Introduction
T cell acute lymphoblastic leukemia (T-ALL) is a
neoplasm derived from T cell lineage-committed lymphoblasts,
accounting for ~15% of childhood and 25% of adult ALL cases. T-ALL
affects bone marrow and peripheral blood, often presenting with a
large thymic tumor, lymphadenopathy (tumor in lymph nodes) and
hepatosplenomegaly (tumor in the liver and spleen). Thymic tumors
are located in the anterior mediastinum and often exhibit rapid
growth, which may manifest as respiratory distress caused by
tracheal compression. T-ALL lymphoblasts have a scant cytoplasm
(bare nucleus), express CD3, which is considered T cell
lineage-specific, and frequently exhibit coexpression of CD4 and
CD8 at the cortical T stage. Genetic alterations in T-ALL result in
aberrant activities of oncogenic transcription factors and the
NOTCH1 pathway, with del(9p) resulting in the loss of the tumor
suppressor gene cyclin-dependent kinase inhibitor 2A
(CDKN2A) (1,2).
Dysregulation of T cell-related transcription
factors results from chromosomal rearrangements, including of T
cell receptor (TCR) genes. Oncogenic transcription factors
implicated in T-ALL include T cell leukemia homeobox protein 1
(TLX1), TLX3, LIM domain only 1 (LMO1),
LMO2, lymphoblastic leukemia-derived sequence 1
(LYL1) and T-ALL 1 (TAL1) (3,4).
TLX1 activation has been associated with a favorable
prognosis in T-ALL, whereas the expression of TAL1,
LYL1 or TLX3 has been associated with lower overall
survival of patients with T-ALL (5). Genome-wide sequencing analyses in
human T-ALL cohorts revealed associations between these oncogenic
transcription factors and 10 pathways that are recurrently mutated
in T-ALL, such as Myc, NOTCH and PI3K/AKT/mTOR (4,6).
Taken together, the dysregulation of T cell-related transcription
factors is implicated in T-ALL development and patient
prognosis.
TAL1 was originally identified as stem cell
leukemia (SCL) gene arising from a translocation between
chromosomes 1 and 14, t(1;14)(p33;qll), involving the regulatory
element of the TCR gene in leukemic stem cells (7). TAL1 protein requires either LMO1 or
LMO2 for its oncogenic capacity (3). As previously reported (8), mice transgenic for full-length
SCL alone do not develop T cell malignancy, whereas 19/20
(95%) of SCL-LMO1 transgenic mice generated by crossing
sil-SCL and lck-LMO1 mice develop aggressive T cell
leukemia/lymphoma by the age of 6 months. The immunophenotype and
clinical manifestations of the disease in SCL-LMO1
transgenic mice are similar to those observed in human patients
with T-ALL. Tremblay et al (9) reported that TAL1 and
LMO1 gain-of-function in SCL-LMO1 transgenic mice
precedes the acquisition of Notch1 mutations for T-ALL
initiation, suggesting that SCL, LMO1 and
Notch1 gain-of-function may represent the minimum set of
complementing events required for the transformation of susceptible
thymocytes.
NOTCH is a transmembrane receptor for cell-cell
communication that functions through three proteolytic cleavages.
Briefly, NOTCH is produced in the endoplasmic reticulum (ER) as a
single protein (pre-NOTCH), cleaved at its heterodimerization
domain (HD) by a furin-like protease in the Golgi apparatus (S1
cleavage), indicating NOTCH maturation, then transported to the
cell membrane as a transmembrane receptor comprising a heterodimer
(mature form), including extracellular and intracellular NOTCH
domains. Upon binding of a ligand, such as Jagged 1-2 or Delta-like
ligands 1, 3 and 4, mature NOTCH undergoes two successive
proteolytic cleavages (S2 and S3). S2 is mediated by a disintegrin
and metalloproteinase. Finally, NOTCH is activated via S3 cleavage
by γ-secretase, which is a hetero-tetrameric protein complex
composed of one aspartic protease subunit, presenilin 1 or 2, and
the three non-proteolytic subunits, namely, nicastrin, anterior
pharynx defective-1 and presenilin enhancer protein 2 (10,11). Once NOTCH is recognized as a
substrate of γ-secretase on the cytoplasmic membrane, presenilin
undergoes endoproteolysis (endoproteolytic cleavage), termed
autoproteolytic presenilin processing, to generate N- and
C-terminal fragments (12-14),
which results in the catalytic activation of γ-secretase for S3
cleavage at the transmembrane domain of the NOTCH receptor
(10,15). Following S3 cleavage, the NOTCH
intracellular domain translocates to the nucleus, resulting in
induction of NOTCH target molecules, such as hairy and enhancer of
split-1 (HES1) and c-Myc (11).
Mammals have four NOTCH receptors (NOTCH 1-4).
Mutation frequencies for these vary between cancer types (11). NOTCH1 gene is expressed in
hematopoietic stem cells and controls several steps in thymocyte (T
cell in the thymus) specification and differentiation (10,15,16). Somatic activating mutations of
NOTCH1 involving the extracellular HD in exons 26 and 27
and/or the C-terminal PEST domain consisting of a polypeptide
enriched in proline (P), glutamic acid (E), serine (S) and
threonine (T) in exon 34 have been identified in >50% of human
T-ALL cases (17). Although
NOTCH1 activating mutations primarily occur in the HD and/or
PEST domains, activating expansion mutations within the
extracellular juxtamembrane domain (JM) in exon 28 have been
identified in human T-ALL (18,19). By contrast, murine T-ALL is often
associated with somatic deletions at the 5′-end of NOTCH1,
resulting in ligand-independent Notch1 activation (20). These NOTCH1 activating
mutations in T-ALL suggest a potential role for inhibition of
NOTCH1 pathway in cancer therapy.
γ-secretase inhibitors (GSIs) are one of the most
extensively studied NOTCH-targeting therapeutics to combat T-ALL
(11,14). In early clinical trials of
broad-spectrum GSIs, the maximum tolerable dose was limited by
gastrointestinal toxicity (primarily diarrhea) resulting from
intestinal secretory metaplasia, which was prevented by combination
with glucocorticoid treatment (21,22). To date, the clinical application
of GSIs to alleviate T-ALL has not been successful due to adverse
events and limited anti-leukemic efficacy (11). It is reported that selective
inhibition of γ-secretase components induces significant
therapeutic efficacy and less toxicity in preclinical T-ALL models;
MRK-560, a selective presenilin 1 inhibitor, suppresses leukemia
development in both mutant Notch1-driven leukemia mouse
models and human patient-derived xenograft (PDX) models, without
any associated pathological changes in the gastrointestinal tract
or major defects in thymocyte development (23,24). The mechanisms of resistance to
NOTCH inhibition have been studied: Mutational loss of PTEN,
a tumor suppressor gene, is a resistance mechanism in human T-ALL,
which induces PI3K/AKT/mTOR pathway activity (25,26). For the successful clinical
development of GSIs for T-ALL, selective targeting of presenilin 1
in γ-secretase and avoiding resistance to NOTCH inhibition by
overcoming mechanisms such as PI3K/AKT/mTOR activation should be
considered.
Our previous study demonstrated that nelfinavir, a
human immunodeficiency virus 1 (HIV1) protease inhibitor,
suppresses the proliferation of non-small cell lung cancer (NSCLC)
cells in vitro as well as that of human NSCLC xenograft
tumors by inhibiting AKT and inducing ER stress/unfolded protein
response (UPR), which subsequently leads to apoptosis (27). Moreover, data from a phase I
clinical trial of nelfinavir in adults with solid tumors shows that
nelfinavir is well-tolerated and exhibits antitumor activity
(28). More recently, we reported
the antitumor effects of nelfinavir in a SCLC PDX mouse model.
Nelfinavir increases levels of sestrin 2 (SESN2), an endogenous
mTOR-negative regulator, via ER stress/UPR induction, resulting in
inhibition of the mTOR pathway (29). To date, nelfinavir has been
repurposed as an anticancer drug for various types of cancer such
as lung, pancreas, head and neck carcinoma (30). Nelfinavir was originally developed
to target an HIV1 aspartic protease. Presenilin in γ-secretase is
also an aspartic protease (31),
therefore it was hypothesized that nelfinavir inhibits NOTCH
pathway via γ-secretase inhibition by blocking presenilin,
suggesting therapeutic efficacy against NOTCH-associated T-ALL. The
present study, we assessed the efficacy of nelfinavir against T-ALL
cells and investigated mechanisms of action in vitro and in
preclinical treatment studies using a SCL-LMO1 transgenic
mouse model.
Materials and methods
Cell culture
Jurkat and Molt4 cell lines were gifts from Dr David
S Chervinsky (Roswell Park Memorial Institute, New York, NY, USA).
HPB-ALL cell line was a gift from Dr A Thomas Look (Dana-Faber
Cancer Institute, Boston, MA, USA), which was authenticated as
previously described (32). The
human T-ALL cell lines were maintained in RPMI-1640 medium
supplemented with 5% fetal bovine serum (both Thermo Fisher
Scientific, Inc.) at 37°C in an incubator with 5%
CO2.
Reagents
Nelfinavir for in vitro experiments was
obtained from the National Institutes of Health (NIH) AIDS Research
and Reference Reagent Program, Division of AIDS, National Institute
of Allergy and Infectious Diseases (Bethesda, MD, USA). Nelfinavir
for in vivo experiments was obtained from Pfizer Inc.
Compound E (γ-secretase inhibitor XXI; cat. no. 565790) and a
fluorogenic γ-secretase substrate (cat. no. 565764) were purchased
from Sigma-Aldrich (Merck KGaA). Rapamycin (an mTOR inhibitor) was
obtained from LC Laboratories. Primary antibodies against cleaved
NOTCH1 (Val1744; cat. no. D3B8; 1:1,000) for NOTCH1 intracellular
domain (NICD), HES1 (cat. no. D6P2U; 1:1,000), c-Myc (cat. no.
D84C12; 1:1,000), α-tubulin (cat. no. 11H10; 1:1,000), presenilin 1
(cat. no. #3622; 1:1,000) to detect the full-length (FL) and
C-terminal fragment (CTF), β-actin (cat. no. 13E5; 1:1,000),
phosphorylated eukaryotic initiation factor 2α (p-eIF2α; Ser51,
cat. no. #3597; 1:1,000), NOTCH1 (cat. no. D6F11; 1:1,000) and p-S6
ribosomal protein (Ser235/236, cat. no. #2211; 1:1,000) were
purchased from Cell Signaling Technology, Inc. ChaC
glutathione-specific γ-glutamylcyclotransferase 1 (CHAC1; clone no.
N116/14; 1:2,000) and CD3 (cat. no. #A0452; 1:100) antibodies were
obtained from UC Davis/NIH NeuroMab Facility and Dako (Agilent
Technologies, Inc.), respectively.
Cell viability (apoptosis and cell death)
assay
T-ALL cells (2×105 cells per well) were
plated in 6-well plates and treated with 1-20 μM nelfinavir
or an equal volume of 0.1% dimethyl sulfoxide (DMSO) for 16 h at
37°C. The cells were harvested and resuspended in fluorescein
isothiocyanate (FITC)-labeled Annexin V in Binding Buffer (FITC
Annexin V Apoptosis Detection kit I, BD Biosciences) for 15 min at
room temperature and stained with 1 mg/ml propidium iodide
(Sigma-Aldrich; Merck KGaA) in phosphate-buffered saline (PBS) at
room temperature. Cells were immediately analyzed via flow
cytometry. FACScan analysis was performed using Becton-Dickinson
and Company FACSort and CellQuest software version 3.3 (BD
Biosciences). FITC Annexin V-positive staining was used to detect
apoptosis (early + late phase). Propidium iodide was used to detect
dead cells. Cells that were negative for both stains were
considered viable.
Immunoblotting analysis
T-ALL cells (5×105 cells/well) were
plated in 6-well plates, treated with nelfinavir, compound E,
rapamycin or an equal volume of 0.1% DMSO at 37°C, then lysed in 2X
lysis buffer [20% glycerol, 4% sodium dodecyl sulfate, 125 mM
Tris-HCl pH 8.0] as previously described (33). Whole-cell protein lysate was
quantified using a Pierce™ BCA Protein Assay kit (Thermo Fisher
Scientific, Inc.). Afterward, 20 μg lysate/lane was loaded
and separated via 7.5-12.0% sodium dodecyl sulfate-polyacrylamide
gel electrophoresis and transferred to nitrocellulose membranes.
The membranes were blocked for 1 h at room temperature in blocking
buffer [1X Tris-buffered saline (TBS), 5% milk and 0.1% Tween-20]
and incubated with primary antibodies overnight at 4°C. Membranes
were washed three times with wash buffer (1X TBS, 0.1% Tween-20).
Primary antibodies were detected by incubation with horseradish
peroxidase-linked secondary antibodies (anti-rabbit and -mouse IgG;
cat. nos. #7074 and #7076, respectively; both 1:2,000; both Cell
Signaling Technology, Inc.) for 1 h at room temperature and
visualized using an enhanced chemiluminescence detection system
(Amersham ECL detection reagent, cat. no. RPN2232, GE HealthCare).
As loading controls, α-tubulin or β-actin was used. Immunoblotting
experiments were performed at least three times. Densitometry was
performed using NIH Image software (version 1.52, National
Institutes of Health).
Cell-free in vitro γ-secretase assay
Cytoplasmic membrane-enriched cell fractions were
prepared using hypotonic buffer A (10 mM Tris pH 7.4, 1 mM EDTA and
1 mM EGTA) and dissolved in hypotonic buffer B (10 mM Tris pH 7.4,
1 mM EDTA, 1 mM EGTA and 0.2% CHAPS). To measure enzyme activity,
20 μg protein was incubated with 8 μmol/l fluorogenic
γ-secretase substrate NMA-GGVVIATVK(DNP)-DRDRDR-NH2,
nelfinavir or compound E for 2 h at 37°C. The degree of fluorogenic
substrate cleavage was measured by emitted fluorescence using an
Infinite 200 PRO microplate reader (Tecan Group, Ltd.) with an
excitation wavelength of 355 nm and an emission wavelength of 440
nm, as previously reported (34,35).
Microarray assay
RNA was isolated from triplicate samples of 0.1%
DMSO or 20 mM nelfinavir-treated HBP-ALL cells using TRIzol (Thermo
Fisher Scientific, Inc.). cDNA was synthesized using SuperScript II
Reverse Transcriptase (Thermo Fisher Scientific, Inc.) and
processed for Affymetrix gene expression analysis (GeneChip Human
Genome U133 Plus 2.0 Array, cat. no. 900466; GeneChip Scanner 3000;
Affymetrix; Thermo Fisher Scientific, Inc.), following the
manufacturer's instructions. Gene expression data were normalized
using DNA-Chip-Analyzer software (softpedia.com/get/Science-CAD/dChip.shtml;
dchip.org/; Build date: Jul 19 2010) and
differentially expressed genes were identified using Comparative
Marker Selection (version 8) GenePattern application (genepattern.org/#) using a non-parametric
P<0.0001.
Animal experiments
SCL-LMO1 transgenic mice that develop T cell
malignancy were generated by crossing sil-SCL transgenic
mice and lck-LMO1 mice, as previously described (8). A total of ~300 mice (>14 weeks of
age, 1:1 of males and females, 18 to 30 g of weight) were prepared
and supplied from Genetics Branch, National Cancer Institute (NCI,
Bethesda, MD) and maintained with ad libitum feeding and
water and clean and comfortable environment (12-14/10-12 h dark
cycle, 18-23°C with 40-60% humidity) in animal facility (NIH,
Bethesda, MD, USA).
Treatment with nelfinavir
Measurable lesions, including thymic masses with or
without peripheral lymphadenopathy, were screened using SkyScan1178
(Bruker Corporation) for whole-body X-ray and micro-computed
tomography (CT) images under general anesthesia using isoflurane
(3-4% for induction and 1-3% for maintenance) once/week. Once the
measurable lesion was detected by imaging, mice with good physical
condition were treated with intraperitoneal 100 mg/kg nelfinavir
dissolved in vehicle (4% DMSO, 5% polyethylene glycol, 5% Tween-80
in saline) daily for 2 weeks, as previously described (29). Additionally, for
nelfinavir-withdrawal study, four mice that had showed the efficacy
of nelfinavir treatment were continuously administered vehicle
until disease progression. The condition of mice was checked daily
before and after nelfinavir treatment to monitor the progression of
internal lesions once/week.
Evaluation of nelfinavir efficacy
Tumor burden (TB) was calculated based on the
maximum tumor area of measurable lesions and the percentage of TB
after nelfinavir treatment was calculated as ratio to TB before the
treatment, which was indicated as 'TB (%) end'. Response rate (RR)
was calculated based on the percent change in TB before and after
nelfinavir treatment, as previously described (36): Complete response (CR),
disappearance of all target lesions; partial response (PR), ≥30%
decrease in target lesion; progressive disease (PD), ≥20% increase
in target lesion and stable disease (SD), <30% decrease and
<20% increase in target lesion. Baseline TB was used as a
reference. Once RR was confirmed in nelfinavir treatment study, the
mice were sacrificed. Additionally, mice with clinical signs such
as hunched posture, rapid/progressive weight loss, or respiratory
distress were prematurely sacrificed as a humane endpoint. Mice
were euthanized by CO2 asphyxiation with a volume
displacement rate of 30-70% of the chamber volume/min based on the
NIH Animal Research Advisory Committee Guidelines (https://oacu.oir.nih.gov/animal-research-advisory-committee-arac-guidelines)
for euthanasia of rodents. Death after CO2 asphyxiation
was verified by lack of response to any stimulation.
To detect CD4+/CD8+ (double-positive) cell
populations, single-cell suspensions from thymic tumor of
SCL-LMO1 transgenic mice were prepared. The cells were
resuspended in calcium- and magnesium-free Hanks' balanced salt
solution buffer (Thermo Fisher Scientific, Inc.) containing 2%
fetal bovine serum and incubated for 30 min on ice with
phycoerythrin-CD4 and FITC-CD8 (BD Pharmingen; BD Biosciences). The
cells were washed twice with PBS and stained with 1 mg/ml propidium
iodide in PBS at room temperature. FACScan analysis was performed
as aforementioned.
To perform hematoxylin and eosin (H&E), terminal
deoxynucleotidyl transferase-mediated deoxyuridine triphosphate
nick-end labeling (TUNEL) and CD3 immunohistochemical analysis,
tissue pieces (thymus and bone marrow) from each mouse (Table SI) were dissected and fixed in
10% neutral buffered formalin for 24 h at room temperature, and
then embedded in paraffin wax. The formaldehyde-fixed and
paraffin-embedded (FFPE) tumor sections on glass slides were
deparaffinized. Hematoxylin and eosin were stained for two minutes,
respectively with rinsing using deionized water at room
temperature, and then mountings were performed using Fisher
Chemical™ Permount™ Mounting Medium (cat. no. SP15-100, Thermo
Fisher Scientific, Inc.). FFPE tumor sections on glass slides were
deparaffinized and hydrated, then stained with Apo-Direct TUNEL
Assay kit (cat. no. 88-6611-88, Thermo Fisher Scientific, Inc.)
according to the manufacturer's instruction. The glass slides were
counter-stained with hematoxylin for 2 min at room temperature,
visualized TUNEL-positive nuclei, and assessed on ten fields using
a light microscopy as previously described (27). For CD3 immunohistochemical
analysis, unstained FFPE tumor sections on glass slides were
deparaffinized and hydrated, then stained with VECTASTAIN Elite ABC
system (Vector Laboratories, Inc.; Maravai LifeSciences) according
to the manufacturer's instruction, in which staining was performed
using 3,3′-Diaminobenzidine (Sigma-Aldrich; Merck KGaA) at room
temperature under monitoring the staining process using a light
microscope, as previously described (36).
Sequencing
Notch1 mutations in nelfinavir-treated
SCL-LMO1 transgenic mice were analyzed. For DNA preparation,
tissue pieces (thymus, spleen, and bone marrow) from each mouse
(Table SI) were dissected,
flash-frozen and stored at -70°C until use. Tissue was chopped into
<0.5 mm3 pieces on dry ice and DNA was extracted
using a Qiagen DNAeasy Blood & Tissue kit (Qiagen Inc.)
according to the manufacturer's instructions. Notch1 exons
26 and 27 (HD), exon 28 (JM) and exon 34 (PEST domain) were
amplified from genomic DNA via PCR using the following primer
sequences: Exon 26 forward, 5′-GCT GAG GGA GGA CCT GAA CTT GG-3′
and reverse, 5′-CCT GAG CTG GAA TGC TGC CTC TA-3′; exon 27 forward,
5′-CAT GGG CCT CAG TGT CCT-3′ and reverse, 5′-TAG CAA CTG GCA CAA
ACA GC-3′; exon 28 forward, 5′-GCG TAG CCG CTG CCT GAT-3′ and
reverse, 5′-CAG ACT CCC GGT GAG GAT GC-3′; exon 34 forward 1,
5′-GCTGGCCTTTGAGACTGG-3′ and reverse 1, 5′-CTC CTG GGG CAG AAT AGT
GT-3′; exon 34 forward 2, 5′-ACA GAT GCA GCA GCA GAA CC-3′ and
reverse 2, 5′-CCT GGG GCC AGA TAA AAC AGT ACA-3′. PCR was performed
as described by Sulis et al (18) and analyzed using direct dideoxy
Sanger sequencing of the PCR products in DNA Sequencing and Gene
Expression Core (NCI). Somatic deletions at the 5′-end of
Notch1 were analyzed via PCR-based detection using the
following primer sequences: Forward, 5′-ATG GTG GAA TGC CTA CTT TGT
A-3′ and reverse, 5′-CGT TTG GGT AGA AGA GAT GCT TTAC-3′, as
described by Ashworth et al (20).
Statistical analysis
Data are presented as the mean ± SD. The statistical
significance of differences between treated and untreated cells was
analyzed using one-way analysis of variance followed by
Bonferroni's post hoc test. All analyses were performed using the
GraphPad Prism software version 9 (GraphPad Software Inc.;
Dotmatics). P<0.05 was considered to indicate a statistically
significant difference.
Results
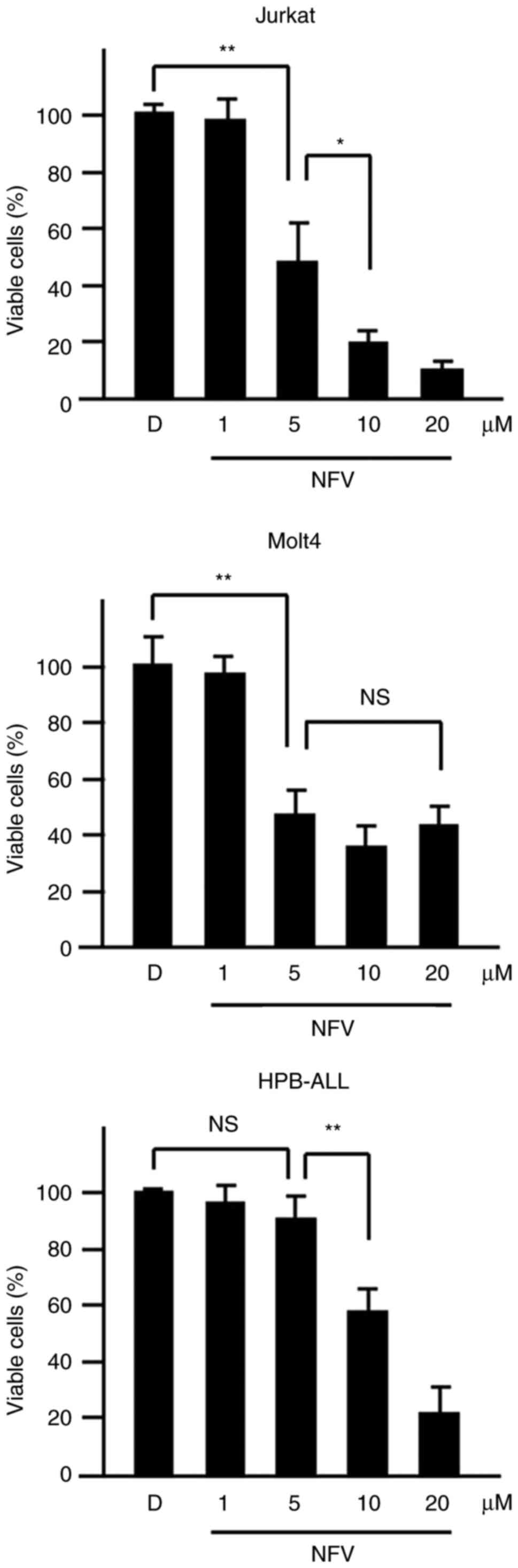
Nelfinavir suppresses T-ALL cell
viability
To evaluate the cytotoxicity of nelfinavir in T-ALL
cells, cell viability assay was performed. T-ALL cell lines with
active NOTCH pathway that harbor either NOTCH1 wild-type
(Jurkat cells) or activating NOTCH1 mutations (Molt4 and
HPB-ALL cells) (17) were treated
with 0.1% DMSO or nelfinavir. Nelfinavir decreased the percentage
of viable cells in a dose-dependent manner compared with DMSO
treatment in activating NOTCH1 mutant T-ALL as well as in
the wild-type cell line (Figs. 1
and S1). Although Molt4 cells
showed a cell death plateau at high concentrations (10 and 20
μM), taken together, these findings suggested that
nelfinavir might inhibit NOTCH activation in T-ALL cells.
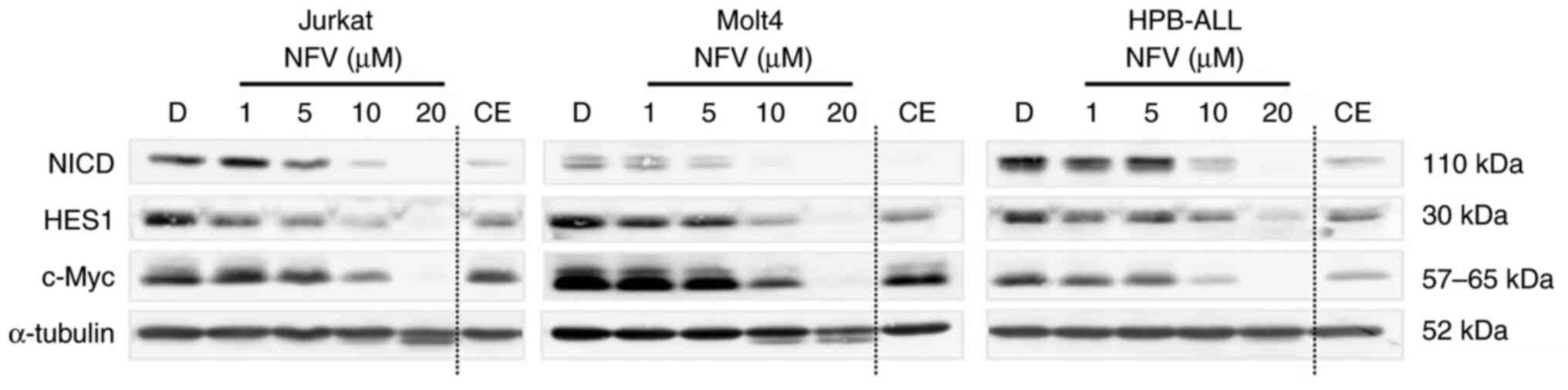
Nelfinavir inhibits the NOTCH1
pathway
To assess whether nelfinavir inhibits NOTCH
activation to exert its cytotoxic effects, NOTCH pathway activity
in nelfinavir-treated T-ALL cells was evaluated. Following 16 h
treatment, nelfinavir downregulated NICD expression in all cell
lines at 5 and 10 μM in a dose-dependent manner (Fig. 2). In addition, nelfinavir
decreased expression of NOTCH1 target molecules (HES1 and c-Myc) at
concentrations ≥10 μM. Cell death-associated decomposition
may affect the results of immunoblotting analyses after 16 h
treatment; therefore, nelfinavir efficacy was assessed at earlier
time points (Fig. S2).
Nelfinavir downregulated expression of NICD and c-Myc in a
time-dependent manner after 4 h treatment, indicating the efficacy
of nelfinavir in the early phase. Taken together, these findings
indicate that inhibition of the NOTCH1 pathway may underlie the
cytotoxicity of nelfinavir against T-ALL.
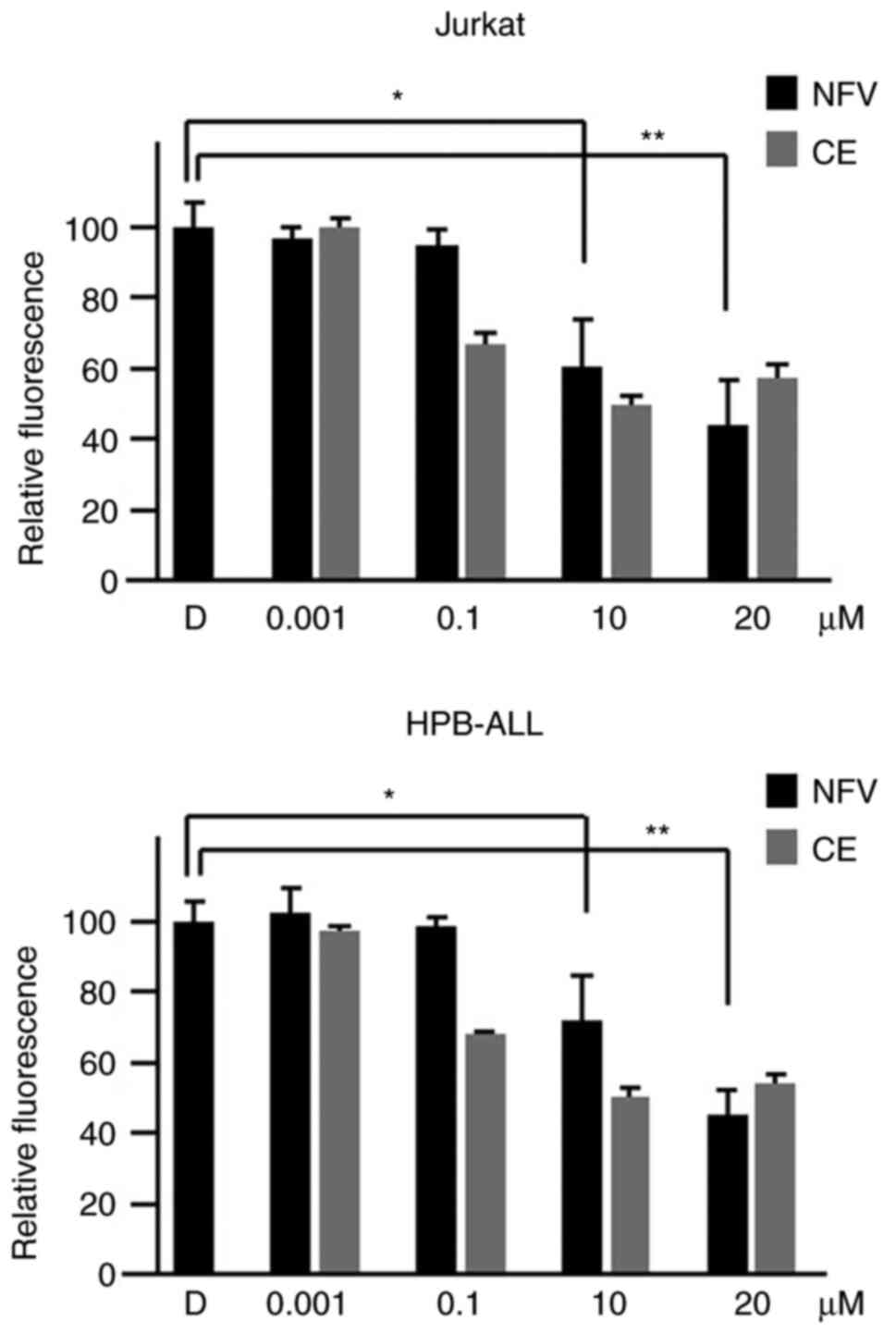
Nelfinavir inhibits γ-secretase
activity
To assess how nelfinavir inhibits the NOTCH1
pathway, cell-free in vitro γ-secretase assays were
performed. When nelfinavir was added to membrane extracts isolated
from Jurkat cells, relative fluorescence was inhibited by 40% at 10
μM and 56% at 20 μM (Fig. 3). These findings indicated that
nelfinavir decreased fluorogenic substrate cleavage by endogenous
γ-secretase, suggesting an inhibitory effect on γ-secretase
activity. In addition, nelfinavir inhibited γ-secretase activity in
membrane extract from HPB-ALL cells. Similar to compound E
treatment as a positive control, nelfinavir inhibited γ-secretase
activity in a dose-dependent manner after 2 h treatment, indicating
that nelfinavir inhibited the NOTCH1 pathway via inhibition of
γ-secretase activity.
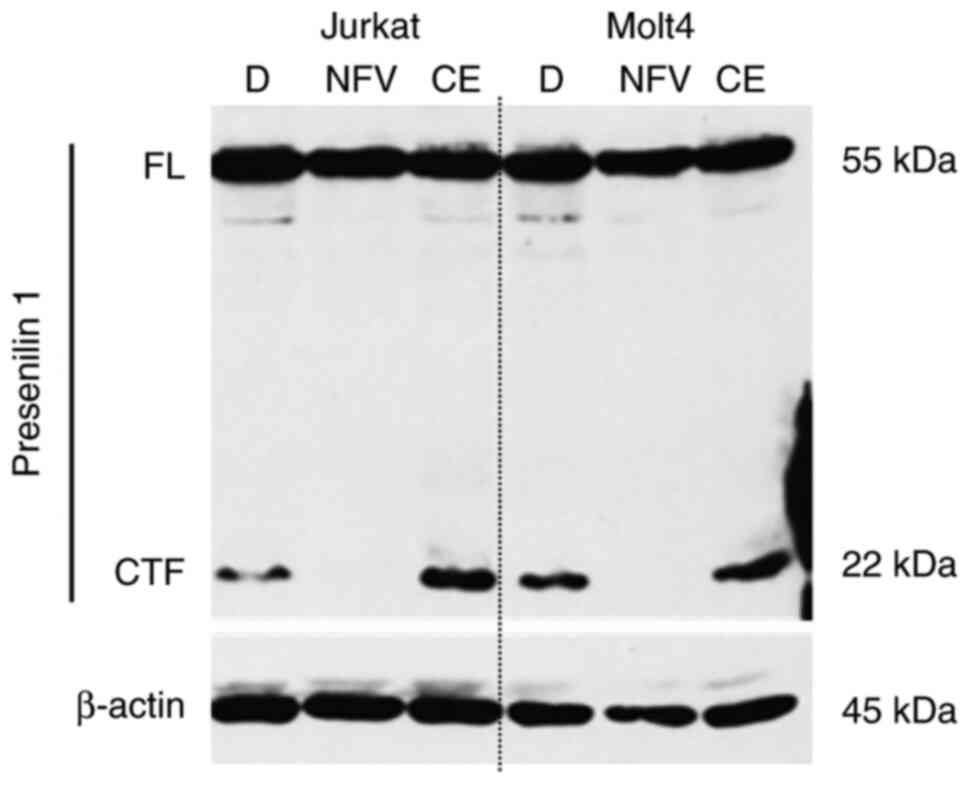
Nelfinavir blocks presenilin 1
processing
To assess how nelfinavir inhibits γ-secretase
activity, the effect of nelfinavir on presenilin processing was
examined. T-ALL cells were treated with nelfinavir and full-length
(FL) and the C-terminal fragment (CTF) of presenilin 1 were
detected via immunoblotting (Fig.
4). Nelfinavir notably decreased expression of presenilin 1
CTF, suggesting that it blocked autoproteolytic processing of FL
presenilin 1. Taken together, these findings suggested that
nelfinavir inhibited γ-secretase activity by blocking presenilin 1
processing.
Nelfinavir increases CHAC1 expression and
inhibits the mTOR pathway
To determine the nelfinavir mechanism of action in
T-ALL cells, HPB-ALL cells were treated with nelfinavir for 4 h
before microarray assays. Among the top 50 differentially expressed
genes, HES family members HES1 and HES4 were
downregulated by nelfinavir compared with DMSO treatment (Fig. 5A). In addition, NOTCH1 mRNA
was moderately decreased by approximately 30%, along with
downregulation of the NOTCH target gene c-Myc (Data S1). Nelfinavir upregulated
CHAC1 (Fig. 5A), a
negative regulator of NOTCH that is induced by ER stress (37) and interferes with NOTCH maturation
by blocking S1 cleavage, which results in NOTCH pathway inhibition
(38). To assess whether CHAC1 is
involved in nelfinavir-induced NOTCH1 inhibition, immunoblotting
analyses were performed for CHAC1 as well as for FL, transmembrane,
and NICD of NOTCH1 (Figs. 5B and
S3A). In agreement with the
microarray data, nelfinavir increased CHAC1 expression in the three
different cell lines. In addition, nelfinavir downregulated NICD as
well as the transmembrane and full-length forms of NOTCH1,
suggesting that CHAC1 interferes with NOTCH1 maturation.
Additionally, nelfinavir enhanced eIF2α phosphorylation, indicative
of ER stress induction. These findings suggest that nelfinavir
inhibited the NOTCH1 pathway by inhibiting γ-secretase activity and
interfering with NOTCH1 maturation through the upregulation of
CHAC1 expression via ER stress induction.
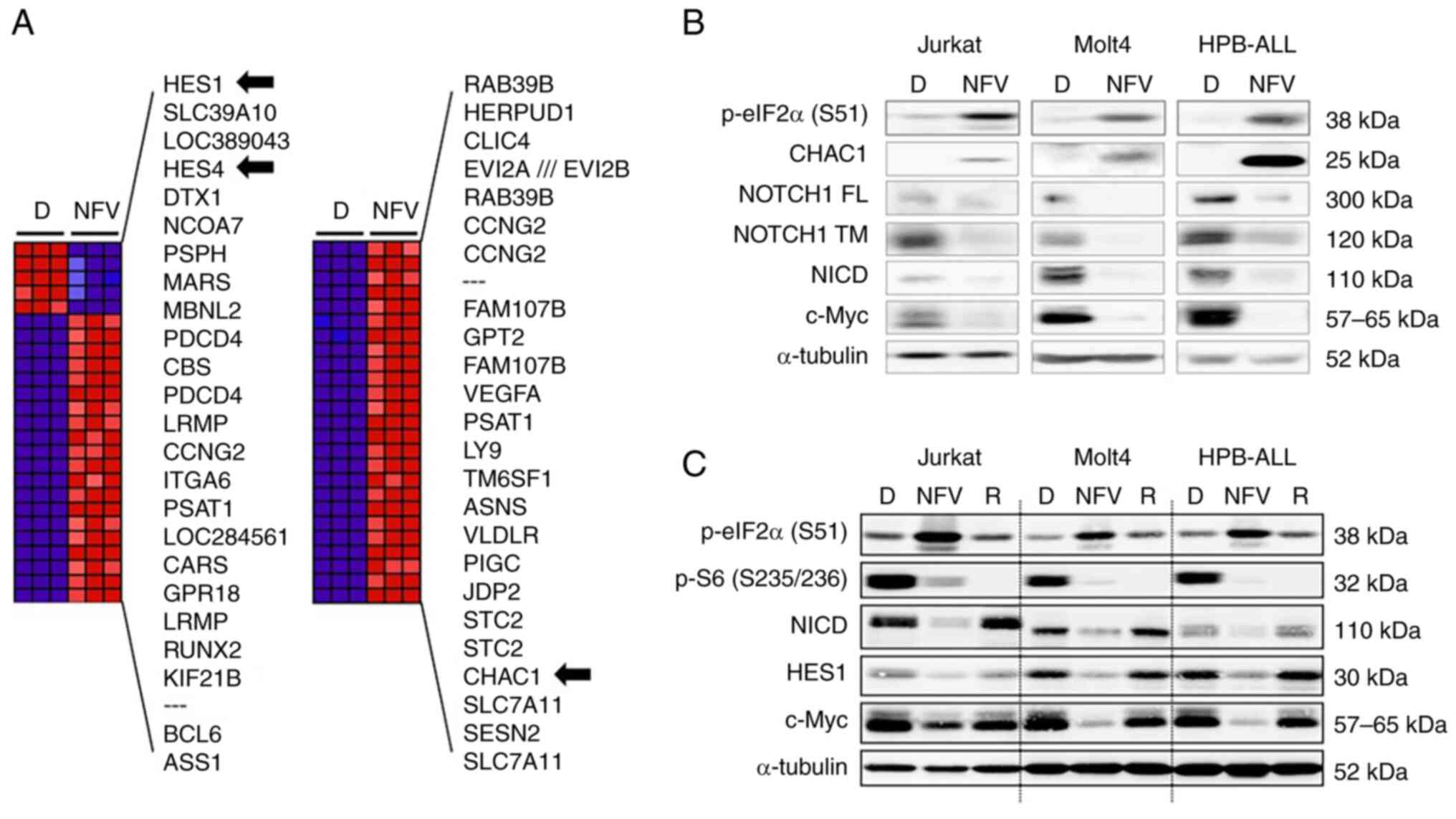 | Figure 5NFV increases CHAC1 expression and
inhibits the mTOR pathway. (A) Expression changes induced by NFV.
Top 50 differentially expressed genes in HPB-ALL cells treated with
20 μM NFV for 4 h compared with 0.1% D treatment. Blue,
relative decrease; red, relative increase; black arrow,
NOTCH-related factors; SESN2, a negative regulator of mTOR. (B)
T-ALL cells were treated with 0.1% D, 20 μM NFV or (C) 100
nM R for 6 h to confirm inhibition of the mTOR pathway.
Immunoblotting analyses were performed. CHAC1, ChaC
glutathione-specific γ-glutamylcyclotransferase 1; HES1, hairy and
enhancer of split-1; p-eIF2α (S51), phosphorylated eukaryotic
initiation factor 2 α subunit at Ser51; p-S6 (S235/236),
phosphorylated S6 ribosomal protein at Ser235/236; FL, full-length;
TM, transmembrane; NICD, NOTCH1 intracellular domain; T-ALL, T cell
acute lymphoblastic leukemia; D, DMSO, NFV, nelfinavir; R,
rapamycin; SESN2, sestrin 2. |
Among the top 50 differentially expressed genes
under nelfinavir treatment, negative mTOR regulator SESN2
was upregulated by nelfinavir (Fig.
5A). To assess whether nelfinavir inhibited the mTOR pathway in
T-ALL cell lines, immunoblotting analysis was performed for S6,
which is an mTOR target (33).
Nelfinavir decreased levels of phosphorylated S6, indicative of
mTOR inhibition, and induced ER stress by enhancing levels of
phosphorylated eIF2α (Figs. 5C
and S3B). These findings
suggested that nelfinavir inhibited the mTOR pathway by increasing
SESN2 expression via the induction of ER stress.
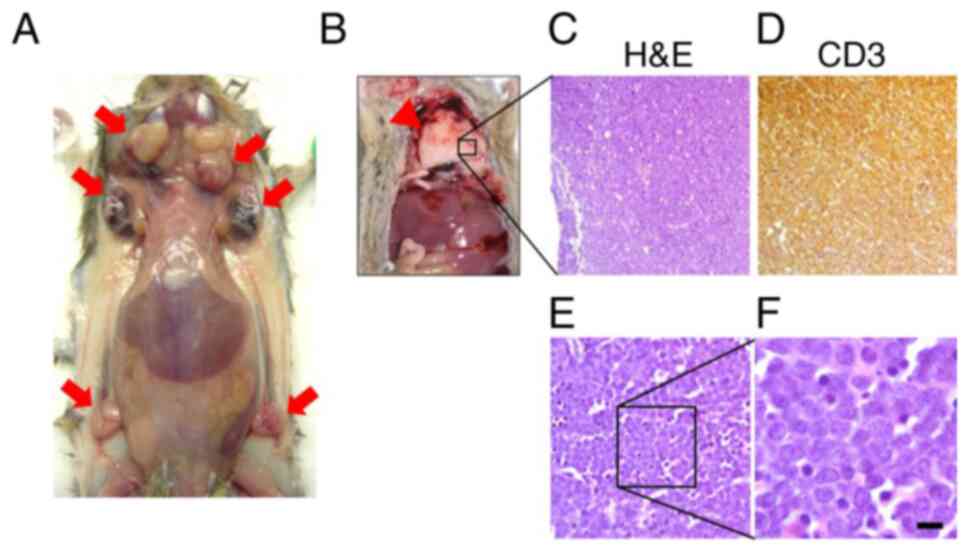
SCL-LMO1 transgenic mice develop T cell
malignancy
Given that SCL-LMO1 transgenic mice that
develop T cell malignancies have a high frequency of Notch1
activation due to acquisition of somatic Notch1 mutations
(8,39), these mice were used to evaluate
the therapeutic potential of nelfinavir in vivo. The mice
presented thymic tumors with or without peripheral lymphadenopathy
between 14 and 25 weeks of age (Fig.
6A and B) and respiratory distress due to tracheal compression
by the enlarged thymus. H&E staining showed no intact thymic
tissue, whereas CD3-positive cells of T cell lineage proliferated
diffusely (Fig. 6C and D). The
proliferating cells were lymphoblasts with scant cytoplasm,
indicative of a high nuclear-cytoplasmic ratio (Fig. 6E and F). These T lymphoblastic
cells were diffusely involved in the bone marrow and enlarged
spleen, comprising 4-10% of leukocytes in the peripheral blood
based on blood smear examination (data not shown). The
immunophenotype of T lymphoblastic cells in the bone marrow,
enlarged thymus and spleen exhibited CD4 and CD8 double-positive
staining in flow cytometry analyses (data not shown). Collectively,
these data supported our previous findings (8) that SCL-LMO1 transgenic mice
develop T cell malignancies and are useful as a mouse model of
human T-ALL.
Nelfinavir decreases T cell malignant TB
in SCL-LMO1 transgenic mice
Mice were intermittently scanned by micro-CT from
the age of 14 weeks to assess disease onset, defined by thymus
enlargement with or without peripheral lymphadenopathy, prior to
nelfinavir treatment. Nelfinavir treatment resulted in shrinkage of
the thymic tumor and peripheral lymphadenopathy, indicating that
malignant tumor cells (T lymphoblastic cells) were decreased
(Fig. 7A). RR in mice nos. 4661
and 4693 was 73.6 and 86.6%, respectively (Table SI). All nine mice treated with
nelfinavir showed PR (Figs. 7B,
S4 and S5; Table
SI) and a 74% reduction in TB (Fig. 7C). Moreover, nelfinavir-treated
mice showed a decrease in the number of CD4+/CD8+ tumor cells in
the thymus as per flow cytometry analyses (Fig. 7D), in addition to more
TUNEL-positive cells, indicative of tumor cell apoptosis enhanced
by nelfinavir treatment. As shown by CD3 staining, malignant T
cells in the bone marrow were decreased by nelfinavir
treatment.
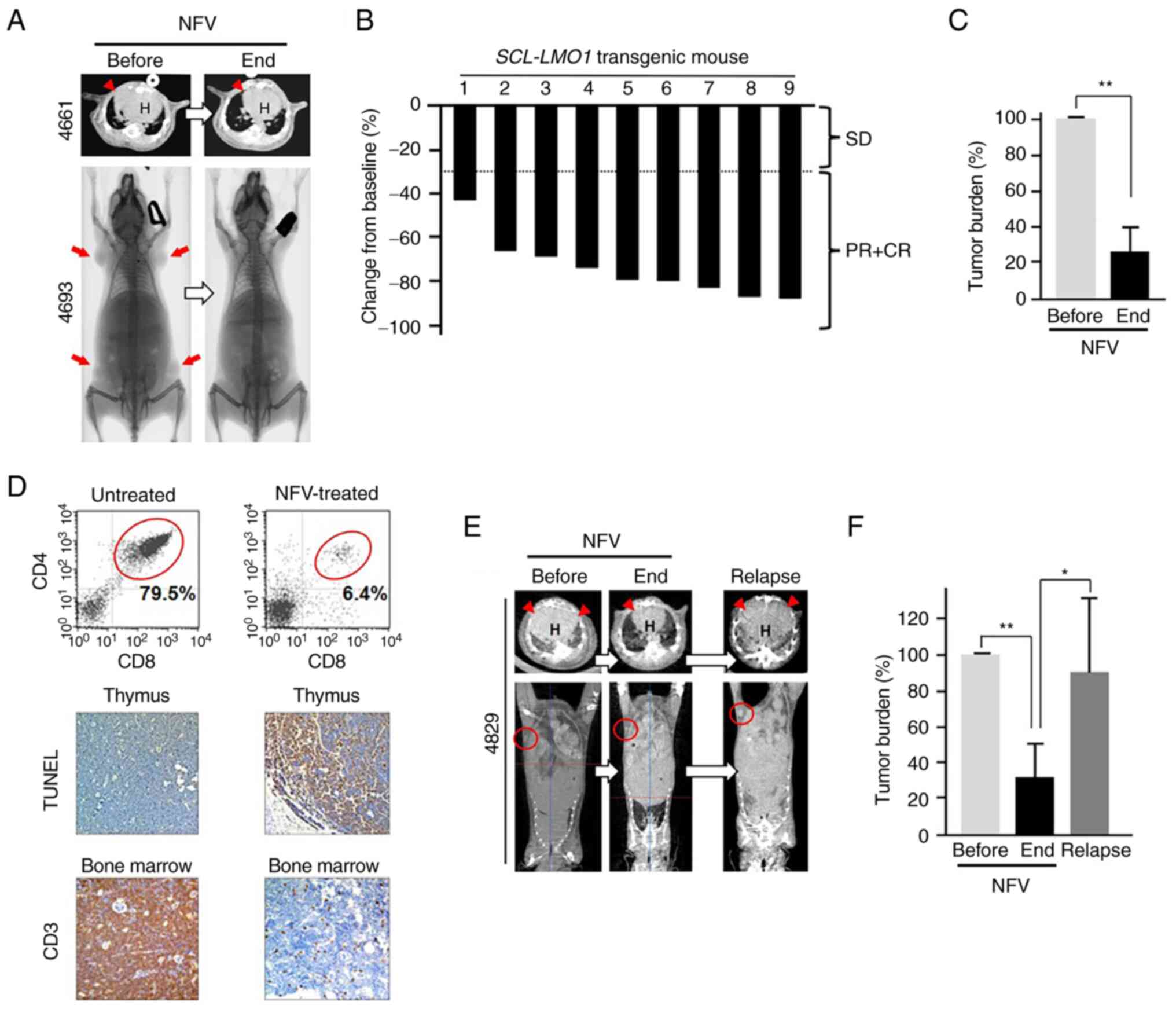 | Figure 7NFV decreases T cell malignant tumor
burden in SCL-LMO1 transgenic mice. (A) Representative
micro-CT image of thymic tumor (red arrowhead) in mouse no. 4661
and whole-body X-ray of lymphadenopathy (tumor in lymph nodes, red
arrow) in mouse no. 4693. Mice treated with 100 mg/kg NFV
intraperitoneally daily for 2 weeks showed tumor regression. The
mice were subjected to whole-body X-ray and micro-CT before and
after NFV treatment. (B) Waterfall plot of change in tumor burden
from baseline, based on response rate to NFV treatment. (C) NFV
decreases tumor burden based on maximum tumor area in
SCL-LMO1 transgenic mice. (D) Efficacy of NFV treatment in
SCL-LMO1 transgenic mice. NFV treatment decreased CD4+/CD8+
(double-positive; red circle) tumor cells in the thymus compared
with those in an untreated mouse, as per flow cytometry. Apoptosis
of tumor cells in the thymus was increased by NFV treatment, as per
TUNEL staining assay. CD3 staining showed population of T malignant
tumor cells in the bone marrow decreased following NFV treatment.
Magnification, ×200. (E) NFV-withdrawal study in SCL-LMO1
transgenic mice. Representative micro-CT images of mouse no. 4829
in panel. Treatment with 100 mg/kg NFV intraperitoneally daily for
2 weeks decreased tumor burden (red arrowhead, thymic tumor; red
circle, lymphadenopathy indicating tumor in a lymph node). (F) NFV
withdrawal induced tumor progression (relapse).
*P<0.05, **P<0.01. H, heart; CR,
complete response; PR, partial response; SD, stable disease; NFV,
nelfinavir. |
To confirm the efficacy of nelfinavir against T cell
malignancy in SCL-LMO1 transgenic mice,
nelfinavir-withdrawal study was performed. Treatment with 100 mg/kg
nelfinavir for 2 weeks decreased TB in the thymus and peripheral
lymph nodes, while nelfinavir withdrawal for 21 days induced tumor
progression (Figs. 7E and
S5). Mice treated with
nelfinavir exhibited a mean 68% reduction in TB, whereas nelfinavir
withdrawal induced tumor progression, indicating relapse (Fig. 7F; Table SII). Collectively, these findings
indicated that nelfinavir exerted an antitumor effect against T-ALL
in vivo.
SCL-LMO1 transgenic mice develop T cell
malignancies, with a frequency of Notch1 mutations similar
to that in humans (39).
Therefore, to confirm an association between nelfinavir efficacy
and Notch mutations, Notch1 exons 26 and 27 (HD), 28
(JM) and 34 (PEST domain), as well as acquired somatic mutations at
the 5′ end, were sequenced in the tumors of treated mice.
Sequencing showed that eight out of nine tissues had a PEST domain
mutation; two out these also had a somatic deletion at the 5′ end
of Notch1 (Table SI).
Mutations in HD and JM were not detected. In total, Notch1
mutations were detected in eight out of nine SCL-LMO1
transgenic mice, indicating that these mice experience a
physiological response to nelfinavir.
Discussion
The present data showed that nelfinavir blocked
presenilin 1 processing and activity of γ-secretase, thus blocking
S3 cleavage, resulting in inhibition of NOTCH1 pathway signaling
and suppression of T-ALL cell viability. These findings support the
hypothesis that nelfinavir suppresses the NOTCH pathway via
γ-secretase inhibition, specifically by blocking presenilin, which
may render it effective against NOTCH-associated T-ALL. Microarray
assays revealed that nelfinavir upregulated CHAC1, which
interfered with NOTCH1 maturation by blocking S1 cleavage. In
addition, nelfinavir downregulated NOTCH1 mRNA expression.
Taken together, these findings suggested that nelfinavir inhibited
the NOTCH1 pathway through multiple mechanisms, including
suppression of NOTCH1 gene expression, upregulation of CHAC1
expression to interfere with NOTCH1 maturation and γ-secretase
inhibition by blocking presenilin 1 processing (Fig. 8).
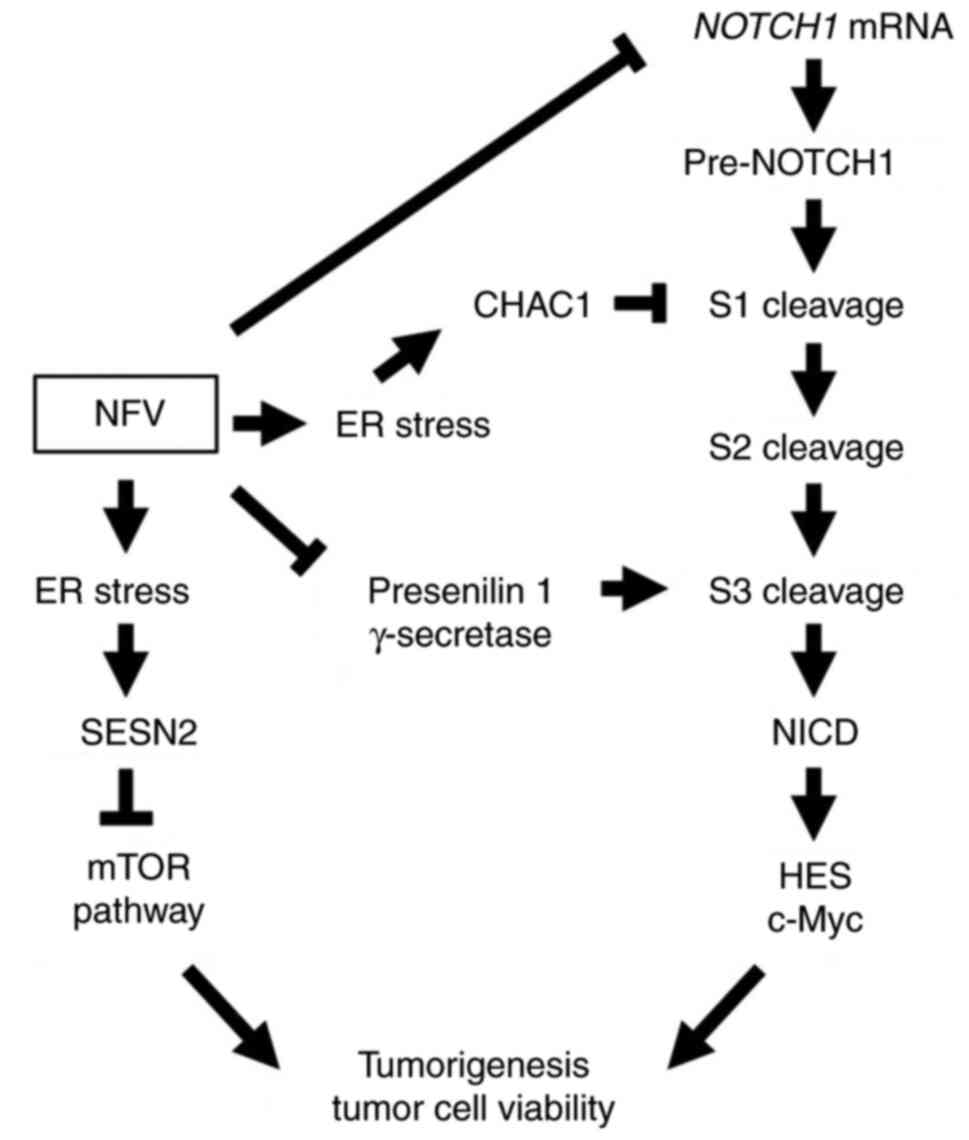 | Figure 8NFV inhibits the NOTCH1 pathway by
downregulating NOTCH1 mRNA, interfering with NOTCH1
maturation, wherein S1 cleavage is blocked by increasing CHAC1
expression via ER stress induction, and γ-secretase inhibition
through blockade of presenilin 1 processing, resulting in blocked
S3 cleavage. NFV inhibits the mTOR pathway by increasing SESN2
expression via ER stress induction. ER, endoplasmic reticulum;
CHAC1, ChaC glutathione-specific γ-glutamylcyclotransferase 1;
NICD, NOTCH1 intracellular domain; HES, hairy and enhancer of
split; SESN2, sestrin 2; NFV, nelfinavir. |
To the best of our knowledge, the present study is
the first to describe that nelfinavir, as an HIV1 aspartic protease
inhibitor, inhibits human aspartic protease presenilin 1, resulting
in NOTCH1 pathway blockade. Aspartic proteases are divided into
five superfamilies (clans): AA, AC, AD, AE and AF. Clan AA includes
the HIV protease and classical aspartic peptidases, such as renin,
pepsin and cathepsin, whereas clan AD includes
intramembrane-cleaving proteases, such as presenilin and signal
peptide peptidase (31,40). Recently, Gu et al (41) reported that nelfinavir inhibits
human aspartic protease DNA damage-inducible 1 homolog 2 (DDI2),
which belongs to Clan AA. Human DDI2 has a retroviral protease-like
domain that is highly conserved in HIV protease, which results in
the direct binding of nelfinavir to the domain for inhibition of
DDI2 activity (41). It remains
unclear whether presenilin has a retroviral protease-like domain
(42,43) and whether nelfinavir could
directly bind to presenilin remains to be solved. Here, nelfinavir
notably decreased the expression of the CTF of presenilin 1, which
indicated inhibition of presenilin 1 activity as an aspartic
protease, because CTF is essential for presenilin 1 activation
(13). Endoproteolytic cleavage
of CTF is mediated by the hydrophobic domain encoded by exon 9 of
presenilin 1 (44).
Nelfinavir-induced downregulation of CTF may be attributed to the
inhibition of presenilin 1 endoproteolytic cleavage. Presenilinase
is the enzyme responsible for endoproteolytic cleavage of
presenilin. Current evidence suggests that presenilinase is
presenilin, making presenilin endoproteolysis an autocleavage event
(45). Further studies are needed
to identify the nelfinavir mechanism responsible for CTF
downregulation. Taken together, the aforementioned studies
demonstrate that nelfinavir inhibits presenilin 1 activity for
γ-secretase inhibition by blocking presenilin 1 processing via CTF
downregulation, rather than by directly binding the catalytic
domain of presenilin 1.
Presenilin is an intramembrane-cleaving protease,
with homologous polytopic proteins presenilin 1 and presenilin 2
forming the presenilin 1- or presenilin 2-containing γ-secretase
complexes, respectively (46). In
a previous study using presenilin-wild-type and knockout mouse
embryonic fibroblasts (MEFs), presenilin 1, 2 or presenilin 1 and 2
double (−/−) MEFs were treated with EDTA to induce
ligand-independent Notch activation (47), showing that presenilin 1 serves a
key role in Notch1 activation. The presenilin 1-containing
γ-secretase complex is primarily localized in the cytoplasmic
membrane, whereas the presenilin 2-containing complex localizes to
a greater extent at endosomes and lysosomes, including the
trans-Golgi network (48).
Moreover, the murine presenilin 1-containing γ-secretase complex
serves a greater role in Notch activation compared with the
presenilin 2-containing complex (49). The aforementioned findings suggest
that presenilin 1 is a more important target molecule for Notch
inhibition than presenilin 2. The present study performed cell-free
in vitro γ-secretase assays by preparing cytoplasmic
membrane-enriched cell fractions and showed that nelfinavir
decreased cleavage of fluorogenic substrates by endogenous
γ-secretase. However, the extent to which presenilin 1 was present
in the endogenous γ-secretase complex was not determined, which is
a limitation of the present study. Nonetheless, given that the
presenilin 1-containing γ-secretase complex is primarily localized
in the cytoplasmic membrane (48), presenilin 1 plays a major role in
Notch1 activation and nelfinavir inhibits presenilin 1 activity by
downregulating CTF, it is hypothesized that nelfinavir inhibits
γ-secretase activity by inhibiting presenilin 1 rather than
presenilin 2 activity. MRK-560 experiments suggest that presenilin
1-selective inhibition is a potential therapeutic strategy for safe
and effective targeting of T-ALL (24). Taken together, nelfinavir may
inhibit γ-secretase activity with less gastrointestinal toxicity
than broad-spectrum GSIs.
Microarray analysis showed that nelfinavir enhanced
CHAC1 gene expression. CHAC1 is a proapoptotic protein that
is upregulated via the eIF2α/activating transcription factor (ATF)
4/ATF3/C/EBP homologous protein pathway during ER stress (50) and functions as a γ-glutamyl
cyclotransferase (GGCT) to degrade glutathione (51). Chi et al (38) reported that CHAC1 inhibits the
NOTCH1 pathway by serving as a GGCT to remove glycine from a
γ-carbon-modified glutamate at position 1,669 of NOTCH1, resulting
in blockade of S1 cleavage by furin-like protease in the Golgi
apparatus. This interferes with NOTCH1 maturation, leading to lower
cell surface expression of FL NOTCH1. Therefore, CHAC1 is also
named blocker of NOTCH (38,52). Here, nelfinavir induced ER stress,
as indicated by upregulation of phosphorylated eIF2α and CHAC1
expression and downregulated NICD, suggesting that nelfinavir
inhibited the NOTCH1 pathway by interfering with NOTCH1 maturation
through increased CHAC1 following ER stress induction. Conversely,
nelfinavir decreased the transmembrane and FL forms of NOTCH1 in
T-ALL cells. The present study did not confirm whether nelfinavir
decreases cell surface expression of FL NOTCH1. Microarray analysis
showed that nelfinavir moderately decreased NOTCH1 mRNA,
suggesting that nelfinavir may interfere with NOTCH1 transcription,
resulting in a decrease in the levels of both transmembrane and FL
forms. Taken together, nelfinavir increases CHAC1 expression to
interfere with NOTCH1 maturation by blocking S1 cleavage and
decreasing NOTCH1 mRNA expression, resulting in NOTCH1
pathway inhibition.
Microarray showed that nelfinavir upregulated
SESN2 gene expression, suggesting that nelfinavir may
inhibit the mTOR pathway in T-ALL cells. mTOR is the catalytic
subunit of mTOR complex 1 (mTORC1) and mTORC2. mTORC1 is activated
via the PI3K/AKT/mTOR pathway to phosphorylate downstream target
molecules, such as S6, promoting cell proliferation. By contrast,
mTORC2 directly phosphorylates AKT at Ser473, indicative of a
feedback loop between AKT, mTORC1 and mTORC2 (53,54). SESN2 is a stress-responsive
gene, also known as hypoxia-induced gene 95 (55), which shares considerable homology
with p53-regulated GADD family member PA26 (56) and was named after the Italian town
Sestri Levante (57). SESN2 is
upregulated by ER stress induction (58) and inhibits mTORC1 activation via
upregulation of nutrient-responsive AMPK and modulation of GAP
activity toward Rags 2 (GATOR2). Both AMPK and GATOR2 negatively
regulate the mTORC1 pathway (59). Here, nelfinavir decreased
phosphorylated S6 expression, indicating mTORC1 inhibition, which
suggests that nelfinavir inhibited the mTOR pathway by upregulating
SESN2 through ER stress induction in T-ALL cells. These findings
are supported by reports in other malignancies, such as breast and
ovarian cancer and SCLC (29,60). As the PI3K/AKT/mTOR pathway is
involved in mechanisms of resistance to NOTCH inhibition in T-ALL
(25,26), nelfinavir may be cytotoxic to
T-ALL cells via NOTCH1 inhibition and suppression of the
mTOR-mediated resistance.
Nelfinavir decreased the T cell malignant TB in
SCL-LMO1 transgenic mice with Notch1 mutation,
indicating that nelfinavir might have therapeutic potential for
NOTCH-associated human T-ALL. The present in vivo study had
several limitations. First, the present study did not include a
control cohort of mice treated with mock/vehicle. Measurable
lesions (thymic masses, with or without peripheral lymphadenopathy)
were screened via micro-CT. Once the lesion was detected,
nelfinavir treatment was started as soon as possible since the
mouse would die from respiratory distress caused by tracheal
compression unless effective treatment were provided. The present
study compared TB before and after nelfinavir treatment. Second,
the present study did not evaluate the mechanisms of action by
which nelfinavir decreased TB because residual tumors following
nelfinavir treatment were used for Notch1 mutation analyses
and immunophenotype evaluation. Further studies of in vivo
pharmacodynamics are needed to confirm nelfinavir mechanisms of
action. Third, one (mouse no. 4661) nelfinavir-treated mouse
experienced diarrhea on day 11 of nelfinavir treatment; however,
the present study did not assess the possibility of
gastrointestinal toxicity following Notch inhibition based on
gastrointestinal tissue specimens. However, the other mice did not
exhibit any gastrointestinal toxicity, indicating that nelfinavir
may selectively target γ-secretase, as in the case of the
presenilin 1-specific inhibitor MRK-560 (24). Taken together, the present in
vivo study using SCL-LMO1 transgenic mice supported the
potential of nelfinavir against T-ALL.
F-box and WD repeat domain containing 7 (FBW7) is a
ubiquitin ligase that plays a role in processes such as cell cycle
progression and signal transduction through ubiquitination and
degradation of its substrates, such as NOTCH and mTOR, suggesting a
key role in both NOTCH and mTOR pathways (61,62). The present study confirmed that
nelfinavir inhibited both pathways in an FBW7-independent manner
using FBW7 knockout DLD1 colorectal cancer cells (data not
shown). ER stress induced by nelfinavir may underlie NOTCH and mTOR
pathway inhibition via the upregulation of CHAC1 and SESN2.
Collectively, the present findings highlight the potential of
nelfinavir as novel therapeutics requiring further validation in
T-ALL clinical trials.
Supplementary Data
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the Figshare repository, figshare.com/(accession no.
10.6084/m9.figshare.24061842).
Authors' contributions
YSC, JJG and SK designed and performed experiments,
analyzed and interpreted data and wrote the manuscript. MO
performed in vivo experiments and analyzed and interpreted
data. GDG performed, analyzed and interpreted the microarray
experiments. AAF supervised the microarray experiments and
interpreted data. PDA provided SCL-LMO1 transgenic mice and
interpreted in vivo data. PAD conceived and supervised the
study. All authors have read and approved the final manuscript. YSC
and SK confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
In vivo experiments were approved by the
NCI-Bethesda Animal Care and Use Committee (approval no. CTB-008,
Bethesda, MD, USA).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Mr Juan Morales
Contreras, Mr Dumas Tarra and Dr John U Dennis (Laboratory Animal
Medicine, NCI, Bethesda, MD, USA) for veterinary services and Ms
Danielle Donahue (Mouse Imaging Facility, National Institute of
Neurological Disorders and Stroke, NIH) for technical assistance
with mouse whole-body X-ray and micro-CT scans. The abstract was
presented at the 102nd annual meeting of the American Association
of Cancer Research [2011 Apr 2-6, Orlando, Florida, USA; Cancer Res
2011;71(8 Suppl):Abstract nr 2603].
Funding
The present study was supported by the Intramural Research
Program of the NIH, Center for Cancer Research, NCI (grant no. ZIA
SC 010378), Johns Hopkins University School of Medicine and Japan
Society for the Promotion of Science KAKENHI (grant no.
JP20K07646).
References
|
1
|
Borowitz MJ, Chan JKC, Béné M and Arber
DA: T-lymphoblastic leukemia/lymphoma. WHO Classification of
Tumours of Haematopoietic and Lymphoid Tissues. Revised 4th
edition. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA,
Stein H and Thiele J: The International Agency for Research on
Cancer; Lyon: pp. 209–213. 2017
|
|
2
|
Czader M, Molina TJ, Choi JK, Leventaki V,
Miles RR, Lin P, Saha V, Tembhare P and Chandy M: T-lymphoblastic
leukaemia/lymphoma NOS. WHO Classification of Tumours Editorial
Board: Haematolymphoid tumours [Internet; beta version ahead of
print]. 11:5th edition. International Agency for Research on
Cancer; Lyon: 2022, https://tumourclassification.iarc.who.int/chapters/63.
Accessed January 17, 2023
|
|
3
|
Tan TK, Zhang C and Sanda T: Oncogenic
transcriptional program driven by TAL1 in T-cell acute
lymphoblastic leukemia. Int J Hematol. 109:5–17. 2019. View Article : Google Scholar
|
|
4
|
Bardelli V, Arniani S, Pierini V, Di
Giacomo D, Pierini T, Gorello P, Mecucci C and La Starza R: T-cell
acute lymphoblastic leukemia: Biomarkers and their clinical
usefulness. Genes (Basel). 12:11182021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ferrando AA, Neuberg DS, Staunton J, Loh
ML, Huard C, Raimondi SC, Behm FG, Pui CH, Downing JR, Gilliland
DG, et al: Gene expression signatures define novel oncogenic
pathways in T cell acute lymphoblastic leukemia. Cancer Cell.
1:75–87. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu Y, Easton J, Shao Y, Maciaszek J, Wang
Z, Wilkinson MR, McCastlain K, Edmonson M, Pounds SB, Shi L, et al:
The genomic landscape of pediatric and young adult T-lineage acute
lymphoblastic leukemia. Nat Genet. 49:1211–1218. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Begley CG, Aplan PD, Davey MP, Nakahara K,
Tchorz K, Kurtzberg J, Hershfield MS, Haynes BF, Cohen DI, Waldmann
TA, et al: Chromosomal translocation in a human leukemic stem-cell
line disrupts the T-cell antigen receptor delta-chain diversity
region and results in a previously unreported fusion transcript.
Proc Natl Acad Sci USA. 86:2031–2035. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Aplan PD, Jones CA, Chervinsky DS, Zhao X,
Ellsworth M, Wu C, McGuire EA and Gross KW: An scl gene product
lacking the transactivation domain induces bony abnormalities and
cooperates with LMO1 to generate T-cell malignancies in transgenic
mice. EMBO J. 16:2408–2419. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tremblay M, Tremblay CS, Herblot S, Aplan
PD, Hébert J, Perreault C and Hoang T: Modeling T-cell acute
lymphoblastic leukemia induced by the SCL and LMO1 oncogenes. Genes
Dev. 24:1093–1105. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Grabher C, von Boehmer H and Look AT:
Notch 1 activation in the molecular pathogenesis of T-cell acute
lymphoblastic leukaemia. Nat Rev Cancer. 6:347–359. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou B, Lin W, Long Y, Yang Y, Zhang H, Wu
K and Chu Q: Notch signaling pathway: Architecture, disease, and
therapeutics. Signal Transduct Target Ther. 7:952022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wolfe MS, Xia W, Ostaszewski BL, Diehl TS,
Kimberly WT and Selkoe DJ: Two transmembrane aspartates in
presenilin-1 required for presenilin endoproteolysis and
gamma-secretase activity. Nature. 398:513–517. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Levitan D, Lee J, Song L, Manning R, Wong
G, Parker E and Zhang L: PS1 N- and C-terminal fragments form a
complex that functions in APP processing and Notch signaling. Proc
Natl Acad Sci USA. 98:12186–12190. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Güner G and Lichtenthaler SF: The
substrate repertoire of γ-secretase/presenilin. Semin Cell Dev
Biol. 105:27–42. 2020. View Article : Google Scholar
|
|
15
|
Steinbuck MP and Winandy S: A review of
Notch processing with new insights into ligand-independent Notch
signaling in T-cells. Front Immunol. 9:12302018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hosokawa H and Rothenberg EV: How
transcription factors drive choice of the T cell fate. Nat Rev
Immunol. 21:162–176. 2021. View Article : Google Scholar
|
|
17
|
Weng AP, Ferrando AA, Lee W, Morris JP IV,
Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT and Aster
JC: Activating mutations of NOTCH1 in human T cell acute
lymphoblastic leukemia. Science. 306:269–271. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sulis ML, Williams O, Palomero T, Tosello
V, Pallikuppam S, Real PJ, Barnes K, Zuurbier L, Meijerink JP and
Ferrando AA: NOTCH1 extracellular juxtamembrane expansion mutations
in T-ALL. Blood. 112:733–740. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ferrando AA: The role of NOTCH1 signaling
in T-ALL. Hematology Am Soc Hematol Educ Program. 353–361. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ashworth TD, Pear WS, Chiang MY, Blacklow
SC, Mastio J, Xu L, Kelliher M, Kastner P, Chan S and Aster JC:
Deletion-based mechanisms of Notch1 activation in T-ALL: Key roles
for RAG recombinase and a conserved internal translational start
site in Notch1. Blood. 116:5455–5464. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Real PJ, Tosello V, Palomero T, Castillo
M, Hernando E, de Stanchina E, Sulis ML, Barnes K, Sawai C,
Homminga I, et al: Gamma-secretase inhibitors reverse
glucocorticoid resistance in T cell acute lymphoblastic leukemia.
Nat Med. 15:50–58. 2009. View Article : Google Scholar
|
|
22
|
López-Nieva P, González-Sánchez L,
Cobos-Fernández MÁ, Córdoba R, Santos J and Fernández-Piqueras J:
More insights on the use of γ-secretase inhibitors in cancer
treatment. Oncologist. 26:e298–e305. 2021. View Article : Google Scholar
|
|
23
|
Baratta MG: Adjusting the focus on
γ-secretase inhibition. Nat Rev Cancer. 19:4192019. View Article : Google Scholar
|
|
24
|
Habets RA, De Bock CE, Serneels L,
Lodewijckx I, Verbeke D, Nittner D, Narlawar R, Demeyer S, Dooley
J, Liston A, et al: Safe targeting of T cell acute lymphoblastic
leukemia by pathology-specific NOTCH inhibition. Sci Transl Med.
11:eaau62462019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Palomero T, Sulis ML, Cortina M, Real PJ,
Barnes K, Ciofani M, Caparros E, Buteau J, Brown K, Perkins SL, et
al: Mutational loss of PTEN induces resistance to NOTCH1 inhibition
in T-cell leukemia. Nat Med. 13:1203–1210. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Martelli AM, Paganelli F, Fazio A,
Bazzichetto C, Conciatori F and McCubrey JA: The key roles of PTEN
in T-cell acute lymphoblastic leukemia development, progression,
and therapeutic response. Cancers (Basel). 11:6292019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gills JJ, Lopiccolo J, Tsurutani J,
Shoemaker RH, Best CJ, Abu-Asab MS, Borojerdi J, Warfel NA, Gardner
ER, Danish M, et al: Nelfinavir, A lead HIV protease inhibitor, is
a broad-spectrum, anticancer agent that induces endoplasmic
reticulum stress, autophagy, and apoptosis in vitro and in vivo.
Clin Cancer Res. 13:5183–5194. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Blumenthal GM, Gills JJ, Ballas MS,
Bernstein WB, Komiya T, Dechowdhury R, Morrow B, Root H, Chun G,
Helsabeck C, et al: A phase I trial of the HIV protease inhibitor
nelfinavir in adults with solid tumors. Oncotarget. 5:8161–8172.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kawabata S, Connis N, Gills JJ, Hann CL
and Dennis PA: Nelfinavir inhibits the growth of small-cell lung
cancer cells and patient-derived xenograft tumors. Anticancer Res.
41:91–99. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Subeha MR and Telleria CM: The anti-cancer
properties of the HIV protease inhibitor Nelfinavir. Cancers
(Basel). 12:34372020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Eder J, Hommel U, Cumin F, Martoglio B and
Gerhartz B: Aspartic proteases in drug discovery. Curr Pharm Des.
13:271–285. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lobbardi R, Pinder J, Martinez-Pastor B,
Theodorou M, Blackburn JS, Abraham BJ, Namiki Y, Mansour M,
Abdelfattah NS, Molodtsov A, et al: TOX regulates growth, DNA
repair, and genomic instability in T-cell acute lymphoblastic
leukemia. Cancer Discov. 7:1336–1353. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kawabata S, Mercado-Matos JR, Hollander
MC, Donahue D, Wilson W III, Regales L, Butaney M, Pao W, Wong KK,
Jänne PA and Dennis PA: Rapamycin prevents the development and
progression of mutant epidermal growth factor receptor lung tumors
with the acquired resistance mutation T790M. Cell Rep. 7:1824–1832.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Farmery MR, Tjernberg LO, Pursglove SE,
Bergman A, Winblad B and Näslund J: Partial purification and
characterization of gamma-secretase from post-mortem human brain. J
Biol Chem. 278:24277–24284. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim SK, Park HJ, Hong HS, Baik EJ, Jung MW
and Mook-Jung I: ERK1/2 is an endogenous negative regulator of the
gamma-secretase activity. FASEB J. 20:157–159. 2006. View Article : Google Scholar
|
|
36
|
Kawabata S, Hollander MC, Munasinghe JP,
Brinster LR, Mercado-Matos JR, Li J, Regales L, Pao W, Jänne PA,
Wong KK, et al: Epidermal growth factor receptor as a novel
molecular target for aggressive papillary tumors in the middle ear
and temporal bone. Oncotarget. 6:11357–11368. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bachhawat AK and Kaur A: Glutathione
degradation. Antioxid Redox Signal. 27:1200–1216. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chi Z, Byrne ST, Dolinko A, Harraz MM, Kim
MS, Umanah G, Zhong J, Chen R, Zhang J, Xu J, et al: Botch is a
γ-glutamyl cyclotransferase that deglycinates and antagonizes
Notch. Cell Rep. 7:681–688. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lin YW, Nichols RA, Letterio JJ and Aplan
PD: Notch1 mutations are important for leukemic transformation in
murine models of precursor-T leukemia/lymphoma. Blood.
107:2540–2543. 2006. View Article : Google Scholar
|
|
40
|
Santos LO, Garcia-Gomes AS, Catanho M,
Sodre CL, Santos ALS, Branquinha MH and d'Avila-Levy CM: Aspartic
peptidases of human pathogenic trypanosomatids: Perspectives and
trends for chemotherapy. Curr Med Chem. 20:3116–3133. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gu Y, Wang X, Wang Y, Wang Y, Li J and Yu
FX: Nelfinavir inhibits human DDI2 and potentiates cytotoxicity of
proteasome inhibitors. Cell Signal. 75:1097752020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
De Strooper B, Iwatsubo T and Wolfe MS:
Presenilins and γ-secretase: Structure, function, and role in
Alzheimer disease. Cold Spring Harb Perspect Med. 2:a0063042012.
View Article : Google Scholar
|
|
43
|
Li X, Dang S, Yan C, Gong X, Wang J and
Shi Y: Structure of a presenilin family intramembrane aspartate
protease. Nature. 493:56–61. 2013. View Article : Google Scholar
|
|
44
|
Fukumori A, Fluhrer R, Steiner H and Haass
C: Three-amino acid spacing of presenilin endoproteolysis suggests
a general stepwise cleavage of gamma-secretase-mediated
intramembrane proteolysis. J Neurosci. 30:7853–7862. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gertsik N, Ballard TE, Am Ende CW, Johnson
DS and Li YM: Development of CBAP-BPyne, a probe for γ-secretase
and presenilinase. Medchemcomm. 5:338–341. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sato T, Diehl TS, Narayanan S, Funamoto S,
Ihara Y, De Strooper B, Steiner H, Haass C and Wolfe MS: Active
gamma-secretase complexes contain only one of each component. J
Biol Chem. 282:33985–33993. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Rand MD, Grimm LM, Artavanis-Tsakonas S,
Patriub V, Blacklow SC, Sklar J and Aster JC: Calcium depletion
dissociates and activates heterodimeric notch receptors. Mol Cell
Biol. 20:1825–1835. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Meckler X and Checler F: Presenilin 1 and
presenilin 2 target γ-secretase complexes to distinct cellular
compartments. J Biol Chem. 291:12821–12837. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Stanga S, Vrancx C, Tasiaux B, Marinangeli
C, Karlström H and Kienlen-Campard P: Specificity of presenilin-1-
and presenilin-2-dependent γ-secretases towards substrate
processing. J Cell Mol Med. 22:823–833. 2018. View Article : Google Scholar
|
|
50
|
Mungrue IN, Pagnon J, Kohannim O,
Gargalovic PS and Lusis AJ: CHAC1/MGC4504 is a novel proapoptotic
component of the unfolded protein response, downstream of the
ATF4-ATF3-CHOP cascade. J Immunol. 182:466–476. 2009. View Article : Google Scholar
|
|
51
|
Kumar A, Tikoo S, Maity S, Sengupta S,
Sengupta S, Kaur A and Bachhawat AK: Mammalian proapoptotic factor
ChaC1 and its homologues function as γ-glutamyl cyclotransferases
acting specifically on glutathione. EMBO Rep. 13:1095–1101. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chi Z, Zhang J, Tokunaga A, Harraz MM,
Byrne ST, Dolinko A, Xu J, Blackshaw S, Gaiano N, Dawson TM and
Dawson VL: Botch promotes neurogenesis by antagonizing Notch. Dev
Cell. 22:707–720. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sathe A, Chalaud G, Oppolzer I, Wong KY,
von Busch M, Schmid SC, Tong Z, Retz M, Gschwend JE, Schulz WA and
Nawroth R: Parallel PI3K, AKT and mTOR inhibition is required to
control feedback loops that limit tumor therapy. PLoS One.
13:e01908542018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yang M, Lu Y, Piao W and Jin H: The
translational regulation in mTOR pathway. Biomolecules. 12:8022022.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Budanov AV, Shoshani T, Faerman A, Zelin
E, Kamer I, Kalinski H, Gorodin S, Fishman A, Chajut A, Einat P, et
al: Identification of a novel stress-responsive gene Hi95 involved
in regulation of cell viability. Oncogene. 21:6017–6031. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Velasco-Miguel S, Buckbinder L, Jean P,
Gelbert L, Talbott R, Laidlaw J, Seizinger B and Kley N: PA26, a
novel target of the p53 tumor suppressor and member of the GADD
family of DNA damage and growth arrest inducible genes. Oncogene.
18:127–137. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Peeters H, Debeer P, Bairoch A, Wilquet V,
Huysmans C, Parthoens E, Fryns JP, Gewillig M, Nakamura Y, Niikawa
N, et al: PA26 is a candidate gene for heterotaxia in humans:
Identification of a novel PA26-related gene family in human and
mouse. Hum Genet. 112:573–580. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Saveljeva S, Cleary P, Mnich K, Ayo A,
Pakos-Zebrucka K, Patterson JB, Logue SE and Samali A: Endoplasmic
reticulum stress-mediated induction of SESTRIN 2 potentiates cell
survival. Oncotarget. 7:12254–12266. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lee JH, Cho US and Karin M: Sestrin
regulation of TORC1: Is sestrin a leucine sensor? Sci Signal.
9:re52016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Brüning A, Rahmeh M and Friese K:
Nelfinavir and bortezomib inhibit mTOR activity via ATF4-mediated
sestrin-2 regulation. Mol Oncol. 7:1012–1018. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Hales EC, Taub JW and Matherly LH: New
insights into Notch1 regulation of the PI3K-AKT-mTOR1 signaling
axis: Targeted therapy of γ-secretase inhibitor resistant T-cell
acute lymphoblastic leukemia. Cell Signal. 26:149–161. 2014.
View Article : Google Scholar
|
|
62
|
Shen W, Zhou Q, Peng C, Li J, Yuan Q, Zhu
H, Zhao M, Jiang X, Liu W and Ren C: FBXW7 and the hallmarks of
cancer: Underlying mechanisms and prospective strategies. Front
Oncol. 12:8800772022. View Article : Google Scholar : PubMed/NCBI
|