-stabilized YTHDF2
promotes cholesterol dysregulation and invasive growth of
glioblastoma. Nat Commun. 12:1772021.
|
|
56
|
Li J, Xie H, Ying Y, Chen H, Yan H, He L,
Xu M, Xu X, Liang Z, Liu B, et al: YTHDF2 mediates the mRNA
degradation of the tumor suppressors to induce AKT phosphorylation
in N6-methyladenosine-dependent way in prostate cancer. Mol Cancer.
19:1522020.
|
|
57
|
Zhang C, Huang S, Zhuang H, Ruan S, Zhou
Z, Huang K, Ji F, Ma Z, Hou B and He X: YTHDF2 promotes the liver
cancer stem cell phenotype and cancer metastasis by regulating OCT4
expression via m6A RNA methylation. Oncogene. 39:4507–4518.
2020.
|
|
58
|
Zhong L, Liao D, Zhang M, Zeng C, Li X,
Zhang R, Ma H and Kang T: YTHDF2 suppresses cell proliferation and
growth via destabilizing the EGFR mRNA in hepatocellular carcinoma.
Cancer Lett. 442:252–261. 2019.
|
|
59
|
Chang G, Shi L, Ye Y, Shi H, Zeng L,
Tiwary S, Huse JT, Huo L, Ma L, Ma Y, et al: YTHDF3 Induces the
translation of m6A-enriched gene transcripts to promote
breast cancer brain metastasis. Cancer Cell. 38:857–871.e7.
2020.
|
|
60
|
Ni W, Yao S, Zhou Y, Liu Y, Huang P, Zhou
A, Liu J, Che L and Li J: Long noncoding RNA GAS5 inhibits
progression of colorectal cancer by interacting with and triggering
YAP phosphorylation and degradation and is negatively regulated by
the m6A reader YTHDF3. Mol Cancer. 18:1432019.
|
|
61
|
Jiang X, Liu B, Nie Z, Duan L, Xiong Q,
Jin Z, Yang C and Chen Y: The role of m6A modification in the
biological functions and diseases. Signal Transduct Target Ther.
6:742021.
|
|
62
|
Li S, Qi Y, Yu J, Hao Y, He B, Zhang M,
Dai Z, Jiang T, Li S, Huang F, et al: Nuclear Aurora kinase A
switches m6A reader YTHDC1 to enhance an oncogenic RNA
splicing of tumor suppressor RBM4. Signal Transduct Target Ther.
7:972022.
|
|
63
|
Li Y, Zheng JN, Wang EH, Gong CJ, Lan KF
and Ding X: The m6A reader protein YTHDC2 is a potential biomarker
and associated with immune infiltration in head and neck squamous
cell carcinoma. PeerJ. 8:e103852020.
|
|
64
|
Sun S, Han Q, Liang M, Zhang Q, Zhang J
and Cao J: Downregulation of m6 A reader YTHDC2 promotes
tumor progression and predicts poor prognosis in non-small cell
lung cancer. Thorac Cancer. 11:3269–3279. 2020.
|
|
65
|
Wang J, Tan L, Jia B, Yu X, Yao R, OUYang
N, Yu X, Cao X, Tong J, Chen T, et al: Downregulation of
m6A reader YTHDC2 promotes the proliferation and
migration of malignant lung cells via CYLD/NF-κB pathway. Int J
Biol Sci. 17:2633–2651. 2021.
|
|
66
|
Müller S, Bley N, Glaß M, Busch B,
Rousseau V, Misiak D, Fuchs T, Lederer M and Hüttelmaier S: IGF2BP1
enhances an aggressive tumor cell phenotype by impairing
miRNA-directed downregulation of oncogenic factors. Nucleic Acids
Res. 46:6285–6303. 2018.
|
|
67
|
Müller S, Glaß M, Singh AK, Haase J, Bley
N, Fuchs T, Lederer M, Dahl A, Huang H, Chen J, et al: IGF2BP1
promotes SRF-dependent transcription in cancer in a m6A- and
miRNA-dependent manner. Nucleic Acids Res. 47:375–390. 2019.
|
|
68
|
Zhang L, Wan Y, Zhang Z, Jiang Y, Gu Z, Ma
X, Nie S, Yang J, Lang J, Cheng W and Zhu L: IGF2BP1 overexpression
stabilizes PEG10 mRNA in an m6A-dependent manner and promotes
endometrial cancer progression. Theranostics. 11:1100–1114.
2021.
|
|
69
|
Hu X, Peng WX, Zhou H, Jiang J, Zhou X,
Huang D, Mo YY and Yang L: IGF2BP2 regulates DANCR by serving as an
N6-methyladenosine reader. Cell Death Differ. 27:1782–1794.
2020.
|
|
70
|
Cui J, Tian J, Wang W, He T, Li X, Gu C,
Wang L, Wu J and Shang A: IGF2BP2 promotes the progression of
colorectal cancer through a YAP-dependent mechanism. Cancer Sci.
112:4087–4099. 2021.
|
|
71
|
Yu D, Pan M, Li Y, Lu T, Wang Z, Liu C and
Hu G: RNA N6-methyladenosine reader IGF2BP2 promotes lymphatic
metastasis and epithelial-mesenchymal transition of head and neck
squamous carcinoma cells via stabilizing slug mRNA in an
m6A-dependent manner. J Exp Clin Cancer Res. 41:62022.
|
|
72
|
Yang Z, Wang T, Wu D, Min Z, Tan J and Yu
B: RNA N6-methyladenosine reader IGF2BP3 regulates cell cycle and
angiogenesis in colon cancer. J Exp Clin Cancer Res.
39:2032020.
|
|
73
|
Huang W, Li Y, Zhang C, Zha H, Zhou X, Fu
B, Guo J and Wang G: IGF2BP3 facilitates cell proliferation and
tumorigenesis via modulation of JAK/STAT signalling pathway in
human bladder cancer. J Cell Mol Med. 24:13949–13960. 2020.
|
|
74
|
Lin H, Wang Y, Wang P, Long F and Wang T:
Mutual regulation between N6-methyladenosine (m6A) modification and
circular RNAs in cancer: Impacts on therapeutic resistance. Mol
Cancer. 21:1482022.
|
|
75
|
Xie F, Huang C, Liu F, Zhang H, Xiao X,
Sun J, Zhang X and Jiang G: CircPTPRA blocks the recognition of RNA
N6-methyladenosine through interacting with IGF2BP1 to suppress
bladder cancer progression. Mol Cancer. 20:682021.
|
|
76
|
Li B, Zhu L, Lu C, Wang C, Wang H, Jin H,
Ma X, Cheng Z, Yu C, Wang S, et al: circNDUFB2 inhibits non-small
cell lung cancer progression via destabilizing IGF2BPs and
activating anti-tumor immunity. Nat Commun. 12:2952021.
|
|
77
|
Hay N: Reprogramming glucose metabolism in
cancer: Can it be exploited for cancer therapy? Nat Rev Cancer.
16:635–649. 2016.
|
|
78
|
Tan YT, Lin JF, Li T, Li JJ, Xu RH and Ju
HQ: LncRNA-mediated posttranslational modifications and
reprogramming of energy metabolism in cancer. Cancer Commun (Lond).
41:109–120. 2021.
|
|
79
|
Vaupel P, Schmidberger H and Mayer A: The
Warburg effect: Essential part of metabolic reprogramming and
central contributor to cancer progression. Int J Radiat Biol.
95:912–919. 2019.
|
|
80
|
Li Z, Peng Y, Li J, Chen Z, Chen F, Tu J,
Lin S and Wang H: N6-methyladenosine regulates
glycolysis of cancer cells through PDK4. Nat Commun.
11:25782020.
|
|
81
|
Chen H, Gao S, Liu W, Wong CC, Wu J, Wu J,
Liu D, Gou H, Kang W, Zhai J, et al: RNA
N6-methyladenosine methyltransferase METTL3 facilitates
colorectal cancer by activating the m6A-GLUT1-mTORC1 axis and is a
therapeutic target. Gastroenterology. 160:1284–1300. e162021.
|
|
82
|
Wang Q, Guo X, Li L, Gao Z, Su X, Ji M and
Liu J: N6-methyladenosine METTL3 promotes cervical
cancer tumorigenesis and Warburg effect through YTHDF1/HK2
modification. Cell Death Dis. 11:9112020.
|
|
83
|
Zhang C, Chen L, Liu Y, Huang J, Liu A, Xu
Y, Shen Y, He H and Xu D: Downregulated METTL14 accumulates BPTF
that reinforces super-enhancers and distal lung metastasis via
glycolytic reprogramming in renal cell carcinoma. Theranostics.
11:3676–3693. 2021.
|
|
84
|
Ou B, Liu Y, Yang X, Xu X, Yan Y and Zhang
J: C5aR1-positive neutrophils promote breast cancer glycolysis
through WTAP-dependent m6A methylation of ENO1. Cell Death Dis.
12:7372021.
|
|
85
|
Yu H, Zhao K, Zeng H, Li Z, Chen K, Zhang
Z, Li E and Wu Z: N6-methyladenosine (m6A)
methyltransferase WTAP accelerates the Warburg effect of gastric
cancer through regulating HK2 stability. Biomed Pharmacother.
133:1110752021.
|
|
86
|
Huang J, Sun W, Wang Z, Lv C, Zhang T,
Zhang D, Dong W, Shao L, He L, Ji X, et al: FTO suppresses
glycolysis and growth of papillary thyroid cancer via decreasing
stability of APOE mRNA in an N6-methyladenosine-dependent manner. J
Exp Clin Cancer Res. 41:422022.
|
|
87
|
Yu H, Yang X, Tang J, Si S, Zhou Z, Lu J,
Han J, Yuan B, Wu Q, Lu Q and Yang H: ALKBH5 inhibited cell
proliferation and sensitized bladder cancer cells to cisplatin by
m6A-CK2α-mediated glycolysis. Mol Ther Nucleic Acids. 23:27–41.
2020.
|
|
88
|
Liu H, Lyu H, Jiang G, Chen D, Ruan S, Liu
S, Zhou L, Yang M, Zeng S, He Z, et al: ALKBH5-mediated m6A
demethylation of GLUT4 mRNA promotes glycolysis and resistance to
HER2-targeted therapy in breast cancer. Cancer Res. 82:3974–3986.
2022.
|
|
89
|
Yao X, Li W, Li L, Li M, Zhao Y, Fang D,
Zeng X and Luo Z: YTHDF1 upregulation mediates hypoxia-dependent
breast cancer growth and metastasis through regulating PKM2 to
affect glycolysis. Cell Death Dis. 13:2582022.
|
|
90
|
Carracedo A, Cantley LC and Pandolfi PP:
Cancer metabolism: Fatty acid oxidation in the limelight. Nat Rev
Cancer. 13:227–232. 2013.
|
|
91
|
Currie E, Schulze A, Zechner R, Walther TC
and Farese RV Jr: Cellular fatty acid metabolism and cancer. Cell
Metab. 18:153–161. 2013.
|
|
92
|
Li Z and Zhang H: Reprogramming of
glucose, fatty acid and amino acid metabolism for cancer
progression. Cell Mol Life Sci. 73:377–392. 2016.
|
|
93
|
Butler LM, Perone Y, Dehairs J, Lupien LE,
de Laat V, Talebi A, Loda M, Kinlaw WB and Swinnen JV: Lipids and
cancer: Emerging roles in pathogenesis, diagnosis and therapeutic
intervention. Adv Drug Deliv Rev. 159:245–293. 2020.
|
|
94
|
Zuo X, Chen Z, Gao W, Zhang Y, Wang J,
Wang J, Cao M, Cai J, Wu J and Wang X: M6A-mediated upregulation of
LINC00958 increases lipogenesis and acts as a nanotherapeutic
target in hepatocellular carcinoma. J Hematol Oncol. 13:52020.
|
|
95
|
Yang Z, Yu GL, Zhu X, Peng TH and Lv YC:
Critical roles of FTO-mediated mRNA m6A demethylation in regulating
adipogenesis and lipid metabolism: Implications in lipid metabolic
disorders. Genes Dis. 9:51–61. 2021.
|
|
96
|
Sun D, Zhao T, Zhang Q, Wu M and Zhang Z:
Fat mass and obesity-associated protein regulates lipogenesis via
m6 A modification in fatty acid synthase mRNA. Cell Biol
Int. 45:334–344. 2021.
|
|
97
|
Duan X, Yang L, Wang L, Liu Q, Zhang K,
Liu S, Liu C, Gao Q, Li L, Qin G and Zhang Y: m6A demethylase FTO
promotes tumor progression via regulation of lipid metabolism in
esophageal cancer. Cell Biosci. 12:602022.
|
|
98
|
Altman BJ, Stine ZE and Dang CV: From
Krebs to clinic: Glutamine metabolism to cancer therapy. Nat Rev
Cancer. 16:619–634. 2016.
|
|
99
|
Wang Y, Bai C, Ruan Y, Liu M, Chu Q, Qiu
L, Yang C and Li B: Coordinative metabolism of glutamine carbon and
nitrogen in proliferating cancer cells under hypoxia. Nat Commun.
10:2012019.
|
|
100
|
Han S, Zhu L, Zhu Y, Meng Y, Li J, Song P,
Yousafzai NA, Feng L, Chen M, Wang Y, et al: Targeting
ATF4-dependent pro-survival autophagy to synergize glutaminolysis
inhibition. Theranostics. 11:8464–8479. 2021.
|
|
101
|
Okazaki A, Gameiro PA, Christodoulou D,
Laviollette L, Schneider M, Chaves F, Stemmer-Rachamimov A,
Yazinski SA, Lee R, Stephanopoulos G, et al: Glutaminase and
poly(ADP-ribose) polymerase inhibitors suppress pyrimidine
synthesis and VHL-deficient renal cancers. J Clin Invest.
127:1631–1645. 2017.
|
|
102
|
Xiao Y, Thakkar KN, Zhao H, Broughton J,
Li Y, Seoane JA, Diep AN, Metzner TJ, von Eyben R, Dill DL, et al:
The m6A RNA demethylase FTO is a HIF-independent
synthetic lethal partner with the VHL tumor suppressor. Proc Natl
Acad Sci USA. 117:21441–21449. 2020.
|
|
103
|
Chen P, Liu XQ, Lin X, Gao LY, Zhang S and
Huang X: Targeting YTHDF1 effectively re-sensitizes
cisplatin-resistant colon cancer cells by modulating GLS-mediated
glutamine metabolism. Mol Ther Oncolytics. 20:228–239. 2021.
|
|
104
|
Ma L, Chen T, Zhang X, Miao Y, Tian X, Yu
K, Xu X, Niu Y, Guo S, Zhang C, et al: The m6A reader
YTHDC2 inhibits lung adenocarcinoma tumorigenesis by suppressing
SLC7A11-dependent antioxidant function. Redox Biol.
38:1018012021.
|
|
105
|
Koppula P, Zhuang L and Gan B: Cystine
transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient
dependency, and cancer therapy. Protein Cell. 12:599–620. 2021.
|
|
106
|
Hu Z, Yin Y, Jiang J, Yan C, Wang Y, Wang
D and Li L: Exosomal miR-142-3p secreted by hepatitis B virus
(HBV)-hepatocellular carcinoma (HCC) cells promotes ferroptosis of
M1-type macrophages through SLC3A2 and the mechanism of HCC
progression. J Gastrointest Oncol. 13:754–767. 2022.
|
|
107
|
Wang X, Chen Y, Wang X, Tian H, Wang Y,
Jin J, Shan Z, Liu Y, Cai Z, Tong X, et al: Stem cell factor SOX2
confers ferroptosis resistance in lung cancer via upregulation of
SLC7A11. Cancer Res. 81:5217–5229. 2021.
|
|
108
|
Wang W, Green M, Choi JE, Gijón M, Kennedy
PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, et al:
CD8+ T cells regulate tumour ferroptosis during cancer
immunotherapy. Nature. 569:270–274. 2019.
|
|
109
|
Xu Y, Lv D, Yan C, Su H, Zhang X, Shi Y
and Ying K: METTL3 promotes lung adenocarcinoma tumor growth and
inhibits ferroptosis by stabilizing SLC7A11 m6A modification.
Cancer Cell Int. 22:112022.
|
|
110
|
Liu L, He J, Sun G, Huang N, Bian Z, Xu C,
Zhang Y, Cui Z, Xu W, Sun F, et al: The N6-methyladenosine
modification enhances ferroptosis resistance through inhibiting
SLC7A11 mRNA deadenylation in hepatoblastoma. Clin Transl Med.
12:e7782022.
|
|
111
|
Fan Z, Yang G, Zhang W, Liu Q, Liu G, Liu
P, Xu L, Wang J, Yan Z, Han H, et al: Hypoxia blocks ferroptosis of
hepato-cellular carcinoma via suppression of METTL14 triggered
YTHDF2-dependent silencing of SLC7A11. J Cell Mol Med.
25:10197–10212. 2021.
|
|
112
|
Ji FH, Fu XH, Li GQ, He Q and Qiu XG: FTO
prevents thyroid cancer progression by SLC7A11 m6A methylation in a
ferroptosis-dependent manner. Front Endocrinol (Lausanne).
13:8577652022.
|
|
113
|
Li W, Huang G, Wei J, Cao H and Jiang G:
ALKBH5 inhibits thyroid cancer progression by promoting ferroptosis
through TIAM1-Nrf2/HO-1 axis. Mol Cell Biochem. 478:729–741.
2023.
|
|
114
|
Ye J, Chen X, Jiang X, Dong Z, Hu S and
Xiao M: RNA demethylase ALKBH5 regulates hypopharyngeal squamous
cell carcinoma ferroptosis by posttranscriptionally activating
NFE2L2/NRF2 in an m6 A-IGF2BP2-dependent manner. J Clin
Lab Anal. 36:e245142022.
|
|
115
|
Yang H, Hu Y, Weng M, Liu X, Wan P, Hu Y,
Ma M, Zhang Y, Xia H and Lv K: Hypoxia inducible lncRNA-CBSLR
modulates ferroptosis through m6A-YTHDF2-dependent modulation of
CBS in gastric cancer. J Adv Res. 37:91–106. 2021.
|
|
116
|
Ma L, Zhang X, Yu K, Xu X, Chen T, Shi Y,
Wang Y, Qiu S, Guo S, Cui J, et al: Targeting SLC3A2 subunit of
system XC-is essential for m6A reader YTHDC2
to be an endogenous ferroptosis inducer in lung adenocarcinoma.
Free Radic Biol Med. 168:25–43. 2021.
|
|
117
|
Han Y, Liu D and Li L: PD-1/PD-L1 pathway:
Current researches in cancer. Am J Cancer Res. 10:727–742.
2020.
|
|
118
|
Xu-Monette ZY, Zhou J and Young KH: PD-1
expression and clinical PD-1 blockade in B-cell lymphomas. Blood.
131:68–83. 2018.
|
|
119
|
Yi M, Zheng X, Niu M, Zhu S, Ge H and Wu
K: Combination strategies with PD-1/PD-L1 blockade: Current
advances and future directions. Mol Cancer. 21:282022.
|
|
120
|
Buchmann K: Evolution of innate immunity:
Clues from Invertebrates via fish to mammals. Front Immunol.
5:4592014.
|
|
121
|
Gajewski TF, Schreiber H and Fu YX: Innate
and adaptive immune cells in the tumor microenvironment. Nat
Immunol. 14:1014–1022. 2013.
|
|
122
|
Yin H, Zhang X, Yang P, Zhang X, Peng Y,
Li D, Yu Y, Wu Y, Wang Y, Zhang J, et al: RNA m6A methylation
orchestrates cancer growth and metastasis via macrophage
reprogramming. Nat Commun. 12:13942021.
|
|
123
|
Li HB, Tong J, Zhu S, Batista PJ, Duffy
EE, Zhao J, Bailis W, Cao G, Kroehling L, Chen Y, et al:
m6A mRNA methylation controls T cell homeostasis by
targeting the IL-7/STAT5/SOCS pathways. Nature. 548:338–342.
2017.
|
|
124
|
Wan W, Ao X, Chen Q, Yu Y, Ao L, Xing W,
Guo W, Wu X, Pu C, Hu X, et al: METTL3/IGF2BP3 axis inhibits tumor
immune surveillance by upregulating N6-methyladenosine
modification of PD-L1 mRNA in breast cancer. Mol Cancer.
21:602022.
|
|
125
|
Ni Z, Sun P, Zheng J, Wu M, Yang C, Cheng
M, Yin M, Cui C, Wang G, Yuan L, et al: JNK signaling promotes
bladder cancer immune escape by regulating METTL3-mediated m6A
modification of PD-L1 mRNA. Cancer Res. 82:1789–1802. 2022.
|
|
126
|
Zheng H, Zheng WJ, Wang ZG, Tao YP, Huang
ZP, Yang L, Ouyang L, Duan ZQ, Zhang YN, Chen BN, et al: Decreased
expression of programmed death ligand-L1 by seven in absentia
homolog 2 in cholangiocarcinoma enhances T-cell-mediated antitumor
activity. Front Immunol. 13:8451932022.
|
|
127
|
Yang S, Wei J, Cui YH, Park G, Shah P,
Deng Y, Aplin AE, Lu Z, Hwang S, He C and He YY: m6A
mRNA demethylase FTO regulates melanoma tumorigenicity and response
to anti-PD-1 blockade. Nat Commun. 10:27822019.
|
|
128
|
Tsuruta N, Tsuchihashi K, Ohmura H,
Yamaguchi K, Ito M, Ariyama H, Kusaba H, Akashi K and Baba E: RNA
N6-methyladenosine demethylase FTO regulates PD-L1 expression in
colon cancer cells. Biochem Biophys Res Commun. 530:235–239.
2020.
|
|
129
|
Qiu X, Yang S, Wang S, Wu J, Zheng B, Wang
K, Shen S, Jeong S, Li Z, Zhu Y, et al: M6A demethylase
ALKBH5 regulates PD-L1 expression and tumor immunoenvironment in
intrahepatic cholangiocarcinoma. Cancer Res. 81:4778–4793.
2021.
|
|
130
|
You Y, Wen D, Zeng L, Lu J, Xiao X, Chen
Y, Song H and Liu Z: ALKBH5/MAP3K8 axis regulates PD-L1+ macrophage
infiltration and promotes hepatocellular carcinoma progression. Int
J Biol Sci. 18:5001–5018. 2022.
|
|
131
|
Hu J, Qiu D, Yu A, Hu J, Deng H, Li H, Yi
Z, Chen J and Zu X: YTHDF1 is a potential pan-cancer biomarker for
prognosis and immunotherapy. Front Oncol. 11:6072242021.
|
|
132
|
Han D, Liu J, Chen C, Dong L, Liu Y, Chang
R, Huang X, Liu Y, Wang J, Dougherty U, et al: Anti-tumour immunity
controlled through mRNA m6A methylation and YTHDF1 in
dendritic cells. Nature. 566:270–274. 2019.
|
|
133
|
Tsuchiya K, Yoshimura K, Inoue Y, Iwashita
Y, Yamada H, Kawase A, Watanabe T, Tanahashi M, Ogawa H, Funai K,
et al: YTHDF1 and YTHDF2 are associated with better patient
survival and an inflamed tumor-immune microenvironment in
non-small-cell lung cancer. Oncoimmunology. 10:19626562021.
|
|
134
|
Yang X and Liu J: Targeting PD-L1
(Programmed death-ligand 1) and inhibiting the expression of
IGF2BP2 (Insulin-like growth factor 2 mRNA-binding protein 2)
affect the proliferation and apoptosis of hypopharyngeal carcinoma
cells. Bioengineered. 12:7755–7764. 2021.
|
|
135
|
Yankova E, Blackaby W, Albertella M, Rak
J, De Braekeleer E, Tsagkogeorga G, Pilka ES, Aspris D, Leggate D,
Hendrick AG, et al: Small-molecule inhibition of METTL3 as a
strategy against myeloid leukaemia. Nature. 593:597–601. 2021.
|
|
136
|
Xu QC, Tien YC, Shi YH, Chen S, Zhu YQ,
Huang XT, Huang CS, Zhao W and Yin XY: METTL3 promotes intrahepatic
cholangiocarcinoma progression by regulating IFIT2 expression in an
m6A-YTHDF2-dependent manner. Oncogene. 41:1622–1633. 2022.
|
|
137
|
Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu
H, Ni T, Zhang ZS, Zhang T, Li C, et al: Small-molecule targeting
of oncogenic FTO demethylase in acute myeloid leukemia. Cancer
Cell. 35:677–691.e10. 2019.
|
|
138
|
Su R, Dong L, Li Y, Gao M, Han L,
Wunderlich M, Deng X, Li H, Huang Y, Gao L, et al: Targeting FTO
suppresses cancer stem cell maintenance and immune evasion. Cancer
Cell. 38:79–96.e11. 2020.
|
|
139
|
Su R, Dong L, Li C, Nachtergaele S,
Wunderlich M, Qing Y, Deng X, Wang Y, Weng X, Hu C, et al: R-2HG
exhibits anti-tumor activity by targeting
FTO/m6A/MYC/CEBPA signaling. Cell. 172:90–105.e23.
2018.
|
|
140
|
Fang Z, Mu B, Liu Y, Guo N, Xiong L, Guo
Y, Xia A, Zhang R, Zhang H, Yao R, et al: Discovery of a potent,
selective and cell active inhibitor of m6A demethylase
ALKBH5. Eur J Med Chem. 238:1144462022.
|
|
141
|
Takahashi H, Hase H, Yoshida T, Tashiro J,
Hirade Y, Kitae K and Tsujikawa K: Discovery of two novel ALKBH5
selective inhibitors that exhibit uncompetitive or competitive type
and suppress the growth activity of glioblastoma multiforme. Chem
Biol Drug Des. 100:1–12. 2022.
|
|
142
|
Selberg S, Seli N, Kankuri E and Karelson
M: Rational design of novel anticancer small-molecule RNA m6A
demethylase ALKBH5 inhibitors. ACS Omega. 6:13310–13320. 2021.
|
|
143
|
Mahapatra L, Andruska N, Mao C, Le J and
Shapiro DJ: A novel IMP1 inhibitor, BTYNB, targets c-Myc and
inhibits melanoma and ovarian cancer cell proliferation. Transl
Oncol. 10:818–827. 2017.
|
|
144
|
Müller S, Bley N, Busch B, Glaß M, Lederer
M, Misiak C, Fuchs T, Wedler A, Haase J, Bertoldo JB, et al: The
oncofetal RNA-binding protein IGF2BP1 is a druggable,
post-transcriptional super-enhancer of E2F-driven gene expression
in cancer. Nucleic Acids Res. 48:8576–8590. 2020.
|
|
145
|
Weng H, Huang F, Yu Z, Chen Z, Prince E,
Kang Y, Zhou K, Li W, Hu J, Fu C, et al: The m6A reader
IGF2BP2 regulates glutamine metabolism and represents a therapeutic
target in acute myeloid leukemia. Cancer Cell. 40:1566–1582.e10.
2022.
|














