Introduction
Gliomas are tumors arising within the brain and the
spinal cord, among which glioblastoma (GBM) is widely acknowledged
as the most malignant tumor (1).
Despite significant advances in understanding of GBM tumorigenesis
and treatment over the past few decades, patients with GBM continue
to face dismal outcomes, characterized by high recurrence rates and
rapid disease progression (2).
Therefore, it is imperative to further explore the underlying
mechanisms of GBM pathogenesis to improve patient prognosis.
Therapeutics targeting kinases have shown promise as
an efficacious treatment for a variety of cancers, given the high
correlation between those kinases and the initiation and
progression of certain cancers (3). To date, several kinase inhibitors
have been approved by the U.S. Food and Drug Administration for
clinical use in cancer treatment (4,5).
Polo-like kinases (PLKs) comprise a protein family that selectively
binds to and phosphorylates substrates on specific motifs
recognized by the POLO box domains. The PLK family consists of five
members, among which PLK2 has been implicated as a potential tumor
suppressor. PLK2 downregulation has been observed in several types
of cancers, including breast cancer, GBM, HPV+ head and
neck squamous cell carcinoma, kidney chromophobe, lung
adenocarcinoma, lung squamous cell carcinoma, prostate
adenocarcinoma and uterine corpus endometrial carcinoma. However,
PLK2 upregulation has also been detected in cholangiocarcinoma,
colon adeno-carcinoma, esophageal carcinoma, kidney renal clear
cell, kidney renal papillary cell carcinoma, pheochromocytoma and
paraganglioma, stomach adenocarcinoma and thyroid cancer on
TIMER2.0 (Fig. 1A; http://timer.cistrome.org/). These data indicate
multifaceted roles of PLK2 in different cancers. Furthermore,
recent findings have identified PLK2 as a novel biomarker for the
prognosis of human GBM (6). The
PLK2/Notch axis may be closely linked to the development of
acquired resistance to temozolomide in GBM (7). Consequently, further investigation is
necessary to comprehensively understand the role of PLK2 in GBM
tumorigenesis.
Dual specificity tyrosine-phosphorylation-regulated
kinases (DYRKs) constitute a group of evolutionarily conserved
kinases that induce phosphorylation on tyrosine, serine and
threonine residues. A total of five members have been identified,
including DYRK1A, DYRK1B, DYRK2, DYRK3 and DYRK4. DYRKs are known
to phosphorylate a broad range of proteins involved in diverse
cellular processes (8). Abnormal
expression or activity of DYRK1A has been implicated in the
development of numerous cancers, including B-cell acute
lymphoblastic leukemia, hepatocellular carcinoma and glioma
(9-11). DYRK1A inhibitors have been approved
for the treatment of certain types of cancer, such as metastatic
breast cancer (12). The role of
DYRK1A is complex, and whether DYRK1A employs tumor suppressive or
oncogenic activities is most likely dependent on its specific
substrates. For instance, DYRK1A exerts a tumor-promoting effect by
phosphorylating various transcription factors, including Gli1 and
STAT3 (13,14). Conversely, DYRK1A may maintain its
antitumor effect by activating ASK1 (15). The precise role of DYRK1A in GBM
pathogenesis has yet to be fully elucidated. In the present study,
it was demonstrated that DYRK1A phosphorylates PLK2 at Ser358.
DYRK1A-mediated phosphorylation of PLK2 enhances both protein
stability and kinase activity. Introduction of PLK2 leads to a
significant decrease in glioma cell malignancy, which is further
weakened in the presence of DYRK1A. These results suggested a
potential contribution of DYRK1A-mediated PLK2 phosphorylation to
glioma pathogenesis.
Materials and methods
Dataset acquisition
The transcriptome sequencing data and corresponding
clinical data of primary GBM were procured from The Cancer Genome
Atlas (TCGA) database (https://portal.gdc.cancer.gov/). The present study
utilized the TCGA GBM cohort comprising 169 tumor samples and 5
normal brain samples to analyze differentially expressed genes
(DEGs) using count data. Additionally, PLK2 expression across
cancers was evaluated using the TIMER2.0 website. The GSE68848,
GSE16011 and GSE4290 datasets downloaded from the Gene Expression
Omnibus database (GEO; https://www.ncbi.nlm.nih.gov/geo/) were employed to
further validate the expression of PLK2 (16-18).
Overall survival (OS) information was also acquired from the
datasets for prognostic evaluation. Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) was used
for multiple sequence alignment.
Identification of differentially
expressed genes and function analysis
Differentially expressed genes were identified as
previously described (19). DEGs
between GBM tissues and normal brain tissues were analyzed using
the 'limma', 'edgeR' and 'DESeq2' R packages with the cutoff
criteria of |log2FC|≥1 and P<0.05. The raw count data of the
TCGA GBM cohort were employed as the input for limma, edgeR and
DESeq2. Volcano plots were generated to display DEG distribution
from the three algorithms mentioned above with the 'tinyarray' R
package. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and
Genomes (KEGG) analyses were performed utilizing the
'clusterProfiler' R package to predict the biological functions and
related pathways. Kaplan-Meier analysis to assess the overall
survival of patients was performed with 'survminer' and 'survival'
R packages. Log-rank test was used to compare the survival curves
between the groups.
Cell culture, vectors and
transfection
The human cell lines 293 (cat. no. CRL-1573), 293T
(cat. no. CRL-3216) and U87MG (cat. no. HTB-14) were obtained from
the American Type Culture Collection (ATCC). Of note, U87MG cells
were cells established likely from GBM of unknown origin. U251MG
cells were purchased from Cell Bank Type Culture Collection,
Chinese Academy of Sciences (Xi'an China) as previously described
(20). 293, 293T, U87MG and U251MG
cells were cultured in high glucose DMEM supplemented with 10% FBS,
100 units/ml penicillin and 0.1 mg/ml streptomycin. All cells were
maintained in a 37°C humidified incubator containing 5%
CO2. All experiments were performed using cells within
20 passages after receipt.
For transfection, cells were seeded into cell
culture dishes or plates. When cell confluency reached ~80% within
24 h, plasmids were transfected into cells by Lipofectamine 3000
transfection reagent at room temperature (for six-well plate, 2.5
µg plasmid in total was used in transfection for each well).
All transfections were carried out with Lipofectamine 3000 (cat.
no. L3000001; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions.
Expression plasmids were generated by inserting the
gene of interest into pCMV6-entry and pcDNA3.1+ C-HA. pCMV6-entry
and pcDNA3.1+ C-HA were purchased from OriGene Technologies, Inc.
(cat. no. PS100001) and Addgene, Inc. (plasmid cat. no. 128034),
respectively. DYRK1A (K179R), PLK2S248A, and PLK2S358A
mutant-expressing plasmids generated by inserting specific
sequences into the pCMV6-entry vector were purchased from Charles
River Laboratories, Inc. pCMV6-entry and pcDNA3.1+ C-HA were used
as negative control vectors in transfection.
Cycloheximide (CHX) chase assay
Cycloheximide was purchased from MedChemExpress
(cat. no. HY-12320). A CHX chase assay was performed as previously
described (21). Briefly, 293
cells were seeded in six-well plates one day before transfection.
293 cells were transfected according to the manufacturer's
instructions. A total of 36 h after transfection, cells were
treated with 150 µg/ml CHX and separately harvested at 0, 1,
2, 4, 6, and 8 h for western blotting (WB).
Cell proliferation and viability
assay
Cell Counting Kit-8 (WST-8/CCK-8) (cat. no. C0043;
Beyotime Institute of Biotechnology) was used as a convenient and
robust way of performing a cell viability assay. Briefly, cells
were suspended adequately and seeded into 96-well plates at a
density of 5×103 cells/well with 3 replicates a day
before the cell viability assay was performed as follows: The
supernatant was removed, and 100 µl fresh medium containing
10% CCK-8 reagent was added as a working solution at 0, 24, 48, and
72 h time points. The OD450 and OD650 values of each well were
measured by a microplate reader after the cells were incubated with
working solution for 1.5 h in a 37°C humidified incubator
containing 5% CO2.
Transwell invasion assay
Transwell plates (8-µm diameter pores;
Corning, Inc.) were used to determine the invasion potential of
U87MG and U251MG cells. Briefly, the upper faces of the membranes
were precoated with Matrigel (cat no. 354234; BD Biosciences) at
37°C for 1 h. A total of 5×104 cells were resuspended in
serum-free medium and transferred into the upper chambers in
triplicate. Complete cell culture media were added to the lower
chambers. After 60 h of incubation at 37°C humidified incubator
containing 5% CO2, the media were removed, and the cells
were fixed with 4% paraformaldehyde for 20 min at room temperature.
A 0.1% (w/v) crystal violet solution was used for cell staining.
The upper side of the filter was gently wiped with cotton swabs,
and the chamber was air-dried. Representative images were captured
by inverted microscopy. The total number of cells on ten individual
fields for each membrane was counted; average numbers and standard
deviation of invading cells were calculated.
Wound healing assay
U251MG and U87MG cells were seeded on a six-well
plate and cultured until the cell confluence reached ~90%. Straight
line wounds were created by scratching a cell monolayer with
sterile 100-µl pipette tips. The medium was gently replaced
for the removal of the non-adherent cells generated during
scratching. Cells were then maintained in serum-free media. The
cells migrated slowly to fill the wound area. Images of the wells
were captured at 0 and 48 h, separately. Wound areas were used to
assess the migration rate of the cells. The results were quantified
and analyzed using ImageJ 1.53t software (National Institutes of
Health).
mRNA extraction and reverse
transcription-quantitative (RT-q) PCR
Total RNA was extracted from cells by TRI Reagent
following the manufacturer's instructions (cat. no. T9424;
Sigma-Aldrich; Merck KGaA). Reverse transcription was performed
using the PrimeScript™ RT reagent kit with gDNA eraser (cat no.
RR047A; Takara Bio, Inc.). RT-qPCR assays were performed as
previously described (22). cDNA
was synthesized from 1 µg of total RNA by HiScript III RT
SuperMix (cat. no. R323-01; Vazyme Biotech Co., Ltd.). qPCR was
performed using a LightCycler480II Real-time PCR system [Roche
Diagnostics (Shanghai) Co., Ltd.] with SYBR® Green-based
gene expression analysis. A comparative CT method
(2−ΔΔCq) was used to analyze the gene expression level
as previously described (23). The
primers targeting PLK2 for qPCR were as follows: forward, 5′-CTA
CGC CGC AAA AAT TAT TCC TC-3′ and reverse, 5′-TCT TTG TCC TCG AAG
TAG TGG T-3′. The internal control of qPCR was beta-actin, with the
following primers: forward, 5′-GAC AGG ATG CAG AAG GAG ATT ACT-3′
and reverse, 5′-TGA TCC ACA TCT GCT GGA AGG T-3′.
Colony formation assay
The colony formation assay is an in vitro
cell survival assay that is used to evaluate the ability of a
single cell to grow into a colony. The colony is defined to consist
of at least 50 cells (24). U251MG
cells were counted and seeded at 8×102 cells per six-cm
plate. Media were changed every four days. After two weeks, cell
colonies were grown, and the media were removed. Cells were washed
with PBS three times, fixed with 4% paraformaldehyde for 15 min at
room temperature, and stained with 0.1% crystal violet for 30 min.
The colonies were counted manually under a microscope and images
were captured.
Immunofluorescence
U251MG cells were fixed with 4% paraformaldehyde at
room temperature for 20 min and immunostained with mouse
anti-DYRK1A (cat. no. WH0001859M1; MilliporeSigma) at a dilution of
1:100 and rabbit anti-PLK2 sequentially at a dilution of 1:100
(cat. no. 14812; Cell Signaling Technology, Inc.).
CoraLite488-conjugated goat anti-rabbit IgG (H+L) (SA00013-2) and
CoraLite594-conjugated goat anti-mouse IgG (H+L) (cat. no.
SA00013-3; Proteintech Group, Inc.) were used to index and display
the immunofluorescent signals both at a dilution of 1:200. DAPI (1
µg/ml; Roche Applied Science) was applied in mounting medium
to indicate the nucleus. The images were captured by a fluorescence
confocal microscope (LSM880; Leica Microsystems GmbH). The
association analysis was achieved by ImageJ 1.53t software.
WB, antibodies, and reagents
Cells were harvested and washed with ice-cold PBS
twice. RIPA lysis buffer (Beyotime Institute of Biotechnology)
mixed with a protease inhibitor cocktail mixture was used for cell
lysis. The protein concentration was determined by a Pierce™ BCA
Protein Assay kit. Protein samples were separated by 10 and 12%
glycine SDS-PAGE. PageRuler pre-stained protein ladder (Thermo
Fisher Scientific, Inc.) was used to indicate protein molecular
weights. Proteins were transferred from the gel to nitrocellulose
membranes. The membranes were blocked with 5% non-fat milk (in 1X
Tris-buffered saline with 0.1% Tween®20) at room
temperature for 1 h. The primary antibodies used in the present
study were as follows: anti-FLAG antibody (cat. no. F1804;
MilliporeSigma), HA-Tag antibody (F-7) (cat. no. sc-7392; Santa
Cruz Biotechnology, Inc.), anti-DYRK1A antibody (7D10) (cat. no.
WH0001859M1; MilliporeSigma), anti-GAPDH antibody (cat. no.
60004-1-Ig; Proteintech Group, Inc.), anti-β-actin antibody (cat.
no. A1978; MilliporeSigma), anti-PLK2 antibody (cat. no. 14812;
Cell Signaling Technology, Inc.), p-Ser/Phosphoserine antibody
(cat. no. sc-81514, Santa Cruz Biotechnology, Inc.),
anti-alpha-synuclein antibody (cat. no. 66412-1-Ig; Cell Signaling
Technology, Inc.) and anti-alpha-synuclein (phospho S129) antibody
(cat. no. ab51253; Abcam). Secondary antibodies
IRDye®680RD goat anti-mouse IgG (cat. no. 926-68070),
IRDye®680RD goat anti-rabbit IgG (cat. no. 926-68071),
IRDye®800CW goat anti-mouse IgG (cat. no. 926-32210) and
IRDye®800CW goat anti-rabbit IgG (cat. no. 926-32211)
were all purchased from LI-COR Biosciences. Images were acquired by
directly scanning the nitrocellulose membranes with LI-COR Odyssey
classic imager from LI-COR Biosciences-U.S. The quantification was
performed by ImageJ 1.53t software. Harmine was purchased from
MedChemExpress (cat. no. HY-N0737A).
Coimmunoprecipitation assay
Coimmunoprecipitation assays were performed as
previously described (22).
Briefly, cells were harvested and lysed in WB and IP cell lysis
buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, and 1% Triton
X-100 in the presence of a protease inhibitor mixture (Roche
Applied Science). The cell lysate was centrifuged at 22,000 × g at
4°C for 15 min. The supernatant was carefully retained. Supernatant
containing 100 µg protein was saved and used as input.
Primary antibodies and protein A/G-agarose beads (Santa Cruz
Biotechnology, Inc.) were added to the supernatant and maintained
on a tube rotator at 4°C for 4 h. Mouse IgG (Beyotime Institute of
Biotechnology) was applied as a negative control. Samples were
analyzed by 10% glycine SDS-PAGE.
Lentivirus production and
transduction
The lentivirus vectors were prepared based on the
2nd generation system. Lentiviruses were produced by transfection
of 293T cells with three plasmids together, pLent-EF1a-FH-CMV-Puro
(pLV100008-OE; WZ Biosciences) carrying the gene of interest, pMD2.
G (cat. no. 12259; Addgene, Inc.), and psPAX2 (cat. no. 12260;
Addgene, Inc.) packaging constructs. 293T cells were plated in
100-mm dishes to reach 70-90% confluency by the time of
transfection. The transfection was performed at room temperature
with the vectors of pLent-EF1a-FH-CMV-Puro (10 µg), pMD2. G
(5 µg) and psPAX2 (10 µg) for each 100-mm cell
culture dish. Media were refreshed after 12 h of transfection (10
ml for each 100-mm dish). A total of 48 h after transfection, the
lentivirus-containing supernatant was collected and filtered with
0.45 µm filters to isolate the lentiviral particles for the
following infection. These lentiviruses were introduced into U87MG
and U251MG cell lines on Day 2 of culture at a volume ratio of 1:5.
The cell culture media were replaced with fresh media within 24 h
of infection and incubated for 5 days before further experiments.
Stable clones transduced with PLK2, DYRK1A, shPLK2 and scramble
control were selected for 10 days by puromycin at the concentration
of 2 µg/ml. Lentiviral particles packaging human PLK2 and
DYRK1A are based on the sequences of Q9NYY3 and Q13627-2,
respectively, in the UniProt database (https://www.uniprot.org/). Lentiviral particles
packaging the shRNA targeted PLK2 (5′-TAG TCA AGT GAC GGT GCT G-3′)
and the scramble control (5′-TTC TCC GAA CGT GTC ACG T-3′).
Dephosphorylation assay
Cell lysate samples containing 100 µg protein
were incubated with thermosensitive alkaline phosphatase (AP; cat.
no. EF0651; Thermo Fisher Scientific, Inc.) at 37°C for 30 min.
Then, the sample was placed into a 75°C metal bath for 5 min to
deactivate AP. Samples were analyzed by WB.
Statistical analysis
Data are presented as the mean ± standard deviation
(SD) from three independent experiments. For immu-noblotting, one
representative picture is shown. Quantifications from three
independent experiments are defined with blot density by ImageJ
1.53t software. Differences between two groups are determined by
unpaired Student's t-test. Two-way ANOVA followed by Tukey's post
hoc test was applied for multiple comparisons of the protein level
change at different time point for CHX assay. The data are
evaluated for statistical significance with analysis of variance or
non-parametric analysis by Prism 7 (Dotmatics). P<0.05 was
considered to indicate a statistically significant difference.
Results
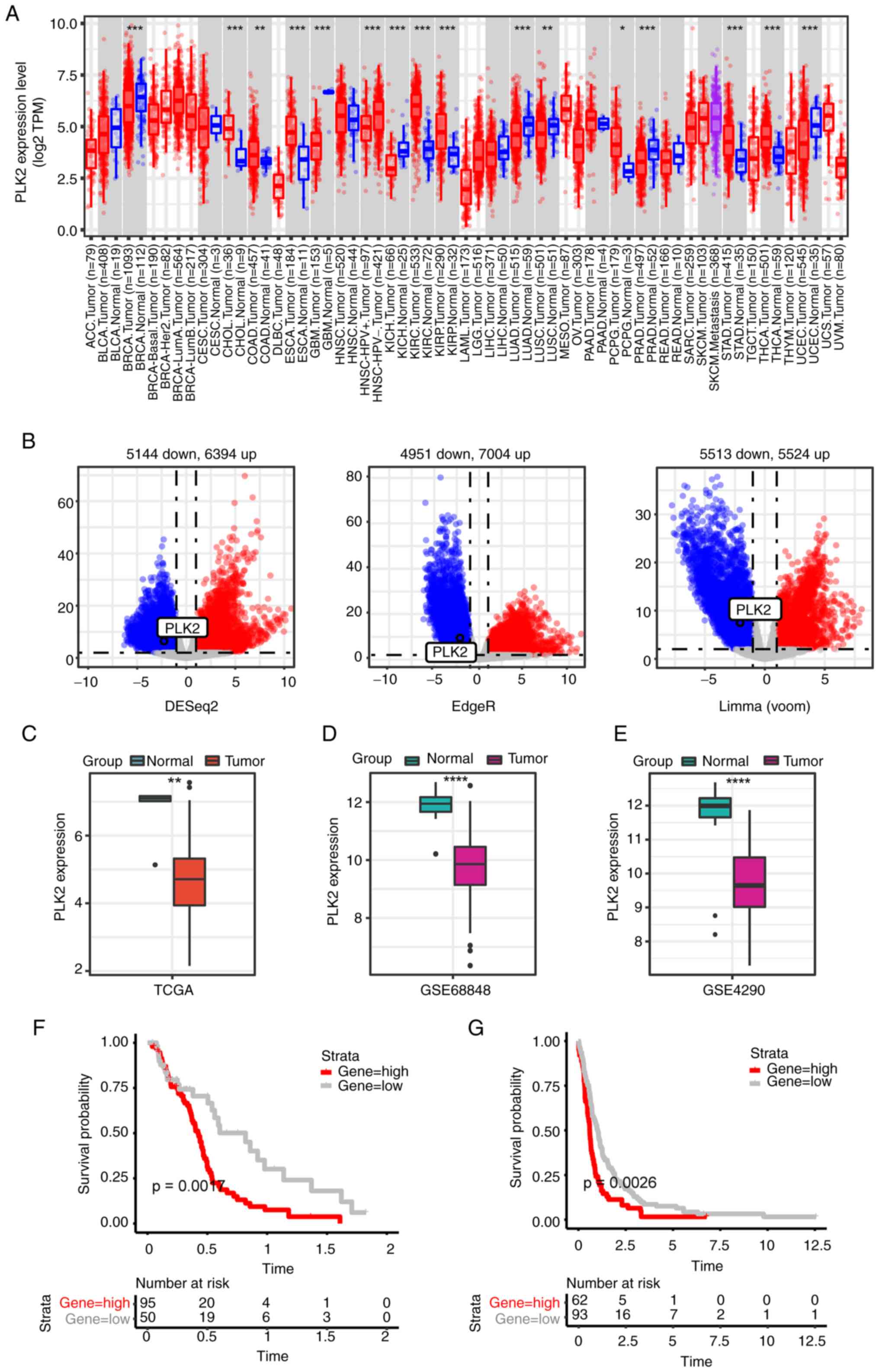
PLK2 is downregulated in GBM and
significantly associated with prognosis
PLK2 exhibits widespread dysregulation across
multiple cancer types. The expression of PLK2 in tumors and normal
tissues was explored on TIMER2.0 (Fig.
1A). To improve understanding of the expression of PLK2 in GBM,
three differential expression analyses were conducted between tumor
tissues and normal brains in the TCGA-GBM cohort. DEGs in the
TCGA-GBM cohort were visualized using Volcano plots (Fig. 1B). As indicated, PLK2 was markedly
downregulated in GBM tissues compared with normal brains (Fig. 1B and C). The downregulation of PLK2
in tumor tissues was further confirmed in the GSE68848 and GSE4290
datasets (Fig. 1D and E,
respectively). In addition, PLK2 expression was considered to be
associated with the OS of GBM patients (6). Function analysis was performed with
common upregulated and downregulated genes (Fig. S1). GO analysis suggested that
features relevant to tumor malignancy are promoted, such as
positive regulation of cell adhesion, focal adhesion and
extracellular matrix structural constituent (Fig. S1A). KEGG analysis revealed
upregulation of classic pathways associated with tumor growth,
including ECM-receptor interaction, cell adhesion molecules, and
transcriptional misregulation in cancer (Fig. S1B). Kaplan-Meier analysis followed
by the log-rank test was performed to assess OS. The OS of patients
evidently exhibited that high expression of PLK2 was strongly
associated with poor prognosis in both the TCGA-GBM cohort and
GSE16011 dataset (Fig. 1F and G).
It should be noted that PLK2 expression is lower in tumor tissues,
but low PLK2 expression predicts favorable prognosis. Due to the
inconsistency of PLK2 expression and prognosis value, pathways and
functions predicted by function analysis may not be markedly
suggestive. This seemingly paradoxical finding suggests that other
unidentified mechanisms may regulate PLK2 in GMB pathogenesis,
warranting further investigation.
DYRK1A modulates PLK2 protein levels in a
kinase activity-dependent manner
PLK2 is mainly involved in cellular biofunctions by
direct phosphorylation of specific substrates. However, the
critical role of phosphorylation in its kinase activity remains
poorly understood (25).
Endogenous PLK2 protein in 293 cells is extremely low and could be
barely detected by WB. Hence, 293 cells were only used for
detection of exogenous PLK2 after transfection. In the present
study, DYRK1A and PLK2 expression vectors were transfected into 293
cells and it was found that overexpression of DYRK1A led to a
significant increase in exogenous PLK2 protein levels by ~5.6-fold
compared with the control, while PLK2 mRNA levels remained
relatively unchanged (Fig. 2A and
B). To confirm that endogenous PLK2 protein is also regulated
by DYRK1A, a DYRK1A-expressing vector was transfected into two GBM
cell lines. The results revealed that PLK2 protein levels were
increased to 180±1.0 and 147±1.3% upon DYRK1A overexpression
compared with the controls in U87MG cells and U251MG cells,
respectively (Fig. 2C and E).
Meanwhile, PLK2 mRNA was relatively unchanged upon DYRK1A
overexpression in both cell lines (Fig. 2D and F). These results suggested
that DYRK1A likely exerts post-translational regulation on PLK2. As
a protein kinase, DYRK1A is commonly involved in cellular processes
by phosphorylation on specific substrates. It was then explored
whether PLK2 is phosphorylated by DYRK1A. As expected, the
phosphorylation and total levels of PLK2 were both upregulated in
the presence of DYRK1A (Fig. 2G).
DYRK1A-mediated PLK2 phosphorylation was mostly eliminated by
treatment with AP (Fig. 2H). A
previous study found that the substituted mutant with Arg in place
of Lys179 (K179R) in DYRK1A disrupts the direct interaction with
ATP. The K179R mutant of DYRK1A is kinase-inactive, and its
autophosphorylation ability is impaired (26). To further validate that the kinase
activity of DYRK1A is essential for PLK2 phosphorylation, the
DYRK1A kinase inactive K179R mutant vector was transfected into
U87MG cells. In contrast to wild-type DYRK1A, the DYRK1A K179R
mutant failed to induce PLK2 protein accumulation (Fig. 2I). These results demonstrated that
DYRK1A increases PLK2 protein levels in a kinase activity-dependent
manner by directly phosphorylating PLK2.
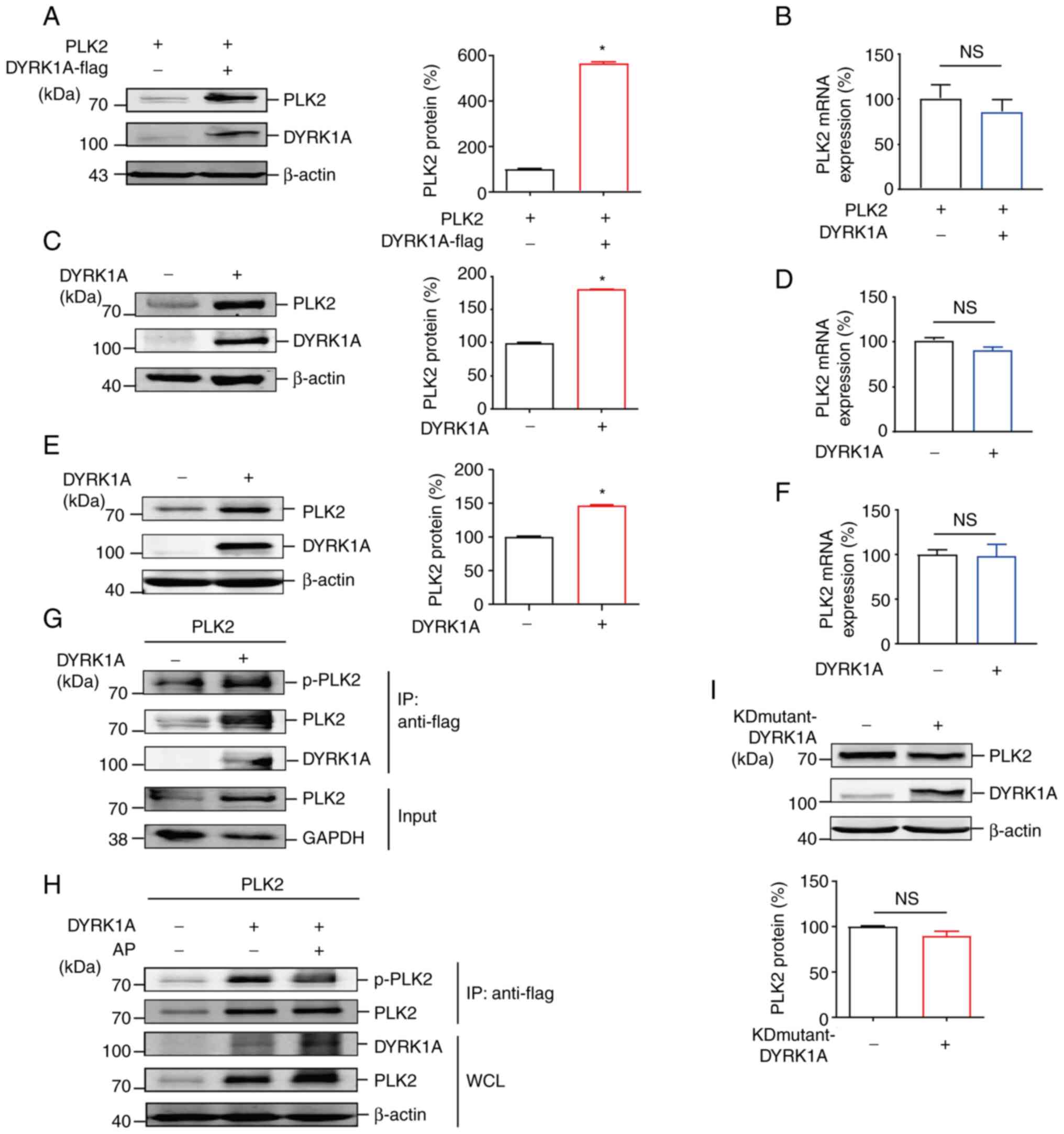 | Figure 2DYRK1A modulates PLK2 protein levels
in a kinase activity-dependent manner. (A) 293 cells were
cotransfected with the PLK2 expression vector together with the
DYRK1A expression vector or the control vector. PLK2 and DYRK1A
were detected by anti-Flag antibody and anti-DYRK1A antibody,
respectively (n=3). (B) Cells and transfection conditions were the
same as in panel A. RNA was isolated 48 h after transfection.
RT-qPCR was performed to examine PLK2 mRNA expression (n=3).
Results were normalized to the control group. (C and E) (C) U87MG
and (E) U251MG cells were transfected with the DYRK1A expression
vector or the control vector. PLK2 and DYRK1A proteins were
examined. (D and F) Cells and transfection conditions were the same
as in panels C and E. PLK2 mRNA expression in (D) U87MG and (F)
U251MG cells was detected by RT-qPCR (n=3). (G)
Co-immunoprecipitation was performed on 293 cells transfected with
PLK2 and DYRK1A/control expression vectors. p-PLK2, PLK2, and
DYRK1A were detected by WB (n=3). (H) A dephosphorylation assay was
performed on 293 cells transfected with PLK2 and DYRK1A expression
vectors. PLK2 was pulled down by immunoprecipitation with an
anti-Flag antibody and blotted with an anti-p-Ser antibody. Cell
lysates were maintained with alkaline phosphatase at 37°C for 30
min for dephosphorylation. p-PLK2, PLK2, and DYRK1A were detected
by WB (n=3). (I) U87MG cells were transfected with a kinase-dead
mutant of DYRK1A (K179R) and the control vector. Endogenous PLK2
and DYRK1A proteins were examined by WB (n=3).
*P<0.05. DYRK1A, dual specificity
tyrosine-phosphorylation-regulated kinase 1A; PLK2, polo-like
kinase 2; RT-qPCR, reverse transcription-quantitative PCR; WB,
western blotting; p-, phosphorylated; NS, not significant. |
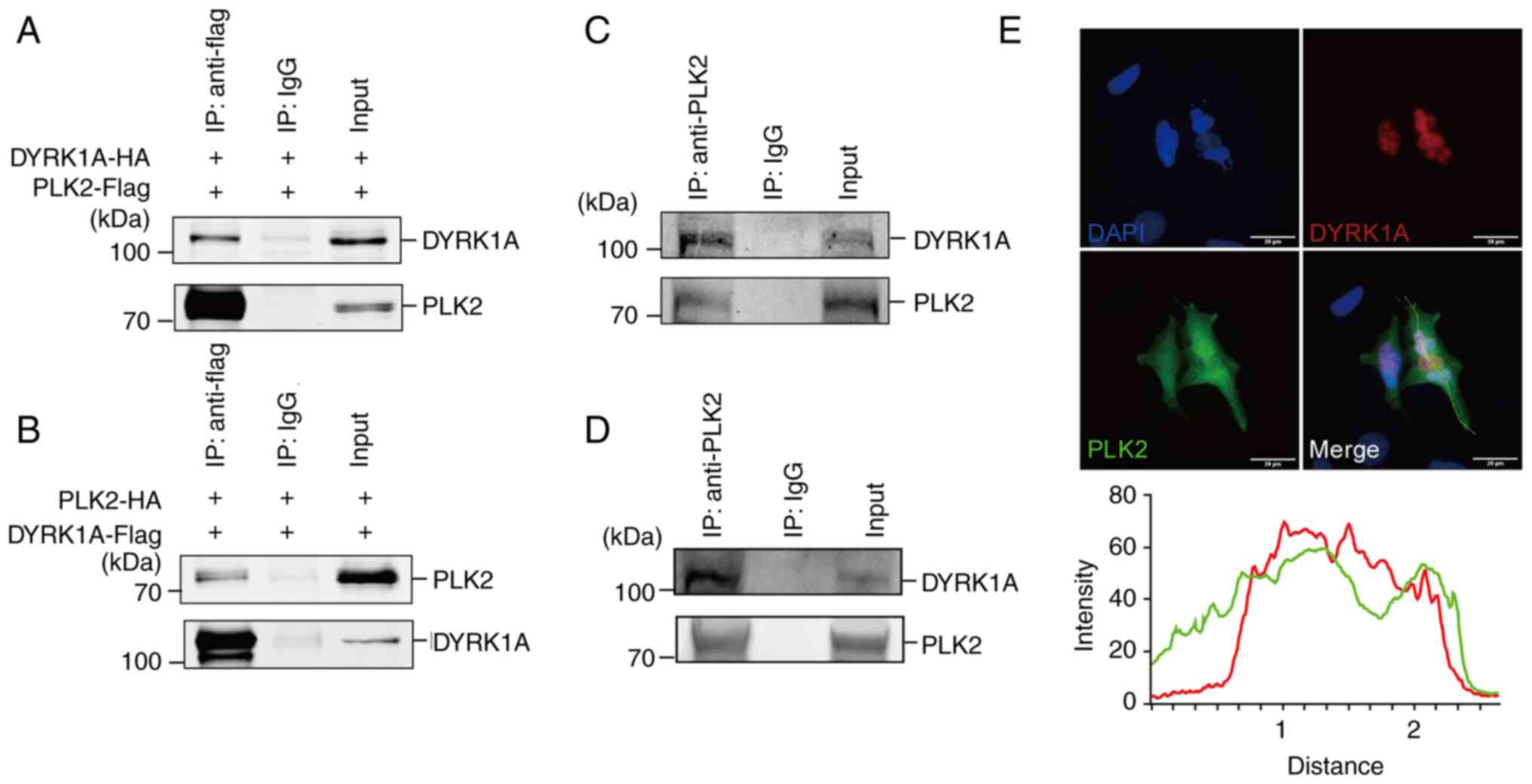
DYRK1A interacts with PLK2 in glioma
cells
To validate whether DYRK1A directly interacts with
PLK2, co-immunoprecipitation was employed. The results demonstrated
that PLK2 can pull down DYRK1A in 293 cells (Fig. 3A). Similarly, DYRK1A was able to
pull down PLK2 as well (Fig. 3B).
To further explore the interaction of endogenous DYRK1A and PLK2,
co-immunoprecipitation was performed in U87MG and U251MG cells. A
significant interaction between DYRK1A and PLK2 was observed in
both cell lines (Fig. 3C and D).
Immunofluorescence was then performed to confirm the intracellular
localization of DYRK1A and PLK2. PLK2 scattered in both the nucleus
and cytoplasm, while DYRK1A mainly stayed in the nucleus (Fig. 3E). Colocalization analysis showed
that they were predominantly colocalized in the U251MG cell
nucleus.
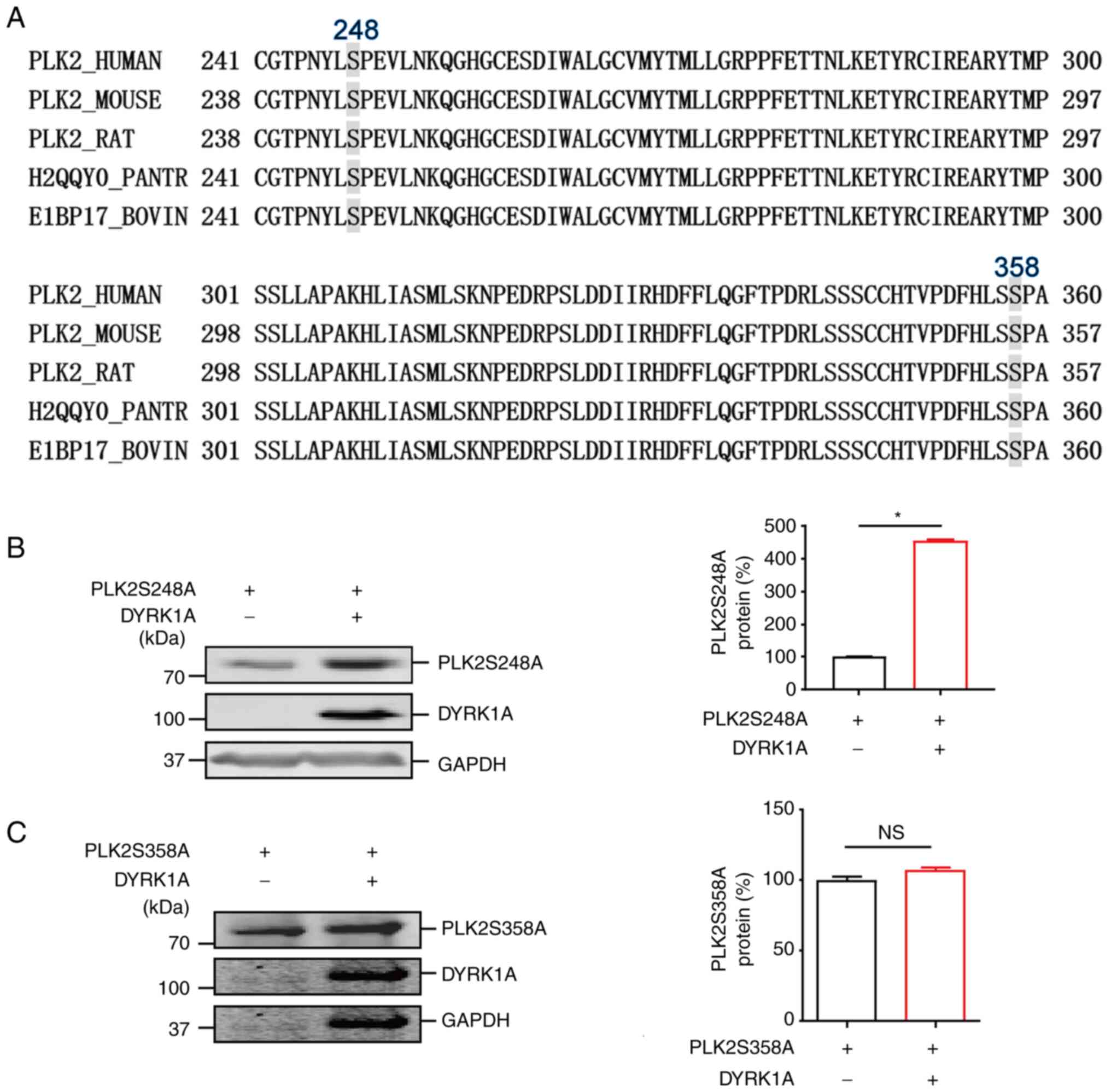
Identification of phosphorylation sites in PLK2 by
DYRK1A. It was previously found that a large proportion of
DYRK1A-recognized substrates contain a consensus RPX(S/T) P motif
(27). To identify the potential
phosphorylation sites on PLK2, sequence alignment with RPX(S/T)P
was performed using the Clustal Omega alignment tool (https://www.ebi.ac.uk/Tools/msa/clustalo/). As shown
in Fig. 4A, two highly conserved
sites, Ser248 and Ser358, were revealed in the PLK2 coding region.
Substituted mutants of PLK2 S248A and S358A were constructed and
employed to further validate whether Ser248 and Ser358 are
phosphorylated by DYRK1A. The results revealed that, consistent
with wild-type PLK2, PLK2 S248A mutant protein was significantly
increased by DYRK1A. However, the PLK2 Ser358A mutant had only a
mild increase upon DYRK1A overexpression (Fig. 4B and C). These results indicated
that Ser358 in PLK2 may be the phosphorylation site induced by
DYRK1A.
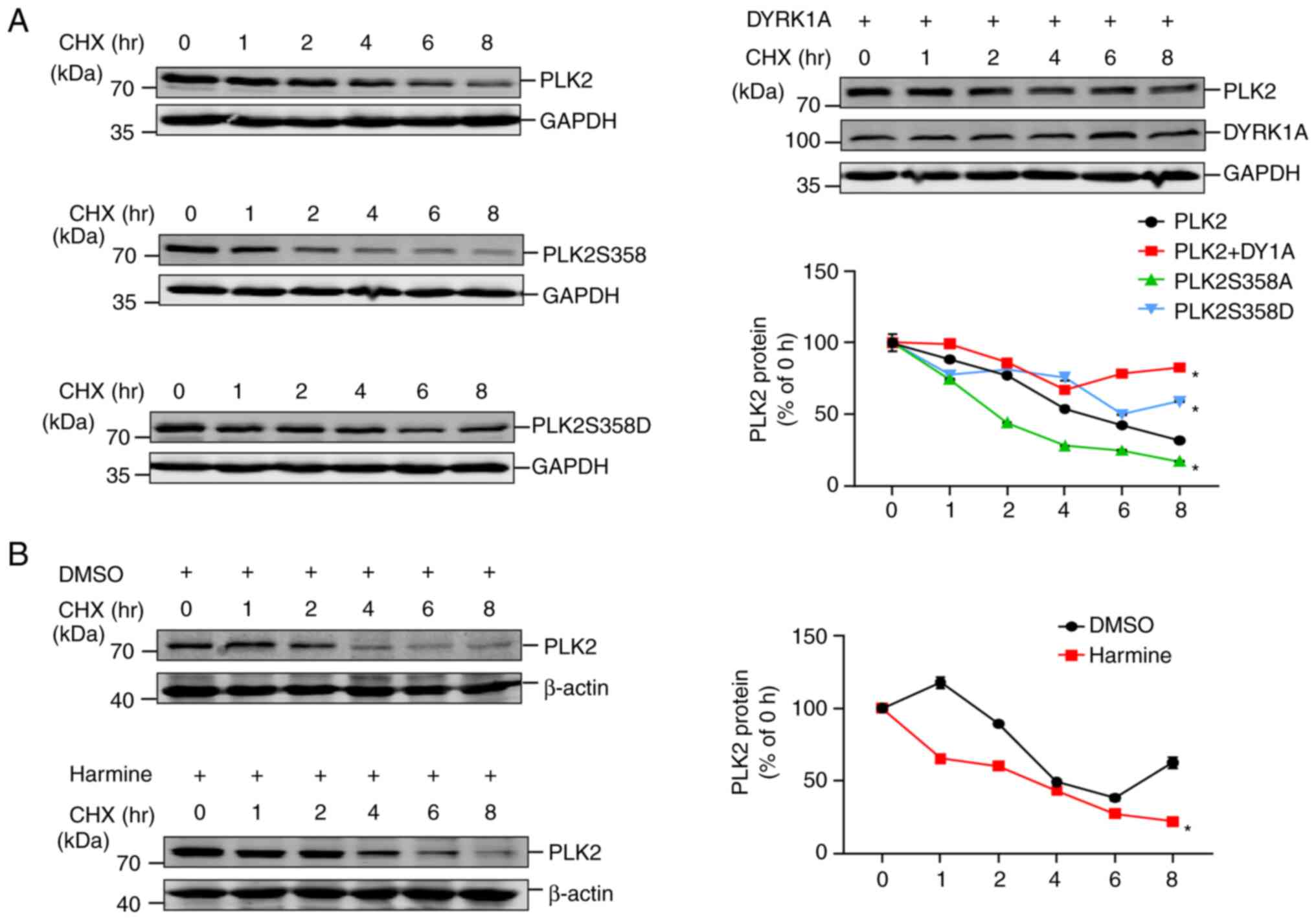
Phosphorylation at Ser358 increases PLK2
protein stability
The impact of phosphorylation on protein stability
is an important regulatory mechanism of post-translational
modifications. To explore whether phosphorylation of PLK2 induced
by DYRK1A affects its protein stability, a CHX assay was conducted.
A previous study revealed that PLK2 is degraded rapidly, with a
half-life of ~15 min (28).
Nevertheless, in the present study it was demonstrated that PLK2
protein is still detectable even after treatment with CHX for 8 h.
Within 8 h, degradation of PLK2 was significantly slower in the
presence of DYRK1A than in the control (Fig. 5A). Similarly, the
phosphorylation-mimicking mutant PLK2S358D also exhibited
decelerated degradation compared with wild-type PLK2. By contrast,
PLK2S358A manifests remarkably accelerated degradation. These data
demonstrated that DYRK1A-mediated PLK2 phosphorylation plays a
crucial role in regulating PLK2 protein stability. Harmine is a
potent and selective natural DYRK inhibitor that is commonly
applied to deactivate DYRK1A kinase activity (29,30).
Harmine was employed to further validate whether DYRK1A kinase
activity is vital for PLK2 protein stability. The results revealed
that treatment with harmine resulted in slower degradation of
endogenous PLK2 compared with DMSO treatment (Fig. 5B). Taken together, the findings of
the present study demonstrated that DYRK1A-mediated phosphorylation
may increase PLK2 protein stability in vitro.
Enhancement of PLK2 kinase activity by
DYRK1A
PLK2 kinase activity is vitally crucial for
substrate phosphorylation. To investigate whether PLK2 kinase
activity was impacted by DYRK1A, PLK2 kinase activity was examined
by detecting α-synuclein Ser129 phosphorylation. α-synuclein is
widely acknowledged as a specific substrate of PLK2. PLK2
overexpression markedly increases phosphorylation of α-synuclein at
Ser129 and promotes abnormal aggregation of α-synuclein (31,32).
Hence, α-synuclein Ser129 phosphorylation could be used as an
indicator of PLK2 kinase activity. Notably, both DYRK1A and PLK2
increased α-synuclein accumulation compared with the control
(Fig. 6A). Consistent with the
previous studies (31,32), PLK2 robustly induces α-synuclein
Ser129 phosphorylation. However, DYRK1A introduction alone was not
able to induce α-synuclein Ser129 phosphorylation (Fig. 6A). Notably, α-synuclein Ser129
phosphorylation was significantly increased in the presence of both
PLK2 and DYRK1A compared with PLK2 alone (Fig. 6B). This result suggested that the
increase in α-synuclein Ser129 phosphorylation may be attributed to
the enhancement of PLK2 activity induced by DYRK1A. Further
analysis using phosphorylation-null mutant PLK2 S358A and
phosphorylation-mimicking mutant PLK2 S358D was applied for
comparison. Compared with wild-type PLK2, PLK2 S358A had
significantly reduced α-synuclein Ser129 phosphorylation.
Conversely, PLK2 S358D significantly elevated α-synuclein Ser129
phosphorylation (Fig. 6C). Taken
together, these data demonstrated that Ser358 of PLK2 is critical
for PLK2 kinase activity and can be modulated by DYRK1A.
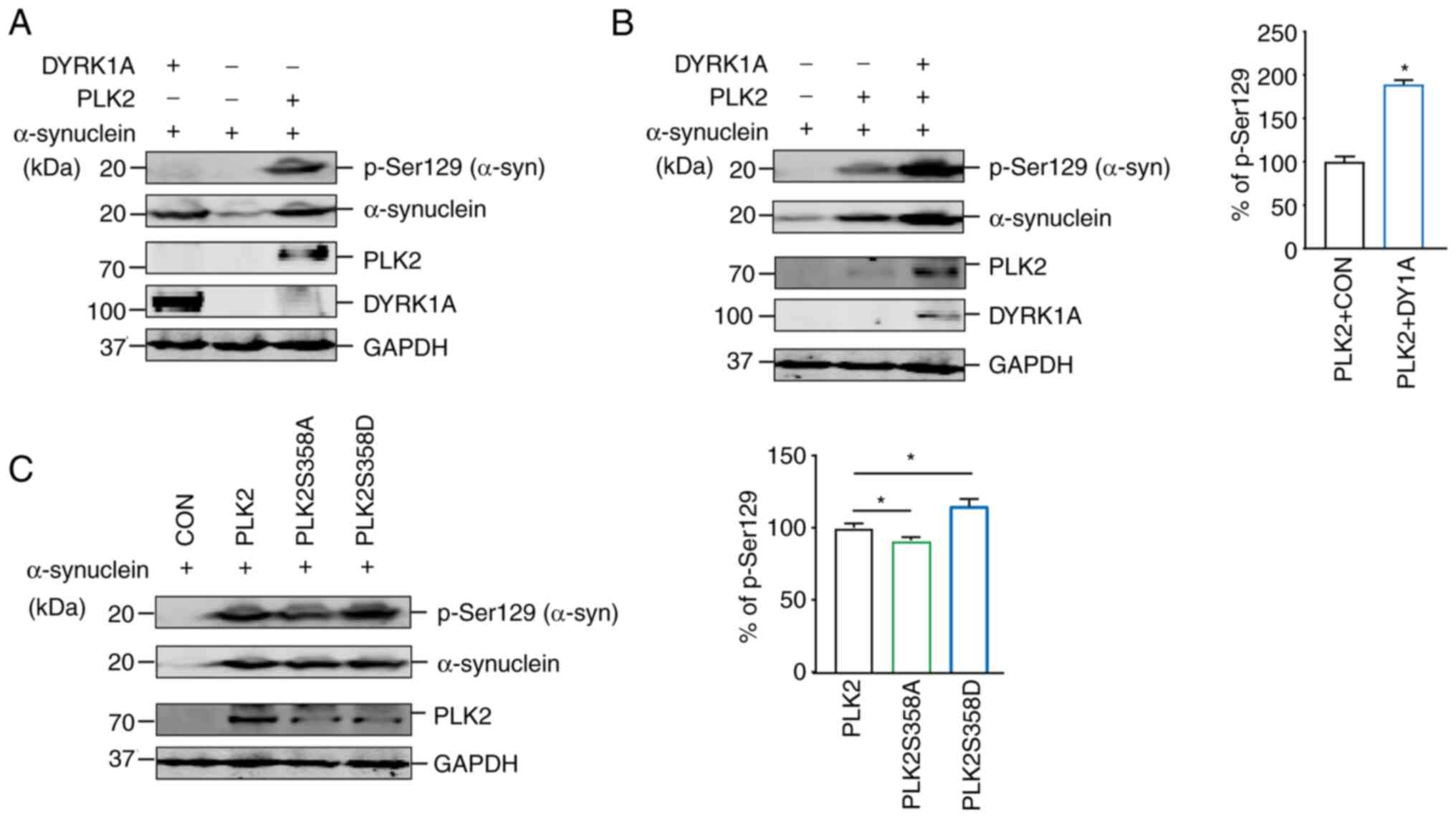 | Figure 6Enhancement of PLK2 kinase activity
by DYRK1A. (A) 293 cells were transfected with α-synuclein
expression vector together with PLK2 or DYRK1A expression vector.
α-synuclein, α-synuclein S129, PLK2 and DYRK1A protein expression
was detected by WB. (n=3). (B) 293 cells were transfected with
α-synuclein expression vector together with PLK2 alone or both PLK2
and DYRK1A expression vectors. α-synuclein, α-synuclein S129, PLK2,
and DYRK1A protein expression was detected by WB (n=3). (C) 293
cells were transfected with α-synuclein expression vector together
with PLK2, PLK2S358A or PLK2S358D expression vectors. α-synuclein,
α-synuclein S129 and PLK2 protein expression were detected by WB
(n=3). *P<0.05. PLK2, polo-like kinase 2; DYRK1A,
dual specificity tyrosine-phosphorylation-regulated kinase 1A; WB,
western blotting. |
DYRK1A-mediated phosphorylation
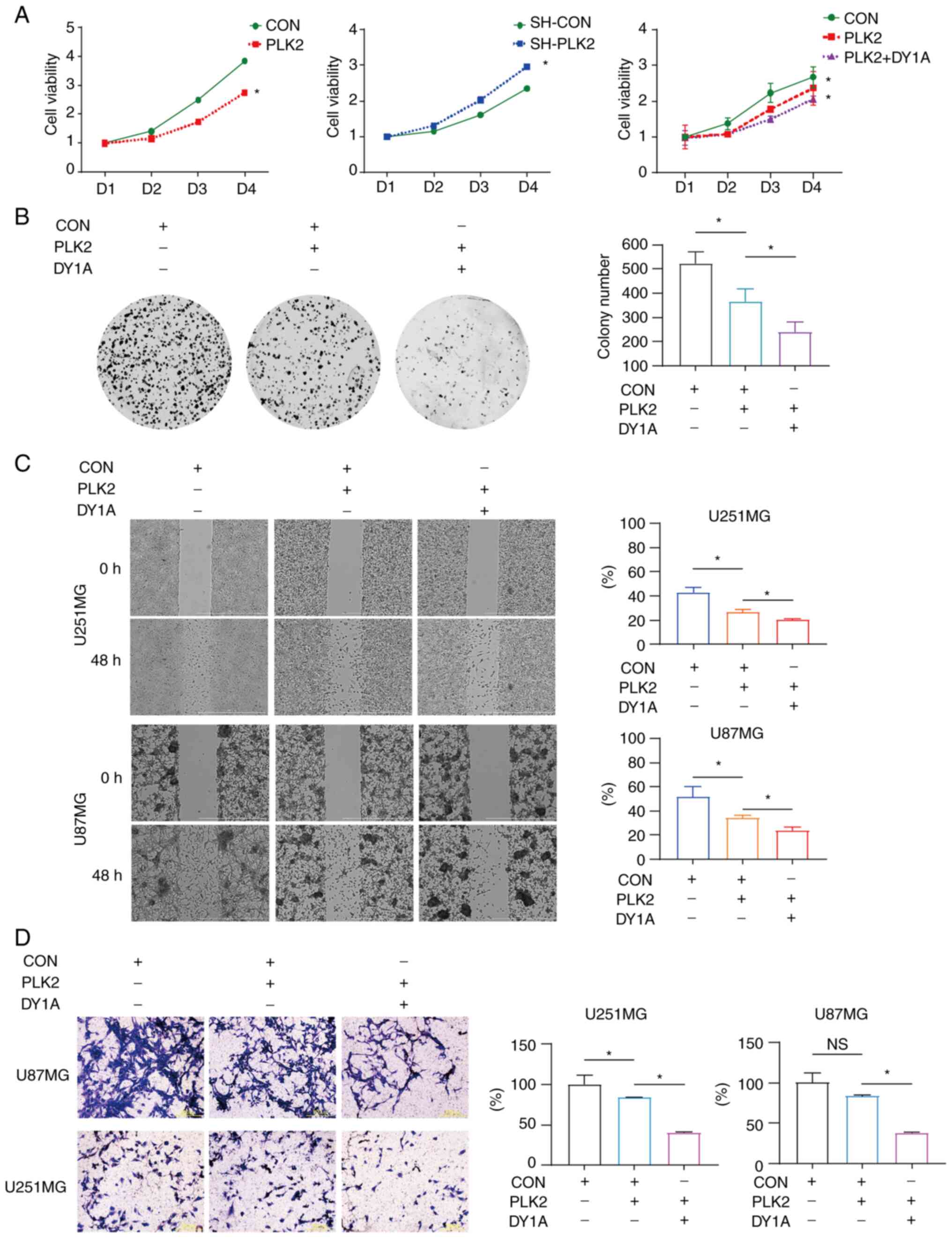
attenuates proliferation and migration/invasion in GBM cells
Previous studies revealed that PLK2 and DYRK1A are
highly correlated with GBM malignancy (7,33).
Whether their interaction contributes to GBM properties has yet to
be uncovered. In the present study, in vitro cell viability
assays as well as colony formation assays were first performed on
U87MG and U251MG stable cells. The results indicated that PLK2
introduction significantly impaired cell viability, while PLK2
silencing had the opposite effect. The introduction of PLK2
together with DYRK1A further suppressed cell viability compared
with introducing PLK2 alone (Fig.
7A). Moreover, PLK2 overexpression significantly decreased cell
self-renewal, which was even weakened in the presence of DYRK1A in
U251MG cells (Fig. 7B). It is
noteworthy that U87MG cells are incapable of growing into colonies
with such few cells for colony formation assay. U87MG cells can
grow colonies only if cell confluency reaches 50% or more.
Migration and invasion are essential processes in GBM progression;
thus, it was then investigated whether the interaction of PLK2 and
DYRK1A affects these processes in GBM cells. The wound healing
assay results demonstrated that the migratory ability of U87MG and
U251MG cells was significantly decreased upon PLK2 overexpression
and further attenuated in the presence of PLK2 together with DYRK1A
(Fig. 7C). Additionally, the
invasion potential of both cell lines was determined by the
Transwell invasion assay. DYRK1A significantly enhanced the
suppression of invasion induced by PLK2 in U87MG and U251MG cells
(Fig. 7D). Collectively, these
data indicated the potential roles of DYRK1A-mediated PLK2
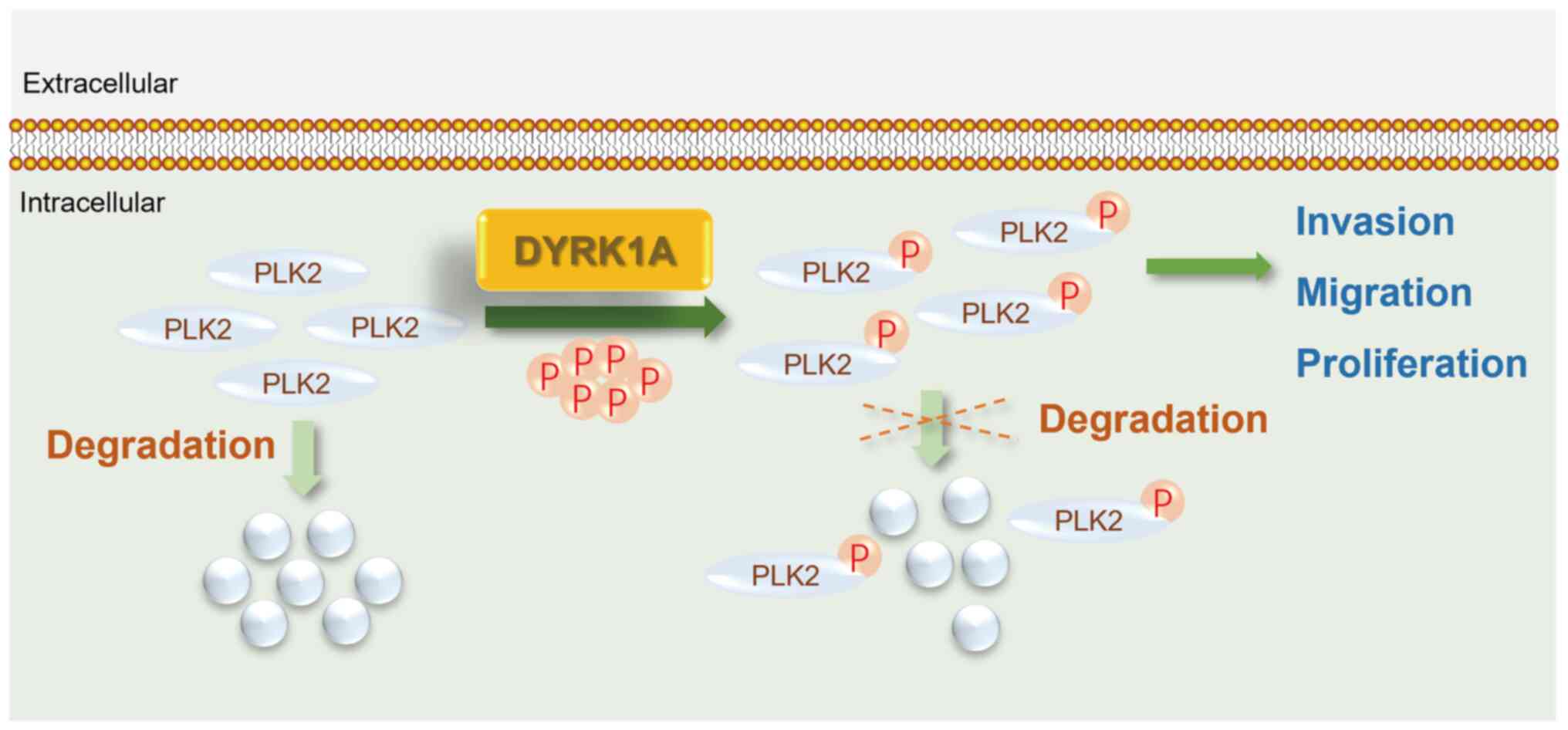
phosphorylation in regulating glioma cell malignancy (Fig. 8).
Discussion
The involvement of PLKs in various cancer types has
been extensively studied, including their potential as therapeutic
targets in GBM, where PLK2 has been identified as a novel
prognostic biomarker (6).
Hypermethylation of PLK2 has been implicated in GBM prognosis
(34). PLK2 commonly serves as a
tumor suppressor, and the expression of PLK2 is frequently lower in
multiple types of cancer, including GBM. Strikingly, it was
identified that a high level of PLK2 was still positively
correlated with poor prognosis. These results indicated that
uncharacterized regulatory mechanisms may be involved.
The tumorigenic role of PLK2 is intricate and
multifaceted. PLK2 dysregulation has been observed in various
cancer types and is considered to play pivotal roles in cancer
pathogenesis. For instance, partial or complete loss of PLK2
expression commonly occurs in colorectal carcinomas and impacts
mTOR signaling (35). Silencing
PLK2 leads to increased cell proliferation and decreased apoptosis
in gastric cancer cells (36).
PLK2 mRNA and protein expression are simultaneously low in
hepatocellular carcinoma and are positively correlated with patient
OS (37). In addition, PLK2 is
hypermethylated in a high percentage of patients with multiple
myeloma and B-cell lymphoma (38).
Of note, PLK2 expression is exceedingly suppressed in GBM samples,
particularly in temozolomide-resistant GBM. Reduced PLK2 expression
enhances temozolomide resistance in GBM by activating Notch
signaling. Meanwhile, upregulation of PLK2 decreased GBM cell
malignancy (7), which is in line
with the present results. In the present study, it was found that
PLK2 could interact with DYRK1A and be phosphorylated by it in
vitro. A previous study showed that four phosphorylation sites,
including Ser497, Ser588, Tyr590 and Ser299, affect PLK2 protein
stability (25). It was observed
that DYRK1A-mediated phosphorylation increased PLK2 protein levels
by decelerating its degradation, further addressing the critical
role of PLK2 phosphorylation in PLK2 protein stability.
Introduction of DYRK1A in the presence of PLK2 further attenuates
proliferation, migration and invasion of GBM cells in vitro,
underscoring the substantial contribution of DYRK1A-mediated PLK2
phosphorylation in GBM cell malignancy.
The functional importance of PLK2 kinase activity
has been extensively studied. PLK2 has been found to phosphorylate
PLK1 at Ser-137, which is sufficient to mediate the survival signal
in colon cancer cells, highlighting the important role of PLK2
kinase activity in cell growth (39). In addition, PLK2 phosphorylates
CPAP at S589 and S595, impacting procentriole formation during the
centrosome cycle (40). In the
central nervous system, PLK2 phosphorylates alpha-synuclein at
Ser129, rendering alpha-synuclein one of the major substrates of
PLK2 (41). In the present study,
it was revealed that the S358A mutant of PLK2 has no effect on
alpha-synuclein Ser129 phosphorylation, whereas the
phospho-mimicking mutant PLK2S358D enhances alpha-synuclein Ser129
phosphorylation. These results demonstrated that the
phosphorylation of Ser358 induced by DYRK1A tightly regulates PLK2
kinase activity.
The role of DYRK1A in the pathogenesis of multiple
types of cancer is controversial. DYRK1A plays a tumor-promoting
role in acute megakaryocytic leukemia in Down's syndrome models
(42), B-cell acute lymphoblastic
leukemia (14), neuroblastoma
(43), pancreatic ductal
adenocarcinoma (44), ovarian
cancer (45), non-small cell lung
cancer (46,47), bladder cancer (48) and head and neck squamous cell
carcinoma (49). Conversely,
DYRK1A exerts a tumor suppressive effect in acute myeloid leukemia
(50). Notably, DYRK1A appears to
act as a double-edge kinase in GBM and osteosarcoma (11,51-54).
In the present study, differential expression of DYRK1A was not
observed in GBM. Nevertheless, an antitumoral capacity of the
DYRK1A/PLK2 axis in regulating the biological processes of GBM
cells was revealed. DYRK1A enhances PLK2-induced cell growth
inhibition by phosphorylating PLK2 at Ser358, a critical site
responsible for kinase activity. DYRK1A-mediated PLK2
phosphorylation attenuates cell migration and invasion. Of note,
introduction of DYRK1A alone may facilitate GBM cell growth (data
not shown). However, it was observed that the co-expression of PLK2
and DYRK1A suppresses tumor cell malignancy. Numerous selective
DYRK inhibitors for cancers, including GBM, have been described in
previous studies (11,51,55,56).
None of the DYRK1A inhibitors have been approved for clinical use
in GBM, although certain of them show promising effects in clinical
studies (51,57). Therefore, the present study
enriched the possible mechanism of DYRK1A in GBM pathogenesis.
Further investigation is required to fully
comprehend the potential therapeutic applications of PLK2.
PLK2-mediated TAp73 phosphorylation prevents TAp73 activity, which
confers an invasive phenotype through activation of POSTN (58,59).
Nevertheless, how the DYRK1A/PLK2/TAp73 axis functions in GBM
remains unknown. In addition, the potential damage to healthy brain
tissue caused by radiation therapy against GBM, such as
inflammation and necrosis, is a major concern. Radiation-induced
necrosis has been reported to affect over 30% of patients with GBM
(60-62). A previous study has shown that
PLK2-mediated phosphorylation and translocation of Nrf2 activates
anti-inflammatory effects via p53/Plk2/p21cip1 signaling
in acute kidney injury (63). Nrf2
is a critical regulatory factor that helps GBM tumors maintain low
immunogenicity and antiapoptotic proliferative phenotypic
characteristics (64). Therefore,
it is worthwhile to investigate whether the DYRK1A/PLK2/Nrf axis is
involved in GBM immunogenicity regulation.
Supplementary Data
Availability of data and materials
All data generated or analyzed during this study are
included in this published article. PLK2 expression in multiple
cancers is available on TIMER2.0 database(http://timer.cistrome.org/).
Authors' contributions
ST and PW conceived and designed the experiments. ST
and JZ performed the experiments. ST and PW performed the
bioinformatics and data analysis. PW reviewed and revised the
manuscript. All authors read and approved the final manuscript. ST
and PW confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank the biological
imaging facility of Shandong University for their support in
immunofluorescence image acquisition and analysis. The authors
would also like to thank Dr Xiulian Sun (Shandong University,
China) for generously providing them with the pCMV6-entry-DYRK1A
vector.
Funding
The present study was supported by the Natural Science
Foundation of Shandong (grant no. ZR2022MH313).
References
|
1
|
Louis DN, Holland EC and Cairncross JG:
Glioma classification: A molecular reappraisal. Am J Pathol.
159:779–786. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Janjua TI, Rewatkar P, Ahmed-Cox A, Saeed
I, Mansfeld FM, Kulshreshtha R, Kumeria T, Ziegler DS, Kavallaris
M, Mazzieri R and Popat A: Frontiers in the treatment of
glioblas-toma: Past, present and emerging. Adv Drug Deliv Rev.
171:108–138. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bhullar KS, Lagarón NO, McGowan EM, Parmar
I, Jha A, Hubbard BP and Rupasinghe HPV: Kinase-targeted cancer
therapies: Progress, challenges and future directions. Mol Cancer.
17:482018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shah NP, Tran C, Lee FY, Chen P, Norris D
and Sawyers CL: Overriding imatinib resistance with a novel ABL
kinase inhibitor. Science. 305:399–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lombardo LJ, Lee FY, Chen P, Norris D,
Barrish JC, Behnia K, Castaneda S, Cornelius LA, Das J, Doweyko AM,
et al: Discovery of
N-(2-chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide
(BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor
activity in preclinical assays. J Med Chem. 47:6658–6661. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ding Y, Liu H, Zhang C, Bao Z and Yu S:
Polo-like kinases as potential targets and PLK2 as a novel
biomarker for the prognosis of human glioblastoma. Aging (Albany
NY). 14:2320–2334. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Alafate W, Xu D, Wu W, Xiang J, Ma X, Xie
W, Bai X, Wang M and Wang J: Loss of PLK2 induces acquired
resistance to temozolomide in GBM via activation of notch
signaling. J Exp Clin Cancer Res. 39:2392020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Boni J, Rubio-Perez C, López-Bigas N,
Fillat C and de la Luna S: The DYRK family of kinases in cancer:
Molecular functions and therapeutic opportunities. Cancers (Basel).
12:21062020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li Y, Xie X, Jie Z, Zhu L, Yang JY, Ko CJ,
Gao T, Jain A, Jung SY, Baran N, et al: DYRK1a mediates
BAFF-induced noncanonical NF-κB activation to promote autoimmunity
and B-cell leukemo-genesis. Blood. 138:2360–2371. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li YL, Zhang MM, Wu LW, Liu YH, Zhang ZY,
Zeng LH, Lin NM and Zhang C: DYRK1A reinforces
epithelial-mesenchymal transition and metastasis of hepatocellular
carcinoma via cooperatively activating STAT3 and SMAD. J Biomed
Sci. 29:342022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Recasens A, Humphrey SJ, Ellis M, Hoque M,
Abbassi RH, Chen B, Longworth M, Needham EJ, James DE, Johns TG, et
al: Global phosphoproteomics reveals DYRK1A regulates CDK1 activity
in glioblastoma cells. Cell Death Discov. 7:812021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kaltheuner IH, Anand K, Moecking J, Düster
R, Wang J, Gray NS and Geyer M: Abemaciclib is a potent inhibitor
of DYRK1A and HIP kinases involved in transcriptional regulation.
Nat Commun. 12:66072021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ehe BK, Lamson DR, Tarpley M, Onyenwoke
RU, Graves LM and Williams KP: Identification of a DYRK1A-mediated
phosphorylation site within the nuclear localization sequence of
the hedgehog transcription factor GLI1. Biochem Biophys Res Commun.
491:767–772. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bhansali RS, Rammohan M, Lee P, Laurent
AP, Wen Q, Suraneni P, Yip BH, Tsai YC, Jenni S, Bornhauser B, et
al: DYRK1A regulates B cell acute lymphoblastic leukemia through
phosphorylation of FOXO1 and STAT3. J Clin Invest. 131:e1359372021.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Choi HK and Chung KC: Dyrk1A positively
stimulates ASK1-JNK signaling pathway during apoptotic cell death.
Exp Neurobiol. 20:35–44. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Madhavan S, Zenklusen JC, Kotliarov Y,
Sahni H, Fine HA and Buetow K: Rembrandt: Helping personalized
medicine become a reality through integrative translational
research. Mol Cancer Res. 7:157–167. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gravendeel LAM, Kouwenhoven MCM, Gevaert
O, de Rooi JJ, Stubbs AP, Duijm JE, Daemen A, Bleeker FE, Bralten
LB, Kloosterhof NK, et al: Intrinsic gene expression profiles of
gliomas are a better predictor of survival than histology. Cancer
Res. 69:9065–9072. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sun L, Hui AM, Su Q, Vortmeyer A,
Kotliarov Y, Pastorino S, Passaniti A, Menon J, Walling J, Bailey
R, et al: Neuronal and glioma-derived stem cell factor induces
angiogenesis within the brain. Cancer Cell. 9:287–300. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tan S, Spear R, Zhao J, Sun X and Wang P:
Comprehensive characterization of a novel E3-related gene signature
with implications in prognosis and immunotherapy of low-grade
gliomas. Front Genet. 13:9050472022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Alafate W, Li X, Zuo J, Zhang H, Xiang J,
Wu W, Xie W, Bai X, Wang M and Wang J: Elevation of CXCL1 indicates
poor prognosis and radioresistance by inducing mesenchymal
transition in glioblastoma. CNS Neurosci Ther. 26:475–485. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu Q, Tang Y, Chen L, Liu N, Lang F, Liu
H, Wang P and Sun X: E3 ligase SCFβTrCP-induced DYRK1A protein
degradation is essential for cell cycle progression in HEK293
cells. J Biol Chem. 291:26399–26409. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang P, Zhao J and Sun X: DYRK1A
phosphorylates MEF2D and decreases its transcriptional activity. J
Cell Mol Med. 25:6082–6093. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Franken NAP, Rodermond HM, Stap J, Haveman
J and van Bree C: Clonogenic assay of cells in vitro. Nat Protoc.
1:2315–2319. 2006. View Article : Google Scholar
|
|
25
|
Rozeboom AM and Pak DTS: Identification
and functional characterization of polo-like kinase 2
autoregulatory sites. Neuroscience. 202:147–157. 2012. View Article : Google Scholar
|
|
26
|
Himpel S, Panzer P, Eirmbter K, Czajkowska
H, Sayed M, Packman LC, Blundell T, Kentrup H, Grötzinger J, Joost
HG and Becker W: Identification of the autophosphorylation sites
and characterization of their effects in the protein kinase DYRK1A.
Biochem J. 359:497–505. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Himpel S, Tegge W, Frank R, Leder S, Joost
HG and Becker W: Specificity determinants of substrate recognition
by the protein kinase DYRK1A. J Biol Chem. 275:2431–2438. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ma S, Liu MA, Yuan YLO and Erikson RL: The
serum-inducible protein kinase Snk is a G1 phase polo-like kinase
that is inhibited by the calcium- and integrin-binding protein CIB.
Mol Cancer Res. 1:376–384. 2003.PubMed/NCBI
|
|
29
|
Göckler N, Jofre G, Papadopoulos C, Soppa
U, Tejedor FJ and Becker W: Harmine specifically inhibits protein
kinase DYRK1A and interferes with neurite formation. FEBS J.
276:6324–6337. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kumar K, Wang P, Sanchez R, Swartz EA,
Stewart AF and DeVita RJ: Development of kinase-selective,
harmine-based DYRK1A inhibitors that induce pancreatic human β-cell
proliferation. J Med Chem. 61:7687–7699. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mbefo MK, Paleologou KE, Boucharaba A,
Oueslati A, Schell H, Fournier M, Olschewski D, Yin G, Zweckstetter
M, Masliah E, et al: Phosphorylation of synucleins by members of
the polo-like kinase family. J Biol Chem. 285:2807–2822. 2010.
View Article : Google Scholar :
|
|
32
|
Waxman EA and Giasson BI: Characterization
of kinases involved in the phosphorylation of aggregated
α-synuclein. J Neurosci Res. 89:231–247. 2011. View Article : Google Scholar
|
|
33
|
Liu H, Sun Q, Chen S, Chen L, Jia W, Zhao
J and Sun X: DYRK1A activates NFATC1 to increase glioblastoma
migration. Cancer Med. 10:6416–6427. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xia X, Cao F, Yuan X, Zhang Q, Chen W, Yu
Y, Xiao H, Han C and Yao S: Low expression or hypermethylation of
PLK2 might predict favorable prognosis for patients with
glioblastoma multiforme. PeerJ. 7:e79742019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Matthew EM, Yang Z, Peri S, Andrake M,
Dunbrack R, Ross E and El-Deiry WS: Plk2 loss commonly occurs in
colorectal carcinomas but not adenomas: Relationship to mTOR
signaling. Neoplasia. 20:244–255. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu LY, Wang W, Zhao LY, Guo B, Yang J,
Zhao XG, Song TS, Huang C and Xu JR: Silencing of polo-like kinase
2 increases cell proliferation and decreases apoptosis in SGC-7901
gastric cancer cells. Mol Med Rep. 11:3033–3038. 2015. View Article : Google Scholar
|
|
37
|
Pellegrino R, Calvisi DF, Ladu S, Ehemann
V, Staniscia T, Evert M, Dombrowski F, Schirmacher P and Longerich
T: Oncogenic and tumor suppressive roles of polo-like kinases in
human hepatocellular carcinoma. Hepatology. 51:857–868.
2010.PubMed/NCBI
|
|
38
|
Benetatos L, Dasoula A, Hatzimichael E,
Syed N, Voukelatou M, Dranitsaris G, Bourantas KL and Crook T:
Polo-like kinase 2 (SNK/PLK2) is a novel epigenetically regulated
gene in acute myeloid leukemia and myelodysplastic syndromes:
Genetic and epigenetic interactions. Ann Hematol. 90:1037–1045.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Matsumoto T, Wang P, Ma W, Sung HJ, Matoba
S and Hwang PM: Polo-like kinases mediate cell survival in
mitochondrial dysfunction. Proc Natl Acad Sci USA. 106:14542–14546.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chang J, Cizmecioglu O, Hoffmann I and
Rhee K: PLK2 phosphorylation is critical for CPAP function in
procentriole formation during the centrosome cycle. EMBO J.
29:2395–2406. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Inglis KJ, Chereau D, Brigham EF, Chiou
SS, Schöbel S, Frigon NL, Yu M, Caccavello RJ, Nelson S, Motter R,
et al: Polo-like kinase 2 (PLK2) phosphorylates alpha-synuclein at
serine 129 in central nervous system. J Biol Chem. 284:2598–2602.
2009. View Article : Google Scholar :
|
|
42
|
Malinge S, Bliss-Moreau M, Kirsammer G,
Diebold L, Chlon T, Gurbuxani S and Crispino JD: Increased dosage
of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic
leukemia in a murine model of down syndrome. J Clin Invest.
122:948–962. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Soppa U, Schumacher J, Florencio Ortiz V,
Pasqualon T, Tejedor FJ and Becker W: The down syndrome-related
protein kinase DYRK1A phosphorylates p27(Kip1) and cyclin D1 and
induces cell cycle exit and neuronal differentiation. Cell Cycle.
13:2084–2100. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Luna J, Boni J, Cuatrecasas M, Bofill-De
Ros X, Núñez-Manchón E, Gironella M, Vaquero EC, Arbones ML, de la
Luna S and Fillat C: DYRK1A modulates c-MET in pancreatic ductal
adenocarcinoma to drive tumour growth. Gut. 68:1465–1476. 2019.
View Article : Google Scholar
|
|
45
|
MacDonald J, Ramos-Valdes Y, Perampalam P,
Litovchick L, DiMattia GE and Dick FA: A systematic analysis of
negative growth control implicates the DREAM complex in cancer cell
dormancy. Mol Cancer Res. 15:371–381. 2017. View Article : Google Scholar
|
|
46
|
Li Y, Zhou D, Xu S, Rao M, Zhang Z, Wu L,
Zhang C and Lin N: DYRK1A suppression restrains Mcl-1 expression
and sensitizes NSCLC cells to Bcl-2 inhibitors. Cancer Biol Med.
17:387–400. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Li YL, Ding K, Hu X, Wu LW, Zhou DM, Rao
MJ, Lin NM and Zhang C: DYRK1A inhibition suppresses STAT3/EGFR/Met
signalling and sensitizes EGFR wild-type NSCLC cells to AZD9291. J
Cell Mol Med. 23:7427–7437. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kottakis F, Polytarchou C, Foltopoulou P,
Sanidas I, Kampranis SC and Tsichlis PN: FGF-2 regulates cell
proliferation, migration, and angiogenesis through an
NDY1/KDM2B-miR-101-EZH2 pathway. Mol Cell. 43:285–298. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Martin CE, Nguyen A, Kang MK, Kim RH, Park
NH and Shin KH: DYRK1A is required for maintenance of cancer
stemness, contributing to tumorigenic potential in
oral/oropharyngeal squamous cell carcinoma. Exp Cell Res.
405:1126562021. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Guard SE, Poss ZC, Ebmeier CC, Pagratis M,
Simpson H, Taatjes DJ and Old WM: The nuclear interactome of DYRK1A
reveals a functional role in DNA damage repair. Sci Rep.
9:65392019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Pozo N, Zahonero C, Fernández P, Liñares
JM, Ayuso A, Hagiwara M, Pérez A, Ricoy JR, Hernández-Laín A,
Sepúlveda JM and Sánchez-Gómez P: Inhibition of DYRK1A destabilizes
EGFR and reduces EGFR-dependent glioblastoma growth. J Clin Invest.
123:2475–2487. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lee SB, Frattini V, Bansal M, Castano AM,
Sherman D, Hutchinson K, Bruce JN, Califano A, Liu G, Cardozo T, et
al: An ID2-dependent mechanism for VHL inactivation in cancer.
Nature. 529:172–177. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Litovchick L, Florens LA, Swanson SK,
Washburn MP and DeCaprio JA: DYRK1A protein kinase promotes
quiescence and senescence through DREAM complex assembly. Genes
Dev. 25:801–813. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Guo X, Williams JG, Schug TT and Li X:
DYRK1A and DYRK3 promote cell survival through phosphorylation and
activation of SIRT1. J Biol Chem. 285:13223–13232. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhang L, Li D and Yu S: Pharmacological
effects of harmine and its derivatives: A review. Arch Pharm Res.
43:1259–1275. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lee Walmsley D, Murray JB, Dokurno P,
Massey AJ, Benwell K, Fiumana A, Foloppe N, Ray S, Smith J,
Surgenor AE, et al: Fragment-derived selective inhibitors of
dual-specificity kinases DYRK1A and DYRK1B. J Med Chem.
64:8971–8991. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhou Q, Reekie TA, Abbassi RH, Venkata DI,
Font JS, Ryan RM, Rendina LM, Munoz L and Kassiou M: Flexible
analogues of azaindole DYRK1A inhibitors elicit cytotoxicity in
glioblastoma cells*. Aust J Chem. 71:789–797. 2018. View Article : Google Scholar
|
|
58
|
Hu ZB, Liao XH, Xu ZY, Yang X, Dong C, Jin
AM and Lu H: PLK2 phosphorylates and inhibits enriched TAp73 in
human osteosarcoma cells. Cancer Med. 5:74–87. 2016. View Article : Google Scholar
|
|
59
|
Landré V, Antonov A, Knight R and Melino
G: p73 promotes glioblastoma cell invasion by directly activating
POSTN (periostin) expression. Oncotarget. 7:11785–11802. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Brandes AA, Tosoni A, Spagnolli F, Frezza
G, Leonardi M, Calbucci F and Franceschi E: Disease progression or
pseudo-progression after concomitant radiochemotherapy treatment:
Pitfalls in neurooncology. Neuro Oncol. 10:361–367. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
DeAngelis LM, Delattre JY and Posner JB:
Radiation-induced dementia in patients cured of brain metastases.
Neurology. 39:789–796. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sheline GE, Wara WM and Smith V:
Therapeutic irradiation and brain injury. Int J Radiat Oncol Biol
Phys. 6:1215–1228. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kim DE, Byeon HE, Kim DH, Kim SG and Yim
H: Plk2-mediated phosphorylation and translocalization of Nrf2
activates anti-inflammation through p53/Plk2/p21cip1
signaling in acute kidney injury. Cell Biol Toxicol. Jul
16–2022.Epub ahead of print.
|
|
64
|
Awuah WA, Toufik AR, Yarlagadda R,
Mikhailova T, Mehta A, Huang H, Kundu M, Lopes L, Benson S, Mykola
L, et al: Exploring the role of Nrf2 signaling in glioblastoma
multiforme. Discov Oncol. 13:942022. View Article : Google Scholar : PubMed/NCBI
|