Introduction
Unlike non-malignant cells, cancer cells utilize a
different program for glucose metabolism. Due to the altered tumor
microenvironment, cancer cells have to elevate glycolytic activity
to generate adequate adenosine-5′-triphosphate (ATP) within a
hypoxic microenvironment to maintain rapid cell proliferation. This
phenomenon of continuously producing high lactate in cancer cells
in the presence of oxygen is known as the Warburg effect (1-3).
During tumor expansion, uncontrollable cell
proliferation and abnormal tumor angiogenesis impede the nutrient
supply for cancer cells (4).
Consequently, hypoxic regions, where oxygen concentration is low,
are frequently found in solid tumors. To adapt to this hypoxic
environment, tumor cells usually adopt glycolysis for their energy
supply to facilitate angiogenesis, which, in turn, contributes to
the continued growth and survival of a tumor (5). Therefore, oxygen homeostasis and
regulating downstream cellular metabolic events are critical during
tumor progression. Members from the family of prolyl hydroxylase
(PHD), a group of well-established oxygen sensors, are potentially
the major participants that sense oxygen and regulate glucose
metabolism in tumors (6-8).
PHDs mediate hypoxia-inducible factor-1α (HIF-1α)
hydroxylation at 2 prolyl residues in the oxygen-dependent
degradation domain, enabling it to be captured, ubiquitinated and
ultimately degraded in the proteasome (9). There are three known PHD isoforms in
humans; PHD1, PHD2 and PHD3. PHD2 is one of the key enzymes
mediating HIF-1α degradation under normoxia (10-12). In addition, PHD2 is downregulated
or even barely detected in numerous cancer cells and tumor tissues
(13). The decreased expression
of PHD2 is significantly correlated with some genetic disorders,
which ultimately develop malignancies (12). Therefore, such findings render
PHD2 a potential tumor suppressor (14). In addition, PHD2 has been
associated with tumor vasculature, which occurs after tumor
glycolysis (15-17). Nevertheless, the role of PHD2 in
tumor glycolysis is still unknown. The present study focused on
investigating glycolysis, including glucose uptake, lactate
production and energetic molecular quantification and confirming
the regulatory effect of PHD2 in glycolysis in colon cancer cells
by gain- and loss-of-functional assays.
Materials and methods
Cell lines
HEK293T and human colorectal carcinoma cell lines
(HT-29, HCT116, Ls174t, LoVo, SW480, SW620, RKO and Caco-2) were
obtained from the American Type Culture Collection. Cells were
maintained in Dulbecco's Modified Eagle's Medium (DMEM) containing
10% fetal bovine serum (FBS), supplemented with 100 U/ml penicillin
and 0.1 mg/ml streptomycin at 37°C in an atmosphere containing 5%
CO2. All reagents for cell culturing were purchased from
Invitrogen (Thermo Fisher Scientific, Inc.).
PHD2 knockdown and overexpression
For PHD2 silencing, the pGCL-GFP expressing shRNA
against PHD2 [short hairpin (sh)PHD2; target sequence: GGA TGG AAT
CCA TGA GCT A] or non-targeting shRNA (Mock; TTC TCC GAA CGT GTC
ACG T) were designed and synthesized by Shanghai GeneChem Co., Ltd.
For PHD2 overexpression, the open reading frame of PHD2 was
constructed into the pGC-FU lentiviral vector (pGC-FU-PHD2)
(Shanghai GeneChem Co., Ltd.), while the empty vector was used as
vehicle control (vehicle).
HEK293T cells were co-transfected with helper
plasmids (pHelper 1.0 and pHelper 2.0; Shanghai GeneChem Co., Ltd.)
and lentiviral plasmids (pGCL-GFP-shPHD2 or pGC-FU-PHD2) using
Lipofectamine®2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) at 37°C for 24 h for lentivirus production.
Lentivirus was harvested at 48 and 72 h following transfection.
Cell debris was removed by centrifugation (300 × g; 10 min; room
temperature) and the virus was then filtered through a 0.45
μm cellulose acetate filter and collected by
ultracentrifugation (100,000 × g; 4 h; 4°C).
For lentivirus infection (37°C for 12 h), Ls174t and
SW480 cells were transduced with PHD2 silencing (PHD2-KD),
overexpressing (PHD2-OE) lentivirus, or control lentivirus
(represented as MOCK for non-targeting shRNA and vehicle for empty
vector), separately, at the multiplicity of infection of 10
(Ls174t) or 20 (SW480). At four days following infection,
GFP-positive cells were isolated by a FACSCalibur flow cytometer
(BD Biosciences). PHD2 overexpression and silencing were confirmed
by western blotting.
HIF-1α lentiviruses transduction
For HIF-1α silencing, the pLKO.1-puro lentiviral
vectors encoding shRNA targeting HIF-1α (HIF-1α-KD) or
non-targeting control shRNA (MOCK; MilliporeSigma) were obtained
for lentiviruses packaging. The cells were transduced with viral
supernatant containing 8 μg/ml of Polybrene (MilliporeSigma)
and then selected with medium containing 0.6 μg/ml of
puromycin (MilliporeSigma) after 12 h (18).
Reverse transcription-quantitative (RT-q)
PCR
Human cancerous and adjacent normal colorectum were
collected from specimens directly after surgery. Total RNA was
extracted using the TRIzol® (Thermo Fisher Scientific,
Inc.) method and the complementary DNA was synthesized using a
commercially available kit (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocols. RT-qPCR was
carried out using an iCycler system (Bio-Rad Laboratories, Inc.)
with SYBR® Green PCR master mix (Applied Biosystems;
Thermo Fisher Scientific, Inc.). PCR amplification was performed
with the following cycling conditions: i) Initial activation at
95°C for 10 min, and subsequently 40 cycles of denaturation at 95°C
for 15 sec; ii) Annealing at 55°C for 30 sec and extension at 72°C
for 30 sec (40 cycles). Primers used were as follows: Phd2: 5′-CGA
TCC GGT GAC TTT TCC CAC-3′ and 5′-GTA GGA GGT CTC CGT CCT-3′;
hexokinease-2 (Hk2): 5′-AAG ATG CTG CCC ACC TAC G-3′ and 5′-TCG CTT
CCC ATT CCT CAC A-3′; PHD kinase 1 (Pdk1): 5′-AAG ATG AGT GAC CGA
GGA GGT-3′ and 5′-CCA TAA CCA AAA CCA GCC AGA G-3′; glucose
transporter 1 (Glut1): 5′-GTG CTC CTG GTT CTG TTC TTC A-3′ and
5′-GCC AGA AGC AAT CTC ATC GAA-3′; β-actin: 5′-ACC CCG TGC TGC TGA
CCG AG-3′ and 5′-TCC CGG CCA GCC AGG TCC A-3′. The expression
levels of detected genes were normalized to the level of β-actin
mRNA. The relative expression of the target genes was calculated
using the following formula: gene
expression=2−(ΔCq-normal-ΔCq-tumor) (19). The experiment was independently
repeated three times.
ELISA assay
Cells were washed with ice-cold PBS, detached and
pelleted by centrifugation (1,000 × g; 5 min; 4°C). The cells were
resuspended in 300 μl of hypotonic buffer (10 mM Hepes; pH
7.6; 60 mM KCl; 1 mM EDTA; 1 mM dithiothreitol) containing
phenylmethylsulfonyl fluoride (PMSF; 1 mM) and protease inhibitor
cocktail (10 μl/ml) and incubated on ice for 15 min. Then,
20 μl of 10% NP-40 was added and the tube was vortexed for
10 sec. The lysates were cleared by centrifugation (13,000 × g; 1
min; 4°C) and supernatants (cytosolic fractions) were collected and
stored at -80°C.
Nuclear contents were obtained from the pellets.
First, the pellets were resuspended in extraction buffer (20 mM
Tris-HCl; pH 8; 420 mM NaCl; 1.5 mM MgCl2; 0.2 mM EDTA)
supplemented with PMSF (0.5 mM), protease inhibitors cocktail (10
μl/ml) and glycerol (25% v/v). After 30 min at 4°C, the
debris was removed by centrifugation (13,000 × g; 15 min; 4°C). The
supernatant containing nuclear proteins was aliquoted and stored at
−80°C for further analysis. Protein concentrations were determined
using a Bradford protein assay (Bio-Rad Laboratories, Inc.).
For the p50 detection, the levels of p50 were
measured using a TransAM NF-κB p50 ELISA kit (cat. no. 31101;
Active Motif, Inc.) according to the manufacturer's instructions
with a microplate reader (Bio-Rad Laboratories, Inc.). The
experiment was conducted in triplicates and was independently
repeated three times.
Western blotting and
co-immunoprecipitation
Western blotting was conducted using lysates
extracted from human tissues or cells as previously described
(20). In general, tissues or
cells were solubilized in RIPA buffer (BioTeke Corporation)
containing a proteinase inhibitor cocktail (1 mM EDTA; 1 mM PMSF; 1
mM iodoacetic acid). After the protein concentration was determined
with the Bradford method, identical amounts of proteins (50
μg) were separated on a 10% SDS-PAGE gel and transferred to
polyvinylidene fluoride membranes. The membranes were then blocked
with 5% skimmed milk for 2 h at room temperature followed by
overnight incubation with antibodies against PHD2 (1:2,000; cat.
no. NBP1-30328; Novus Biologicals, LLC), glucose transporter 1
(GLUT1; 1:500; cat. no. sc-377228; Santa Cruz Biotechnology, Inc.),
PDK1 (1:500; cat. no. sc-515944; Santa Cruz Biotechnology, Inc.),
hexokinase (HK) 2 (1:1,000; cat. no. sc-374091; Santa Cruz
Biotechnology, Inc.), p65 (1:1,000; cat. no. sc-8008; Santa Cruz
Biotechnology, Inc.) and GAPDH (1:1,000; cat. no. sc-47724; Santa
Cruz Biotechnology, Inc.) at 4°C. The following day, membranes were
rinsed and incubated with HRP-conjugated secondary antibodies
(1:2,000, cat. no. BA1082; Wuhan Boster Biological Technology,
Ltd.) at room temperature for 1 h. SuperSignal West Femto Maximum
Sensitivity Substrate (Pierce; Thermo Fisher Scientific, Inc.) was
used for development and images were captured using a Gel Doc XR
system (Bio-Rad Laboratories, Inc.). β-actin was used as the
loading control. ImageJ software (NIH; version 1.8.0) was used for
densitometry.
For immunoprecipitation assay, fresh cell lysates
using RIPA buffer (cat. no. PP1901; BioTek Instruments, Inc.)
containing a proteinase inhibitor cocktail (1 mM EDTA; 1 mM PMSF; 1
mM iodoacetic acid) (500 μg) were incubated with primary
antibody (1 μg) at 4°C for 3 h. Then, 20 μl of
protein A/G PLUS agarose beads (cat. no. sc-2003; Santa Cruz
Biotechnology, Inc.) was supplemented for overnight incubation. The
following day, the beads were pelleted and collected by
centrifugation (10,000 × g; 15 sec; 4°C), followed by washing with
RIPA buffer as aforementioned. Then, immobilized proteins were
eluted in the boiled SDS buffer for electrophoresis and
immunoblotting. Primary antibodies were used to detect the HA-tag
(1:1,000; cat. no. sc-805; Santa Cruz Biotechnology, Inc.),
Flag-tag (1:4,000; cat. no. F3165; MilliporeSigma), IKKα (1:1,000;
cat. no. 2682; Cell Signaling Technology, Inc.), anti-IKKβ
(1:1,000; cat. no. 2684; Cell Signaling Technology, Inc.).
HRP-conjugated anti-rabbit (1:5,000; cat. no. 7074; Cell Signaling
Technology, Inc.), -mouse (1:5,000; cat. no. 7076; Cell Signaling
Technology, Inc.), or -goat (1:5,000; cat. no. 7075; Cell Signaling
Technology, Inc.) secondary antibodies were used for incubation for
1 h at room temperature.
Electrophoretic mobility shift assay
An oligonucleotide containing the IκB-binding site
was radiolabeled with [γ−32p] dATP using T4
polynucleotide kinase at 37°C for 1 h. The labeled oligonucleotide
was purified with the QIAquick Nucleotide Removal Kit (cat. no.
28304; Qiagen GmbH) according to the manufacturer's instruction.
The binding between probe (1 ng) and nuclear extracts (5 ng) was
allowed to form in 20 μl reaction buffer (1 μg poly
(dI-dC); 0.1 μg BSA; 1 mM DTT; 60 mM KCl; 10% glycerol and
20 mM HEPES, pH 8.4) at 30°C for 30 min. Then, 1 μg of p65
antibody (cat. no. 05-361; Upstate Biotechnology, Inc.) was added
to immobilize the oligonucleotide/protein complexes, which were
then separated by native electrophoresis in a 6% polyacrylamide gel
that was exposed to an X-ray film (Amersham; Cytiva) for 96 h at
-80°C.
2-deoxy-D-glucose uptake assay
Cells (1×107 cells/ml) were washed twice
with PBS and resuspended and incubated in 100 μl PBS
supplemented with 5 mM of 2-deoxy-D-glucose [containing 2
μCi/ml 2-deoxy-(1-3H) glucose] at 37°C for 30
min. The glucose uptake was terminated by adding 100 μl of
ice-cold PBS containing 100 μM phloretin. The cells were
subjected to brief centrifugation (20,000 × g; 30 sec; room
temperature). Then, the cell pellets were washed once and
transferred into a scintillation vial containing 10 ml of Optifluor
(Perkin-Elmer Inc.). The incorporated radioactivity was measured by
a liquid scintillation counter (Beckman Coulter, Inc.). The uptaken
2-deoxy-[1-3H] glucose concentrations were presented as
nmol/104 cells.
L-lactate assay
The day before the assay, cells were seeded
(1.2×105 per well) in 12-well plates in complete medium
in quadruplicate. The medium was replaced with starve medium (DMEM
containing 2% FBS) on the next day for 48 h. Then, the medium was
collected and the lactate concentration was determined using a
fluorometric lactate assay kit (cat. no. K607; BioVision, Inc.).
Finally, the results were normalized to protein content.
Measurement of intracellular adenine
nucleotide levels
For ATP and adenosine diphosphate (ADP) measurement,
cell lysate samples were deproteinized with 5% trichloroacetic acid
at various times before and after the glucose addition. Following
centrifugation (14,000 × g; 10 sec; 4°C), the supernatants were
neutralized by ether extraction, lyophilized and stored at -80°C
until further analysis. Then, the nucleotide extracts were analyzed
by high-performance liquid chromatography using an anion exchange
hypersil C18 column (4.6×250 mm) at a flow rate of 0.9 ml/min over
a 10-min buffer gradient (0.1 mol/l KH2PO4,
pH 6.0). The eluting flow was continuously monitored by UV
absorption at 254 nm and the peaks of nucleoside triphosphates were
quantified by electronic integration with external standards used
as reference. Intracellular ATP contents were quantified and
normalized to cell number. The results are presented as the ATP/ADP
ratio, which is unaffected by sample volume loss during ether
extraction or cell numbers.
Human colorectal cancer xenografts in
immunodeficient mice
A total of 48 combined immunodeficient (SCID) mice
(male, 6-8 weeks old, 20-25 g) were purchased from the Army Medical
University Institute of Experimental Animal. Mice were fed at an
ambient of 45-55% relative humidity with the constant temperature
of 21±2°C under a 12-h light/dark cycle. Lentivirus for
PHD2-silencing or -overexpression and their corresponding controls
were used to transduce SW480 (for PHD2 overexpression) or Ls174t
cells (for PHD2 depletion). SCID mice were randomly divided into 3
groups (n=16) for PHD2 overexpression, silencing and control.
Stably transduced SW480 and Ls174t cells or the corresponding
parental cells (~2×106) in 200 μl of PBS were
admixed with 200 μl of Matrigel (BD Biosciences). The cells
were delivered to the mice through subcutaneous injection. At three
days after injection, the mice were subjected to measurements of
tumor size, body weight and other side effects twice a week. Tumor
volume (mm3) was calculated by the formula: 0.52 ×
length × width × thickness. After 40 days, mice were sacrificed
(n=6) and tumors were excised for further analysis. The rest of the
mice were subjected to monitoring until they reached the humane
endpoint for survival analysis. The humane endpoint was defined as
weight loss of more than 20% and mice were sacrificed by
CO2 asphyxiation (at the rate of 30-70% of the chamber
volume per minute) followed by cervical dislocation. Survival rate
was analyzed using the Kaplan-Meier method. Animal studies were
performed under a protocol approved by the Army Medical University
Institutional Animal Care and Use Committee (approval no.
AMUWEC20232707).
Ki-67 proliferation index
Tumor tissues harvested from the xenografts were
fixed in 10% formalin for 24 h at room temperature, gradual
dehydration in increasing alcohol concentrations and then embedded
within paraffin for 8 h at room temperature as previously described
(21). Tissue sections were then
incubated with antibodies against Ki67 (1:100; cat. no. NB500-170;
Novus Biologicals, LLC). Nuclei were counterstained with Mayer's
hematoxylin (MilliporeSigma) for 5 min at room temperature.
Ki-67-positive cell from 6-8 random views of each section was
counted and images captured under a light inverted microscope
(XSZ-D2; Olympus Corporation) by a researcher who was blinded to
this study. ImageJ software (NIH; v1.8.0) was used to process the
results and the data were presented as the average percentage of
Ki-67-positive cells per view (22).
Statistical analysis
The results were analyzed using SPSS 13.0 software
(v13.0; SPSS, Inc.). One-way analysis of variance with Tukey's post
hoc test was performed to compare results from different groups.
The quantitative data were expressed as the mean ± SEM. All
experiments were repeated three to five times independently. The
survival analysis was performed using the log-rank test. Pearson's
correlation coefficient was used to evaluate the relationship
between the expression of PHD2 and glycolysis-related genes in
cancerous and adjacent normal colon tissues. P<0.05 was
considered to indicate a statistically significant difference.
Results
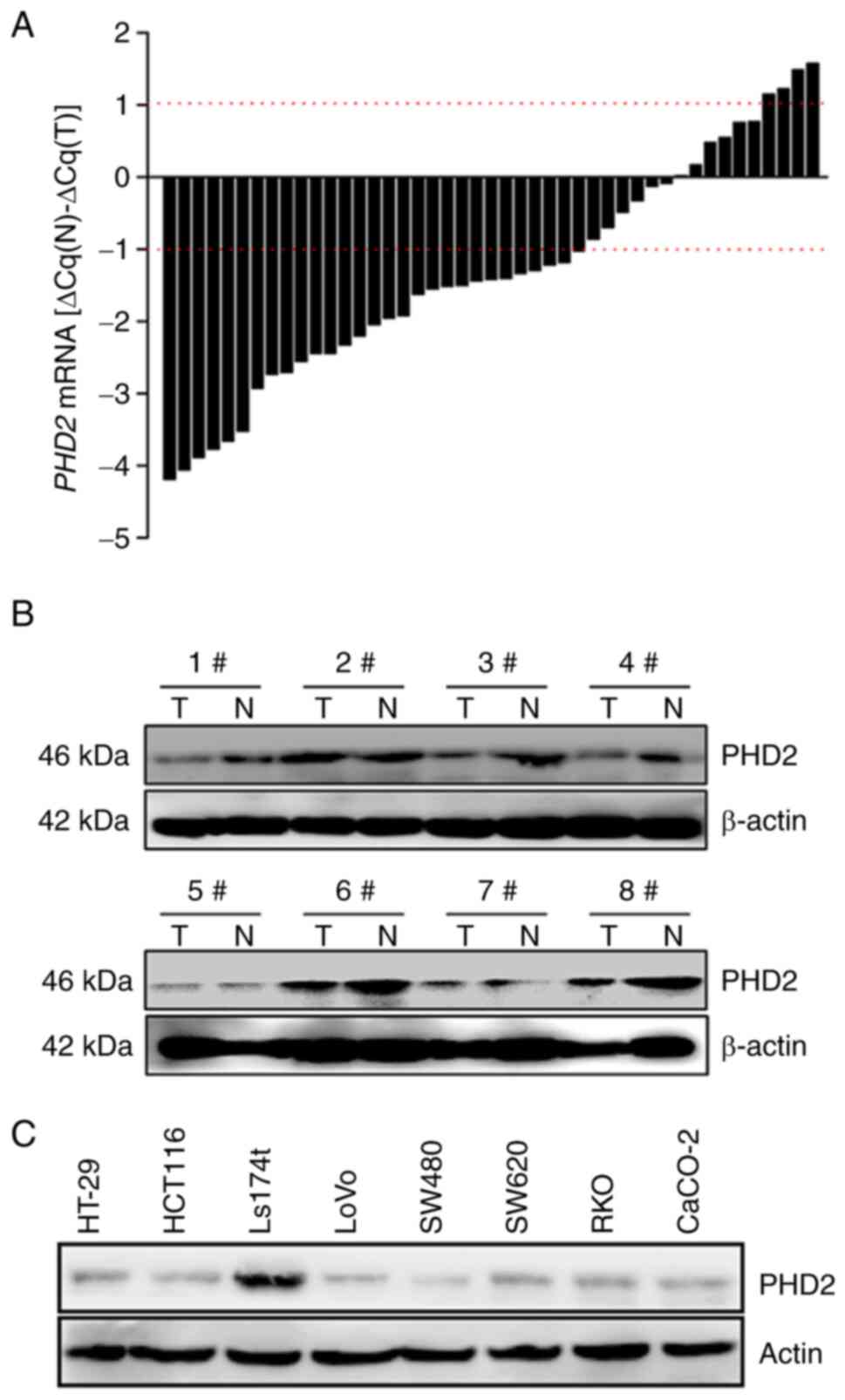
PHD2 is decreased in colorectal
tumors
A total of 45 paired human colorectal tumors and
adjacent normal tissues were interrogated for PHD2 expression. PHD2
reduction was observed in 29 of 45 (64.4%) tumors compared with the
paired normal colorectal tissues (Fig. 1A). In addition, univariate
analysis revealed that PHD2 expression is significantly different
in tumor and paired normal tissues at the transcriptional level
(P<0.001). Notably, our previous study demonstrated that the
decreased PHD2 expression in tumor tissue favors higher tumor
grades and poorer overall survival of colorectal cancer patients
(23). In agreement with the
findings at the mRNA level, decreased PHD2 expression at the
protein level was also observed in all tested paired samples
(Fig. 1B). In addition, results
from our cell-based immunoblotting assay also revealed that PHD2
was barely detected in most tested colon cancer cells, except for
Ls174t cells (Fig. 1C).
Low expression levels of PHD2 correlate
with high expression levels of glycolysis-related genes
Genes closely related to glycolysis, such as HK2,
PDK1 and GLUT1, were also evaluated in tumor and paired normal
adjacent tissues. Consistent with previous reports (24-26), the mRNA levels of those genes,
including HK2 (P<0.01), PDK1 (P<0.05) and GLUT1 (P<0.001),
were significantly higher in tumor tissues than in paired normal
tissues (Fig. 2A). To elucidate
the potential relationships between the expression levels of PHD2
and glycolysis-related genes, we plotted PHD2 mRNA levels against
HK2, PDK1 and GLUT1 mRNA levels in colorectal tissues from both
tumor and adjacent normal colorectum. As a result, the expression
of HK2 (P<0.00001), PDK1 (P=0.00217) and GLUT1 (P<0.0019)
were significantly inversely-correlated with that of PHD2 in
colorectal tumors (Fig. 2B).
Together, these findings suggested that PHD2 is downregulated in
colorectal tumor tissues and may be involved in tumor-associated
glycolysis.
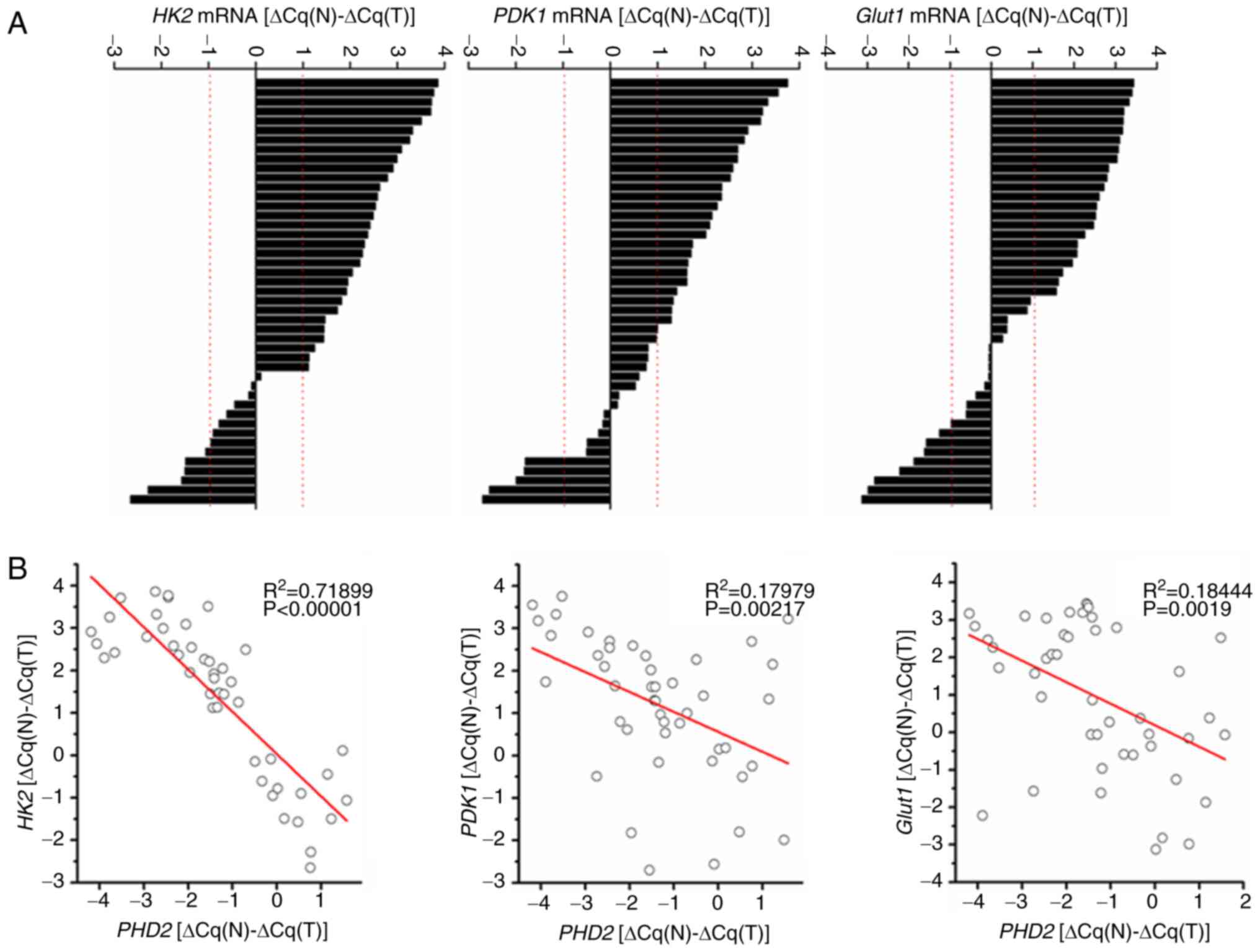 | Figure 2PHD2 is negatively correlated with
glycolysis-related genes in human colorectal cancers. (A) Results
of RT-qPCR detecting the expression of HK2, PDK1, or
GLUT1. ΔCq(N) and ΔCq(T) indicate the Cq value
of GAPDH was subtracted from Cq value of HK2, PDK1,
or GLUT1 in normal tissue or tumor tissue, respectively. Bar
value: (ΔCq(N)-ΔCq(T)) depicts the fold-changes of
HK2, PDK1, or GLUT1 mRNA in tumor tissues vs.
normal tissues and the difference is expressed in log2 scale. (B)
Significant negative correlation between the relative mRNA
expression level of PHD2 (x-axis) and HK2,
PDK1, or GLUT1 (y-axis) in the colorectal cancer
samples. PHD2, prolyl hydroxylase domain protein 2; RT-qPCR,
reverse transcription-quantitative PCR. |
Inhibition of PHD2 stimulates glycolytic
activity in colorectal cancer cells
To confirm the role of PHD2 in tumor-associated
glycolysis, in vitro glycolytic activity was examined in
different human colorectal cancer cells with altered PHD2
expression levels. Among the tested colon cancer cell lines, SW480
cells presented the lowest, while Ls174t cells expressed the
highest PHD2 protein expression (Fig.
1C). Therefore, PHD2 was overexpressed in SW480 cells (PHD2-OE)
and PHD2 silenced in Ls174t cells (PHD2-KD) via lentiviral-based
modification, respectively. Transduction efficiency and PHD2
expression were evaluated by immunoblotting analysis (Fig. 3A). As the data indicated a
negative correlations between PHD2 and GLUT1, HK2 and PDK1, the
protein level of these proteins was assessed in PHD2-OE cells and
control cells (Fig. 3B). Compared
with control counterparts, PHD2 overexpression significantly
suppressed the expression of GLUT1, HK2 and PDK1 proteins.
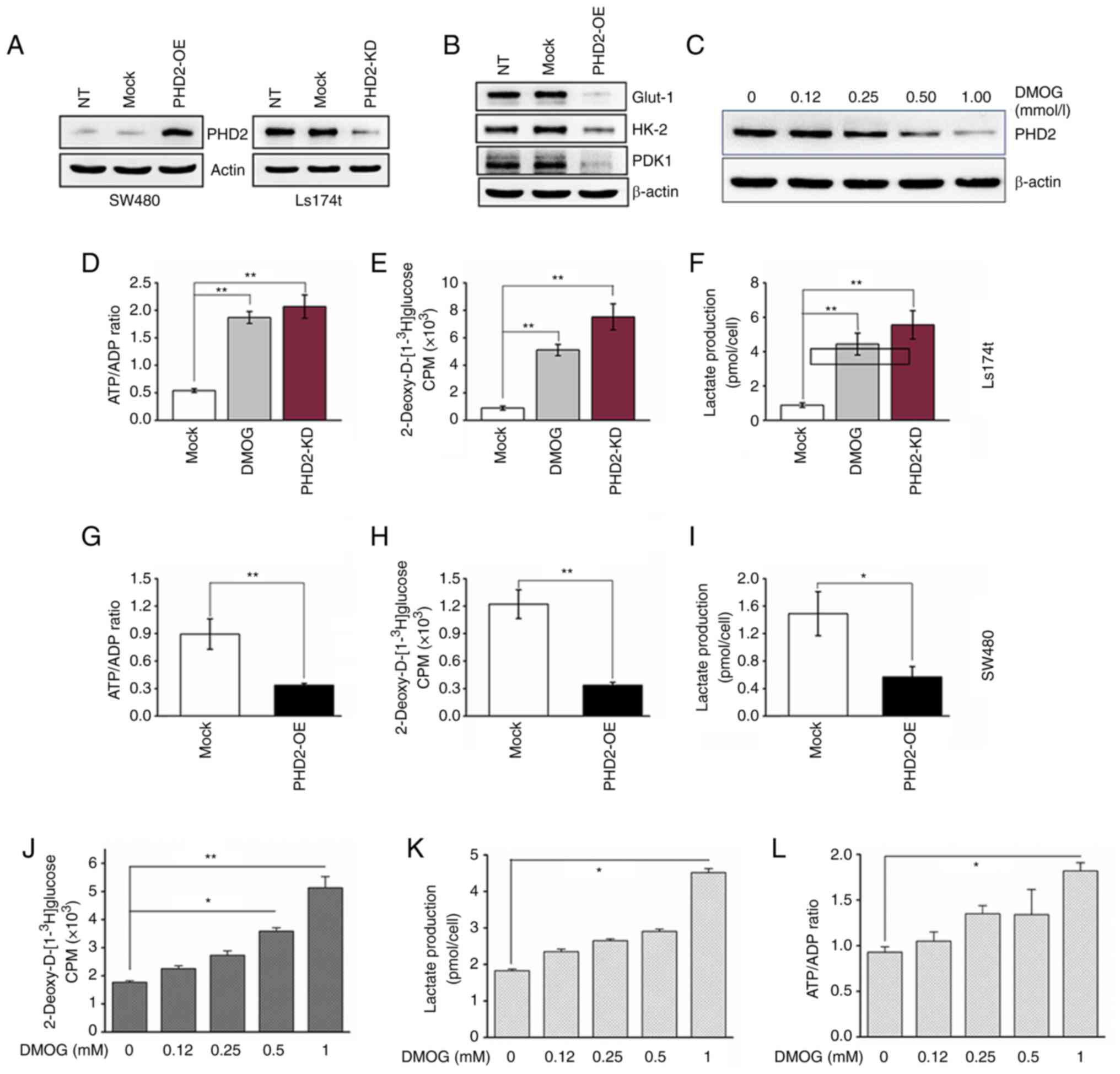 | Figure 3Inhibition of PHD2 promotes
glycolysis in colorectal cancer cells. (A) PHD2 overexpression and
silencing were confirmed by western blot analysis. (B) Expression
of GLUT1, HK2 and PDK1 in SW480 cells with altered PHD2 expression.
(C) Expression of PHD2 in Ls174t treated with DMOG at indicated
concentrations for 24 h. (D and G) Quantitative analysis of the
ATP/ADP ratio, (E and H) glucose uptake and (F and I) extracellular
lactate in control, (0.5 mM) DMOG treated and PHD2-KD colon cancer
cells. (J) Glucose uptake, (K) lactate production and (L) the ratio
of ATP/ADP, were quantitatively analyzed in Ls174t cells
preincubated in the medium containing 0-1 mM DMOG for 24 h (n=3;
*P<0.05 and **P<0.01 vs. Mock). PHD2,
prolyl hydroxylase domain protein 2; GLUT1, glucose transporter 1;
HK2, hexokinease-2; PDK1, PHD kinase 1; DMOG,
dimethyloxalylglycine; ATP, adenosine-5′-triphosphate; ADP,
adenosine diphosphate; NT, non-transduced; Mock, non-targeting
shRNA control (for PHD2 knockdown) or vehicle control (for PHD2
overexpression); PHD2-OE, PHD2 overexpression; PHD2-KD,
PHD2-knockdown. |
Notably, PHD2 can also be inhibited in vitro
by the competitive inhibitor dimethyloxalylglycine (DMOG), a
cell-permeable 2-oxoglutarate analog (27). It was observed that 24 h exposure
to DMOD led to a dose-dependent PHD2 reduction in colon cancer
cells (0, 0.125, 0.25, 0.50, or 1.00 mM DMOG; Fig. 3C). Then, the role of PHD2 in
glycolytic activity was further investigated by evaluating glucose
uptake, lactate production and intracellular ATP/ADP ratio. It was
found the glycolytic activity increased greatly in PHD2-KD cells
and DMOG (0.5 mM) treated cells compared with the control group
(Fig. 3D-F). By contrast,
opposite effects could be observed in PHD2-OE cells (Fig. 3G-I). In addition, not only was a
dose-dependent inhibitory effect of DMOG on PHD2 expression
discovered but it was also noted that DMOG promoted glucose uptake,
lactate production and intracellular ATP/ADP ratio in colon cancer
cells in the same manner (Fig. 3J and
K). Taken together, the results suggested that PHD2 may be
involved in regulating glycolysis in colon cancer cells.
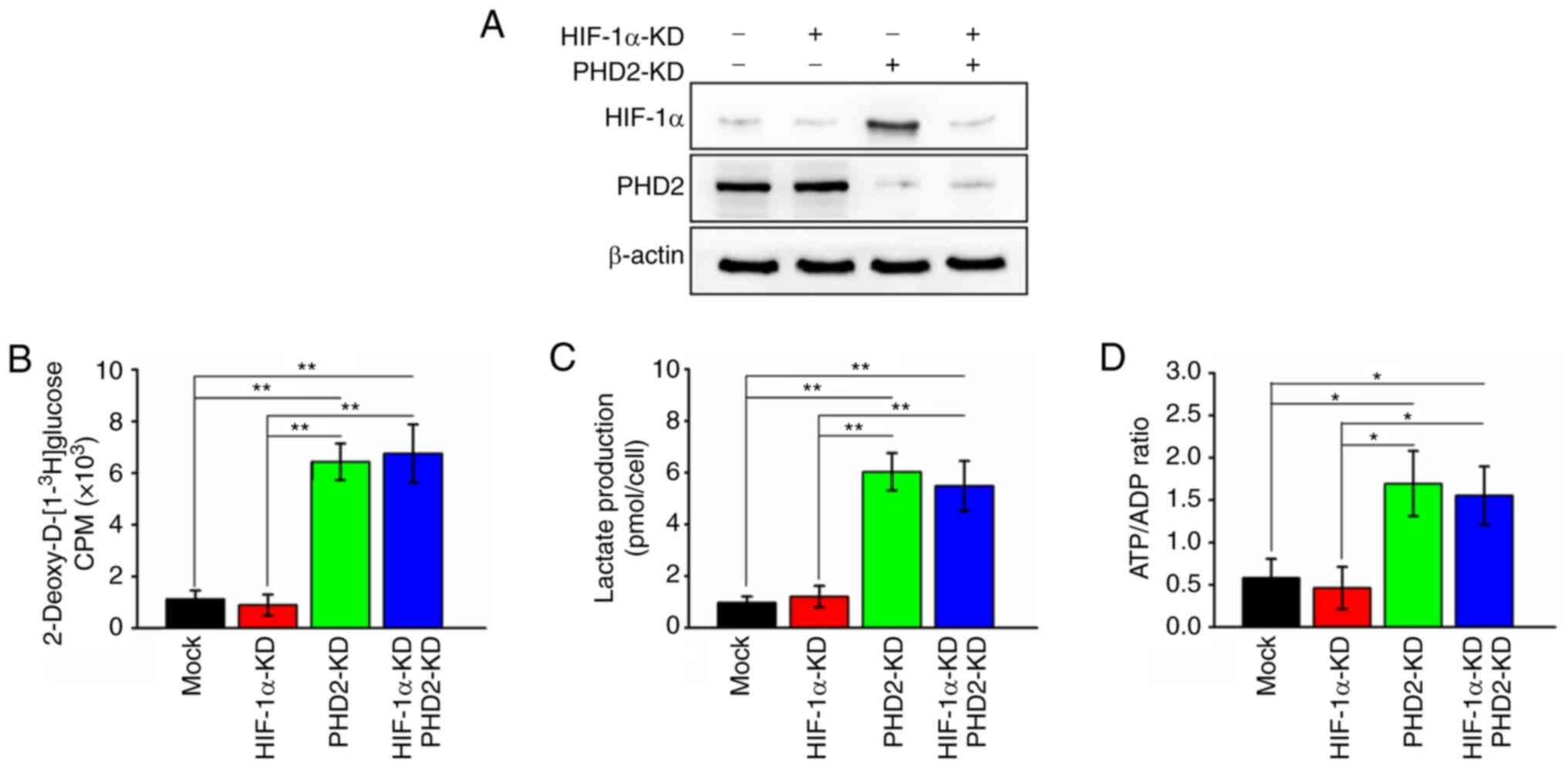
Regulation of glycolysis by PHD2 is
independent of HIF-1α
As HIF-1α is the functionally characterized target
of PHD2 (28), it was next
determined whether the effects of PHD2 on glycolysis were dependent
on HIF-1α. For this purpose, the present study generated HIF-1α
deficient and HIF-1α/PHD2 double deficient colon cancer cells and
the expression of HIF-1α and PHD2 was evaluated. HIF-1α expression
was significantly suppressed by lentivirus-mediated silencing and
PHD2 depletion elevated the expression of HIF-1α (Fig. 4A). In addition, it was noticed
that cells with HIF-1α silenced did not promote glycolysis activity
as reflected by no significant change in glucose uptake, lactate
production and intracellular ATP/ADP ratio compared with parental
cells (Fig. 4B-D). It also found
that further knockdown of HIF-1α in colon cancer cells did not
affect the PHD2 depletion-mediated promotive effects on colon
cancer glycolysis activity (Fig.
4B-D). These findings prompted the hypothesis that
PHD2-promoted glycolysis is independent of HIF-1α in colon cancer
cells.
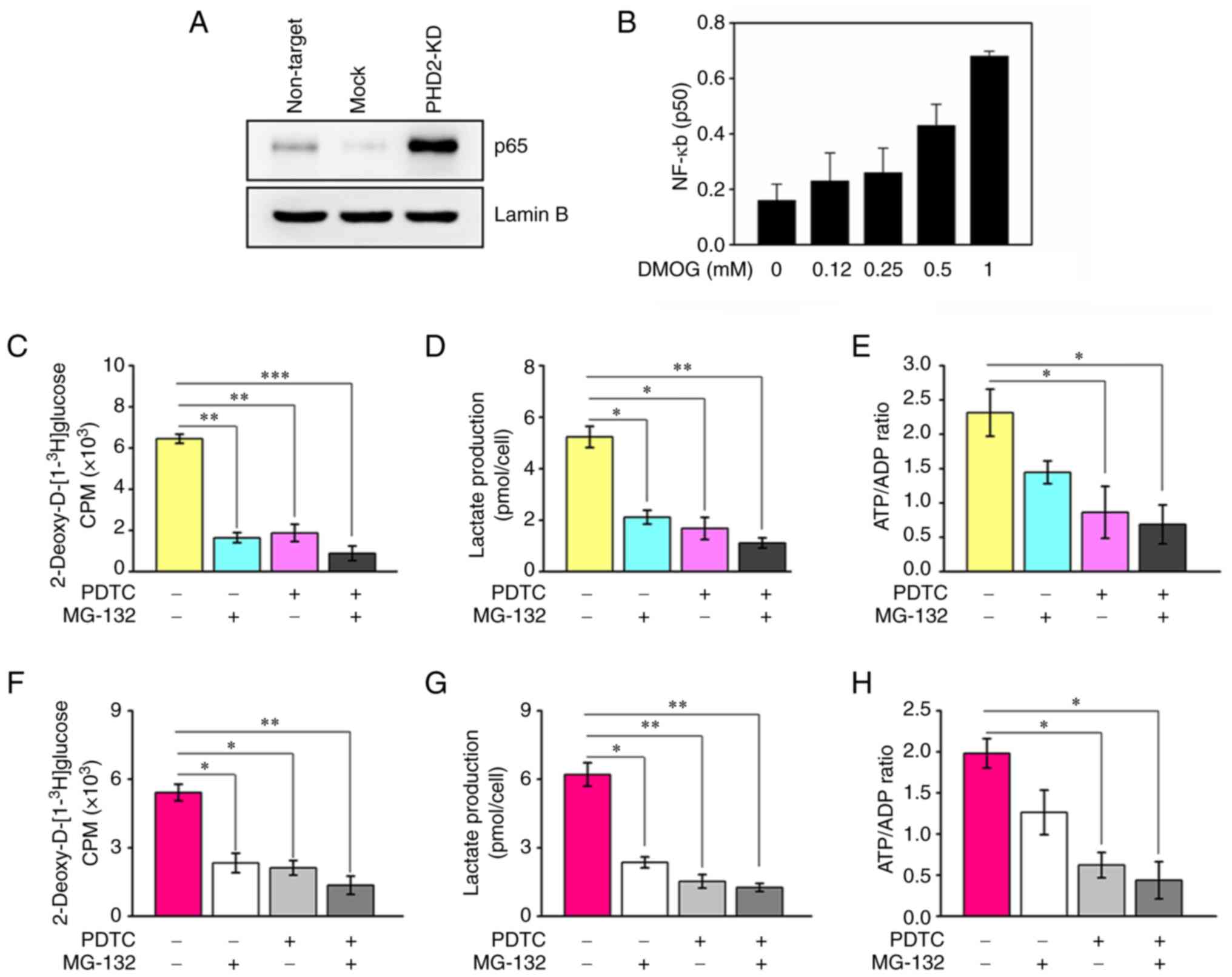
PHD2 inhibits glycolytic activity through
IKKβ/NF-κB
Accumulating evidence has revealed that PHD2 has
novel targets other than HIF-1α (29). NF-κB signaling components are
notable candidate targets (30,31). Therefore, the present study
determined how PHD2 affects NF-κB signaling activity and found that
depletion of PHD2 led to upregulated nuclear p65 subunit (Fig. 5A). Also, DMOG-mediated PHD2
suppression increased nuclear translocation of the p50 subunit
(Fig. 5B). Notably, employing the
IKKβ inhibitor (PDTC) or preventing the proteasomal degradation of
IκB significantly attenuated the promotive effects of PHD2
knockdown (Fig. 5C-E) or PHD2
inhibition (Fig. 5F-H) on
glycolysis in colon cancer cells. These findings indicated that
PHD2 inhibits glycolytic activity through the IKKβ/NF-κB
pathway.
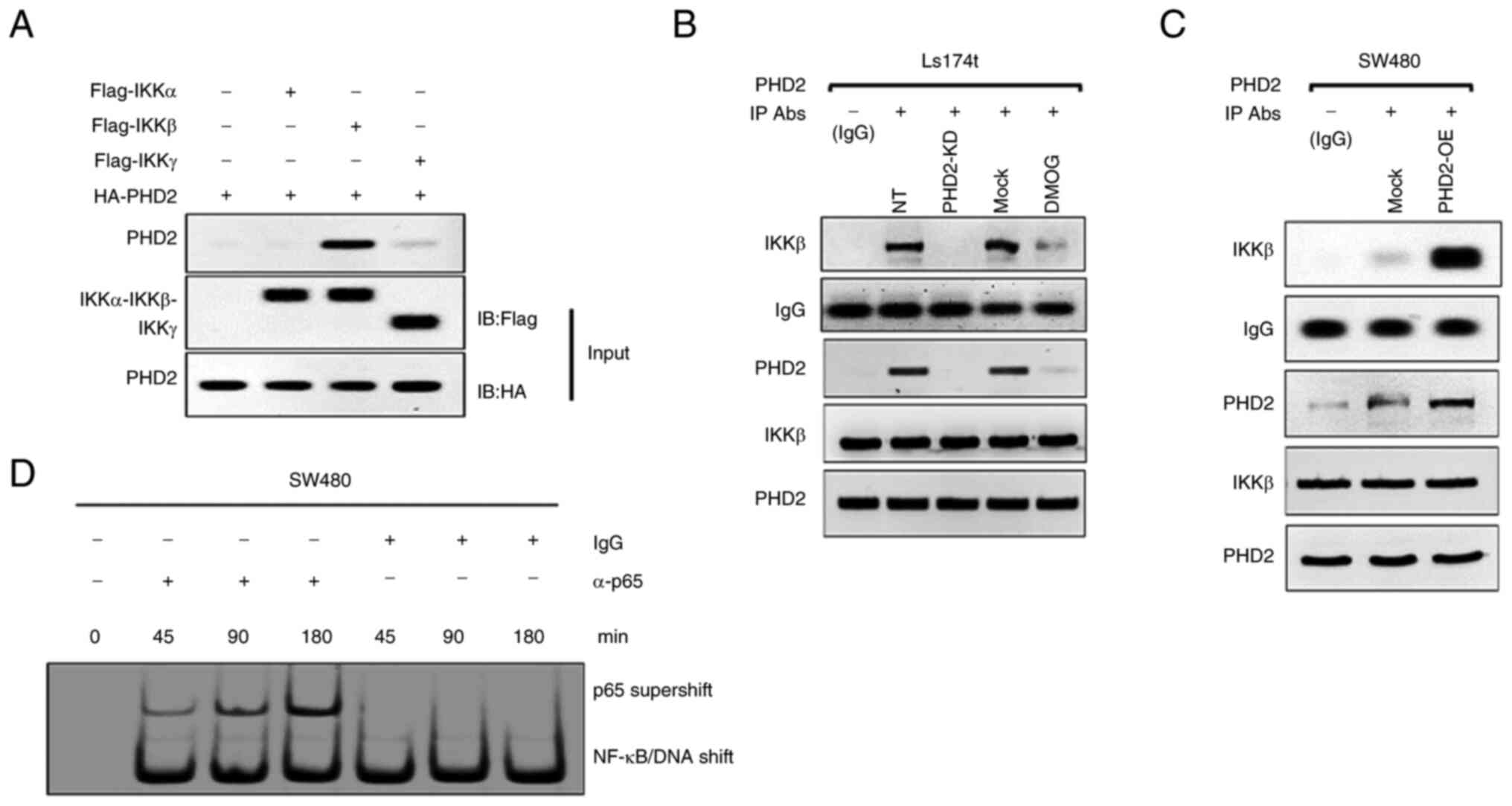
PHD2 regulates the DNA binding properties
of NF-κB
Prior studies have shown that NF-κB relies on IκB
degradation to enter the nucleus and initiate transcription
(32,33). In addition, NF-κB-dependent IκB
synthesis is critical for NF-κB signaling termination (34). Furthermore, IKKβ has been reported
to be the target of PHD2 (30,31). Thus, PHD2 may modulate NF-κB
signaling in an IKKβ-dependent manner. Through immunoprecipitation
assay, it was noted that, among the IKK isoforms, PHD2 preferably
interacts with IKKβ (Fig. 6A).
The present study further confirmed the interaction between PHD2
and IKKβ in cells with altered PHD2 expression. As shown, silencing
or inhibiting PHD2 attenuated the PHD2-IKKβ interaction, while
enhanced binding between PHD2 and IKKβ was observed in PHD2-OE
cells (Fig. 6B and C). Meanwhile,
the expression of p65 in the cytosol and nucleus was influenced by
PHD2 overexpression (Fig. 6D),
suggesting that PHD2 exerts its effect dose-dependently on IKKβ,
presumably after p65 nuclear translocation.
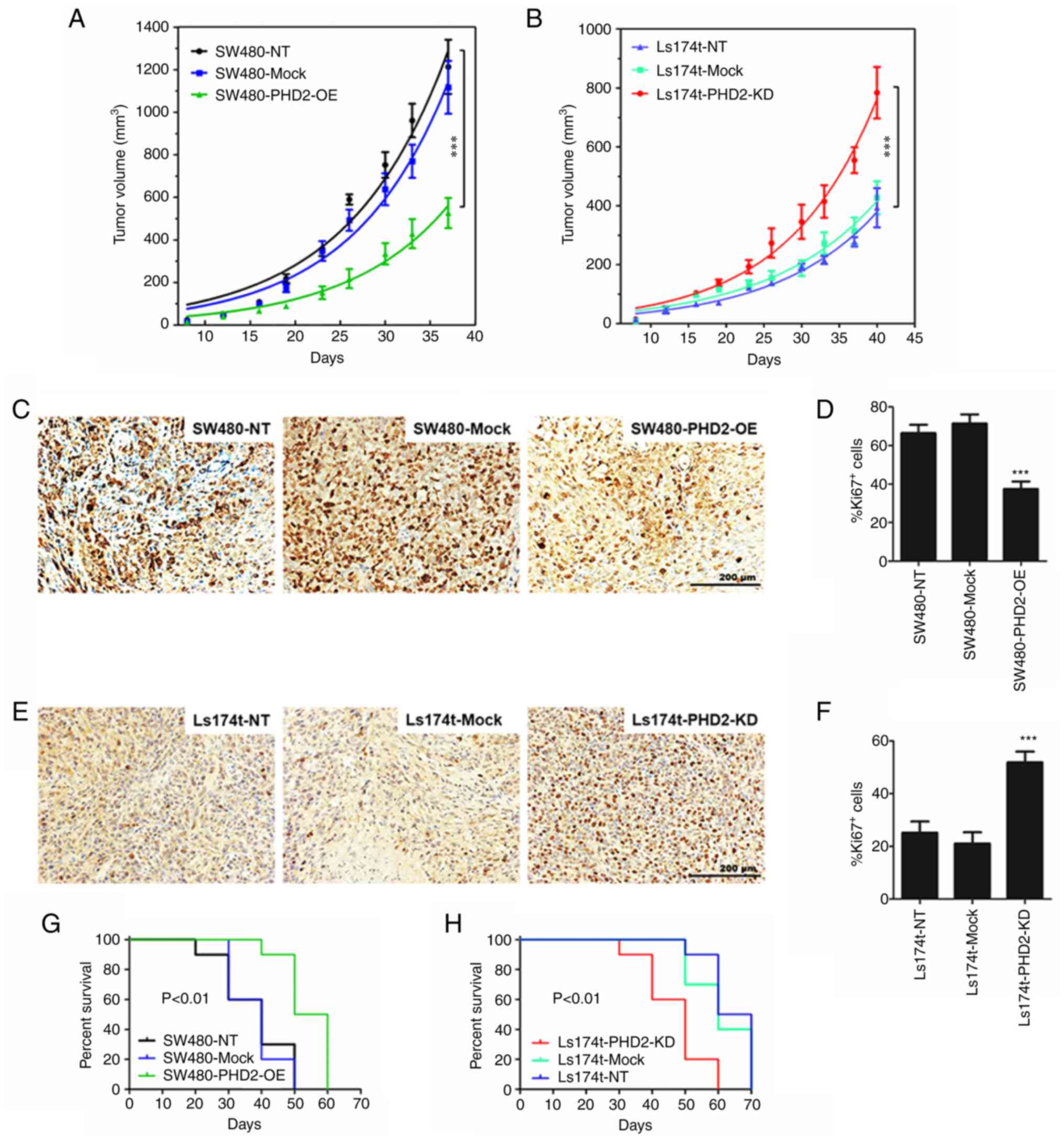
PHD2 inhibits colorectal tumor growth in
vivo and prolongs animal survival
To further characterize the functional significance
of PHD2 on tumor growth in vivo, a subcutaneous xenograft
model of colorectal cancer was established in SCID mice. Parental
colon cancer cells (SW480 cells or Ls174t) or parental cells with
or without PHD2 expression modulation (PHD2-OE, PHD2-KD, or Mock
control) were subcutaneously inoculated into SCID mice. The data
showed that colon cancer cells expressing control vectors grow into
sizable tumors and were comparable to non-transduced cells
(Fig. 7A and B). In addition,
mice transplanted with Ls174t cells, which express a higher level
of PHD2, developed smaller tumors than those implanted with SW480
cells (Fig. 7A and B). Notably,
the data also demonstrated that tumor growth was significantly
inhibited by PHD2 overexpression (Fig. 7A) but was dramatically promoted by
PHD2 silencing (Fig. 7B). The
result suggests a suppressive effect of exogenous PHD2 on human
colorectal cancer progression in vivo. Additionally, the
findings further revealed that Ki-67-positive cell proportion,
representing the tumor proliferation index, was significantly
reduced in mice transplanted with PHD2 overexpressing cancer cells
and significantly higher in mice inoculated with PHD2 silencing
cancer cells compared with the corresponding control mice (Fig. 7C-F). Kaplan-Meier curves were used
to determine survival in each mouse group. The log-rank test
demonstrated that PHD2 overexpression significantly prolonged the
survival of tumor-bearing mice compared with the wild-type or mock
control groups (P<0.01) (Fig.
7G). Again, as shown in Fig.
7H, PHD2 deficiency reduced the survival of tumor-bearing mice
(P<0.01). Taken together, the data indicated that PHD2 markedly
delayed tumor growth and increases survival of mice in a model of
colorectal cancer.
Discussion
A promising therapeutic target selective for cancer
cells may focus on their dependence on aerobic glycolysis for
energy metabolism (35). An
increasing body of evidence has revealed that elevated glycolysis
can enhance the O-glycosylation of IKKβ that subsequently
triggers the activation of NF-κB signaling (36-38). Other studies have implicated
IKKβ/NF-κB signaling in cancer cell metabolic stress adaption and
energy metabolism, including colorectal cancer (39-42). In addition, PHD2-mediated
activation of NF-κB signaling has been reported via the
IκB-dependent pathway (31).
However, whether PHD2 regulates glycolysis by modulating NF-κB
signaling in colorectal cancer has not been well studied. The
present study explored the crucial role of PHD2 in colon cancer
cell glycolysis and investigated the potential regulatory effects
of PHD2 in NF-κB signaling during glycolysis both in vitro
and in vivo.
By interrogating PHD2 expression in human tissues
and cell lines, it was found that PHD2 expression was significantly
downregulated in cancerous tissues and cells, in agreement with
previous findings (29,43). In association with PHD2
downregulation, the present study also observed elevated critical
glycolytic enzymes, HK2, PDK1 and GLUT1, in human colorectal cancer
tissues, negatively correlated with PHD2 expression level. The
present in vitro study consistently reported that PHD2
overexpression decreased, while PHD2 depletion increased, the
expression of GLUT1, HK2 and PDK1 in colon cancer cells. Similar
findings have also been observed by other researchers (44). These findings highlight the
importance of PHD2 in cancer glucose metabolism and shed light on
the tumor-suppressive role of PHD2.
PHD2 has been reported to regulate HIF-1α
proteasomal degradation via hydroxylation in normoxia (9). Meanwhile, in the presence of DMOG,
or during hypoxia, PHD2 is suppressed. In turn, HIF-1α enters
nuclear and mediates glycolysis (45). Similarly, upregulated HIF-1α in
PHD2 depleted cells was observed. However, in contrast to the
expectation that silencing HIF-1α in PHD2 deficient cells would
affect the glucose metabolism, it was found that the
PHD2-deficiency-enhanced glycolysis did not depend on the absence
of HIF-1α. Notably, a previous study has demonstrated that
depleting PHD2 in colon cancer cells (HCT116) promotes tumor
progression in a HIF-1α-independent but NF-κB-dependent manner
(31). A recent study has shown
that PHD2 activates NF-κB signaling independent of HIF-1 signaling
(46). These studies, at least
partially, support the findings of the present study. They also
prompted the discovery of the 'talk' between PHD2 and NF-κB
signaling. Notably, the results of the present study showed that
PHD2 inhibitor DMOG induced NF-κB activation. These findings
indicated that the HIF-hydroxylase activity in PHD2 is not
necessary for NF-κB inhibition. A recent study revealed that PHD2
exerts an inhibitory effect on NF-κB signaling in colon cancer
(47). In addition, PHD2 has been
demonstrated to hydroxylate Ikkβ within a conserved motif (LXXLAP)
in malignant cells from different organs, such as colon and cervix
(30). Thus, targeting the
PHD2/Ikkβ/NF-κB axis may be a promising therapeutic strategy for
colorectal cancer.
To further validate the findings of the present
study, an in vivo xenograft model was generated. In mice
that received PHD2 overexpressing cells, a smaller tumor burden,
extended survival and reduced tumor cell proliferation were
observed. By contrast, mice receiving PHD2 deficient cells
developed a larger tumor burden, shorter survival and enhanced cell
proliferation. These data were consistent with findings in a recent
study on colon cancer (47).
Together, these data highlighted a tumor-suppressive role of PHD2
in colorectal cancer.
However, it has to be mentioned that a variety of
molecules have been identified as PHD2 targets apart from Ikkβ and
HIF-1α. For example, PHD2 has been reported to hydroxylate NDRG3
(48) and eukaryotic elongation
factor 2 kinase (49), linked to
tumor progression (50).
Therefore, PHD2 may inhibit tumor progression through different
pathways synergistically and these molecules and other
un-identified participants may be involved in this process.
However, due to the limited resources, the current study only
focused on dissecting the role of the PHD2/Ikkβ/NF-κB axis during
the glycolysis of colon cancer. The results would be more
comprehensive if the crosstalk among these potential candidates
could be explored simultaneously.
PHD3 competes with HSP90 for IKKβ binding,
explaining why PHD modulation does not require its hydroxylase
activity (51). PHDs share a
conserved C-terminal domain with hydroxylase activity. It was
hypothesized that PHD2 may modulate NF-κB signaling by means of
IκκB hydroxylation.
NF-κB activation in colon cancer cells has been well
documented in previous studies (52,53). The results from the current study
revealed a decreased PHD2 in colorectal cancer. The present study
also demonstrated that PHD2 terminated NF-κB signaling in
colorectal cancer cells. Together, these findings shed light on the
potential significance of PHD2 during colorectal cancer
progression. In addition, the crosstalk between NF-κB and IKK
connected cancer progression and glycolysis. The present study
revealed that PHD2 depletion in colorectal cancer cells enhanced
glycolysis (Fig. 3). These
findings indicated a suppressive effect of PHD2 on colorectal
cancer cells, at least to a certain degree, via the IKK/NF-κB axis.
Furthermore, as aforementioned, PHD2 targets multiple proteins; it
is possible that other proteins downstream of PHD2 also participate
in this process. In addition to the associations between PHD2
expression level and colorectal tumor stage and overall survival of
patients (23), the present study
indicated the suppressive function of PHD2 in colorectal
cancer.
NF-κB signaling-induced cancer cell survival and
therapy resistance have been reported in numerous malignancies,
including colorectal cancer. Elevated NF-κB activity has been
observed in colorectal cancer cells treated with chemotherapeutics
(54); therefore, targeting NF-κB
is expected to potentiate conventional therapeutic regimens in
comprehensive strategies. Furthermore, an increasing number of
studies indicate that metabolic pathways may be favorable targets
(55,56). For example, the present study
found that PHD2 inhibited NF-κB and knockdown of PHD2 led to
upregulation of glycolysis in colorectal cancer cells, which
essentially increases tumor cell proliferation and reduces animal
survival. Taken together, evaluating PHD2 level may facilitate the
selection of a targeted therapeutic regime for colorectal cancer
with elevated NF-κB activity.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GX and LX designed the study and wrote the
manuscript. HW, LX, WY, SW and GX performed the experiments. LX, HW
and SW analyzed the data. LX and GX advised on experimental
procedures and revision of the paper. GX and LX confirm the
authenticity of all the raw data. All authors contributed to this
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The experimental protocol was established, according
to the ethical guidelines of the 1964 Declaration of Helsinki. The
protocol of immunohistochemistry for patient tissues was approved
by the Ethics Committee of the First Affiliated Hospital (Southwest
Hospital), the Third Military Medical University [Army Medical
University; approval no. 2012 (12)] and all patients or family members
involved provided written informed consent. Animal studies were
performed under a protocol approved by the Army Medical University
Institutional Animal Care and Use Committee (approval no.
AMUWEC20232707).
Patient consent for publication
Written informed consent for publication was
obtained from all participants.
Competing interests
The authors declare that they have no competing
interests.
Authors' information
Dr Ganfeng Xie: ORCID: 0000-0003-0466-7710.
Acknowledgments
The authors gratefully acknowledge Dr Xin Liu from
the Department of Central Laboratory, Third Military Medical
University (Army Medical University), for their kind help in
providing RKO and LoVo cell lines. The authors would like to
express their sincere gratitude to Pro Houjie Liang from the Third
Military Medical University (Army Medical University) for
generously providing the HT-29 cells used in the present study.
Funding
The present study was supported by the Sichuan Science and
Technology Program (grant no. 2023YFS0318) and the Chengdu Science
and Technology Program (grant no. 2022-YF05-01438-SN).
References
|
1
|
Dang CV: Cancer metabolism: The known,
unknowns. Biochim Biophys Acta Rev Cancer. 1870:12018.
|
|
2
|
Martinez-Reyes I and Chandel NS: Cancer
metabolism: Looking forward. Nat Rev Cancer. 21:669–680. 2021.
|
|
3
|
Trefts E and Shaw RJ: AMPK: Restoring
metabolic homeostasis over space and time. Mol Cell. 81:3677–3690.
2021.
|
|
4
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011.
|
|
5
|
Zecchin A, Kalucka J, Dubois C and
Carmeliet P: How endothelial cells adapt their metabolism to form
vessels in tumors. Front Immunol. 8:17502017.
|
|
6
|
Losman JA, Koivunen P and Kaelin WG Jr:
2-Oxoglutarate-dependent dioxygenases in cancer. Nat Rev Cancer.
20:710–726. 2020.
|
|
7
|
Liao C and Zhang Q: Understanding the
oxygen-sensing pathway and its therapeutic implications in
diseases. Am J Pathol. 190:1584–1595. 2020.
|
|
8
|
Semenza GL: The Genomics and genetics of
oxygen homeostasis. Annu Rev Genomics Hum Genet. 21:183–204.
2020.
|
|
9
|
Koivunen P and Kietzmann T:
Hypoxia-inducible factor prolyl 4-hydroxylases and metabolism.
Trends Mol Med. 24:1021–1035. 2018.
|
|
10
|
Wong BW, Kuchnio A, Bruning U and
Carmeliet P: Emerging novel functions of the oxygen-sensing prolyl
hydroxylase domain enzymes. Trends Biochem Sci. 38:3–11. 2013.
|
|
11
|
Maxwell PH and Eckardt KU: HIF prolyl
hydroxylase inhibitors for the treatment of renal anaemia and
beyond. Nat Rev Nephrol. 12:157–168. 2016.
|
|
12
|
Singh L, Aldosary S, Saeedan AS, Ansari MN
and Kaithwas G: Prolyl hydroxylase 2: A promising target to inhibit
hypoxia-induced cellular metabolism in cancer cells. Drug Discov
Today. 23:1873–1882. 2018.
|
|
13
|
Kato H, Inoue T, Asanoma K, Nishimura C,
Matsuda T and Wake N: Induction of human endometrial cancer cell
senescence through modulation of HIF-1alpha activity by EGLN1. Int
J Cancer. 118:1144–1153. 2006.
|
|
14
|
Kaelin WG Jr: The VHL tumor suppressor
gene: Insights into oxygen sensing and cancer. Trans Am Clin
Climatol Assoc. 128:298–307. 2017.
|
|
15
|
Chen F, Chen J, Yang L, Liu J, Zhang X,
Zhang Y, Tu Q, Yin D, Lin D, Wong PP, et al: Extracellular
vesicle-packaged HIF-1α-stabilizing lncRNA from tumour-associated
macrophages regulates aerobic glycolysis of breast cancer cells.
Nat Cell Biol. 21:498–510. 2019.
|
|
16
|
Sadiku P, Willson JA, Dickinson RS, Murphy
F, Harris AJ, Lewis A, Sammut D, Mirchandani AS, Ryan E, Watts ER,
et al: Prolyl hydroxylase 2 inactivation enhances glycogen storage
and promotes excessive neutrophilic responses. J Clin Invest.
127:3407–3420. 2017.
|
|
17
|
Guentsch A, Beneke A, Swain L, Farhat K,
Nagarajan S, Wielockx B, Raithatha K, Dudek J, Rehling P, Zieseniss
A, et al: PHD2 is a regulator for glycolytic reprogramming in
macrophages. Mol Cell Biol. 37:e00236–16. 2016.
|
|
18
|
Xiang L, Gilkes DM, Hu H, Takano N, Luo W,
Lu H, Bullen JW, Samanta D, Liang H and Semenza GL:
Hypoxia-inducible factor 1 mediates TAZ expression and nuclear
localization to induce the breast cancer stem cell phenotype.
Oncotarget. 5:12509–12527. 2014.
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
|
|
20
|
Farooq SM, Hou Y, Li H, O'Meara M, Wang Y,
Li C and Wang JM: Disruption of GPR35 exacerbates dextran sulfate
sodium-induced colitis in mice. Dig Dis Sci. 63:2910–2922.
2018.
|
|
21
|
Wang S, Wu Y, Hou Y, Guan X, Castelvetere
MP, Oblak JJ, Banerjee S, Filtz TM, Sarkar FH, Chen X, et al: CXCR2
macromolecular complex in pancreatic cancer: A potential
therapeutic target in tumor growth. Transl Oncol. 6:216–225.
2013.
|
|
22
|
Ruifrok AC and Johnston DA: Quantification
of histochemical staining by color deconvolution. Anal Quant Cytol
Histol. 23:291–299. 2001.
|
|
23
|
Xie G, Zheng L, Ou J, Huang H, He J, Li J,
Pan F and Liang H: Low expression of prolyl hydroxylase 2 is
associated with tumor grade and poor prognosis in patients with
colorectal cancer. Exp Biol Med (Maywood). 237:860–866. 2012.
|
|
24
|
Zhou L, Wang Y, Zhou M, Zhang Y, Wang P,
Li X, Yang J, Wang H and Ding Z: HOXA9 inhibits HIF-1α-mediated
glycolysis through interacting with CRIP2 to repress cutaneous
squamous cell carcinoma development. Nat Commun. 9:14802018.
|
|
25
|
Dupuy F, Tabariès S, Andrzejewski S, Dong
Z, Blagih J, Annis MG, Omeroglu A, Gao D, Leung S, Amir E, et al:
PDK1-dependent metabolic reprogramming dictates metastatic
potential in breast cancer. Cell Metab. 22:577–589. 2015.
|
|
26
|
Denko NC: Hypoxia, HIF1 and glucose
metabolism in the solid tumour. Nat Rev Cancer. 8:705–713.
2008.
|
|
27
|
Jaakkola P, Mole DR, Tian YM, Wilson MI,
Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji
M, Schofield CJ, et al: Targeting of HIF-alpha to the von
Hippel-Lindau ubiquitylation complex by O2-regulated prolyl
hydroxylation. Science. 292:468–472. 2001.
|
|
28
|
Vidimar V, Licona C, Cerón-Camacho R,
Guerin E, Coliat P, Venkatasamy A, Ali M, Guenot D, Le Lagadec R,
Jung AC, et al: A redox ruthenium compound directly targets PHD2
and inhibits the HIF1 pathway to reduce tumor angiogenesis
independently of p53. Cancer Lett. 440-441:145–155. 2019.
|
|
29
|
Meneses AM and Wielockx B: PHD2: From
hypoxia regulation to disease progression. Hypoxia (Auckl).
4:53–67. 2016.
|
|
30
|
Cummins EP, Berra E, Comerford KM,
Ginouves A, Fitzgerald KT, Seeballuck F, Godson C, Nielsen JE,
Moynagh P, Pouyssegur J and Taylor CT: Prolyl hydroxylase-1
negatively regulates IkappaB kinase-beta, giving insight into
hypoxia-induced NFkappaB activity. Proc Natl Acad Sci USA.
103:18154–18159. 2006.
|
|
31
|
Chan DA, Kawahara TLA, Sutphin PD, Chang
HY, Chi JT and Giaccia AJ: Tumor vasculature is regulated by
PHD2-mediated angiogenesis and bone marrow-derived cell
recruitment. Cancer Cell. 15:527–538. 2009.
|
|
32
|
Hoesel B and Schmid JA: The complexity of
NF-κB signaling in inflammation and cancer. Mol Cancer.
12:862013.
|
|
33
|
Birbach A, Gold P, Binder BR, Hofer E, de
Martin R and Schmid JA: Signaling molecules of the NF-kappa B
pathway shuttle constitutively between cytoplasm and nucleus. J
Biol Chem. 277:10842–10851. 2002.
|
|
34
|
Rückert F, Grützmann R and Pilarsky C:
Feedback within the inter-cellular communication and tumorigenesis
in carcinomas. PLoS One. 7:e367192012.
|
|
35
|
Stine ZE, Walton ZE, Altman BJ, Hsieh AL
and Dang CV: MYC, metabolism, and cancer. Cancer Discov.
5:1024–1039. 2015.
|
|
36
|
Obacz J, Pastorekova S, Vojtesek B and
Hrstka R: Cross-talk between HIF and p53 as mediators of molecular
responses to physiological and genotoxic stresses. Mol Cancer.
12:932013.
|
|
37
|
Johnson RF and Perkins ND: Nuclear
factor-κB, p53, and mitochondria: Regulation of cellular metabolism
and the Warburg effect. Trends Biochem Sci. 37:317–324. 2012.
|
|
38
|
Wilde L, Roche M, Domingo-Vidal M, Tanson
K, Philp N, Curry J and Martinez-Outschoorn U: Metabolic coupling
and the reverse Warburg effect in cancer: Implications for novel
biomarker and anticancer agent development. Semin Oncol.
44:198–203. 2017.
|
|
39
|
Wang X, Liu R, Qu X, Yu H, Chu H, Zhang Y,
Zhu W, Wu X, Gao H, Tao B, et al: α-Ketoglutarate-activated NF-κB
signaling promotes compensatory glucose uptake and brain tumor
development. Mol Cell. 76:148–162.e7. 2019.
|
|
40
|
Ishak Gabra MB, Yang Y, Lowman XH, Reid
MA, Tran TQ and Kong M: IKKβ activates p53 to promote cancer cell
adaptation to glutamine deprivation. Oncogenesis. 7:932018.
|
|
41
|
Ruf M, Pitzen T, Meyer C and Drumm J:
Atlantoaxial rotatory dislocation: Delayed diagnose will result in
more invasive treatment options. J Neurol Surg A Cent Eur
Neurosurg. 82:1–8. 2021.
|
|
42
|
Taniguchi K and Karin M: NF-κB,
inflammation, immunity and cancer: Coming of age. Nat Rev Immunol.
18:309–324. 2018.
|
|
43
|
Gaete D, Rodriguez D, Watts D, Sormendi S,
Chavakis T and Wielockx B: HIF-prolyl hydroxylase domain proteins
(PHDs) in cancer-potential targets for anti-tumor therapy? Cancers
(Basel). 13:9882021.
|
|
44
|
Chen T, Zhou Q, Tang H, Bozkanat M, Yuan
JX, Raj JU and Zhou G: miR-17/20 controls prolyl hydroxylase 2
(PHD2)/hypoxia-inducible factor 1 (HIF1) to regulate pulmonary
artery smooth muscle cell proliferation. J Am Heart Assoc.
5:e0045102016.
|
|
45
|
Lee G, Won HS, Lee YM, Choi JW, Oh TI,
Jang JH, Choi DK, Lim BO, Kim YJ, Park JW, et al: Oxidative
dimerization of PHD2 is responsible for its inactivation and
contributes to metabolic reprogramming via HIF-1alpha activation.
Sci Rep. 6:189282016.
|
|
46
|
Li J, Yuan W, Jiang S, Ye W, Yang H,
Shapiro IM and Risbud MV: Prolyl-4-hydroxylase domain protein 2
controls NF-κB/p65 transactivation and enhances the catabolic
effects of inflammatory cytokines on cells of the nucleus pulposus.
J Biol Chem. 290:7195–7207. 2015.
|
|
47
|
Wang L, Niu Z, Wang X, Li Z, Liu Y, Luo F
and Yan X: PHD2 exerts anti-cancer and anti-inflammatory effects in
colon cancer xenografts mice via attenuating NF-κB activity. Life
Sci. 242:1171672020.
|
|
48
|
Lee DC, Sohn HA, Park ZY, Oh S, Kang YK,
Lee KM, Kang M, Jang YJ, Yang SJ and Hong YK: A lactate-induced
response to hypoxia. Cell. 161:595–609. 2015.
|
|
49
|
Romero-Ruiz A, Bautista L, Navarro V,
Heras-Garvín A, March-Díaz R, Castellano A, Gómez-Díaz R, Castro
MJ, Berra E, López-Barneo J and Pascual A: Prolyl
hydroxylase-dependent modulation of eukaryotic elongation factor 2
activity and protein translation under acute hypoxia. J Biol Chem.
287:9651–9658. 2012.
|
|
50
|
Wang X, Xie J and Proud CG: Eukaryotic
elongation factor 2 kinase (eEF2K) in cancer. Cancers.
9:1622017.
|
|
51
|
Xue J, Li X, Jiao S, Wei Y, Wu G and Fang
J: Prolyl hydroxylase-3 is down-regulated in colorectal cancer
cells and inhibits IKKbeta independent of hydroxylase activity.
Gastroenterology. 138:606–615. 2010.
|
|
52
|
Lind DS, Hochwald SN, Malaty J, Rekkas S,
Hebig P, Mishra G, Moldawer LL, Copeland EM III and Mackay S:
Nuclear factor-kappa B is upregulated in colorectal cancer.
Surgery. 130:363–369. 2001.
|
|
53
|
Wang Z, Sheng C, Kan G, Yao C, Geng R and
Chen S: RNAi screening identifies that TEX10 promotes the
proliferation of colorectal cancer cells by increasing NF-κB
activation. Adv Sci (Weinh). 7:20005932020.
|
|
54
|
Goka ET, Chaturvedi P, Lopez DTM, Garza A
and Lippman ME: RAC1b overexpression confers resistance to
chemotherapy treatment in colorectal cancer. Mol Cancer Ther.
18:957–968. 2019.
|
|
55
|
Gyamfi J, Kim J and Choi J: Cancer as a
metabolic disorder. Int J Mol Sci. 23:11552022.
|
|
56
|
Ge T, Gu X, Jia R, Ge S, Chai P, Zhuang A
and Fan X: Crosstalk between metabolic reprogramming and
epigenetics in cancer: Updates on mechanisms and therapeutic
opportunities. Cancer Commun (Lond). 42:1049–1082. 2022.
|