Liver cancer is the sixth most frequently diagnosed
cancer worldwide and the third most common cause of cancer-related
death, with ~906,000 new cases and 830,000 deaths worldwide in 2020
(1). In addition, the incidence
and mortality rates of liver cancer are generally higher in men
than in women (1). Hepatocellular
carcinoma (HCC) accounts for the largest proportion of cases among
all liver cancer types, is heterogeneous and imposes a large global
socio-economic burden (2). HCC
can occur either due to the dedifferentiation of hepatocytes or due
to the development of intrahepatic stem cells (3-5),
is characterized by difficulties in early detection, is prone to
metastasis and is associated with poor prognosis (6). The aggressiveness of HCC is closely
related to its degree of differentiation, microvascular
infiltration, intrahepatic metastases and satellite lesions, and
poorly differentiated HCC is associated with earlier recurrence and
poorer prognosis compared with well-differentiated HCC (7).
HCC therapy options include surgical excision, liver
transplantation, radiofrequency ablation and microwave ablation for
early-stage HCC (8-10). However, 40% of patients already
have advanced HCC when they are first diagnosed (11), resulting in limited treatment
options, and even radical treatment can still result in recurrence
or transformation within a short period, commonly within 1-3 years
(12). Although treatments with
multi-tyrosine kinase inhibitors (TKIs) have been demonstrated to
prolong the overall survival (OS) of patients with HCC (13,14), drug resistance and side effects
limit the effect of TKIs (15,16).
The rise in immune checkpoint inhibitors (ICIs) and
breakthroughs in immunology studies have sparked an expanding
interest in cancer immunotherapy, especially in antibodies against
programmed cell death protein 1 (PD-1) and programmed cell death
ligand 1 (PD-L1) (17). Anti-PD-1
drugs, such as nivolumab and pembrolizumab, are efficient and
well-tolerated in patients with advanced HCC and are currently
recognized as second-line treatment options for patients with HCC
(18,19). A clinical study, IMBrave150,
revealed that atezolizumab (anti-PD-L1) in combination with
bevacizumab (anti-vascular endothelial growth factor) was more
beneficial than sorafenib (SOR) in patients with advanced HCC, and
prolonged the median OS time of patients with unresectable HCC
(uHCC) (20,21). Compared with SOR, the combination
immunotherapy of durvalumab (anti-PD-L1) and tremelimumab
(anti-cytotoxic T-lymphocyte associated protein 4) administered in
the HIMALAYA study, which likely exerted antitumor effects by
activating T cells, exhibited an improved therapeutic effect for
uHCC (17,22). In addition, immunotherapy drugs
have the advantage that they do not need to be metabolized by the
liver, which marks a significant advancement in the management of
advanced HCC (23,24). Nevertheless, ICI therapy for HCC
still has shortcomings such as a low response rate, high tumor
tolerance to ICI therapy and multiple side effects (25-28), leading to the use of ICI therapy
in combination with various other therapies.
It has been confirmed that, during the development
and progression of HCC, the balance between regulatory cell death
(RCD) and cell survival serves a crucial function, and resistance
to apoptosis and evasion of cell death is one of the hallmarks of
HCC (29). Overcoming or delaying
TKI resistance increases tumor cell death (30,31), and inducing inflammatory forms of
cell death may enhance the tumor response to ICI treatment
(32). Therefore, cell death has
emerged as a popular research topic in the treatment of HCC.
Notably, considering that resistance to apoptotic RCD is a general
characteristic of cancer, non-apoptotic RCD (NARCD) serves a more
crucial role during the development of HCC and its response to
therapy (29). TKIs and ICIs are
both tightly associated with the regulation of NARCD pathways
(33,34).
At present, ferroptosis, pyroptosis and necroptosis
are three highly studied types of NARCD in HCC development and
treatment, and these influence the fate of cells in the liver
(35-40). There are some differences and
similarities among these three types of NARCD, and the key features
of ferroptosis, pyroptosis and necroptosis, including the
morphological and biochemical features, key regulators, and related
drugs are shown in Table I.
Furthermore, the roles of these NARCD pathways in the tumor
microenvironment (TME) and tumor immune microenvironment (TIME) are
gradually being recognized. Treatments targeting these NARCD
pathways in combination with TKI treatments (41-43) or ICI therapies (44-46) exhibit synergistically enhanced
anticancer activity compared with single treatment. Treatments
targeting these NARCD pathways could exert anticancer effects even
in cancer types resistant to TKIs and ICIs (34,47,48). Only a few patients exhibit a
response to TKI or ICI treatment alone, while triggering
ferroptosis, necroptosis or pyroptosis can alter this response
status and improve the response rate of therapy (32,49). Furthermore, ferroptosis,
pyroptosis and necroptosis, as three potential novel mechanisms of
immunogenic cell death (ICD) (50,51), have been suggested to transform
immune 'cold' tumors into immune 'hot' tumors, increasing the
sensitivity to ICI therapy, activating CD8+ T cell
adaptive immunity and maintaining durable immune memory so that the
body gains long-term antitumor immunity (52). Thus, ferroptosis, pyroptosis and
necroptosis are considered three novel potential therapeutic
targets to improve the treatment outcomes of HCC (49).
The present review first investigates the role of
ferroptosis, pyroptosis and necroptosis in the TME and TIME of HCC
and summarizes the related novel targets and signaling pathways.
Subsequently, the current status of targeting ferroptosis,
pyroptosis and necroptosis in combination with multiple HCC
treatment modalities is described. In particular, the potential
applications of targeting ferroptosis, pyroptosis and necroptosis
in combination with ICIs to enhance immune efficacy are
discussed.
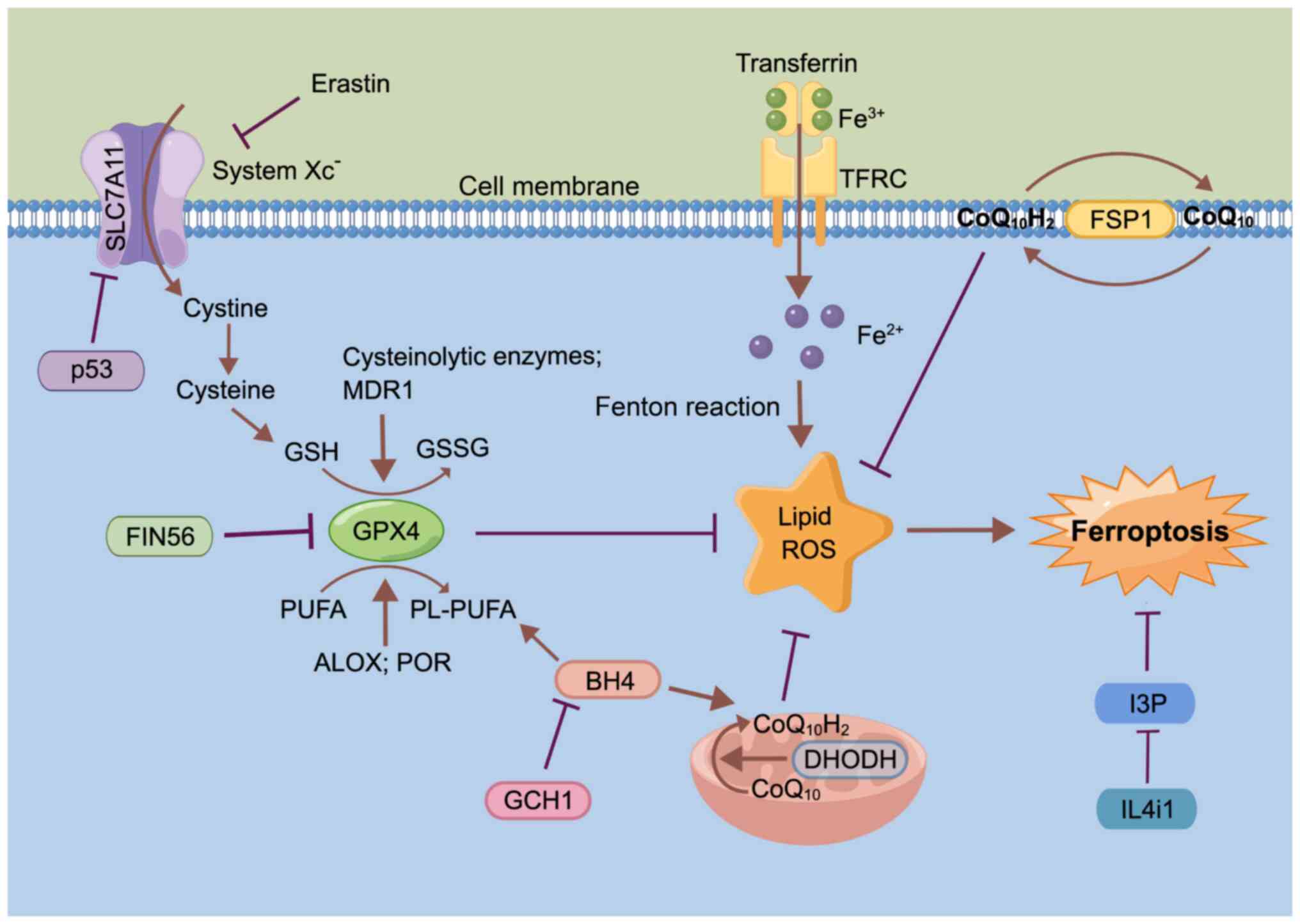
Originally conceptualized in 2012, ferroptosis is
considered a lipid peroxidation-driven, iron-dependent and
non-apoptotic form of cell death (53). Morphologically, the cellular
microstructure after ferroptosis is characterized by organelle
expansion, rupture of the plasma membrane and moderate chromatin
condensation, and the mitochondrial ultrastructure showing
abnormalities including contraction, fracture, enlargement of
cristae, increased membrane density and rupture of the outer
membrane (53-56). Mechanically, ferroptosis is driven
by iron-dependent phospholipid (PL) peroxidation, and regulated by
multiple cellular metabolic pathways, including redox homeostasis,
iron handling, mitochondrial activity, and metabolism of amino
acids, lipids and sugars (Fig. 1)
(53,57).
Unsaturated fatty acids in cell membranes, such as
polyunsaturated fatty acids (PUFAs), are mainly affected by lipid
peroxidation driven by free radicals (62). The key characteristic of PUFAs
driving ferroptosis is their ability to bind to PLs in membranes
upon PUFA activation (62).
Arachidonate lipoxygenase and cytochrome P450 oxidoreductase act as
regulators of lipid peroxidation, mediating PUFA peroxidation to
promote ferroptosis (54,63).
GSH peroxidase 4 (GPX4) acts as a key inhibitor of
ferroptosis and is regulated via several mechanisms. For example,
ferroptosis inducing 56 can induce degradation of GPX4 to induce
ferroptosis (Fig. 1) (64) or covalently binds to the
selenocysteine (Sec) site of GPX4 to induce GPX4 inactivation
(65). GSH depletion also
inactivates GPX4, and thus, drives ferroptosis, while both
GSH-related enzymes and the multidrug resistance protein 1, promote
GSH depletion (66,67). As research has progressed, a
number of novel ferroptosis-inducing mechanisms independent of GPX4
have been identified, such as ferroptosis suppressor protein 1 and
dihydroorotate dehydrogenase inhibiting ferroptosis by producing
ubiquinol (CoQ10H2) in the cell cytomembrane
and inner mitochondrial membranes, respectively (68,69).
Mitochondria serve a diversified role in the process
of ferroptosis, iron metabolism and the oxidative phosphorylation
pathway, and reactive oxygen species (ROS) are highly involved in
ferroptosis (70). It has been
demonstrated that the typical metabolic activities of the
mitochondria, including tricarboxylic acid cycle and electron
transport chain activities, are required for cellular lipid
peroxide production in ferroptosis induced by cysteine (Cys)
deprivation, but not in that induced by inhibiting GPX4 (57,71).
GTP cyclic hydrolase 1 improves ferroptosis
sensitivity by inhibiting generation of the antioxidant
tetrahydrobiopterin and increasing the abundance of
CoQ10H2 and PL-PUFA (73,74). IL-4-induced-1 stimulates
ferroptosis by inhibiting production of the metabolite
indole-3-pyruvate and limiting the activation of cytoprotective
gene expression programs (75).
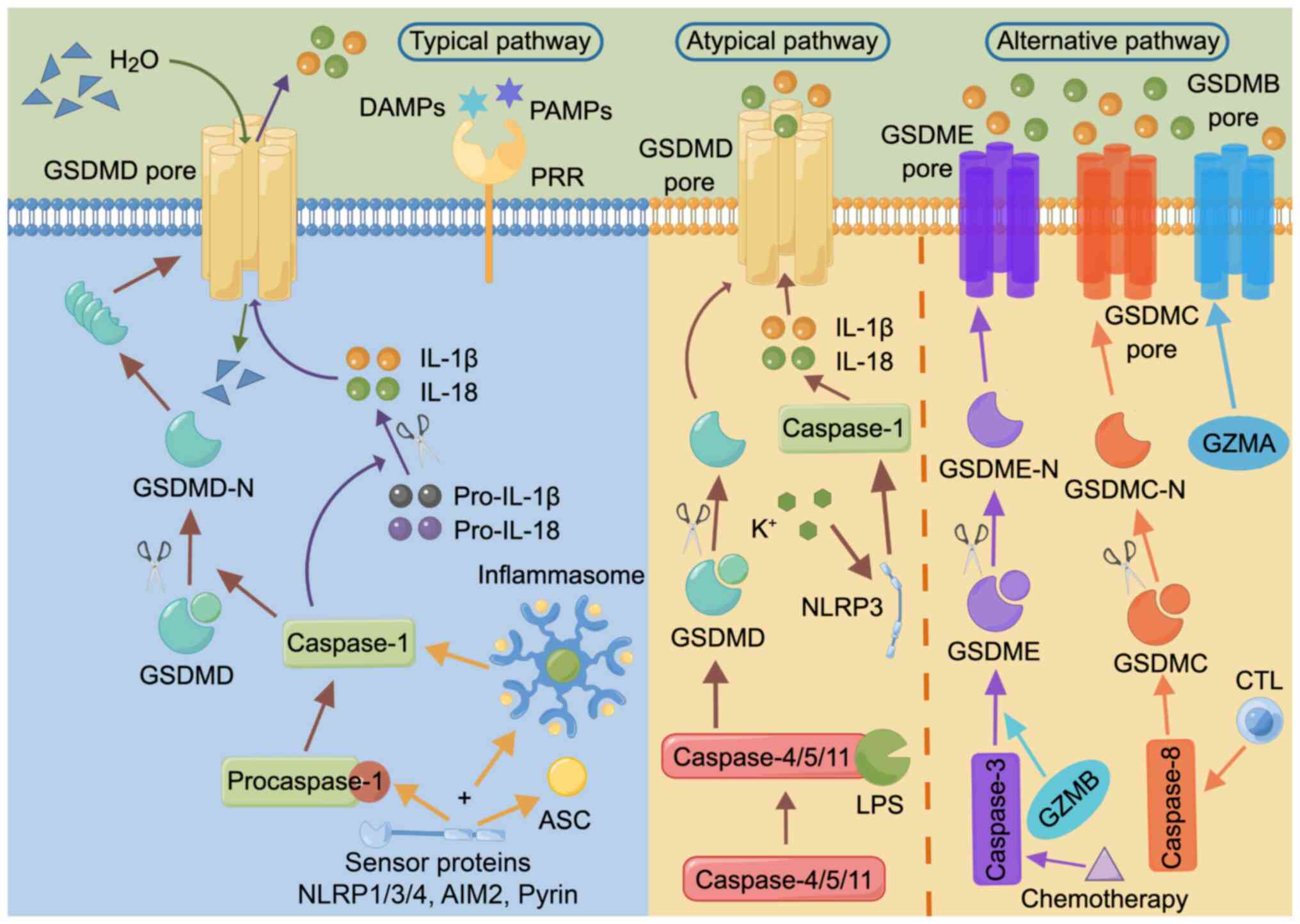
The typical pathway of pyroptosis is that, upon host
stimulation, GSDMD is cleaved by caspase-1, followed by
oligomerization and the formation of functional pores in the cell
membrane, leading to the release of inflammatory molecules such as
IL-1β and IL-18, disruption of osmotic pressure, water influx, and
thus, cell swelling, formation of membrane vesicles with
bubble-like protrusions (also known as scorch bodies) and plasma
membrane cleavage, and ultimately pyroptosis (80-83).
The atypical pathway of pyroptosis is a process not
reliant on inflammasome sensors, and involves the lytic apoptosis
of GSDMD cleaved by caspase-4/5/11 interacting with stimulators
such as lipopolysaccharide (LPS). The subsequently released
K+ activates NOD-like receptor family pyrin domain
containing 3 (NLRP3), which in turn activates caspase-1, and thus,
indirectly induces IL-1β and IL-18 production (84-86).
In addition, there are alternative pathways of
pyroptosis, such as via activated caspase-3/8, that can mediate the
cleavage of GSDME or GSDMC, releasing the N-terminal PFD and
eventually inducing pyroptosis (87-91). Alternatively, granzyme B from
lymphocytes induces pyroptosis by activating caspase-3 and
subsequent GSDME cleavage or by directly cleaving GSDME (92,93). Most GSDMs (except DFNB59) have an
N-terminal PFD and a C-terminal RD (81), and thus, also have the potential
to undergo cellular scorching, with GSDMB, GSDMC and GSDME being
the executors of cancer cell pyroptosis (CCP) (78). For example, GSDME can be cleaved
by small molecule kinase inhibitors or following the
chemotherapy-induced activation of caspase-3, resulting in
pyroptosis and the improvement of lung cancer and melanoma
treatments (87,94). GSDMC can be cleaved by caspase-8
to trigger CCP, including in lung and liver cancer (88). GSDMB can be cleaved directly by
granzyme A released from natural killer (NK) cells and cytotoxic T
lymphocytes (CTLs) independently of caspases, contributing to the
necrosis of murine cancer cells (95).
In the induction of pyroptosis, inflammasomes are
oligomeric complexes composed of sensor proteins, bridging proteins
and effector caspases (96,97), and similarly serve as molecular
platforms for the activation of inflammatory caspases (97). Pattern recognition receptors
(PRRs) can act as sensor proteins, recognizing the relevant
molecular patterns that initiate inflammasome activation (98). Among them, NLRP1/3/4, absent in
melanoma 2 and pyrin can recruit the adapter apoptosis-associated
speck-like protein containing a caspase recruitment domain and/or
pro-caspase-1 to assemble into inflammasomes and thereby activate
caspase-1, which in turn induces IL-1β and IL-18 processing
(99,100).
The initiation of necroptosis relies on specific
death receptors (DRs), including FAS, tumor necrosis factor
receptor 1 (TNFR1) and TNF-related apoptosis-inducing ligand
(TRAIL) receptors TRAIL-R1 and TRAIL-R2, or PRRs, such as toll-like
receptor (TLR)3, to recognize unfavorable signals from the intra-
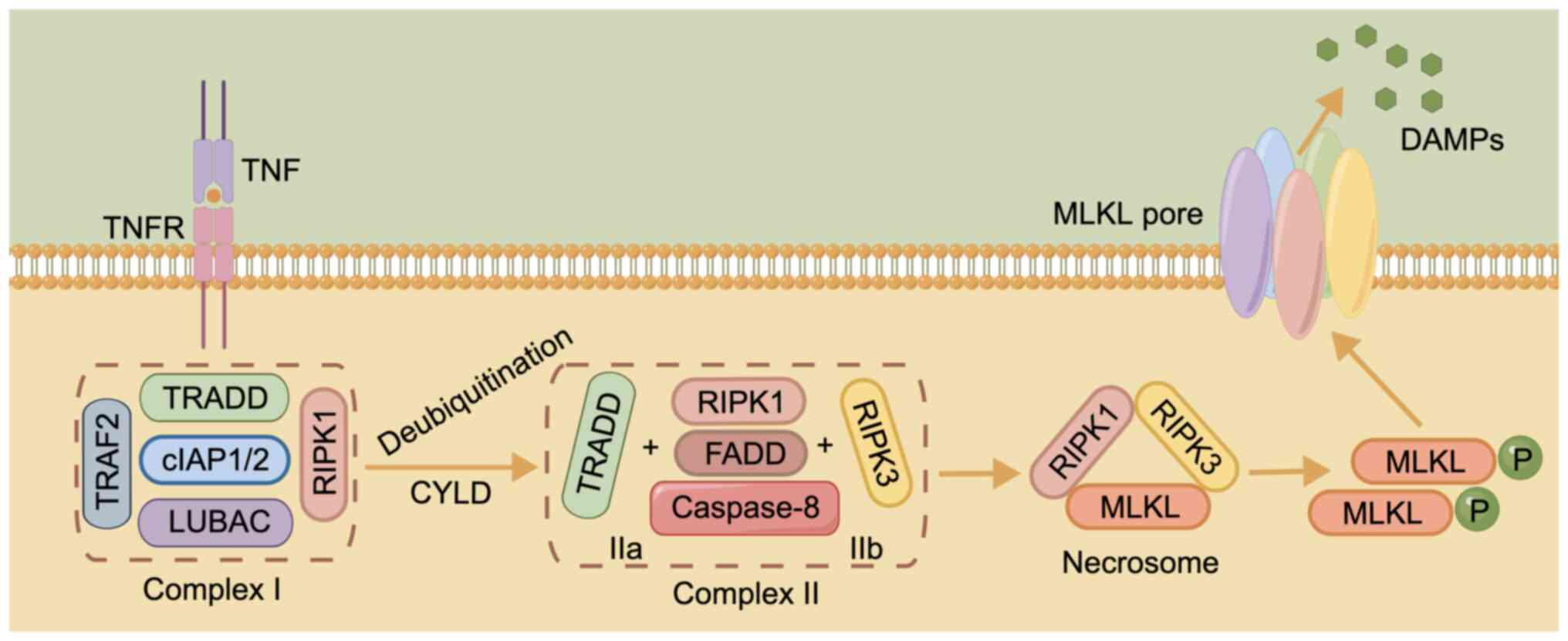
and extracellular microenvironment (101). TNF is a major stimulus in
necroptosis, delivering cell death signals via binding to DRs
(102). This process is
associated with complex I, consisting of TNFR1-associated death
domain protein (TRADD), linear ubiquitin chain assembly complex,
TNF receptor-associated factor 2 (TRAF2), cellular inhibitor of
apoptosis protein (cIAP)1/2 and receptor-interacting protein kinase
(RIPK)1 (103). When
cylindromatosis deubiquitinates RIPK1, A20, ubiquitin-specific
peptidase (USP)21 or USP20, complex I becomes unstable, and TRADD
and RIPK1 are isolated and assembled with Fas-associated death
domain (FADD) protein and caspase-8 to form complex IIa, or RIPK3
replaces TRADD to form complex IIb with the other components
(104,105). Inactivation of caspase-8 and
cIAP induces the conversion of complex IIb into a necrosome,
triggering DR-induced necroptosis (106,107).
The necrosome is formed by binding of RIPK3 and
RIPK1 via the RIP homotypic-interacting motif (RHIM) domain at
first, followed by recruitment of mixed lineage kinase domain-like
pseudokinase (MLKL) after the formation of the complex (108). Phosphorylated MLKL, the main
executor of necroptosis, later oligomerizes and migrates to the
plasma membrane, damaging it and releasing potential
damage-associated molecular patterns to trigger necroptosis
(Fig. 3) (109,110). Another TLR containing RHIM, TIR
domain-containing adapter-inducing interferon-β enables direct
RHIM-dependent signaling, initiating necrosis via receptor
interacting protein 3 and MLKL (111). A recent study found that
extracellular osmotic pressure was also a stimulus for the
induction of necroptosis and that the activation of RIPK3 by the
Na+/H+ exchanger solute carrier family 9
member A1 increased the cytosolic pH, and this is a pathway that
does not depend on the RHIM structural domain to activate the
downstream effector MLKL (112).
As research regarding ferroptosis progresses, more
ferroptosis-related drugs and targets are being identified for HCC
treatment. As a type of TKI, SOR also induces ferroptosis to
augment anti-HCC benefits (113,114). Furthermore, SOR attenuates the
binding of beclin-1 to MCL1 by modulating the Src homology region 2
domain-containing phosphatase-1/STAT3 axis, whilst enhancing the
binding to SLC7A11 and thereby inhibiting system Xc-
activity (115). In addition,
glutaminase 2 exerts anti-HCC effects by depleting glutamine, and
thus, promoting ferroptosis (116). Using CRISPR screening,
phosphatidylserine-transfer RNA kinase depletion has been
identified to interfere with Sec and Cys synthesis, leading to GSH
depletion and GPX4 inactivation, thus inducing ferroptosis and
enhancing the sensitivity to targeted chemotherapy in HCC treatment
(117). The long non-coding RNA
HEPFA promotes erastin-induced ferroptosis by mediating the
destabilization of SLC7A11 through ubiquitination, causing its
depletion and the consequent accumulation of ROS and iron (118). In addition, suppressor of
cytokine signaling 2 can enhance the degradation of K48-linked
polyubiquitinated SLC7A11, and thus, promote ferroptosis to enhance
the sensitivity of HCC radiotherapy (119). Ferroptosis inducer erastin and
photosensitizer are ultrasonically treated with CD47-transfected
donor cells to form engineered exosomes to cause ferroptosis in HCC
and avoid phagocytosis by the mononuclear phagocyte system for
improved anticancer effects (120). In a previous study, since
arsenic trioxide (ATO) could induce ferroptosis, the therapeutic
effect of ATO was enhanced by the construction of arsenic-loaded
mimetic iron oxide nanoparticles, specifically magnetic
nanoparticles containing ATO that were camouflaged with HCC cell
membranes (121). In another
study, a novel cascade of copper-based nanocatalysts, which can
result in ferroptosis alone, also enhanced the HCC treatment
effects of cyclooxygenase-2 inhibitor meloxicam and SOR (122). It has also been reported that
the pH sensitivity of liposomal vesicles is enhanced following
incubation with amphiphilic dendrimers, thereby improving the
delivery of the anticancer drug SOR and the ferroptosis inducer
hemin in the acidic TME, synergistically treating the induction of
ferroptosis and apoptosis (123).
In addition to the previous understanding that SOR
induces the death of hepatoma cells, investigations have also
revealed that SOR could trigger ferroptosis in hepatic stellate
cells (HSCs), as demonstrated in an analysis of liver tissue HSCs
from patients with advanced fibrotic HCC treated with SOR
monotherapy (124,125). Liver fibrosis is a frequent
pathological process that numerous chronic liver diseases undergo
before progressing to cirrhosis and HCC, and the conversion of
quiescent, vitamin A-storing cells into proliferating, fibrotic
myofibroblasts by activated HSCs is a critical step in the
development of hepatic fibrosis; therefore, targeted removal of
HSCs is of great therapeutic significance (126). ELAV-like RNA binding protein 1,
zinc finger protein 36 and N6-methyladenosine can act as regulators
of SOR-induced ferroptosis in HSCs (124,125,127). In addition, artesunate reduces
hepatic fibrosis by triggering ferroptosis in activated HSCs
(128). Furthermore, SOR and
artesunate induce ferroptosis through different pathways, and their
combined use greatly improves the therapeutic effect in HCC
(128). With the rising
development of ICIs, SOR has often been used to assess the
effectiveness of ICIs in treating HCC, such as in the phase III
IMbrave150 and HIMALAYA trials, which demonstrated that the
efficacy of ICI combination therapy was comparable to or better
than that of SOR in patients with advanced HCC (21). Furthermore, clinical trials of
ICIs in combination with SOR (such as NCT03439891 and NCT02988440)
are underway and may provide novel perspectives on the
administration of ICIs in combination with TKIs.
Given the notable anticancer effect of SOR, there
has been an increasing number of studies on the association between
ferroptosis and SOR resistance in HCC. For instance, it has been
found that Yes1 associated transcriptional regulator
(YAP)/transcriptional coactivator with PDZ-binding motif (TAZ) and
activating transcription factor 4 (ATF4) drive resistance to SOR in
HCC by increasing SLC7A11 expression and preventing ferroptosis,
and knockdown of YAP/TAZ expression helped to overcome the SOR
resistance in HCC (30).
Additionally, in SOR-resistant HCC cells, ETS proto-oncogene 1
increases the transcription of microRNA (miR)-23a-3p, which could
directly target the 3'-untranslated region of acyl-CoA synthetase
long chain family member 4 (ACSL4), the key positive regulator of
ferroptosis (47). The
co-delivery of ACSL4 small interfering RNA and miR-23a-3p inhibitor
abolishes the SOR response (47).
Furthermore, metallothionein-1G has been found to facilitate SOR
resistance through inhibition of ferroptosis (129). The genetic and pharmacological
inhibition of MT-1G enhances the anticancer activity of SOR by
inducing ferroptosis of HCC cells (129). A novel photoactive SOR-ruthenium
(II) complex, is irradiated to reduce SOR resistance in HCC by
inducing ferroptosis and disrupting purine metabolism (31). It has also been found that
treatment of HCC with SOR induces macropinocytosis, which replaces
ferroptosis-depleted Cys, and thus, promotes resistance to SOR,
while amiloride can target micropinocytosis (130).
The proliferation of cancer cells depends on lactic
acid, which is produced following glucose consumption via aerobic
glycolysis to provide energy and is closely associated with
ferroptosis (131). For
instance, the glycolytic enzyme α-enolase 1 protects cancer cells
from ferroptosis by reducing mitochondrial iron accumulation
through inhibition of the iron regulatory protein 1/mitoferrin-1
pathway (132). Monocarboxylate
transporter 1 (MCT1)-mediated lactic acid uptake promotes ATP
production and AMP-activated protein kinase inactivation in HCC,
which in turn upregulates downstream stearoyl-coenzyme A
desaturase-1 to enhance the production of anti-ferroptosis
monounsaturated fatty acids (133). Hypoxia-inducible factor-1α also
drives resistance to ferroptosis in solid tumors by promoting
lactate production (134).
Nuclear factor erythroid 2-related factor 2 (NRF2)
has long been known to influence tumor progression as a pivotal
regulator of the antioxidant response (135,136). Nuclear accumulation of NRF2 has
been found to activate ferroptosis-associated proteins, while
Kelch-like ECH-associated protein 1 (Keap1), an adaptor of the
Cul3-ubiquitin E3 ligase complex, is responsible for the
degradation of NRF2; in turn, phosphorylated p62 binds Keap1 with
high affinity, thus targeting the p62/Keap1/NRF2 pathway increases
the anticancer activity of elastin and SOR, a process that is
facilitated by disulfiram/Cu (137-139). The GSH S-transferase
Z1/NRF2/GPX4 axis and the leukemia inhibitory factor
receptor/NF-κB/lipocalin-2 axis can also be targeted to promote
ferroptosis, thereby enhancing the sensitivity to SOR in HCC
treatment (140,141).
Necroptosis serves an important role in HCC
development. According to a previous study, the hepatic
microenvironment epigenetically shapes lineage commitment of liver
tumorigenesis, and abnormal activation of hepatocyte oncogenes can
lead to cholangiocarcinoma if adjacent hepatocytes undergo
necroptosis, but hepatocytes regulated by the same oncogenic
factors can develop into HCC cells if they are surrounded by
apoptotic hepatocytes (151).
Deletion of RIPK1, a central element of necroptosis, in hepatocytes
induces downregulation of TRAF2, leading to impaired caspase-8
activation and NF-κB activation; therefore, the
RIPK1/TRAF2/caspase-8 pathway has a notable influence on the
development of HCC (152). As
confirmed by a previous study, the NF-κB signaling pathway is an
important player in necroptosis (153). Apigetrin and deferasirox exert
anti-HCC effects by inhibiting the NF-κB signaling pathway to
induce necroptosis (43,154). HSP90α has been found to bind
with the necrosome complex and promotes chaperone-mediated
autophagy degradation, which leads to necroptosis blocking and
results in SOR resistance (48).
The HSP90 inhibitor 17-allylamino demethoxygeldanamycin could
inhibit HSP90α activity and reverse SOR resistance in HCC by
activating necroptosis (48).
FADD, RIPK1, RIPK3 and MLKL are key signaling molecules in
necroptosis, and miR-675 could target FADD to induce necroptosis
and inhibit HCC progression via the RIPK3/MLKL axis (155), and rapamycin could induce HCC
cell necroptosis via the RIPK1/RIPK3/MLKL signaling pathway
(156). A recent study has found
that, as the liver progressively ages, necroptosis increases, which
in turn continuously exacerbates chronic liver inflammation, thus
exacerbating the HCC transition (157). This necroptosis-mediated liver
inflammation process may be prevented by β-carotene and
heterogeneous nuclear ribonucleoprotein A1 (158,159). In addition, excess sorbitol
dehydrogenase (SORD) in HCC cells could inhibit tumor growth and
stemness by enhancing necroptosis signaling, and treatment with
human recombinant SORD controlled HCC cell growth and regulated
macrophage polarization in the tumor microenvironment (160). Nuclear protein 1 (NUPR1) and
Linc00176 are also associated with HCC cell necroptosis, and
inhibition of NUPR1 with small compound ZZW-115 and deletion of
Linc00176 could induce necroptosis and exert anti-HCC effects
(161,162). In addition, connexin 32 has been
found to bind to Src and then mediate the inactivation of caspase 8
to trigger necroptosis in HCC cells, which could be used as an
anticancer target to enhance the function of necroptosis inducers
(163).
During the treatment of tumors, ferroptosis,
pyroptosis and necroptosis are closely associated with the immune
response (50,95,164). It has been found that
immunotherapy-activated CD8+ T cells could induce
ferroptosis of tumor cells by downregulating the expression of
SLC3A2 and SLC7A11, and ferroptosis inducers in combination with
checkpoint blockade synergistically enhanced antitumor efficacy
(164). Similarly,
CD8+ T cells have also been found to trigger tumor
clearance through activation of the GSDM granzyme axis to induce
pyroptosis (95,165), as have NK cells (93,95). Furthermore, necroptotic tumor
cells can release damage-associated molecular patterns such as heat
shock proteins, being more immunogenic than naïve tumor cells, to
activate the immune system with the participation of
CD8+ T cells, NK cells and dendritic cells (DCs)
(166-168), which may be related to the
activation of NF-κB (50).
Necrotic tumor cells, as well as fibroblast vaccination, contribute
to the induction of antitumor immunogenicity by necrotic apoptotic
cells (50,167,169), which enhances the antitumor
efficacy of ICI treatments (167).
The relationship between ferroptosis and the tumor
immune response is complicated. It has been demonstrated that
GPX4-associated ferroptotic hepatocyte death could cause a
HCC-suppressive immune response, characterized by a C-X-C motif
chemokine ligand 10-dependent infiltration of cytotoxic
CD8+ T cells that is counterbalanced by PD-L1
upregulation on tumor cells, as well as by a marked high mobility
group box 1-mediated myeloid derived suppressor cell (MDSC)
infiltration (170). A triple
combination of the ferroptosis-inducing natural compound withaferin
A, the C-X-C motif chemokine receptor 2 inhibitor SB225002 and
α-PD-1 contributed to improved treatment of HCC compared with
single treatment or dual combinations (170). In addition, inhibition of
phosphoglycerate mutase 1 promotes ferroptosis and downregulates
PD-L1 expression in HCC cells, further enhancing the infiltration
of CD8+ T cells, and thereby exerting antitumor effects
(36). Furthermore, TLR2 agonist
Pam3CSK4 could promote polarization of MDSCs into DCs and
macrophages and recovery of T cell function by downregulating the
expression of RUNX family transcription factor 1 (RUNX1), and TLR2
and RUNX1 may exert a regulatory effect on MDSCs by increasing ROS,
which is related to the ferroptosis pathway (171).
Tumor-associated macrophages (TAMs), a key
tumor-infiltrating immune cell type in the TME, encourage tumor
invasion and metastasis by switching from the pro-inflammatory and
antitumor type M1 TAMs to the anti-inflammatory and pro-tumor type
M2 TAMs (172). Previous studies
have demonstrated that TAMs generally exhibit a pro-tumor phenotype
and inhibit CTLs by expressing PD-L1, thereby inducing immune
escape and tolerance (173-175). Inhibition of apolipoprotein C1
can reverse the M2 phenotype to the M1 phenotype in TAMs via the
ferroptosis pathway, enhancing the effect of anti-PD-1
immunotherapy and exerting anti-HCC effects (176). Injection of dextran chitosan
hydrogel can induce the repolarization of macrophages to the
M1-like phenotype, and promote the maturation and activation of
DCs, and combined treatment with PD-1 immunotherapy can effectively
treat the peritoneal dissemination of advanced HCC and malignant
ascites (37). A recent study
also revealed that inhibition of the heavy chain subunit from
system Xc- encoded by the SLC7A11 gene (xCT) could
induce ferroptosis of TAMs via the GPX4/ribonucleotide reductase
regulatory subunit M2 signaling pathway and inhibit M2-type
polarization via the suppressor of cytokine signaling
3/STAT6/peroxisome proliferator-activated receptor (PPAR)-γ pathway
(35). Furthermore, this xCT
inhibition-mediated macrophage ferroptosis increased PD-L1
expression in macrophages and improved the antitumor efficacy of
anti-PD-L1 therapy (35).
TKIs can remodel the TME of HCC and alter the
immunosuppressive microenvironment, which is closely connected to
pyroptosis, NK cells, T cells and regulatory T cells (177). Among them, SOR is a multi-target
kinase inhibitor that directly modulates immunity, causing HCC cell
death by inducing macrophage pyroptosis and activating cytotoxic NK
cells (41). A study has found
that the pyroptosis-score (PYS) could be used to assess the
prognosis of patients with HCC due to hepatitis B virus (HBV-HCC),
as a higher PYS in patients with HBV-HCC was associated with a
worse prognosis, and these patients were more likely to receive
anti-PD-L1 therapy (178). In
addition, a risk score model related to pyroptosis can be
constructed to predict the prognosis and immunological
characteristics of HCC (38).
The interaction of necroptosis and the immune
response is considered to be important in HCC development and
treatment because necroptosis is a primary cause of liver
inflammation and upregulates not only proinflammatory M1 TAMs, but
also proinflammatory cytokines and chemokines, ultimately leading
to chronic liver disease (179).
However, a study has reported that RIPK3 is not expressed in
hepatocytes, and MLKL does not depend on RIPK3 alone to regulate
endoplasmic reticulum (ER)-mitochondrial Mg2+ dynamics
(108). Defective MLKL inhibits
mitochondrial Mg2+ absorption and ER Mg2+
release, and the resulting metabolic stress, in turn, induces
parthanatos [a form of ICD associated with antitumor immunity
(180)], activates anticancer
immune responses and increases the therapeutic impact of ICIs,
inhibiting HCC progression and immune escape (181). Similarly, RIPK3 defects in TAMs
activate PPAR and promote fatty acid metabolism, which in turn
induces the accumulation and polarization of M2 TAMs and ultimately
promotes HCC progression (182).
In addition, higher expression levels of RIPK1, RIPK3 and MLKL, are
associated with good prognosis in HCC and are especially positively
associated with CD3+ and CD8+ T cells in HCC
(183). However, monoclonal
antibodies targeting CD147 structural domain 1 can induce atypical
necroptosis independent of RIPK (184). Furthermore, both senescence and
SORD induce necroptosis in HCC cells and promote M1 TAM
polarization (157,160).
In conclusion, induction of ferroptosis, pyroptosis
and necroptosis is a novel avenue for killing HCC cells, providing
novel targets and signaling pathways for drug discovery, increasing
treatment effectiveness of immunotherapy, and possibly addressing
drug resistance. The combination of ferroptosis, pyroptosis and
necroptosis targets with ICIs may improve the therapeutic effect
and prognosis in patients with advanced HCC. However, some problems
still remain. First, although several compounds or drugs that
induce ferroptosis, pyroptosis and necroptosis have been
identified, clinical studies to assess their feasibility and safety
are lacking. Therefore, more research should be performed in the
future to improve the safety and personalized treatment options of
this treatment avenue. Second, clinical studies of novel drugs or
therapies for HCC are usually long and costly, which makes it
difficult to develop novel medicines and treatments based on
ferroptosis, pyroptosis and necroptosis. Third, the significance
and mechanisms of ferroptosis, pyroptosis and necroptosis in HCC
still need to be elaborated in further detail.
Not applicable.
RJL and XDY wrote the manuscript. RJL drew the
figures. SSY and ZWG participated in the conception of the
manuscript. XBZ and YSZ revised the manuscript. Data authentication
is not applicable. All authors read and approved the final version
of the manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present study was supported by the Fundamental Research
Funds for the Central Universities, Beijing University of Chinese
Medicine (grant no. 2021-JYB-XJSJJ-055), Beijing Traditional
Chinese Medicine 'Torch Inheritance 3+3 Project'-the Wang Pei
Famous Doctor Inheritance Workstation-Dongzhimen Hospital Branch
(grant no. 405120602) and 'Talent development program of Dongzhimen
Hospital, Beijing University of Chinese Medicine'.
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
|
|
2
|
Singh D, Vignat J, Lorenzoni V, Eslahi M,
Ginsburg O, Lauby-Secretan B, Arbyn M, Basu P, Bray F and
Vaccarella S: Global estimates of incidence and mortality of
cervical cancer in 2020: A baseline analysis of the WHO global
cervical cancer elimination initiative. Lancet Glob Health.
11:e197–e206. 2023.
|
|
3
|
van Malenstein H, van Pelt J and Verslype
C: Molecular classification of hepatocellular carcinoma anno 2011.
Eur J Cancer. 47:1789–1797. 2011.
|
|
4
|
Park YN: Update on precursor and early
lesions of hepatocellular carcinomas. Arch Pathol Lab Med.
135:704–715. 2011.
|
|
5
|
Trevisani F, Cantarini MC, Wands JR and
Bernardi M: Recent advances in the natural history of
hepatocellular carcinoma. Carcinogenesis. 29:1299–1305. 2008.
|
|
6
|
Choi JY, Lee JM and Sirlin CB: CT and MR
imaging diagnosis and staging of hepatocellular carcinoma: Part I.
Development, growth, and spread: Key pathologic and imaging
aspects. Radiology. 272:635–654. 2014.
|
|
7
|
Komuta M: Histological heterogeneity of
primary liver cancers: Clinical relevance, diagnostic pitfalls and
the pathologist's role. Cancers (Basel). 13:28712021.
|
|
8
|
Berardi G, Igarashi K, Li CJ, Ozaki T,
Mishima K, Nakajima K, Honda M and Wakabayashi G: Parenchymal
sparing anatomical liver resections with full laparoscopic
approach: Description of technique and short-term results. Ann
Surg. 273:785–791. 2021.
|
|
9
|
Clavien PA, Lesurtel M, Bossuyt PM, Gores
GJ, Langer B and Perrier A; OLT for HCC Consensus Group:
Recommendations for liver transplantation for hepatocellular
carcinoma: An international consensus conference report. Lancet
Oncol. 13:e11–e22. 2012.
|
|
10
|
Pan T, Xie QK, Lv N, Li XS, Mu LW, Wu PH
and Zhao M: Percutaneous CT-guided radiofrequency ablation for
lymph node oligometastases from hepatocellular carcinoma: A
propensity score-matching analysis. Radiology. 282:259–270.
2017.
|
|
11
|
Cabibbo G, Enea M, Attanasio M, Bruix J,
Craxì A and Cammà C: A meta-analysis of survival rates of untreated
patients in randomized clinical trials of hepatocellular carcinoma.
Hepatology. 51:1274–1283. 2010.
|
|
12
|
Xing R, Gao J, Cui Q and Wang Q:
Strategies to improve the antitumor effect of immunotherapy for
hepatocellular carcinoma. Front Immunol. 12:7832362021.
|
|
13
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al: Sorafenib in advanced hepatocellular carcinoma. New Engl J
Med. 359:378–390. 2008.
|
|
14
|
Kudo M, Finn RS, Qin S, Han KH, Ikeda K,
Piscaglia F, Baron A, Park JW, Han G, Jassem J, et al: Lenvatinib
versus sorafenib in first-line treatment of patients with
unresectable hepatocellular carcinoma: A randomised phase 3
non-inferiority trial. Lancet. 391:1163–1173. 2018.
|
|
15
|
Reig M, Torres F, Rodriguez-Lope C, Forner
A, LLarch N, Rimola J, Darnell A, Ríos J, Ayuso C and Bruix J:
Early dermatologic adverse events predict better outcome in HCC
patients treated with sorafenib. J Hepatol. 61:318–324. 2014.
|
|
16
|
Chen S, Cao Q, Wen W and Wang H: Targeted
therapy for hepatocellular carcinoma: Challenges and opportunities.
Cancer Lett. 460:1–9. 2019.
|
|
17
|
Greten TF, Lai CW, Li G and
Staveley-O'Carroll KF: Targeted and immune-based therapies for
hepatocellular carcinoma. Gastroenterology. 156:510–524. 2019.
|
|
18
|
El-Khoueiry AB, Sangro B, Yau T, Crocenzi
TS, Kudo M, Hsu C, Kim TY, Choo SP, Trojan J, Welling TH Rd, et al:
Nivolumab in patients with advanced hepatocellular carcinoma
(CheckMate 040): An open-label, non-comparative, phase 1/2 dose
escalation and expansion trial. Lancet. 389:2492–2502. 2017.
|
|
19
|
Zhu AX, Finn RS, Edeline J, Cattan S,
Ogasawara S, Palmer D, Verslype C, Zagonel V, Fartoux L, Vogel A,
et al: Pembrolizumab in patients with advanced hepatocellular
carcinoma previously treated with sorafenib (KEYNOTE-224): A
non-randomised, open-label phase 2 trial. Lancet Oncol. 19:940–952.
2018.
|
|
20
|
Galle PR, Finn RS, Qin S, Ikeda M, Zhu AX,
Kim TY, Kudo M, Breder V, Merle P, Kaseb A, et al: Patient-reported
outcomes with atezolizumab plus bevacizumab versus sorafenib in
patients with unresectable hepatocellular carcinoma (IMbrave150):
An open-label, randomised, phase 3 trial. Lancet Oncol.
22:991–1001. 2021.
|
|
21
|
Cheng AL, Qin S, Ikeda M, Galle PR,
Ducreux M, Kim TY, Lim HY, Kudo M, Breder V, Merle P, et al:
Updated efficacy and safety data from IMbrave150: Atezolizumab plus
bevacizumab vs. sorafenib for unresectable hepatocellular
carcinoma. J Hepatol. 76:862–873. 2022.
|
|
22
|
Kelley RK, Sangro B, Harris W, Ikeda M,
Okusaka T, Kang YK, Qin S, Tai DW, Lim HY, Yau T, et al: Safety,
efficacy, and pharmacodynamics of tremelimumab plus durvalumab for
patients with unresectable hepatocellular carcinoma: Randomized
expansion of a phase I/II study. J Clin Oncol. 39:2991–3001.
2021.
|
|
23
|
Giannini EG, Aglitti A, Borzio M, Gambato
M, Guarino M, Iavarone M, Lai Q, Levi Sandri GB, Melandro F,
Morisco F, et al: Overview of immune checkpoint inhibitors therapy
for hepatocellular carcinoma, and the ITA.LI.CA cohort derived
estimate of amenability rate to immune checkpoint inhibitors in
clinical practice. Cancers (Basel). 11:16892019.
|
|
24
|
Greten TF, Abou-Alfa GK, Cheng AL, Duffy
AG, El-Khoueiry AB, Finn RS, Galle PR, Goyal L, He AR, Kaseb AO, et
al: Society for immunotherapy of cancer (SITC) clinical practice
guideline on immunotherapy for the treatment of hepatocellular
carcinoma. J Immunother Cancer. 9:e0027942021.
|
|
25
|
Finn RS, Ikeda M, Zhu AX, Sung MW, Baron
AD, Kudo M, Okusaka T, Kobayashi M, Kumada H, Kaneko S, et al:
Phase Ib study of lenvatinib plus pembrolizumab in patients with
unresectable hepatocellular carcinoma. J Clin Oncol. 38:2960–2970.
2020.
|
|
26
|
Wang Z, Wang Y, Gao P and Ding J: Immune
checkpoint inhibitor resistance in hepatocellular carcinoma. Cancer
Lett. 555:2160382023.
|
|
27
|
Dolladille C, Ederhy S, Sassier M, Cautela
J, Thuny F, Cohen AA, Fedrizzi S, Chrétien B, Da-Silva A, Plane AF,
et al: Immune checkpoint inhibitor rechallenge after immune-related
adverse events in patients with cancer. JAMA Oncol. 6:865–871.
2020.
|
|
28
|
Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X,
Li Z, Traugh N, Bu X, Li B, et al: Signatures of T cell dysfunction
and exclusion predict cancer immunotherapy response. Nat Med.
24:1550–1558. 2018.
|
|
29
|
Nyiramana MM, Cho SB, Kim EJ, Kim MJ, Ryu
JH, Nam HJ, Kim NG, Park SH, Choi YJ, Kang SS, et al: Sea hare
hydrolysate-induced reduction of human non-small cell lung cancer
cell growth through regulation of macrophage polarization and
non-apoptotic regulated cell death pathways. Cancers (Basel).
12:7262020.
|
|
30
|
Gao R, Kalathur RKR, Coto-Llerena M, Ercan
C, Buechel D, Shuang S, Piscuoglio S, Dill MT, Camargo FD,
Christofori G and Tang F: YAP/TAZ and ATF4 drive resistance to
Sorafenib in hepatocellular carcinoma by preventing ferroptosis.
EMBO Mol Med. 13:e143512021.
|
|
31
|
Lai Y, Lu N, Luo S, Wang H and Zhang P: A
photoactivated sorafenib-ruthenium(II) prodrug for resistant
hepatocellular carcinoma therapy through ferroptosis and purine
metabolism disruption. J Med Chem. 65:13041–13051. 2022.
|
|
32
|
Rosenbaum SR, Wilski NA and Aplin AE:
Fueling the fire: Inflammatory forms of cell death and implications
for cancer immunotherapy. Cancer Discov. 11:266–281. 2021.
|
|
33
|
Hadian K and Stockwell BR: The therapeutic
potential of targeting regulated non-apoptotic cell death. Nat Rev
Drug Discov. 22:723–742. 2023.
|
|
34
|
Gao W, Wang X, Zhou Y, Wang X and Yu Y:
Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor
immunotherapy. Signal Transduct Target Ther. 7:1962022.
|
|
35
|
Tang B, Zhu J, Wang Y, Chen W, Fang S, Mao
W, Xu Z, Yang Y, Weng Q, Zhao Z, et al: Targeted xCT-mediated
ferroptosis and protumoral polarization of macrophages is effective
against HCC and enhances the efficacy of the anti-PD-1/L1 response.
Adv Sci (Weinh). 10:e22039732023.
|
|
36
|
Zheng Y, Wang Y, Lu Z, Wan J, Jiang L,
Song D, Wei C, Gao C, Shi G, Zhou J, et al: PGAM1 inhibition
promotes HCC ferroptosis and synergizes with anti-PD-1
immunotherapy. Adv Sci (Weinh). 10:e23019282023.
|
|
37
|
Meng J, Yang X, Huang J, Tuo Z, Hu Y, Liao
Z, Tian Y, Deng S, Deng Y, Zhou Z, et al: Ferroptosis-enhanced
immunotherapy with an injectable dextran-chitosan hydrogel for the
treatment of malignant ascites in hepatocellular carcinoma. Adv Sci
(Weinh). 10:e23005172023.
|
|
38
|
Wang H, Zhang B, Shang Y, Chen F, Fan Y
and Tan K: A novel risk score model based on pyroptosis-related
genes for predicting survival and immunogenic landscape in
hepatocellular carcinoma. Aging (Albany NY). 15:1412–1444.
2023.
|
|
39
|
Peng YL, Wang LX, Li MY, Liu LP and Li RS:
Construction and validation of a prognostic signature based on
necroptosis-related genes in hepatocellular carcinoma. PLoS One.
18:e2797442023.
|
|
40
|
Wang Y, Wang Y, Pan J, Gan L and Xue J:
Ferroptosis, necroptosis, and pyroptosis in cancer: Crucial cell
death types in radiotherapy and post-radiotherapy immune
activation. Radiother Oncol. 184:1096892023.
|
|
41
|
Hage C, Hoves S, Strauss L, Bissinger S,
Prinz Y, Pöschinger T, Kiessling F and Ries CH: Sorafenib induces
pyroptosis in macrophages and triggers natural killer cell-mediated
cytotoxicity against hepatocellular carcinoma. Hepatology.
70:1280–1297. 2019.
|
|
42
|
Li Y, Yang W, Zheng Y, Dai W, Ji J, Wu L,
Cheng Z, Zhang J, Li J, Xu X, et al: Targeting fatty acid synthase
modulates sensitivity of hepatocellular carcinoma to sorafenib via
ferroptosis. J Exp Clin Canc Res. 42:62023.
|
|
43
|
Bhosale PB, Abusaliya A, Kim HH, Ha SE,
Park MY, Jeong SH, Vetrivel P, Heo JD, Kim JA, Won CK, et al:
Apigetrin promotes TNFα-induced apoptosis, necroptosis, G2/M phase
cell cycle arrest, and ROS generation through inhibition of NF-κB
pathway in Hep3B liver cancer cells. Cells. 11:27342022.
|
|
44
|
Wang Q, Wang Y, Ding J, Wang C, Zhou X,
Gao W, Huang H, Shao F and Liu Z: A bioorthogonal system reveals
antitumour immune function of pyroptosis. Nature. 579:421–426.
2020.
|
|
45
|
Xu C, Sun S, Johnson T, Qi R, Zhang S,
Zhang J and Yang K: The glutathione peroxidase Gpx4 prevents lipid
peroxidation and ferroptosis to sustain Treg cell activation and
suppression of antitumor immunity. Cell Rep. 35:1092352021.
|
|
46
|
Wang W, Marinis JM, Beal AM, Savadkar S,
Wu Y, Khan M, Taunk PS, Wu N, Su W, Wu J, et al: RIP1 kinase drives
macrophage-mediated adaptive immune tolerance in pancreatic cancer.
Cancer Cell. 34:757–774.e7. 2018.
|
|
47
|
Lu Y, Chan YT, Tan HY, Zhang C, Guo W, Xu
Y, Sharma R, Chen ZS, Zheng YC, Wang N and Feng Y: Epigenetic
regulation of ferroptosis via ETS1/miR-23a-3p/ACSL4 axis mediates
sorafenib resistance in human hepatocellular carcinoma. J Exp Clin
Cancer Res. 41:32022.
|
|
48
|
Liao Y, Yang Y, Pan D, Ding Y, Zhang H, Ye
Y, Li J and Zhao L: HSP90α mediates sorafenib resistance in human
hepatocellular carcinoma by necroptosis inhibition under hypoxia.
Cancers (Basel). 13:2432021.
|
|
49
|
Tang R, Xu J, Zhang B, Liu J, Liang C, Hua
J, Meng Q, Yu X and Shi S: Ferroptosis, necroptosis, and pyroptosis
in anticancer immunity. J Hematol Oncol. 13:1102020.
|
|
50
|
Aaes TL, Kaczmarek A, Delvaeye T, De
Craene B, De Koker S, Heyndrickx L, Delrue I, Taminau J, Wiernicki
B, De Groote P, et al: Vaccination with necroptotic cancer cells
induces efficient anti-tumor immunity. Cell Rep. 15:274–287.
2016.
|
|
51
|
Krysko DV, Garg AD, Kaczmarek A, Krysko O,
Agostinis P and Vandenabeele P: Immunogenic cell death and DAMPs in
cancer therapy. Nat Rev Cancer. 12:860–875. 2012.
|
|
52
|
Davola ME, Cormier O, Vito A, El-Sayes N,
Collins S, Salem O, Revill S, Ask K, Wan Y and Mossman K: Oncolytic
BHV-1 is sufficient to induce immunogenic cell death and synergizes
with low-dose chemotherapy to dampen immunosuppressive T regulatory
cells. Cancers (Basel). 15:12952023.
|
|
53
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012.
|
|
54
|
Tang D, Chen X, Kang R and Kroemer G:
Ferroptosis: Molecular mechanisms and health implications. Cell
Res. 31:107–125. 2021.
|
|
55
|
Friedmann Angeli JP, Schneider M, Proneth
B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch
A, Eggenhofer E, et al: Inactivation of the ferroptosis regulator
Gpx4 triggers acute renal failure in mice. Nat Cell Biol.
16:1180–1191. 2014.
|
|
56
|
Doll S, Proneth B, Tyurina YY, Panzilius
E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A,
et al: ACSL4 dictates ferroptosis sensitivity by shaping cellular
lipid composition. Nat Chem Biol. 13:91–98. 2017.
|
|
57
|
Stockwell BR: Ferroptosis turns 10:
Emerging mechanisms, physiological functions, and therapeutic
applications. Cell. 185:2401–2421. 2022.
|
|
58
|
Stockwell BR, Jiang X and Gu W: Emerging
mechanisms and disease relevance of ferroptosis. Trends Cell Biol.
30:478–490. 2020.
|
|
59
|
Shah R, Shchepinov MS and Pratt DA:
Resolving the role of lipoxygenases in the initiation and execution
of ferroptosis. ACS Central Sci. 4:387–396. 2018.
|
|
60
|
Patel SJ, Protchenko O, Shakoury-Elizeh M,
Baratz E, Jadhav S and Philpott CC: The iron chaperone and nucleic
acid-binding activities of poly(rC)-binding protein 1 are separable
and independently essential. Proc Natl Acad Sci USA.
118:e21046661182021.
|
|
61
|
Bloomer SA and Brown KE: Hepcidin and iron
metabolism in experimental liver injury. Am J Pathol.
191:1165–1179. 2021.
|
|
62
|
Zhang HL, Hu BX, Li ZL, Du T, Shan JL, Ye
ZP, Peng XD, Li X, Huang Y, Zhu XY, et al: PKCβII phosphorylates
ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat Cell
Biol. 24:88–98. 2022.
|
|
63
|
Zou Y, Li H, Graham ET, Deik AA, Eaton JK,
Wang W, Sandoval-Gomez G, Clish CB, Doench JG and Schreiber SL:
Cytochrome P450 oxidoreductase contributes to phospholipid
peroxidation in ferroptosis. Nat Chem Biol. 16:302–309. 2020.
|
|
64
|
Shimada K, Skouta R, Kaplan A, Yang WS,
Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ and
Stockwell BR: Global survey of cell death mechanisms reveals
metabolic regulation of ferroptosis. Nat Chem Biol. 12:497–503.
2016.
|
|
65
|
Hassannia B, Vandenabeele P and Vanden
Berghe T: Targeting ferroptosis to iron out cancer. Cancer Cell.
35:830–849. 2019.
|
|
66
|
Cao JY, Poddar A, Magtanong L, Lumb JH,
Mileur TR, Reid MA, Dovey CM, Wang J, Locasale JW, Stone E, et al:
A genome-wide haploid genetic screen identifies regulators of
glutathione abundance and ferroptosis sensitivity. Cell Rep.
26:1544–1556.e8. 2019.
|
|
67
|
Hao S, Yu J, He W, Huang Q, Zhao Y, Liang
B, Zhang S, Wen Z, Dong S, Rao J, et al: Cysteine dioxygenase 1
mediates erastin-induced ferroptosis in human gastric cancer cells.
Neoplasia. 19:1022–1032. 2017.
|
|
68
|
Bersuker K, Hendricks JM, Li Z, Magtanong
L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al:
The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit
ferroptosis. Nature. 575:688–692. 2019.
|
|
69
|
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee
H, Koppula P, Wu S, Zhuang L, Fang B, et al: DHODH-mediated
ferroptosis defence is a targetable vulnerability in cancer.
Nature. 593:586–590. 2021.
|
|
70
|
Liu Y, Lu S, Wu LL, Yang L, Yang L and
Wang J: The diversified role of mitochondria in ferroptosis in
cancer. Cell Death Dis. 14:5192023.
|
|
71
|
Gao M, Yi J, Zhu J, Minikes AM, Monian P,
Thompson CB and Jiang X: Role of mitochondria in ferroptosis. Mol
Cell. 73:354–363.e3. 2019.
|
|
72
|
Jiang L, Kon N, Li T, Wang SJ, Su T,
Hibshoosh H, Baer R and Gu W: Ferroptosis as a p53-mediated
activity during tumour suppression. Nature. 520:57–62. 2015.
|
|
73
|
Kraft VAN, Bezjian CT, Pfeiffer S,
Ringelstetter L, Müller C, Zandkarimi F, Merl-Pham J, Bao X,
Anastasov N, Kössl J, et al: GTP cyclohydrolase
1/tetrahydrobiopterin counteract ferroptosis through lipid
remodeling. ACS Central Sci. 6:41–53. 2020.
|
|
74
|
Soula M, Weber RA, Zilka O, Alwaseem H, La
K, Yen F, Molina H, Garcia-Bermudez J, Pratt DA and Birsoy K:
Metabolic determinants of cancer cell sensitivity to canonical
ferroptosis inducers. Nat Chem Biol. 16:1351–1360. 2020.
|
|
75
|
Zeitler L, Fiore A, Meyer C, Russier M,
Zanella G, Suppmann S, Gargaro M, Sidhu SS, Seshagiri S, Ohnmacht
C, et al: Anti-ferroptotic mechanism of IL4i1-mediated amino acid
metabolism. Elife. 10:e648062021.
|
|
76
|
Shi J, Zhao Y, Wang K, Shi X, Wang Y,
Huang H, Zhuang Y, Cai T, Wang F and Shao F: Cleavage of GSDMD by
inflammatory caspases determines pyroptotic cell death. Nature.
526:660–665. 2015.
|
|
77
|
Cookson BT and Brennan MA:
Pro-inflammatory programmed cell death. Trends Microbiol.
9:113–114. 2001.
|
|
78
|
Hou J, Hsu JM and Hung MC: Molecular
mechanisms and functions of pyroptosis in inflammation and
antitumor immunity. Mol Cell. 81:4579–4590. 2021.
|
|
79
|
Liu Z, Wang C, Yang J, Zhou B, Yang R,
Ramachandran R, Abbott DW and Xiao TS: Crystal structures of the
full-length murine and human gasdermin D reveal mechanisms of
autoinhibition, lipid binding, and oligomerization. Immunity.
51:43–49.e4. 2019.
|
|
80
|
Ding J, Wang K, Liu W, She Y, Sun Q, Shi
J, Sun H, Wang DC and Shao F: Pore-forming activity and structural
autoinhibition of the gasdermin family. Nature. 535:111–116.
2016.
|
|
81
|
Liu X, Zhang Z, Ruan J, Pan Y, Magupalli
VG, Wu H and Lieberman J: Inflammasome-activated gasdermin D causes
pyroptosis by forming membrane pores. Nature. 535:153–158.
2016.
|
|
82
|
Aglietti RA and Dueber EC: Recent insights
into the molecular mechanisms underlying pyroptosis and gasdermin
family functions. Trends Immunol. 38:261–271. 2017.
|
|
83
|
Fink SL and Cookson BT: Pillars article:
Caspase-1-dependent pore formation during pyroptosis leads to
osmotic lysis of infected host macrophages. Cell Microbiol.
2006.8:1812–1825
J Immunol. 202:1913–1926. 2019.
|
|
84
|
Kayagaki N, Stowe IB, Lee BL, O'Rourke K,
Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT,
et al: Caspase-11 cleaves gasdermin D for non-canonical
inflammasome signalling. Nature. 526:666–671. 2015.
|
|
85
|
Kayagaki N, Warming S, Lamkanfi M, Vande
Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, et al:
Non-canonical inflammasome activation targets caspase-11. Nature.
479:117–121. 2011.
|
|
86
|
Yang D, He Y, Muñoz-Planillo R, Liu Q and
Núñez G: Caspase-11 requires the pannexin-1 channel and the
purinergic P2X7 pore to mediate pyroptosis and endotoxic shock.
Immunity. 43:923–932. 2015.
|
|
87
|
Wang Y, Gao W, Shi X, Ding J, Liu W, He H,
Wang K and Shao F: Chemotherapy drugs induce pyroptosis through
caspase-3 cleavage of a gasdermin. Nature. 547:99–103. 2017.
|
|
88
|
Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu
JM, Nie L, Chen Y, Wang YC, Liu C, et al: PD-L1-mediated gasdermin
C expression switches apoptosis to pyroptosis in cancer cells and
facilitates tumour necrosis. Nat Cell Biol. 22:1264–1275. 2020.
|
|
89
|
Rogers C, Fernandes-Alnemri T, Mayes L,
Alnemri D, Cingolani G and Alnemri ES: Cleavage of DFNA5 by
caspase-3 during apoptosis mediates progression to secondary
necrotic/pyroptotic cell death. Nat Commun. 8:141282017.
|
|
90
|
Orning P, Weng D, Starheim K, Ratner D,
Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA, et al: Pathogen
blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin
D and cell death. Science. 362:1064–1069. 2018.
|
|
91
|
Sarhan J, Liu BC, Muendlein HI, Li P,
Nilson R, Tang AY, Rongvaux A, Bunnell SC, Shao F, Green DR and
Poltorak A: Caspase-8 induces cleavage of gasdermin D to elicit
pyroptosis during Yersinia infection. Proc Natl Acad Sci USA.
115:E10888–E10897. 2018.
|
|
92
|
Liu Y, Fang Y, Chen X, Wang Z, Liang X,
Zhang T, Liu M, Zhou N, Lv J, Tang K, et al: Gasdermin E-mediated
target cell pyroptosis by CAR T cells triggers cytokine release
syndrome. Sci Immunol. 5:eaax79692020.
|
|
93
|
Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu
X, Junqueira C, Meza-Sosa KF, Mok TMY, Ansara J, et al: Gasdermin E
suppresses tumour growth by activating anti-tumour immunity.
Nature. 579:415–420. 2020.
|
|
94
|
Erkes DA, Cai W, Sanchez IM, Purwin TJ,
Rogers C, Field CO, Berger AC, Hartsough EJ, Rodeck U, Alnemri ES
and Aplin AE: Mutant BRAF and MEK inhibitors regulate the tumor
immune microenvironment via pyroptosis. Cancer Discov. 10:254–269.
2020.
|
|
95
|
Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y,
Wang Y, Li D, Liu W, Zhang Y, et al: Granzyme A from cytotoxic
lymphocytes cleaves GSDMB to trigger pyroptosis in target cells.
Science. 368:eaaz75482020.
|
|
96
|
Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li
P, Hu L and Shao F: Inflammatory caspases are innate immune
receptors for intracellular LPS. Nature. 514:187–192. 2014.
|
|
97
|
Deets KA and Vance RE: Inflammasomes and
adaptive immune responses. Nat Immunol. 22:412–422. 2021.
|
|
98
|
Guo H, Callaway JB and Ting JP:
Inflammasomes: Mechanism of action, role in disease, and
therapeutics. Nat Med. 21:677–687. 2015.
|
|
99
|
Wang K, Sun Q, Zhong X, Zeng M, Zeng H,
Shi X, Li Z, Wang Y, Zhao Q, Shao F and Ding J: Structural
mechanism for GSDMD targeting by autoprocessed caspases in
pyroptosis. Cell. 180:941–955.e20. 2020.
|
|
100
|
Loveless R, Bloomquist R and Teng Y:
Pyroptosis at the forefront of anticancer immunity. J Exp Clin Canc
Res. 40:2642021.
|
|
101
|
Degterev A, Huang Z, Boyce M, Li Y, Jagtap
P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA and Yuan J:
Chemical inhibitor of nonapoptotic cell death with therapeutic
potential for ischemic brain injury. Nat Chem Biol. 1:112–119.
2005.
|
|
102
|
Frank D and Vince JE: Pyroptosis versus
necroptosis: Similarities, differences, and crosstalk. Cell Death
Differ. 26:99–114. 2019.
|
|
103
|
Choi ME, Price DR, Ryter SW and Choi AMK:
Necroptosis: A crucial pathogenic mediator of human disease. JCI
Insight. 4:e1288342019.
|
|
104
|
Lork M, Verhelst K and Beyaert R: CYLD,
A20 and OTULIN deubiquitinases in NF-κB signaling and cell death:
So similar, yet so different. Cell Death Differ. 24:1172–1183.
2017.
|
|
105
|
Priem D, van Loo G and Bertrand MJM: A20
and cell death-driven inflammation. Trends Immunol. 41:421–435.
2020.
|
|
106
|
Ye K, Chen Z and Xu Y: The double-edged
functions of necroptosis. Cell Death Dis. 14:1632023.
|
|
107
|
Feoktistova M, Geserick P, Kellert B,
Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Häcker G
and Leverkus M: cIAPs block Ripoptosome formation, a RIP1/caspase-8
containing intracellular cell death complex differentially
regulated by cFLIP isoforms. Mol Cell. 43:449–463. 2011.
|
|
108
|
Mompeán M, Li W, Li J, Laage S, Siemer AB,
Bozkurt G, Wu H and McDermott AE: The structure of the necrosome
RIPK1-RIPK3 core, a human hetero-amyloid signaling complex. Cell.
173:1244–1253.e10. 2018.
|
|
109
|
Sun L, Wang H, Wang Z, He S, Chen S, Liao
D, Wang L, Yan J, Liu W, Lei X and Wang X: Mixed lineage kinase
domain-like protein mediates necrosis signaling downstream of RIP3
kinase. Cell. 148:213–227. 2012.
|
|
110
|
Kaczmarek A, Vandenabeele P and Krysko DV:
Necroptosis: The release of damage-associated molecular patterns
and its physiological relevance. Immunity. 38:209–223. 2013.
|
|
111
|
Kaiser WJ, Sridharan H, Huang C, Mandal P,
Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J and Mocarski ES:
Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J
Biol Chem. 288:31268–31279. 2013.
|
|
112
|
Zhang W, Fan W, Guo J and Wang X: Osmotic
stress activates RIPK3/MLKL-mediated necroptosis by increasing
cytosolic pH through a plasma membrane Na+/H+
exchanger. Sci Signal. 15:eabn58812022.
|
|
113
|
Coriat R, Nicco C, Chéreau C, Mir O,
Alexandre J, Ropert S, Weill B, Chaussade S, Goldwasser F and
Batteux F: Sorafenib-induced hepatocellular carcinoma cell death
depends on reactive oxygen species production in vitro and in vivo.
Mol Cancer Ther. 11:2284–2293. 2012.
|
|
114
|
Louandre C, Ezzoukhry Z, Godin C, Barbare
JC, Mazière JC, Chauffert B and Galmiche A: Iron-dependent cell
death of hepatocellular carcinoma cells exposed to sorafenib. Int J
Cancer. 133:1732–1742. 2013.
|
|
115
|
Huang CY, Chen LJ, Chen G, Chao TI and
Wang CY: SHP-1/STAT3-signaling-axis-regulated coupling between
BECN1 and SLC7A11 contributes to sorafenib-induced ferroptosis in
hepatocellular carcinoma. Int J Mol Sci. 23:110922022.
|
|
116
|
Suzuki S, Venkatesh D, Kanda H, Nakayama
A, Hosokawa H, Lee E, Miki T, Stockwell BR, Yokote K, Tanaka T and
Prives C: GLS2 is a tumor suppressor and a regulator of ferroptosis
in hepatocellular carcinoma. Cancer Res. 82:3209–3222. 2022.
|
|
117
|
Chen Y, Li L, Lan J, Cui Y, Rao X, Zhao J,
Xing T, Ju G, Song G, Lou J and Liang J: CRISPR screens uncover
protective effect of PSTK as a regulator of chemotherapy-induced
ferroptosis in hepatocellular carcinoma. Mol Cancer. 21:112022.
|
|
118
|
Zhang B, Bao W, Zhang S, Chen B, Zhou X,
Zhao J, Shi Z, Zhang T, Chen Z, Wang L, et al: LncRNA HEPFAL
accelerates ferroptosis in hepatocellular carcinoma by regulating
SLC7A11 ubiquitination. Cell Death Dis. 13:7342022.
|
|
119
|
Chen Q, Zheng W, Guan J, Liu H, Dan Y, Zhu
L, Song Y, Zhou Y, Zhao X, Zhang Y, et al: SOCS2-enhanced
ubiquitination of SLC7A11 promotes ferroptosis and
radiosensitization in hepatocellular carcinoma. Cell Death Differ.
30:137–151. 2023.
|
|
120
|
Du J, Wan Z, Wang C, Lu F, Wei M, Wang D
and Hao Q: Designer exosomes for targeted and efficient ferroptosis
induction in cancer via chemo-photodynamic therapy. Theranostics.
11:8185–8196. 2021.
|
|
121
|
Liu J, Li X, Chen J, Zhang X, Guo J, Gu J,
Mei C, Xiao Y, Peng C, Liu J, et al: Arsenic-loaded biomimetic iron
oxide nanoparticles for enhanced ferroptosis-inducing therapy of
hepatocellular carcinoma. ACS Appl Mater Interfaces. 15:6260–6273.
2023.
|
|
122
|
Tian H, Zhao S, Nice EC, Huang C, He W,
Zou B and Lin J: A cascaded copper-based nanocatalyst by modulating
glutathione and cyclooxygenase-2 for hepatocellular carcinoma
therapy. J Colloid Interface Sci. 607:1516–1526. 2022.
|
|
123
|
Su Y, Zhang Z, Lee LTO, Peng L, Lu L, He X
and Zhang X: Amphiphilic dendrimer doping enhanced ph-sensitivity
of liposomal vesicle for effective co-delivery toward synergistic
ferroptosis-apoptosis therapy of hepatocellular carcinoma. Adv
Healthc Mater. 12:e22026632023.
|
|
124
|
Zhang Z, Yao Z, Wang L, Ding H, Shao J,
Chen A, Zhang F and Zheng S: Activation of ferritinophagy is
required for the RNA-binding protein ELAVL1/HuR to regulate
ferroptosis in hepatic stellate cells. Autophagy. 14:2083–2103.
2018.
|
|
125
|
Zhang Z, Guo M, Li Y, Shen M, Kong D, Shao
J, Ding H, Tan S, Chen A, Zhang F and Zheng S: RNA-binding protein
ZFP36/TTP protects against ferroptosis by regulating autophagy
signaling pathway in hepatic stellate cells. Autophagy.
16:1482–1505. 2020.
|
|
126
|
Tsuchida T and Friedman SL: Mechanisms of
hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol.
14:397–411. 2017.
|
|
127
|
Shen M, Li Y, Wang Y, Shao J, Zhang F, Yin
G, Chen A, Zhang Z and Zheng S: N6-methyladenosine
modification regulates ferroptosis through autophagy signaling
pathway in hepatic stellate cells. Redox Biol. 47:1021512021.
|
|
128
|
Li ZJ, Dai HQ, Huang XW, Feng J, Deng JH,
Wang ZX, Yang XM, Liu YJ, Wu Y, Chen PH, et al: Artesunate
synergizes with sorafenib to induce ferroptosis in hepatocellular
carcinoma. Acta Pharmacol Sin. 42:301–310. 2021.
|
|
129
|
Sun X, Niu X, Chen R, He W, Chen D, Kang R
and Tang D: Metallothionein-1G facilitates sorafenib resistance
through inhibition of ferroptosis. Hepatology. 64:488–500.
2016.
|
|
130
|
Byun JK, Lee S, Kang GW, Lee YR, Park SY,
Song IS, Yun JW, Lee J, Choi YK and Park KG: Macropinocytosis is an
alternative pathway of cysteine acquisition and mitigates
sorafenib-induced ferroptosis in hepatocellular carcinoma. J Exp
Clin Cancer Res. 41:982022.
|
|
131
|
Byun JK: Tumor lactic acid: A potential
target for cancer therapy. Arch Pharm Res. 46:90–110. 2023.
|
|
132
|
Zhang T, Sun L, Hao Y, Suo C, Shen S, Wei
H, Ma W, Zhang P, Wang T, Gu X, et al: ENO1 suppresses cancer cell
ferroptosis by degrading the mRNA of iron regulatory protein 1. Nat
Cancer. 3:75–89. 2022.
|
|
133
|
Zhao Y, Li M, Yao X, Fei Y, Lin Z, Li Z,
Cai K, Zhao Y and Luo Z: HCAR1/MCT1 regulates tumor ferroptosis
through the lactate-mediated AMPK-SCD1 activity and its therapeutic
implications. Cell Rep. 33:1084872020.
|
|
134
|
Yang Z, Su W, Wei X, Qu S, Zhao D, Zhou J,
Wang Y, Guan Q, Qin C, Xiang J, et al: HIF-1α drives resistance to
ferroptosis in solid tumors by promoting lactate production and
activating SLC1A1. Cell Rep. 42:1129452023.
|
|
135
|
Ma Q: Role of nrf2 in oxidative stress and
toxicity. Annu Rev Pharmacol Toxicol. 53:401–426. 2013.
|
|
136
|
Sporn MB and Liby KT: NRF2 and cancer: The
good, the bad and the importance of context. Nat Rev Cancer.
12:564–571. 2012.
|
|
137
|
Ichimura Y, Waguri S, Sou YS, Kageyama S,
Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, et
al: Phosphorylation of p62 activates the Keap1-Nrf2 pathway during
selective autophagy. Mol Cell. 51:618–631. 2013.
|
|
138
|
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R
and Tang D: Activation of the p62-Keap1-NRF2 pathway protects
against ferroptosis in hepatocellular carcinoma cells. Hepatology.
63:173–184. 2016.
|
|
139
|
Ren X, Li Y, Zhou Y, Hu W, Yang C, Jing Q,
Zhou C, Wang X, Hu J, Wang L, et al: Overcoming the compensatory
elevation of NRF2 renders hepatocellular carcinoma cells more
vulnerable to disulfiram/copper-induced ferroptosis. Redox Biol.
46:1021222021.
|
|
140
|
Wang Q, Bin C, Xue Q, Gao Q, Huang A, Wang
K and Tang N: GSTZ1 sensitizes hepatocellular carcinoma cells to
sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis.
Cell Death Dis. 12:4262021.
|
|
141
|
Yao F, Deng Y, Zhao Y, Mei Y, Zhang Y, Liu
X, Martinez C, Su X, Rosato RR, Teng H, et al: A targetable
LIFR-NF-κB-LCN2 axis controls liver tumorigenesis and vulnerability
to ferroptosis. Nat Commun. 12:73332021.
|
|
142
|
Hu J, Dong Y, Ding L, Dong Y, Wu Z, Wang
W, Shen M and Duan Y: Local delivery of arsenic trioxide
nanoparticles for hepatocellular carcinoma treatment. Signal
Transduct Target Ther. 4:282019.
|
|
143
|
Shangguan F, Zhou H, Ma N, Wu S, Huang H,
Jin G, Wu S, Hong W, Zhuang W, Xia H and Lan L: A novel mechanism
of cannabidiol in suppressing hepatocellular carcinoma by inducing
GSDME dependent pyroptosis. Front Cell Dev Biol. 9:6978322021.
|
|
144
|
Dai X, Sun F, Deng K, Lin G, Yin W, Chen
H, Yang D, Liu K, Zhang Y and Huang L: Mallotucin D, a clerodane
diterpenoid from croton crassifolius, suppresses HepG2 cell growth
via inducing autophagic cell death and pyroptosis. Int J Mol Sci.
23:142172022.
|
|
145
|
Shen Z, Zhou H, Li A, Wu T, Ji X, Guo L,
Zhu X, Zhang D and He X: Metformin inhibits hepatocellular
carcinoma development by inducing apoptosis and pyroptosis through
regulating FOXO3. Aging (Albany NY). 13:22120–22133. 2021.
|
|
146
|
Chen Z, He M, Chen J, Li C and Zhang Q:
Long non-coding RNA SNHG7 inhibits NLRP3-dependent pyroptosis by
targeting the miR-34a/SIRT1 axis in liver cancer. Oncol Lett.
20:893–901. 2020.
|
|
147
|
Kofahi HM, Taylor NGA, Hirasawa K, Grant
MD and Russell RS: Hepatitis C virus infection of cultured human
hepatoma cells causes apoptosis and pyroptosis in both infected and
bystander cells. Sci Rep. 6:374332016.
|
|
148
|
Wei Q, Zhu R, Zhu J, Zhao R and Li M:
E2-induced activation of the NLRP3 inflammasome triggers pyroptosis
and inhibits autophagy in HCC cells. Oncol Res. 27:827–834.
2019.
|
|
149
|
Zhang Y, Yang H, Sun M, He T, Liu Y, Yang
X, Shi X and Liu X: Alpinumisoflavone suppresses hepatocellular
carcinoma cell growth and metastasis via NLRP3
inflammasome-mediated pyroptosis. Pharmacol Rep. 72:1370–1382.
2020.
|
|
150
|
Wang F, Xu C, Li G, Lv P and Gu J:
Incomplete radiofrequency ablation induced chemoresistance by
up-regulating heat shock protein 70 in hepatocellular carcinoma.
Exp Cell Res. 409:1129102021.
|
|
151
|
Seehawer M, Heinzmann F, D'Artista L,
Harbig J, Roux PF, Hoenicke L, Dang H, Klotz S, Robinson L, Doré G,
et al: Necroptosis microenvironment directs lineage commitment in
liver cancer. Nature. 562:69–75. 2018.
|
|
152
|
Schneider AT, Gautheron J, Feoktistova M,
Roderburg C, Loosen SH, Roy S, Benz F, Schemmer P, Büchler MW,
Nachbur U, et al: RIPK1 suppresses a TRAF2-dependent pathway to
liver cancer. Cancer Cell. 31:94–109. 2017.
|
|
153
|
Hoesel B and Schmid JA: The complexity of
NF-κB signaling in inflammation and cancer. Mol Cancer.
12:862013.
|
|
154
|
Jomen W, Ohtake T, Akita T, Suto D, Yagi
H, Osawa Y and Kohgo Y: Iron chelator deferasirox inhibits NF-κB
activity in hepatoma cells and changes sorafenib-induced programmed
cell deaths. Biomed Pharmacother. 153:1133632022.
|
|
155
|
Harari-Steinfeld R, Gefen M, Simerzin A,
Zorde-Khvalevsky E, Rivkin M, Ella E, Friehmann T, Gerlic M,
Zucman-Rossi J, Caruso S, et al: The lncRNA H19-derived
MicroRNA-675 promotes liver necroptosis by targeting FADD. Cancers
(Basel). 13:4112021.
|
|
156
|
Zheng Y, Kong F, Liu S, Liu X, Pei D and
Miao X: Membrane protein-chimeric liposome-mediated delivery of
triptolide for targeted hepatocellular carcinoma therapy. Drug
Deliv. 28:2033–2043. 2021.
|
|
157
|
Mohammed S, Thadathil N, Selvarani R,
Nicklas EH, Wang D, Miller BF, Richardson A and Deepa SS:
Necroptosis contributes to chronic inflammation and fibrosis in
aging liver. Aging Cell. 20:e135122021.
|
|
158
|
Hammerich L and Tacke F: Eat more carrots?
Dampening cell death in ethanol-induced liver fibrosis by
β-carotene. Hepatobil Surg Nutr. 2:248–251. 2013.
|
|
159
|
Zhao B, Lv X, Zhao X, Maimaitiaili S,
Zhang Y, Su K, Yu H, Liu C and Qiao T: Tumor-promoting actions of
HNRNP A1 in HCC are associated with cell cycle, mitochondrial
dynamics, and necroptosis. Int J Mol Sci. 23:102092022.
|
|
160
|
Lee SY, Kim S, Song Y, Kim N, No J, Kim KM
and Seo HR: Sorbitol dehydrogenase induction of cancer cell
necroptosis and macrophage polarization in the HCC microenvironment
suppresses tumor progression. Cancer Lett. 551:2159602022.
|
|
161
|
Lan W, Santofimia-Castaño P, Xia Y, Zhou
Z, Huang C, Fraunhoffer N, Barea D, Cervello M, Giannitrapani L,
Montalto G, et al: Targeting NUPR1 with the small compound ZZW-115
is an efficient strategy to treat hepatocellular carcinoma. Cancer
Lett. 486:8–17. 2020.
|
|
162
|
Tran DDH, Kessler C, Niehus SE, Mahnkopf
M, Koch A and Tamura T: Myc target gene, long intergenic noncoding
RNA, Linc00176 in hepatocellular carcinoma regulates cell cycle and
cell survival by titrating tumor suppressor microRNAs. Oncogene.
37:75–85. 2018.
|
|
163
|
Xiang YK, Peng FH, Guo YQ, Ge H, Cai SY,
Fan LX, Peng YX, Wen H, Wang Q and Tao L: Connexin32 activates
necroptosis through Src-mediated inhibition of caspase 8 in
hepatocellular carcinoma. Cancer Sci. 112:3507–3519. 2021.
|
|
164
|
Wang W, Green M, Choi JE, Gijón M, Kennedy
PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, et al:
CD8+ T cells regulate tumour ferroptosis during cancer
immunotherapy. Nature. 569:270–274. 2019.
|
|
165
|
Xi G, Gao J, Wan B, Zhan P, Xu W, Lv T and
Song Y: GSDMD is required for effector CD8+ T cell
responses to lung cancer cells. Int Immunopharmacol.
74:1057132019.
|
|
166
|
Yatim N, Jusforgues-Saklani H, Orozco S,
Schulz O, Barreira da Silva R, Reis e Sousa C, Green DR, Oberst A
and Albert ML: RIPK1 and NF-κB signaling in dying cells determines
cross-priming of CD8+ T cells. Science. 350:328–334. 2015.
|
|
167
|
Kang T, Huang Y, Zhu Q, Cheng H, Pei Y,
Feng J, Xu M, Jiang G, Song Q, Jiang T, et al: Necroptotic cancer
cells-mimicry nanovaccine boosts anti-tumor immunity with tailored
immune-stimulatory modality. Biomaterials. 164:80–97. 2018.
|
|
168
|
Snyder AG, Hubbard NW, Messmer MN, Kofman
SB, Hagan CE, Orozco SL, Chiang K, Daniels BP, Baker D and Oberst
A: Intratumoral activation of the necroptotic pathway components
RIPK1 and RIPK3 potentiates antitumor immunity. Sci Immunol.
4:eaaw20042019.
|
|
169
|
Galluzzi L, Buqué A, Kepp O, Zitvogel L
and Kroemer G: Immunogenic cell death in cancer and infectious
disease. Nat Rev Immunol. 17:97–111. 2017.
|
|
170
|
Conche C, Finkelmeier F, Pešić M, Nicolas
AM, Böttger TW, Kennel KB, Denk D, Ceteci F, Mohs K, Engel E, et
al: Combining ferroptosis induction with MDSC blockade renders
primary tumours and metastases in liver sensitive to immune
checkpoint blockade. Gut. 72:1774–1782. 2023.
|
|
171
|
Li S, Li F, Xu L, Liu X, Zhu X, Gao W and
Shen X: TLR2 agonist promotes myeloid-derived suppressor cell
polarization via Runx1 in hepatocellular carcinoma. Int
Immunopharmacol. 111:1091682022.
|
|
172
|
Li Z, Wu T, Zheng B and Chen L:
Individualized precision treatment: Targeting TAM in HCC. Cancer
Lett. 458:86–91. 2019.
|
|
173
|
Loeuillard E, Yang J, Buckarma E, Wang J,
Liu Y, Conboy C, Pavelko KD, Li Y, O'Brien D, Wang C, et al:
Targeting tumor-associated macrophages and granulocytic
myeloid-derived suppressor cells augments PD-1 blockade in
cholangiocarcinoma. J Clin Invest. 130:5380–5396. 2020.
|
|
174
|
DeNardo DG and Ruffell B: Macrophages as
regulators of tumour immunity and immunotherapy. Nat rev immunol.
19:369–382. 2019.
|
|
175
|
Farhood B, Najafi M and Mortezaee K:
CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A
review. J Cell Physiol. 234:8509–8521. 2019.
|
|
176
|
Hao X, Zheng Z, Liu H, Zhang Y, Kang J,
Kong X, Rong D, Sun G, Sun G, Liu L, et al: Inhibition of APOC1
promotes the transformation of M2 into M1 macrophages via the
ferroptosis pathway and enhances anti-PD1 immunotherapy in
hepatocellular carcinoma based on single-cell RNA sequencing. Redox
Biol. 56:1024632022.
|
|
177
|
Chen R, Li Q, Xu S, Ye C, Tian T, Jiang Q,
Shan J and Ruan J: Modulation of the tumour microenvironment in
hepatocellular carcinoma by tyrosine kinase inhibitors: From
modulation to combination therapy targeting the microenvironment.
Cancer Cell Int. 22:732022.
|
|
178
|
Li J, Yu J, Zhang T, Pu X, Li Y and Wu Z:
Genomic analysis quantifies pyroptosis in the immune
microenvironment of HBV-related hepatocellular carcinoma. Front
immunol. 13:9323032022.
|
|
179
|
Mohammed S, Nicklas EH, Thadathil N,
Selvarani R, Royce GH, Kinter M, Richardson A and Deepa SS: Role of
necroptosis in chronic hepatic inflammation and fibrosis in a mouse
model of increased oxidative stress. Free Radical Bio Med.
164:315–328. 2021.
|
|
180
|
Tang D, Kang R, Berghe TV, Vandenabeele P
and Kroemer G: The molecular machinery of regulated cell death.
Cell Res. 29:347–364. 2019.
|
|
181
|
Jiang X, Deng W, Tao S, Tang Z, Chen Y,
Tian M, Wang T, Tao C, Li Y, Fang Y, et al: A RIPK3-independent
role of MLKL in suppressing parthanatos promotes immune evasion in
hepatocellular carcinoma. Cell Discov. 9:72023.
|
|
182
|
Wu L, Zhang X, Zheng L, Zhao H, Yan G,
Zhang Q, Zhou Y, Lei J, Zhang J, Wang J, et al: RIPK3 orchestrates
fatty acid metabolism in tumor-associated macrophages and
hepatocarcinogenesis. Cancer Immunol Res. 8:710–721. 2020.
|
|
183
|
Nicolè L, Sanavia T, Cappellesso R,
Maffeis V, Akiba J, Kawahara A, Naito Y, Radu CM, Simioni P,
Serafin D, et al: Necroptosis-driving genes RIPK1, RIPK3 and MLKL-p
are associated with intratumoral CD3+ and
CD8+ T cell density and predict prognosis in
hepatocellular carcinoma. J Immunother Cancer. 10:e0040312022.
|
|
184
|
Pomlok K, Pata S, Kulaphisit M, Pangnuchar
R, Wipasa J, Smith DR, Kasinrerk W and Lithanatudom P: An IgM
monoclonal antibody against domain 1 of CD147 induces non-canonical
RIPK-independent necroptosis in a cell type specific manner in
hepatocellular carcinoma cells. Biochim Biophys Acta Mol Cell Res.
1869:1192952022.
|