Introduction
Gene amplification is defined as an increase in the
copy number of a restricted region of a chromosome arm and is one
of the hallmarks of genomic instability (1). Highly amplified genes manifest
themselves as either of two cytogenetically identifiable
structures: Intrachromosomal homogeneously staining regions (HSRs)
and extrachromosomal double minutes (DMs)/extrachromosomal DNA
(ecDNA) (2). Previously, Benner
et al (3) analyzed a
number of primary human cancer types and cancer cell lines and
observed that most cancer cells contained ecDNAs only and a few
contained HSRs or both, which indicated that ecDNAs are the
predominant cytogenetic marker for gene amplification in cancer
cells. ecDNA-based gene amplification drives elevated copy numbers
and promotes intratumoral genetic heterogeneity, suggesting a
pivotal role for ecDNAs in cancer evolution (4). Previously, ecDNAs have usually been
ignored in the standard analytic approaches of high-throughput
short-read DNA sequencing. With the development of sequencing and
analytic technologies, the re-discovery that oncogenes/drug
resistance genes can be amplified through ecDNAs and the newfound
importance of ecDNAs in cancer suggest that eliminating ecDNAs in
cancer cells might be a way to deal with cancer. The underlying
molecular mechanism of the formation of ecDNAs remains to be
elucidated.
A large body of evidence points to double-strand
breaks (DSBs), tandem duplication, breakage-fusion-bridge cycles
and chromothripsis as key intermediates leading to gene
amplification (5-7). Thus, it was hypothesized that the
molecular mechanism of the formation of ecDNAs may involve DSBs and
the subsequent repair pathways. Homologous recombination (HR),
classic non-homologous end joining (c-NHEJ) and alternative
non-homologous end joining (a-NHEJ) are classic repair pathways to
process DSBs utilized by cells. Our previous studies revealed that
HR, c-NHEJ and a-NHEJ (data not shown) were involved in the
formation of ecDNAs in methotrexate (MTX)-resistant colorectal
cancer cells (8,9).
Mismatch repair (MMR) is a highly conserved cellular
process. In addition to its role in the repair of replication
errors, MMR has also been implicated in the repair of DSBs via
classic DSBs repair pathways. Studies have demonstrated that key
components of MMR, particularly MutS homolog (MSH) 3, are involved
in the cellular response to DSBs. For example, MSH3 accumulates
rapidly at sites of DSBs generated by laser micro-irradiation
(10). MSH2-MSH3 binds branched
recombination intermediates and promotes removal of nonhomologous
DNA at DSB ends during single-strand annealing and gene conversion
(two pathways of HR) in Saccharomyces cerevisiae (11-13). MSH3 may cooperate with proteins of
c-NHEJ to recognize and repair platinum drug-induced interstrand
cross-links (13). In addition,
Dillon et al (14)
reported that inhibition of MSH3 could decrease the abundance of
microDNA (a type of small-size ecDNA with no amplified genes),
which may originate from DSBs. Since MSH3 can be involved in the
process of DSB repair and DMs (as a type of large-size ecDNA with
amplified genes) contained amplified genes derived from DSBs, it
was hypothesized that ecDNAs (DMs) may also be regulated by
MSH3-related DNA repair pathways.
It has been found that the elimination of amplified
oncogenes from tumor cells can reverse the tumor phenotype. Studies
have suggested that specific incorporation of ecDNAs into the
cytoplasmic micronuclei (MN)/nuclear buds (NBUDs) participates in
oncogene elimination after treatment with hydroxyurea (HU) and
other chemotherapy drugs (15-17). Our previous studies also revealed
expelled ecDNAs by MN/NBUDs in protein kinase (DNA-PKcs) or
BRCA1-depleted MTX-resistant cancer cells (8,9).
However, to the best of our knowledge, the influence of MMR
depletion on the elimination of ecDNAs by MN/NBUDs has not yet been
elucidated.
In the present study, MTX-resistant HT29 human
colorectal cancer cells were used to investigate the formation
mechanism of ecDNAs in the process of MTX resistance development,
hoping to provide a basis for targeting MSH3 to effectively reverse
tumor drug resistance caused by ecDNAs.
Materials and methods
Cell lines and cell culture
The HT29 human colorectal cancer cell line was
purchased from the Chinese Academy of Sciences and was
authenticated by the Beijing Microread Genetics Co., Ltd. HT29
MTX-resistant cells were generated by continuous culture of
parental HT29 cells in high-glucose Dulbecco's modified Eagle's
medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.) containing 15%
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and
supplemented with increasing concentration of MTX (Calbiochem
Biochemicals; Merck KGaA). COLO 320DM cells were cultured in RPMI
1640 medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 10% FBS. Cells were maintained in a humidified incubator at
37°C supplied with 5% CO2. In the present study,
HSR-containing (containing HSRs only) and DM-containing (containing
more ecDNAs and less HSRs) cells indicate cells resistant to
10−5 and 10−4 mol/l MTX, respectively.
Western blot analysis
Whole-cell extracts were prepared using the RIPA
lysis buffer (Applygen Technologies, Inc.) and protein
concentrations were measured using BCA Protein Assay Kit (Applygen
Technologies, Inc.). Proteins (30 µg per lane) were resolved
on 6.5-15% SDS-PAGE and transferred onto polyvinylidene fluoride
(PVDF) membranes (MilliporeSigma). The PVDF membranes were blocked
with 5% nonfat milk in Tris-buffered saline with 0.05% Tween 20
(TBST) for 1 h at room temperature, primary antibodies were diluted
in 5% BSA (MilliporeSigma) and incubated overnight at 4°C and
fluorochrome-labelled secondary antibodies (Rockland
Immunochemicals Inc.) were incubated for 1 h at room temperature.
Immunoreactivity was detected by Odyssey fluorescence scanning
system (LI-COR Biosciences) at wavelengths of 800 nm or 700 nm.
Protein expression was quantified utilizing ImageJ software version
1.53c (http://imagej.nih.gov/ij/). The
antibodies used in the present study are listed in Table SI.
Stable short hairpin (sh)RNA
transfection
The shRNA lentiviral expression vectors and control
vectors (GeneCopoeia, Inc.) were transfected into HSR- and
DM-containing MTX-resistant HT29 cells and COLO 320DM cells at a
MOI of 10, and incubated for 12 h at 37°C according to the
manufacturer's protocol. The target sequences of shRNAs for MSH3
were as follows: 5′-CTT CTA CCA GCT ATC TTC T-3′ and 5′-GGA CAG GAG
TTT ATG ATA GAA-3′. The target sequence of shRNA for control was
5′-GCT TCG CGC CGT AGT CTT A-3′. Puromycin was added to the medium
at 72 h after transfection to obtain stable transfected clones.
Rescue assay
For MSH3 rescue, DM-sh-MSH3-1 cells were infected
with lentivirus particles containing MSH3 overexpression construct
or negative control (Shanghai GeneChem Co., Ltd.) at a MOI of 5
according to the protocol of lentiviral transfection. The cells
were named DM-sh-MSH3-overexpression (ov)-MSH3 and
DM-sh-MSH3-ov-negative control (NC), respectively. Verification was
performed using reverse transcription-quantitative (RT-q) PCR and
western blot analysis.
RT-qPCR
Genomic DNA was extracted using a QIAmp DNA Mini Kit
(Qiagen GmbH). Total RNA was extracted from cultured cells at a
density of 3×106 using TRIzol® (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacture's
protocol. cDNA synthesis was performed using an All-In-One
First-Strand cDNA Synthesis Kit (GeneCopoeia, Inc.) according to
the manufacturer's protocol. qPCR was performed using the Light
Cycler 480 SYBR Green Kit (Roche Applied Science) according to the
manufacture's protocol. The expression levels of target genes were
normalized to those of β-Actin. Thermocycling conditions
were 95°C for 15 sec, 60°C for 30 sec, 72°C for 30 sec and 40
cycles). 2−ΔΔCq quantification was performed as
previously described (18). The
DNA primers were as follows: DHFR-F: 5′-ATT TTG TTC AGT GCC
TAC CAC A-3′ and DHFR-R: 5′-GCC TGA ATG ATA TCT ACA AGC
TG-3′, ZFYVE16-F: 5′-AGG AAG CAA CCA CCA CAAC-3′ and
ZFYVE16-R: 5′-CAG CAC CAC CAA CAG A TACA-3′, MSH3-F:
5′-TGT CTG GTG TTT CGC CTG AT-3′ and MSH3-R: 5′-TTA GCC AAT
AAC CGC TCT AC-3′, POLK-F: 5′-GCG GTG TTG GTT AGG TTC TC-3′
and POLK-R: 5′-AAT AAG CAA AAG GGC TAC TG-3′,
XRCC4-F: 5′-AAC TCC ACA ATG CGA GAA TC-3′ and
XRCC4-R: 5′-AAT GCT CAA ACA GCC TAC TC-3′, GLRX-F:
5′-CCC ACA TTG TAG GGA ATC AT-3′ and GLRX-R: 5′-CCC ACA GTC
TAT TCG TAG CA-3′, CAST-F: 5′-TTG ACT CCA TAG CCA ACC TT-3′
and CAST-R: 5′-GT CAC TTT TCC CAG AAT CCG-3′, CCNH-F:
5′-GTA TTG CAG CAC TGA TTA TGT CC-3′ and CCNH-R: 5′-TCA TGA
AAA TAG CCA TAG GTG A-3′, c-Myc-F: 5′-GAT TCT CTG CTC TCC
TCG AC-3′ and c-Myc-R: 5′-GCC CGT TAA ATA AGC TGC-3′,
ACTB-F: 5′-CTT CTA CAA TGA GCT GCG TG-3′ and ACTB-R:
5′-AAG CAA ATA GAA CCT GCA GAG-3′. The cDNA primers were as
follows: MSH3-F: 5′-CTG CCA AAG TTG GGG ATA AA-3′ and
MSH3-R: 5′-AAA TGC ATT CGG ATC TCG TC-3′, ACTB-F:
5′-GGG AAA TCG TGC GTG ACA TT-3′ and ACTB-R: 5′-GGA ACC GCT
CAT TGC CAA T-3′.
DNA Fluorescence in situ hybridization
(FISH)
Cells were incubated with colcemid (MilliporeSigma)
before cytogenetic preparation by KCl treatment and fixation. The
BAC clone RP11-90A9 (chr5: 80551016-80731513) was used as template
for synthesis of FISH probes for the DHFR gene (BACPAC
Resource Center). Cells were hybridized with FISH probes as
previously described (8). In
short, the BAC clones were extracted using a Genopure Plasmid
MidiKit (Roche Applied Science) according to the manufacturer's
protocol and labelled with Cy3-dUTP or Green-dUTP using a BioPrime
DNA Labelling System Kit (Invitrogen; Thermo Fisher Scientific,
Inc.). The slides with interphase or metaphase spreads were
digested in RNaseA (10 mg/ml, 40 min at 37°C) (Thermo Fisher
Scientific), washed in 2X saline sodium citrate (SSC), dehydrated
through an ethanol series, then digested in pepsin-HCl (15 min at
37°C), fixed in 1% paraformaldehyde (10 min at room temperature),
dehydration (gradient dehydration with 75, 85, and 100% ethanol for
3 min each), treated in 70% formamide (3 min at 75°C), washed in 2X
SSC and dehydrated. Probe and slides were incubated for 48 h at
37°C. The slides were then immersed in 50% formamide (15 min at
44°C) and washed in 2X SSC. Following dehydration, the slides were
counterstained with 4', 6'-diamidino-2-phenylindole (DAPI).
High-quality images were captured using a fluorescence microscope
(Leica DM6 B; Leica Microsystems GmbH) and analyzed using the Leica
Application Suite X (version 2.0.0.14332; Leica Microsystems GmbH).
Interphase cells (~100) were evaluated for each group. The amount
of MN/NBUDs in each karyotype was also observed, among which
MN/NBUDs with DHFR fluorescence signal were scored as
MN/NBUDsDHFR+, whereas MN/NBUDs without DHFR
fluorescence signal were scored as MN/NBUDsDHFR−.
Repair assays
HR, c-NHEJ and a-NHEJ reporter plasmids pHPRT-DRGFP,
pimEJ5GFP and EJ2GFP-puro (Addgene, Inc.) were transfected into
DM-containing MTX-resistant cells using Lipofectamine®
2000 (Invitrogen; Thermo Fisher Scientific, Inc.). Puromycin was
added to select stable clones. Reverse transfection was performed
by plating DSB reporter-containing cells into 6-well plates that
already contained preformed small interfering (si) RNA transfection
complexes. In the MSH3 rescue assay, MSH3 overexpression lentivirus
was added 12 h after siMSH3 transfection. DSBs were induced 48 h
after the siRNA transfection by a second transfection with 4
µg pCBASceI which expressing I-SceI mixed with siRNA duplex
using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). Green fluorescent protein (GFP)-positive cells
were quantified by flow cytometric analysis (FACS-LSR II; BD
Biosciences) at room temperature 3 days after transfection as
previously described (19,20).
Drug sensitivity assay
Cells were plated into 96-well plates at a density
of 5,000 cells/well and incubated in the presence of MTX for 72-96
h. A CellTiter 96 AQueous One Solution Cell Proliferation Assay
(Promega Corporation) was used to measure the cell viability. The
optical density value was read on a microplate reader (Tecan Group,
Ltd.) at a wavelength of 490 nm and the IC50 values were
calculated.
Statistical analysis
All experiments were performed at least three times
independently. For western blot, quantitative PCR, ecDNA number,
DSB relative repair efficiency and IC50 value, unpaired
Student's t-test was used to determine statistically significant
differences between 2 groups, one- or two-way analysis of variance
(ANOVA) followed by the Dunnett's test was used for comparing >2
groups (one control group). Differences in the amount of MN/NBUDs,
as well as dihydrofolate reductase (DHFR)-containing
MN/NBUDs and HSR frequency, between different groups were evaluated
using the χ2 test. P<0.05 was considered to indicate
a statistically significant difference.
Results
Increased MSH3 expression is associated
with gene amplification in MTX-resistant HT29 cells
For an improved understanding of the mechanisms
contributing to the resistance to cytotoxic drugs via gene
amplification, HT29 cell lines that were resistant to MTX were
generated as previously described (21). HSRs and ecDNAs (DMs) are the main
amplified forms of DHFR in MTX-resistant cells. When
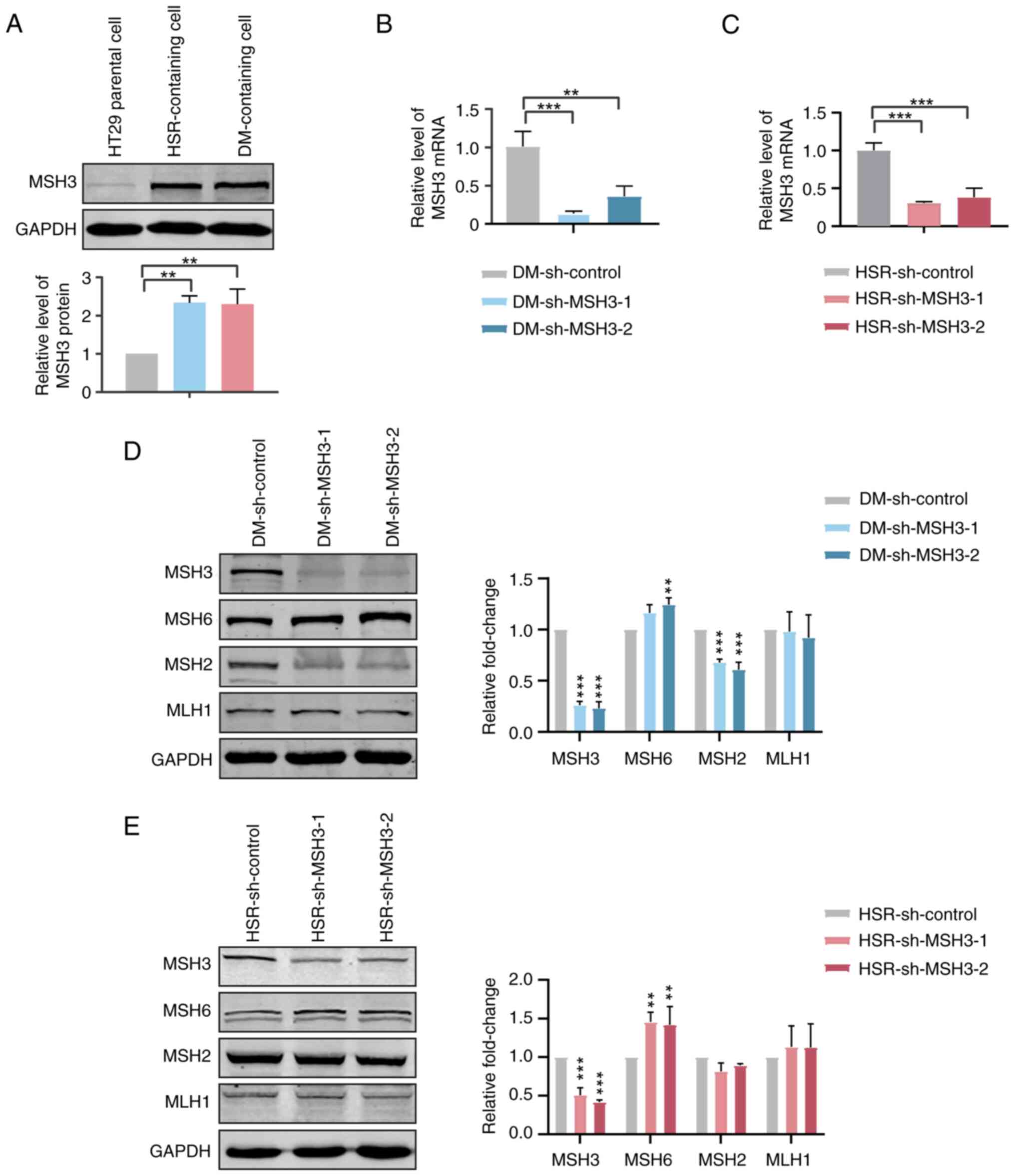
measuring the expression levels of MSH3 in parental, HSR- and
DM-containing MTX-resistant HT29 cells, it was observed that the
expression levels of MSH3 in MTX-resistant cells were ~ twice as
high as those in HT29 parental cells (Fig. 1A). Thus, the present results
suggested that MSH3 might be associated with MTX resistance and
gene amplification.
To further investigate the association between MSH3
and gene amplification, MSH3 was stably depleted in MTX-resistant
HT29 cells by shRNA transfection (Fig. 1B and C). The expression levels of
other key proteins of MMR, including MSH2, MSH6 and MutL homolog 1
(MLH1), were examined to determine the influence of MSH3 depletion
on MMR activity. The expression levels of MSH6 were increased,
those of MSH2 were decreased and those of MLH1 were unchanged in
MSH3-depleted MTX-resistant cells (Fig. 1D and E). This was consistent with
the fact that MSH2 is relatively unstable when not forming a
heterodimer complex with MSH3 or MSH6 and MSH6 is transcriptionally
upregulated to compensate for the MSH3 deficiency (22).
Inhibition of MSH3 decreases gene
amplification in DM-containing cells
Based on the result of a previous comparative
genomic hybridization array, DHFR, MSH3, zinc finger
FYVE-type containing 16 (ZFYVE16), DNA polymerase κ
(POLK), X-ray repair cross complementing (XRCC) 4,
glutaredoxin (GLRX), calpastatin (CAST) and cyclin H
(CCNH) were co-localized within the same HSRs of chromosome
5 in HSR-containing MTX-resistant cells. Furthermore, DHFR,
MSH3 and ZFYVE16 were co-localized within the same
ecDNAs and POLK, XRCC4, GLRX, CAST and
CCNH were co-localized within the same HSRs in DM-containing
MTX-resistant cells. Our previous study also revealed that polo
like kinase 2 (PLK2) is not amplified on chromosome 5,
following the development of MTX resistance in HT-29 cells, meaning
this can be used as a negative control of gene amplification
(8,9). To determine the influence of MSH3 on
gene amplification, the changes in gene copy numbers were examined
in MSH3-depleted DM- and HSR-containing MTX-resistant cells. The
present results demonstrated that the copy numbers of all the
amplified genes within ecDNAs and HSRs were decreased in
DM-containing MTX-resistant cells. In particular, those within HSRs
were decreased by >50% in DM-containing MTX-resistant cells
(Fig. 2A). The copy numbers of
amplified genes were not changed in HSR-containing HT29 cells
(Fig. 2B). In addition, to verify
whether MSH3 could affect the type of gene amplification, the
DHFR gene was labeled and the karyotype was compared between
control and MSH3-depleted DM-containing cells using FISH. Not only
was the amount of ecDNAs decreased, but HSR numbers were also
decreased by 75% in MSH3-depleted DM-containing cells (Fig. 2C-E).
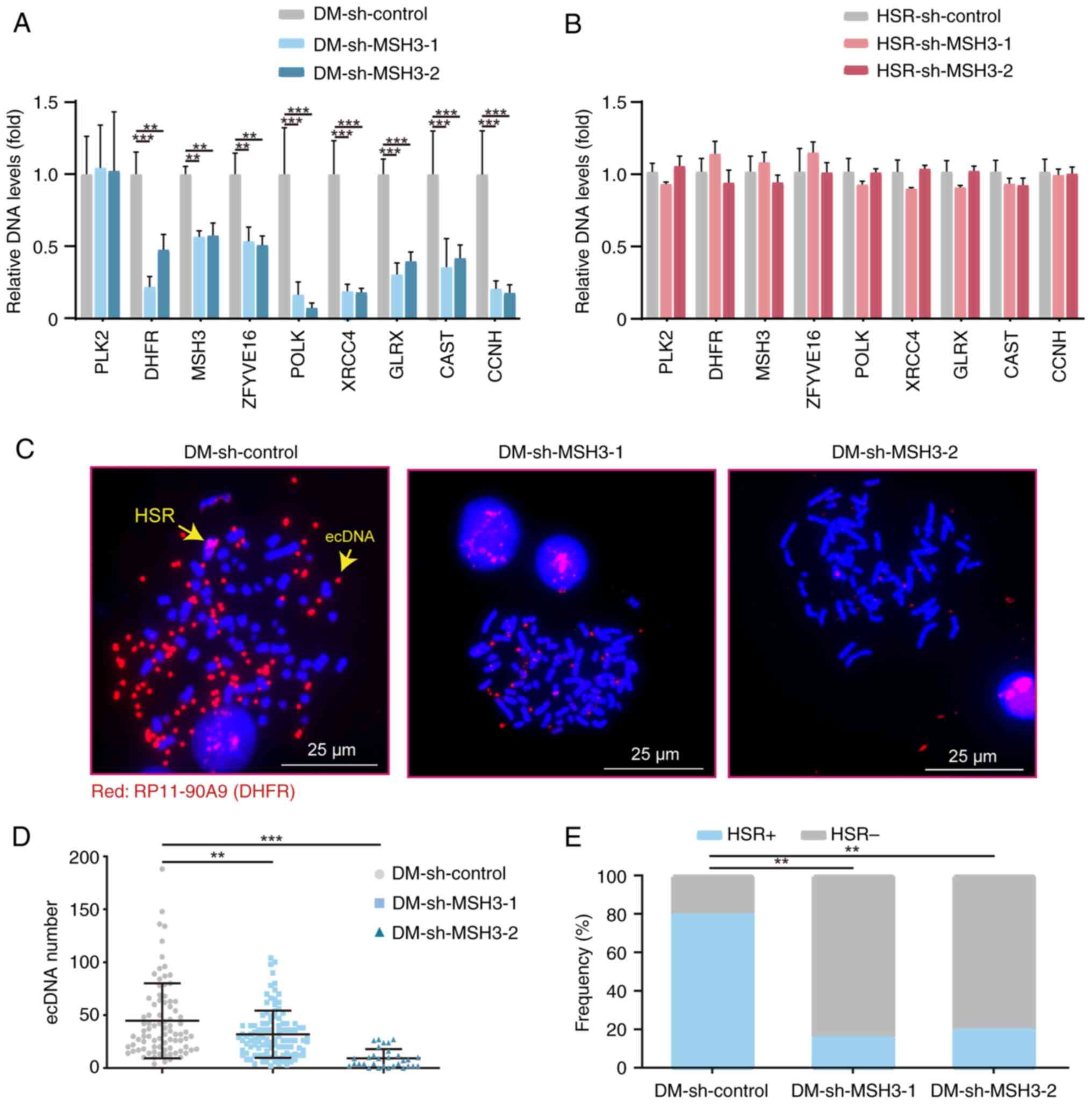 | Figure 2Inhibition of MSH3 decreases gene
amplification in DM- but not HSR-containing MTX-resistant cells. (A
and B) Quantitative PCR analysis of PLK2, DHFR,
MSH3, ZFYVE16, POLK, XRCC4,
GLRX, CAST and CCNH amplification in control
and MSH3-depleted clones of (A) DM- and (B) HSR-containing cells
(n=3; **P<0.01, ***P<0.001). (C)
Fluorescence in situ hybridization analysis of metaphase
nuclei in control and MSH3-depleted clones of DM-containing cells
using bacterial artificial chromosome containing human DHFR
as the probe. DHFR signals are shown in red and nuclei
stained by DAPI are shown in blue. Yellow arrows indicate HSR and
ecDNA. (D) Quantification of ecDNAs in control and MSH3-depleted
clones. (**P<0.01, ***P<0.001). (E)
Frequency of HSRs in control and MSH3-depleted clones (P<0.01).
DHFR, MSH3, ZFYVE16, POLK,
XRCC4, GLRX, CAST and CCNH were
detected to be co-localized within the same HSRs of chromosome 5 in
HSR-containing MTX-resistant cells. DHFR, MSH3 and
ZFYVE16 were co-localized within the same ecDNAs and
POLK, XRCC4, GLRX, CAST and CCNH
were co-localized within the same HSRs in DM-containing
MTX-resistant cells. PLK2 is not amplified on chromosome 5
following the development of MTX resistance in HT-29 cells and this
can be used as a negative control of gene amplification. MSH3, MutS
homolog 3; DM, double minute; HSR, homogeneously staining region;
MTX, methotrexate; ecDNA, extrachromosomal DNA. |
Furthermore, MSH3 was rescued in DM-sh-MSH3 cells
(Fig. 3A). The copy numbers of
amplified genes on both ecDNAs and HSRs were recovered (Fig. 3B). Also, the changes in the amount
of ecDNAs and corresponding gene amplification in MSH3 depleted
human COLO 320DM cells was examined. A previous study found that
ecDNAs in COLO 320DM cells carry a large number of amplified
c-Myc (MYC proto-oncogene, bHLH transcription factor)
oncogene (15). The results of
the present study showed that the amount of ecDNAs and the copy
number of the c-Myc gene were significantly reduced in COLO
320DM cells with MSH3 depletion compared to controls (Fig. 3C-F). Thus, it was concluded that
MSH3 was indeed involved in the formation of ecDNAs in tumor
cells.
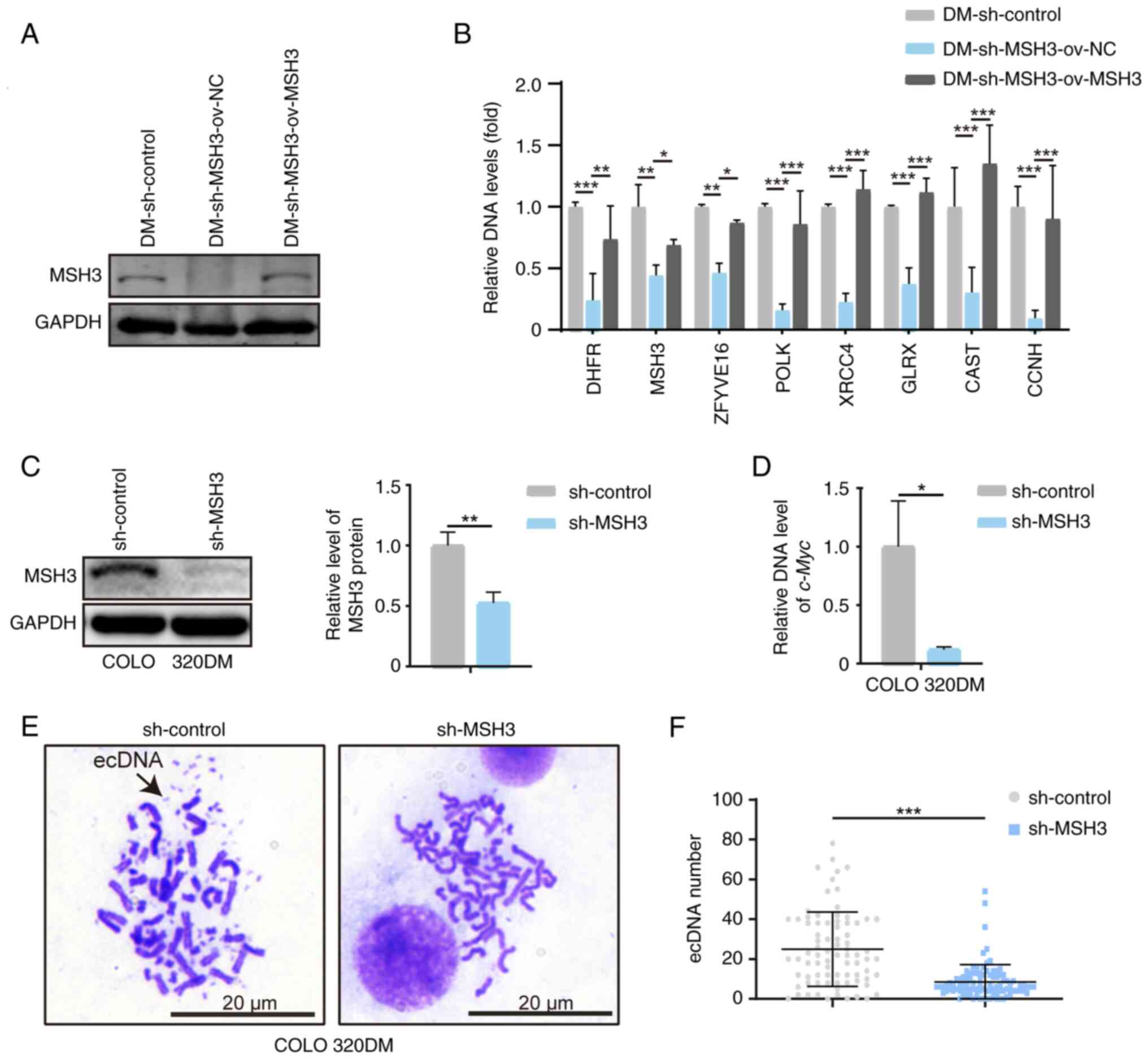 | Figure 3Inhibition of MSH3 decreases gene
amplification in DM-containing cells. (A) Western blot analysis of
MSH3 in DM-containing MTX-resistant HT29 cells: Control,
MSH3-depleted and MSH3-rescued clones. (B) Quantitative PCR
analysis of amplified genes located on ecDNAs and HSRs in
DM-containing MTX-resistant HT29 cells: Control, MSH3-depleted and
MSH3-rescued clones. (C) Western blot analysis of MSH3 in COLO
320DM cells: Control and MSH3-depleted clones. (D) Quantitative PCR
analysis of amplified oncogenes c-Myc located on ecDNAs in
COLO 320DM cells: Control and MSH3-depleted clones. Data are
presented as the mean ± SD (n=3; *P<0.05,
**P<0.01, ***P<0.001). (E)
Representative metaphase nuclei of COLO 320DM cells: Control and
MSH3-depleted clones. Black arrow indicates ecDNA. (F)
Quantification of ecDNAs in COLO 320DM cells: Control and
MSH3-depleted clones (***P<0.001). MSH3, MutS homolog
3; DM, double minute; MTX, methotrexate; ecDNA, extrachromosomal
DNA; HSR, homogeneously staining region; c-Myc, MYC
proto-oncogene, bHLH transcription factor. |
Inhibition of MSH3 affects the formation
of ecDNAs by regulating the HR, c-NHEJ and a-NHEJ pathways
MSH3 has been indicated to participate in the repair
of DSBs (10). HR, c-NHEJ and
a-NHEJ are considered to be the most classical DSB repair pathways
in mammalian cells. HR is a type of repair pathway with high
fidelity, whose DSB repair nuclease-RAD50 DSB repair protein-nibrin
[MRX complex in yeast, (MRN)] complex can sense the DSBs and
promote the loading of RAD51 recombinase (RAD51) to initiate
synapsis (23). However, a
previous study demonstrated that HR is also an important mechanism
of structural rearrangement contributing to human genomic
variability (24). c-NHEJ is a
fast but error-prone DSB repair mechanism. The heterodimer formed
by XRCC6 (KU70) and XRCC5 (KU86) recognizes the DSBs
and recruits DNA-PKcs kinase to activate downstream DNA ligase 4,
which completes end processing. a-NHEJ, a type of back-up pathway,
is a more error-prone repair pathway characterized by deletions and
unbalanced translocations. a-NHEJ proteins include poly(ADP-ribose)
polymerase 1 (PARP1), MRN complex, XRCC1 and DNA ligase 3
(LIG3) and participate in the binding, cleavage and ligation of DNA
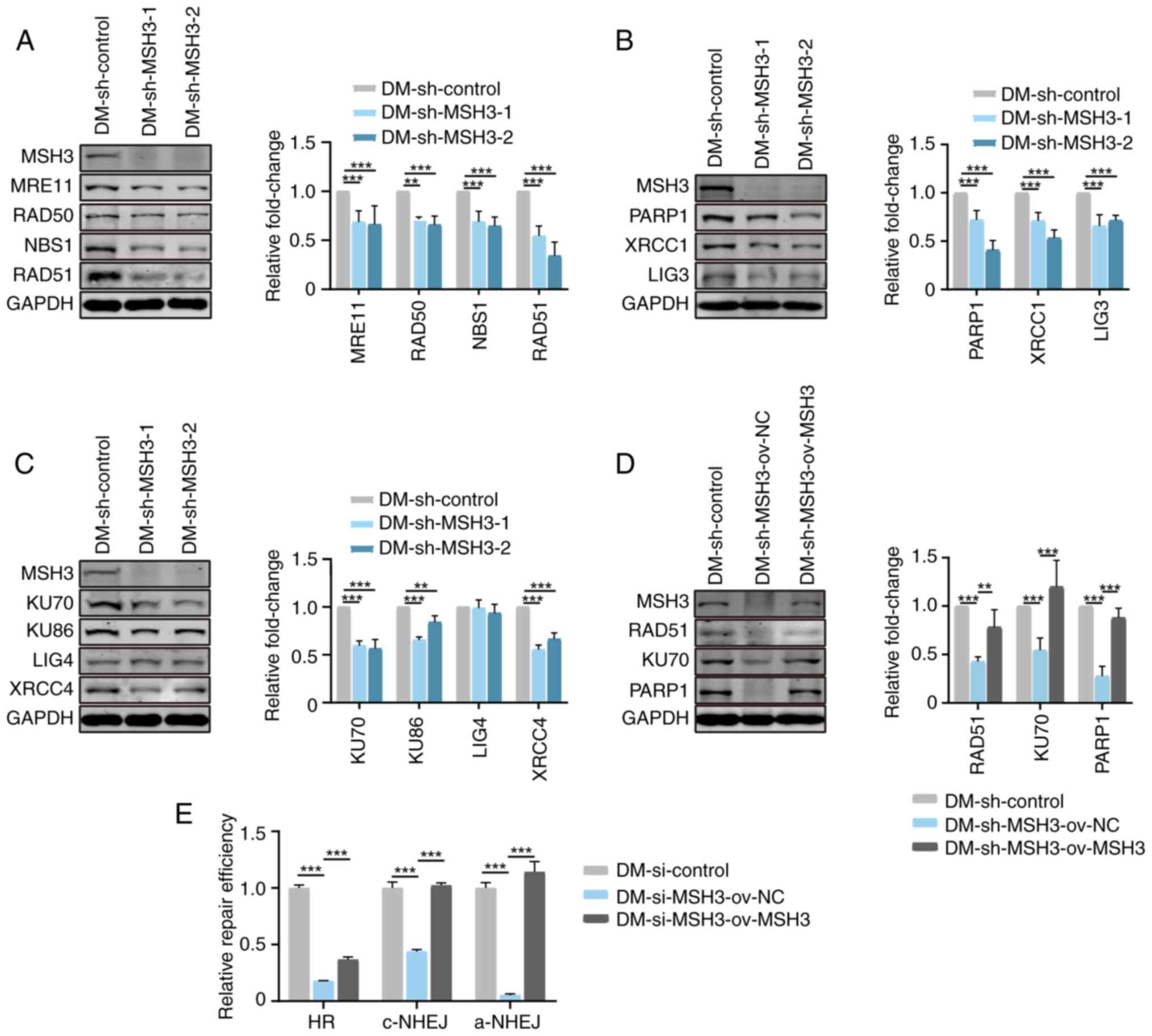
cleavage ends to complete the repair of DSBs (25,26). To explore the mechanism by which
MSH3 affects the formation of ecDNAs, the expression levels of key
proteins of these DSB repair pathways were further detected in
DM-containing cells pretreated with 10−3 mol/l MTX (a
type of DSB inducer). As shown in Fig. 4A-C, key proteins of HR (MRN
complex and RAD51), a-NHEJ (PARP1, XRCC1 and LIG3) and c-NHEJ
(KU70, KU86 and XRCC4) were markedly decreased in MSH3-depleted
DM-containing cells. Furthermore, the expression levels of key
proteins (RAD51, KU70 and PARP1) of these three repair pathways
were markedly restored after MSH3 recovery (Fig. 4D). Simultaneously, reporter cell
lines were established to measure the efficiency of these pathways.
In these DM-containing MTX-resistant cells, each of HR, c-NHEJ,
a-NHEJ events triggered by a DSB digested by I-SceI endonuclease
can restore a functional GFP, which can be quantified by
fluorescence-activated cell sorting (19,20). Subsequently, the present study
examined the repair efficiency of reporter cell lines. The repair
efficiencies of HR, c-NHEJ and a-NHEJ were decreased by 82.1, 56.2
and 94.7%, respectively, in MSH3-depleted cells and rescued in
MSH3-recovered cells (Fig. 4E),
indicating that MSH3 was required for efficient repair of DSBs via
the HR, c-NHEJ and a-NHEJ pathways. Overall, it was hypothesized
that MSH3 may participate in the formation of ecDNAs via
recruitment of key proteins of repair pathways (HR, c-NHEJ and
a-NHEJ) to DSBs.
Inhibition of MSH3 eliminates ecDNAs
through MN/NBUDs in DM-containing MTX-resistant cells
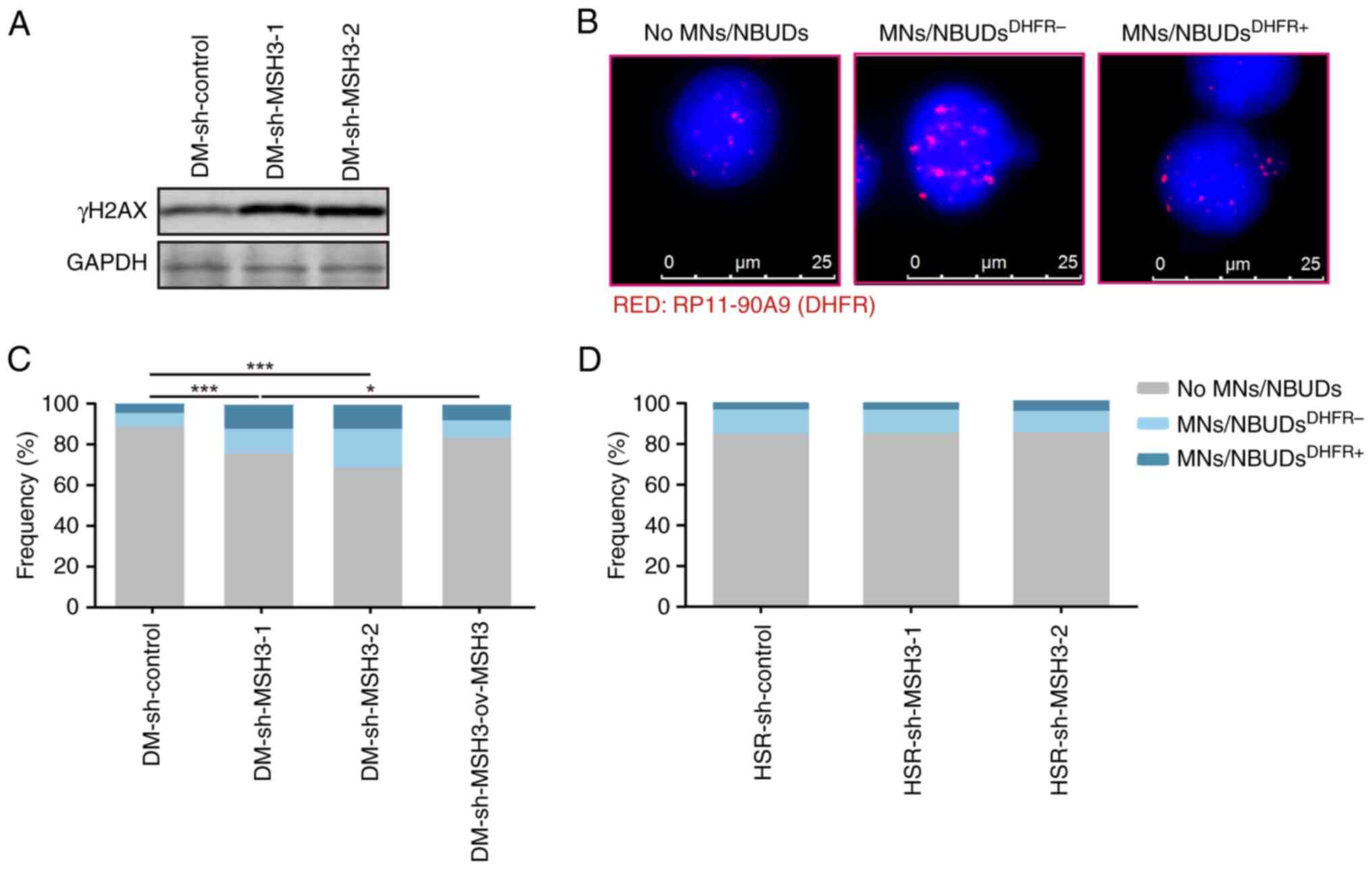
The presence of MN/NBUDs in a cell is an indicator
of DNA damage and genetic instability. MN/NBUDs are small
DNA-containing structures localized separately from the main
nucleus of the cell. Amplified genes or ecDNAs have been observed
to be eliminated by MN/NBUDs (27). To clarify whether the decreased
amplified gene is eliminated by MN/NBUDs, DHFR was labelled
using FISH to detect the formation of MN/NBUDs and the expulsion of
MN/NBUDs containing amplified genes. Fig. 5B shows the nuclei with no
MN/NBUDs, nuclei with MN/NBUDs containing no DHFR signals
and nuclei with MN/NBUDs containing DHFR signals. The
present results demonstrated that the MN/NBUD amount was increased
by >85.8% in MSH3-depleted cells (P<0.001), which was
consistent with the expression of nuclear γ-H2AX (Fig. 5A), indicating the accumulation of
DSBs. Simultaneously, compared with that in control cells, the
amount of MN/NBUDs with DHFR signals was more than twice as
high in MSH3-depleted DM-containing cells and the expulsion of
MN/NBUDs was restored after MSH3 was rescued (Fig. 5C). However, depletion of MSH3 had
no effect on the formation of MN/NBUDs in HSR-containing cells
(Fig. 5D). The present results
indicated that inhibition of MSH3 may promote the formation of
MN/NBUDs and then the efflux of MN/NBUDs containing amplified
genes, thus reducing ecDNAs or HSRs in the nuclei of DM-containing
MTX-resistant cells.
Inhibition of MSH3 increases the
sensitivity to MTX in DM-containing MTX-resistant cells
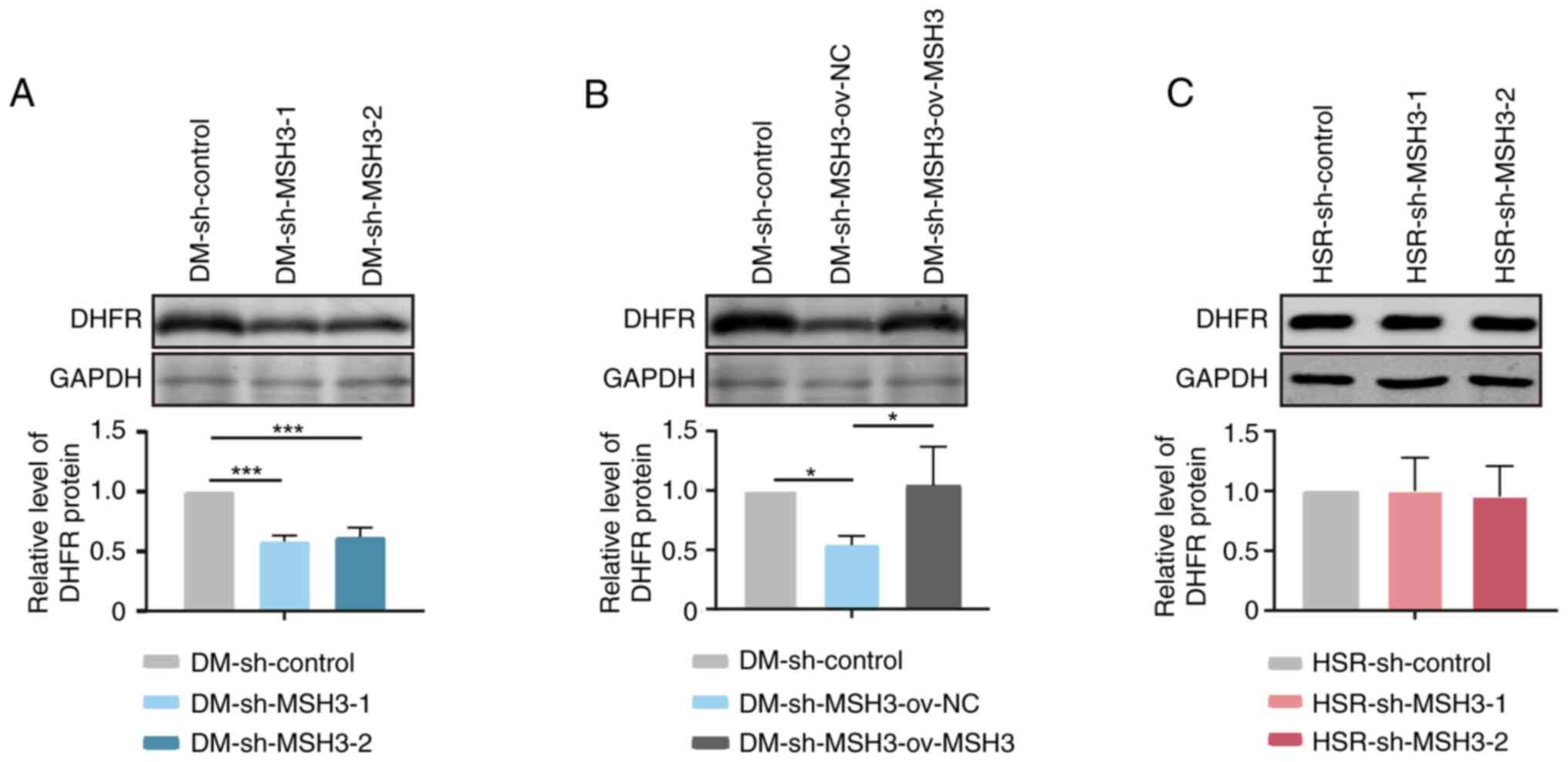
MTX is a cytotoxic drug widely used in cancer
therapy. High DHFR expression originating from DHFR
amplification is the most important mechanism to induce MTX
resistance in cancers (28). In
the present study, markedly decreased DHFR copy numbers and
DHFR expression were observed in MSH3-depleted DM-containing cells
(Figs. 2A and 6A), while no changes were observed in
MSH3-depleted HSR-containing cells (Figs. 2B and 6C). Furthermore, restored DHFR
amplification and expression were found in DM-sh-MSH3-ov-MSH3 cells
(Figs. 3B and 6B). To address the role of MSH3 in drug
resistance, the present study examined the IC50 value of
both DM- and HSR-containing MTX-resistant cells. Compared with
control cells, 1.98 and 1.93 times decreased IC50 values
were observed in MSH3-depleted DM-containing cells (P<0.01),
while this was 1.22 and 1.38 times increased in MSH3-depleted
HSR-containing cells (P>0.05). Furthermore, compared with that
of control cells, the IC50 value of DM-sh-MSH3-ov-NC
cells was decreased by 1.58 times and the IC50 value of
DM-sh-MSH3-ov-NC cells was increased by 1.75 times after the
restoration of MSH3, indicating that the resistance was
re-established after the restoration of MSH3 expression (Table I). These results indicated that
inhibition of MSH3 could sensitize DM-containing MTX-resistant
cells to MTX and suggested the promising potential for a
combination therapy of MTX and MSH3 inhibitors in DM-containing
MTX-resistant cancers.
 | Table IIC50 values of DM- and
HSR-containing cells. |
Table I
IC50 values of DM- and
HSR-containing cells.
| HT-29 cell
line | IC50
(mol/l) | Fold change |
|---|
| DM-sh-control |
2.15×10−3±2.11×10−4 | 1 |
| DM-sh-MSH3-1 |
1.09×10−3±1.90×10−4 | 1.98b vs. DM-sh-control |
| DM-sh-MSH3-2 |
1.11×10−3±2.82×10−4 | 1.93a vs. DM-sh-control |
|
DM-sh-MSH3-ov-NC |
1.37×10−3±2.52×10−4 | 1.58a vs. DM-sh-control |
|
DM-sh-MSH3-ov-MSH3 |
2.39×10−3±4.19×10−4 | 1.75a vs. DM-sh-MSH3-ov-NC |
| HSR-sh-control |
2.65×10−4
±1.06×10−5 | 1 |
| HSR-sh-MSH3-1 |
3.24×10−4
±6.31×10−5 | 1.22 vs.
HSR-sh-control |
| HSR-sh-MSH3-2 |
3.65×10−4±9.03×10−5 | 1.38 vs.
HSR-sh-control |
Discussion
In recent years, ecDNAs have reattracted attention
in the field of cancer research. As they lack centromeres, ecDNAs
are subject to non-equal segregation to daughter cells, which can
rapidly lead to heterogeneity of ecDNA amounts in cells within a
tumor and enable daughter cells to achieve higher ecDNA copy
numbers than mother cells (4).
The presence of oncogenes or drug resistance genes on ecDNAs
enhances the fitness of cells containing ecDNAs and promote the
malignant phenotype of tumors (29). Thus, exploring the molecular
mechanism of ecDNA formation and then effectively eliminating
ecDNAs in cancer cells is becoming a promising strategy for cancer
treatment (30). Previous studies
have demonstrated that DSBs and canonical repair mechanisms may be
involved in the formation of ecDNAs. For example, depleting BRCA1,
DNA-PKcs or PARP1 (data not shown; the core proteins of HR, c-NHEJ
or a-NHEJ pathways), can decrease the amount of ecDNAs and reverse
MTX resistance in DM-containing MTX-resistant HT29 cells (8,9).
MMR mostly occurs at the post-replication stage and
is responsible for repairs of mismatches and small stranded DNA
loops that are important in stabilizing the genome. In recent
years, MMR has also been suggested to be involved in various
aspects of DNA metabolism, such as the DNA damage response and HR,
which mediates DSB repair. Thus, the present study examined whether
MSH3, a key DNA MMR protein, may participate in the formation of
ecDNAs or HSRs associated with DSB repair mechanisms in cancer
cells.
In the present study, HT29 human colorectal cancer
cells and stepwise induced MTX-resistant cancer cells, which
contain amplified DHFR gene with different cytogenetic
manifestations, HSRs or ecDNAs, respectively, were selected as
research objects. The present study revealed increased protein
expression levels of MSH3 in MTX-resistant cells. The markedly
altered expression of other MMR core proteins (increased MSH6 and
decreased MSH2) in MSH3-depleted MTX-resistant cells demonstrated
that MSH3 could affect the MMR system. Furthermore, the amounts of
ecDNAs, HSRs and corresponding amplified genes were markedly
decreased by depletion of MSH3 and could be rescued by
overexpression of MSH3 in DM-containing cells. By contrast, the
amount of HSRs and amplified genes were not altered in
HSR-containing cells. The present results suggested that MSH3 may
function differently in various stages of drug resistance with
different types of gene amplification. Although both types of cells
(DM- and HSR-containing cells) contain HSRs, the environments in
which HSRs exist are different. Whether the presence of ecDNAs or
the different degree of drug resistance affect the function of MSH3
in HSRs, or whether there are some other reasons, needs further
study. Nevertheless, to the to the best of the authors' knowledge,
the present study was the first to demonstrate that MSH3 not only
contributes to microsatellite stability, meiotic and mitotic
recombination, DNA-damage signaling, apoptosis, class-switch
recombination, somatic hypermutation and triplet-repeat expansion
as previously reported but also to the formation of ecDNAs
(31-33).
To the best of the authors' knowledge, the main
function of MSH3 is the repair of a few base pair mismatches, while
ecDNAs usually exhibit severe DSBs repair (34,35). Thus, it was hypothesized that MSH3
may promote the formation of ecDNAs via several classical DSB
repair pathways, such as the HR, c-NHEJ or a-NHEJ pathways. By
further examining the core protein expression and activity of HR,
c-NHEJ and a-NHEJ pathways in MSH3-depleted DM-containing cells,
the present study revealed that MSH3 may affect the formation of
ecDNAs by regulating these canonical DSB repair pathways.
Furthermore, the present study confirmed the function of MSH3 in
the formation of ecDNAs by detecting the activity and protein
expression of such pathways in MSH3 rescue experiments. HR is
pivotal to maintain replication fidelity and employs an intact
sister chromatid as a template for information exchange and
faithful repair (21). Abnormal
elevation of HR activity can also lead to an increased rate of
mutation and progressive accumulation of genetic variation in
multiple myeloma cells (36). It
has been reported that the MMR mechanism can interact directly with
HR (37,38). The present results further
demonstrated that MSH3 may contribute to the dysfunctional HR
activity, resulting in the formation of ecDNAs. c-NHEJ is the major
DSB repair system in higher eukaryotes, particularly during phases
of the cell cycle when a homologous sister chromatid is absent
(39). c-NHEJ often results in
error-prone outcomes, with partial loss of genome information at
the site of the DSBs. A previous report indicated that
MSH2-deficient cells exhibit an increase in c-NHEJ-mediated DSB
repair compared with normal mouse cells (40). Conversely, MSH6 has been reported
to be involved in the repair of DSBs through a direct physical
interaction with KU70, a core factor of c-NHEJ (41). Thus, the function of MSH3 in
c-NHEJ remains to be elucidated. In the present study, depletion of
MSH3 could inhibit the protein expression and activity of the
c-NHEJ pathway in DM-containing cells. It was hypothesized that
MSH3 may promote the formation of ecDNAs by increasing the
frequency of blunt or nearly blunt double-stranded DNA breaks that
can be joined by c-NHEJ factors (42-44). The a-NHEJ pathway appears to have
evolved as a back-up mechanism for c-NHEJ (45). Since alternative end-joining is
more error-prone than c-NHEJ, it is considered to serve a role in
driving genomic instability and thus, the tumorigenic process.
Eccleston et al (46)
reported that MLH1, exonuclease 1 and MSH2 are important for
efficient a-NHEJ-mediated antibody gene class switch recombination,
by converting DNA nicks and point mutations into double-strand DNA
breaks. Similarly, the present study indicated a novel function of
MSH3, which could promote the formation of ecDNAs via the a-NHEJ
pathway. It is difficult to say which repair pathway MSH3 mainly
affects. It was hypothesized that MSH3 may serve a role in
different cell states through different pathways, such as in
different cell cycle stages and in the absence/presence of abnormal
c-NHEJ pathway. Overall, the present study revealed that MSH3 may
be a focal mechanism to promote the formation of ecDNAs by
regulating canonical DSB repair pathways.
Since MN/NBUDs are the biomarkers of DNA damage in
cells (47), to verify whether
depletion of MSH3 could induce DSBs accumulation, the present study
examined the amount of MN/NBUDs in MSH3-depleted DM-containing
cells. Markedly increased numbers of MN/NBUDs, combined with
increased gH2AX levels, demonstrated that inhibition of MSH3 could
induce more DSBs accumulation. Previous studies have also suggested
that specific incorporation of ecDNAs into the cytoplasmic MN
participated in oncogene elimination (15-17,48). For example, low-dose HU treatment
could induce the generation of ecDNA-type MN and the extrusion of
amplified genes (49,50). Amplified EGFR in
glioblastoma, amplified MYCN in neuroblastoma and amplified
CDK4 in liposarcoma can be eliminated by ecDNA-type MN
(51,52). The present study further revealed
markedly increased amounts of MN/BUDs with amplified DHFR in
MSH3-depleted DM-containing cells. Our previous studies indicated
effective expelling of ecDNAs with MN/NBUDs in DM-containing cells
following inhibition of the HR and c-NHEJ pathways but not the
a-NHEJ pathway (8,9). Combined with the observation that
MSH3 may regulate the activity of HR and c-NHEJ, it was
hypothesized that inhibition of MSH3 may promote the expulsion of
ecDNAs via HR and c-NHEJ pathways. Notably, most studies reported
that expulsion of amplified genes can be detected in the form of
ecDNAs only but not in the form of HSRs (15,52). It is difficult to distinguish
ecDNAs and HSRs from MN/NBUDs under the present experimental
conditions. Due to the observation of markedly decreased HSRs and
ecDNA amounts in MSH3-depleted DM-containing cells, it was
hypothesized that not only ecDNAs but also HSRs have the potential
to be expelled by MN/BNUBs, which was supported by the observation
of expelled HSRs with MYCN amplification by MN in NBC cells
treated with HU or cisplatin (53).
MMR has been demonstrated to participate in the DNA
damage response after treatment with certain chemotherapeutic
agents. Inhibition of MSH3 has also been required for the
sensitization to cisplatin and oxaliplatin, in which MSH3 is
involved in the repair of DSBs (13). In addition, another study
demonstrated that lack of MSH3 did not affect the cellular response
to cisplatin within their experimental conditions (54). Thus, the exact role of MSH3 in
modulating drug resistance remains to be determined. In the present
study, the markedly increased sensitivity to MTX combined with the
decreased amplification and corresponding expression of the
DHFR gene in MSH3-depleted DM-containing cells, all of which
could be rescued by overexpression of MSH3, suggested that MSH3 may
participate in the regulation of sensitivity to MTX by changing the
amount of ecDNAs. The present study also has the limitation that it
is not yet possible to explain why MSH3 depletion does not affect
drug resistance and gene amplification in cells containing HSRs.
Further studies are needed for improved understanding of the role
of MSH3 on gene amplification at different stages of drug
resistance.
In conclusion, the present study revealed that MSH3
could regulate ecDNA formation via DSB repair pathways (HR, c-NHEJ
and a-NHEJ) and ecDNA efflux by MN/NBUDs and affected the
sensitivity to MTX in cancer cells (Fig. 7). Thus, MSH3 is expected to become
a novel target for the treatment of MTX-resistant tumors and other
tumors containing ecDNAs.
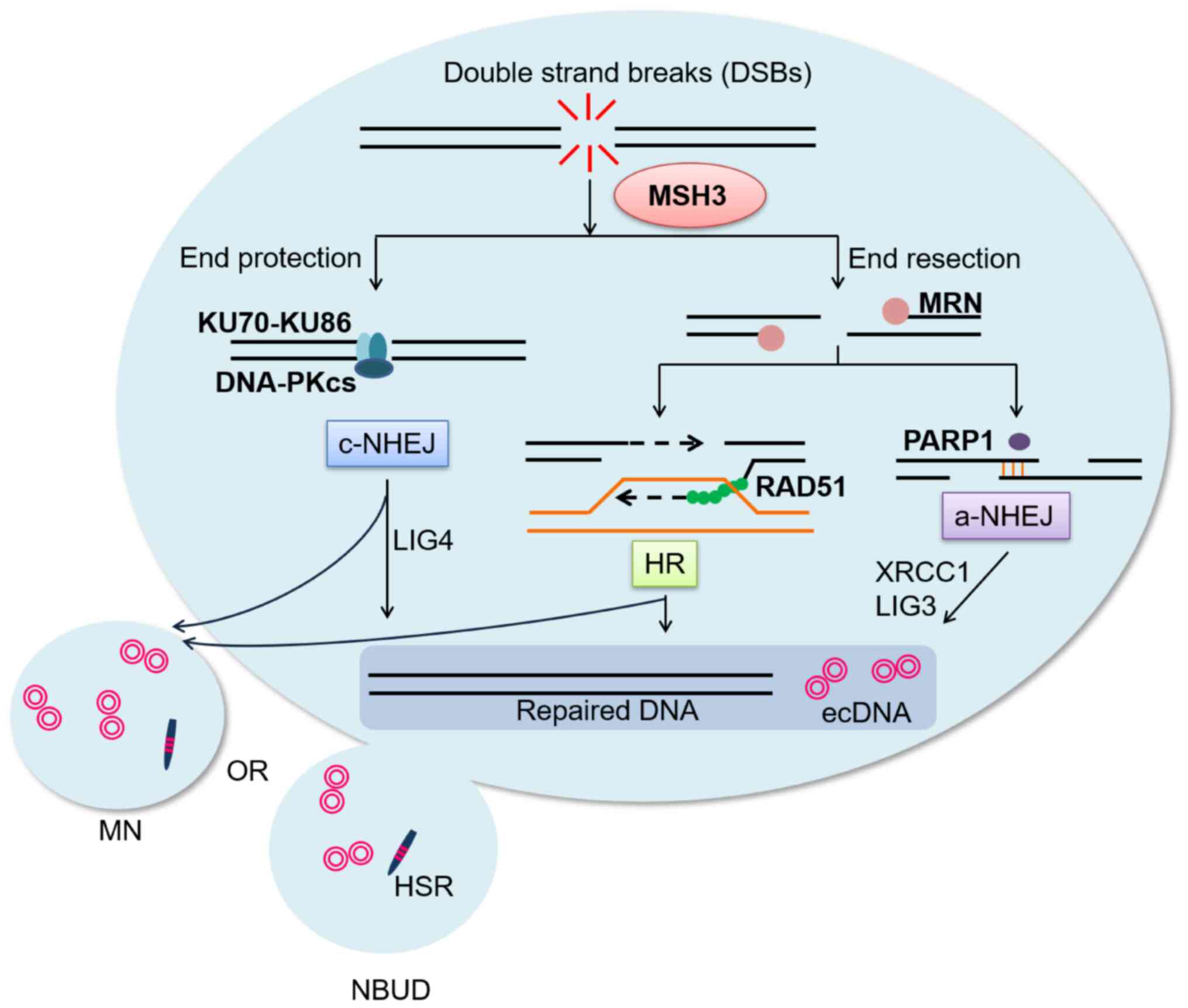 | Figure 7Schematic diagram summarizing the
mechanism. MSH3 can regulate ecDNA formation via double-strand
break repair pathways (homologous recombination, classic
non-homologous end joining and alternative non-homologous end
joining) and mediate ecDNA efflux by micronuclei/nuclear buds.
MSH3, MutS homolog 3; ecDNA, extrachromosomal DNA; DSBs,
double-strand breaks; MRN, MRX complex in yeast; PARP1,
poly(ADP-ribose) polymerase 1; RAD51, RAD51 recombinase; HR,
homologous recombination; c-NHEJ, classic non-homologous end
joining; a-NHEJ, alternative non-homologous end joining; ecDNA,
extrachromosomal DNA; MN, micronuclei; HSR, homogeneously staining
region; NBUD, nuclear buds; MN, micronuclei. |
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
XM and SF conceived and designed the study. XW, YQ,
RX and JZ performed experiments and analyzed the data. HZ, JZ, XC,
GJ, PL and WS provided advice and technical assistance. JM, TS, YL
and SX performed experiments. XM and XW confirm the authenticity of
all the raw data. XM and XW wrote the paper. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 81572915), HMU Marshal Initiative
Funding (grant no. HMUMIF-21007) and Natural Science Foundation of
Heilongjiang Province (grant no. LH2020H015).
References
|
1
|
Albertson DG: Gene amplification in
cancer. Trends Genet. 22:447–455. 2006.
|
|
2
|
Storlazzi CT, Lonoce A, Guastadisegni MC,
Trombetta D, D'Addabbo P, Daniele G, L'Abbate A, Macchia G, Surace
C, Kok K, et al: Gene amplification as double minutes or
homogeneously staining regions in solid tumors: Origin and
structure. Genome Res. 20:1198–1206. 2010.
|
|
3
|
Benner SE, Wahl GM and Von Hoff DD: Double
minute chromosomes and homogeneously staining regions in tumors
taken directly from patients versus in human tumor cell lines.
Anticancer Drugs. 2:11–25. 1991.
|
|
4
|
Turner KM, Deshpande V, Beyter D, Koga T,
Rusert J, Lee C, Li B, Arden K, Ren B, Nathanson DA, et al:
Extrachromosomal oncogene amplification drives tumour evolution and
genetic heterogeneity. Nature. 543:122–125. 2017.
|
|
5
|
Menghi F, Barthel FP, Yadav V, Tang M, Ji
B, Tang Z, Carter GW, Ruan Y, Scully R, Verhaak RGW, et al: The
tandem duplicator phenotype is a prevalent genome-wide cancer
configuration driven by distinct gene mutations. Cancer Cell.
34:197–210.e5. 2018.
|
|
6
|
Gisselsson D, Jin Y, Lindgren D, Persson
J, Gisselsson L, Hanks S, Sehic D, Mengelbier LH, Øra I, Rahman N,
et al: Generation of trisomies in cancer cells by multipolar
mitosis and incomplete cytokinesis. Proc Natl Acad Sci USA.
107:20489–20493. 2010.
|
|
7
|
Korbel JO and Campbell PJ: Criteria for
inference of chromothripsis in cancer genomes. Cell. 152:1226–1236.
2013.
|
|
8
|
Meng X, Qi X, Guo H, Cai M, Li C, Zhu J,
Chen F, Guo H, Li J, Zhao Y, et al: Novel role for non-homologous
end joining in the formation of double minutes in
methotrexate-resistant colon cancer cells. J Med Genet. 52:135–144.
2015.
|
|
9
|
Cai M, Zhang H, Hou L, Gao W, Song Y, Cui
X, Li C, Guan R, Ma J, Wang X, et al: Inhibiting homologous
recombination decreases extrachromosomal amplification but has no
effect on intrachromosomal amplification in methotrexate-resistant
colon cancer cells. Int J Cancer. 144:1037–1048. 2019.
|
|
10
|
Hong Z, Jiang J, Hashiguchi K, Hoshi M,
Lan L and Yasui A: Recruitment of mismatch repair proteins to the
site of DNA damage in human cells. J Cell Sci. 121:3146–3154.
2008.
|
|
11
|
Surtees JA and Alani E: Mismatch repair
factor MSH2-MSH3 binds and alters the conformation of branched DNA
structures predicted to form during genetic recombination. J Mol
Biol. 360:523–536. 2006.
|
|
12
|
Lyndaker AM and Alani E: A tale of tails:
Insights into the coordination of 3' end processing during
homologous recombination. Bioessays. 31:315–321. 2009.
|
|
13
|
Takahashi M, Koi M, Balaguer F, Boland CR
and Goel A: MSH3 mediates sensitization of colorectal cancer cells
to cisplatin, oxaliplatin, and a poly(ADP-ribose) polymerase
inhibitor. J Biol Chem. 286:12157–12165. 2011.
|
|
14
|
Dillon LW, Kumar P, Shibata Y, Wang YH,
Willcox S, Griffith JD, Pommier Y, Takeda S and Dutta A: Production
of extrachromosomal MicroDNAs is linked to mismatch repair pathways
and transcriptional activity. Cell Rep. 11:1749–1759. 2015.
|
|
15
|
Von Hoff DD, McGill JR, Forseth BJ,
Davidson KK, Bradley TP, Van Devanter DR and Wahl GM: Elimination
of extrachromosomally amplified MYC genes from human tumor cells
reduces their tumorigenicity. Proc Natl Acad Sci USA. 89:8165–8169.
1992.
|
|
16
|
Eckhardt SG, Dai A, Davidson KK, Forseth
BJ, Wahl GM and Von Hoff DD: Induction of differentiation in HL60
cells by the reduction of extrachromosomally amplified c-myc. Proc
Natl Acad Sci USA. 91:6674–6678. 1994.
|
|
17
|
Shimizu N, Shimura T and Tanaka T:
Selective elimination of acentric double minutes from cancer cells
through the extrusion of micronuclei. Mutat Res. 448:81–90.
2000.
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
|
|
19
|
Pierce AJ, Hu P, Han M, Ellis N and Jasin
M: Ku DNA end- binding protein modulates homologous repair of
double-strand breaks in mammalian cells. Genes Dev. 15:3237–3242.
2001.
|
|
20
|
Bennardo N, Cheng A, Huang N and Stark JM:
Alternative-NHEJ is a mechanistically distinct pathway of mammalian
chromosome break repair. PLoS Genet. 4:e10001102008.
|
|
21
|
Morales C, García MJ, Ribas M, Miró R,
Muñoz M, Caldas C and Peinado MA: Dihydrofolate reductase
amplification and sensitization to methotrexate of
methotrexate-resistant colon cancer cells. Mol Cancer Ther.
8:424–432. 2009.
|
|
22
|
Chang DK, Ricciardiello L, Goel A, Chang
CL and Boland CR: Steady-state regulation of the human DNA mismatch
repair system. J Biol Chem. 275:18424–18431. 2000.
|
|
23
|
Syed A and Tainer JA: The MRE11-RAD50-NBS1
complex conducts the orchestration of damage signaling and outcomes
to stress in DNA replication and repair. Annu Rev Biochem.
87:263–294. 2018.
|
|
24
|
Startek M, Szafranski P, Gambin T,
Campbell IM, Hixson P, Shaw CA, Stankiewicz P and Gambin A:
Genome-wide analyses of LINE-LINE-mediated nonallelic homologous
recombination. Nucleic Acids Res. 43:2188–2198. 2015.
|
|
25
|
Simsek D and Jasin M: Alternative
end-joining is suppressed by the canonical NHEJ component
Xrcc4-ligase IV during chromosomal translocation formation. Nat
Struct Mol Biol. 17:410–416. 2010.
|
|
26
|
Sallmyr A and Tomkinson AE: Repair of DNA
double-strand breaks by mammalian alternative end-joining pathways.
J Biol Chem. 293:10536–10546. 2018.
|
|
27
|
Kisurina-Evgenieva OP, Sutiagina OI and
Onishchenko GE: Biogenesis of Micronuclei. Biochemistry (Mosc).
81:453–464. 2016.
|
|
28
|
Morales C, Ribas M, Aiza G and Peinado MA:
Genetic determinants of methotrexate responsiveness and resistance
in colon cancer cells. Oncogene. 24:6842–6847. 2005.
|
|
29
|
Zhang Y, Dong K, Jia X, Du S, Wang D, Wang
L, Qu H, Zhu S, Wang Y, Wang Z, et al: A novel extrachromosomal
circular DNA related genes signature for overall survival
prediction in patients with ovarian cancer. BMC Med Genomics.
16:1402023.
|
|
30
|
Hung KL, Yost KE, Xie L, Shi Q, Helmsauer
K, Luebeck J, Schöpflin R, Lange JT, Chamorro González R, Weiser
NE, et al: ecDNA hubs drive cooperative intermolecular oncogene
expression. Nature. 600:731–736. 2021.
|
|
31
|
Jiricny J: The multifaceted
mismatch-repair system. Nat Rev Mol Cell Biol. 7:335–346. 2006.
|
|
32
|
Jiricny J: Postreplicative mismatch
repair. Cold Spring Harb Perspect Biol. 5:a0126332013.
|
|
33
|
Chatterjee N, Lin Y and Wilson JH:
Mismatch repair enhances convergent transcription-induced cell
death at trinucleotide repeats by activating ATR. DNA Repair
(Amst). 42:26–32. 2016.
|
|
34
|
Bannister LA, Waldman BC and Waldman AS:
Modulation of error-prone double-strand break repair in mammalian
chromosomes by DNA mismatch repair protein Mlh1. DNA Repair (Amst).
3:465–474. 2004.
|
|
35
|
Zhu J, Yu Y, Meng X, Fan Y, Zhang Y, Zhou
C, Yue Z, Jin Y, Zhang C, Yu L, et al: De novo-generated small
palindromes are characteristic of amplicon boundary junction of
double minutes. Int J Cancer. 133:797–806. 2013.
|
|
36
|
Shammas MA, Shmookler Reis RJ, Koley H,
Batchu RB, Li C and Munshi NC: Dysfunctional homologous
recombination mediates genomic instability and progression in
myeloma. Blood. 113:2290–2297. 2009.
|
|
37
|
Marti TM, Kunz C and Fleck O: DNA mismatch
repair and mutation avoidance pathways. J Cell Physiol. 191:28–41.
2002.
|
|
38
|
Li GM: Mechanisms and functions of DNA
mismatch repair. Cell Res. 18:85–98. 2008.
|
|
39
|
Lieber MR: The mechanism of double-strand
DNA break repair by the nonhomologous DNA end-joining pathway. Annu
Rev Biochem. 79:181–211. 2010.
|
|
40
|
Smith JA, Waldman BC and Waldman AS: A
role for DNA mismatch repair protein Msh2 in error-prone
double-strand-break repair in mammalian chromosomes. Genetics.
170:355–363. 2005.
|
|
41
|
Shahi A, Lee JH, Kang Y, Lee SH, Hyun JW,
Chang IY, Jun JY and You HJ: Mismatch-repair protein MSH6 is
associated with Ku70 and regulates DNA double-strand break repair.
Nucleic Acids Res. 39:2130–2143. 2011.
|
|
42
|
Stavnezer J and Schrader CE: Mismatch
repair converts AID-instigated nicks to double-strand breaks for
antibody class-switch recombination. Trends Genet. 22:23–28.
2006.
|
|
43
|
Schrader CE, Guikema JE, Linehan EK,
Selsing E and Stavnezer J: Activation-induced cytidine
deaminase-dependent DNA breaks in class switch recombination occur
during G1 phase of the cell cycle and depend upon mismatch repair.
J Immunol. 179:6064–6071. 2007.
|
|
44
|
Eccleston J, Schrader CE, Yuan K,
Stavnezer J and Selsing E: Class switch recombination efficiency
and junction microhomology patterns in Msh2-, Mlh1-, and
Exo1-deficient mice depend on the presence of mu switch region
tandem repeats. J Immunol. 183:1222–1228. 2009.
|
|
45
|
Della-Maria J, Zhou Y, Tsai MS, Kuhnlein
J, Carney JP, Paull TT and Tomkinson AE: Human Mre11/human
Rad50/Nbs1 and DNA ligase IIIalpha/XRCC1 protein complexes act
together in an alternative nonhomologous end joining pathway. J
Biol Chem. 286:33845–33853. 2011.
|
|
46
|
Eccleston J, Yan C, Yuan K, Alt FW and
Selsing E: Mismatch repair proteins MSH2, MLH1, and EXO1 are
important for class-switch recombination events occurring in B
cells that lack nonhomologous end joining. J Immunol.
186:2336–2343. 2011.
|
|
47
|
Terradas M, Martín M and Genescà A:
Impaired nuclear functions in micronuclei results in genome
instability and chromothripsis. Arch Toxicol. 90:2657–2667.
2016.
|
|
48
|
Shimizu N, Kanda T and Wahl GM: Selective
capture of acentric fragments by micronuclei provides a rapid
method for purifying extrachromosomally amplified DNA. Nat Genet.
12:65–71. 1996.
|
|
49
|
Shimizu N, Itoh N, Utiyama H and Wahl GM:
Selective entrapment of extrachromosomally amplified DNA by nuclear
budding and micronucleation during S phase. J Cell Biol.
140:1307–1320. 1998.
|
|
50
|
Shimizu N, Misaka N and Utani K:
Nonselective DNA damage induced by a replication inhibitor results
in the selective elimination of extrachromosomal double minutes
from human cancer cells. Genes Chromosomes Cancer. 46:865–874.
2007.
|
|
51
|
Canute GW, Longo SL, Longo JA, Winfield
JA, Nevaldine BH and Hahn PJ: Hydroxyurea accelerates the loss of
epidermal growth factor receptor genes amplified as double-minute
chromosomes in human glioblastoma multiforme. Neurosurgery.
39:976–983. 1996.
|
|
52
|
Ambros IM, Rumpler S, Luegmayr A,
Hattinger CM, Strehl S, Kovar H, Gadner H and Ambros PF:
Neuroblastoma cells can actively eliminate supernumerary MYCN gene
copies by micronucleus formation-sign of tumour cell revertance?
Eur J Cancer. 33:2043–2049. 1997.
|
|
53
|
Prochazka P, Hrabeta J, Vícha A and
Eckschlager T: Expulsion of amplified MYCN from homogenously
staining chromosomal regions in neuroblastoma cell lines after
cultivation with cisplatin, doxorubicin, hydroxyurea, and
vincristine. Cancer Genet Cytogenet. 196:96–104. 2010.
|
|
54
|
Sawant A, Kothandapani A, Zhitkovich A,
Sobol RW and Patrick SM: Role of mismatch repair proteins in the
processing of cisplatin interstrand cross-links. DNA Repair (Amst).
35:126–136. 2015.
|