Introduction
It is predicted that in 2023, >66,200 women will
be diagnosed with endometrial cancer (EC) and >13,030 women will
die of the disease in the United States; this is one of few cancers
for which incidence and mortality is steadily increasing, which is
thought to be due, at least in part, to the worsening obesity
epidemic in the United States (1).
Although most endometrial cancer is detected early and is curable
with surgery, a substantial proportion of patients are diagnosed
with recurrent or metastatic disease (2). Advanced EC has a poor prognosis with
a five-year survival of ~17% and median overall survival from time
of diagnosis of <12 months (3).
Cytotoxic chemotherapy is generally less effective in patients with
advanced and recurrent disease and until recently, few other
treatment options were available (2,4,5). In
fact, prior to the approval of pembrolizumab for the treatment of
recurrent or metastatic mismatch repair deficient EC in 2018, no
new treatments had been approved by the U.S. Food and Drug
Administration for this disease since progestin therapy in the
1970s (4). As tumor genomics has
gained traction, better understanding of the molecular drivers of
solid tumors has opened the door to the use of newer, targeted
therapies in EC (2,6).
A previous genomic analysis reported that the
majority of patients with EC have alterations in the PI3K/AKT/mTOR
pathway, and this was the case for both endometrioid and serous
histologies, as well as for all four EC molecular subtypes in The
Cancer Genome Atlas (POLE ultramutated, microsatellite instability
hypermutated, copy-number low and copy-number high) (7). Mutations which affect the
PI3K/AKT/mTOR pathway cause its activation and allow for unchecked
cell proliferation, migration and survival (7). Clinical trials for recurrent or
advanced ECs have been previously attempted using mTOR inhibitors
both alone and in combination with progestins, selective estrogen
receptor modulators and chemotherapy (6,8).
Unfortunately, these agents exhibited far lower efficacy than
expected, which was thought to be due to lack of target selectivity
and the activation of alternative pathways or feedback loops that
compensated for the mTOR pathway (6,8).
Given this limited success, the use of novel AKT targeting
inhibitors, dual inhibitors of AKT and mTOR, or combinations of
AKT/mTOR inhibitors with other antineoplastic agents such as DNA
repair agents may be an effective treatment strategy for
endometrial cancer.
Ipatasertib (IPAT) is an orally administered direct
inhibitor of all three isoforms of phosphorylated AKT, upstream
from mTOR, and has been reported to show promise in the treatment
of certain solid tumors in pre-clinical and clinical trials
(9-17). Recent studies have reported that
IPAT has anti-proliferative activity and synergy with
chemotherapeutic agents in multiple different types of cancer cell
lines (7,14,18).
In vivo, IPAT has been reported to demonstrate efficacy in
inhibiting tumor growth alone and in combination with certain
chemotherapeutic agents [docetaxel, carboplatin and paclitaxel
(PTX)] in mouse models of breast, gastric and prostate cancer
(7,14,18).
Our previous, recent study demonstrated that IPAT significantly
inhibited cell proliferation, induced cell cycle arrest and
apoptosis, reduced cellular invasion and increased sensitivity to
PTX in uterine serous carcinoma (USC) cells (19).
IPAT has been well-tolerated in phase I and II
trials performed in patients including those with breast, lung,
gastric and prostate cancer, and is currently under investigation
in numerous phase II trials of various solid tumors, including in
endometrial cancer combined with megestrol acetate (9,11-13,15-17).
IPAT in combination with paclitaxel improved progression-free
survival in a phase II trial of triple-negative breast cancer
(20). A phase III trial of
castrate resistant prostate cancer reported improved
progression-free survival (PFS); however, overall survival data is
still maturing (21). Given that
the PI3K/AKT/mTOR alterations are highly prevalent in endometrioid
histology ECs (8), to the present
study investigated the anti-tumorigenic properties of IPAT and its
synergistic anti-proliferative effect with PTX in human
endometrioid EC cell lines, primary cultures of endometrioid EC and
a genetically engineered mouse model of endometrioid EC.
Materials and methods
Cell culture and reagents
The EC, HEC-1A and ECC-1 cell lines, were used in
the present study. HEC-1A cells were maintained in McCoy's 5A
medium with 10% fetal bovine serum (FBS; Thermo Fisher Scientific,
Inc.). ECC-1 cells were cultured in 1640 medium with 5% FBS. Both
cell lines were authenticated annually by Labcorp using short
tandem repeat profiling. Mycoplasma detection kit (Thermo Fisher
Scientific, Inc.) was used to test both cell lines for mycoplasma
every six months. All media were supplemented with 100 U/ml of
penicillin and 100 ug/ml of streptomycin. The cells were cultured
in a humidified 5% CO2 environment at 37°C. IPAT was
donated from Genentech, Inc. PTX was purchased from MedChemExpress.
All antibodies were purchased from Cell Signaling Technology, Inc.
and ABclonal Biotech Co., Ltd. MTT, crystal violet, DMSO and
propidium iodide were purchased from MilliporeSigma.
MTT assay
HEC-1A and ECC-1 cells were plated in 96-well plates
at a concentration of 3×103 and 5×105
cells/well, respectively, and cultured at 37°C for 24 h. The cells
were then treated with a range of concentrations of IPAT and PTX
for 72 h. MTT (5 mg/ml) was added at a dose of 5 µl/well at
the end of treatment. After 1 h of incubation, the MTT reaction was
terminated through the addition of DMSO at 100 µl/well. The
results were calculated by measuring absorption at 575 nm using a
microplate reader (Tecan Group, Ltd.). The effect of IPAT was
calculated as a percentage of control cell growth obtained from
DMSO-treated cells grown in the same 96-well plates.
IC50 values were calculated using an IC50
Calculator (AAT Bioquest, Inc.). Each experiment was performed in
triplicate to assess the consistency of the results.
Colony formation assay
HEC-1A and ECC-1 cells were seeded (1,000
cells/well) in 6 cm culture dishes and cultured overnight, and
medium was then replaced with fresh growth medium containing the
indicated concentrations of IPAT for 72 h. Cells were cultured at
37°C for 14 days with medium changes every three to four days.
Cells were stained with 0.5% crystal violet and colonies were
counted under the microscope.
Cleaved Caspase 3, 8 and assays
HEC-1A and ECC-1 cells were plated in 6-well plates
at a concentration of 3.5×105 and 5×105
cells/well, respectively. After treating with different doses of
IPAT and in combination with PTX for 14 h, 150-180 µl of 1×
caspase lysis buffer was added into each well. The BCA assay
(Thermo Fisher Scientific, Inc.) was used to determine the
concentration of protein. Lysates (10-15 ug) in black clear bottom
96-well plates were incubated with reaction buffer and 200
µM of caspase 3, caspase 8 or caspase 9 substrate (AAT
Bioquest, Inc.) for 30 min. The fluorescence intensity for cleaved
caspase 3 (excitation, 400 nm; emission, 505 nm), cleaved caspase 8
(excitation, 376 nm; emission, 482 nm) and cleaved caspase 9
(excitation, 341 nm; emission 441 nm) was assessed using a Tecan
microplate reader. Assays were performed in triplicate and repeated
three times.
Cell cycle assay
For cell cycle analysis, HEC-1A and ECC-1 cells were
treated with different concentrations of IPAT for 36 h. The cells
were harvested using 0.25% trypsin at 37°C and then fixed in 90%
ice-cold methanol solution in a cold room for 1 h. The cells were
resuspended in RNase A solution for 30 min, followed by staining
with propidium iodide for 10 min at room temperature. All samples
were assessed using a Cellometer (Nexcelom Bioscience LLC) to
identify cell cycle progression. The results were analyzed using
FCS4 express software (Molecular Devices LLC).
Reactive oxygen species (ROS) assay
HEC-1A and ECC-1 cells were plated and grown in
96-well plates at a concentration of 8×103 cells/well
for 24 h. The cells were then treated with a range of doses of IPAT
for an additional 16 h. To measure intracellular ROS production,
cells were exposed to the oxidation sensitive probe
2′,7′-dichlorofluorescin diacetate (DCFH-DA) at 10 µM for 1
h at room temperature. Fluorescence was then quantified at 575 nm
using a microplate reader (Tecan Group, Ltd.) and normalized to
corresponding MTT measurements of the same plate.
Mitochondrial membrane potential
assays
Mitochondrial membrane potential was analyzed using
the specific fluorescent probes JC-1 and tetramethylrhodamine ethyl
ester (TMRE; AAT Bioquest, Inc.). HEC-1A and ECC-1 cells were
plated in 96-well plates at the concentration of 4×103
cells/well for the TMRE assay and 1×104 cells/well for
the JC-1 assay. After 24 h, cells were exposed to a range of doses
of IPAT for 16 h. JC-1 (2 µM) or TMRE (80 µM) were
added into each well for 30 min at 37°C. The fluorescence intensity
was measured for JC-1 (excitation, 480 nm; emission 590 nm) and
TMRE (excitation, 549 nm; emission 575 nm). Each assay was repeated
at least three times.
Western blotting
HEC-1A and ECC-1 cells were plated in 6-well plates
at a concentration of 5×104 cells/well and cultured for
24 h or until 70-80% confluence as assessed using an inverted light
microscope. Cells were then treated with different concentrations
of IPAT at 37°C for different times, as determined for each
experiment. Cell lysates were prepared using RIPA buffer
(MilliporeSigma) and isolated protein solutions were maintained on
ice. The BCA assay was used to determine the protein concentration.
Equal amounts of protein (30 µg/well) were separated by 12%
gel electrophoresis and transferred onto a polyvinylidene
difluoride (PVDF) membrane. The membranes were blocked using a 5%
nonfat milk solution for 1 h at room temperature and then incubated
with different primary antibodies overnight at 4°C. All antibodies
were from Cell Signaling Technology, Inc. and diluted 1:1,000
against phosphorylated (p)-AKT (cat. no. 9271), p-S6 (cat. no.
4858), CDK4 (cat. no. 12790), CDK6 (cat. no. 3136), Cyclin D1 (cat.
no. 2978), Bcl-2 (cat. no. 4223), myeloid cell leukemia-1 (Mcl-1;
cat. no. 5453), Bip (cat. no. 3177), PRKR-like endoplasmic
reticulum kinase (PERK; cat. no. 5683), Calnexin (cat. no. 2679),
protein disulfide isomerase (PDI; cat. no. 3501) and Bcl-xL (cat.
no. 2764). The antibodies against α-tubulin (cat. no. 3873) were
diluted 1:3,000. Membranes were then washed with TBST solution with
0.1% Tween 20 and incubated with a secondary peroxidase-conjugated
antibody for 1 h. Antibody levels were assessed using an enhanced
chemiluminescence detection system on the ChemiDoc System (Bio-Rad
Laboratories, Inc.). After developing for p-AKT or p-S6, the
membranes were stripped and re-probed using pan-AKT (1:1,000; cat.
no. 4685) and pan-S6 (1:1,000; cat. no. 2217) antibodies overnight
at 4°C, and then incubated with peroxidase-conjugated goat
anti-rabbit secondary antibodies (1:4,000; cat. no. 7074) for 1 h
at room temperature. Intensity for each band was measured and
normalized to either β-actin or α-tubulin as an internal control.
Experiments were performed in triplicate.
Lkb1fl/flp53fl/fl
transgenic mouse model of EC
A Lkb1fl/flp53fl/fl
genetically engineered mouse model of EC created previously by our
laboratory, has been previously reported for the study of the
effects of metabolism and targeted therapy on EC tumor growth
(22-24). All mice were handled according to
protocols approved by University of North Carolina at Chapel Hill
(UNC-CH) Institutional Animal Care and Use Committee (approval no.
20-219). In the UNC animal facility, the mice were housed with a 12
h light/dark cycle at 22±2°C and were given free access to food and
water. At six to eight weeks of age,
Lkb1fl/flp53fl/fl mice were injected
with one-sided intrauterine Adenovirus Cre (AdCre, University of
Iowa Transfer Vector Core; titer of
1011-1012) to induce EC in the UNC animal
facility. Mice were anesthetized using intraperitoneal injection of
75 mg/kg ketamine and 1 mg/kg medetomidine. Immediately after
surgery, mice were injected intraperitoneally with antisedan (1
mg/kg) and buprenorphine (0.1 mg/kg) subcutaneously. Buprenorphine
(0.1 mg/kg) was administered subcutaneously to the mice every 8 h
for two consecutive days as an analgesic. Eight weeks after tumor
induction, the mice were divided into four groups and treated with
either IPAT (50 mg/kg, oral gavage, daily), PTX (10 mg/kg,
intraperitoneal, weekly), a combination of IPAT (50 mg/kg, oral
gavage, daily) with PTX (10 mg/kg, intraperitoneal, weekly), or
placebo for 4 weeks. Each group included 25-30 mice. The animals
were weighed weekly throughout the study. After 4 weeks of
treatment, all mice were anaesthetized using carbon dioxide
inhalation (45% volume displacement/min) and once fully
anesthetized, euthanized by cervical. The endometrial tumors were
then collected, measured and weighed. No tumor >1.5 cm in
diameter or 2 g in weight were found. Half of the endometrial tumor
was snap-frozen and stored at -80°C, and the other half was fixed
in 10% neutral-buffered formalin for 24-48 h at room temperature
and paraffin embedded.
Immunohistochemistry (IHC)
After the mouse endometrial tumor tissues were
formalin-fixed for 24-48 h at room temperature. The tumor tissues
were paraffin-embedded at the Animal Histopathology Core Facility
at UNC-CH. Slides (tissue thickness, 5 µm) were first
incubated with protein block solution (Dako; Aglient Technologies,
Inc.) for 1 h at room temperature and then with the primary
antibodies against Ki-67 (1:400; cat. no. 34330; Cell Signaling
Technologies, Inc.), p-S6 (1:500; cat. no. 4858; Cell Signaling
Technologies, Inc.), p-H2AX (1:100; cat. no. AP0687; ABclonal
Biotech Co., Ltd.) and KIF14 (1:150; cat. no. A10275; ABclonal
Biotech Co., Ltd.) for 2 h at 37°C. The slides were then washed
with TBST with 0.1% Tween 20 and incubated with a biotinylated goat
anti-rabbit secondary antibodies (1:250; cat. no. NC9256157, Vector
Laboratories, Inc.) at room temperature for 1 h. Further processing
was performed using ABC-Staining Kits (Vector Laboratories, Inc.)
for 10-15 min at room temperature, and hematoxylin was used for
counterstain for 1 min at room temperature. IHC slides were scanned
using a Motic EasyScan instrument (Motic Instruments) and scored
using ImagePro software (version 2.1.8; Media Cybernetics,
Inc.).
Primary cell cultures of EC
Fourteen tumor specimens were collected from EC
patients undergoing hysterectomy at N.C. Women's Hospital. All
patients provided written informed consent and approval was
obtained from the UNC Institutional Review Board committee
(approval no. 20-3013). Tissues were gently washed with PBS and
then minced using scissors in DMEM/F12 medium (Gibco; Thermo Fisher
Scientific, Inc.). Tissues cells were digested in 0.1% collagenase
IA solution (MilliporeSigma) containing 100 U/ml penicillin and
streptomycin for 0.5 to 1 h at 37°C. The cells were plated in
96-well plates at 8×105 cells/well and 6-well plates at
5×104 cells/well, and treated with IPAT or PTX for
different time periods at 37°C. Cell proliferation was assessed by
MTT assay after 72 h of treatment and western blotting and cleaved
caspase 3 assays were performed after 24 h of treatment, all
according to the aforementioned methods.
Statistical analysis
All measurement data were presented as mean ±
standard deviation. The differences between two groups were
analyzed using an unpaired Student's t-test. Statistical comparison
of multiple groups was performed using one-way ANOVA with Tukey's
multiple comparison test. Linear regression analysis was performed
to analyze the relationship between the expression levels of AKT
and S6 phosphorylation and the sensitivity of EC cells to IPAT.
Statistical analysis was performed using both SAS (version 9.4; SAS
Institute Inc.) and GraphPad Prism (version 8; GraphPad Software;
Dotmatics) statistical software packages. All tests were two-sided
with P<0.05 considered to indicate a statistically significant
difference.
Results
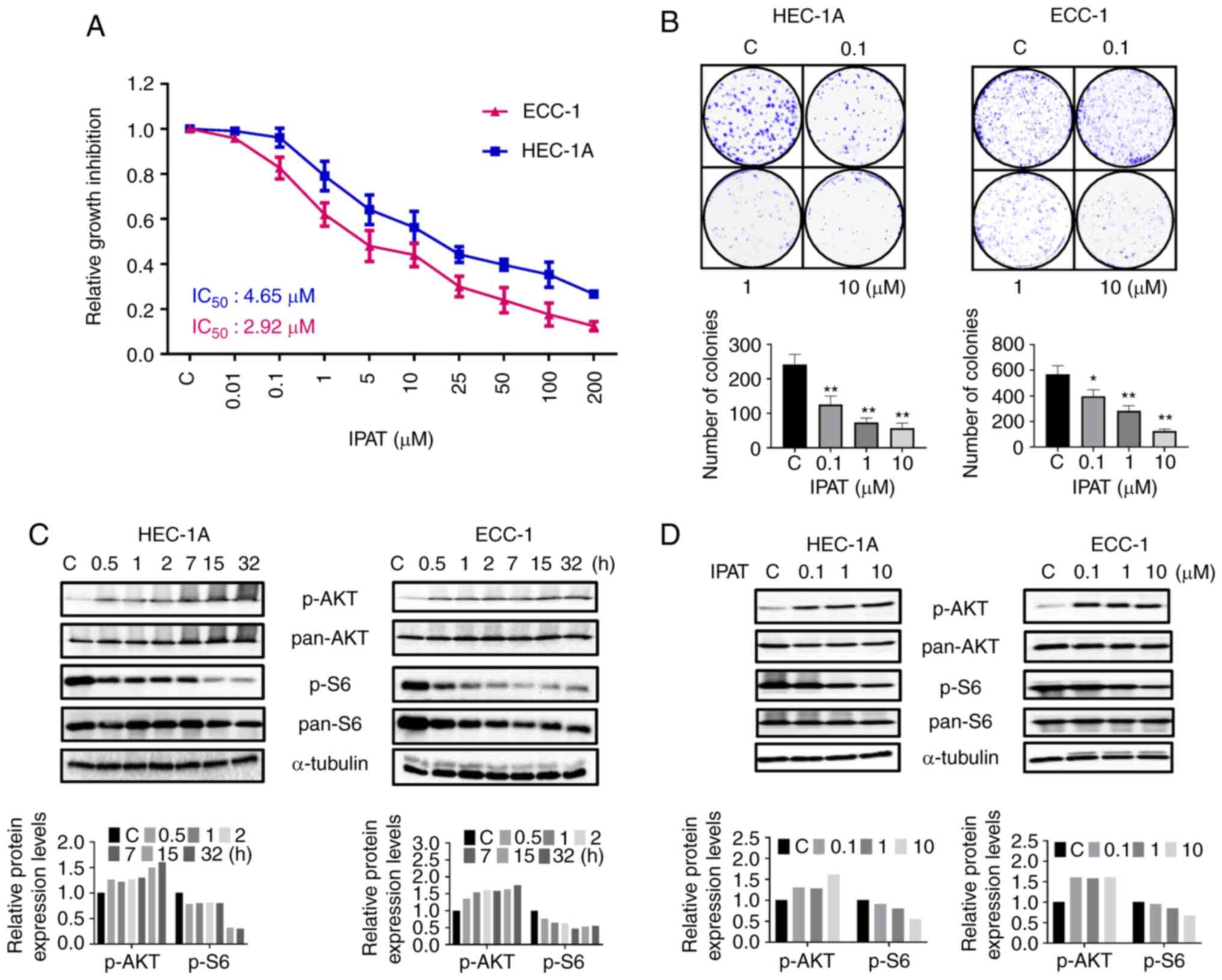
Effect of IPAT on cell proliferation in
EC cells
The HEC-1A and ECC-1 EC cell lines were treated with
IPAT at different concentrations for 72 h. Cell proliferation was
assessed using the MTT assay. IPAT demonstrated a dose dependent
reduction in cell proliferation in both cell lines. The
IC50 dose was 4.65 µM in HEC-1A and 2.92
µM in ECC-1 cells, respectively (Fig. 1A). Colony formation assays were
performed to evaluate the long-term effects of IPAT on cell growth
in these two cell lines. HEC-1A and ECC-1 cells were treated with
0.1, 1 and 10 µM IPAT for 48 h and then the culture media
was changed every 3 days until day 14 of incubation. Colony
formation was significantly inhibited at 10 µM IPAT, in both
cell lines. The colony-forming ability of HEC-1A and ECC-1 was
reduced by 76.34 and 79.92% respectively, after exposure to 10
µM of IPAT for 48 h (Fig.
1B). These results suggested that IPAT has a potent
anti-proliferative activity in EC cells.
The effect of IPAT on the AKT/mTOR signaling pathway
was evaluated using western blotting analysis in the HEC-1A and
ECC-1 cell lines. The results demonstrated that treatment with 5
µM IPAT for 30 min markedly increased the expression of
phosphorylated AKT and markedly decreased the expression of
phosphorylated S6 in both cell lines. Furthermore, these changes
were maintained in a time-dependent manner after increasing the
duration of exposure to at least 30 h (Fig. 1C). Increasing IPAT concentrations
increased the expression of phosphorylated AKT (ser473) in HEC-1A
cells and decreased phosphorylated expression of S6 (ser235/236) in
both cell lines after 24 h of treatment (Fig. 1D). Together, these results
demonstrated that IPAT inhibits cell proliferation via the
AKT/mTOR/S6 signaling pathway in EC cells.
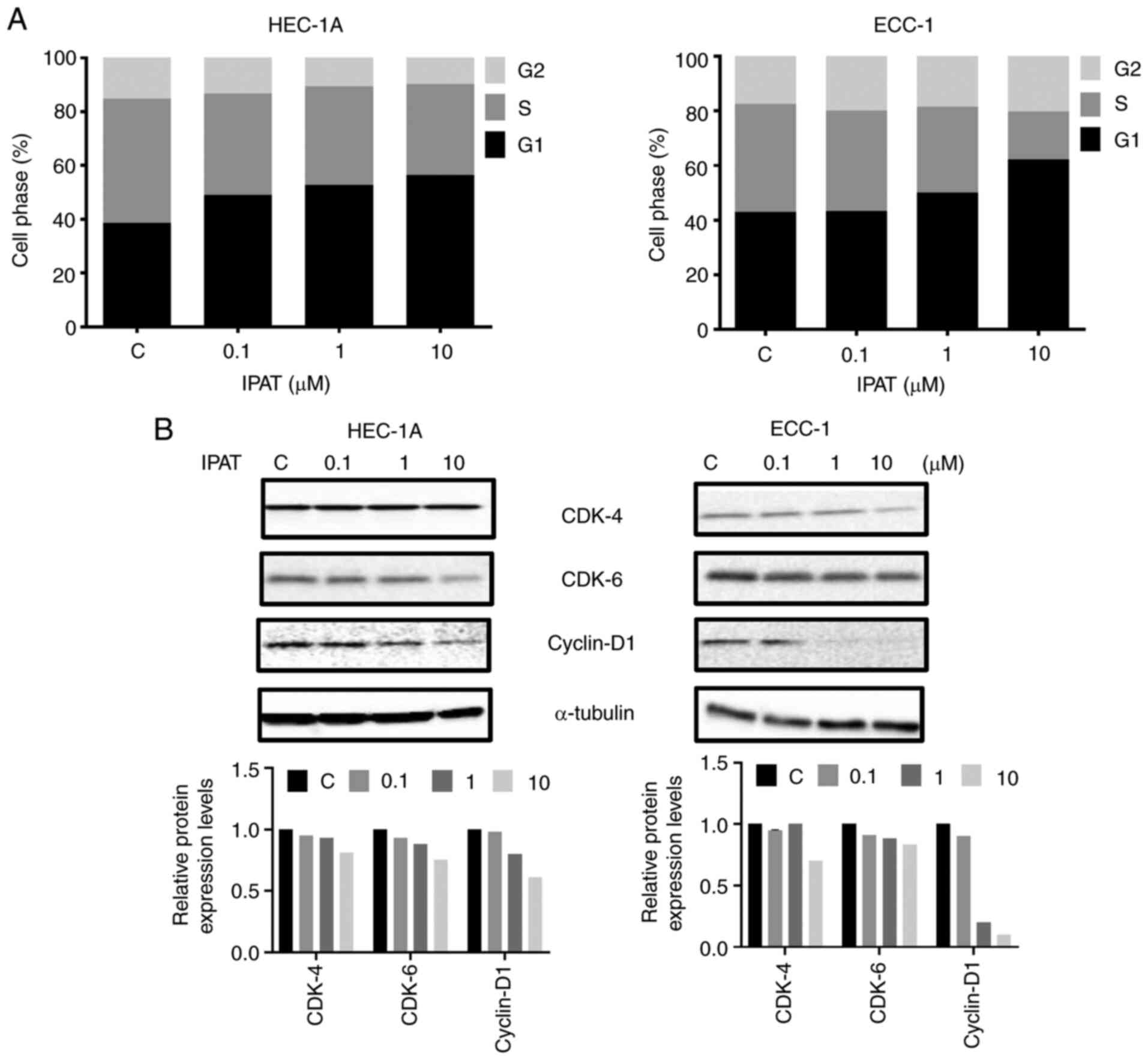
Effect of IPAT on cell cycle progression
in EC cells
To evaluate the effect of IPAT on cell cycle in both
cell lines, the cell cycle profile was analyzed after treating the
HEC-1A and ECC-1 cells with a range of doses (0.1-10 uM) of IPAT
for 36 h. IPAT induced marked G1 phase arrest with an accompanying
decrease in the number of cells in S phase in a dose-dependent
fashion in both cell lines. Treatment of HEC-1A and ECC-1 cells
with 10 µM IPAT markedly increased the G1 phase, by 17.81
and 19.4% respectively, and decreased the S phase by 12.28 and
22.16%, respectively (Fig. 2A).
Western blotting demonstrated that IPAT markedly downregulated the
protein expression levels of cell cycle associated proteins cyclin
D1, CDK-4 and CKD-6 with increased doses of IPAT in both cell lines
after 24 h of treatment (Fig.
2B).
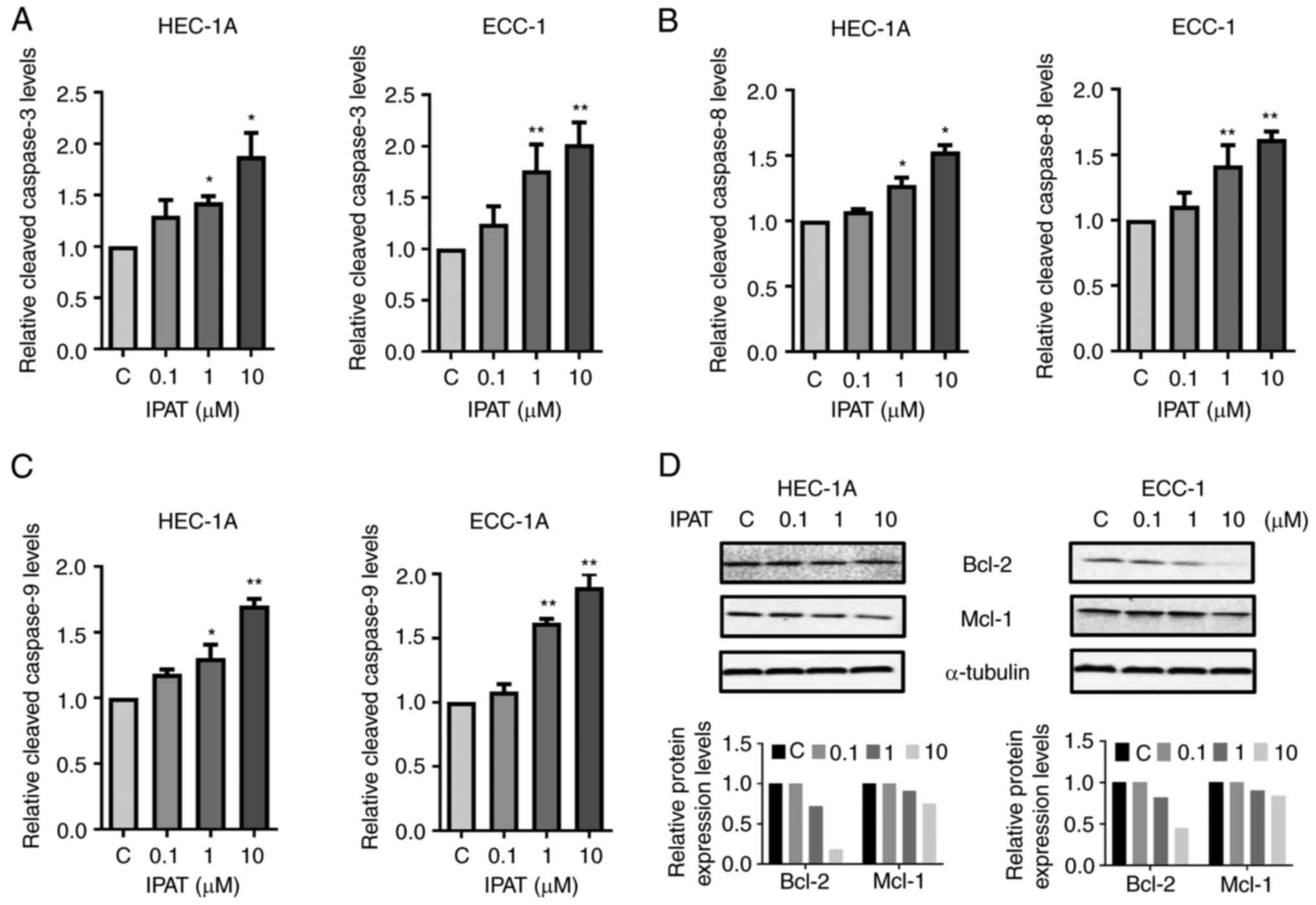
Effect of IPAT on apoptosis in EC
cells
To evaluate whether growth inhibition by IPAT was
related to apoptosis, its apoptotic effect on HEC-1A and ECC-1
cells was evaluated using cleaved caspase 3, 8 and 9 assays. HEC-1A
and ECC-1 cells were treated with varying concentrations of IPAT
for 18 h and a significant dose-dependent increase in the
activities of cleaved caspase-3, 8 and 9 was demonstrated when
compared with controls (Fig.
3A-C). Treatment of ECC-1 cells with 10 µM IPAT
significantly increased the activities of cleaved caspase-3,
cleaved caspase-8 and cleaved caspase-9 by 98.9, 61.8 and 89.8%,
respectively. In the HEC-1A cells, 10 µM IPAT increased the
activities of cleaved caspase 3 by 87.6%, cleaved caspase 8 by
53.5% and cleaved caspase 9 by 69.4%, respectively. Furthermore,
western blotting for the apoptosis-associated proteins Bcl-2 and
Mcl-1 demonstrated a marked dose-dependent decrease with increasing
doses of IPAT after 24 h of treatment (Fig. 3D). These results suggested that the
inhibition of cell growth and the induction of apoptosis by IPAT
depend on both extrinsic and intrinsic apoptotic pathways in EC
cells.
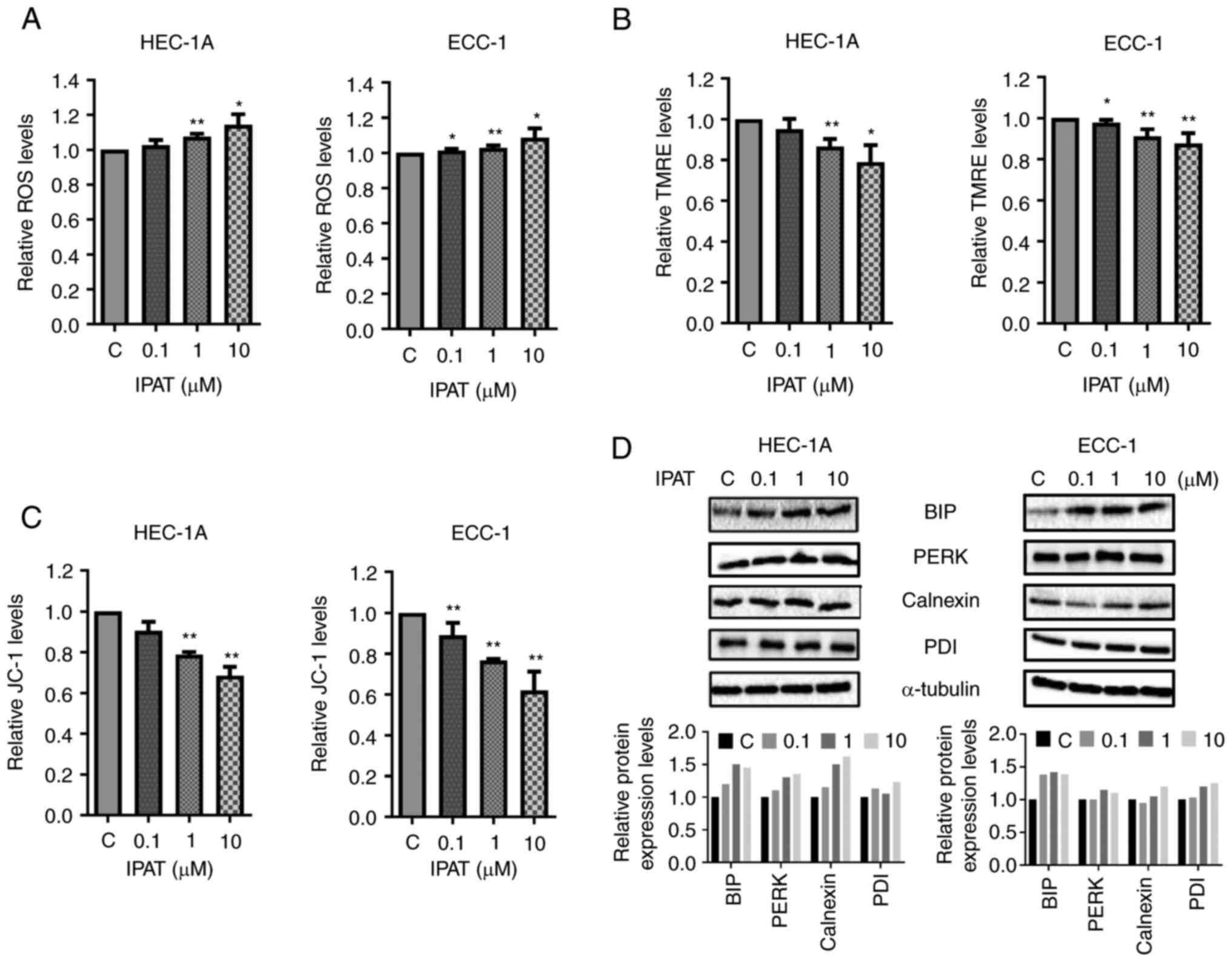
Effect of IPAT on cellular stress in EC
cells
ROS have been implicated in cellular responses to
stress and are involved in the mediation of apoptosis via
mitochondrial DNA damage (25). To
evaluate the involvement of oxidative stress in the
anti-proliferative effect of IPAT in EC cells, intracellular ROS
levels were assessed using the ROS fluorescence indicator DCHF-DA.
Treatment with different doses of IPAT for 16 h significantly
increased cellular ROS production in a dose-dependent fashion in
the HEC-1A and ECC-1 cells (Fig.
4A). At a dose of 10 µM, IPAT significantly increased
ROS production by 21.1 and 23.4% in HEC-1A and ECC-1 cells compared
with control cells, respectively. Changes in markers for
mitochondrial membrane potential were assessed after 16 h of IPAT
treatment in both cell lines. Both TMRE and JC-1 assays
demonstrated that IPAT reduced mitochondrial membrane potential
with increasing doses of IPAT in both cell lines. IPAT (10
µM) decreased JC1 production by 31.5% and TMRE production by
21.1% in HEC-1A cells and decreased JC1 production by 38.1% and
TMRE production by 12.5% in ECC-1 cells (Fig. 4B and C). Western blotting
demonstrated that IPAT induced the expression of PDI, Calnexin, BiP
and PERK which are all cellular stress associated proteins
(Fig. 4D). These results suggested
that an increase in cellular stress may be one mechanism by which
IPAT inhibits cellular proliferation in EC cells.
IPAT demonstrated synergistic activity
with paclitaxel in EC cells
As the combination of IPAT and PTX was previously
reported to synergistically reduce cell proliferation compared to
either agent alone in uterine serous carcinoma cells (19), the present study evaluated whether
IPAT could increase the sensitivity of EC cell lines to PTX. PTX
effectively inhibited cell proliferation in a dose dependent manner
in both cell lines after 72 h of treatment, with an IC50
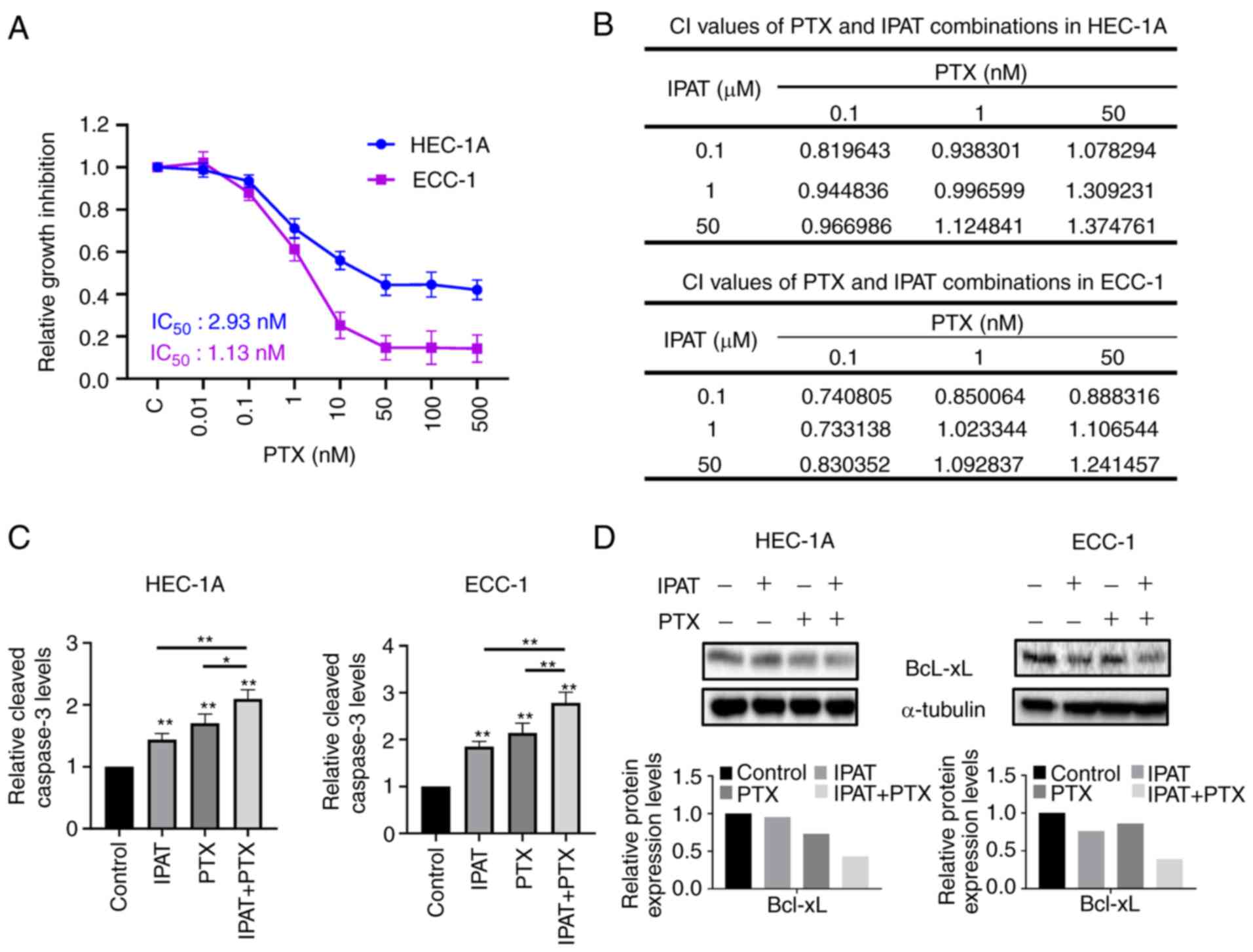
of 2.93 nM in HEC-1A and 1.13 nM in ECC-1 cells (Fig. 5A). Depending on the effect of IPAT
or PTX on cell proliferation, both cell lines were treated with
three different doses of IPAT alone, PTX alone or the combination
for 72 h. MTT results demonstrated that the combination of low dose
IPAT with PTX produced synergistic activity (combination index
<1) compared with monotherapy alone in both cell lines (Fig. 5B). Apoptosis in the cell lines was
then assessed, in the presence of either agent alone or in
combination, using the ELISA cleaved caspase-3 assay. HEC-1A and
ECC-1 cells were treated with IPAT, PTX and the combination of IPAT
and PTX for 16 h. Cleaved caspase-3 activity was significantly
increased in the combination treatment compared with either agent
alone (Fig. 5C). IPAT (1
µM) increased caspase-3 activity by 84.4 and 43.6%, and 1 nM
PTX enhanced caspase-3 activity by 114 and 71.2% in the ECC-1 and
HEC-1AA cells, respectively. The combination of 1 µM IPAT
and 1 nM PTX was demonstrated to be more potent than each either
agent alone in inducing cleaved caspase-3 activity, with a
significant 178.2 and 96.6% increase in ECC-1 and HEC-1A cells,
respectively. Furthermore, results from the western blotting assay
demonstrated that the combination 1 µM IPAT and 1 nM PTX had
a greater ability to reduce the expression of Bcl-xL in HEC-1A and
ECC-1 cells compared with control, PTX or IPAT alone (Fig. 5D). These results indicated that the
synergistic effect of the combination treatment was due in part to
effects on apoptotic pathways in EC cells.
IPAT inhibited tumor growth and increased
sensitivity to PTX in vivo
Given that IPAT significantly inhibited EC cell
proliferation and induced apoptosis in vitro, the effect of
IPAT, PTX and the combination of IPAT and PTX on tumor growth in
the Lkb1fl/flp53fl/fl mouse model of
EC was evaluated. The mice were divided into four groups (25-30
mice per group) and treated with IPAT (50 mg/kg, oral gavage,
daily), PTX (10 mg/kg, intraperitoneal, weekly), the combination of
IPAT and PTX, or vehicle for 4 weeks, starting 10 weeks after tumor
induction with AdCre. During treatment, the daily oral dose of IPAT
was well tolerated without significant toxicity or body weight
changes compared with control mice (data not shown). Tumor weights
were significantly reduced by 52.2% in the IPAT group compared
placebo after 4 weeks of treatment. PTX demonstrated similar
anti-tumor activity compared with IPAT, and the combination of IPAT
and PTX effectively inhibited tumor growth and exhibited
significantly increased anti-tumor efficacy compared with IPAT
alone or PTX alone (Fig. 6A).
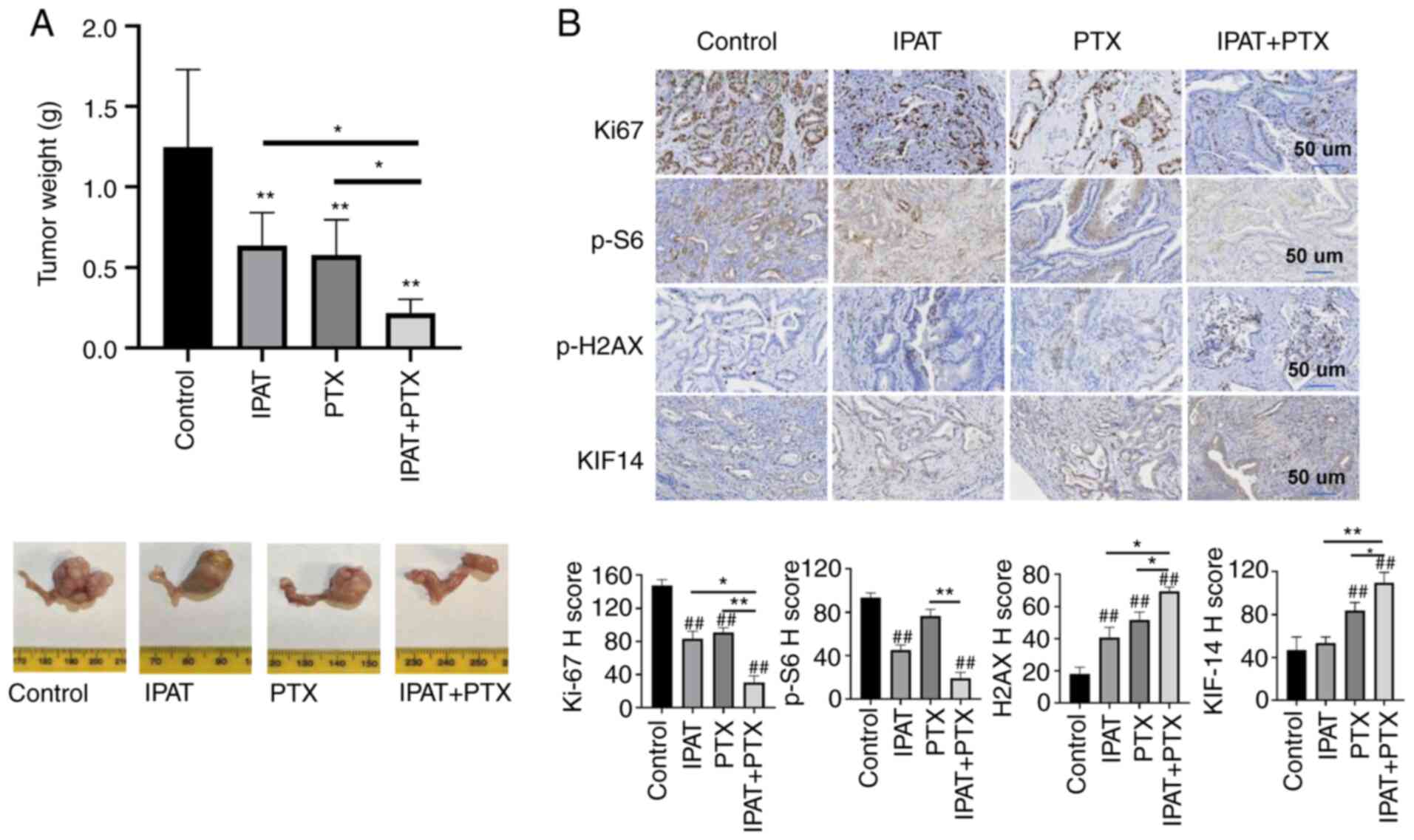 | Figure 6Effect of IPAT and PTX on tumor
growth in Lkb1fl/flp53fl/fl mice.
Lkb1fl/flp53fl/fl mice were divided
into four groups: control, IPAT, PTX and IPAT + PTX. The mice were
treated with IPAT (50 mg/kg, oral gavage, daily), PTX (10 mg/kg,
intraperitoneal, weekly), the combination and vehicle for 4 weeks.
(A) Average tumor weights from each group at the end of treatment
were compared and analyzed. IPAT or PTX significantly inhibited
tumor growth, and the combination treatment synergistically further
inhibited tumor growth in the mice. (B) The expression of Ki67,
p-S6, p-H2AX and KI14 was assessed using immunohistochemical
staining in the EC tissues. IHC results were scored by multiplying
the percentage of positive cells (P) by the intensity (I).
*P<0.05 and **P<0.01.
##P<0.01 vs. control. IPAT, ipatasertib; PTX,
paclitaxel. |
To explore the mechanism of action underlying
synergistic anti-tumor effects of IPAT in combination with PTX, the
expression of Ki67, p-S6, p-H2AX and KIF-14 in the endometrial
cancer tissues from Lkb1fl/flp53fl/fl
mouse model was assessed using immunohistochemistry. IPAT treatment
significantly inhibited the expression of p-S6, and the protein
expression level of Ki67 was significantly reduced in IPAT, PTX and
the combination groups compared with the control group (Fig. 6B). Furthermore, EC tissues from
each treatment group and the control group were stained for the DNA
damage marker p-H2AX and the microtubule motor protein KIF-14. The
protein expression levels of p-H2AX and KIF14 in PTX and PTX + IPAT
groups were significantly higher compared with those in the control
and IPAT groups. Compared with the other groups, the combination of
IPAT and PTX had the greatest effect on the protein expression
levels of Ki67, p-S6, p-H2AX and KIF-14. These results suggested
that IPAT increased sensitivity to PTX through DNA damage pathways
in EC in vivo.
Effect of IPAT and PTX on cell
proliferation in primary cultures of human EC
Given that primary cell cultures from human tumors
have the potential capacity to predict the sensitivity of cytotoxic
agents, which established cancer cell lines are not able to do
(26), the effect of IPAT on cell
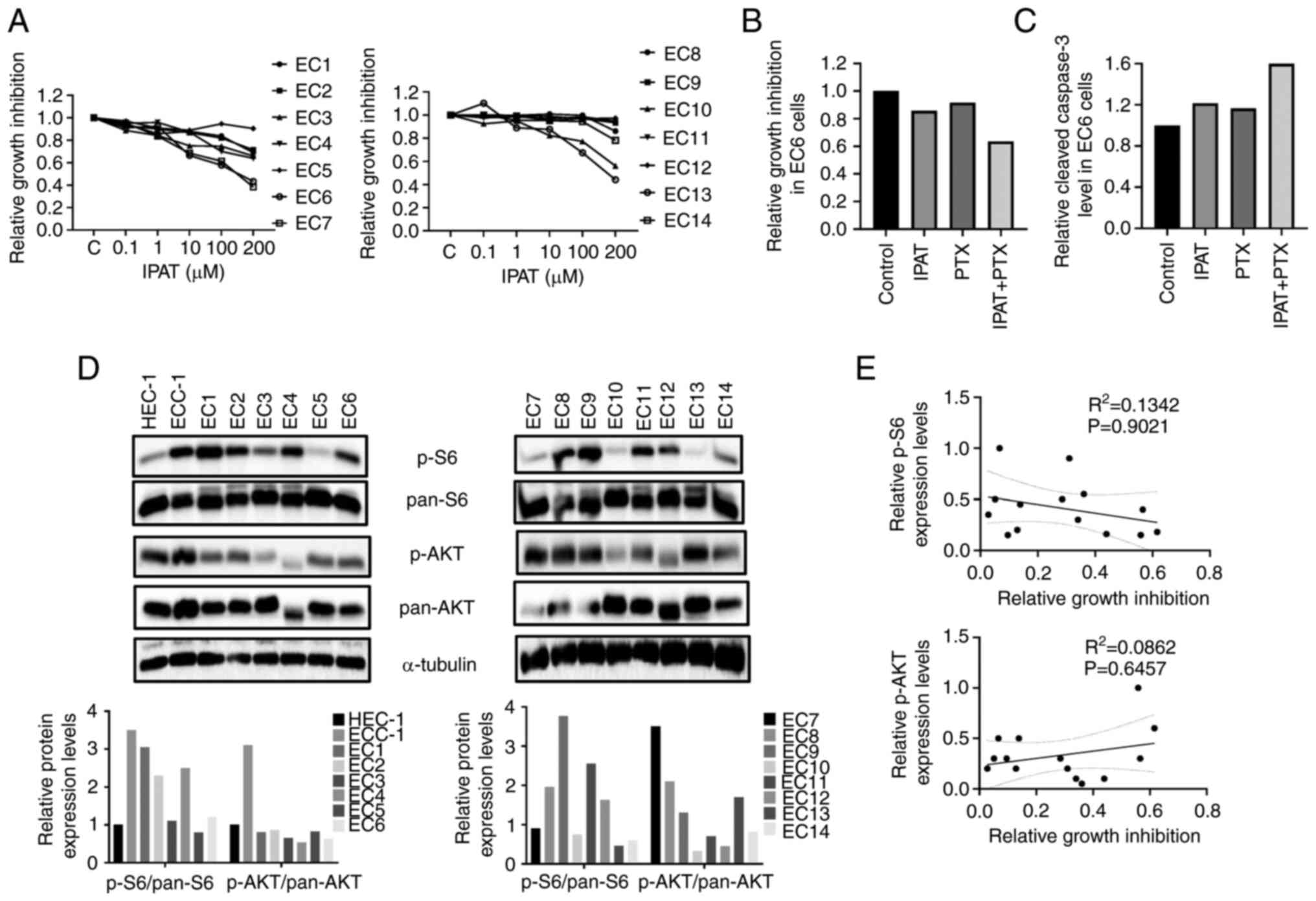
proliferation was evaluated in primary cell cultures of human EC. A
total of fourteen primary cell cultures of EC were isolated and
cultured for the present study (Table
I). Each cell culture was treated with different concentrations
of IPAT, PTX and the combination for 24 and 72 h. Treatment with
IPAT for 72 h resulted in reduced cell viability in 11/14 primary
cultures, while all primary cultures exhibited diverse responses to
PTX (Fig. 7A). The combination of
low doses of IPAT and PTX demonstrated synergistic responses in
8/14 primary cultured cells. The representative EC6 sample,
demonstrated that the combined treatment increased inhibition of
cell proliferation compared with PTX or IPAT alone (Fig. 7B). Three unresponsive IPAT primary
cultures (EC9, EC11 and EC12) did not demonstrate any synergistic
inhibition of cell proliferation in combination with PTX. Four of
the 11 primary cultures which demonstrated an inhibitory response
to IPAT were assayed for cleaved caspase 3 activity. The results
demonstrated that the combination treatment had a markedly greater
effect on inducing the activity of cleaved caspase 3 in three
cases. The combined treatment increased the activity of cleaved
caspase 3 in the representative EC6 sample, (Fig. 7C).
 | Table IClinical and pathological
characteristics of 14 patients with endometrial carcinoma. |
Table I
Clinical and pathological
characteristics of 14 patients with endometrial carcinoma.
| Case | Age | Race | Figo stagea | Tumor size, cm | Histology | HER2 |
|---|
| EC1 | 54 | Hispanic
(White) | IA | 3.5 | Endometrioid, grade
2 | Negative |
| EC2 | 73 | White | IA | 4.5 | Endometrioid, grade
1 | Negative |
| EC3 | 46 | Hispanic
(White) | IA | 2.2 | Endometrioid, grade
1 | N/A |
| EC4 | 46 | Black | IA | 3.5 | Endometrioid, grade
3 | N/A |
| EC5 | 63 | Black | IA | 4.5 | Endometrioid, grade
1 | N/A |
| EC6 | 43 | Black | IIIC1 | 0.5 | Endometrioid, grade
1 | N/A |
| EC7 | 80 | White | IB | 5.3 | Endometrioid, grade
2 | N/A |
| EC8 | 40 | White | IA | 3.7 | Endometrioid, grade
1 | N/A |
| EC9 | 67 | White | IA | 4.8 | Endometrioid, grade
2 | N/A |
| EC10 | 74 | White | IB | 6 | Endometrioid, grade
1 | N/A |
| EC11 | 54 | Hispanic
(White) | IA | 3.3 | Endometrioid, grade
1 | N/A |
| EC12 | 64 | White | IB | 5 | Dedifferentiated
endometrial adenocarcinoma | N/A |
| EC13 | 69 | White | IA | 3.3 | Endometrioid, grade
1 | N/A |
| EC14 | 73 | White | IA | 2.6 | Uterine
carcinosarcoma | N/A |
To evaluate the relationship between the expression
of p-AKT and p-S6, and sensitivity to IPAT, the protein expression
levels of p-AKT and S6 were assessed using western blotting in the
14 untreated primary cell cultures, HEC-1A and ECC-1 cells. Marked
differential expression of p-AKT and p-S6 were demonstrated in
primary cultures and EC cells (Fig.
7D). Sensitivity to IPAT was not associated with the protein
expression levels of p-AKT or p-S6 through comparison of expression
levels and sensitivity to IPAT using linear regression models in
primary cultures and EC cell lines (Fig. 7E).
Discussion
More than 80% of patients with EC have ≥1 genetic
alterations that affect the activity and function of the
PI3K/AKT/mTOR signaling pathway and these alterations result in
this pathway being the most commonly dysregulated signaling pathway
in EC (27,28). IPAT is an orally administered
direct inhibitor of all three isoforms of p-AKT that has been
reported to have anti-tumor efficacy in numerous pre-clinical
models (9-12). Recent phase I-II clinical trials
have reported that oral daily IPAT was well tolerated with few
adverse events and resulted in clinical activity (target engagement
with a tolerable safety profile and limited cancer control) in
advanced solid tumors (12,16,17).
To generate pre-clinical data on the effect of IPAT on EC, the
anti-proliferative and anti-tumorigenic effects of IPAT were
assessed in human EC cell lines, primary cultures of EC and a
transgenic mouse model of EC. Treating EC cell lines with IPAT led
to marked inhibition of the AKT/mTOR signaling pathway, markedly
reduced cellular proliferation, and induced cellular stress, cell
cycle G1 arrest and apoptosis. The combination of IPAT and PTX
synergistically inhibited cell viability and induced apoptosis in
EC cells and primary cultures of EC tumors, compared with treatment
with either drug alone. Additionally, IPAT significantly reduced
tumor growth and increased sensitivity to PTX in the
Lkb1fl/flp53fl/fl mouse model of EC,
accompanied by a marked decrease in Ki67 and p-S6 protein
expression levels and a marked increase in the protein expression
levels of p-H2AX and KIF14 in endometrial tumor tissues. These data
suggested that the targeting of AKT by IPAT not only effectively
inhibited cell proliferation and tumor growth and but also enhanced
the efficacy of PTX in the treatment of EC in vitro and
in vivo.
Previous studies reported that IPAT increased the
phosphorylation of AKT and decreased the phosphorylation of the
proline rich AKT substrate PRAS40 in a dose dependent manner in
cancer cells (18,29). Treatment with effective doses of
IPAT with xenograft mouse cancer models can inhibit p-PRAS40
activity and target p-S6 in tumor tissue, which suggested that IPAT
is a potent inhibitor of AKT/mTOR signaling and that inhibition of
AKT/mTOR signaling is essential for a robust anticancer response
in vivo (18,29). The results of our previous study
demonstrated that IAPT increased phosphorylation of AKT (ser473)
and decreased phosphorylation of S6 (ser235/236) in a dose- and
time-dependent manner in USC cells, which provided further evidence
that IPAT inhibits the AKT/mTOR pathway (19). Growing evidence has suggested that
the activation of AKT/mTOR and its downstream signaling pathways is
responsible for cell survival, angiogenesis, cell cycle
progression, apoptosis and cellular stress, and subsequently,
inhibition of the AKT/mTOR pathway results in inhibition of cell
proliferation, cell cycle arrest, and the induction of apoptosis
and cellular stress in EC cells (28,30).
Previous studies of IPAT in numerous tumor types reported that IPAT
inhibited the activity of the AKT/mTOR signaling pathway, induced
cell cycle G1 arrest and increased apoptosis in cancer cells, which
ultimately resulted in a robust anti-tumorigenic response in
xenograft models with a hyperactive PI3K/AKT/mTOR signaling pathway
(18,31). Previous studies reported that IPAT
inhibited tumor cell growth through numerous mechanisms, including
activation of PUMA, FoxO3a and NF-κB regulated apoptosis, as well
as induction of autophagy signaling and ROS-mediated caspase
activation (32-34). Importantly, compared with other
allosteric AKT inhibitors such as MK-2206, IPAT was able to
overcome MK-2206 resistance in prostate cancer cells and xenograft
mouse models, thereby expanding the practicability of IPAT for
clinical applications (35).
In the present study, similar effects of IPAT on
phosphorylation of AKT (ser473) and S6 (ser235/236) were
demonstrated in EC cells, which provided further evidence that IPAT
is a potent inhibitor of the AKT/mTOR signaling pathway in EC.
Treatment of EC cell lines with different concentrations of IPAT
also reduced cell proliferation, caused cell cycle G1 phase arrest
and induced mitochondrial apoptosis in a dose-dependent manner.
Furthermore, IPAT was effective in inhibiting cell proliferation in
11/14 primary cultures and decreasing tumor growth with reduced
protein expression levels of Ki67 and p-S6 in the
Lkb1fl/flp53fl/fl mouse model of EC.
Taken together, these findings demonstrated the inhibitory effect
of IPAT on the AKT/mTOR signaling pathway, and that the targeting
of AKT by IPAT enables concomitant downregulation of p-S6 and
suppresses tumor growth through a combination of increased cellular
stress and induction of apoptosis and cell cycle arrest in EC.
Aberrant PI3K/AKT signaling regulates ROS production
in cancer cells through multiple molecular mechanisms that
contribute to the regulation of mitochondrial oxidative metabolism
and NADPH oxidase activation (36). Elevated cellular ROS levels
contribute to the loss of mitochondrial membrane potential and
subsequently directly cause DNA damage, activate apoptotic and
autophagy pathways, ultimately resulting in increased efficacy of
anti-tumor agents or the reversal of chemotherapeutic resistance in
cancer cells (37,38). IPAT significantly increased
intracellular ROS level and activated apoptotic signaling pathways
in TRAIL-resistant HT-29 cells, with reversal of TRAIL-resistance
being dependent on ROS generation, DNA damage and p53-mediated PUMA
up-regulation (32). The results
of the present study support the role of IPAT in inducing ROS
production as we found that IPAT decreased mitochondrial membrane
potential in a dose-dependent manner in EC cells, which indicated
that induction of ROS may be a critical early step in the
anti-tumorigenic activity of IPAT. Due to the role of cellular
stress in IPAT-induced growth inhibition, further investigation of
how IPAT-induced cellular stress affects apoptosis and cell growth
inhibition in EC cells and
Lkb1fl/flp53fl/fl mouse model is
required.
The PI3K/AKT signaling pathway participates in the
regulation of oncogenic activity and the development of resistance
in cancer cells to various chemotherapeutics drugs. Each AKT
isoform exhibits different biologic functions and has a distinct
impact on chemoresistance to chemotherapeutic agents including PTX,
cisplatin and doxorubicin in EC cell lines (39,40).
AKT inhibition enhances the effect of cytotoxic agents, and the AKT
inhibitor AZD5363, in combination with chemotherapeutic drugs,
markedly reduces cell viability and induces apoptosis in EC cells
(41). Alteration of AKT activity
induced by chemotherapeutic agents may be an intrinsic or adaptive
resistance mechanism for cancer survival; therefore, targeting AKT
might be used to improve sensitivity to chemotherapeutic agents and
promote therapeutic efficacy (12). Two randomized phase II clinical
trials have reported that the combination of IPAT with PTX was well
tolerated and significantly improved PFS compared with PTX alone in
inoperable locally advanced/metastatic triple-negative breast
cancer (13,20). Although a recent phase III clinical
trial reported no improvement in efficacy for IPAT + PTX in
PIK3CA/AKT1/PTEN-altered hormone receptor-positive HER2-negative
advanced breast cancer, the safety profile for the combination
treatment was consistent with known adverse effects of IPAT alone
and PTX alone (42). Although PTX
has been reported to display its anticancer effects by promoting
microtubule polymerization and stabilization and arresting cells in
the metaphase of the bipolar spindle, its ability to cause DNA
damage and induce oxidative stress and apoptosis which suggested
that PTX may have other cellular cytotoxic modes of action
(43,44). In the present study, the possible
synergistic effect of the combination of IPAT and PTX was evaluated
and it was demonstrated that the combination of IPAT and PTX
significantly enhanced the cytotoxic and apoptotic properties of
either drug alone in EC cells or primary cultures of EC.
Furthermore, IPAT + PTX demonstrated potent inhibition of tumor
growth with increased expression of DNA damage and microtubule
markers and markedly decreased the expression of p-S6, compared
with IPAT alone or PTX alone in the
Lkb1fl/flp53fl/fl mouse model, which
indicated that IPAT may potentiate the efficacy of PTX. Thus, it
was hypothesized that increased DNA damage, inhibited AKT/mTOR/S6
pathway and altered microtubule function; moreover, altered
microtubule function may be potential mechanisms by which the
combination of IPAT and PTX synergistically inhibits EC tumor
growth.
Inhibition of p-AKT enhances the effect of cytotoxic
agents, which indicated that p-AKT levels could be associated with
sensitivity to cytotoxic agents (12,45).
High expression of p-AKT strongly predicted the sensitivity to IPAT
in multiple pre-clinical cancer models (18,31).
In the double-blind randomized phase II FAIRLANE trial, cancer
patients with high p-AKT scores, but not mTOR scores, demonstrated
significantly higher objective response rates with IPAT treatment,
which indicated that baseline functional protein levels of p-AKT
might have predictive value for IPAT (46). However, our previous study
demonstrated that baseline expression of p-AKT and S6 was
independent of cell sensitivity to inhibition by IPAT in primary
cultures of USC and USC cell lines (19). Similar results were demonstrated in
the present study. By analyzing the baseline protein expression
levels of p-AKT and S6 and the sensitivity of IPAT to inhibit cell
growth, it was demonstrated that the expression levels of p-AKT and
p-S6 in EC cell lines and primary cultures were not correlated with
the sensitivity of IAPT to inhibit cell proliferation. Due to its
potential clinical importance, further work will be needed to
identify the relationship between IPAT sensitivity and the
expression of p-AKT and S6 in future EC clinical trials.
In summary, the present study evaluated the
potential effects of IPAT and the combination of IPAT + PTX on cell
proliferation and tumor growth in human endometrioid EC cells,
primary cultures of endometrioid EC tumors and the
Lkbfl/flp53fl/fl mouse model of
endometrioid EC. Our results demonstrated that IPAT inhibited cell
viability and tumor growth through the induction of cellular
stress, cell cycle G1 arrest and apoptosis, and that IPAT
synergistically potentiated the effect of PTX on tumor growth
inhibition. These results provided a strong biological rationale
for clinical trials investigating IPAT alone and the combination of
IPAT and PTX in patients with EC.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JO, ZZ, LB, TH, XZ, YF, HS and WS performed the
experiments. JO, ZZ, TH, CJ, BD, XS and CZ analyzed and interpreted
the data. JO, ZZ, and CZ wrote the manuscript. AAS, CZ and VB
designed experiments, revised the manuscript and obtained funding.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All patients provided written informed consent and
were approved by the Institutional Review Board of the University
of North Carolina at Chapel Hill (approval no. 20-3013). Animal
study was approved by the UNC-CH Institutional Animal Care and Use
Committee (approval no. 20-219).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This work was supported by The Endometrial Cancer Molecularly
Targeted Therapy Consortium and The National Institutes of
Health/National Cancer Institute (grant no. R37CA226969).
References
|
1
|
Siegel RL, Miller KD, Wagle NS and Jemal
A: Cancer statistics, 2023. CA Cancer J Clin. 73:17–48. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Crosbie EJ, Kitson SJ, McAlpine JN,
Mukhopadhyay A, Powell ME and Singh N: Endometrial cancer. Lancet.
399:1412–1428. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tronconi F, Nero C, Giudice E, Salutari V,
Musacchio L, Ricci C, Carbone MV, Ghizzoni V, Perri MT, Camarda F,
et al: Advanced and recurrent endometrial cancer: State of the art
and future perspectives. Crit Rev Oncol Hematol. 180:172022.
View Article : Google Scholar
|
|
4
|
Makker V, Green AK, Wenham RM, Mutch D,
Davidson B and Miller DS: New therapies for advanced, recurrent,
and metastatic endometrial cancers. Gynecol Oncol Res Pract.
4:192017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nero C, Tronconi F, Giudice E, Scambia G
and Lorusso D: Management of stage III and IVa uterine cancer. Int
J Gynecol Cancer. 32:316–322. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
van den Heerik A, Horeweg N, de Boer SM,
Bosse T and Creutzberg CL: Adjuvant therapy for endometrial cancer
in the era of molecular classification: Radiotherapy,
chemoradiation and novel targets for therapy. Int J Gynecol Cancer.
31:594–604. 2021. View Article : Google Scholar :
|
|
7
|
LoRusso PM: Inhibition of the
PI3K/AKT/mTOR pathway in solid tumors. J Clin Oncol. 34:3803–3815.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Roncolato F, Lindemann K, Willson ML,
Martyn J and Mileshkin L: PI3K/AKT/mTOR inhibitors for advanced or
recurrent endometrial cancer. Cochrane Database Syst Rev.
10:CD0121602019.PubMed/NCBI
|
|
9
|
Bang YJ, Kang YK, Ng M, Chung HC, Wainberg
ZA, Gendreau S, Chan WY, Xu N, Maslyar D, Meng R, et al: A phase
II, randomised study of mFOLFOX6 with or without the Akt inhibitor
ipatasertib in patients with locally advanced or metastatic gastric
or gastroesophageal junction cancer. Eur J Cancer. 108:17–24. 2019.
View Article : Google Scholar
|
|
10
|
Chan JJ, Tan TJY and Dent RA: Novel
therapeutic avenues in triple-negative breast cancer: PI3K/AKT
inhibition, androgen receptor blockade, and beyond. Ther Adv Med
Oncol. 11:17588359198804292019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
de Bono JS, De Giorgi U, Rodrigues DN,
Massard C, Bracarda S, Font A, Arranz Arija JA, Shih KC, Radavoi
GD, Xu N, et al: Randomized phase II study evaluating akt blockade
with ipatasertib, in combination with abiraterone, in patients with
metastatic prostate cancer with and without PTEN loss. Clin Cancer
Res. 25:928–936. 2019. View Article : Google Scholar
|
|
12
|
Isakoff SJ, Tabernero J, Molife LR, Soria
JC, Cervantes A, Vogelzang NJ, Patel MR, Hussain M, Baron A,
Argilés G, et al: Antitumor activity of ipatasertib combined with
chemotherapy: Results from a phase Ib study in solid tumors. Ann
Oncol. 31:626–633. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim SB, Dent R, Im SA, Espié M, Blau S,
Tan AR, Isakoff SJ, Oliveira M, Saura C, Wongchenko MJ, et al:
Ipatasertib plus paclitaxel versus placebo plus paclitaxel as
first-line therapy for metastatic triple-negative breast cancer
(LOTUS): A multicentre, randomised, double-blind,
placebo-controlled, phase 2 trial. Lancet Oncol. 18:1360–1372.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Morgillo F, Della Corte CM, Diana A, Mauro
CD, Ciaramella V, Barra G, Belli V, Franzese E, Bianco R, Maiello
E, et al: Phosphatidylinositol 3-kinase (PI3Kα)/AKT axis blockade
with taselisib or ipatasertib enhances the efficacy of
anti-microtubule drugs in human breast cancer cells. Oncotarget.
8:76479–76491. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Oliveira M, Saura C, Nuciforo P, Calvo I,
Andersen J, Passos-Coelho JL, Gil Gil M, Bermejo B, Patt DA,
Ciruelos E, et al: FAIRLANE, a double-blind placebo-controlled
randomized phase II trial of neoadjuvant ipatasertib plus
paclitaxel for early triple-negative breast cancer. Ann Oncol.
30:1289–1297. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Saura C, Roda D, Roselló S, Oliveira M,
Macarulla T, Pérez-Fidalgo JA, Morales-Barrera R, Sanchis-García
JM, Musib L, Budha N, et al: A First-in-Human phase I study of the
ATP-competitive AKT inhibitor ipatasertib demonstrates robust and
safe targeting of AKT in patients with Solid tumors. Cancer Discov.
7:102–113. 2017. View Article : Google Scholar :
|
|
17
|
Shapiro GI, LoRusso P, Cho DC, Musib L,
Yan Y, Wongchenko M, Chang I, Patel P, Chan IT, Sanabria-Bohorquez
S, et al: A phase Ib open-label dose escalation study of the
safety, pharmacokinetics, and pharmacodynamics of cobimetinib
(GDC-0973) and ipatasertib (GDC-0068) in patients with locally
advanced or metastatic solid tumors. Invest New Drugs. 39:163–174.
2021. View Article : Google Scholar
|
|
18
|
Lin J, Sampath D, Nannini MA, Lee BB,
Degtyarev M, Oeh J, Savage H, Guan Z, Hong R, Kassees R, et al:
Targeting activated Akt with GDC-0068, a novel selective Akt
inhibitor that is efficacious in multiple tumor models. Clin Cancer
Res. 19:1760–1772. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Buckingham L, Hao T, O'Donnell J, Zhao Z,
Zhang X, Fan Y, Sun W, Zhang Y, Suo H, Secord AA, et al:
Ipatasertib, an oral AKT inhibitor, inhibits cell proliferation and
migration, and induces apoptosis in serous endometrial cancer. Am J
Cancer Res. 12:2850–2862. 2022.PubMed/NCBI
|
|
20
|
Dent R, Oliveira M, Isakoff SJ, Im SA,
Espié M, Blau S, Tan AR, Saura C, Wongchenko MJ, Xu N, et al: Final
results of the double-blind placebo-controlled randomized phase 2
LOTUS trial of first-line ipatasertib plus paclitaxel for
inoperable locally advanced/metastatic triple-negative breast
cancer. Breast Cancer Res Treat. 189:377–386. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sweeney C, Bracarda S, Sternberg CN, Chi
KN, Olmos D, Sandhu S, Massard C, Matsubara N, Alekseev B, Parnis
F, et al: Ipatasertib plus abiraterone and prednisolone in
metastatic castration-resistant prostate cancer (IPATential150): A
multi-centre, randomised, double-blind, phase 3 trial. Lancet.
398:131–142. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guo H, Kong W, Zhang L, Han J, Clark LH,
Yin Y, Fang Z, Sun W, Wang J, Gilliam TP, et al: Reversal of
obesity-driven aggressiveness of endometrial cancer by metformin.
Am J Cancer Res. 9:2170–2193. 2019.PubMed/NCBI
|
|
23
|
Staley A, Tucker K, Yin Y, Zhang X, Fan Y,
Zhang Y, Fang Z, Sun W, Suo H, Zhao X, et al: Highly potent
dopamine receptor D2 antagonist ONC206 demonstrates
anti-tumorigenic activity in endometrial cancer. Am J Cancer Res.
11:5374–5387. 2021.PubMed/NCBI
|
|
24
|
Pierce SR, Fang Z, Yin Y, West L, Asher M,
Hao T, Zhang X, Tucker K, Staley A, Fan Y, et al: Targeting
dopamine receptor D2 as a novel therapeutic strategy in endometrial
cancer. J Exp Clin Cancer Res. 40:612021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kaminskyy VO and Zhivotovsky B: Free
radicals in cross talk between autophagy and apoptosis. Antioxid
Redox Signal. 21:86–102. 2014. View Article : Google Scholar
|
|
26
|
Kodack DP, Farago AF, Dastur A, Held MA,
Dardaei L, Friboulet L, von Flotow F, Damon LJ, Lee D, Parks M, et
al: Primary patient-derived cancer cells and their potential for
personalized cancer patient care. Cell Rep. 21:3298–3309. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cheung LW, Hennessy BT, Li J, Yu S, Myers
AP, Djordjevic B, Lu Y, Stemke-Hale K, Dyer MD, Zhang F, et al:
High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer
elucidates a novel mechanism for regulation of PTEN protein
stability. Cancer Discov. 1:170–185. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mazloumi Gavgani F, Smith Arnesen V,
Jacobsen RG, Krakstad C, Hoivik EA and Lewis AE: Class I
Phosphoinositide 3-Kinase PIK3CA/p110α and PIK3CB/p110β Isoforms in
Endometrial Cancer. Int J Mol Sci. 19:39312018. View Article : Google Scholar
|
|
29
|
Lin K, Lin J, Wu WI, Ballard J, Lee BB,
Gloor SL, Vigers GP, Morales TH, Friedman LS, Skelton N and
Brandhuber BJ: An ATP-site on-off switch that restricts phosphatase
accessibility of Akt. Sci Signal. 5:ra372012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pavlidou A and Vlahos NF: Molecular
alterations of PI3K/Akt/mTOR pathway: A therapeutic target in
endometrial cancer. Scientific World Journal. 2014:7097362014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Blake JF, Xu R, Bencsik JR, Xiao D, Kallan
NC, Schlachter S, Mitchell IS, Spencer KL, Banka AL, Wallace EM, et
al: Discovery and preclinical pharmacology of a selective
ATP-competitive Akt inhibitor (GDC-0068) for the treatment of human
tumors. J Med Chem. 55:8110–8127. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yu L, Liu Z, Qiu L, Hao L and Guo J:
Ipatasertib sensitizes colon cancer cells to TRAIL-induced
apoptosis through ROS-mediated caspase activation. Biochem Biophys
Res Commun. 519:812–818. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cocco S, Leone A, Roca MS, Lombardi R,
Piezzo M, Caputo R, Ciardiello C, Costantini S, Bruzzese F, Sisalli
MJ, et al: Inhibition of autophagy by chloroquine prevents
resistance to PI3K/AKT inhibitors and potentiates their antitumor
effect in combination with paclitaxel in triple negative breast
cancer models. J Transl Med. 20:2902022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sun L, Huang Y, Liu Y, Zhao Y, He X, Zhang
L, Wang F and Zhang Y: Ipatasertib, a novel Akt inhibitor, induces
transcription factor FoxO3a and NF-κB directly regulates
PUMA-dependent apoptosis. Cell Death Dis. 9:9112018. View Article : Google Scholar
|
|
35
|
Savill KMZ, Lee BB, Oeh J, Lin J, Lin E,
Chung WJ, Young A, Chen W, Miś M, Mesh K, et al: Distinct
resistance mechanisms arise to allosteric vs. ATP-competitive AKT
inhibitors. Nat Commun. 13:20572022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Koundouros N and Poulogiannis G:
Phosphoinositide 3-Kinase/Akt signaling and redox metabolism in
cancer. Front Oncol. 8:1602018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Aggarwal V, Tuli HS, Varol A, Thakral F,
Yerer MB, Sak K, Varol M, Jain A, Khan MA and Sethi G: Role of
reactive oxygen species in cancer progression: Molecular mechanisms
and recent advancements. Biomolecules. 9:7352019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Asaduzzaman Khan M, Tania M, Zhang DZ and
Chen HC: Antioxidant enzymes and cancer. Chin J Cancer Res.
22:87–92. 2010. View Article : Google Scholar
|
|
39
|
Girouard J, Lafleur MJ, Parent S, Leblanc
V and Asselin E: Involvement of Akt isoforms in chemoresistance of
endometrial carcinoma cells. Gynecol Oncol. 128:335–343. 2013.
View Article : Google Scholar
|
|
40
|
Gagnon V, Van Themsche C, Turner S,
Leblanc V and Asselin E: Akt and XIAP regulate the sensitivity of
human uterine cancer cells to cisplatin, doxorubicin and taxol.
Apoptosis. 13:259–271. 2008. View Article : Google Scholar
|
|
41
|
Fabi F, Adam P, Parent S, Tardif L, Cadrin
M and Asselin E: Pharmacologic inhibition of Akt in combination
with chemotherapeutic agents effectively induces apoptosis in
ovarian and endometrial cancer cell lines. Mol Oncol. 15:2106–2119.
2021. View Article : Google Scholar :
|
|
42
|
Turner N, Dent RA, O'Shaughnessy J, Kim
SB, Isakoff SJ, Barrios C, Saji S, Bondarenko I, Nowecki Z, Lian Q,
et al: Ipatasertib plus paclitaxel for PIK3CA/AKT1/PTEN-altered
hormone receptor-positive HER2-negative advanced breast cancer:
Primary results from cohort B of the IPATunity130 randomized phase
3 trial. Breast Cancer Res Treat. 191:565–576. 2022. View Article : Google Scholar :
|
|
43
|
Veerabhadrappa B, Subramanian S, S J S and
Dyavaiah M: Evaluating the genetic basiss of anti-cancer property
of Taxol in Saccharomyces cerevisiae model. FEMS Microbiol Lett.
368:fnab0772021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhao X, Kong W, Tucker K, Staley A, Fan Y,
Sun W, Yin Y, Huang Y, Fang Z, Wang J, et al: SPR064, a pro-drug of
paclitaxel, has anti-tumorigenic effects in endometrial cancer cell
lines and mouse models. Am J Transl Res. 12:4264–4276.
2020.PubMed/NCBI
|
|
45
|
Shariati M and Meric-Bernstam F: Targeting
AKT for cancer therapy. Expert Opin Investig Drugs. 28:977–988.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shi Z, Wulfkuhle J, Nowicka M, Gallagher
RI, Saura C, Nuciforo PG, Calvo I, Andersen J, Passos-Coelho JL,
Gil-Gil MJ, et al: Functional mapping of AKT signaling and
biomarkers of response from the FAIRLANE trial of neoadjuvant
ipatasertib plus paclitaxel for triple-negative breast cancer. Clin
Cancer Res. 28:993–1003. 2022. View Article : Google Scholar
|
|
47
|
Kasius JC, Pijnenborg JMA, Lindemann K,
Forsse D, van Zwol J, Kristensen GB, Krakstad C, Werner HMJ and
Amant F: Risk stratification of endometrial cancer patients: FIGO
stage, biomarkers and molecular classification. Cancers.
13:58482021. View Article : Google Scholar : PubMed/NCBI
|